


Abstract
To gain a better understanding of the complexity of gene expression in normal and diseased tissues it is important to account for the spatial context and identity of cells in situ. State-of-the-art spatial profiling technologies, such as the Nanostring GeoMx Digital Spatial Profiler (DSP), now allow quantitative spatially resolved measurement of the transcriptome in tissues. However, the bioinformatics pipelines currently used to analyse GeoMx data often fail to successfully account for the technical variability within the data and the complexity of experimental designs, thus limiting the accuracy and reliability of the subsequent analysis. Carefully designed quality control workflows, that include in-depth experiment-specific investigations into technical variation and appropriate adjustment for such variation can address this issue. Here, we present standR, an R/Bioconductor package that enables an end-to-end analysis of GeoMx DSP data. With four case studies from previously published experiments, we demonstrate how the standR workflow can enhance the statistical power of GeoMx DSP data analysis and how the application of standR enables scientists to develop in-depth insights into the biology of interest.
Graphical Abstract
Graphical Abstract.
Introduction
Quantitative gene expression analysis of disease systems using technologies such as bulk RNA-seq have led to many biomarker discoveries and mechanistic insights through the application of differential expression, transcriptional network and pathway analysis methods (1,2). Single-cell RNA sequencing added further resolution to transcriptomics studies by enabling the investigation of the whole transcriptome at a single cell level, fueling the identification of many novel cell states (3,4). New generation spatial molecular measurement platforms that incorporate spatial information with existing imaging and sequencing technologies allow in-depth and fine-grained analyses, such as cell–cell interactions, cellular neighbourhood analysis and cell type deconvolution (5,6). Further, these technologies enable spatially resolved questions, such as the identification of differential expression between different parts of a tumour, between tissues with and without a particular cellular infiltrate, or of tissues adjacent to and distant from certain anatomical features.
Amongst the spatial platforms, Nanostring's GeoMx Digital Spatial Profiler (DSP) (7) is one of the more robust platforms for Formal-Fixed Paraffin-Embedded (FFPE) tissues (8), providing regions of interest (ROIs) level-selection methods, with ROIs ranging from tens to hundreds of cells. The FFPE compatibility allows the GeoMx DSP to be applicable to clinical and pathological investigations using banked FFPE archival tissues, thus enabling retrospective clinical cohort studies. However, the generation of DSP data involves placing tissue samples on glass slides, where different slides may introduce technical variations to the data, becoming a source of batch effects. Batch effects can dominate the variation in the data, hindering the identification of biological variations of interest, leading to false discovery (9). Besides the sampling biases, like unbalanced cell count and segment (area of interest) size, other technical factors may also include variability of FFPE materials or tissue segments from patients, including factors such as the age of materials as well as other technical variation during tissue fixation, such as fixation time and tissue preparation, all of these could lead to unwanted sample-to-sample variations. Taking these factors together with other technical variations that are commonly seen in bulk or mini-bulk RNA-seq experiments (e.g. sequencing errors and sequencing depth or library size) (10), it is necessary to perform data quality control (QC) and filtering and appropriate normalization or batch correction when analysing GeoMx DSP data. Moreover, it has been long-established that linear-based methods such as Limma (11,12) and edgeR (13) are more appropriate for carrying out differential expression (DE) analysis compared to traditional T-test (14), especially for datasets with limited sample size. Methods must also correctly account for the complexity of experimental designs in spatial data, where multiple samples may be taken from one patient, or adjacent regions in a single tissue. Taken together, it is essential to construct a computational workflow that can carry out comprehensive QC, data normalization and can be compatible with complex experimental designs and sophisticated DE methods.
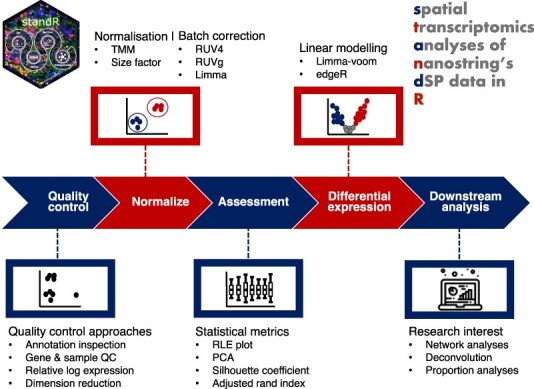
Based on our literature review of publications with GeoMx DSP transcriptome datasets from 2020 to July 2022 (Figure 1A), the current most generic approaches rely heavily on the default quality control of the platform as well as standard paired T-tests (Figure 1D), which may be inadequate to handle the complexity of the experiment. To this end, we have developed a Bioconductor R package standR (Spatial transcriptomics analyses and decoding in R) to assist the QC, normalization and batch correction, differential expression analysis, and downstream analysis of Nanostring GeoMx transcriptomics data. Here we introduce standR and describe the package's workflow and utility in analysing GeoMx DSP datasets. We have also performed a comprehensive comparison of results from each stage between the standR and the current generic GeoMx DSP workflow on four publicly available GeoMx datasets.
Figure 1.

Literature Review and the standR workflow. (A) Bar plot shows the increasing trend of publications with Nanostring GeoMx DSP datasets. (B–D) Bar plots show the preferential choices of QC, normalization and differential expression methods in publications. (E) Diagram demonstrates the comparison between standR and generic workflows. (F) Flow diagram shows the standR workflow.
