

Abstract
Neuroblastoma (NB) is the most common cancer in infancy with an urgent need for more efficient targeted therapies. The development of novel (combinatorial) treatment strategies relies on extensive explorations of signaling perturbations in neuroblastoma cell lines, using RNA-Seq or other high throughput technologies (e.g. phosphoproteomics). This typically requires dedicated bioinformatics support, which is not always available. Additionally, while data from published studies are highly valuable and raw data (e.g. fastq files) are nowadays released in public repositories, data processing is time-consuming and again difficult without bioinformatics support. To facilitate NB research, more user-friendly and immediately accessible platforms are needed to explore newly generated as well as existing high throughput data. To make this possible, we developed an interactive data centralization and visualization web application, called CLEAN (the Cell Line Explorer web Application of Neuroblastoma data; https://ccgg.ugent.be/shiny/clean/). By focusing on the regulation of the DNA damage response, a therapeutic target of major interest in neuroblastoma, we demonstrate how CLEAN can be used to gain novel mechanistic insights and identify putative drug targets in neuroblastoma.
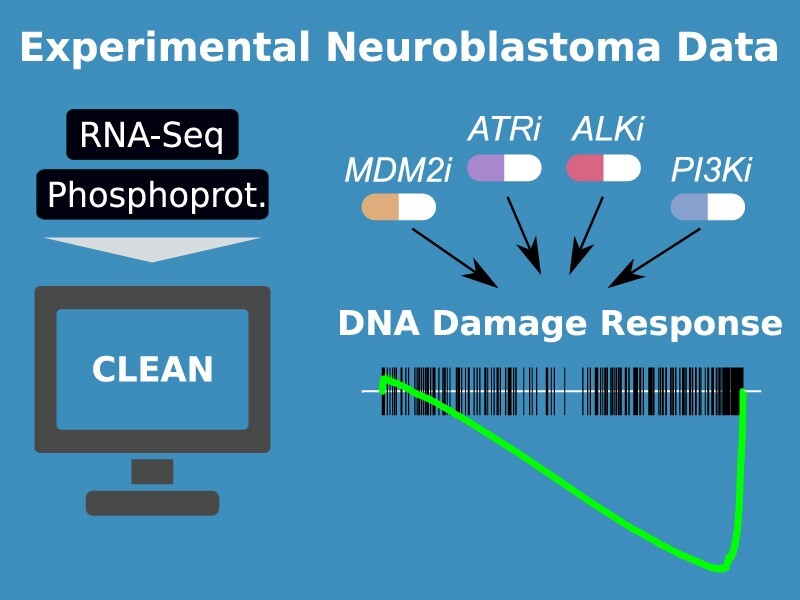
Graphical Abstract
Graphical Abstract.
Introduction
Neuroblastoma (NB) is the most common cancer in infancy and accounts for approximately 15% of pediatric cancer-related deaths (1). Despite initial positive treatment response, high-risk NB frequently leads to relapse, resulting in a survival rate as low as 35% (2). Our groups and others have recently demonstrated in different preclinical NB animal models that targeting the DNA damage response (DDR) pathway is a promising treatment strategy for high-risk NB (3–6).
Successful investigations of new or combinatorial drug targets are often based on extensive in vitro experiments in cell lines, followed by high throughput experiments such as RNA-Seq and/or phosphoproteomics (3,7,8). Over the past few decades, generation of these types of data has become increasingly efficient, leading to a strong reduction in price as well as increase in accuracy. This has led to a surge in omics data abundance in the NB, as well as most other pre-clinical fields (9)—a development that has brought both opportunities and challenges (10). While each omics study generates tens of thousands of data points, only a fraction of this information is used to draw conclusions and an even smaller proportion is addressed in the final manuscript of any study. A single perturbation, even in a relatively homogenous system like immortal cell lines, causes major, complex downstream effects at several regulatory levels (3). A complete understanding of the cellular response is a task that goes beyond the scope of a single study. Providing real access to these datasets would let us deepen our understanding of each experiment and move the entire field of NB research forward substantially. Despite attempts to increase reproducibility, by mandating upload of raw data to online repositories and encouraging code sharing (11–13), proper accessibility to this excess information remains a problem.
