

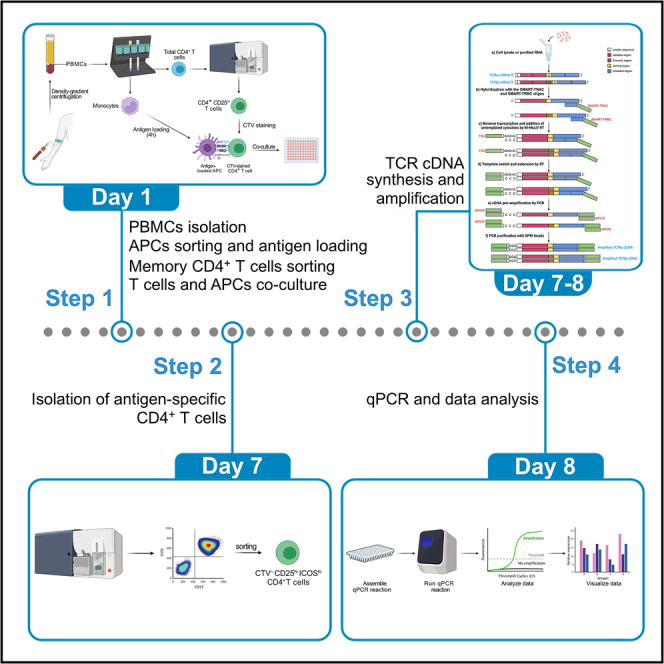
Summary
Identifying defined T cell clones within a polyclonal population is key to clarifying their phenotype and function. Here, we present a protocol for detecting specified T cell clones in a heterogeneous cell population. We describe steps for stimulating human CD4+ T cells isolated from blood with a protein antigen, sorting antigen-specific cells by fluorescence-activated cell sorting, and detecting among these the presence of predefined T cell clones, based on their T cell receptor (TCR). TCR cDNA is amplified through 5′-RACE (TCR-SMART) and detected by qPCR.
For complete details on the use and execution of this protocol, please refer to Notarbartolo et al. (2021).1
Subject areas: Bioinformatics, Cell isolation, Flow Cytometry, Gene Expression, Immunology, Molecular Biology, Sequence analysis, Single Cell
Graphical abstract
Highlights
-
•
In vitro stimulation of human CD4+ T cells with a protein antigen
-
•
Antigen-specific human CD4+ T cells identification and isolation by FACS
-
•
Unbiased targeted amplification of TCR cDNA from low input samples
-
•
Detection of defined T cell clones within a heterogeneous population by qPCR
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Identifying defined T cell clones within a polyclonal population is key to clarifying their phenotype and function. Here, we present a protocol for detecting specified T cell clones in a heterogeneous cell population. We describe steps for stimulating human CD4+ T cells isolated from blood with a protein antigen, sorting antigen-specific cells by fluorescence-activated cell sorting, and detecting among these the presence of predefined T cell clones, based on their T cell receptor (TCR). TCR cDNA is amplified through 5′-RACE (TCR-SMART) and detected by qPCR.
Before you begin
This protocol describes the steps to isolate human CD4+ T cells from peripheral blood, stimulate them with a recombinant protein antigen, identify and FACS sort antigen-specific T cells based on cell proliferation and the upregulation of activation-induced markers, and detect the presence of pre-defined T cell clones in a population of interest, based on their T cell receptor (TCR) sequence.
Using this protocol, we demonstrated, for instance, that moderately expanded CD4+ T cell clones identified by scTCR-seq in PBMCs from patients with COVID-19 included clones specific to SARS-CoV-2 antigens.1
Before starting, you need to identify the complementarity-determining region (CDR) sequence of the TCR(s) you want to detect and design target-specific qPCR primers. Also, verify to have all the reagents and tools described in the protocol.
The protocol is versatile and can be adapted to different experimental settings. Plan all the steps in advance and adjust them to your purpose.
Identification of TCR sequences in single-cell TCR-seq data
Timing: 3 h
This step describes how to extract information on T cell clone-specific TCRα and TCRβ sequences from V(D)J-sequencing datasets generated with the Chromium Single Cell Immune Profiling kit (10× Genomics).
