

Abstract
Diabetes Mellitus Type 2 (T2D) is an emerging health burden in the USand worldwide, impacting approximately 15% of Americans. Current front-line therapeutics for T2D patients include sulfonylureas that act to reduce A1C and/or fasting blood glucose levels, or Metformin that antagonizes the action of glucagon to reduce hepatic glucose production. Next generation glucomodulatory therapeutics target members of the high-affinity glucose transporter Sodium-Glucose-Linked-Transporter (SGLT) family. SGLT1 is primarily expressed in intestinal epithelium, whose inhibition reduces dietary glucose uptake, whilst SGLT2 is highly expressed in kidney - regulating glucose reabsorption. A number of SGLT2 inhibitors are FDA approved whilst SGLT1 and dual SGLT1 & 2 inhibitor are currently in clinical trials. Here, we discuss and compare SGLT2, SGLT1, and dual inhibitors’ biochemical mechanism and physiological effects.
Keywords: Type-2 Diabetes Mellitus, SGLT, Flozin, Clinical trials
Graphical Abstract
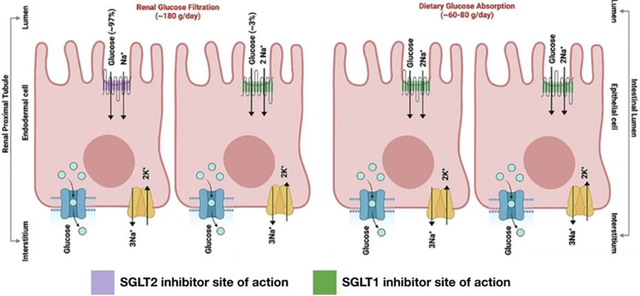
Here the biochemical mechanism and physiological effects of the SGLT2 and SGLT1 selective, and dual SGLT1/2 inhibitors effects are studied and compared to understand their therapeutic potential in treating type-2 Diabetes Mellitus and describe the evolution of advanced therapeutics against SGLTs.
1. Introduction
Diabetes Mellitus (DM) is characterized by pancreatic β-cells insufficiency in either inadequate synthesis and release of insulin (type 1, T1D) or target cells/tissue becoming resistant to glucose uptake driven by insulin (type 2, T2D). Insulin plays a pivotal role in glucose homeostasis by lowering elevated blood glucose to homeostatic levels via stimulating cellular glucose intake in adipose tissue and skeletal muscle fibers and promoting glycogenesis in the liver. However, in diabetics this dysregulation of insulin-to-glucagon ratio leads to hyperglycemia, and advanced-glycation end products that significantly contribute to cardiovascular and renal complications. [1]
Genetic and environmental factors such as obesity, alcohol use and inactivity are driving increasing prevalence of T2D worldwide. [2] Amongst the several strategies proposed for the management of T2D, Metformin emerged as one of the leading medications and acts by sensitizing cells to insulin and preventing gluconeogenesis in the liver. [3] Some 150 million people worldwide are prescribed Metformin, including 45 to 55% of diabetic patients (with no contraindications), and often prescribed in combination with other therapeutics dictated by patient-specific physiology and regimen. [4] Complimentarily, there are many other classes of anti-diabetes medications that treat T2D with varied mechanisms: preventing glucose secretion from liver cells, stimulating the release of insulin from pancreatic β cells, sensitizing to current insulin, inhibiting Dipeptidyl peptidase-4 (DPP-4), stimulating Glucagon-like peptide-1 (GLP-1) signaling to modulate digestion or glucose reabsorption in the kidneys. [5] Herein we discuss these, and their related mechanisms of action.
Sodium-Glucose-Linked-Transporters (SGLT) function as plasma membrane sodium and glucose symporters involved in glucose uptake from the lumen into the intestinal epithelium, in and renal nephrons. SGLT1 is primarily expressed in the epithelial lining of the intestines while SGLT2 is primarily located in the lining of the nephron in the kidneys. SGLT inhibitors have become popular second-line treatments for T2D management due to their glucomodulatory, reno-protective and cardiovascular effects. [6] Additionally, approximately 94% of T2D patients develop at least one cardiovascular, metabolic or renal comorbidity with traditional medications, further promoting the use of SGLT inhibitors. [7] Some current SGLT2 inhibitors include Canagliflozin, Dapagliflozin, and Empagliflozin, which decrease renal glucose reabsorption and consequently increase excretion in the urine. [8] More recently, SGLT2 has been studied at a biochemical/molecular level, elucidating its structure, analyzing interactions with inhibitors and other proteins. [9] Although SGLT2 inhibitors are an established therapy for diabetic patients with cardiovascular complications and diabetic nephropathy, their use has been associated with a higher risk for urinary tract infections, fractures, and limb amputations. [10, 11] SGLT1 inhibition presents itself as a potential alternative given its intestinal prevalence and prevention of dietary glucose uptake with a number of drugs such as LX2761, TP0438836, and Mizagliflozin in development. Here we will begin by discussing the benefits of targeting each SGLT receptor with respect to inhibition mechanisms and their physiologic effects.
2. Background
2.1. Biological significance of SGLT1/2
The expression profiles of SGLT1 and SGLT2 isoforms are crucial in understanding their physiological differences. SGLTs are expressed on the apical membrane, bringing sodium ions down their concentration gradient into the epithelial cell and allowing the glucose molecule to enter the cell against its concentration gradient. [12]
2.1.1. Role in renal glucose transport
In the kidney, approximately 900 mmol of glucose per day is reabsorbed in the proximal convoluted tubule of the nephron in healthy adults. [13] 97% of this reabsorption is mediated by SGLT2 which appears in the early S1/S2 segments of the tubule. SGLT1 is more predominant in the late S2/S3 segments and accounts for approximately 3% reabsorption. [14] For SGLT to function in transporting glucose, active transport of Na+ ions out of the tubule lining is necessary. This transport is driven by basolateral Na+/K+-ATPase, bringing 3 Na+ out of the tubule into the interstitium and 2 K+ into the tubule from the interstitium, while keeping Na+ at a low intracellular concentration. [15] Therefore, when glucose is present in the lumen of the nephron, SGLT2 transporters on the apical membrane function as symporters where they adopt different outward and inward conformations to take one Na+ and glucose from the lumen and bring them inside the tubule cell (Figure 1). Overall, the glucose reabsorption machinery in kidneys is not saturated under normal conditions. The glucose concentration in plasma at which the filtered glucose load reaches the Tmax (2.08 mmol/min) is known as the theoretical plasma glucose threshold. However, glucosuria begins at ~10 mmol plasma glucose concentration in healthy individuals and this deviation of the “actual threshold” from the “theoretical threshold” is due to the nonlinear transition in glucose reabsorption and excretion curves as the Tmax is approached. This “curve rounding” have been explained by the Tmax heterogeneity of individual nephrons. As such, low levels of glucose start getting excreted into the urine at modestly elevated glucose levels (10–11 mmol) in the plasma of healthy adults and increase linearly when glucose levels exceed the 15–16 mmol concentration range. However, in conditions where the GFR is diminished such as kidney diseases, glucosuria may require high plasma glucose levels whereas lower plasma glucose levels are needed when GFR is increased as during pregnancy or Diabetes. As with most transporters, SGLTs also reach their maximum renal transport at concentrations of 200 mg dL−1 above which glucose can be found in the urine (glycosuria). [16] In diabetes, the renal transport maximum of SGLTs increases due to an unknown mechanism, and SGLT mRNA is affected: in rodent models, both SGLT1 and 2 expression increases while in human T2D patients, only SGLT1 significantly increases. [17, 18] In either case, the natural physiological response is to overexpress glucose transporters due to the tissue receiving low levels of glucose. Therefore, studies knocking out SGLT2 (the predominant SGLT transporter in the kidneys) or employing an SGLT2 inhibitor result in significantly smaller a fraction of glucose reabsorbed mitigating hypergylcemia. Complementing physiologic transport studies, recent protein structure determination via NMR and cryo-EM in conjugation with computational homology modeling has allowed visualization of the SGLT1/2 structure, how macromolecules and ions bind to them, understanding the residues implicated and intra and intermolecular interactions, and domain-specific conformational changes. [19, 20]
Figure 1. Cellular localization and biological activity of SGLT1 and SGLT2 in sodium-glucose co-transport.

SGLT1 (green) along with SGLT2 (purple) localized on the luminal side of the renal proximal tubule, and mainly SGLT1 on epithelial cells of the small intestine shown schematically. GLUT transporters (teal) and Sodium-Potassium pumps (yellow) are localized on the interstitium side and mainly assist in the transport of glucose and sodium-potassium ions across the renal cells and intestinal epithelial cells. The overall glucose absorbed is ~180 g day−1 in kidneys and ~60–80 g day-1.
2.1.2. Role in gastrointestinal glucose transport
While SGLT2 is primarily expressed in renal proximal tubule cells, SGLT1 transporters are more widely expressed in the intestinal epithelium, heart, central nervous system (CNS), kidneys, and other tissues. [21] The intestinal epithelial cells (enterocytes) that line the lumen allow for the transport of glucose after they have been broken down into monomeric units along the alimentary canal. These enterocytes, like nephron epithelium, also have an apical membrane facing the lumen where the SGLT1 receptor is localized, and a basolateral membrane facing the circulation (Figure 1). Dietary glucose (200–300 grams day−1) is drawn into the enterocyte with two Na+ ions going down their concentration gradient (Figure 1). Glucose transporters (GLUT) located on the basolateral membranes are directly involved in glucose transport into the blood (Figure 1). [22] This passive transporter, specifically GLUT2 found on pancreatic, intestinal, liver, and kidney cells work with high KM, allowing it to act as a glucose sensor for managing the release of blood glucose modulatory molecules such as insulin. [23] Intestinal glucose transport also mediates intestinal hormone secretion that regulates blood glucose concentration, including GLP-1 and glucose-dependent insulinotropic peptide (GIP). [24] These incretin hormones are released after nutrient intake and are mainly involved in modulating the release of insulin. All this works in concert with the SGLT1 receptor, as supported by the knockout studies which show that eliminating the SGLT1 (SLC5A1) gene results in lower levels of these incretin hormones and GLUT2 expression. [25]
There are many different gastrointestinal hormones released by distinct types of cells. Cholecystokinin (CCK) and GIP are released by I and K cells in the proximal gut respectively while Oxyntomodulin (OXM), GLP-1, and Peptide YY (PYY) are released by L cells in the distal gut. [26] These hormones play a preventative role in the progression of diabetes by indirectly managing blood glucose as well as alimentary canal signaling that manages digestion. Through SGLT1 modulation, diabetes can be treated not only by preventing dietary absorption but also by regulating hormones that promote satiety in diabetic patients. GLP-1 functions in decreasing pancreatic α-cell glucagon secretion, a hormone responsible for promoting glycogenolysis in the liver and releasing glucose into the bloodstream. [27] GLP-1 modulation plays a pivotal role in the SGLT1 inhibition pathway, and GLP-1 receptor agonists that do not function directly through an SGLT1 mechanism have already been approved by the FDA for T2D treatment. Sustained GLP-1 release via SGLT1 inhibition arises when glucose continues down the alimentary canal to later intestinal segments where the gut contains bacteria that metabolize glucose and release short-chain fatty acids (SCFAs) that trigger GLP-1 release. [25, 28] This allows for a different hyperglycemia inhibition pathway as consumed carbohydrates can directly be prevented from entering the bloodstream and excreted in the stool. Small intestines are largely responsible for the absorption of nutrients, and this is mainly mediated through Brush Border Enzymes (BBE) that function in digestion.
