

Abstract
Peptide-Centric Chimeric Antigen Receptors (PC-CARs), which recognize oncoprotein epitopes displayed by human leukocyte antigens (HLAs) on the cell surface, offer a promising strategy for targeted cancer therapy (1). We have previously developed a PC-CAR targeting a neuroblastoma-associated PHOX2B peptide, leading to robust tumor cell lysis restricted by two common HLA allotypes (2). Here, we determine the 2.1 Å structure of the PC-CAR:PHOX2B/HLA-A*24:02/β2m complex, which reveals the basis for antigen-specific recognition through interactions with CAR complementarity-determining regions (CDRs). The PC-CAR adopts a diagonal docking mode, where interactions with both conserved and polymorphic HLA framework residues permit recognition of multiple HLA allotypes from the A9 serological cross-reactivity group, covering a combined American population frequency of up to 25.2%. Comprehensive characterization using biochemical binding assays, molecular dynamics simulations, and structural and functional analyses demonstrate that high-affinity PC-CAR recognition of cross-reactive pHLAs necessitates the presentation of a specific peptide backbone, where subtle structural adaptations of the peptide are critical for high-affinity complex formation and CAR-T cell killing. Our results provide a molecular blueprint for engineering CARs with optimal recognition of tumor-associated antigens in the context of different HLAs, while minimizing cross-reactivity with self-epitopes.
One-Sentence Summary:
This work demonstrates the structure and molecular mechanism of a peptide-centric CAR that recognizes a targeted cancer epitope on polymorphic HLA allotypes with restricted antigenic peptide cross-reactivity.
Introduction
Chimeric Antigen Receptor (CAR) T-cell therapy (1, 2) has achieved remarkable efficacy for liquid tumors such as B-cell leukemias (3, 4). On the other hand, while therapeutics for some solid tumors have seen anti-tumor activity (5–7), they have been plagued by issues such as cytokine release syndrome and the lack of a sustained response. Thus, identifying tumor-specific antigens and developing highly selective CARs is critical for clinical applications (8). While several promising cancer-specific antigens have been discovered, most proteins are expressed intracellularly and thus are undruggable by traditional CAR-T therapy (9, 10). These inaccessible targets, however, are naturally degraded into peptides by the proteasome, which can be presented on the cell surface via major histocompatibility complex (MHC) class I molecules as peptide/MHC-I (pMHC) complexes, especially when aberrantly overexpressed in cancer cells (11). Thus, CARs targeting endogenous peptides offer a new avenue for cancer immunotherapy (12).
Neuroblastoma is an extracranial solid tumor responsible for 11% of pediatric cancer deaths (13). With the completion of a phase 1–2 clinical trial, the success of a third generation GD2-CAR T cell underscores the potential for CAR-T therapy for treating this malignancy (6). To this end, we recently reported the development of a peptide-centric CAR (PC-CAR) 10LH targeting a neuroblastoma-enriched, unmutated peptide (QYNPIRTTF) derived from the PHOX2B oncoprotein (14). While the single-chain fragment variable (scFv) based CAR displayed exquisite specificity for the PHOX2B antigen, its application was limited by HLA restriction to HLA-A*24:02 and HLA-A*23:01.
Here, we leverage insights from our 2.1 Å crystal structure of a 10LH:PHOX2B/HLA-A*24:02/β2 microglobulin (β2m) complex to expand the patient cohort amenable to CAR-T therapy. We characterize the molecular basis for peptide-centric recognition and explore the effect of HLA polymorphisms on 10LH binding. The PC-CAR 10LH can recognize the PHOX2B antigen presented by the HLA alleles from the A9 serological group, suggesting that it can be applied to treat up to a quarter of American, high-risk neuroblastoma patients. Through molecular dynamics (MD) simulations, biochemical assays, and in situ experiments, we report on the conformational plasticity of the peptide, enabling high-affinity complex formation and slow dissociation. Finally, we show that cytokine release by 10LH necessitates proper presentation of the R6 side chain and a conserved backbone, narrowing the cross-reactivity profile of the PC-CAR compared to naturally occurring TCR and TCR-like molecules. Our study uncovers structural principles required for tumor-associated HLA recognition, informing future PC-CAR T cell therapy development.
Results
The PC-CAR 10LH engages its PHOX2B/HLA-A*24:02 antigen through a prominent peptide selectivity filter
To understand the molecular basis of peptide-centric HLA recognition by 10LH, we determined the crystal structure of the 10LH:PHOX2B/HLA-A*24:02/β2m complex at a resolution of 2.1 Å (Fig. 1A, fig. S1, fig. S2A, table S1, movie S1). The PC-CAR engages the N- and C- termini of the MHC-I peptide binding groove via its complementarity-determining region 3 (CDR3) loops from the heavy and light chain, respectively (Fig. 1B). As reported in prior studies (15), AlphaFold (16) was unable to capture both the peptide interactions and the receptor docking orientation in its top 25 predictions of the 10LH:PHOX2B/HLA-A*24:02/β2m complex (movie S2). We compared our 10LH structure to existing TCR:pHLA-I complexes and found that the CAR employed a diagonal binding mode, with a relatively shallow docking angle (32.2°; Fig. 1C), which buried a moderate interface area on the peptide (fig. S3A). In contrast, the HLA/receptor interface was comparatively high relative to naturally occurring TCRs (fig. S3B), with interactions between the CDR loops and the α1 and α2 helices of HLA-A*24:02. HLA residues computationally identified as stabilizing the complex with 10LH were skewed towards one end of the MHC-I peptide-binding groove (fig. S3C), while TCRs displayed a more uniform footprint. Overall, the PC-CAR engages the PHOX2B/HLA-A*24:02/β2m complex in a manner resembling TCR:pHLA-I complexes, albeit with notable differences in its precise molecular interactions and extent of molecular surfaces, which can contribute to the formation of a high-affinity complex with a nanomolar equilibrium dissociation constant (14), KD.
Fig. 1. Structural basis for high-affinity 10LH:PHOX2B/HLA-A*24:02/β2m complex formation.