Note: the Cell Ranger software (10× Genomics) requires a workstation with a minimum of an 8-core processor, 64 GB RAM, and 1 TB of free storage space to function. The Python script, instead, is computationally not demanding and can be run on a personal computer. The timing indicated at the beginning of the paragraph refers to the analysis of a single V(D)J-sequencing dataset, where the Cell Ranger analysis was performed on a computer node equipped with a 64-core processor and 256 GB RAM, and the Python script was run in an interactive session on a machine with a 128-core processor and 2 TB RAM.
-
1.To extract the information about T cell clones TCR sequences from single-cell TCR-seq data follow the sub-steps described below.
-
a.Process the single-cell V(D)J-sequencing data with Cell Ranger using the following parameters: cellranger vdj --id=<sample id> --fastqs=<path to fastqs folder> --sample=<sample name> --reference=/path/to/refdata-cellranger-vdj-GRCh38-alts-ensembl-4.0.0 --localcores=8 --localmem=64.Note: the Cellranger vdj script requires that fastq files have already been demultiplexed and that your computational platform meets the minimum hardware requirements previously described. For an accurate description of Cell Ranger use and output refer to the official guide at https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/using/vdj.
 CRITICAL: We recommend using Cell Ranger v6.0 or above as these versions automatically generate a file containing the required information on CDR1, CDR2, and CDR3 sequences leaving to the researcher only the need of filtering the sequences of interest. Use barcodes to filter the output files containing the annotated information.
CRITICAL: We recommend using Cell Ranger v6.0 or above as these versions automatically generate a file containing the required information on CDR1, CDR2, and CDR3 sequences leaving to the researcher only the need of filtering the sequences of interest. Use barcodes to filter the output files containing the annotated information. -
b.Among the Cell Ranger output files, select the “filtered_contig_annotation.csv” file, a tabular file of productive contig sequences for each cell with columns containing framework (fwr) and CDR annotation.CRITICAL: use this file instead of the “clonotypes.csv” file, because the latter includes the clonotypes defined only based on CDR3 sequences, while the information on CDR1 and CDR2 sequences is not retained.
-
c.Run the following scripts to select the T cell clones of interest and obtain their CDR sequences.Note: execution of the Python scripts requires that the cell barcodes of interest have already been selected and stored in a text file. The script assumes that all files are in the same directory and the use of CDR nucleotide sequences instead of amino acidic sequences.#!/usr/bin/env pythonimport pandas as pd#Import the file containing cell contigscontig_path="/cellranger/outs/filtered_contig_annotations.csv"contig=pd.read_csv(contig_path)#Import the file containing cell barcodesbarcodes = pd.read_csv("./barcodes.txt", header=None)#Convert it in a listbarcodes = barcodes[0].tolist()#Filter out csv lines not containing the desired barcodescontig = contig[contig.barcode.isin(barcodes)].copy()#Reconstruct all the nt sequencescontig['sequence_nt'] = (contig['fwr1_nt'] +contig['cdr1_nt'] +contig['fwr2_nt'] +contig['cdr2_nt'] +contig['fwr3_nt'] +contig['cdr3_nt'] +contig['fwr4_nt'])#Reconstruct all the aa sequencescontig['sequence'] = (contig['fwr1'] +contig['cdr1'] +contig['fwr2'] +contig['cdr2'] +contig['fwr3'] +contig['cdr3'] +contig['fwr4'])#Eliminate duplicates using nt sequencesdeduplicated = contig[['barcode','chain','sequence_nt']].copy()#Reshape data structurededuplicated_pv = deduplicated.pivot(index='barcode',columns='chain',values='sequence_nt')#Keep only one occurrence for duplicates with the same 'TRA' or 'TRB' valuesdeduplicated_df = (deduplicated_pv.drop_duplicates(subset=['TRA', 'TRB'],keep='first'))#Keep only TCR sequences of interestcontig = contig[contig.barcode.isin(deduplicated_df.index)]#Write resultscontig.to_csv('./unique_selected_contig.csv')Note: examples of the file formats used in this step are reported in the supplemental information (Data S1).
-
a.
qPCR primer design
-
2.Design qPCR primers specific for the identification of the T cell clone(s) of interest. You can use Primer3 (v4.1.0)2 with default parameters, except for the “Product Size Range” which is set at 80–150 bases to be used in combination with a fast qPCR protocol.Note: other primer design tools or a manual design can be employed based on the user’s experience.
-
a.Set the “Targets” parameter to design qPCR primers binding the CDR1 (forward primer) and CDR3 (reverse primer) regions of the TCR of interest to maximize their specificity.