Materials and methods
Nanostring GeoMx DSP data pre-processing
The Nanostring GeoMx datasets used in this study are publicly available and were downloaded from Nanostring's Spatial Organ Atlas (https://nanostring.com/products/geomx-digital-spatial-profiler/spatial-organ-atlas/). As the default read-out from the GeoMx experiment, probeQC count is used as the input for both the generic and standR workflow. Initial data processing and sample-based QC were conducted using standR, any ROIs that were assigned with QC flags indicating low qualities (including ‘Low Percent Aligned Reads’, ‘Low Percent Stitched Reads’, ‘Low Surface Area’, ‘Low Nuclei Count’) were excluded from further analysis. Negative probes are also removed from the dataset in the analyses since they were used in the generation of probeQC count with an outlier-detection strategy.
Count data transformation
ProbeQC count is transformed into log-transformed counts-per-million (logCPM) count. Compared to raw count, the logCPM count offers several advantages, such as stabilizing the variance across different expression levels and simplifying the interpretation of fold changes between samples, and it is required for the statistical modelling of limma voom to allow standard linear modelling (11,12), which is used in the standR pipeline. The transformation is done by using the cpm function from the edgeR package (13) with a default prior count of 0.25 to avoid log of zero.
Quality control
For the gene filtering, the generic workflow removes genes with raw count smaller than the ROI-specific limitation of quantification (LOQ) in all ROIs, while the standR workflow first calculates a threshold by taking the logarithm of the sum of a minimal count (default is 5) divided by the median of library size and 2 divided by the mean of library size within the standR package's addPerROIQC function. This function removes genes which have logCPM count smaller than the calculated threshold in 90% of the ROIs. The default criterium for filtering used is 90% of all ROIs which aims to identify genes that are not expressed in most of the ROIs. The assumption is that these low expressing genes makes minimal contributions to the downstream statistical analyses (e.g. DE), and removing them would improve the statistical power of the analyses. The QC results were assessed by visualizing the mean-variance distribution of genes (Figure 2B), generated using voom from the limma R package with the linear modelling equation: model.matrix(∼0 + TissueGroups).
Figure 2.

Gene and sample filtering by standR retains tissue relevant genes while removing low quality samples. Gene filtering (A, B), sample filtering (C, D). (A) Mean expression-variance of the genes across all the samples in each GeoMx dataset, colors denote if genes are removed by the gene filtering process of the standR or generic workflow. Blue contour lines indicate data density. (B) limma-voom mean–variance relationship plots of genes across different biological groups in four GeoMx datasets (see methods). red line: lowess regression, legend as per in (A). (C) plot of cell count against total detection for each ROI in the diabetic kidney dataset, colors depicting the distributions of ROIs for individual slide annotation of the ROIs. ROIs with less than 50 000 total detection count (red dotted line) filtered out. (D) After sample filtering, the mean expression-variance plots of gene distribution between standR-filtered or retained samples (right) for either diabetic kidney (top) and colon (bottom) datasets were shown. The linear regression of genes of filtered (blue), retained (green) or unfiltered samples were plotted showing the retained samples maintaining the mean–variance relationship in the data while those samples filtered are different.
For sample filtering: In the standR workflow, the distribution of both library size and cell count were taken into account, we used a common library size threshold of 50 000 and cell count threshold of 50 so that lower-quality ROIs with very small library sizes and very small cell count in the distribution histograms can be removed. To quantify the differences between the original ROIs, filtered ROIs and retained ROIs, we fit a linear model between log-scaled mean expression and the variance of the gene expressions, and the fitted data were then used to calculate the residual sum of squares (RSS). To verify the genes filtered by the generic workflow in the brain dataset, brain tissue-specific (including cerebellum, cerebral, hippocampal and midbrain) regionally elevated genes were obtained from the Human Protein Atlas (15).
Normalization
Different normalisation methods are available in the standR package via the geomxNorm and geomxBatchCorrection functions. When performing RUV4 batch correction, we first identified 200 negative control genes using the findNCGs function from the standR package. The geomxBatchCorrection function was then applied where the parameter k, which indicates the unwanted factor to be used, was set to 3 for the diabetic kidney and brain data, and 2 for the lymph node data. The weight matrices from RUV4 were then included in the design matrix of the linear model as covariates when performing DE analysis.
Assessing normalisation performance
To perform PCA and RLE analysis, the drawPCA and plotRLExpr functions from the standR package were used with default settings. To calculate the similarity statistics for assessing normalizations, adjusted rand index, jaccard index, mirkin distance and silhouette coefficient were calculated using the plotClusterEvalStats function from the standR package between the first two principal components of the data and their slide and tissue type annotations, which indicate batch effect and biological effect, respectively.
Differential expression analysis
In the generic workflow, we performed differential expression (DE) using either the paired T-test from the R package stats, or the linear-mixed model from the R package lmerTest (16) (with model matrix: expression ∼ Group + (1 | SlideName)). We follow DE with a multiple testing adjustment using the Benjamini-Hochberg correction to identify statistically significant (FDR < 0.05) DE genes. In the standR workflow, duplicateCorrelation from the limma R package were first used to calculate the consensus correlation across patients to account for patient variation as a random effect. The linear model was then fitted to the appropriate experimental design containing the biological factors of interest. DE was then performed for specific contrasts of interest, including comparing abnormal glomeruli in diabetic kidney (n = 60) to glomeruli in normal kidney (n = 12); comparing B cell zone (n = 24) to T cell zone (n = 24) in lymph node; comparing longitudinal muscle layer (n = 8) to circular muscle layer (n = 20) in colon; and comparing cortical layer II/III (n = 18) to hippocampus CA1 areas (n = 13) in brain tissues. The resulting statistic was an empirical Bayes moderated t-statistic, followed by multiple testing adjustment was carried out with the Benjamini–Hochberg procedure to identify statistically significant (FDR < 0.05) DE genes.