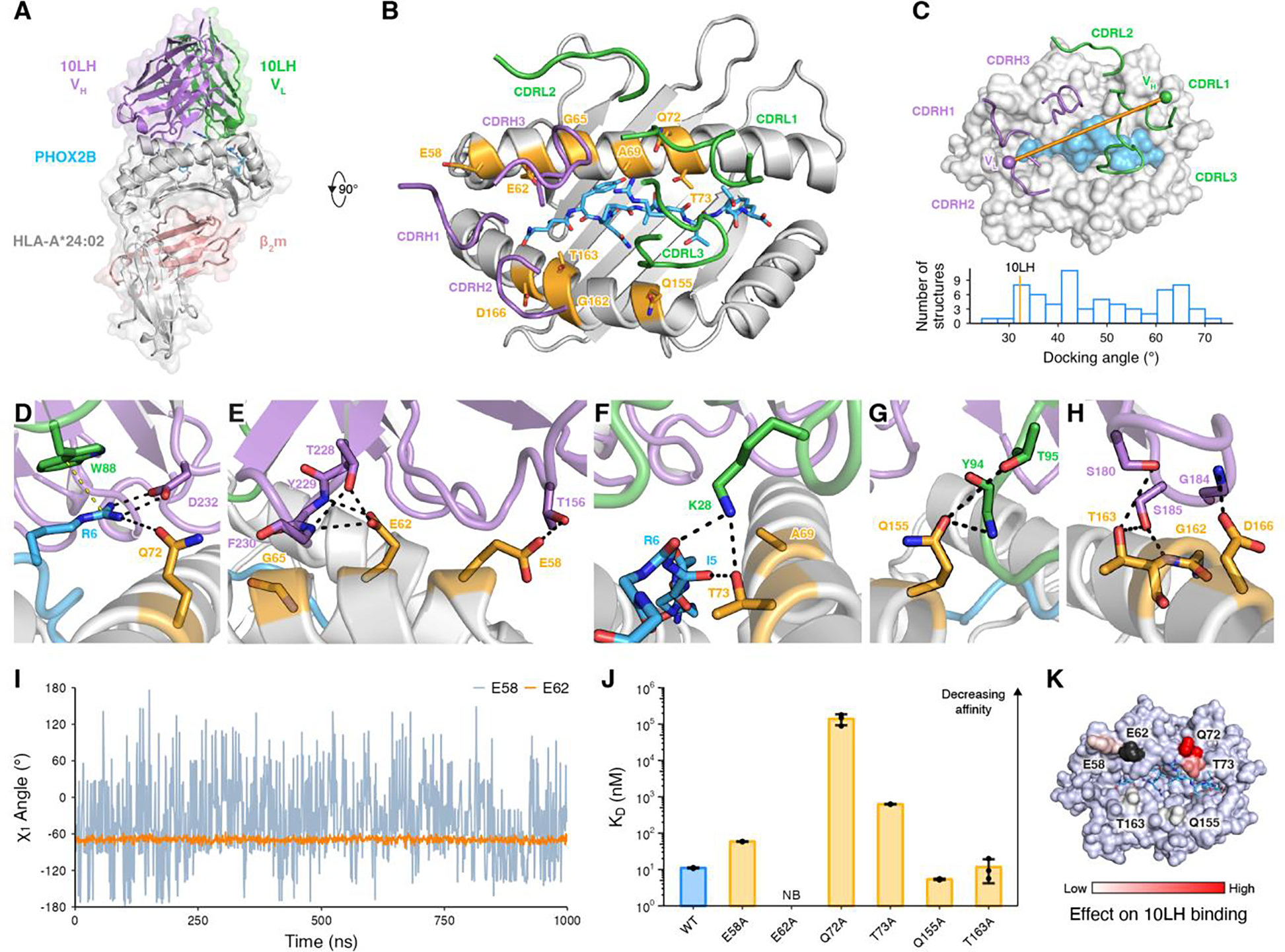
(A) Crystal structure of the 10LH:PHOX2B/HLA-A*24:02/β2m complex (PDB ID 8EK5). Surface representation is shown in lighter shades. The PHOX2B peptide is shown as sticks. (B) Top-down view of the structure shown in (A). The 10LH CDR loops are shown as cartoon tubes. The HLA framework residues (orange) and the PHOX2B peptide (blue) are shown in sticks. (C) Top-down view of 10LH:PHOX2B/HLA-A*24:02/β2m shown in surface representation. The 10LH interdomain vector is drawn between the centroids of the conserved disulfide bonds in the heavy (purple) and light (green) chains. A histogram of TCR docking angles is calculated using the TCR interdomain vectors. The corresponding value for 10LH is indicated by an orange line at 32.2°. (D-H) Close-up view of 10LH and PHOX2B/HLA-A*24:02/β2m interactions for the R6 in PHOX2B (D) and the framework residues along the α1 (E, F) and α2 (G, H) HLA helices. Hydrogen bonds and cation-π interactions are indicated by black and yellow dashed lines, respectively. (I) The χ1 side chain torsion angle from MD simulations of the 10LH:PHOX2B/HLA-A*24:02 complex for E58 and E62. Data are mean across n = 3 independent 1 μs runs. (J) SPR determined KD values for 10LH:PHOX2B/HLA-A*24:02/β2m with alanine mutated framework residues. Data are mean ± SD for n = 3 technical replicates. KD, equilibrium dissociation constant; NB, no binding. (K) The fold change of KD for the alanine mutated 10LH:PHOX2B/HLA-A*24:02/β2m compared to the WT mapped on the HLA-A*24:02 surface. E62A (no binding) is colored black.
We next focused on interactions in the PC-CAR:PHOX2B/HLA-A*24:02/β2m complex, which conferred antigen specificity. Q72 of HLA-A*24:02 interacts with R6 from PHOX2B, orienting the Arg side chain for salt bridge formation with D232 from CDRH3 (Fig. 1D). This network of contacts is stabilized further by a cation-π interaction with W88 from CDRL3, forming an exquisite peptide selectivity filter. Stable docking of the PC-CAR to the pHLA complex is mediated by multiple contacts with amino acids along the α1 and α2 helices, termed framework residues. On the α1 helix, E58 forms a hydrogen bond with T156 of 10LH CDRH1, and E62 forms three hydrogen bonds with CDRH3 residues (Fig. 1E). Moreover, K28 from the 10LH CDRL3 bridges both the peptide and HLA through a network of contacts with the R6 backbone carbonyl and the T73 side chain hydroxyl group (Fig. 1F). Four framework residues on the α2 helix form additional contacts with the CDRH2 and CDRL3 loops (Fig. 1G, H). To further investigate the stability of identified contacts between HLA-A*24:02 framework residues and 10LH, we conducted molecular dynamics (MD) simulations of the PC-CAR:pMHC platform at the 1 μs timescale, followed by experimental surface plasmon resonance (SPR) binding. Overall, our MD simulations showed a stable ternary complex, with relatively low mean root mean square fluctuation (RMSF) values compared to the crystallographic coordinates (fig. S4A). However, analysis of χ1 side chain torsion angles for the HLA framework residues identified in our structure revealed a range of dynamic effects (fig. S4B). For instance, while the E62 side chain is rigid along the simulation trajectory, E58 samples a wide range of χ1 angles suggesting a more transient interaction with the CDRH3 loop (Fig. 1I). Using a panel of HLA-A*24:02 point mutants, we observed a range of effects on 10LH binding by SPR (Fig. 1J, fig. S5, fig. S6). Consistent with our MD simulations and analysis of the structure, E62A completely abrogated binding while E58A, Q155A, and T163A had a more subtle effect. Mapping these effects on the surface of HLA-A*24:02 revealed interaction hotspots (Fig. 1K) formed by E62 as well as Q72 and T73, residues that are adjacent to R6 of PHOX2B. Thus, these results demonstrate that high-affinity PC-CAR:PHOX2B/HLA-A*24:02/β2m complex formation emerges from the specific recognition of both R6 on PHOX2B, in addition to key framework residues on HLA-A*24:02. This is reminiscent of how TCRs engage their pMHC-I antigens (17, 18), albeit with a strict requirement for a precise network of contacts formed by the Q72-R6-D232-W88 tetrad, which bridges elements from all three components of the molecular complex.
10LH interactions with polymorphic HLA framework residues enable recognition of multiple allotypes
Our structural analysis showed that a network of interactions with HLA framework residues was critical for 10LH complex formation. Thus, sequence divergence of HLA allotypes from the optimal HLA-A*24:02 interface is likely to disrupt antigen-specific recognition, despite maintaining PHOX2B presentation. We hypothesized that 10LH could recognize the PHOX2B peptide presented by members of the A9 serological cross-reactivity group (CREG) (19), including multiple allotypes beyond the previously identified (14) HLA-A*24:02 and HLA-A*23:01 (Fig. 2A). Upon prediction of PHOX2B binders among common (global freq. > 0.1%) HLAs by NetMHCpan 4.1 (20) (n = 219), we computed the sequence conservation for different framework residues (Fig. 2B). While Q72 is completely conserved across PHOX2B binding alleles, other positions including E62, G65, and A69 are highly polymorphic (Fig. 2C). These residues cluster on one face of the α1 helix, and form an epitope previously implicated in HLA alloreactivity (21). Guided by the observed polymorphisms, we evaluated the effect of introducing specific HLA-A*24:02 substitutions on binding to 10LH by SPR (Fig. 2D, fig. S7, fig. S8). Our results show that G65 substitutions to the arginine or glutamine polymorphism found in different HLAs abrogate binding, likely due to steric clashes with CDRH3. The E62Q and E62R substitution also resulted in a loss of binding, consistent with our previous alanine mutagenesis results. Substitution of A69 and G162 with bulkier, charged amino acids led to an increase of KD values by two to three orders of magnitude, likely due to disruption of hydrophobic interactions. The D166E substitution had a more subtle effect. Thus, point mutations of polymorphisms found in PHOX2B-presenting alleles provide a strong basis for the HLA restriction of PHOX2B antigen recognition by 10LH.
Fig. 2. Key framework residues on HLA molecules confer allelic interactions with 10LH.