2.2. Biological activity of SGLT1/2/Dual inhibitors
2.2.1. History of SGLT inhibitors
In 1885, von Mering published a Phlorizin molecule isolated from the root bark of apple trees that causes glycuresis and diuresis in human patients, suggesting application in nephritis treatment. [29] [30] In 1962, Phlorizin was shown to inhibit sugar transport in kidneys and small intestines. [31] A follow up showed that Phlorizin acts at the level of the proximal convoluted tubule for renal reabsorption of the luminal glucose. [32] In vivo studies of diabetic rats found that Phlorizin normalizes blood glucose and promotes insulin sensitivity for peripheral tissue glucose uptake, suggest potential use as a treatment for hyperglycemia. [33] However, concerns with solubility, low bioavailability, and unselective isoform binding, limiting development (Table 1). [34] Further, T-1095 was a synthetic Phlorizin derivative that had fewer problems in development but still faced problems with isoform selectivity (Table 1). [35] Subsequent targeting focussed on C-glucoside-containing molecules for SGLT2 inhibition as opposed to O-glucosides (Figure 2). This led to increasing the half-life of novel selective SGLT2 inhibitors, evading degradation from β-glucosidase enzymes that mainly target the O-glucosides. [36] Additionally, there were notable differences in selecting SGLT2 over SGLT1 (2700:1 for Empagliflozin), leading to the desired renal effects (Table 1). [37] Other drugs that fall under this selective SGLT2 inhibition category are Canagliflozin, Dapagliflozin, and Ertugliflozin. The three approved by the FDA include Canagliflozin, Dapagliflozin, and Empagliflozin, and combination drugs with Metformin exist. [38] Recently, dual inhibitors as well as selective SGLT1 inhibitors are being investigated. Sotagliflozin is a dual inhibitor that has been observed in lowering glucose and is in phase 3 clinical trials for T2D. Due to the broad expression profile of SGLT1, Sotagliflozin exhibits many cardiovascular, renal, and intestinal functions. Adverse effects include diarrhea, which is similar to α-glucosidase inhibitors (AGIs). Mizagliflozin is a selective SGLT1 inhibitor currently being investigated as a non-absorbable, rapidly degrading molecule that can also potentially improve chronic constipation. [39, 40] The beneficial effects include modulation of GLP-1 and other incretin hormones that remain crucial in managing glucose concentrations. As the SGLT inhibition field grows and more clinical trials are performed, the advantages of SGLT isoform inhibition are slowly being unveiled and adverse effects are better understood.
Table 1.
Summary of binding affinities for SGLT inhibitors.[144]
| Molecule | Classification | SGLT1 IC50 (nM) | SGLT2 IC50 (nM) | SGLT2/1 Selectivity ratio | Structure |
|---|---|---|---|---|---|
|
| |||||
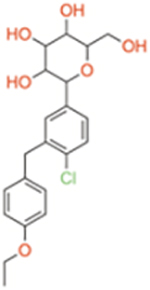
| Ertugliflozin | Selective SGLT2 | ~1960 | ~0.877 | ~2235 |
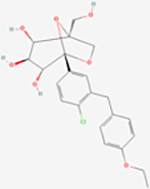
|
|
| |||||
| Empagliflozin | Selective SGLT2 | ~8300 | ~3.1 | ~2700 |
|
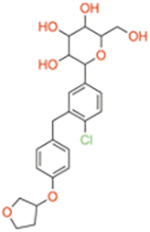
| Dapagliflozin | Selective SGLT2 | ~1400 | ~1.2 | ~1200 |
|
| Canagliflozin | Selective SGLT2 | ~710 | ~2.7 | ~260 |
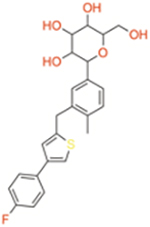
|
| Sotagliflozin | Dual SGLT1/2 | ~36 | ~1.8 | ~20 |
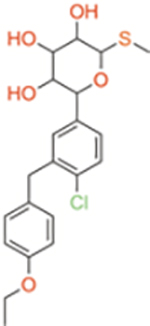
|
| Phlorizin | Dual SGLT1/2 | ~400 | ~65 | ~6.2 |
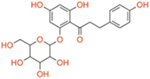
|
| T-1095 | Dual SGLT1/2 | ~200 | ~50 | ~4 |
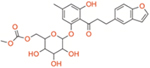
|
| LX2761 | Selective SGLT1 | ~2.2 | ~2.7 | ~0.8 |
|
| TP0438836 | Selective SGLT1 | ~7 | ~28 | ~0.25 |
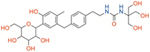
|
| Mizagliflozin | Selective SGLT1 | ~27 | ~8170 | ~0.003 |
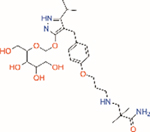
|
Figure 2. Chemical and functional evolution of the SGLT inhibitors.

Phlorizin is a natural O-glucoside that was shown to inhibit sugar transport in kidneys and small intestine in 1962. Since then, the molecules used to treat T2D have faced evolution in structure and function to avoid β-glucosidase action, for better selectivity towards SGLT isoforms, and enhance the number of beneficial side effects. The merits (green boxes) and disadvantages (red boxes) of respective classes of molecules.
2.2.2. SGLT2 inhibitors
Canagliflozin, Empagliflozin, Dapagliflozin and Ertugliflozin are have extensive preclinical and clinical research and are currently, FDA approved as a T2D treatment, branded as Invokana, Jardiance, Farxiga, and Steglatro. Canagliflozin was originally discovered by a research group in Japan investigating the transition to C-glucosides for SGLT2 inhibition after it was found that O-glucosides were susceptible to hydrolysis via β-glucosidase in the intestine. [41] In that study, many different heteroatom rings and substituent groups were added to a phenyl molecule attached to glucose at C1. [41] The IC50s of these molecules were used to determine their inhibitory activities against the human SGLT2 receptor, and urinary glucose excretion (UGE) in male Sprague-Dawley (SD) rats to understand the extent of their biological glycosuria effect. Results showed that a phenylthiophene derivative had the lowest IC50 and further modifications led to a fluorine substituent attachment, reporting 2.2 nM IC50 on hSGLT2, 910 nM on hSGLT1 (Table 1). [41] Dapagliflozin is a C-aryl glucoside discovered similarly through testing different R substituent groups and finding an EC50 of 1.1 nM to hSGLT2 and 1390 nM to hSGLT1, showing approximately 1200-fold selectivity for SGLT2 than SGLT1 in comparison to Canagliflozin having a 260-fold selectivity (Table 1). [42] Empagliflozin was discovered by Boehringer Ingelheim using similar methods of C-glucoside variation and was reported to have even higher SGLT2 selectivity (2700-fold). [43] Ertugliflozin falls in a novel subclass of SGLT2 selective inhibitors integrating a unique dioxa-bicyclo [3.2.1] octane (bridged ketal) ring system. Ertugliflozin exhibits high selectivity (>2000 fold) for SGLT2 over SGLT1 and demonstrates an IC50 of 0.877 nM for human SGLT2 while 1960 nM for human SGLT1. Thus, Ertugliflozin and Empagliflozin show the highest selectivity for SGLT2 over SGLT1 (> 2000-fold) in comparison with other SGLT2 selective inhibitors. [44] Further studies into SGLT2 inhibitors showed a significant blood glucose reduction after injection in hyperglycemic ZDF rats and very slight blood glucose changes against normoglycemic rats, avoiding potential hypoglycemic complications. [41, 45, 46] Further solidifying SGLT2 inhibition as a major diabetic medication target is its high oral availability of 65%, 78%, 78%, and 100% for Canagliflozin, Dapagliflozin, Empagliflozin, and Ertugliflozin respectively as well as rapid absorption and > 10 hour half-lives, unlike the initial SGLT inhibitor such as Phlorizin. [47–49] In addition to the renal SGLT2 binding, intestinal SGLT1 inhibition is seen in Canagliflozin clinical trials, and this is likely since Canagliflozin, despite being classified as a highly selective SGLT2 inhibitor, still has a higher affinity (approximately 650 +/− 150 nM) than Dapagliflozin and Empagliflozin (approximately 1400 and 8300 nM respectively), allowing for the physiological effects of SGLT inhibition to arise (Table 1). One clinical trial (NCT01173549) investigated intestinal absorption of glucose as well as incretin GIP, PYY, and GLP-1 levels, finding along with increased UGE, there was 31% reduced intestinal absorption at 1 hour and 20% at 2 hours post-meal, 50% reduction in GIP, 60% PYY increase, and 35% higher GLP-1 (Figure 3). [50] The study was able to conclude that an SGLT1 intestinal inhibition mechanism is an additional component of Canagliflozin’s success as a therapeutic.
Figure 3. Biological activity of SGLT1 and SGLT2 inhibitors.

The major mechanism of action and related physiological effects (yellow box), the beneficial side effects (green box), and the adverse side effects (red box) of the SGLT1 and SGLT2 selective inhibitors.