Gene-set over representation analysis
The Molecular Signatures Database (MSigDB) gene-sets (17,18) data was obtained via the R package msigdbr. C5 and the Hallmark gene-sets was then used in the over representation analysis. The enricher function from the R package clusterProfiler (19) was then used to perform the over representation analysis. Gene-sets with adjusted P-value smaller than 0.05 were considered as significantly enriched gene-sets.
Implementation and availability
The vignette includes a quick introduction of the standR package is available in Bioconductor. A more complete guide of using functions standR to analyse GeoMx dataset is hosted on Github via: https://davislaboratory.github.io/GeoMXAnalysisWorkflow/.
Results
A comprehensive analysis workflow for Nanostring GeoMx DSP data: standR
From a review of the published studies from January 2020 to July 2022, we observed that there is a trend to use the combination of ProbeQC and limitation of quantification (LOQ) filtering strategy to conduct data quality control (QC) (Figure 1B). ProbeQC is the default data processing method provided with GeoMx DSP data, where negative probes are used to detect and remove outliers in the dataset. LOQ is a metric calculated based on the distribution of negative probes and is used as a proxy of the quantifiable limit of gene expression for each tissue fragment (7). After QC, the data is typically scaled using third quantile (Q3) normalization to account for technical variation in the dataset (Figure 1C). Most commonly, differential expression (DE) analysis is performed using standard t-test or linear mixed model (LMM) (Figure 1D). Based on these and for ease of comparison in this study, we define a generic workflow composing these commonly used analysis steps: probeQC and LOQ filtering for data QC, then a Q3 normalisation of the data, followed by identification of differentially expressed genes using a t-test or a LMM (Figure 1E).
In this study we proposed a refined analysis workflow for Nanostring GeoMx DSP data, which we believe is more suitable for spatial contexture analysis and the complex experimental designs typically found in Nanostring GeoMx DSP experiments. Here we present the standR analysis workflow which consists of recommended strategies for each step (Figure 1F) in a sequential manner.
The workflow is built upon the standard data infrastructure for spatial data within the R/Bioconductor community, namely SpatialExperiment (20). This offers several distinct advantages. Firstly, adopting the SpatialExperiment infrastructure provides users with a robust and reliable framework for managing and analysing spatial transcriptomics data. By leveraging the comprehensive functionalities of SpatialExperiment, standR offer users the ability to explore the complex relationships between gene expression and spatial location. Secondly, using the SpatialExperiment data structure ensures compatibility with a wide range of Bioconductor tools, allowing researchers to incorporate state-of-the-art algorithms and analytical approaches into their spatial transcriptomics workflows, enabling comprehensive exploration of different spatial transcriptomic landscapes of interest.
First up in the workflow is QC. In this stage, our approach aims to identify genes that are lowly expressed in over 90% of the regions of interest (ROIs), such genes are then removed from the analysis because genes with constantly low expression are unlikely to be determined as significantly differential expressed genes given their inadequate significance power (21). Subsequently, ROIs with low cell count and/or low total detection count are considered as low-quality tissue fragments and filtered from the analysis to avoid bias due to sample quality in the downstream comparisons.
After QC, suitable normalization method is required due to variation within the Nanostring GeoMx data can be driven by various complex factors, including the desired biological factor such as diseased and control groups or different tissue/cell type groups, or unwanted technical factors such as slide variations (datasets may have each slide containing individual or multiple patient samples), tissue microarray cores differences, different experiment runs or sequencing depth variation (Supplementary figure S1). In such cases where batch effects are observed, it is recommended to apply an appropriate batch effect correction method in the workflow to remove unwanted variation so that fair comparisons between biological groups can be established. Finally, in the standR workflow, DE workflows such as limma-voom (11,12) or edgeR (13) are preferred instead of standard t-test or LMM, as these methods have been shown to be more appropriate for obtaining accurate DE results from complex experimental designs (14).
Comparison between standR and a generic workflow of commonly used analytical processes
To demonstrate the advantages of using the standR analysis workflow, here we applied both standR and generic workflows to analyse four publicly available Nanostring GeoMx DSP datasets from the Spatial Organ Atlas (22). Results generated at corresponding stages from two workflows are systematically compared. The public datasets used are from human diabetic kidney, lymph node, colon and brain tissues, respectively, using the whole transcriptome atlas (WTA) panel for GeoMX DSP (> 18000 genes).