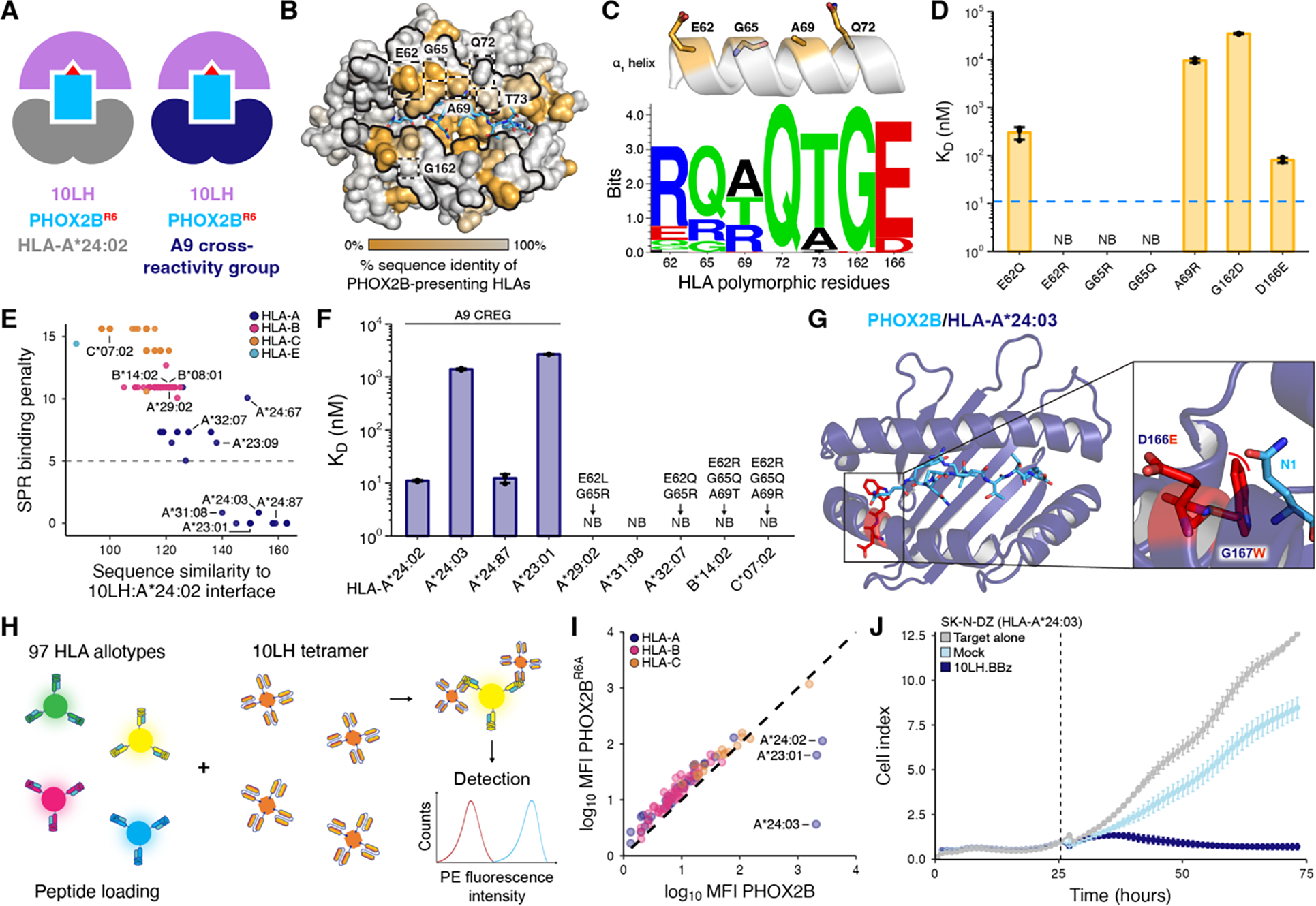
(A) Schematic depicting cross-HLA interactions by 10LH. (B) Sequence identity across 219 HLA-I alleles computationally predicted to present the PHOX2B peptide mapped onto the HLA-A*24:02 surface (PDB ID 8EK5). The α1 and α2 helices and HLA framework residues are denoted by the black outlines and dashed boxes, respectively, on the HLA-A*24:02 surface. (C) Schematic depicting the HLA framework residues on the α1 helix. Below, a sequence motif of the HLA framework residues across common alleles predicted to bind the PHOX2B peptide (n = 219). Created using Seq2Logo. (D) SPR determined KD values for PHOX2B/HLA-A*24:02/β2m with site-directed mutations of the framework residues. Data are mean ± SD for n = 3 technical replicates. The blue dashed line depicts the KD of the WT PHOX2B/HLA-A*24:02/β2m to 10LH. (E) Scatter plot of sequence similarity of HLA interface residues (BLOSUM62 score) against the SPR binding penalty for the predicted 10LH binders. The SPR binding penalty was calculated by adding the log base 10 ratio of the mean KD value for each polymorphism in a given allele to the wild-type KD. The gray dashed line indicates the presence of at least one non-binding polymorphism. (F) SPR determined KD values for selected HLA allotypes. Polymorphisms among the framework residues are listed above non-binding alleles. Data are mean ± SD for n = 3 technical replicates. NB, no binding. (G) Structural model of PHOX2B/HLA-A*24:03 with polymorphisms relative to HLA-A*24:02 denoted in red sticks. The zoom-in details the steric clash (red curve) between the polymorphic residue (red) and the PHOX2B peptide (blue). (H) A summary of SAB screening, where beads are pre-incubated over weekend (o/w) with PHOX2B or PHOX2BR6A peptides, and stained by 10LH PE-tetramers for a shift in MFI. (I) Correlation of log10(MFI) for 10LH stained HLA allotypes pre-incubated with PHOX2B and PHOX2BR6A. Data are mean for n = 3 technical replicates with SD and individual data points are shown in fig. S12. The black dashed line represents a conceptual 1:1 correlation (no difference between PHOX2B- and PHOX2BR6A-dependent 10LH interactions). (J) In vitro cytotoxic activity of 10LH.BBz CAR T cells against HLA-A*24:03 neuroblastoma cell line SK-N-DZ. 10LH.BBz CAR T cells were co-cultured (black dashed line indicates addition of effector cells to target cells) at a 3:1 E:T ratio with SK-N-DZ target cells. Target cell viability was determined using the cell index of the xCELLigence Real-Time Cell Analysis (RTCA) system. Target cell index was normalized to the timepoint immediately before addition of effector cells. Values represent mean ± SD using effector cells from n = 2 biological donors, in triplicates.
We used our measured KD values for individual framework residue substitutions on HLA-A*24:02 to calculate a SPR binding penalty for all PHOX2B-presenting alleles and plotted these values against their BLOSUM62 (22) sequence similarity to HLA-A*24:02 with respect to interface residues in our 10LH complex structure as determined by PDBePISA (23). We observed a partitioning of the allelic landscape into two distinct sets, with a clear cluster of HLA allotypes having a high sequence identity to HLA-A*24:02 and low binding penalty (Fig. 2E). We then conducted SPR binding experiments for a subset of HLAs from both sets (Fig. 2F, fig. S2B, fig. S9, fig. S10), confirming interactions with 10LH for members of the A9 CREG, defined by the A23 and A24 serological splits. Notably, our identified 10LH binders included HLA-A*31:08, which was separate from the A9 cross-reactivity group. Analysis of this allotype’s sequence relative to HLA-A*24:02 revealed extensive polymorphisms in the HLA groove (fig. S11A), likely resulting in an altered conformation of the PHOX2B antigen and, therefore, impaired binding to 10LH as shown by our SPR data (fig. S11B). For alleles from the A9 CREG, an approximately 100-fold increase in KD (micromolar range, as opposed to 11 nM for HLA-A*24:02) was observed. This could be attributed to the altered presentation of the PHOX2B antigen resulting from specific MHC-I groove polymorphisms as shown by our structural modeling for HLA-A*24:03 and HLA-A*23:01 (Fig. 2G and fig. S11C, D). Conversely, HLA-A*23:09 and HLA-A*24:67 from the A9 CREG were predicted to not bind to 10LH due to the deleterious G65R framework residue polymorphism. Other non-binding alleles outside of the A9 CREG, such as HLA-B*14:02, showed polymorphisms within multiple framework residues, leading to steric hindrance with the 10LH CDRs (fig. S11E, F). One selected allele, HLA-B*08:01, did not form stable complexes with the PHOX2B peptide despite the prediction by NetMHCpan 4.1 (20) (fig. S10). Thus, our structural modeling and biochemical analysis of framework residue polymorphisms identifies a set of PHOX2B-presenting HLA alleles that can cross-react with 10LH.