-
b.If Primer3 does not provide any primer as output, you can modify some parameters, such as “Primer size”, “Primer Tm”, and “Primer GC%”, to make them less stringent. As an alternative, try to partially shift the target region.CRITICAL: validate the quality of designed qPCR primers by verifying they generate a unique PCR product. For instance, you can check that the dissociation curve after the qPCR run has a single peak or visualize the PCR product, which should appear as a single band, on an agarose gel.
-
a.
Institutional permissions
The human blood samples analyzed in this protocol were collected according to the guidelines of a study protocol approved by the Institutional Review Board Milano Area 2. We remind readers to obtain all the necessary permissions from the relevant institutions before starting the experiment.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD4-BUV395 (clone SK3) (dilution used 1:100) | BD Biosciences | Cat. Num. 563550; RRID:AB_2738273 |
| Anti-human CD8-FITC (clone RPA-T8) (dilution used 1:20) | BD Biosciences | Cat. Num. 561947; RRID: AB_10894003 |
| Anti-human CD14-Alexa Fluor 488 (clone MφP9) (dilution used 1:100) | BD Biosciences | Cat. Num. 562689; RRID: AB_2737723 |
| Anti-human CD19-BB515 (clone HIB19) (dilution used 1:100) | BD Biosciences | Cat. Num. 564456; RRID: AB_2744309 |
| Anti-human CD56-BB515 (clone B159) (dilution used 1:100) | BD Biosciences | Cat. Num. 564488; RRID: AB_2744428 |
| Anti-human CD45RA-Qdot655 (clone MEM-56) (dilution used 1:400) | Thermo Fisher Scientific | Cat. Num. Q10069; RRID: AB_2556451 |
| Anti-human CCR7-BV711 (clone 150503) (dilution used 1:100) | BD Biosciences | Cat. Num. 566602; RRID: AB_2739758 |
| Anti-human CD25-PE (clone BC96) (dilution used 1:200) | BioLegend | Cat. Num. 302605; RRID: AB_314275 |
| Anti-human ICOS-PE-Cy7 (clone DX29) (dilution used 1:100) | BD Biosciences | Cat. Num. 567395; RRID: AB_2916579 |
| Biological samples | ||
| Human blood draws from patients with mild COVID-19 | Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Infectious Diseases Unit, Milan, Italy. Notarbartolo et al. | N/A |
| Human serum | Swiss Blood Center Basel | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Ficoll-Paque PLUS | Cytiva | Cat. Num. 17144003 |
| DPBS 1X, no calcium, no magnesium | Thermo Fisher Scientific Gibco | Cat. Num. 14190094 |
| RPMI 1640 medium, no glutamine | Thermo Fisher Scientific Gibco | Cat. Num. 31870025 |
| RPMI 1640 medium, HEPES, no glutamine | Thermo Fisher Scientific Gibco | Cat. Num. 42401018 |
| GlutaMAX supplement (100X) | Thermo Fisher Scientific Gibco | Cat. Num. 35050038 |
| MEM non-essential amino acids (NEAA) solution (100X) | Thermo Fisher Scientific Gibco | Cat. Num. 11140035 |
| Sodium pyruvate (100 mM) | Thermo Fisher Scientific Gibco | Cat. Num. 11360039 |
| Penicillin-Streptomycin (5,000 U/mL) | Thermo Fisher Scientific Gibco | Cat. Num. 15070063 |
| Kanamycin sulfate | Thermo Fisher Scientific Gibco | Cat. Num. 15160054 |
| 2-Mercaptoethanol (50 mM) | Thermo Fisher Scientific Gibco | Cat. Num. 31350010 |
| Ethylenediaminetetraacetic acid (EDTA) disodium salt solution, 0.5 M | Merck Sigma-Aldrich | Cat. Num. E7889-100ML |
| Recombinant SARS-CoV-2 RBD and N proteins | Fondazione Istituto Nazionale Genetica Molecolare, Milan, Italy. Notarbartolo et al. | N/A |
| Recombinant human interleukin-2 (IL-2) | Miltenyi Biotec | Cat. Num. 130-097-744 |
| Phytohemagglutinin (PHA) | Thermo Fisher Scientific Remel | Cat. Num. R30852801 |
| BD Horizon brilliant stain buffer | BD Biosciences | Cat. Num. 566349 |
| Nuclease-free water (not DEPC-treated) | Thermo Fisher Scientific Invitrogen | Cat. Num. AM9937 |
| Triton X-100 | Merck Sigma-Aldrich | Cat. Num. X100-100ML |
| RiboLock RNase inhibitor (40 U/μL) | Thermo Fisher Scientific | Cat. Num. EO0381 |