Clinical trials were performed to determine beneficial side effects which mainly consisted of ameliorating cardiovascular disease and improving renal function (Figure 3). The Canagliflozin Treatment and Trial Analysis (CANTATA) studies and other trials studying Dapagliflozin, Empagliflozin, and Ertugliflozin found that there was a significant reduction in hemoglobin A1c levels following treatment, showing proof of blood glucose clearance. [51–53] To understand the cardiovascular effects, the Canagliflozin Cardiovascular Assessment Study (CANVAS) program, the EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) trial, and the DECLARE-TIMI 58 (Dapagliflozin Effects on Cardiovascular Events) were the largest clinical trials held. [54–56] CANVAS found that with Canagliflozin, there was a 14% reduction in major adverse cardiac events (MACE) which includes cardiovascular-related death, non-fatal myocardial infarction and stroke, and heart failure hospitalization risk was reduced by 33%. [54] EMPA-REG Empagliflozin trials also found a 14% reduction in MACE and a 35% heart failure hospitalization risk reduction, which was similar to the cardiovascular benefits of Canagliflozin. [55] On the other hand, the DELCARE-TIMI 58 trials found MACE rates of 8.8% in the Dapagliflozin group vs. 9.8% in the placebo (HR of 0.93). [93] These results concluded that Dapagliflozin does not result in a statistically significant lower chance of MACE compared to placebo and is associated with only a 17% cardiovascular risk reduction. [56] A reduction of systolic blood pressure has been observed in patients treated with SGLT2 inhibitors by approximately 3.93 mmHg for Canagliflozin, 2.7 mmHg for Dapagliflozin, and 3.44 mmHg for 10mg Empagliflozin and 4.16 mmHg for 25mg. [54, 56, 57] This effect is likely due to the increased UGE and natriuresis resulting in osmotic diuresis and reduced intravascular volume when higher osmotic pressure in the filtrate leads to water being drawn into the lumen of the nephron for excretion. [58] In addition to the direct glucose-lowering effect, SGLT2 inhibitors have been observed to play a role in raising high density lipids (HDLs) levels, lowering triglyceride levels, and decreasing body weight. [59] A more recent study performed in mice attributed these changes in body fat to the upregulation of crucial mitochondrial genes for fatty acid oxidation via a Peroxisome proliferator-activated receptor α (PPARα) mechanism in the liver. [60] The increase in fatty acid oxidation is associated with the generation of ketone bodies to sustain the energy demands of cardiac muscle. This makes SGLT2 inhibitors effective in T2D patients with cardiovascular comorbidities. [61] The renal benefits of these inhibitors arise from significant reductions in albuminuria progression and slowed glomerular filtration rate (GFR). [54–56] The glomerulus functions in separating large proteins such as albumin from the filtrate that continues through the nephron to produce urine. Both albumin presence in the urine, as well as reduced filtration rate due to decreased glomerular pressure, are signs of kidney damage, and SGLT2 inhibitors likely improve kidney function through an anti-inflammatory mechanism, lowering Tumor necrosis factor (TNF) receptor 1 and Interleukin 6 (IL6) levels in the plasma. [62] However, the mechanisms through which they exert their effects are not fully understood.
Adverse side effects of SGLT2 inhibitors include female genital mycotic infections (GMI), urinary tract infections (UTI), and abnormalities in volume depletion (Figure 3). [63] In randomized controlled clinical trials, there is a notable 3 to 4-fold increase in GMIs, especially in females, with 13.6%, 10.6%, 8.4%, and 5.4% incidences in low-dose Ertugliflozin, Canagliflozin, Dapagliflozin, and Empagliflozin groups, respectively. [64] This is because pharmacologically induced glucosuria creates warm and moist genitalia environments in which bacteria and fungi can grow. More specifically, studies have found that Candida glabrata were mostly present in mild to moderate vulvovaginal candidiasis, and the excessive presence of glucose from glucosuria causes the fungus to secrete a protein that results in vaginal epithelial cells adhesion and impaired phagocytic recognition. [65] However, it is unknown why Canagliflozin has a notably higher incidence of female GMIs. A similar mechanism occurs in males taking SGLT2 inhibitors which results in UTIs from Candida albicans when the natural synthesis of a glucose-inducible protein allows for the fungus to adhere to uroepithelium. [66] Meta-analysis data suggests a 6.8% incidence of UTIs in patients consuming Dapagliflozin vs. a 5.3% incidence in the placebo group. [67] A 6.0% incidence of UTIs was observed in patients taking Canagliflozin vs. 5.4% in the placebo group. A 13.5% incidence of UTIs in patients taking Empagliflozin vs. 14.7% in the placebo group was reported. A 6.3% incidence of UTI was observed in patients consuming Ertugliflozin in comparison to a 6.4% in the placebo group. [68] Dapagliflozin was the only drug associated with UTIs while the other SGLT2 inhibitors only showed increased risk. These infections were quickly treated with antifungals and rarely led to severe sepsis. Issues with volume depletion arise from osmotic diuresis since increased UGE results in increased water excretion. CANVAS trials for Canagliflozin found significantly higher rates of volume depletion events with 26.0 events per 1000 patients compared to 18.5 events in the placebo group, while the other drugs have similar adverse events compared to the placebo. [54]
Despite the renoprotective effects, less common side effects of SGLT2 inhibitors, in general, include acute renal failure (ARF), diabetic ketoacidosis (DKA), fractures, and amputations (Figure 3). An analysis of the FDA adverse event report system (FAERS) database indicates that SGLT2 inhibitors are associated with 6.5% of the reported adverse event cases. [69] Canagliflozin specifically was linked to ARF 7.3% of the time, which was significantly greater than other FDA-approved Empagliflozin and Dapagliflozin SGLT2 inhibitors (4.7% and 4.8% respectively). [69] While the exact mechanism for ARF from Canagliflozin and SGLT2 inhibitors is unknown, a combination of reduced glomerular pressure, volume depletion, and increased medullary oxygen consumption are potential reasons to believe that hypoxic kidney damage occurs more often with these drugs. [70] Additionally, increased plasma erythropoietin and reticulocytosis indicate hypoxia, especially in the medulla of the kidney, as a result of SGLT2 inhibition. [71] A more indirect mechanism consists of downstream renal SGLT1. When renal SGLT2 inhibition occurs, renal SGLT1 facilitates the transport of glucose at a higher frequency and activates the aldose reductase gene, making sorbitol and fructose. Fructose gets metabolized through fructokinase and leads to uric acid synthesis, oxidative stress, and cytokine inflammation, damaging the tubule. [72] While it is unclear why Canagliflozin causes ARF significantly higher than other FDA-approved SGLT2 inhibitors, and all inhibitors are being monitored by the FDA for these effects. The risk of DKA when taking SGLT2 inhibitors is warned by the FDA. In a systematic review of published case reports, euglycemic DKA was observed more commonly in Canagliflozin patients than in patients taking other FDA-approved SGLT2 inhibitors. [73] While the exact mechanism behind this is unclear, it may be due to Canagliflozin’s ability to simultaneously increase glucagon secretion causing gluconeogenesis while increasing UGE, resulting in the excessive formation of ketones. [74] In addition to this, it is shown that the excessive reabsorption of ketone bodies from the kidney is likely because of the positive electrochemical gradient created when SGLT2 is inhibited. [75] Increased fractures have only been observed in the CANVAS trials, with a balanced spread of upper and lower limb fractures occurring in 4% of patients taking Canagliflozin vs. 2.6% of those taking placebo. [76] Other Canagliflozin clinical trials have not noted significant increases in fractures and neither do other SGLT2 inhibitors. This is indicated through increased levels of type 1 β-carboxytelopeptide and osteocalcin which are resorption and formation markers, respectively, and a statistically significant reduction in hip bone mineral density. [77] When sodium does not undergo facilitated transport through SGLT, the excess luminal sodium increases the activity of sodium-dependent phosphate cotransporters 2A and 2C around the same proximal renal tubule area, allowing for increased reabsorption of phosphate. This leads to increased FGF23 protein followed by decreased expression of a crucial enzyme responsible for converting Vitamin D to its active form. Without this, calcium is not absorbed in the gastrointestinal (GI) tract and parathyroid hormone (PTH) is secreted, which degrades the bone matrix to increase the plasma calcium. [78]
Amputations are controversial adverse effects associated with SGLT2 inhibitors. They occur significantly more per 1000 patients in CANVAS trials with the worst amputations occurring at the level of toes and metatarsal. [54] Similar to fractures, this occurs only in Canagliflozin with significance. Although the exact mechanism for this is not known, it is speculated that the hypovolumia side effect of SGLT2 inhibitors may contribute to decreased perfusion to the lower extremity. [79] Other mechanisms when considering amputations as a potential side effect of all SGLT2 inhibitors (since there is not enough data to determine whether it is unique to Canagliflozin) include increased blood viscosity and a link between diuretics and amputation, both major contributors to the deterioration of tissue, ischemia and limb salvage. [80, 81] When considering amputations as a Canagliflozin concern, differences between Canagliflozin and Dapagliflozin/Empagliflozin include more SGLT1 selectivity as previously mentioned, and complex 1 inhibition through AMPK activation. [82] This leads to greater AMP and ADP ratios and subsequently lowers oxygen uptake in cells. Additionally, Canagliflozin has the potential to inhibit endothelial cell proliferation contributing to peripheral artery disease, limb ischemia, and subsequently amputations. [83] Therefore, physicians must be aware of these and other adverse effects as well as more recent meta-analyses that have studied large scale Canagliflozin use. [84]
2.2.3. SGLT1 inhibitors
Selective SGLT1 inhibition shows a lot of promise but have lagged behind SGLT2 inhibitors. The reduced risk of MACE and heart failure in general from the use of SGLT2 selective inhibitors is mainly in patients with established atherosclerotic disease and does not necessarily address diabetics with multiple risk factors. In addition to this, inhibition of SGLT1 presents a strategy of preventing dietary glucose from being absorbed in the intestines before the glucose makes its way to the kidneys, providing the opportunity to prevent the SGLT2 adverse effects. Current SGLT1 selective inhibitors in development are Mizagliflozin (DSP-3235, KGA-3235 or GSK-1614235 alias names), KGA-2727, LX2761, and TP0438836. KGA-2727 was first developed in 2012 as the first selective SGLT1 inhibitor with a pyrazole-O-glucoside structure by adding different R substituent groups onto the O-glucoside. [85] The study calculated inhibitory constants as 97.4 nM and 13600 nM for hSGLT1 and hSGLT2 respectively, which was more selective for SGLT1 than previously described Phlorizin. LX2761 was discovered as a C-glucoside that mimicked the structure of Sotagliflozin, a dual SGLT1/2 inhibitor, and IC50s were screened for greater SGLT1 selectivity. [86] The resulting LX2761 structure had a 2.2 nM and 2.7 nM IC50 toward hSGLT1 and hSGLT2, respectively (Table 1). TP0438836 is a C-phenyl D-glucitol derivative with 7 nM and 28 nM IC50 toward hSGLT1 and hSGLT2 respectively (Table 1). [87] GSK-1614235 was developed as an analog to KGA-2727 with the addition of two methyl groups on the carbon before the terminal amide and has inhibitory constants of 27 nM and 8170 nM for hSGLT1 and hSGLT2 respectively, making the drug more selective for SGLT1 to be used in clinical trials (Table 1). [88] Glucose/fructose UV testing was performed with rat small intestines to confirm the mechanism for SGLT1 inhibitors in preventing glucose absorption, finding a significantly decreased absorption rate than Phlorizin and control at two different KGA-2727 concentrations. [85] LX2761 studies tested cecal (large intestine) glucose levels in mice as well as blood glucose levels, finding a significant increase in the amount of intestinal glucose passing through the small intestines into the large intestines and a decrease in blood glucose 1 hour after a meal challenge. [86] Rat studies with TP0438836 show decreasing plasma glucose dose-dependently 120 minutes after an oral glucose test. [87] GSK-1614235 resulted in reduced glucose absorption when given to patients before rather than 30 minutes after a meal. [88] Rodent models further confirmed the incretin mechanism of SGLT1 selective inhibitors with KGA-2727 reporting increased plasma GLP-1 dose-dependently in ZDF mice, GSK-1614235 finding increased GLP-1 and PYY in humans, and LX2761 showing higher total GLP-1 with low cecal pH to indicate the production of SCFA as proof of concept. [85, 86, 88] TP0438836 is a relatively new molecule with no incretin effect studies. Along with Canagliflozin and other dual inhibitors, these SGLT1 selective inhibitors exert incretin molecule effects that are crucial in managing diabetes and present a potential avenue for continued development.