We further evaluated the cross-reactivity with the common HLA alleles using a single-antigen bead (SAB) assay (24) (Fig. 2H). Inspection of mean fluorescence intensity (MFI) for the positive versus control experiments identified PHOX2B-dependent HLA interactions with 10LH for allotypes from the A9 CREG, in agreement with our SPR results (Fig. 2I, fig. S12). Using an exemplar, novel allele from this group, we demonstrated 10LH CAR-T cell cytotoxicity and cytokine release against an HLA-A*24:03 neuroblastoma cell line (Fig. 2J, fig. S13, fig. S14). Overall, our results provide strong support that 10LH can bind to several additional allotypes from the A9 CREG. Thus, up to 25.2% and 46.7% of the American and global neuroblastoma patient cohort, respectively, is amenable to 10LH CAR-T therapy, not considering linkage disequilibrium among HLA alleles (table S2 and S3).
Peptide backbone plasticity enables high-affinity complex formation with 10LH
To examine the role of PHOX2B structural adaptations in specific recognition by 10LH, we compared the peptide conformations presented by HLA-A*24:02 in the apo and 10LH-bound states (Fig. 3A). A prominent backbone change, highlighted by a 1.7 Å displacement of the P4 carbonyl oxygen, could contribute to an optimal presentation of the R6 side chain rotamer for interaction with the 10LH CDRs. While the peptide in the apo state was involved in two contacts through the R6 carbonyl group and the T8 amine group to D228 of the α3 domain due to the crystal packing (fig. S15), its structural rearrangement at P4 is relatively distant from position 6 and 8, likely induced by 10LH binding. To further characterize this conformational transition, we performed MD simulations of PHOX2B/HLA-A*24:02 in its apo and PC-CAR-bound states. Simulations starting from the coordinates of the apo structure sampled a bimodal distribution of Ψ4 backbone dihedral angles, where one mode overlapped with the distribution of angles sampled in MD simulations of the 10LH-bound complex (Fig. 3B). These data suggest that the PHOX2B peptide can adopt two conformational states in the HLA-A*24:02 groove, where one of the states presents R6 in an optimal conformation for nanomolar-range binding to 10LH.
Fig. 3. PHOX2B conformational adaptations enable high affinity complex formation with 10LH.
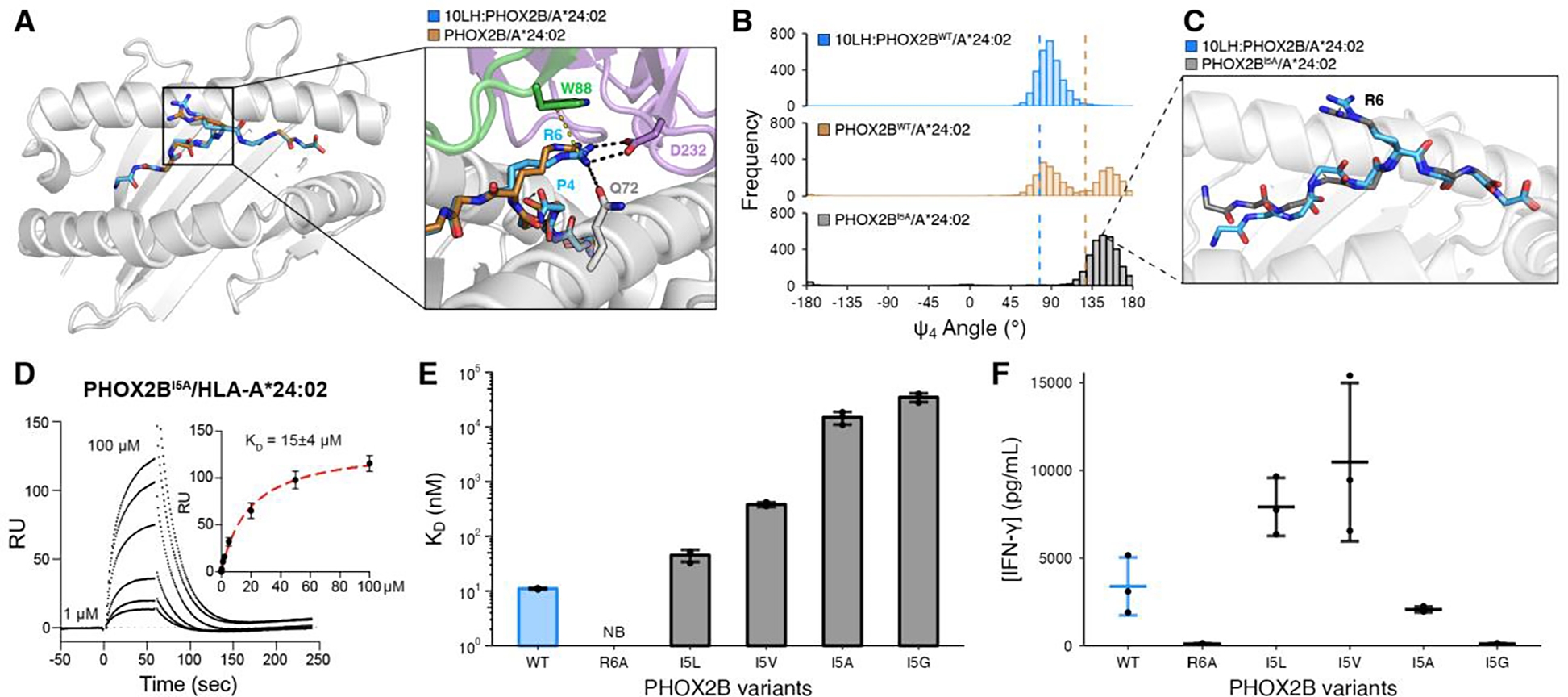
(A) Structural superimposition of PHOX2B/HLA-A*24:02 in the bound (blue; PDB ID 8EK5) and unbound (brown; PDB ID 7MJA) states. The peptides were aligned using their backbone heavy atoms (RMSD = 0.52 Å). The zoom-in details the difference in the R6 side chain conformation and the peptide backbone. (B) Histogram depicting the frequency of Ψ4 backbone dihedral angle values for MD simulations of the bound (blue) and unbound (brown) PHOX2B/HLA-A*24:02 structures as well as the PHOX2BI5A/HLA-A*24:02 unbound variant (black). The dashed lines correspond to the values in the crystal structures (10LH:PHOX2B/HLA-A*24:02/β2m, PDB ID 8EK5; PHOX2B/HLA-A*24:02, PDB ID 7MJA). Data represent 3000 equally-spaced frames from a sum of n = 3 independent 1 μs runs. (C) Representative structure from a frame of PHOX2BI5A/HLA-A*24:02 (black) MD simulations superimposed onto the 10LH:PHOX2B/HLA-A*24:02/β2m (blue) crystal structure. The peptides were aligned using all atoms (RMSD = 1.19 Å). (D) Representative SPR sensorgram of PHOX2BI5A/HLA-A*24:02 flowed over a streptavidin chip coupled with 10LH-biotin. The concentrations of analyte for the top and the bottom sensorgrams are noted. Data are mean ± SD for n = 3 technical replicates. Fits from the kinetic analysis are shown with red lines. KD, equilibrium constant; RU, resonance units. (E) SPR determined KD values for HLA-A*24:02 loaded with I5 mutational scanned peptides. Data are mean ± SD for n = 3 technical replicates. NB, no binding. (F) IFN-γ levels in supernatant of 10LH.BBz CAR T cells co-cultured with HLA-A*24:02 colorectal adenocarcinoma cell line SW620 pulsed with PHOX2B peptide variants. Exogenous peptide was added to SW620 target cells (15 μM final concentration). After four hours of incubation, 10LH.BBz CAR T cells were added to target cells at a 3:1 ET ratio. Supernatant was collected 24 hours post effector cell addition and IFN-γ levels were measured by ELISA. Values represent mean ± SD using effector cells from n = 3 biological donors, in triplicates.