standR gene filtering approach retains tissue relevant genes
The basic principles of gene filtering in both workflows are the same: a gene is removed when its expression is smaller than a certain threshold. However, the generic workflow uses the distinct LOQ, which is calculated based on the geometric mean of the negative probes measured in the tissue fragments of each ROI separately, while standR calculates an overall expression threshold based on both the library size and the minimum count requirement for all genes. Comparing the filtering results for all four datasets tested, the generic workflow tends to remove more genes from the analysis than standR (Figure 1A and B, supplementary figure S2 and supplementary file 1). In the diabetic kidney and lymph node datasets, standR removed markedly fewer genes than the standard filtering, though the genes it did remove were largely also removed by the generic workflow. However, in the other two datasets (colon and brain), the generic removed a substantial proportion of genes, (5.94% and 37.33% respectively), while standR did not remove any genes (Supplementary figure S2B). Our comparison also shows that genes filtered by standR are outliers across all ROIs for the mean expression-variance distribution while the generic workflow may also remove genes with medium level of mean expression and variance (Figure 2A). For example, in the brain dataset, the generic workflow removed some tissue-relevant genes, such as MDGA1 and CLMP (Supplementary figure S3), which may lead to loss of meaningful biological insight. Based on the brain-specific RNA-seq data from the Genotype-Tissue Expression (GTEx) database (23), both MDGA1 and CLMP are expected to be expressed in the cerebral region of human brain (Supplementary figure S3). Notably, CLMP is a membrane protein coding gene where the expression of the CLMP gene was reported in the developing cerebral neocortex and other brain areas and might regulate aspects of synapse development and function in the brain (24,25). Similarly, MDGA1 gene encodes a membrane protein, which has a role in cell adhesion, migration, and axon guidance and, in the developing brain, neuronal migration (26,27). We used linear models to investigate if the standR-filtered genes are biologically significant (Figure 2B). By taking into account biological factors as covariates in the model, it can be seen that genes filtered by both methods (including standR-filtered) are not highly variable between the groups. However, in the brain dataset specifically, there are genes filtered by generic only which are highly variable and potentially DE while no genes were filtered by the standR workflow. Not unexpectedly, MDGA1 and CLMP are amongst them (Supplementary figure S4B labelled), indicating that these two generic-filtered brain-related genes might be differentially expressed between the biological groups in the data. Furthermore, we used the Human Protein Atlas Brain dataset (15) to identify genes that are removed by the generic workflow in the brain dataset, resulting in a set of 231 (out of 413) genes that are brain-specific regionally elevated genes. This reinforce the strength of the standR workflow to be able to retain tissue-relevant genes for downstream analysis.
standR sample filtering is able to remove low-quality samples
During Nanostring GeoMx experiments, low-quality ROIs, such as those with low cell count, might be acquired during the tissue sampling. In order to detect and flag such ROIs, the Nanostring GeoMx NGS pipeline has pre-set cut-offs, such as sequencing read count, sequencing saturation, minimal nuclei count and minimal size of segment area (7,22). As such, the generic workflow does not apply any further sample filtering. However, some low-quality ROIs might not be captured by these pre-defined cut-offs, we therefore included a ROI QC step in the standR workflow, which uses the relationship between the cell count and the total detection distribution of each ROI to identify low-quality ROIs (Figure 2C and supplementary figure S5). In this study we applied a common threshold of 50000 total detection counts and 50 cells to identify low-quality ROIs for the four datasets, removing 11 ROIs in both the diabetic kidney and colon datasets (Supplementary file 2). The mean-variance distribution of the genes for the ROIs that were filtered suggests that genes within them are lowly expressed and less variable compared to the retained ROIs (Figure 2D). Additionally, the residual sum of squares (RSS) (see Materials and methods) between the filtered ROIs and the unfiltered data (36341.41 and 96879.5) is much higher than the RSS between the retained ROIs and the unfiltered data (0.1185 and 4.371) (Figure 2D), indicating that standR filtered ROIs that are very different from the other ROIs in these two datasets. Taken together, this suggests that the standR ROI QC strategy provides an additional filter for low-quality ROIs, supporting a more-accurate downstream analysis.
Comparison of normalization results
Data normalization can adjust data to a comparable scale by removing undesired biases, such as library size differences, batch variations and other technical factors, allowing a better estimation of the data. In the case of GeoMx data, the experiments are usually composed of multiple slides and patient samples, which can lead to batch effects caused by differences between slides. Furthermore, the heterogeneity and density of cells in the selected ROIs can also lead to variation in library size. Other factors including the age of samples, or sample preparation steps can also introduce variation. It is therefore crucial to perform suitable normalization to allow comparative analysis, such as differential expression analysis, between groups. Technical variations can be visualised in QC plots, such as relative log expression (RLE) plots, which are sensitive to technical variations (28), and principal component analysis (PCA) plots, which visualises the variation in the data by dimension reduction and investigate how these variations are related to the factors in the experiment. In the standR package, we provide implementations of different normalization methods, including the ‘trimmed mean of the M values’ (TMM) from the edgeR (13) and the ‘median of ratios’ from DESeq2 (29), both of which are established data normalization method for bulk RNA-seq data. Similarly, established batch correction methods including ‘Removal of Unwanted Variation 4″ (RUV4) (30), ‘Remove Unwanted Variation Using Control Genes’ (RUVg) (31) and ‘Remove Batch Effect’ function in limma (11) are also implemented in standR.
PCA was performed on the raw data of the four GeoMx dataset tested. We identified confounding batch effects due to slide differences in the brain, lymph node and diabetic kidney datasets, while no batch effects were identified in the colon (Supplementary figure S6). The generic workflow uses Q3 normalization, which is a method using the 75th quantile as normalised factor for each ROI. In the batch-confounded datasets (i.e. brain, lymph node and diabetic kidney), the PCA of Q3-normalised data suggest that the batch effect due to the different slides has not been removed, nor is the variation explained by tissue types (Figure 3A, left and supplementary figure S7).