While numerous SGLT2 inhibitors have been or are currently being clinically evaluated, there are significantly fewer trials of selective SGLT1 inhibitors. One crossover study administered GSK-1614235 (selective SGLT1 inhibitor) pre- or post-meal vs. placebo to 12 healthy volunteers to evaluate its effects on glucose absorption and GIP secretion (NCT01607385). [88] In addition to pre-meal dosing of GSK-1614235 producing delayed and reduced intestinal glucose absorption, the selective SGTL1 inhibitor also elevated GIP for 4 hours post-meal. However, some participants reported mild gastrointestinal symptoms: diarrhea (n=4), abdominal pain (n=1), and flatulence (n=1). The mechanism for diarrhea in SGLT1 selective inhibition involves the increase in osmotic pressure due to high amounts of cecal glucose and short chain fatty acids (SCFA), resulting in water being drawn into the large intestines. However, further studies with larger numbers of healthy patients are needed to know certain GI effects. The diarrhea side effect seen with GSK-1614235 has recently led to the repurposing of the drug for chronic constipation under the name Mizagliflozin. [89] Its effect in increasing plasma GLP-1 levels is being used to alleviate abdominal pain and has potential application in patients with irritable bowel syndrome. Despite the low bioavailability of Mizagliflozin, the field of SGLT1 selective inhibitors is mainly centered around the discovery of more stable molecules and trying to understand all the potential benefits and risks of targeting the receptor. [39]
The benefits of SGLT1 inhibition mainly revolve around the modulation of postprandial glucose absorption and incretin hormone levels such as GLP-1, PYY, and GIP (Figure 3). Promoting satiety in the individual and stimulating insulin secretion after a meal is crucial in diabetes management long-term. Success in doing this has been seen with Canagliflozin which, unlike other SGLT2 selective inhibitors, is still strong in SGLT1 binding. Renal SGLT1 inhibition can also prevent the nephrotoxic effects of acute kidney injury (AKI) with SGLT2 selective inhibitors by preventing the fructose metabolism pathway previously mentioned. [72] The sodium myoinositol cotransporter 1 (SMIT1) has been linked to SGLT1, and this interaction contributes to the production of reactive oxygen species (ROS) in myocardial cells under hyperglycemic conditions. [90] Both SMIT1 and SGLT1 are expressed in the heart, and the import of glucose under hyperglycemic conditions can lead to ischemic cell death and heart failure. SGLT1 inhibitors show potential in preventing type 2 DM-related heart failure. [91–93] Potential neuronal benefits to inhibiting SGLT1 has been observed in slowing the development of cerebral ischemic neuronal damage. [94] When sodium rushes into the cell through widely expressed SGLT1 receptors in neurons and myocardial cells, an accumulation of calcium ions can result in mitochondrial dysfunction in those cells. In hyperglycemia, more sodium is going down its concentration gradient and glucose follows going into the target cell, making SGLT1 inhibition a potential avenue to prevent myocardial and cerebral ischemia. In tumor growth, overexpression of SGLT1 expression allows tumor cells to proliferate and leads to poor prognosis in ovarian carcinoma and other types of cancer, giving another example of how SGLT1 inhibition can be beneficial outside of diabetes. [95] Finally, SGLT1 plays a role in the expression of matrix metalloproteinases during hyperglycemic states in human cardiac fibroblasts. Meng et al. found that SGLT1 inhibitors significantly reduce the expression of matrix metalloproteinases and decrease the risk of diabetic cardiomyopathy (Figure 3). [96] These beneficial effects for patients with potential cardiovascular and renal comorbidities make SGLT1 inhibition as promising as SGLT2 inhibition with the added benefit.
Problems that arise when focusing on SGLT1 inhibition mainly consist of the wide expression profile leading to energy-deficient neurons and myocytes, especially in hypoglycemic or hypoxic states. [97] Osmotic diarrhea has already been observed in the Mizagliflozin clinical trial and is a concern due to potential hypotensive and volume depletion-related events. This is similar to the potential adverse effects of SGLT2 selective inhibitors since both involve the excretion of glucose. Although SGLT1 inhibitors have hardly been assessed, the volume depletion events can be seen with SGLT2 inhibitors. Canagliflozin, which has been previously shown to exhibit some SGLT1 inhibitory activity, had significant occurrences of osmotic diuresis (34.5% vs 13.3% in drug vs placebo) and volume depletion events (26.0% vs 18.5%) whereas Empagliflozin did not in the EMPA-REG OUTCOME trial. [54, 55] Notably, SGLT1 deficiency has been linked to elevated bacterial loads, and this is either due to increased glucose moving to the large intestines allowing gut microbiome proliferation, or changes in immune cells such as macrophages impairing the immune system (Figure 3). [98] Finally, SGLT1 has been observed to function in enteric inflammation, specifically by preventing damage from toll-like receptors (TCRs), and in the intestinal epithelium, glucose inhibits cell death by Nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB). [99] Until more SGLT1 selective inhibitors go into clinical trials and more data is retrieved, the potential mechanisms and beneficial/adverse effects remain a mystery.
2.2.4. Dual SGLT1/2 inhibitors
Recently, there has been a push for the application of dual SGLT1/2 inhibitors, which are typically only slightly more selective for SGLT2. After the discovery of Phlorizin and T-1095, there was proof of concept for medications that induce glycosuria as well as increased incretin hormone responses. However, when these drugs failed due to poor selectivity and bioavailability, there was a switch toward only inhibiting SGLT2. Today, with the development of selective SGLT1 inhibitors, dual inhibitors controlling the selectivity for either receptor carve the path to mitigate the detrimental side effects of inhibiting each receptor. The most developed and studied SGLT1/2 dual inhibitor currently being investigated is Sotagliflozin, also known as LX4211. The drug was developed by Lexicon Pharmaceuticals and is derived from an L-xylose instead of the classic D-glucose as seen in the other SGLT inhibitors in hopes of preventing degradation and cross-reactivity with other glucose-binding enzymes. [100] Through a series of different aromatic substitutions, the Sotagliflozin molecule was found to have IC50s of 36 nM and 1.8 nM to hSGLT1 and hSGLT2 respectively (Table 1). [101] In vivo studies to determine the SGLT1 and SGLT2 mechanisms of Sotagliflozin, found significantly greater GLP-1 levels, PYY levels, and cecal glucose levels as well as lower cecal pH, supporting the SGLT1 inhibition mechanism. Additionally, 24 hour UGE studies in mice found 696 +/− 103 mg day−1 after LX4211 administration compared with 1.1 +/− 0.3 mg day−1 measured before the drug. [101]
Clinical trials with Sotagliflozin show promising results that suggest interplay from both SGLT1 and SGLT2 inhibition. A recent meta-analysis of these trials shows 0.42% reduced hemoglobin A1c levels vs placebo groups in nine pooled studies. [102] Changes in body weight were seen by an average of 1.33 kg vs placebo groups. Reductions in systolic and diastolic were 2.4 mmHg and 0.81 mmHg, respectively, and this was not significantly different from the selective SGLT2 inhibitor Empagliflozin. Although Sotagliflozin showed a 5% reduction in the incidence of hypoglycemia (OR 0.95), a 6% increase in the incidence of UTIs (OR 1.06), and a 68% increase in the incidence of DKA (OR 1.68) compared to the placebo group, all these results were not considered statistically significant. Therefore, compared to the placebo group, Sotagliflozin was considered safe in terms of hypoglycemic incidence, UTIs, and DKA. Diarrhea was the main adverse effect, with a 47% increased incidence in the Sotagliflozin group compared to the placebo (OR 1.47). Other adverse events include a 201% increase in the incidence of genital infections (OR 3.01) and a 26% increase in the incidence of volume depletion (OR 1.26) in the Sotagliflozin group compared to the placebo.
Results from the SCORED (10,584 patients with T2D and chronic kidney disease randomized to Sotagliflozin or placebo) and SOLOIST (122 patients with T2D admitted with worsening heart failure randomized to Sotagliflozin or placebo) trials suggest that combined SGLT1/2 inhibition reduces myocardial infarction (MI) and stroke more than SGLT2 inhibitors alone. [103] While the SCORED trial suggests Sotagliflozin reducing both total (fatal and nonfatal) MI and stroke by 32% and 34%, respectively, a meta-analysis of SGLT2 inhibitors fails to show a significant reduction of either MI or stroke.