To meet the need for more user-friendly, simpler and faster ways to explore and access this goldmine of experimental data, we developed a highly interactive data centralization and visualization web application, called CLEAN (the Cell Line Explorer web Application of Neuroblastoma data; https://ccgg.ugent.be/shiny/clean/). We illustrate the CLEAN functionality and its added value for translational NB research by providing new mechanistic insights in the DDR regulation and identifying novel therapeutic targets for future studies.
Materials and methods
Data collection
To find relevant RNA-Seq data for CLEAN, we selected all studies from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (14) that fulfilled the following search criteria: ‘Neuroblastoma [Title]’, ‘Homo sapiens’, ‘Expression profiling by high throughput sequencing’, ‘Cell line’, ‘Sample count from 4 to 10 000’ (implying minimally two replicates for each condition and control). The selected studies were manually curated and studies not focusing on NB, with a focus on comparing cell lines to xenografts, without any form of perturbation or without biological replicates were excluded. Additionally, data from submissions with no associated, published study were also excluded. When multiple submissions were found for a single study, data were merged. Samples were organized into either baseline (control) or perturbation (condition), where all perturbations could be categorized into one of the following: compound treatment (e.g. retinoic acid), drug inhibition, chemotherapy, knock-down, knock-out or overexpression. We added information about concentration and treatment duration if this information was available.
A similar strategy was followed to find relevant phosphoproteomics data in the ProteomeXchange (https://www.proteomexchange.org/) (15). We applied a simple search for ‘neuroblastoma’ and selected only studies where phosphoproteomics data were utilized, making no discrimination on mass spectrometry method. Datasets were excluded and samples organized in the same fashion as for RNA-Seq.
Data processing
We used HISAT2 (16) to align reads from the GEO-derived fastq files to the GRCh38 reference genome. We excluded samples with an alignment rate below 75%. If this exclusion resulted in a lack of replicates, the study was excluded altogether. Subsequent quantification and annotation were performed with HTSeq (17) using GENCODE 29. For normalization of counts and differential expression analysis of RNA-Seq, we used the DESeq2 package (18).
Raw phosphoproteomics intensity data were quantified using MaxQuant (19). Differential expression/phosphorylation was performed using the DEP package (20).
Shiny web application and structure of CLEAN
The current version of CLEAN runs inside a Conda environment, on R (21) and and is built in R Shiny (22). Tables utilize the DT package (23), plots use ggplot2 (24) and plotly (25). See Supplementary file 3 for all packages and versions used for the Conda environment. It is hosted at a webserver at Ghent University.
Gene set enrichment analysis (GSEA)
CLEAN provides both a classical GSEA (also referred to as an overrepresentation analysis) and a preranked GSEA.
The classical GSEA is performed using a one-sided Fisher's exact test on a contingency table that compares the number of genes/proteins/phosphosites below and above the user-defined DE/DP thresholds to their presence in a specified gene set. Odds ratios (OR, indicative of the extent of the enrichment), P values and Padj values (multiple testing correction using the Benjamini-Hochberg method) are reported by CLEAN.
Preranked GSEA was performed using the R fgsea package (fgseaMultilevel function, default parameters) with ranking based on the DESeq2 statistic for RNA-Seq data or –log10(P) * Sign(log2FC) for phosphoproteomics data.
Relevant gene sets were derived from the Molecular Signatures Database (MSigDB) v2023.1 (26). For phosphoproteomics analyses, we also added protein kinases—phosphosite motif dataset. These data were derived from the supplementary information provided by Johnson et al. (27), using the top 1% predicted protein kinases for each phosphomotif and converted protein names to gene names for both kinases and target sites.
Integrated data and links
We added links to other resources frequently used in the field: GeneCards (https://www.genecards.org/) for general gene information (28), ChEMBL (https://www.ebi.ac.uk/chembl/) for up-to-date, target-specific drug information (29), Ensembl (https://www.ensembl.org/) for detailed genomic information (30) and the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) for Kaplan-Meier analysis on the Kocak cohort (31). We also downloaded the CRISPR/cas9 generated gene scores from the DepMap database (https://depmap.org/portal/download/all/, (22Q2)) and integrated them directly in CLEAN (32).
Results
CLEAN is an explorable and standardized neuroblastoma cell line data repository.