| Ethanol, for molecular biology | Merck Sigma-Aldrich | Cat. Num. 51976-500ML-F |
| Tris buffer, 1.0 M, pH 8.0, molecular biology grade | Merck Millipore | Cat. Num. 648314-100ML |
| Critical commercial assays | ||
| CD14 MicroBeads UltraPure, human | Miltenyi Biotec | Cat. Num. 130-118-906 |
| CD4 MicroBeads, human | Miltenyi Biotec | Cat. Num. 130-045-101 |
| MS columns | Miltenyi Biotec | Cat. Num. 130-042-201 |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | Cat. Num. C34557 |
| Maxima H minus reverse transcriptase (200 U/μL) | Thermo Fisher Scientific | Cat. Num. EP0752 |
| KAPA HiFi HotStart PCR Kit (250 U) | Roche | Cat. Num. 07958897001 |
| SPRIselect beads | Beckman Coulter | Cat. Num. B23317 |
| Qubit dsDNA high-sensitivity (HS) kit | Thermo Fisher Scientific Invitrogen | Cat. Num. Q32854 |
| Oligonucleotides | ||
| Oligo(dT)20 primer | Thermo Fisher Scientific Invitrogen | Cat. Num. 18418020 |
| Template-switch oligo (TSO): 5′-AAGCAGTGG TATCAACGCAGAGTACATrGrG+G-3’. Note: at the 3′ end of the oligo, there are two riboguanosines (rG) and one LNA-modified guanosine (+G) to facilitate template switching. The TSO bears the same adapter sequence present in the SMART-TRAC, SMART-TRBC, and ISPCR oligos. |
QIAGEN; Picelli et al. | N/A |
| SMART-TRAC oligo: AAGCAGTGG TATCAACGCAGAGTACACACATCA GAATCCTTACTTTG. Note: this oligo specifically anneals to the TRAC gene, which codes for the constant region of the TCR alpha chain. It also bears the same adapter sequence present in the TSO and the ISPCR oligos. |
QIAGEN; This paper | N/A |
| SMART-TRBC oligo: AAGCAGTGGTAT CAACGCAGAGTACCAGTATCTGG AGTCATTGA. Note: this oligo specifically anneals to the TRBC1 and TRBC2 genes, which code for the constant region of the TCR beta chain. It also bears the same adapter sequence present in the TSO and the ISPCR oligos. |
QIAGEN; This paper | N/A |
| ISPCR oligo: 5′-AAGCAGTGGTAT CAACGCAGAGT-3’. Note: this oligo guides the unbiased amplification of the TCR cDNA. It works both as a forward and reverse PCR primer by binding to the adapter sequence present in the TSO, SMART-TRAC, and SMART-TRBC oligos. |
Eurofins; Picelli et al. | N/A |
| qPCR primers. See Table 1. | Eurofins; This paper | N/A |
| Software and algorithms | ||
| Cell Ranger v7.1 | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger; RRID: SCR_017344 |
| Primer3web v4.1.0 | Untergasser et al. | https://primer3.ut.ee/; RRID: SCR_003139 |
| FlowJo v10.8 | BD Biosciences | https://www.flowjo.com/solutions/flowjo; RRID: SCR_008520 |
| Prism v10 | GraphPad Software | https://www.graphpad.com/; RRID: SCR_000306 |
| Python v3.10.5 | Python Software Foundation |
https://www.python.org/psf-landing/; RRID: SCR_008394 |
| Other | ||
| MACS MultiStand | Miltenyi Biotec | Cat. Num. 130-042-303 |
| MiniMACS separator | Miltenyi Biotec | Cat. Num. 130-090-312 |
| MACSmix tube rotator | Miltenyi Biotec | Cat. Num. 130-090-753 |
| 40 μm cell strainer | Corning | Cat. Num. 352340 |
| BD FACSAria III SORP cell sorter | BD Biosciences | RRID: SCR_016695 |
| PCR thermal cycler (e.g., Biometra TRIO) | Analytik Jena GmbH | Cat. Num. 846-2-070-724 |
| DNA LoBind tubes, 1.5 mL | Eppendorf | Cat. Num. 0030108051 |
| DynaMag-2 magnet | Thermo Fisher Scientific Invitrogen | Cat. Num. 12321D |
| Qubit fluorometer (2.0 or later) | Thermo Fisher Scientific Invitrogen | Cat. Num. Q32866; RRID: SCR_020553 |
| Axygen 96-well PCR microplates | Corning | Cat. Num. PCR-96-LP-AB-C |
| Axygen 70 μm ultra clear pressure sensitive sealing film for real-time PCR | Corning | Cat. Num. UC-500 |
| Fast SYBR green master mix | Thermo Fisher Scientific Applied Biosystems | Cat. Num. 4385612 |
| Real-time PCR system (e.g., QuantStudio 5 or StepOnePlus) | Thermo Fisher Scientific Applied Biosystems | RRID: SCR_020240; RRID: SCR_015805 |
Table 1.