Figure 3.

Normalization and batch correction using standR and generic workflows in the brain dataset. (A) PCA plots of data normalised using either the generic (left) or standR (right) workflows. Panels denotes annotations by either ROIs (top) and tissue structures (bottom). standR normalisation and batch correction was able to reduce slide effects while improving separation of biology. (B) RLE plots of the raw (top), Q3-normalised (middle) and RUV4-normalised (bottom) data show RUV4 gives the best removal of technical variations. (C) Summarised statistics of raw, Q3-normalised and RUV4-normalised data comparing performance in terms of the biology (top) or batch factor (bottom). RUV4 performed the best across the statistics in terms of biology and batch (in general).
Using the standR implemented normalisation functions, RUV4 normalization was applied to batch affected datasets (i.e. diabetic kidney, lymph node and brain), while TMM normalization was applied to the colon dataset. Results as shown in Figure 3A (right) suggested improved grouping based on tissue type (bottom, biological) in the brain dataset while reducing the grouping based on slide (top). Results for RUV4 normalisation on diabetic kidney and lymph node datasets are shown in Supplementary figure S7. These suggest that by using appropriate methods provided by standR, batch effects can be appropriately addressed. Further evidence of appropriate normalisation outcomes can be found in the RLE plots, which shows less technical variations for standR-normalised data as compared to Q3-normalised data from the generic workflow (Figure 3B). This applies to all four-dataset tested, including the colon dataset, where batch effects are not observed (Supplementary figure S8).
To quantify the performance of the normalization methods, we calculated similarity statistics between the first two principal components of the data and data annotation (Figure 3C and Supplementary figure S9, see Materials and methods). It is clear that normalised data from the standR workflow consistently score high in the statistics comparing biology (i.e. tissue types) and consistently score low when comparing batch (i.e. slide differences). This suggests that application of the appropriate normalization strategy based on the standR workflow is able to adjust the data to retain the biological variations in the data, while minimising unwanted technical batch variations.
Comparison of DE results
DE analysis aims to detect statistically significant genes that are differentially expressed between groups of interest, which are used for the biological interpretation of the GeoMx DSP data and downstream analysis, such as pathway enrichment analysis and network analysis. Instead of applying a traditional paired T-test and LMM in the generic workflow to identify DE genes (which assumes that all genes are independent and can be strongly influenced by outliers), the standR workflow recommends the limma-voom DE pipeline, which borrows information between genes to allows a more precise estimation of biological variation (11,12). Moreover, the limma-voom pipeline (12) uses linear modelling which allows greater degrees of freedom and more statistical power and is more useful in the analysis of data from the complex experimental designs typical of GeoMx experiments.
Comparing the DE results between the generic and standR workflows, we define one comparison for each of the four datasets: kidney—comparing abnormal glomerulus in diabetic kidney to abnormal glomerulus in normal kidney; lymph node—comparing B cell zone to T cell zone in lymph node; colon—comparing longitudinal muscle layer to circular muscle layer; and brain—comparing cortical layer II/III to hippocampus CA1 areas (Supplementary file 3). By comparing the DE genes from the three methods, we observed that amongst the four datasets, the DE genes from the lymph node dataset analysis are substantially similar across the three methods (Supplementary Figures S10 and S11). However, varying degrees of overlaps were observed for the DE genes from the other datasets, suggesting many of the DE genes identified were method-specific (Supplementary Figure S10). Notably, the performance of LMM was found to be less robust in the kidney dataset, potentially due to the significantly imbalanced sample sizes between the two groups (n = 60 versus n = 12). Overall, the standR workflow detected a higher number of DE genes compared to the generic workflow either using t-test (for all comparisons) or using LMM (all comparisons except for colon dataset) (Supplementary Figure S11).
By plotting the fold-change and mean expression of genes in a comparison (i.e. MA plots) (32), even dispersion relative to the fold-change are observed in the genes with low average expression in all four comparisons, with the dispersions becoming tighter with higher expression genes (Figure 4A and Supplementary Figure S12). A skewness of the overall distribution toward negative (in the diabetic kidney data) or positive (in the colon data) log-fold-change can also be seen. This trend is more obvious in the results generated from the generic workflow, suggesting that the log-fold-change is not independent to the expression of the genes, i.e. higher expression comes with higher/lower log-fold-change, which may indicate false positive outcomes.
Figure 4.

Differential expression analysis results using standR or generic workflows. (A) MA plots visualising differential expressing genes in the diabetic kidney, lymph node, colon and brain dataset, respectively from the top to bottom using either generic with t-test (left), generic with LMM (middle) or standR with limma-voom (right). Differential expression genes generated using the voom-limma pipeline with duplicationCorrelation and applying t-tests relative to a threshold (TREAT) criterion with absolute fold change >1.2 with adjusted P-value <0.05. (B) Upset plots of biologically relevant gene-sets that were identified for each workflow and each dataset. Overall, standR was able to identify more biological relevant gene-sets.