The aim of the CLEAN initiative is to make all experimental NB cell line data that involve RNA-Seq and/or phosphoproteomics experiments publicly available in an easy-to-use and interactive format. We selected all available studies from the Gene Expression Omnibus (GEO) (14) and the ProteomeXchange (PRX) (15) that fulfilled the following inclusion criteria: (1) NB cell lines; (2) minimally two replicates per condition and (3) some kind of experimental perturbation (e.g. gene knockout, drug treatment; see Materials and methods for full criteria and data processing). All data were reprocessed using standardized pipelines, resulting in the availability of 66 RNA-Seq and 8 phosphoproteomics studies, with 6 chemotherapeutic drug treatments (chemo), 29 compound treatments, 49 drug inhibitions, 40 gene knockdowns (KD), 15 gene knockouts (KO) and 27 gene overexpressions (OE; Figure 1).
Figure 1.

Overview of data processing and availability in CLEAN. Overview of default data processing in CLEAN (left). Pie chart showing the different experimental perturbations that are currently implemented in CLEAN with precise number of studies indicated between parentheses (top-mid). CLEAN screenshot of the default RNA-Seq tab, indicating the 3 main components: (1) the sidebar takes user input and updates what's in the main panel; (2) a searchable table with detailed differential gene expression or related information and (3) an interactive plot (bottom-right). External data sources that are directly (left) or indirectly (via hyperlinks) (top-right), integrated in CLEAN.
Interactively exploring high throughput data derived from neuroblastoma cell lines using CLEAN
The CLEAN web application contains three main tabs: a search tab and two data tabs (i.e. RNA-Seq and Phosphoproteomics). Each tab contains a side bar on the left that accepts user input to query studies and data and a main panel on the right containing a series of downloadable tables and related interactive plots (Figure 1).
The search tab can be used to quickly check (1) whether RNA-Seq or phosphoproteomics data are available for a specified cell line and/or perturbation/treatment of interest (using a studies overview table in the main panel); (2) which previous studies contain significantly up- or downregulated genes/phosphosites, based on user-defined search criteria (i.e. log2 foldchange (FC) and P value; Sidebar: Search for DE/DP Genes/PP-Sites); (3) which previous studies contain specific enrichments of user defined gene sets (Sidebar: GSEA).
The two data tabs have identical features and functionality. They all take user-specified inputs, either indirectly from the search tab or directly after selecting the data of interest in the sidebar (i.e. which studies, cell line, condition, time point and gene(s) of interest). Based on these inputs, standardized differential gene/protein expression information is provided for all studies (i.e. log2FC and P values), both in an easily searchable data table format and an interactive volcano plot. The data table contains gene-specific links to complementary resources, such as GeneCards (28), ChEMBL (29), Ensembl (30) and the R2: Genomics Analysis and Visualization Platform. Relatedly, all findings can be easily correlated to CRISPR/cas9 generated gene effect scores from the DepMap database (32), potentiating the identification of essential genes affected by the selected perturbation easy (Figure 1).
CLEAN provides different forms of existing and user defined gene set enrichment functionality
CLEAN also contains extensive GSEA functionalities, both in the form of a preranked GSEA and an overrepresentation analysis. The former first ranks the genes based on their P values and considers the type of differential expression (up- or downregulated) and the enrichment results (i.e. normalized enrichment scores and P values) are then visualized in an interactive volcano plot at the dataset level, dynamically linked with a running score plot at the individual gene set level. The latter, overrepresentation analysis is performed using Fisher's exact test, considering a set of genes that are defined based on user-determined cut-off criteria for differential expression. Enrichment results (i.e. odds ratios and P values) are again visualized in tables and volcano plots. CLEAN also allows the direct cross-study and -condition comparisons of up to 4 different studies using interactive Venn diagram, allowing for quick enrichment analyses on intersecting or unique gene sets.
The most common gene sets from MSigDB are available (26) and a user also has the option to upload any custom gene set of interest. For phosphoproteomics data analysis, a protein kinase data set from the recently published atlas of serine/threonine substrate specificities is provided (27).
Searching the CLEAN repository for previous experiments that altered the DDR pathway in neuroblastoma cell lines
Given the emerging evidence of a therapeutically targetable role of the DDR in NB (e.g. using AURKA, ATR or CHK1 inhibitors) (3,5,33), we used the CLEAN search tab (available at the home page) to identify other known drugs that inhibit the DDR and could be used as putative combinatorial treatments.