qPCR primers
| TRAC_Fwd | AGAACCCTGACCCTGCCG |
| TRAC_Rev | ATCAAAATCGGTGAATAGGCAGA |
| TRBC_Fwd | AAGCAGAGATCTCCCACACC |
| TRBC_Rev | CTCCTTCCCATTCACCCACC |
| Clone_X_TCRa_Fwd | TGAGTATGTGTATTGGTATCGACA |
| Clone_X_TCRa_Rev | TACCTCCTCCGACTCTGACG |
| Clone_X_TCRb_Fwd | GCCGTTCCCTGGACTTTCA |
| Clone_X_TCRb_Rev | AACTCCAGCACTGCAGATGT |
| Clone_Y_TCRa_Fwd | CAGTCTCTGGAAACCCTTATCTT |
| Clone_Y_TCRa_Rev | TGAAGCCTCCAGTGTTGTCTC |
| Clone_Y_TCRb_Fwd | GCCGTTCCCTGGACTTTCA |
| Clone_Y_TCRb_Rev | ATTATCTCTAGCACTGCAGATGT |
Materials and equipment
Alternatives: This protocol describes a validated custom procedure to isolate PBMCs from blood based on density-gradient centrifugation with Ficoll-Paque Plus. As an alternative, you can refer to the Ficoll-Paque Plus manufacturer’s protocol. (https://cdn.cytivalifesciences.com/api/public/content/digi-16156-pdf).
Alternatives: This protocol uses the Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific) for the full-length cDNA synthesis. You can use any other retrotranscriptase tested to be able to add untemplated cytosines at the 3′ end of cDNA and to perform the template switch. For instance, the Superscript II reverse transcriptase (Thermo Fisher Scientific Invitrogen, cat. no. 18064–014) has been successfully used by others in similar applications.3
Alternatives: This protocol uses 1.5 mL DNA low-binding tubes to perform the SPRI-based purification of the amplified DNA. PCR plates or 0.2 mL tubes can also be used depending on the throughput of the experiment and the type of magnetic stand available.
Alternatives: This protocol uses the Fast SYBR Green Master Mix (Thermo Fisher Scientific Applied Biosystems) for the qPCR reaction. Any other qPCR kit based on a fluorescent DNA intercalating agent, such as SYBR Green, can be used paying attention to adjusting the thermocycler protocol according to the requirements of the kit used.
Alternatives: This protocol runs the qPCR reaction in a 96-well qPCR plate. If a qPCR machine with a 384-well block is available, the qPCR reaction can be run in this format to increase the throughput and cut the cost of the experiment.
Wash buffer: RPMI 1640 medium with HEPES supplemented with 0.5% human serum.
Filter through a 0.22 μm PES membrane. Store at 4°C for up to 2 months.
FACS buffer: DPBS 1× supplemented with 0.5% human serum.
Filter through a 0.22 μm PES membrane. Store at 4°C for up to 2 months.
MACS buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DPBS | 1× | 241.5 mL |
| Human serum | 3% (v/v) | 7.5 mL |
| 0.5 M EDTA | 2 mM | 1 mL |
| Total | 250 mL |
Filter through a 0.22 μm PES membrane. Store at 4°C for up to 2 months.