Based on the results of the trial, dual SGLT1/2 inhibitors avoid AKI as a side effect yet are still a causative factor in genital mycotic infections, volume depletion, and diarrhea. The SCORED trial showed that there was no significant risk of fractures or amputations, unlike Canagliflozin. [104] This is potentially because dual SGLT1/2 inhibitors such as Phlorizin have been implicated in protecting against endothelial dysfunction by increasing levels of NO and promoting the consumption of glucose in palmitic acid-affected endothelial cells. [105] The risk of hypotension in clinical trials was also significant with 2.6% of patients taking Sotagliflozin vs 1.9% of placebo patients. [104] Sotagliflozin trials in type 1 DM show mild to moderate diarrhea in 4.1% of drug patients vs 2.3% in the placebo group, yet cases of volume depletion were significant (1.9% to 0.5% for drug and placebo groups respectively). [106] Due to these severe adverse effects more studies will be needed to better understand the effects of these therapeutics. For SGLT1 inhibition to be a viable focus for DM management, the volume depletion must be mitigated potentially through lower SGLT1 affinity while keeping the GLP-1 and incretin effect. Drugs with lower SGLT1 affinity, in general, can also have enhanced GLP-1 effects through combination therapy with DPP-4 inhibitors, as observed with Sotagliflozin and Canagliflozin. [107]
Licoglifozin (LIK066), another dual SGLT1/2 inhibitor, is currently in phase 2 clinical trials and has demonstrated promise for a dual inhibition role in the management of Τ2Δ (NCT02470403). When administered to obese patients with or without diabetes, it has led to significant weight loss and optimization of levels of glucose and incretin hormones, providing further evidence of the effectiveness of dual inhibition. [108] One clinical trial (125 patients with Τ2D and heart failure randomized to either Licoglifozin, Empagliflozin, or placebo) showed an apparent reduction in N-terminal pro-brain natriuretic peptide (NT-proBNP) when dosing 10 mg once daily of Licoglifozin vs. placebo at week 12. [109] NT-proBNP is a biomarker measured to indicate the severity and prognosis of heart failure, which is a common cardiovascular complication associated with T2D. [110, 111] These findings may suggest dual inhibition can play a role in alleviating heart failure in T2D patients, however larger studies are needed to confirm this. The specific mechanisms of how SGLT1/2 inhibition contributes to the reduction of NT-proBNP have not yet been elucidated; however, it is proposed that the enhancement of renal sodium and glucose handling, changes in cardiac metabolism, oxygen supply, and sodium-hydrogen exchange may play a role. [109] Licoglifozin is considered generally safe and well-tolerated, with the most common adverse event being diarrhea (90% with Licoglifozin 150 mg once per day vs. 25% with placebo in a 12 week randomized study). [108] Although larger and longer-duration studies are needed to confirm other side effects, preliminary clinical data suggest low incidences of bone fracture (1.6%), genital mycotic infection (1.6%), and UTIs (3.3%) of patients with T2D and heart failure taking Licoglifozin (2.5 mg, 10 mg, or 20 mg once daily) for 12 weeks. [109] These findings may suggest that dual inhibition may mitigate the side effects commonly seen in SGLT2 inhibitors alone.
YG1699 is another dual SGLT1/2 inhibitor undergoing phase 1 clinical trials with results still being analyzed (NC05089617). While it intends to achieve similar goals to other dual inhibitors (enhancing glycemic control and weight loss in T2D patients), further studies may elucidate more of the unique benefits and adverse events associated with this and other dual inhibitors.
2.2.5. Other Classes of Diabetes Medication
Current diabetes therapeutics revolve around the modulation of the incretin and enteroendocrine hormones discussed previously or act at the level of insulin /glucagon. Due to adverse effects, a number of other targets have been studied for glucose modulation.
GLP-1 Receptor agonists
GLP-1 receptor agonists are one avenue for hyperglycemic diabetes treatment and current therapeutics include Exenatide, Lixisenatide, Liraglutide, Dulaglutide and Semaglutide[112]. These are insulinotropic peptide agonists that mimic GLP-1 biological activity at picomolar concentrations. [113] They increase insulin and decrease glucagon secretion mainly through a cAMP/protein kinase A (PKA) intracellular mechanism that promotes insulin gene transcription in pancreatic beta cells and a cAMP/PKA mechanism that blocks P/Q-type Ca2+ channels and prevents glucagon exocytosis in pancreatic α-cells. [114, 115] This promotes satiety in the individual. GLP-1 receptor agonists are associated with nausea and vomitting resulting in 5–15% of patients discontinuing treatment. [112] Additionally, GLP-1 as well as its receptor agonists are susceptible to degradation via dipeptidyl peptidase 4 (DPP-4) so as this avenue for diabetes medication grows, it is crucial to develop receptor agonists that slow degradation. [112] The currently approved receptor agonists avoid this problem and are typically separated into short-acting and long-acting groups based on the patient’s specific diabetes management plan.
DPP-4 inhibitors
Inhibiting DPP-4 to maintain elevated levels of GLP-1 in hyperglycemic states is another potential avenue for treating diabetes. Some of the FDA-approved drugs include Sitagliptin, Vildagliptin, Linagliptin, Saxagliptin, and Alogliptin which are small molecule peptide mimetics that block the proteolytic activity at the active site of DPP-4. DPP-4 inhibitors are not a homogenous class of molecules even though they are classified as “-gliptin” molecules. Subclassifications of DPP-4 inhibitors are named based on what subunits of DPP-4 they interact with. [116] Studies show that postprandial GLP-1 concentrations can increase 2–3 fold and hemoglobin A1c levels lower as a result of DPP-4 inhibitors. [117] However, nasopharyngitis and skin lesions concerns but a lower risk of acute pancreatitis, due to the increase of GLP-1, has been reported. [116] The additive effects of GLP-1 receptor agonists and DPP-4 are not expected to have added clinical benefit. [116]
α-glucosidase inhibitors (AGIs)
AGIs competitively and reversibly bind α-glucosidase which is a BBE found in the small intestines. [118] The enzyme functions in cleaving α-1,4 glycosidic linkages and reduces postprandial blood glucose levels through delayed carbohydrate absorption in the small intestines. [119] Current AGIs include Acarbose, Miglitol, and Voglibose which are structurally similar to sugars and have a nitrogen group either in their pyranose-like ring (iminosugars) or have a nitrogen covalently attached to the ring (carbasugars). [120] These serve to mimic smaller tri- and disaccharides that bind to the active site of the enzyme. Due to their biological function at the intestinal level, AGIs work to lower postprandial glucose levels but not fasting glucose levels. Side effects are remarkably similar to that of SGLT1 inhibition which also works at the intestinal level. As short-chain carbohydrates stay in the intestinal lumen, water flowing into the intestines following these macromolecules results in flatulence, diarrhea, bloating, and abdominal cramping. [119] However, protective cardiovascular effects are seen with AGIs such as a lower risk of myocardial infarction and stroke, lower blood pressure, and improvements in triglyceride levels. [121, 122]
Peroxisome proliferator-activated receptor- γ (PPAR-γ) agonists
PPAR-γ agonists are called thiazolidinediones and work by increasing insulin sensitivity and glucose uptake in muscle cells. [123] These nuclear receptors regulate carbohydrate metabolism, cell growth, differentiation, apoptosis, and lipid metabolism. [124] Isoforms of this receptor have many different expression profiles. PPAR-γ1 is found in a broad range of tissue types for energy metabolism while PPAR-γ2 is mainly found in adipose tissue for lipid metabolism. [125] When thiazolidinediones bind to PPAR-γ, they cause heterodimerization of PPAR-γ with retinoid X receptor (RXR), and both act as transcription factors for genes involved in carbohydrate and lipid metabolism. [126] The increase in insulin sensitivity arises from the fact that thiazolidinediones can enhance the lipid-storing capacity of adipose tissue and increases the number of adipocytes, allowing for less adverse lipid accumulation and therefore reducing insulin resistance in muscle and liver cells. [127] Troglitazone, Rosiglitazone, and Pioglitazone are the three main drugs that fall in this category and contain a thiazolidine-2,4-dione sulfur group that makes the class distinct from others. Severe adverse effects, such as hepatotoxicity, have resulted in Troglitazone being removed from the market after FDA approval, compared to Rosiglitazone and Pioglitazone with lower heaptotoxicity,[128] along with a number of other side effects of consideration. [129]
Biguanides and Sulfonylurea
Metformin is an antidiabetic drug in the biguanides drug class. [130] It serves as a first line of defense for diabetes medication and works through many mechanisms of action. The main anti-diabetic mechanism is reducing liver gluconeogenesis through the increase in AMP-activated protein kinase (AMPK) stimulation, a key protein in inhibiting transcription factors for reduced gluconeogenesis enzyme activity. [130] Although it is not fully known how metformin and other biguanides increase AMPK stimulation, there are multiple possible mechanisms: increasing AMP/ATP ratio, activating liver kinase B1, cAMP reduction, and glucagon secretion blockage, glycerophosphate dehydrogenase inhibition in the mitochondria, and fructose-1–6-bisphosphatase inhibition. [131–135] Biguanides also increase skeletal muscle glucose uptake through an unclear AMPK-dependent pathway. [130] Other drugs in the family include Phenformin and Imeglimin. Phenformin was approved as a diabetic treatment but was later canceled in many countries due to lactic acidosis. [136] Side effects are minimal and dual oral therapies are constantly being developed, especially with SGLT inhibitors, for additive glucose-lowering effects. Sulfonylureas structurally consist of a phenyl group attached to a sulfonyl group which is attached to a urea group, allowing it to bind to the sulfonylurea receptor (SUR) on pancreatic beta cell ATP-sensitive potassium channels. [137, 138] This results in the decrease of potassium efflux and cell depolarization, triggering Ca2+ release and insulin secretion. [139] These drugs also function by decreasing the liver-mediated metabolism of insulin, decreasing pancreatic α-cell glucagon secretion, and increasing insulin sensitivity in peripheral tissue. [140] SURs have two different subtypes (SUR1 and SUR2) with different expression profiles and different binding affinities for sulfonylureas. SUR1 is mainly expressed in the brain and pancreatic β-cells while SUR2 is predominant in cardiac and smooth muscle. [137] This means that sulfonylureas with a greater affinity to SUR2 can have cardiotoxic side effects. Drugs under the sulfonylurea class include Chlorpropamide and Tolbutamide as first-generation and Glyburide, Glipizide, Glimepiride, and Gliclazide as second-generation, mainly differing in pharmacokinetics and degradation. [141] Due to the increased risk of cardiovascular symptoms, Tolbutamide has been labeled with a warning by the FDA and requires cardiovascular safety studies. [142] Other drugs in this class either have no change in cardiovascular risk or even beneficial cardiovascular effects. Patients are typically not administered sulfonylureas as a second-line medication because of these potential side effects as well as hypoglycemia since insulin secretion is stimulated with no regard to blood glucose levels. [143] If used, they are usually short-acting second-generation which reduces the risk of medication-induced hypoglycemia.