We employed a curated set of 276 DDR-related genes derived from Knijnenburg et al. (34) and uploaded these genes as a custom gene set in CLEAN (Search tab, Sidebar: GSEA). Querying the CLEAN repository for enrichments of these DDR genes in significantly downregulated genes (default settings: log2FC < –1.5 at 5% FDR) resulted in several RNA-Seq studies with significant (Padj < 0.05) enrichments. As expected, DDR enrichments were found after treating NB cells with the ATR inhibitor elimusertib (CLB-GE cells, 48 h treatment; Padj= 3.1e-14). Interestingly and in line with our recent experimental findings (6), strong enrichments were also observed after treating NB cells for 24h with the ALK inhibitor lorlatinib (NB1 cells; Padj= 7.1e-14), for 48h with the PI3K/mTOR inhibitor dactolisib (NB1 cells; Padj= 1.2e-13) or for 24 h with the MDM2 inhibitor idasanutlin (NB1691 cells; Padj= 1.1e-30). Background information on all these studies is available in CLEAN and after selecting the latter study (35) for further exploration, CLEAN redirects the user to the RNA-Seq tab.
A more detailed differential gene expression analysis of the selected study in the RNA-Seq tab indeed confirms a strong DDR signature (P = 1.1e-25), characterized by a downregulation of RRM1/2, EXO1, BRCA1, FANCI, BRIP1, CDC25A, POLQ, CHEK1 and many other genes (Figure 2A, B, Supplementary file 1). We then used the Venn diagram functionality in CLEAN to determine where the downstream signaling pathways upon ATR inhibition (elimusertib), ALK inhibition (lorlatinib), PI3K inhibition (dactolisib) and MDM2 inhibition (idasanutlin) converge. We found 109 commonly downregulated genes that were strongly enriched for the DDR (as expected) and many cell cycle-related processes such as G2M checkpoints and E2F targets, as can be observed when selecting the Canonical Pathways as a data resource for the GSEA (Figure 2C, D). While some of these genes (e.g. RRM2, AURKB, CDK2) have previously been shown to have potential as a drug target in neuroblastoma (33,36,37), many of them are currently underexplored and could serve as an interesting source of further research.
Figure 2.

CLEAN-based identification of previously studied drugs that result in a reduced DNA Damage Response transcriptomic signature in neuroblastoma. CLEAN was used to search for previous studies that resulted in significantly downregulated genes (default settings; i.e. logFC < –1.5 at 5% FDR) that were enriched (at 5% FDR) for a custom DNA Damage Response (DDR) gene set (276 genes, as described by Knijnenburg et al. (34)). Four retrieved studies were then further explored in the RNA-Seq tab. Running score (A) and volcano plot (B) for the DDR gene set in the study of Chen et al, 2019 (35). DDR genes indicated in blue in the volcano plot with labelling of genes that are discussed in the main text. (C) Venn diagram showing the number of unique and intersecting genes between the 4 selected studies. (D) A GSEA using canonical pathways was then performed in the 109 common genes. Bar plots indicate –log10(Padj) values for a selection of enriched pathways as indicated. All plots were generated based on downloaded data from CLEAN. See Supplementary file 1 for an illustration of this use case.
Motif enrichment analyses on previously published phosphoproteomics studies suggest a role for ribosomal S6 kinases in mediating ALK and ATR inhibition
Apart from the large amount of RNA-Seq data, CLEAN contains phosphoproteomics data that have been previously generated upon drug treatment of NB cell lines (accessible via the phosphoproteomics tab). In contrast to RNA-Seq, phosphoproteomics data provide information on earlier and more upstream signal transduction pathways in response to different stimuli. Using this technology, we previously demonstrated that elimusertib-induced ATR inhibition results in an expected dephosphorylation of ATR targets and an increased, compensatory phosphorylation response of ATM targets (3), in keeping with the observations of others (38). Like the RNA-Seq functionality, these data are easily explorable using available interactive tables and volcano plots in CLEAN.