To assess how well the DE genes identified in either workflow are representative of biological systems and processes, we perform gene-sets over-representation analysis for the up and down-regulated DE genes identified by both or unique to either workflow (Supplementary File 4). In all four datasets, more biologically relevant gene-sets were found by the standR workflow using the limma-voom method (Figure 4B) compared to those identified by the other two methods. For both kidney and lymph node datasets, all the relevant genesets were identified from DE genes from both limma and t-test methods. For the colon and brain datasets, 12 and 11 tissue-specific gene-sets (respectively) were found to be enriched only in the DE genes from the standR workflow with the limma-voom method and not from the other methods. Taken together, these findings suggest that the standR workflow is able to identify specific and biologically relevant DE genes that might otherwise have been missed.
Discussion
Spatial transcriptomics analysis allows for a greater understanding of the cellular context of disease biology (33). As one of the key pioneering platforms of this technology, the Nanostring GeoMx DSP offers the ability to study whole-genome spatial transcriptome, with over 18 000 genes for human (22 000 genes for mouse) in a high-throughput manner from both Formalin-Fixed Paraffin-Embedded (FFPE) and fresh frozen materials (7,22). It is crucial to process and analyse the Nanostring GeoMx transcriptome data carefully to identify differential expressed genes with high confidence, leading to a better understanding of spatial transcriptome profiles in the tissues of interest. Here we described standR, a Bioconductor package providing quality control, normalization and assessment, and visualization functions for GeoMx transcriptomic data, and recommended a workflow incorporating the well-established limma-voom differential expression pipeline to identify DE genes from GeoMx experiments.
There are key issues that differentiates the standR workflow from generic workflow, one of which is the gene filtering approach. In the generic workflow, the LOQ was meant for modelling the expression background in each tissue segments to allow removing genes with false signals. However, because LOQ is calculated based on the expression of negative control probes, it will be affected by the cell count and size of each ROI, as well as stickiness or other physical features related to the tissue. In our investigations of the brain dataset, the cell count per segment is negative correlated with the segment area, while the LOQ per segments are positively correlated with the area (Supplementary Figure S12). In this case, filtering genes based on LOQ threshold will remove genes with medium expression level and variance (Figure 2A and B, generic only), which may be of biological relevance. This was found in the analysed brain dataset where brain tissue-related genes such as MDGA1 and CLMP were removed by generic only (LOQ). On the other hand, for the standR workflow, the gene filtering threshold is more targeted, using direct calculation based on the expression, while accounting for the library size variation of each ROI. As such, this threshold is relatively stable, and genes with extremely low expression and variation can be accurately detected (Figure 2A, B).
GeoMx data are typically acquired in batches based on equipment throughput, a comprehensive understanding of potential biological and technical variation factors within the dataset is crucial in order to determine appropriate normalization strategies. To facilitate this exploration, the standR package provides the plotSampleInfo function, which allows users to visualize the variation factors using an alluvial plot (see Supplementary Figure S1). Using this tool, researchers can interrogate the distribution of samples across the different batches, enabling the identification of any batch-related variation in the expression data. The standR workflow further recommends using PCA in tandem with the alluvial plot to visualize data variation contributed by different factors both before and after normalization. This approach maximises the chance that the appropriate normalization techniques are employed, thereby preserving the integrity of downstream analyses. These systematic approaches encourage informed decision-making in selecting the most suitable normalization method for GeoMx data.
In this paper, we have established that the use of paired t-tests and LMM approaches on GeoMx data is inadequate to handle the complexity of the spatial dataset experimental designs. There are currently several Bioconductor packages available analysis of GeoMx spatial data, including GeoMxTools, GeoDiff and GeoMxWorkflows (34–36). These packages perform DE analysis using LMM-based methods to allow for modelling the individual as a random effect. This is a useful approach in cross-individual experiments, and in particular, GeoMxTools have been used in a recent study on breast cancer GeoMx data to identify DE genes (37). However, as shown in our benchmarking, the LMM-based approach is still limited to singular gene comparisons similar to the paired t-test, and requires a large number of replicates in the experiment to increase the degree of freedom and statistical power (38). To address the issue with cross-individual comparisons, the standR workflow recommends using the limma-voom pipeline with the duplicateCorrelation strategy, which not only borrows information from all genes using an empirical Bayes method to improve the statistical comparison, but also account for the correlation between individuals by computing consensus correlation between replicates for each gene using restricted maximum likelihood (REML) (39).
While standR leverages on the use of limma-voom pipeline for its performance in bulk RNA-seq data analyses, particular for scenarios with limited samples, there are other approaches that may also be applicable based on the data and project team preference and thus a thorough evaluation is recommended to determine the optimal approach. One other potential approach of note is to use a negative binomial mixed effect regression model, such can that implemented in the R package glmmTMB (40) which is originally designed for single-cell RNA-seq analyses. These existing alternatives emphasize the need for comprehensive investigations to select the appropriate methodology tailored to the specific characteristics of individual datasets and research objectives. It can also be beneficial to refer to positive control data from relevant experiments if they are available, for example micro-dissected bulk RNA-seq data, if available, can serve as a valuable benchmark for a particular experimental system, enabling the evaluation of different analysis choices in QC, normalisation, batch correction, and so on. Resources like the MSigDB (18), also contain centralized archives of differential expression results from diverse tissues, cells, and stimuli in the form of gene expression signatures. Integrating MSigDB for downstream functional analysis post-differential expression is typical, and usually highly informative, enhancing the interpretation of research findings.