3. Significance of study
SGLT2 inhibitors are well-established therapeutics for T2D management.
SGLT2 selective inhibitors mainly consist of Canagliflozin, Empagliflozin, Ertugliflozin, and Dapagliflozin with Empagliflozin and Dapagliflozin having high selectivity for SGLT2 and Canagliflozin having more selectivity for SGLT1 than the others. Clinical trials show that SGLT2 selective inhibitors have renoprotective and cardiovascular protective effects consisting of reduced albuminuria, slowing decline in GFR, lower chance of MACE, lower systolic blood pressure, and lower fat counts in adipocytes. While Dapagliflozin is not as effective as other SGLT2 inhibitors in alleviating cardiovascular complications, Canagliflozin is associated with a higher incidence of amputations, fractures, female GMIs, ARF, and euglycemic DKA. It is not clear whether Canagliflozin’s side effects are due to its more split selectivity for SGLT1 and 2 or if these adverse events are possibly generalized for all SGLT2 inhibitors (Table 1). Of note amputations and fractures were only found to be significant in one trial (CANVAS). Additionally, SGLT1 selective inhibitor, Mizagliflozin, as well as dual inhibitor Sotagliflozin did not note these adverse effects, suggesting some potential off-target mechanism inherent in Canagliflozin’s structure may exist.
SGLT1 inhibitors’ increase in intestinal glucose results in diarrhea and volume depletion / hypotensive events, however, the wide expression profile for SGLT1 has resulted in numerous studies to understand its role in myocytes and neurons, providing potential mechanisms for how SGLT1 inhibition can be used to not only treat diabetes in a postprandial state but also treat other risk factors associated with hyperglycemia.
The well-characterized SGLT1/2 dual inhibitor, Sotagliflozin (SGLT2/SGLT1 selectivity ratio ~20), shows clinical data supporting the strategy of dual inhibition for the treatment of T2D (Table 1). While Sotagliflozin results in similar A1c level and systolic blood pressure reduction as SGLT2 inhibitors, it appears to show promise in reducing the incidence of side effects commonly seen in SGLT2 inhibitors, such as bone fractures, UTIs, and GMIs. However, larger studies are needed to fully support these benefits. Additionally, Sotagliflozin may reduce the risk of heart failure, myocardial infarction, and stroke in type 2 DM patients more than SGLT2 inhibitors alone suggesting a synergistic effect of dual SGLT1 and SGLT2 inhibition; similar to SGLT1 inhibitors, the primary side effects associated with dual inhibition include diarrhea and volume depletion. More clinical trials of SGLT1-only inhibitors are needed to determine whether dual inhibition provides any reduction in the incidence of these side effects compared to SGLT1 inhibitors alone. Overall, SGLT1 and SGLT2 targeting has significant potential given their expression profiles, yet dual inhibitors may offer a combination of benefits for T2D management due to their effects on UGE and incretin hormones. The delicate interplay with receptor selectivity is pivotal in allowing GLP-1 secretion while preventing adverse GI SGLT1-mediated symptoms from arising.
Funding Sources
V.A.K. acknowledges support from the NIH NEI R15 EY029504, NIH NIDCR R01DE031812, NIH NIAMS R21AR079708, NIH NIAMS R01AR080895, NIH NIDCR R01DE029321, NIH NCATS UL1TR003017, and the Undergraduate Research and Innovation program at NJIT.
Biography

Vivek A. Kumar, PhD.
Vivek Kumar is an Associate Professor of Biomedical Engineering at the New Jersey Institute of Technology as an with co-appointments in Chemical and Materials Engineering, Biology and Endodontics at the Rutgers School of Dental Medicine. We explore computational peptide materials design, drug discovery, development and delivery, biomaterials and tissue engineering. We have broad interests in inflammation modulation and angiogenesis, especially in understanding the role of small growth factor or cytokine mimics’ ability to signal. Vivek teaches Biomaterials and Biomedical Translation and Entrepreneurship. He strives to encourage research involvement with undergraduates, graduates and post-doctoral scientists @ the KumarLab (kumarlab.njit.edu).
Footnotes
Conflict of interest disclosure
The authors declare no financial or commercial Conflict of Interest.
Contributor Information
Abdul-Rahman Azizogli, Department of Biological Sciences, New Jersey Institute of Technology, Newark, NJ, 07102.
Michael R Vitti, University of Virginia School of Medicine, Charlottesville, VA, 22903.
Richa Mishra, Department of Biomedical Engineering, New Jersey Institute of Technology, Newark, NJ, 07102.
Laura Osorno, Department of Biomedical Engineering, New Jersey Institute of Technology, Newark, NJ, 07102.
Corey Heffernan, Department of Biomedical Engineering, New Jersey Institute of Technology, Newark, NJ, 07102.
Vivek A Kumar, Department of Biomedical Engineering, New Jersey Institute of Technology, Newark, NJ, 07102; Department of Biological Sciences, New Jersey Institute of Technology, Newark, NJ, 07102; Department of Chemical and Materials Engineering, New Jersey Institute of Technology, Newark, NJ, 07102; Department of Endodontics, Rutgers School of Dental Medicine, Newark, NJ, 07103.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- [1].Wang AY-M, Journal of Diabetes 2011, 3, 119. [DOI] [PubMed] [Google Scholar]
- [2].Olokoba AB, Obateru OA, Olokoba LB, Oman Med J 2012, 27, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bailey CJ, Turner RC, New England Journal of Medicine 1996, 334, 574. [DOI] [PubMed] [Google Scholar]
- [4].Drzewoski J, Hanefeld M, Pharmaceuticals (Basel) 2021, 14, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].American Diabetes A, Diabetes Care 2017, 41, S73. [Google Scholar]
- [6].Giorgino F, Vora J, Fenici P, Solini A, Cardiovascular Diabetology 2020, 19, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arnold SV, Kosiborod M, Wang J, Fenici P, Gannedahl G, LoCasale RJ, Diabetes, Obesity and Metabolism 2018, 20, 2000. [DOI] [PubMed] [Google Scholar]
- [8].Tsimihodimos V, Filippas-Ntekouan S, Elisaf M, European Journal of Pharmacology 2018, 838, 153. [DOI] [PubMed] [Google Scholar]
- [9].Niu Y, Liu R, Guan C, Zhang Y, Chen Z, Hoerer S, Nar H, Chen L, Nature 2022, 601, 280. [DOI] [PubMed] [Google Scholar]
- [10].Tsimihodimos V, Filippatos T, Elisaf M, Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2018, 12, 1117. [DOI] [PubMed] [Google Scholar]
- [11].Lupsa BC, Inzucchi SE, Diabetologia 2018, 61, 2118. [DOI] [PubMed] [Google Scholar]
- [12].Poulsen SB, Fenton RA, Rieg T, Curr Opin Nephrol Hypertens 2015, 24, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gerich JE, Diabet Med 2010, 27, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, Thomson SC, Koepsell H, Vallon V, American Journal of Physiology-Renal Physiology 2014, 306, F188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dennis VW, Brazy PC, in Homeostasis of Phosphate and Other Minerals, Springer, 1978, 79. [Google Scholar]
- [16].DeFronzo R, Davidson J, Del Prato S, Diabetes, Obesity and Metabolism 2012, 14, 5. [DOI] [PubMed] [Google Scholar]
- [17].Freitas HS, Anhê GF, Melo KFS, Okamoto MM, Oliveira-Souza M, Bordin S, Machado UF, Endocrinology 2008, 149, 717. [DOI] [PubMed] [Google Scholar]
- [18].Norton L, Shannon CE, Fourcaudot M, Hu C, Wang N, Ren W, Song J, Abdul-Ghani M, DeFronzo RA, Ren J, Jia W, Diabetes, Obesity and Metabolism 2017, 19, 1322. [DOI] [PubMed] [Google Scholar]
- [19].Paz A, Claxton Derek P, Kumar Jay P, Kazmier K, Bisignano P, Sharma S, Nolte Shannon A, Liwag Terrin M, Nayak V, Wright Ernest M, Grabe M, McHaourab Hassane S, Abramson J, Proceedings of the National Academy of Sciences 2018, 115, E2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Han L, Qu Q, Aydin D, Panova O, Robertson MJ, Xu Y, Dror RO, Skiniotis G, Feng L, Nature 2022, 601, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Song P, Onishi A, Koepsell H, Vallon V, Expert Opinion on Therapeutic Targets 2016, 20, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Navale AM, Paranjape AN, Biophys Rev 2016, 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thorens B, in International Review of Cytology, Vol. 137 (Eds: Friedlander M, Mueckler M), Academic Press, 1992, 209. [DOI] [PubMed] [Google Scholar]
- [24].Moriya R, Shirakura T, Ito J, Mashiko S, Seo T, American Journal of Physiology-Endocrinology and Metabolism 2009, 297, E1358. [DOI] [PubMed] [Google Scholar]
- [25].Gorboulev V, Schürmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H, Diabetes 2012, 61, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gutierrez-Aguilar R, Woods SC, Curr Opin Endocrinol Diabetes Obes 2011, 18, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vilsbøll T, Diabetes, Obesity and Metabolism 2009, 11, 11. [DOI] [PubMed] [Google Scholar]