To maximize the efficacy of CLEAN in integrating phosphoproteomics datasets we have implemented a feature based on the recently published atlas of substrate specificities for the human serine/threonine kinome (Sidebar: GSEA Settings—S/T-Kinases) (27). This feature allows the analysis of protein kinase enrichments based on differentially phosphorylated phosphosite motifs within the proteins of interest. When performing this analysis on elimusertib-treated NB cells (3), we could indeed confirm a (non-significant) decreased activity of ATR (NES = –1.2, Padj = 0.12) and a strong significant increase in ATM activity (NES = 2, Padj = 4.1e-9) upon treatment (Figure 3A). Additionally, this feature identified decreased activity of the related kinases CHEK1, CHEK2 and AURKA. Given the higher described DDR inhibition upon treatment with lorlatinib, we then hypothesized that lorlatinib would result in similar protein kinase responses. Using the search tab (Studies table—condition column, search: Lorlatinib), CLEAN was able to identify two studies that previously explored the phosphoproteomic response to lorlatinib treatment: Emdal et al. (NB1 cells) (39) and Van den Eynden et al. (CLB-BAR cells) (40). While a motif enrichment analysis did not indicate any significant enrichment for ATM and ATR itself in any of those studies, reduced protein kinase activity was predicted in both studies for the ATR target CHEK1, in agreement with our recent wet-lab experimental findings (6), as well as the ATM target CHEK2 and AURKA (Figure 3B). Strikingly, mirrored responses were observed upon stimulation of NB1 cells with the ALK ligand ALKAL2 (study of Borenäs et al. (41)), strongly suggesting that these responses are ALK-specific (Figure 3B, Supplementary file 2).
Figure 3.

Comparison of phosphomotif-based protein kinase enrichments after ATR and ALK inhibition and/or stimulation. Four studies were selected in CLEAN that had available phosphoproteomics data after drug treatment with elimusertib, lorlatinib or the ALK ligand ALKAL2. The preranked GSEA functionality was then used to search for protein kinase enrichments and results were downloaded. (A) Volcano plot showing protein kinase enrichment results after elimusertib treatment in the study of Szydzik et al. (3). Protein kinases that are discussed in the main text are labelled. (B) Heatmap showing normalized enrichment scores (color scale) and P values (size) of the four selected studies as indicated on the bottom. See Supplementary file 2 for an illustration of this use case. NES, normalized enrichment scores.
The strongest reductions in predicted protein kinase activity upon ATR or ALK inhibition were found for several members of the Ribosomal S6 kinases: RPS6KA2, RPS6KA4, RPS6KA6 (also known as RSK4), P90RSK (also known as MAPKAPK1), MAPKAPK2 and P70S6K (Figure 3A-B, Supplementary file 2). Several of these kinases are activated by the MAPK/ERK pathway (42), and phosphorylate the ribosome protein S6 (RPS6) (43). Further exploration in CLEAN indicates that this protein is indeed strongly dephosphorylated upon lorlatinib treatment, mainly at S235 (Padj = 4.2e-05; log2FC = –1.3). Strikingly, this protein is also highly essential for NB cell lines, as can be observed from the very low DepMap gene effect scores (gene effect score in NB1 cells = –2.47). Interestingly, using the links provided to the R2: Genomics Analysis and Visualization Platform, it can be easily shown that high RPS6KA6 expression is strongly correlated with worse survival in neuroblastoma patients (P = 2.07e-12).
Discussion
CLEAN aims to centralize all available RNA-Seq and phosphoproteomics data derived from NB cell line experiments in a standardized and easily accessible format. It is an ongoing open data science initiative that was developed by a multidisciplinary group of scientists with experience in computational biology, NB and signal transduction. We are planning regular updates (every 3 months) of the available cell line and related gene set data. Additionally, extensions are foreseen for data generated from organoids or NB model organisms (e.g. mouse models). New data sets will be selected from literature or at a user's request (a contact tab is included in the application). While the CLEAN scope is restricted to NB, its functionality can also be used for other (cancer) cell lines. To facilitate this broader applicability, an option is provided for users to upload other (unpublished) DE analysis results and use all the features provided by CLEAN and compare with other available data. These data are not saved and are deleted after the user session expires.
The available data in CLEAN were reprocessed using state-of-the-art methods. While this standardized approach makes the data highly comparable and results were mostly identical to the original studies, differences with the originally used methods (e.g. aligners, genome annotations, differential expression methods, …) could result in deviations from the original findings. While we experienced no bottlenecks in downloading, processing or data storage, the manual curation of the metadata was a time-consuming task. This is illustrated by reference samples originally annotated as ‘untreated’, ‘control’, ‘DMSO’, ‘empty vector’, ‘timepoint 0’ or simply as ‘sample 1’, sometimes with little to no further explanation in the manuscript and lack of source code, rendering automation of this curation step impossible. Open data initiatives like CLEAN as well as research reproducibility in general would strongly benefit from mandating of code sharing for any data processing, showing how the researcher started at a and ended at z and/or stricter guidelines for annotation of uploaded data in public repositories (11–13).