The standR package and workflow is designed for the analysis of the GeoMx transcriptome data. For GeoMx protein data, the quality control and normalization strategies will be different as the protein panel often contains fewer than 100 markers, with additional housekeeping and IgG markers as negative control markers. Considering the usage of Nanostring GeoMx protein data in the long term and its potential to aid in therapeutics and screening, there is a necessity to develop a comprehensive workflow for analysing both protein and transcript data. With the rapid development of higher plex and finer resolution spatial technologies, specialised analysis workflows and packages, such as standR, are essential for ensuring appropriate data QC and processing. While Nanostring GeoMx DSP is reaching maturity as a technology platform, more complex and data rich technologies such as the Nanostring CosMx single molecular imaging platform (CosMx SMI) (41) are now being released. These platforms will also require specialised analysis pipelines and software in order to fully harness the power of spatial location and neighbourhood at the single cell level.
In conclusion, we describe our GeoMx analysis package, standR. We analyse the literature describing GeoMx experiments in order to identify common analytical steps and construct from the most common of these a generic analysis workflow. Then we compare the results from each step between the standR workflow and the generic workflow for four publicly available GeoMx WTA datasets. We provided evidence that standR’s application improves on the detection of biologically meaningful and nuanced results within spatial datasets in comparison to the generic workflow. Overall, we show that the standR workflow provides a comprehensive and reasonable quality control process, a better normalization strategy, and a more sophisticated differential expression analysis pipeline.
Supplementary Material
Acknowledgements
The authors thank Nanostring Technologies for releasing the publicly available GeoMx datasets, Professor Terry Speed (WEHI) for assistance in the implementation of the RUV-4 function in the standR package and James Monkman and Tony Blick of the University of Queensland for their assistance with reviewing the manuscript.
Contributor Information
Ning Liu, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia; Department of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia; South Australian immunoGENomics Cancer Institute (SAiGENCI), Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5005, Australia.
Dharmesh D Bhuva, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia; Department of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia; South Australian immunoGENomics Cancer Institute (SAiGENCI), Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5005, Australia.
Ahmed Mohamed, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia; Department of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia.
Micah Bokelund, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia.
Arutha Kulasinghe, Frazer Institute, Faculty of Medicine, The University of Queensland, Brisbane, Queensland 4102, Australia.
Chin Wee Tan, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia; Department of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia; Frazer Institute, Faculty of Medicine, The University of Queensland, Brisbane, Queensland 4102, Australia.
Melissa J Davis, Bioinformatics Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria 3052, Australia; Department of Medical Biology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia; South Australian immunoGENomics Cancer Institute (SAiGENCI), Faculty of Health and Medical Sciences, The University of Adelaide, Adelaide, SA 5005, Australia; Frazer Institute, Faculty of Medicine, The University of Queensland, Brisbane, Queensland 4102, Australia; Department of Clinical Pathology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville, VIC 3010, Australia.
Data availability
Supplementary Data are available at NAR online. The GeoMx DSP datasets used in this paper are available in the Nanostring's Spatial Organ Atlas (https://nanostring.com/products/geomx-digital-spatial-profiler/spatial-organ-atlas/). The standR package is available in Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/standR.html).
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Australian Academy of Sciences (AAS): Regional Collaborations Programme COVID-19 Digital Grants scheme for Chin Wee Tan and Arutha Kulasinghe; Dharmesh Bhuva and Melissa Davis are supported by the Grant-in-Aid Scheme administered by Cancer Council Victoria; Australian Lions Childhood Cancer Foundation; Melissa Davis is funded by the Betty Smyth Centenary Fellowship in Bioinformatics and the Cure Brain Cancer Foundation and National Breast Cancer Foundation [CBCNBCF-19-009]; WEHI acknowledges the support of the Operational Infrastructure Program of the Victorian Government. Funding for open access charge: Davis lab funds.
Conflict of interest statement. None declared.