T cell medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | 224.75 mL | |
| 100 × GlutaMAX Supplement | 1% (v/v) | 2.5 mL |
| 100 mM MEM NEAA | 1 mM | 2.5 mL |
| 100 mM Sodium pyruvate | 1 mM | 2.5 mL |
| 50 mM 2-Mercaptoethanol | 50 μM | 250 μL |
| 5,000 U/mL Penicillin 5,000 μg/mL Streptomycin |
50 U/mL 50 μg/mL |
2.5 mL |
| 100 × Kanamycin | 1% (v/v) | 2.5 mL |
| Human serum | 5% (v/v) | 12.5 mL |
| Total | 250 mL |
Filter through a 0.22 μm PES membrane. Store at 4°C for up to 1 month.
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Nuclease-free H2O | 95.5 μL | |
| 10% (v/v) Triton X-100 | 0.2% (v/v) | 2 μL |
| (40 U/μL) RiboLock RNase inhibitor | 1 U | 2.5 μL |
| Total | 100 μL |
Prepare a larger stock solution without the RNase inhibitor. Filter through a 0.22 μm PES membrane. Store at 4°C for up to 3 months. Add the RNase inhibitor to the working aliquot just before use.
Elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Nuclease-free H2O | 9.9 mL | |
| 1 M Tris-Cl pH 8.0 | 10 mM | 0.1 mL |
| Total | 10 mL |
Filter through a 0.22 μm PES membrane. Store at 4°C for up to 3 months.
Step-by-step method details
Isolation of PMBCs
This step allows you to isolate PBMCs from blood.
-
1.
Take the RPMI 1640 medium (with HEPES) and the wash buffer out from the fridge and let them reach 20°C–25°C.
-
2.Isolate PBMCs from blood by density-gradient centrifugation with Ficoll-Paque Plus.
-
a.Dilute blood with RPMI 1640 medium 1:2 (e.g., 20 mL blood with 40 mL RPMI 1640 medium) and mix by gently pipetting.
-
b.Aliquot 10 mL of Ficoll-Paque Plus in two 50 mL conical tubes.
-
c.Slowly add 30 mL of diluted blood on top of the Ficoll-Paque Plus in each tube by pipetting it at a 2–3 s/mL rate, while tilting the tube to maximize the Ficoll-Paque Plus surface.CRITICAL: too fast pipetting results in the mixing of blood and Ficoll-Paque Plus solution and impairs the efficiency of PBMC separation.
-
d.Centrifuge at 800 × g for 30 min at 18°C–20°C in a swing-out rotor centrifuge with minimum acceleration and brake.Note: only this centrifugation is performed with minimal acceleration and brake; use full acceleration and brake in the next steps. At the end of this centrifugation step, PBMCs form a ring (buffy-coat) in the middle of the tube, between the plasma and the semi-transparent Ficoll-Paque Plus layers.
-
e.Collect the buffy-coat by slowly aspirating with a 1 mL pipette from the top, trying to minimize the carry-over of Ficoll-Paque Plus from the lower layer. Combine the buffy-coats from the two tubes in a new 50 mL conical tube.
-
f.Thoroughly wash the PBMCs by adding 20 mL of RPMI 1640 medium and pipetting up and down 3–4 times, while avoiding bubbling.Note: this step serves to remove the residual Ficoll-Paque Plus.
-
g.Centrifuge at 450 × g for 15 min at 18°C–20°C.
-
h.Aspirate the supernatant and resuspend the pellet in 10 mL of wash buffer.
-
i.Transfer the cell suspension into a 15 mL conical tube and centrifuge at 300 × g for 10 min at 18°C–20°C.
-
j.Aspirate the supernatant, resuspend the isolated PBMCs in 10 mL of wash buffer, and count them. You should get 1–2 × 106 cells per mL of blood and, thus, 20–40 × 106 PBMCs starting from a 20 mL blood draw. Troubleshooting 1.CRITICAL: when erythrocytes in whole blood are aggregated, some mononuclear cells are trapped in the clumps and sediment with the erythrocytes. This tendency to trap mononuclear cells is reduced by diluting the blood. Aggregation of erythrocytes is enhanced at higher temperatures (37°C), which consequently decreases the yield of mononuclear cells. At lower temperatures (4°C) the rate of aggregation is decreased but the time of separation is increased, which also decreases the yield of mononuclear cells. A compromise temperature of 18°C–20°C gives optimal results.