CLEAN potentiates the interactive exploration of previous studies using datasets that were generated at later time points. As we have demonstrated with the 2 use cases, this functionality can be employed to directly compare the findings of different studies or to perform GSEA using gene sets that that were published later. Recent studies, as well as our ongoing work, suggest that genomic alterations in MYCN and ALK, which are characteristic for high-risk NB, result in specific vulnerabilities to DDR inhibition (3,5). Using CLEAN we identified several drugs (i.e. lorlatinib, dactolisib and elumusertib) with different targets that resulted in 109 commonly differentially expressed genes in different NB cell lines. The identification of lorlatinib is in agreement with our recent wet lab experiments (6). Further, several of these genes have been previously investigated as putative therapeutic targets (e.g. RRM2, CDK2, CHEK1 (33,36)), illustrating how CLEAN can be used to identify novel targets that can be selected for further experimental validation.
We further illustrated the strength of CLEAN in applying newly published GSEA datasets on older studies by performing a protein kinase S/T phosphosite GSEA on existing phosphoproteomics datasets, using the recently published atlas of substrate specificities for the human serine/threonine kinome (27). This analysis confirmed a previously suggested and strong compensatory activity of ATM upon ATR inhibition (3,6,38). Furthermore, a role for several Ribosome S6 Kinase (RSK) proteins was suggested in mediating the effects of both ATR and ALK inhibition. Interestingly, RSK has been previously demonstrated to suppress ATM activity (43). As phosphomotifs of protein kinases from similar phylogenies are highly similar, there's a risk of mispredictions for related protein kinases. This could explain why the reduced ATR activity upon ATR inhibition was rather weak and non-significant (i.e. overlapping hyperphosphorylated ATM S/TQ sites weaken the signal) and also makes it hard to distinguish between the different RSKs based on these results.
In conclusion, CLEAN is a highly interactive and easy to use web application that centralizes all NB cell line data in a standardized format, providing a rich resource for future preclinical neuroblastoma research. We demonstrated with two use cases that CLEAN can be used to independently generate new hypothesis and identify novel putative drug targets. Additionally, CLEAN is also suitable to orthogonally validate findings derived from newly developed computational approaches, as an alternative to costly and time-consuming wet-lab experimental validation experiments.
Supplementary Material
Acknowledgements
The authors thank all members from the Van den Eynden lab, Palmer lab and Hallberg lab for extensively testing CLEAN and providing valuable input.
Contributor Information
Jonatan L Gabre, Department of Human Structure and Repair, Anatomy and Embryology Unit, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium; Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Peter Merseburger, Department of Human Structure and Repair, Anatomy and Embryology Unit, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium; Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Arne Claeys, Department of Human Structure and Repair, Anatomy and Embryology Unit, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium.
Joachim Siaw, Department of Human Structure and Repair, Anatomy and Embryology Unit, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium; Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Sarah-Lee Bekaert, Cancer Research Institute Ghent, Ghent, Belgium; Department of Biomolecular Medicine, Ghent University, Ghent, Belgium.
Frank Speleman, Cancer Research Institute Ghent, Ghent, Belgium; Department of Biomolecular Medicine, Ghent University, Ghent, Belgium.
Bengt Hallberg, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Ruth H Palmer, Department of Medical Biochemistry and Cell Biology, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Jimmy Van den Eynden, Department of Human Structure and Repair, Anatomy and Embryology Unit, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent, Ghent, Belgium.
Data availability
All results provided in this manuscript are based on previously published, publicly available data. All these data are accessible in CLEAN at https://ccgg.ugent.be/shiny/clean/.
Supplementary data
Supplementary Data are available at NAR Cancer Online.
Funding
Ghent University Special Research Fund Starting Grant [BOF.STG.2019.0073.01 to J.V.d.E.]; Research Foundation Flanders [FWO.OPR.2023.0063.01 to F.S., J.V.d.E., R.H.P. and V424522N to J.V.d.E.]; Belgian Foundation Against Cancer [STI.STK.2023.0017.02 to F.S., J.V.d.E.]; Stand up to Cancer, the Flemish Cancer Society [STI.VLK.2022.0013.01 to A.C.]; Swedish Cancer Society [CAN21/01549 to R.H.P.]; Swedish Childhood Cancer Foundation [2019-0078 to R.H.P. and 2021-0068 to J.S.].