The observed structural plasticity at position 4 of PHOX2B also led to the hypothesis that presentation of the R6 sidechain could be influenced by the adjacent Ile residue at position 5, which forms hydrophobic contacts with the floor of the HLA-A*24:02 groove. MD simulations of the PHOX2BI5A/HLA-A*24:02 complex sampled a narrower distribution of Ψ4 angles relative to wild-type, limited to conformations that largely exclude those found in simulations of the 10LH-bound structure (Fig. 3B). Analysis of representative MD snapshots revealed a 1.4 Å shift of the P4 Cα atom, causing the peptide backbone to sink deeper into the HLA groove (Fig. 3C). This led to a 2.1 Å shift in the Cζ atom of R6, away from the optimal geometry for interactions with the 10LH CDRH3s. In agreement, the I5A substitution reduced binding to 10LH by approximately 1000-fold as determined by SPR (Fig. 3D, fig. S16, fig. S17). These results suggest that peptide backbone structural adaptations, as sampled by the apo pHLA state, can potentially impact CAR-T recognition by affecting R6 rotamer selection and its priming for interactions with the scFv.
Next, we conducted MD simulations followed by SPR binding experiments, introducing Leu, Val, or Gly substitutions at position 5. We observed a negative correlation between the frequency of sampling Ψ4 angles, which resembled the scFv bound state (fig. S18A), and 10LH binding affinity, where conservative substitutions of I5 to Leu or Val maintained nanomolar-range KD values (Fig. 3E, fig. S18B–E, fig. S19). In contrast, I5A and I5G perturbation of R6 sidechain placement led to approximately 1,500- and 3,500-fold reduction, respectively, in 10LH binding affinity by sampling an altered peptide backbone conformation. Finally, we measured cytokine release triggered on 10LH CAR-T cells by peptide pulsing on HLA-A*24:02 positive antigen presenting cells (APCs) (fig. S20). Noticeably, although the SPR-determined KD values of I5L and I5V were more than 4- and 30-fold lower than the WT, respectively, pulsed APCs with either mutant peptide demonstrated enhanced T cell activation relative to the WT peptide (Fig. 3F). This enhancement could be contributed by both a difference in peptide loading and by our observed 10LH dissociation rate constants from the pHLAs, with kd values of 0.0007 sec−1 and 0.0026 sec−1 versus 0.0022 sec−1 measured for I5L, I5V, and I5, respectively (table S4), in agreement with kinetic proofreading models for T cell receptor activation (25–27). In an analogous manner to TCRs, both factors antigen density and pHLA dissociation rate for effective PHOX2B antigen discrimination by 10LH CAR-T cells may explain how HLA-A*23:01, which has approximately two orders of magnitude reduced binding to 10LH relative to HLA-A*24:02 (2.6 μM vs. 11 nM KD) shows potent CAR-T killing of tumor cells (14), since both interactions show similar kd values by SPR (0.0085 sec−1 versus 0.0022 sec−1). These results allow us to establish a relationship between pHLA dissociation rate from the scFv, known as CAR dwell time, and CAR-T cell activation, cytokine release, and cytotoxicity, established by previous studies focusing on both TCRs and CARs (28–30).
Molecular mimicry of the PHOX2B antigen elicits 10LH cross-reactivity with self-epitopes
To characterize the peptide cross-reactivity range of 10LH experimentally, we first applied a peptide X-scan (31) approach to explore all amino acid (excluding Cys) substitutions of non-anchor PHOX2B residues and measured 10LH binding in vitro using a high-throughput assay, which leverages micro-refolding to generate fluorophore-labeled pMHC complexes (32) (Fig. 4A). In agreement with our structural and SPR data (fig. S16, fig. S18), R6 is required while the I5L and I5V mutations can be tolerated for binding to 10LH, respectively (fig. S21A). While position 1 and 8 are solvent exposed, they are out of reach from the 10LH CDRs and thus can tolerate a wide range of amino acids. Additionally, position 3 and 7 display a preference for hydrophobic residues and given their location and orientation in the HLA groove, they could facilitate a proper backbone conformation for peptide loading and CAR recognition. We then generated a peptide pattern to capture the per-position amino acid binding preferences established from our X-scan assay, and obtained a set of 38 potentially cross-reactive peptide sequences found in the normal human proteome after cross-referencing with ScanProsite (33) (fig. S21B). We repeated micro-refoldings with these peptides and confirmed binding to 10LH by SPR for three peptides in our set (ATG2A, CNGB3, and GLB1) in addition to two cross-reactive peptides identified previously by our selective cross-reactive antigen presentation (sCRAP) algorithm (ABCA8 and MYO7B) (14). All peptides bind 10LH with KD values ranging from 1 μM to 27 μM (fig. S22, fig. S23), however, the ATG2A, CNGB3, and GLB1 peptides exhibit a slow dissociation rate, within the same profile as the cognate PHOX2B antigen. Focusing on the highest affinity peptide (ATG2A, LYLPVRVLI), we determined two complementary ATG2A/HLA-A*24:02 structures at 1.8 Å and 3.1 Å resolution (table S1). The 1.8 Å resolution structure showed one pHLA molecule in the asymmetric unit, where the key R6 side chain was buried in the HLA groove and inaccessible for 10LH recognition (fig. S24A). However, in the 3.1 Å resolution structure, one of the four pHLA complexes in the asymmetric unit adopted a peptide backbone which resembles PHOX2B with a solvent-exposed R6 side chain (Fig. 4C and fig. S24B–E). These results suggest that ATG2A/HLA-A*24:02 can sample a minor conformational state with a PHOX2B-like backbone structure as a potential basis for driving strong cross-reactivity with 10LH in vitro.
Fig. 4. Mapping the 10LH/peptide cross-reactivity landscape.
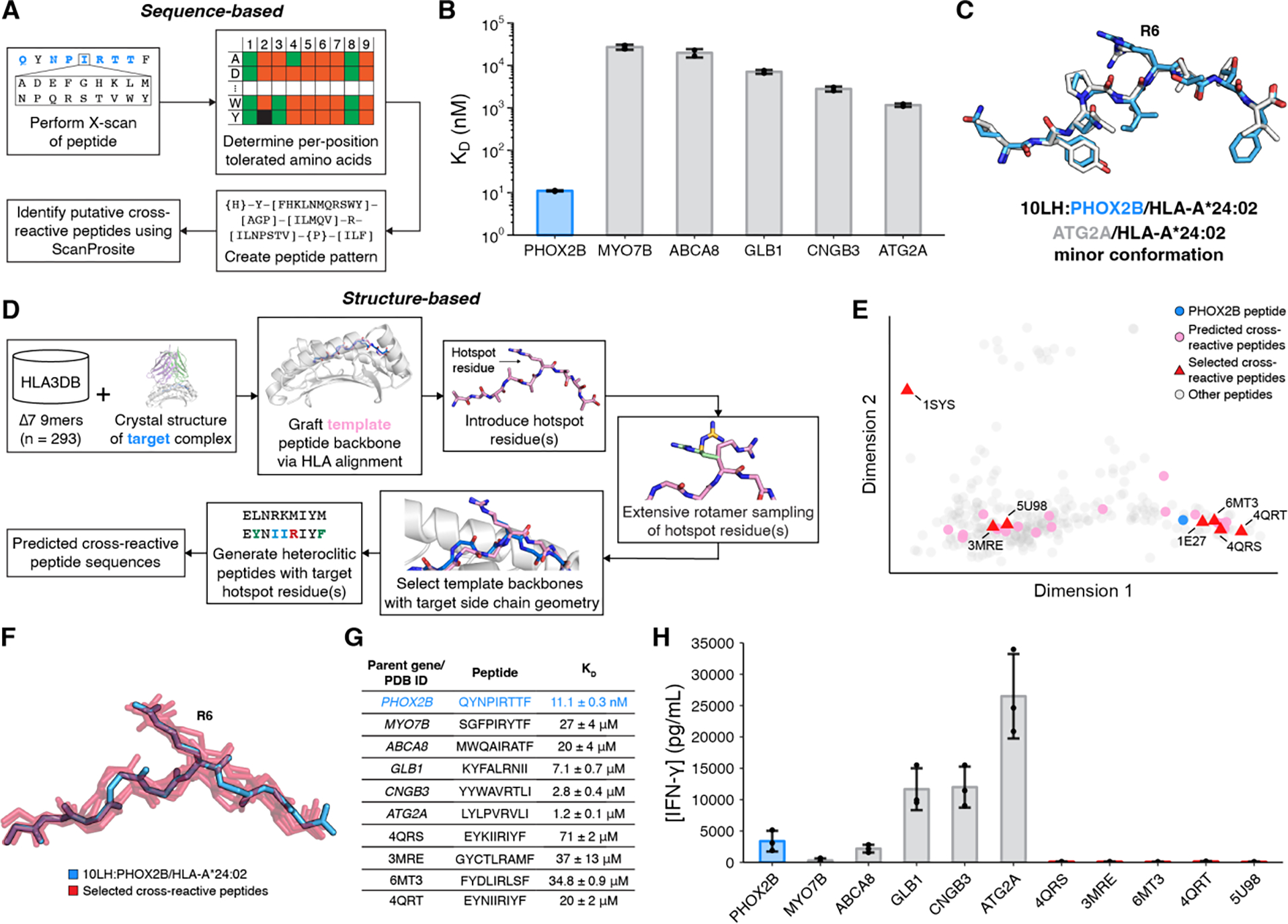
(A) Summary of sequence-based prediction of cross-reactive peptides by X-scan/ScanProsite: (1) perform X-scan on target peptide; (2) determine per-position tolerated amino acids; (3) create peptide pattern; (4) identify putative cross-reactive peptide using ScanProsite. (B) SPR determined KD values for cross-reactive peptides. Data are mean ± SD for n = 2 technical replicates. (C) Structural superimposition of the 10LH:PHOX2B/HLA-A*24:02/β2m peptide (blue; PDB ID 8EK5) with the minor conformation of the 3.1 Å structure of ATG2A/HLA-A*24:02 (white; PDB ID 8SBL). The peptides were aligned using their backbone heavy atoms (RMSD = 0.86 Å). (D) Summary of structure-based prediction of cross-reactive peptides: (1) graft template peptide backbones from HLA3DB; (2) introduce hotspot residues; (3) conduct extensive rotamer sampling; (4) select template backbones with target side chain geometry; (5) generate heteroclitic peptides with target hotspot residues and conserved backbone; (6) obtain a set of predicted cross-reactive peptides. (E) PCA plot of the conformational landscape of peptide backbones. The PCA transformation explains 66% of the variation. (F) Structural superimposition of selected, predicted cross-reactive peptide backbones (red) to the PHOX2B backbone (blue) with the side chain of R6 shown. The peptides were aligned using HLA Cα atoms. (G) Table of cross-reactive peptide sequences with their origin and SPR-determined KD values. Data are mean ± SD for n = 2 or 3 technical replicates. (H) IFN-γ levels in supernatant of 10LH.BBz CAR T cell co-cultured with HLA-A*24:02 colorectal adenocarcinoma cell line SW620 pulsed with cross-reactive peptides. Exogenous peptide was added to SW620 target cells (15 μM final concentration). After four hours of incubation, 10LH.BBz CAR T cells were added to target cells at a 3:1 ET ratio. Supernatant was collected 24 hours post effector cell addition and IFN-γ levels were measured by ELISA. Values represent mean ± SD using effector cells from n = 3 biological donors, in triplicate.