- [28].Powell DR, Smith M, Greer J, Harris A, Zhao S, DaCosta C, Mseeh F, Shadoan MK, Sands A, Zambrowicz B, Ding ZM, J Pharmacol Exp Ther 2013, 345, 250. [DOI] [PubMed] [Google Scholar]
- [29].Tian L, Cao J, Zhao T, Liu Y, Khan A, Cheng G, Int J Mol Sci 2021, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chasis H, Jolliffe N, Smith HW, The Journal of clinical investigation 1933, 12, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Alvarado F, Crane RK, Biochim Biophys Acta 1962, 56, 170. [DOI] [PubMed] [Google Scholar]
- [32].Vick H, Diedrich DF, Baumann K, Am J Physiol 1973, 224, 552. [DOI] [PubMed] [Google Scholar]
- [33].Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA, J Clin Invest 1987, 79, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Crespy V, Aprikian O, Morand C, Besson C, Manach C, Demigné C, Rémésy C, J Nutr 2001, 131, 3227. [DOI] [PubMed] [Google Scholar]
- [35].Oku A, Ueta K, Arakawa K, Ishihara T, Nawano M, Kuronuma Y, Matsumoto M, Saito A, Tsujihara K, Anai M, Asano T, Kanai Y, Endou H, Diabetes 1999, 48, 1794. [DOI] [PubMed] [Google Scholar]
- [36].Choi CI, Molecules 2016, 21.28035966 [Google Scholar]
- [37].Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P, Diabetes Obes Metab 2012, 14, 83. [DOI] [PubMed] [Google Scholar]
- [38].Hsia DS, Grove O, Cefalu WT, Curr Opin Endocrinol Diabetes Obes 2017, 24, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ohno H, Kojima Y, Harada H, Abe Y, Endo T, Kobayashi M, Xenobiotica 2019, 49, 463. [DOI] [PubMed] [Google Scholar]
- [40].Inoue T, Takemura M, Fushimi N, Fujimori Y, Onozato T, Kurooka T, Asari T, Takeda H, Kobayashi M, Nishibe H, Isaji M, Eur J Pharmacol 2017, 806, 25. [DOI] [PubMed] [Google Scholar]
- [41].Nomura S, Sakamaki S, Hongu M, Kawanishi E, Koga Y, Sakamoto T, Yamamoto Y, Ueta K, Kimata H, Nakayama K, Tsuda-Tsukimoto M, Journal of Medicinal Chemistry 2010, 53, 6355. [DOI] [PubMed] [Google Scholar]
- [42].Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN, Journal of Medicinal Chemistry 2008, 51, 1145. [DOI] [PubMed] [Google Scholar]
- [43].Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P, Diabetes, Obesity and Metabolism 2012, 14, 83. [DOI] [PubMed] [Google Scholar]
- [44].Fediuk DJ, Nucci G, Dawra VK, Cutler DL, Amin NB, Terra SG, Boyd RA, Krishna R, Sahasrabudhe V, Clin Pharmacokinet 2020, 59, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Steven S, Oelze M, Hanf A, Kröller-Schön S, Kashani F, Roohani S, Welschof P, Kopp M, Gödtel-Armbrust U, Xia N, Redox biology 2017, 13, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Han S, Hagan DL, Taylor JR, Xin L, Meng W, Biller SA, Wetterau JR, Washburn WN, Whaley JM, Diabetes 2008, 57, 1723. [DOI] [PubMed] [Google Scholar]
- [47].Devineni D, Murphy J, Wang SS, Stieltjes H, Rothenberg P, Scheers E, Mamidi RN, Clinical Pharmacology in Drug Development 2015, 4, 295. [DOI] [PubMed] [Google Scholar]
- [48].Kasichayanula S, Liu X, LaCreta F, Griffen SC, Boulton DW, Clinical Pharmacokinetics 2014, 53, 17. [DOI] [PubMed] [Google Scholar]
- [49].Ndefo UA, Anidiobi NO, Basheer E, Eaton AT, P T 2015, 40, 364. [PMC free article] [PubMed] [Google Scholar]
- [50].Polidori D, Sha S, Mudaliar S, Ciaraldi TP, Ghosh A, Vaccaro N, Farrell K, Rothenberg P, Henry RR, Diabetes care 2013, 36, 2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stenlöf K, Cefalu WT, Kim K-A, Jodar E, Alba M, Edwards R, Tong C, Canovatchel W, Meininger G, Current Medical Research and Opinion 2014, 30, 163. [DOI] [PubMed] [Google Scholar]
- [52].Zhang L, Feng Y, List J, Kasichayanula S, Pfister M, Diabetes, Obesity and Metabolism 2010, 12, 510. [DOI] [PubMed] [Google Scholar]
- [53].Liakos A, Karagiannis T, Athanasiadou E, Sarigianni M, Mainou M, Papatheodorou K, Bekiari E, Tsapas A, Diabetes, Obesity and Metabolism 2014, 16, 984. [DOI] [PubMed] [Google Scholar]
- [54].Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, New England Journal of Medicine 2017, 377, 644.28605608 [Google Scholar]
- [55].Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, N Engl J Med 2015, 373, 2117.26378978 [Google Scholar]
- [56].Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS, N Engl J Med 2019, 380, 347. [DOI] [PubMed] [Google Scholar]
- [57].Tikkanen I, Narko K, Zeller C, Green A, Salsali A, Broedl UC, Woerle HJ, on E-REGBPI behalf of the, Diabetes Care 2014, 38, 420. [DOI] [PubMed] [Google Scholar]
- [58].Weir MR, Januszewicz A, Gilbert RE, Vijapurkar U, Kline I, Fung A, Meininger G, The Journal of Clinical Hypertension 2014, 16, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Storgaard H, Gluud LL, Bennett C, Grøndahl MF, Christensen MB, Knop FK, Vilsbøll T, PLoS One 2016, 11, e0166125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wei D, Liao L, Wang H, Zhang W, Wang T, Xu Z, Life Sciences 2020, 247, 117414. [DOI] [PubMed] [Google Scholar]
- [61].Budoff MJ, Wilding JPH, International Journal of Clinical Practice 2017, 71, e12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Heerspink HJL, Perco P, Mulder S, Leierer J, Hansen MK, Heinzel A, Mayer G, Diabetologia 2019, 62, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Flores E, Santos-Gallego CG, Diaz-Mejía N, Badimon JJ, Cardiovasc Drugs Ther 2018, 32, 213. [DOI] [PubMed] [Google Scholar]
- [64].Engelhardt K, Ferguson M, Rosselli JL, Annals of Pharmacotherapy 2020, 55, 543. [DOI] [PubMed] [Google Scholar]
- [65].Goswami R, Dadhwal V, Tejaswi S, Datta K, Paul A, Haricharan RN, Banerjee U, Kochupillai NP, Journal of Infection 2000, 41, 162. [DOI] [PubMed] [Google Scholar]
- [66].Geerlings S, Fonseca V, Castro-Diaz D, List J, Parikh S, Diabetes Research and Clinical Practice 2014, 103, 373. [DOI] [PubMed] [Google Scholar]
- [67].Puckrin R, Saltiel M-P, Reynier P, Azoulay L, Yu OHY, Filion KB, Acta Diabetologica 2018, 55, 503. [DOI] [PubMed] [Google Scholar]
- [68].Kovacich N, Chavez B, P t 2018, 43, 736. [PMC free article] [PubMed] [Google Scholar]
- [69].Perlman A, Heyman SN, Matok I, Stokar J, Muszkat M, Szalat A, Nutrition, Metabolism and Cardiovascular Diseases 2017, 27, 1108. [DOI] [PubMed] [Google Scholar]
- [70].O’Neill J, Fasching A, Pihl L, Patinha D, Franzén S, Palm F, American journal of physiology-renal physiology 2015, 309, F227. [DOI] [PubMed] [Google Scholar]
- [71].Heyman SN, Khamaisi M, Rosenberger C, Szalat A, Abassi Z, Journal of Clinical Medicine Research 2016, 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hahn K, Ejaz AA, Kanbay M, Lanaspa MA, Johnson RJ, Nature Reviews Nephrology 2016, 12, 711. [DOI] [PubMed] [Google Scholar]
- [73].Dutta S, Kumar T, Singh S, Ambwani S, Charan J, Varthya SB, J Family Med Prim Care 2022, 11, 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wibawa K, Kuhuwael F, Putra C, Widiastuti S, Suciadi L, Clinical Diabetology 2021, 10, 204. [Google Scholar]
- [75].Taylor SI, Blau JE, Rother KI, J Clin Endocrinol Metab 2015, 100, 2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Watts NB, Bilezikian JP, Usiskin K, Edwards R, Desai M, Law G, Meininger G, The Journal of clinical endocrinology and metabolism 2016, 101, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bilezikian JP, Watts NB, Usiskin K, Polidori D, Fung A, Sullivan D, Rosenthal N, J Clin Endocrinol Metab 2016, 101, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Blau JE, Bauman V, Conway EM, Piaggi P, Walter MF, Wright EC, Bernstein S, Courville AB, Collins MT, Rother KI, Taylor SI, JCI Insight 2018, 3, e99123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chatterjee S, Bandyopadhyay D, Ghosh RK, Majumdar U, Aneja A, Lavie CJ, Deedwania P, Current Problems in Cardiology 2019, 44, 207. [DOI] [PubMed] [Google Scholar]
- [80].Imprialos KP, Boutari C, Stavropoulos K, Doumas M, Karagiannis AI, Journal of Neurology, Neurosurgery & Psychiatry 2017, 88, 249. [DOI] [PubMed] [Google Scholar]
- [81].Potier L, Roussel R, Velho G, Saulnier P-J, Bumbu A, Matar O, Schneider F, Ragot S, Marre M, Mohammedi K, Hadjadj S, Diabetologia 2019, 62, 939. [DOI] [PubMed] [Google Scholar]
- [82].Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, Day EA, Salt IP, Steinberg GR, Hardie DG, Diabetes 2016, 65, 2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Behnammanesh G, Durante ZE, Peyton KJ, Martinez-Lemus LA, Brown SM, Bender SB, Durante W, Frontiers in Pharmacology 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Papadokostaki E, Rizos E, Tigas S, Liberopoulos EN, The International Journal of Lower Extremity Wounds 2019, 19, 21. [DOI] [PubMed] [Google Scholar]
- [85].Shibazaki T, Tomae M, Ishikawa-Takemura Y, Fushimi N, Itoh F, Yamada M, Isaji M, J Pharmacol Exp Ther 2012, 342, 288. [DOI] [PubMed] [Google Scholar]
- [86].Goodwin NC, Ding Z-M, Harrison BA, Strobel ED, Harris AL, Smith M, Thompson AY, Xiong W, Mseeh F, Bruce DJ, Diaz D, Gopinathan S, Li L, O’Neill E, Thiel M, Wilson AGE, Carson KG, Powell DR, Rawlins DB, Journal of Medicinal Chemistry 2017, 60, 710. [DOI] [PubMed] [Google Scholar]