Conflict of interest statement. None declared.
References
- 1. Matthay K.K., Maris J.M., Schleiermacher G., Nakagawara A., Mackall C.L., Diller L., Weiss W.A.. Neuroblastoma. Nat. Rev. Dis. Primers. 2016; 2:16078. [DOI] [PubMed] [Google Scholar]
- 2. Ladenstein R., Pötschger U., Pearson A.D.J., Brock P., Luksch R., Castel V., Yaniv I., Papadakis V., Laureys G., Malis J.et al.. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol. 2017; 18:500–514. [DOI] [PubMed] [Google Scholar]
- 3. Szydzik J., Lind D.E., Arefin B., Kurhe Y., Umapathy G., Siaw J.T., Claeys A., Gabre J.L., Van den Eynden J., Hallberg B.et al.. ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat. Commun. 2021; 12:6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Keller K.M., Krausert S., Gopisetty A., Luedtke D., Koster J., Schubert N.A., Rodríguez A., van Hooff S.R., Stichel D., Dolman M.E.M.et al.. Target Actionability Review: a systematic evaluation of replication stress as a therapeutic target for paediatric solid malignancies. Eur. J. Cancer. 2022; 162:107–117. [DOI] [PubMed] [Google Scholar]
- 5. Roeschert I., Poon E., Henssen A.G., Dorado Garcia H., Gatti M., Giansanti C., Jamin Y., Ade C.P., Gallant P., Schülein-Völk C.et al.. Combined inhibition of aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Cancer. 2021; 2:312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borenäs M., Umapathy G., Lind D.E., Lai W.-Y., Guan J., Johansson J., Jennische E., Schmidt A., Kurhe Y.V., Gabre J.L.et al.. ALK signalling primes the DNA damage response sensitizing ALK-driven neuroblastoma to therapeutic ATR inhibition. Proc. Natl. Acad. Sci. U.S.A. 2024; 121:e2315242121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Y., Volegova M., Nasholm N., Das S., Kwiatkowski N., Abraham B.J., Zhang T., Gray N.S., Gustafson C., Krajewska M.et al.. Synergistic anti-tumor effect of combining selective CDK7 and BRD4 inhibition in neuroblastoma. Front. Oncol. 2022; 11:773186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang L., Zhang X.-O., Rozen E.J., Sun X., Sallis B., Verdejo-Torres O., Wigglesworth K., Moon D., Huang T., Cavaretta J.P.et al.. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022; 13:3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katz K., Shutov O., Lapoint R., Kimelman M., Brister J.R., O’Sullivan C. The sequence read Archive: a decade more of explosive growth. Nucleic Acids Res. 2021; 50:D387–D390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hong M., Tao S., Zhang L., Diao L.-T., Huang X., Huang S., Xie S.-J., Xiao Z.-D., Zhang H.. RNA sequencing: new technologies and applications in cancer research. J. Hematol. Oncol. 2020; 13:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker M. 1,500 scientists lift the lid on reproducibility. Nature. 2016; 533:452–454. [DOI] [PubMed] [Google Scholar]
- 12. Nosek B.A., Alter G., Banks G.C., Borsboom D., Bowman S.D., Breckler S.J., Buck S., Chambers C.D., Chin G., Christensen G.et al.. SCIENTIFIC STANDARDS. Promoting an open research culture. Science. 2015; 348:1422–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stodden V., Seiler J., Ma Z.. An empirical analysis of journal policy effectiveness for computational reproducibility. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S.et al.. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017; 45:D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Putri G.H., Anders S., Pyl P.T., Pimanda J.E., Zanini F.. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics. 2022; 38:2943–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 20. Zhang X., Smits A.H., van Tilburg G.B.A., Ovaa H., Huber W., Vermeulen M.. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018; 13:530–550. [DOI] [PubMed] [Google Scholar]
- 21. Team, R.C. R: A Language and Environment for Statistical Computing. 2022;
- 22. Chang W., Cheng J., Allaire J., Sievert C., Schloerke B., Xie Y., Allen J., McPherson J., Dipert A., Borges B.. shiny: web Application Framework for R. 2022;
- 23. Xie Y., Cheng J., Tan X.. DT: a wrapper of the JavaScript library ‘DataTables’. 2023;
- 24. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2016; New York: Springer-Verlag. [Google Scholar]
- 25. Sievert C. Interactive Web-based Data Visualization with R, plotly, and shiny. Chapman and Hall/CRC. 2020; [Google Scholar]