To further explore the conformational landscape of possible cross-reacting peptides from a structural perspective, we leveraged our annotated database of high-resolution pHLA structures, HLA3DB (34) to search for complementarity with the 10LH recognition mode. We used our high-resolution structure of the 10LH:PHOX2B/HLA-A*24:02/β2m complex as a modeling template, aligned the HLA Cα atoms from all existing pHLA complexes to the scFv-bound state of PHOX2B, and performed extensive rotamer sampling of an introduced R6 residue, aiming to identify backbone configurations which allowed optimal side chain placement for interactions with 10LH CDRs (Fig. 4D). We identified 29 potentially cross-reactive peptide backbone scaffolds spanning the observed conformational landscape (Fig. 4E, F). We selected 7 representative peptides on the basis of the PCA plot, and further optimized for binding to HLA-A*24:02 and 10LH through the introduction of anchor residue substitutions and the I5/R6 recognition motif, respectively. SPR experiments showed that 10LH could bind to 6 peptides in our set with KD values in the range of 20 μM to 200 μM, yet exhibited a moderate to rapid dissociation rate (Fig. 4G, fig. S23, fig. S25, table S4). These data indicate that 10LH can engage antigens with a non-PHOX2B-like backbone, albeit with a significant reduction in binding affinity.
To evaluate the functional consequences of the in vitro identified peptide cross-reactivity, we measured cytokine release upon CAR-T stimulation with soluble peptides pulsed on HLA-A*24:02 positive APCs. We observed enhanced peptide-dependent cytokine release relative to the WT peptide for three out of five antigens obtained from our sequence-based approaches (Fig. 4H, fig. S26). Consistently, this can be rationalized by an improvement in the dissociation rate (table S4), in accordance with kinetic proofreading models for T cell receptor activation (25–30). The differences observed were not a result of pMHC complex stability (fig. S23). While peptide cross-reactivity was observed in this cytokine release assay, our prior study (14) indicated that these antigens (ATG2A, CNGB3, and GLB1) are not of clinical concern; cell lines expressing the putative cross-reactive proteins showed no cytotoxicity or IFN-γ release, suggesting that the cross-reactive peptides were not presented on the cell surface. Meanwhile, antigens derived from our orthogonal, structure-guided approach had a moderate to fast dissociation rate and thus did not present a cross-reactivity risk as shown by a lack of sufficient cytokine release (table S4). These data suggest that while in vitro identified cross-reactive peptides can form an initial encounter complex (35) with 10LH, guided by the R6 electrostatic interaction and contacts with HLA framework residues, only a subset can transition to an ideal conformation that produces downstream signaling as measured by IFN-γ release. One possible mechanism for this transition is through intrinsic peptide dynamics, enabling R6 structural adaptations to drive high-affinity complex formation and ultimately leading to slow dissociation rate constant, which triggers signaling. This possible mechanism can provide an additional fidelity checkpoint which limits off-target CAR-T cell activation by self-antigens. Taken together, these findings establish that 10LH triggering on CAR-T cells necessitates not only a specific peptide backbone conformation, thereby achieving sufficient CAR engagement, but also a slow dissociation rate achieved through peptide dynamics, which leads to a narrow cross-reactivity profile for 10LH. Moreover, the only strong cross-reactivity identified from our comprehensive search did not correspond to a risk as the putative peptide was not presented in healthy tissue.
Discussion
Through a combination of structural, biochemical, and functional approaches, we have characterized the molecular basis for peptide-centric recognition of HLA molecules by the 10LH PC-CAR. Our work delineates important structural principles. We find that the formation of a high-affinity complex necessitates a set of conserved HLA framework residues, restricting clinical application to patients expressing an HLA allotype from the A9 serological CREG. Additionally, conformational plasticity of the peptide highlights the importance of sampling a specific backbone structure for CAR-T triggering and cytokine release. We identify three putative cross-reactive peptides whose lack of expression in cell lines suggests that they do not pose a real-world risk. Nevertheless, further, in-depth analysis is required, including rigorous peptide/HLA complex stability and CAR:pHLA binding measurements. Taken together, our analysis reveals significant advantages of PC-CARs over traditional T-cell receptor immunotherapies. As TCRs are inherently cross-reactive (36, 37) their micromolar-range affinity allows them to recognize different epitopes through a variety of strategies (38) not limited to differential docking (39) and accepting structural adjustments (40). In contrast, by adopting a restricted, converged binding geometry with a specific peptide backbone for high-affinity complex formation, 10LH allows for a more restricted antigen cross-reactivity profile, which mitigates off-target toxicity liabilities relative to conventional TCRs.
In contrast to TCR engineering approaches (41), PC-CAR development can yield nanomolar-range, peptide-specific scFv binding without the need for further affinity enhancement. Thus, our binding mechanism is well-poised for future development of bispecific T cell engager constructs (BiTEs) (42, 43). In addition, our results provide a roadmap for repurposing (44) 10LH to target the PHOX2B antigen displayed by other common HLA alleles that are not part of the A9 CREG, such as HLA-B*14:02. Alternatively, as shown recently for other MHC-I antibodies (44), our structure provides a basis for expanding the scope of peptide targets presented by HLA-A*24:02 through the rational design of the CDR3 loops. Thus, the structural principles established through our study can be leveraged to develop a library of nanomolar-range binders that address multiple tumor-associated peptides and HLA specificities, providing optimal modalities for personalized cancer immunotherapy.