- [87].Kuroda S, Kobashi Y, Oi T, Amada H, Okumura-Kitajima L, Io F, Yamamto K, Kakinuma H, Bioorganic & Medicinal Chemistry Letters 2018, 28, 3534. [DOI] [PubMed] [Google Scholar]
- [88].Dobbins RL, Greenway FL, Chen L, Liu Y, Breed SL, Andrews SM, Wald JA, Walker A, Smith CD, American Journal of Physiology-Gastrointestinal and Liver Physiology 2015, 308, G946. [DOI] [PubMed] [Google Scholar]
- [89].Fukudo S, Endo Y, Hongo M, Nakajima A, Abe T, Kobayashi H, Nakata T, Nakajima T, Sameshima K, Kaku K, Lancet Gastroenterol Hepatol 2018, 3, 603. [DOI] [PubMed] [Google Scholar]
- [90].Van Steenbergen A, Balteau M, Ginion A, Ferté L, Battault S, Ravenstein C. d. M. d., Balligand J-L, Daskalopoulos E-P, Gilon P, Despa F, Despa S, Vanoverschelde J-L, Horman S, Koepsell H, Berry G, Hue L, Bertrand L, Beauloye C, Scientific Reports 2017, 7, 41166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Brown DI, Griendling KK, Circulation research 2015, 116, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jorissen W, Wouters E, Bogie JF, Vanmierlo T, Noben J-P, Sviridov D, Hellings N, Somers V, Valcke R, Vanwijmeersch B, Scientific reports 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Van Steenbergen A, Balteau M, Ginion A, Ferté L, Battault S, Ravenstein C. d. M. d., Balligand J-L, Daskalopoulos E-P, Gilon P, Despa F, Scientific reports 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yamazaki Y, Harada S, Wada T, Hagiwara T, Yoshida S, Tokuyama S, European Journal of Pharmacology 2017, 799, 103. [DOI] [PubMed] [Google Scholar]
- [95].Lai B, Xiao Y, Pu H, Cao Q, Jing H, Liu X, Archives of Gynecology and Obstetrics 2012, 285, 1455. [DOI] [PubMed] [Google Scholar]
- [96].Meng L, Uzui H, Guo H, Tada H, Molecular Medicine Reports 2018, 17, 6887. [DOI] [PubMed] [Google Scholar]
- [97].Koepsell H, Pharmacology & therapeutics 2017, 170, 148. [DOI] [PubMed] [Google Scholar]
- [98].Sharma P, Khairnar V, Madunić IV, Singh Y, Pandyra A, Salker MS, Koepsell H, Sabolić I, Lang F, Lang PA, Cellular Physiology and Biochemistry 2017, 42, 1358. [DOI] [PubMed] [Google Scholar]
- [99].Palazzo M, Gariboldi S, Zanobbio L, Selleri S, Dusio GF, Mauro V, Rossini A, Balsari A, Rumio C, The Journal of Immunology 2008, 181, 3126. [DOI] [PubMed] [Google Scholar]
- [100].Goodwin NC, Mabon R, Harrison BA, Shadoan MK, Almstead ZY, Xie Y, Healy J, Buhring LM, DaCosta CM, Bardenhagen J, Mseeh F, Liu Q, Nouraldeen A, Wilson AGE, Kimball SD, Powell DR, Rawlins DB, Journal of Medicinal Chemistry 2009, 52, 6201. [DOI] [PubMed] [Google Scholar]
- [101].Powell DR, Smith M, Greer J, Harris A, Zhao S, DaCosta C, Mseeh F, Shadoan MK, Sands A, Zambrowicz B, Ding Z-M, Journal of Pharmacology and Experimental Therapeutics 2013, 345, 250. [DOI] [PubMed] [Google Scholar]
- [102].Avgerinos I, Karagiannis T, Kakotrichi P, Michailidis T, Liakos A, Matthews DR, Tsapas A, Bekiari E, Diabetes, Obesity and Metabolism 2022, 24, 106. [DOI] [PubMed] [Google Scholar]
- [103].Pitt B, Steg G, Leiter LA, Bhatt DL, Cardiovasc Drugs Ther 2022, 36, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Bhatt DL, Szarek M, Pitt B, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Inzucchi SE, Kosiborod MN, Cherney DZI, Dwyer JP, Scirica BM, Bailey CJ, Díaz R, Ray KK, Udell JA, Lopes RD, Lapuerta P, Steg PG, New England Journal of Medicine 2020, 384, 129.33200891 [Google Scholar]
- [105].Li C-Y, Wang L-X, Dong S-S, Hong Y, Zhou X-H, Zheng W-W, Zheng C, Medical Science Monitor Basic Research 2018, 24, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Garg SK, Henry RR, Banks P, Buse JB, Davies MJ, Fulcher GR, Pozzilli P, Gesty-Palmer D, Lapuerta P, Simó R, New England Journal of Medicine 2017, 377, 2337. [DOI] [PubMed] [Google Scholar]
- [107].Takebayashi K, Inukai T, Journal of Clinical Medicine Research 2017, 9, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].He Y-L, Haynes W, Meyers CD, Amer A, Zhang Y, Mahling P, Mendonza AE, Ma S, Chutkow W, Bachman E, Diabetes, Obesity and Metabolism 2019, 21, 1311. [DOI] [PubMed] [Google Scholar]
- [109].de Boer RA, Núñez J, Kozlovski P, Wang Y, Proot P, Keefe D, British Journal of Clinical Pharmacology 2020, 86, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Panagopoulou V, Deftereos S, Kossyvakis C, Raisakis K, Giannopoulos G, Bouras G, Pyrgakis V, Cleman MW, Curr Top Med Chem 2013, 13, 82. [DOI] [PubMed] [Google Scholar]
- [111].Shah AD, Langenberg C, Rapsomaniki E, Denaxas S, Pujades-Rodriguez M, Gale CP, Deanfield J, Smeeth L, Timmis A, Hemingway H, Lancet Diabetes Endocrinol 2015, 3, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Nauck MA, Quast DR, Wefers J, Meier JJ, Molecular Metabolism 2021, 46, 101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Mojsov S, Weir GC, Habener JF, J Clin Invest 1987, 79, 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].MacDonald PE, El-kholy W, Riedel MJ, Salapatek AMF, Light PE, Wheeler MB, Diabetes 2002, 51, S434. [DOI] [PubMed] [Google Scholar]
- [115].Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, González A, Moritoh Y, Shigeto M, Zhang Q, Braun M, Clark A, Johnson PR, Rorsman P, Briant LJB, Physiol Rep 2018, 6, e13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Gallwitz B, Front Endocrinol (Lausanne) 2019, 10, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Deacon CF, Front Endocrinol (Lausanne) 2019, 10, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bischoff H, Clin Invest Med 1995, 18, 303. [PubMed] [Google Scholar]
- [119].Hedrington MS, Davis SN, Expert Opinion on Pharmacotherapy 2019, 20, 2229. [DOI] [PubMed] [Google Scholar]
- [120].Trapero A, Llebaria A, Journal of Medicinal Chemistry 2012, 55, 10345. [DOI] [PubMed] [Google Scholar]
- [121].Hanefeld M, Cagatay M, Petrowitsch T, Neuser D, Petzinna D, Rupp M, Eur Heart J 2004, 25, 10. [DOI] [PubMed] [Google Scholar]
- [122].Hsu PF, Sung SH, Cheng HM, Shin SJ, Lin KD, Chong K, Yen FS, Yu BH, Huang CT, Hsu CC, J Clin Endocrinol Metab 2018, 103, 3611. [DOI] [PubMed] [Google Scholar]
- [123].Takada I, Makishima M, Expert Opinion on Therapeutic Patents 2020, 30, 1. [DOI] [PubMed] [Google Scholar]
- [124].Dubois V, Eeckhoute J, Lefebvre P, Staels B, J Clin Invest 2017, 127, 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tontonoz P, Spiegelman BM, Annu Rev Biochem 2008, 77, 289. [DOI] [PubMed] [Google Scholar]
- [126].Chiarelli F, Di Marzio D, Vasc Health Risk Manag 2008, 4, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Okuno A, Tamemoto H, Tobe K, Ueki K, Mori Y, Iwamoto K, Umesono K, Akanuma Y, Fujiwara T, Horikoshi H, Yazaki Y, Kadowaki T, J Clin Invest 1998, 101, 1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Floyd JS, Barbehenn E, Lurie P, Wolfe SM, Pharmacoepidemiology and Drug Safety 2009, 18, 1238. [DOI] [PubMed] [Google Scholar]
- [129].Henry RR, Endocrinology and Metabolism Clinics of North America 1997, 26, 553. [DOI] [PubMed] [Google Scholar]
- [130].Yendapally R, Sikazwe D, Kim SS, Ramsinghani S, Fraser-Spears R, Witte AP, La-Viola B, Drug Development Research 2020, 81, 390. [DOI] [PubMed] [Google Scholar]
- [131].Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE, J Clin Invest 2001, 108, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC, Science 2005, 310, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ, Nature 2013, 494, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez J-P, Lee H-Y, Cline GW, Samuel VT, Kibbey RG, Shulman GI, Nature 2014, 510, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, Sicheri F, Jessen N, Wasserman DH, Sakamoto K, Nat Med 2018, 24, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bailey CJ, Diabetologia 2017, 60, 1566. [DOI] [PubMed] [Google Scholar]
- [137].Costello RA, Nicolas S, Shivkumar A, Sulfonylureas, StatPearls Publishing, Treasure Island (FL), 2021. [PubMed] [Google Scholar]
- [138].Colagiuri S, Matthews D, Leiter LA, Chan SP, Sesti G, Marre M, Diabetes Res Clin Pract 2018, 143, 1. [DOI] [PubMed] [Google Scholar]
- [139].Proks P, Reimann F, Green N, Gribble F, Ashcroft F, Diabetes 2002, 51 Suppl 3, S368. [DOI] [PubMed] [Google Scholar]
- [140].Lv W, Wang X, Xu Q, Lu W, Curr Top Med Chem 2020, 20, 37. [DOI] [PubMed] [Google Scholar]
- [141].Skillman TG, Feldman JM, The American Journal of Medicine 1981, 70, 361. [DOI] [PubMed] [Google Scholar]
- [142].Meinert CL, Knatterud GL, Prout TE, Klimt CR, Diabetes 1970, 19, Suppl:789. [PubMed] [Google Scholar]
- [143].Landgraf R, Aberle J, Birkenfeld AL, Gallwitz B, Kellerer M, Klein H, Müller-Wieland D, Nauck MA, Reuter HM, Siegel E, Exp Clin Endocrinol Diabetes 2019, 127, S73. [DOI] [PubMed] [Google Scholar]
- [144].Dominguez Rieg JA, Rieg T, Diabetes Obes Metab 2019, 21 Suppl 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.