References
- 1. The Cancer Genome Atlas (TCGA) Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ballouz S., Verleyen W., Gillis J.. Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics. 2015; 31:2123–2130. [DOI] [PubMed] [Google Scholar]
- 3. Saliba A.-E., Westermann A.J., Gorski S.A., Vogel J.. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014; 42:8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu H., Kirita Y., Donnelly E.L., Humphreys B.D.. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol. 2019; 30:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ji A.L., Rubin A.J., Thrane K., Jiang S., Reynolds D.L., Meyers R.M., Guo M.G., George B.M., Mollbrink A., Bergenstrahle J.et al.. Multimodal analysis of composition and spatial architecture in Human squamous cell carcinoma. Cell. 2020; 182:1661–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang S., Chan C.N., Rovira-Clave X., Chen H., Bai Y., Zhu B., McCaffrey E., Greenwald N.F., Liu C., Barlow G.L.et al.. Combined protein and nucleic acid imaging reveals virus-dependent B cell and macrophage immunosuppression of tissue microenvironments. Immunity. 2022; 55:1118–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merritt C.R., Ong G.T., Church S.E., Barker K., Danaher P., Geiss G., Hoang M., Jung J., Liang Y., McKay-Fleisch J.et al.. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020; 38:586–599. [DOI] [PubMed] [Google Scholar]
- 8. Moses L., Pachter L.. Museum of spatial transcriptomics. Nat. Methods. 2022; 19:534–546. [DOI] [PubMed] [Google Scholar]
- 9. Peixoto L., Risso D., Poplawski S.G., Wimmer M.E., Speed T.P., Wood M.A., Abel T.. How data analysis affects power, reproducibility and biological insight of RNA-seq studies in complex datasets. Nucleic Acids Res. 2015; 43:7664–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., Szczesniak M.W., Gaffney D.J., Elo L.L., Zhang X.et al.. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seyednasrollah F., Laiho A., Elo L.L.. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief Bioinform. 2015; 16:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ponten F., Jirstrom K., Uhlen M.. The Human Protein Atlas–a tool for pathology. J. Pathol. 2008; 216:387–393. [DOI] [PubMed] [Google Scholar]
- 16. Kuznetsova A., Brockhoff P.B., Christensen R.H.. lmerTest package: tests in linear mixed effects models. J. Stat. Softw. 2017; 82:1–26. [Google Scholar]
- 17. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Righelli D., Weber L.M., Crowell H.L., Pardo B., Collado-Torres L., Ghazanfar S., Lun A.T.L., Hicks S.C., Risso D. SpatialExperiment: infrastructure for spatially-resolved transcriptomics data in R using bioconductor. Bioinformatics. 2022; 38:3128–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y., Lun A.T., Smyth G.K.. From reads to genes to pathways: differential expression analysis of RNA-seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016; 5:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zollinger D.R., Lingle S.E., Sorg K., Beechem J.M., Merritt C.R.. GeoMx RNA assay: high multiplex, digital, spatial analysis of RNA in FFPE tissue. Methods Mol. Biol. 2020; 2148:331–345. [DOI] [PubMed] [Google Scholar]
- 23. The Genotype-Tissue Expression Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang S., Yang E., Kim D., Kim H., Kim E. Clmp regulates AMPA and kainate receptor responses in the neonatal hippocampal CA3 and kainate seizure susceptibility in mice. Front. Synaptic Neurosci. 2020; 12:567075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen A., Liao S., Cheng M., Ma K., Wu L., Lai Y., Qiu X., Yang J., Xu J., Hao S.et al.. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022; 185:1777–1792. [DOI] [PubMed] [Google Scholar]
- 26. Takeuchi A., O’Leary D.D. Radial migration of superficial layer cortical neurons controlled by novel ig cell adhesion molecule MDGA1. J. Neurosci. 2006; 26:4460–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J., Kim S., Kim H., Hwang I.W., Bae S., Karki S., Kim D., Ogelman R., Bang G., Kim J.Y.et al.. MDGA1 negatively regulates amyloid precursor protein-mediated synapse inhibition in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2115326119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gandolfo L.C., Speed T.P.. RLE plots: visualizing unwanted variation in high dimensional data. PLoS One. 2018; 13:e0191629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gagnon-Bartsch J.A., Jacob L., Speed T.P.. Removing Unwanted Variation from High Dimensional Data with Negative Controls. 2013; Berkeley: Tech Reports from Dep Stat Univ California; 1–112. [Google Scholar]
- 31. Risso D., Ngai J., Speed T.P., Dudoit S.. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014; 32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McDermaid A., Monier B., Zhao J., Liu B., Ma Q.. Interpretation of differential gene expression results of RNA-seq data: review and integration. Brief Bioinform. 2019; 20:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marx V. Method of the Year: spatially resolved transcriptomics. Nat. Methods. 2021; 18:9–14. [DOI] [PubMed] [Google Scholar]
- 34. Ortogero N., Y.Z. V.R., Griswold M., Henderson D. 2022; Bioconductor, Vol. R package version 3.2.0.
- 35. Yang L., Yang Z., Danaher P., Zimmerman S., Hether T., Henderson D., Beechem J.. Background modeling, Quality Control and Normalization for GeoMx RNA data with GeoDiff. 2022; bioRxiv doi:29 May 2022, preprint: not peer reviewed 10.1101/2022.05.26.493637. [DOI]
- 36. Reeves J., D.P. O.N., Griswold M., Yang Z., Zimmerman S., Vitancol R., David H. 2022; R package version 1.5.0. ed. Bioconductor 3.16.
- 37. Bergholtz H., Carter J.M., Cesano A., Cheang M.C.U., Church S.E., Divakar P., Fuhrman C.A., Goel S., Gong J., Guerriero J.L.et al.. Best practices for spatial profiling for breast cancer research with the GeoMx((R)) Digital Spatial Profiler. Cancers (Basel). 2021; 13:4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004; 3:Article3. [DOI] [PubMed] [Google Scholar]
- 39. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brooks M.E., Kristensen K., Van Benthem K.J., Magnusson A., Berg C.W., Nielsen A., Skaug H.J., Machler M., Bolker B.M.. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. R Journal. 2017; 9:378–400. [Google Scholar]
- 41. Lewis Z.R., Phan-Everson T., Geiss G., Korukonda M., Bhatt R., Brown C., Dunaway D., Phan J., Rosenbloom A., Filanoski B.. Subcellular characterization of over 100 proteins in FFPE tumor biopsies with CosMx Spatial Molecular Imager. Cancer Res. 2022; 82:3878–3878. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplementary Data are available at NAR online. The GeoMx DSP datasets used in this paper are available in the Nanostring's Spatial Organ Atlas (https://nanostring.com/products/geomx-digital-spatial-profiler/spatial-organ-atlas/). The standR package is available in Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/standR.html).