Supplementary Material
Acknowledgments:
We are grateful to Dr. Gino Cingolani for assistance with the crystallographic refinement of the 10LH:PHOX2B/HLA-A*24:02/β2m complex. This research used resources [AMX, 17-ID-1] of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This research also used resource of Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory supported by the DOE, Contract No. DE-AC02-76SF00515.
Funding:
This work was supported in part by a St. Baldrick’s Foundation-Stand Up to Cancer (SU2C) Dream Team Translational Research Grant (SU2C-AACR-DT-27-17). The St. Baldrick’s Foundation collaborates with SU2C. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. SU2C is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research (JMM). This work was also supported by NIH grants U54CA232568 as part of the Beau Biden Cancer Moonshot Program (JMM) and NIH R35CA220500 (JMM), R01AI143997 (NGS), R35GM125034 (NGS), Science Center Quod Erat Demonstrandum (QED) program at Philadelphia Science Center (JMM and MY), Children’s Hospital of Philadelphia Cell and Gene Therapy Collaborative (JMM and MY), Giulio D’Angio Endowed Chair (JMM), and Next Generation T Cell Therapies for Childhood Cancers – Cancer Grand Challenges (JMM and NGS).
Footnotes
Competing interests: GFP, MY, QFM, BRK, MDB, JMM, and NGS are listed as co-inventors in provisional patent applications related to this work. JMM, MY, QFM, NLC, SJ, MDB, and BRK have interests in companies involved in the commercialization of 10LH.
Data and materials availability:
All data are available in the main text or the supplementary materials. Code used for the structure-based prediction of cross-reactive peptides can be accessed via https://zenodo.org/badge/latestdoi/633557366.
References
- 1.Labanieh L, Mackall CL, CAR immune cells: design principles, resistance and the next generation. Nature. 614, 635–648 (2023). [DOI] [PubMed] [Google Scholar]
- 2.June CH, Sadelain M, Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA, Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasquini MC, Hu Z-H, Curran K, Laetsch T, Locke F, Rouce R, Pulsipher MA, Phillips CL, Keating A, Frigault MJ, Salzberg D, Jaglowski S, Sasine JP, Rosenthal J, Ghosh M, Landsburg D, Margossian S, Martin PL, Kamdar MK, Hematti P, Nikiforow S, Turtle C, Perales M-A, Steinert P, Horowitz MM, Moskop A, Pacaud L, Yi L, Chawla R, Bleickardt E, Grupp S, Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 4, 5414–5424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narayan V, Barber-Rotenberg JS, Jung I-Y, Lacey SF, Rech AJ, Davis MM, Hwang W-T, Lal P, Carpenter EL, Maude SL, Plesa G, Vapiwala N, Chew A, Moniak M, Sebro RA, Farwell MD, Marshall A, Gilmore J, Lledo L, Dengel K, Church SE, Hether TD, Xu J, Gohil M, Buckingham TH, Yee SS, Gonzalez VE, Kulikovskaya I, Chen F, Tian L, Tien K, Gladney W, Nobles CL, Raymond HE, Hexner EO, Siegel DL, Bushman FD, June CH, Fraietta JA, Haas NB, PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat. Med. 28, 724–734 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Bufalo F, De Angelis B, Caruana I, Del Baldo G, De Ioris MA, Serra A, Mastronuzzi A, Cefalo MG, Pagliara D, Amicucci M, Li Pira G, Leone G, Bertaina V, Sinibaldi M, Di Cecca S, Guercio M, Abbaszadeh Z, Iaffaldano L, Gunetti M, Iacovelli S, Bugianesi R, Macchia S, Algeri M, Merli P, Galaverna F, Abbas R, Garganese MC, Villani MF, Colafati GS, Bonetti F, Rabusin M, Perruccio K, Folsi V, Quintarelli C, Locatelli F, GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 388, 1284–1295 (2023). [DOI] [PubMed] [Google Scholar]
- 7.Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V, Mancusi R, Geraghty AC, Good Z, Mochizuki AY, Gillespie SM, Toland AMS, Mahdi J, Reschke A, Nie EH, Chau IJ, Rotiroti MC, Mount CW, Baggott C, Mavroukakis S, Egeler E, Moon J, Erickson C, Green S, Kunicki M, Fujimoto M, Ehlinger Z, Reynolds W, Kurra S, Warren KE, Prabhu S, Vogel H, Rasmussen L, Cornell TT, Partap S, Fisher PG, Campen CJ, Filbin MG, Grant G, Sahaf B, Davis KL, Feldman SA, Mackall CL, Monje M, GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 603, 934–941 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearlman AH, Hwang MS, Konig MF, Hsiue EH-C, Douglass J, DiNapoli SR, Mog BJ, Bettegowda C, Pardoll DM, Gabelli SB, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S, Targeting public neoantigens for cancer immunotherapy. Nat. Cancer. 2, 487–497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crews CM, Targeting the Undruggable Proteome: The Small Molecules of My Dreams. Chem. Biol. 17, 551–555 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Souza JES, Galante PAF, de Almeida RVB, da Cunha JPC, Ohara DT, Ohno-Machado L, Old LJ, de Souza SJ, SurfaceomeDB: a cancer-orientated database for genes encoding cell surface proteins. Cancer Immun. 12, 15 (2012). [PMC free article] [PubMed] [Google Scholar]
- 11.Rock KL, Reits E, Neefjes J, Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 37, 724–737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poorebrahim M, Mohammadkhani N, Mahmoudi R, Gholizadeh M, Fakhr E, Cid-Arregui A, TCR-like CARs and TCR-CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther. 28, 581–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, Weiss WA, Neuroblastoma. Nat. Rev. Dis. Primer. 2, 1–21 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Yarmarkovich M, Marshall QF, Warrington JM, Premaratne R, Farrel A, Groff D, Li W, di Marco M, Runbeck E, Truong H, Toor JS, Tripathi S, Nguyen S, Shen H, Noel T, Church NL, Weiner A, Kendsersky N, Martinez D, Weisberg R, Christie M, Eisenlohr L, Bosse KR, Dimitrov DS, Stevanovic S, Sgourakis NG, Kiefel BR, Maris JM, Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature. 599, 477–484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Yin R, Feng BY, Varshney A, Pierce BG, Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. Publ. Protein Soc. 31, e4379 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature. 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudolph MG, Stanfield RL, Wilson IA, How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J, T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Yu N, Ohashi M, Alosco S, Granja C, Salazar M, Hegland J, Yunis E, Accurate typing of HLA-A antigens and analysis of serological deficiencies. Tissue Antigens. 50, 380–386 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M, NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. Baltim. Md 1950. 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noun G, Reboul M, Abastado JP, Kourilsky P, Sigaux F, Pla M, Strong alloantigenicity of the alpha-helices residues of the MHC class I molecule. J. Immunol. Baltim. Md 1950. 161, 148–153 (1998). [PubMed] [Google Scholar]
- 22.Henikoff S, Henikoff JG, Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89, 10915–10919 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krissinel E, Henrick K, Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Papadaki GF, Devlin CA, Danon JN, Young MC, Winters TJ, Burslem GM, Procko E, Sgourakis NG, Xeno interactions between MHC-I proteins and molecular chaperones enable ligand exchange on a broad repertoire of HLA allotypes. Sci. Adv. 9, eade7151 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lever M, Lim H-S, Kruger P, Nguyen J, Trendel N, Abu-Shah E, Maini PK, van der Merwe PA, Dushek O, Architecture of a minimal signaling pathway explains the T-cell response to a 1 million-fold variation in antigen affinity and dose. Proc. Natl. Acad. Sci. 113, E6630–E6638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKeithan TW, Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. 92, 5042–5046 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A, Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature. 375, 148–151 (1995). [DOI] [PubMed] [Google Scholar]