- 26. Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P.. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011; 27:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson J.L., Yaron T.M., Huntsman E.M., Kerelsky A., Song J., Regev A., Lin T.-Y., Liberatore K., Cizin D.M., Cohen B.M.et al.. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023; 613:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stelzer G., Rosen N., Plaschkes I., Zimmerman S., Twik M., Fishilevich S., Stein T.I., Nudel R., Lieder I., Mazor Y.et al.. The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinformatics. 2016; 54:1.30.31–31.30.33. [DOI] [PubMed] [Google Scholar]
- 29. Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Félix E., Magariños M.P., Mosquera J.F., Mutowo P., Nowotka Met al.. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2018; 47:D930–D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R.et al.. Ensembl 2022. Nucleic Acids Res. 2021; 50:D988–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kocak H., Ackermann S., Hero B., Kahlert Y., Oberthuer A., Juraeva D., Roels F., Theissen J., Westermann F., Deubzer H.et al.. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death. Dis. 2013; 4:e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S.et al.. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017; 49:1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nunes C., Depestel L., Mus L., Keller K.M., Delhaye L., Louwagie A., Rishfi M., Whale A., Kara N., Andrews S.R.et al.. RRM2 enhances MYCN-driven neuroblastoma formation and acts as a synergistic target with CHK1 inhibition. Sci. Adv. 2022; 8:eabn1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Knijnenburg T.A., Wang L., Zimmermann M.T., Chambwe N., Gao G.F., Cherniack A.D., Fan H., Shen H., Way G.P., Greene C.S.et al.. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018; 23:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen L., Pastorino F., Berry P., Bonner J., Kirk C., Wood K.M., Thomas H.D., Zhao Y., Daga A., Veal G.J.et al.. Preclinical evaluation of the first intravenous small molecule MDM2 antagonist alone and in combination with temozolomide in neuroblastoma. Int. J. Cancer. 2019; 144:3146–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Z., Wang Z., Pang J.C., Yu Y., Bieerkehazhi S., Lu J., Hu T., Zhao Y., Xu X., Zhang H.et al.. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci. Rep. 2016; 6:29090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bogen D., Wei J.S., Azorsa D.O., Ormanoglu P., Buehler E., Guha R., Keller J.M., Mathews Griner L.A., Ferrer M., Song Y.K.et al.. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. 2015; 6:35247–35262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schlam-Babayov S., Bensimon A., Harel M., Geiger T., Aebersold R., Ziv Y., Shiloh Y.. Phosphoproteomics reveals novel modes of function and inter-relationships among PIKKs in response to genotoxic stress. EMBO J. 2021; 40:e104400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emdal K.B., Pedersen A.K., Bekker-Jensen D.B., Lundby A., Claeys S., De Preter K., Speleman F., Francavilla C., Olsen J.V.. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci. Signal. 2018; 11:eaap9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van den Eynden J., Umapathy G., Ashouri A., Cervantes-Madrid D., Szydzik J., Ruuth K., Koster J., Larsson E., Guan J., Palmer R.H.et al.. Phosphoproteome and gene expression profiling of ALK inhibition in neuroblastoma cell lines reveals conserved oncogenic pathways. Sci. Signal. 2018; 11:eaar5680. [DOI] [PubMed] [Google Scholar]
- 41. Borenäs M., Umapathy G., Lai W.Y., Lind D.E., Witek B., Guan J., Mendoza-Garcia P., Masudi T., Claeys A., Chuang T.P.et al.. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021; 40:e105784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frödin M., Gammeltoft S.. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 1999; 151:65–77. [DOI] [PubMed] [Google Scholar]
- 43. Chen C., Zhang L., Huang N.J., Huang B., Kornbluth S.. Suppression of DNA-damage checkpoint signaling by rsk-mediated phosphorylation of Mre11. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:20605–20610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All results provided in this manuscript are based on previously published, publicly available data. All these data are accessible in CLEAN at https://ccgg.ugent.be/shiny/clean/.