- 28.McAffee DB, O’Dair MK, Lin JJ, Low-Nam ST, Wilhelm KB, Kim S, Morita S, Groves JT, Discrete LAT condensates encode antigen information from single pMHC:TCR binding events. Nat. Commun. 13, 7446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettmann J, Huhn A, Abu Shah E, Kutuzov MA, Wilson DB, Dustin ML, Davis SJ, van der Merwe PA, Dushek O, The discriminatory power of the T cell receptor. eLife. 10, e67092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burton J, Siller-Farfán JA, Pettmann J, Salzer B, Kutuzov M, van der Merwe PA, Dushek O, Inefficient exploitation of accessory receptors reduces the sensitivity of chimeric antigen receptors. Proc. Natl. Acad. Sci. 120, e2216352120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Border EC, Sanderson JP, Weissensteiner T, Gerry AB, Pumphrey NJ, Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: strategy for selection of an optimal candidate. Oncoimmunology. 8, e1532759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferré H, Ruffet E, Blicher T, Sylvester-Hvid C, Nielsen LLB, Hobley TJ, Thomas ORT, Buus S, Purification of correctly oxidized MHC class I heavy-chain molecules under denaturing conditions: A novel strategy exploiting disulfide assisted protein folding. Protein Sci. Publ. Protein Soc. 12, 551–559 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gattiker A, Gasteiger E, Bairoch A, ScanProsite: a reference implementation of a PROSITE scanning tool. Appl. Bioinformatics. 1, 107–108 (2002). [PubMed] [Google Scholar]
- 34.Gupta S, Nerli S, Kandy SK, Mersky GL, Sgourakis NG, HLA3DB: comprehensive annotation of peptide/HLA complexes enables blind structure prediction of T cell epitopes (2023), p. 2023.03.20.533510, doi: 10.1101/2023.03.20.533510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozakov D, Li K, Hall DR, Beglov D, Zheng J, Vakili P, Schueler-Furman O, Paschalidis IC, Clore GM, Vajda S, Encounter complexes and dimensionality reduction in protein–protein association. eLife. 3, e01370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mason D, A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today. 19, 395–404 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Sewell AK, Why must T cells be cross-reactive? Nat. Rev. Immunol. 12, 669–677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin Y, Mariuzza RA, The multiple mechanisms of T cell receptor cross-reactivity. Immunity. 31, 849–851 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, Garcia KC, How a single T cell receptor recognizes both self and foreign MHC. Cell. 129, 135–146 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Mazza C, Auphan-Anezin N, Gregoire C, Guimezanes A, Kellenberger C, Roussel A, Kearney A, van der Merwe PA, Schmitt-Verhulst A-M, Malissen B, How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? EMBO J. 26, 1972–1983 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poole A, Karuppiah V, Hartt A, Haidar JN, Moureau S, Dobrzycki T, Hayes C, Rowley C, Dias J, Harper S, Barnbrook K, Hock M, Coles C, Yang W, Aleksic M, Lin AB, Robinson R, Dukes JD, Liddy N, Van der Kamp M, Plowman GD, Vuidepot A, Cole DK, Whale AD, Chillakuri C, Therapeutic high affinity T cell receptor targeting a KRASG12D cancer neoantigen. Nat. Commun. 13, 5333 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsiue EH-C, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, Paul S, DiNapoli SR, Konig MF, Wang Q, Schaefer A, Miller MS, Skora AD, Azurmendi PA, Murphy MB, Liu Q, Watson E, Li Y, Pardoll DM, Bettegowda C, Papadopoulos N, Kinzler KW, Vogelstein B, Gabelli SB, Zhou S, Targeting a neoantigen derived from a common TP53 mutation. Science. 371, eabc8697 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zinn S, Vazquez-Lombardi R, Zimmermann C, Sapra P, Jermutus L, Christ D, Advances in antibody-based therapy in oncology. Nat. Cancer. 4, 165–180 (2023). [DOI] [PubMed] [Google Scholar]
- 44.Yang X, Nishimiya D, Löchte S, Jude KM, Borowska M, Savvides CS, Dougan M, Su L, Zhao X, Piehler J, Garcia KC, Facile repurposing of peptide–MHC-restricted antibodies for cancer immunotherapy. Nat. Biotechnol, 1–12 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W, XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD, Murshudov GN, Papiz MZ, Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Joosten RP, Long F, Murshudov GN, Perrakis A, The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 1, 213–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bricogne G, Blanc E, Brandl M, Flensberg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart O, Vonrhein C, Womack T, BUSTER version XYZ (2017). [Google Scholar]
- 53.Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D, Protein complex prediction with AlphaFold-Multimer (2022), p. 2021.10.04.463034, doi: 10.1101/2021.10.04.463034. [DOI] [Google Scholar]
- 54.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C, Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 65, 712–725 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, Christie CH, Dalenberg K, Di Costanzo L, Duarte JM, Dutta S, Feng Z, Ganesan S, Goodsell DS, Ghosh S, Green RK, Guranović V, Guzenko D, Hudson BP, Lawson CL, Liang Y, Lowe R, Namkoong H, Peisach E, Persikova I, Randle C, Rose A, Rose Y, Sali A, Segura J, Sekharan M, Shao C, Tao Y-P, Voigt M, Westbrook JD, Young JY, Zardecki C, Zhuravleva M, RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49, D437–D451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gowthaman R, Pierce BG, TCR3d: The T cell receptor structural repertoire database. Bioinforma. Oxf. Engl. 35, 5323–5325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 1–2, 19–25 (2015). [Google Scholar]
- 59.McShan AC, Natarajan K, Kumirov VK, Flores-Solis D, Jiang J, Badstübner M, Toor JS, Bagshaw CR, Kovrigin EL, Margulies DH, Sgourakis NG, Peptide exchange on MHC-I by TAPBPR is driven by a negative allostery release cycle. Nat. Chem. Biol. 14, 811–820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bussi G, Donadio D, Parrinello M, Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Humphrey W, Dalke A, Schulten K, VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 (1996). [DOI] [PubMed] [Google Scholar]
- 62.Schrödinger LLC, The PyMOL Molecular Graphics System, Version 1.8 (2015). [Google Scholar]
- 63.Li H, Natarajan K, Malchiodi EL, Margulies DH, Mariuzza RA, Three-dimensional structure of H-2Dd complexed with an immunodominant peptide from human immunodeficiency virus envelope glycoprotein 120. J. Mol. Biol. 283, 179–191 (1998). [DOI] [PubMed] [Google Scholar]
- 64.Coles CH, Mulvaney RM, Malla S, Walker A, Smith KJ, Lloyd A, Lowe KL, McCully ML, Martinez Hague R, Aleksic M, Harper J, Paston SJ, Donnellan Z, Chester F, Wiederhold K, Robinson RA, Knox A, Stacey AR, Dukes J, Baston E, Griffin S, Jakobsen BK, Vuidepot A, Harper S, TCRs with Distinct Specificity Profiles Use Different Binding Modes to Engage an Identical Peptide–HLA Complex. J. Immunol. 204, 1943–1953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A, ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Overall SA, Toor JS, Hao S, Yarmarkovich M, O’Rourke Sara M., Morozov GI, Nguyen S, Japp AS, Gonzalez N, Moschidi D, Betts MR, Maris JM, Smibert P, Sgourakis NG, High throughput pMHC-I tetramer library production using chaperone-mediated peptide exchange. Nat. Commun. 11, 1909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez-Galarza FF, McCabe A, dos Santos EJM, Jones J, Takeshita L, Ortega-Rivera ND, Cid-Pavon GMD, Ramsbottom K, Ghattaoraya G, Alfirevic A, Middleton D, Jones AR, Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, D783–D788 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE, IPD-IMGT/HLA Database. Nucleic Acids Res. 48, D948–D955 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gragert L, Madbouly A, Freeman J, Maiers M, Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum. Immunol. 74, 1313–1320 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Huang P-S, Ban Y-EA, Richter F, Andre I, Vernon R, Schief WR, Baker D, RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design. PLoS ONE. 6, e24109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y-EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P, ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pei R, Lee J, Shih N-J, Chen M, Terasaki PI, Single human leukocyte antigen flow cytometry beads for accurate identification of human leukocyte antigen antibody specificities. Transplantation. 75, 43 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Wittenbrink N, Herrmann S, Blazquez-Navarro A, Bauer C, Lindberg E, Wolk K, Sabat R, Reinke P, Sawitzki B, Thomusch O, Hugo C, Babel N, Seitz H, Or-Guil M, A novel approach reveals that HLA class 1 single antigen bead-signatures provide a means of high-accuracy pre-transplant risk assessment of acute cellular rejection in renal transplantation. BMC Immunol. 20, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Altman JD, Moss PAH, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM, Phenotypic Analysis of Antigen-Specific T Lymphocytes. Science. 274, 94–96 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Code used for the structure-based prediction of cross-reactive peptides can be accessed via https://zenodo.org/badge/latestdoi/633557366.