 Pause point: if you isolate PBMCs from a large blood donation, you can store the PBMCs in wash buffer for 12–16 h at 4°C.Note: if you isolate PBMCs from a small blood draw, we recommend proceeding directly to the next steps.
Pause point: if you isolate PBMCs from a large blood donation, you can store the PBMCs in wash buffer for 12–16 h at 4°C.Note: if you isolate PBMCs from a small blood draw, we recommend proceeding directly to the next steps.
-
a.
Magnetic sorting of CD14+ monocytes
This step enriches CD14+ monocytes by positive selection with magnetic CD14 MicroBeads.
Note: use cold MACS buffer and perform centrifugations at 4°C throughout this step.
-
3.Change the buffer of PBMC suspension from wash buffer to MACS buffer:
-
a.Centrifuge PBMCs at 300 × g for 10 min.
-
b.Resuspend cells in 5 mL of MACS buffer.
-
c.Centrifuge again at 300 × g for 10 min.
-
d.Resuspend 20–25 × 106 PBMCs in 160 μL of MACS buffer.
-
a.
-
4.Label PBMCs with CD14 MicroBeads:
-
a.Vortex CD14 MicroBeads for 20 s to resuspend them well.
-
b.Add 40 μL of CD14 MicroBeads and mix.
-
c.Incubate at 4°C–8°C for 20 min.
-
d.Add 4 mL of MACS buffer.
-
e.Centrifuge at 300 × g for 10 min.
-
f.Resuspend cells in 3 mL of MACS buffer.
-
a.
-
5.Isolate CD14+ cells (monocytes):
-
a.Place an MS column on a MACS magnetic separator.
-
b.Prime the MS column with 1 mL of MACS buffer.
-
c.Apply the magnetically labeled cells on top of the MS Column.
-
d.Collect the flow-through (unlabeled CD14– cells) into a 15 mL conical tube, do not discard.
-
e.Wash the MS column twice with 2 mL of MACS buffer.
-
f.Collect the effluent as CD14– cell fraction into a 15 mL conical tube, as described in sub-step d.
-
g.Temporarily store the CD14– cell fraction on ice.Note: the CD14– cell fraction will be used to isolate CD4+ cells (see step 8).
-
h.Remove the MS column from the magnetic field of the MACS separator and place it on a suitable collection tube (e.g., a 15 mL conic tube).
-
i.Pipette 3 mL of MACS buffer and firmly flush out the CD14+ cell fraction using the plunger supplied with the column.
-
j.Remove the plunger, pipette an additional 3 mL of MACS buffer, and repeat the flushing to collect all the CD14+ cells.
-
k.Count the CD14+ cells and proceed to the next step.Note: monocytes are expected to be 10%–20% of the total PBMCs.
-
a.
Irradiation of CD14+ cells and antigen loading
This step causes DNA damage that prevents cells from dividing and selectively kills residual lymphocytes, which are more sensitive than monocytes to ionizing radiations. The irradiated monocytes are still alive and can produce cytokines and other soluble factors. In addition, irradiation can induce the upregulation of some co-stimulatory molecules on antigen-presenting cells (APCs).4,5
Note: use cold wash buffer and perform centrifugations at 4°C until step 6 g; then, use warm T cell medium and perform centrifugations at 20°C–23°C.
-
6.Irradiate CD14+ cells:
-
a.Centrifuge CD14+ cells at 300 × g for 10 min.
-
b.Resuspend cells in 5 mL of wash buffer.
-
c.Irradiate with 45 Gy of gamma-ray.Note: it also works in the range of 30–60 Gy.
-
d.Centrifuge irradiated CD14+ cells at 300 × g for 10 min.
-
e.Resuspend cells in 5 mL of wash buffer.
-
f.Repeat the sub-steps d. and e. once again.
-
g.Centrifuge irradiated CD14+ cells at 300 × g for 10 min.
-
h.Resuspend cells in 2 mL of T cell medium and count them.
-
a.
-
7.Load irradiated CD14+ cells with a recombinant protein antigen:
-
a.Centrifuge CD14+ cells at 300 × g for 10 min.
-
b.Resuspend cells at a concentration of 1 × 106 cells/mL in warm T cell medium.
-
c.Add the recombinant protein antigen at a concentration of 5 μg/mL.Note: leave an aliquot of monocytes unloaded as a negative control for the antigen-specific stimulation.
-
d.Incubate at 37°C under slow rotation for 4 h.Note: during this incubation step the recombinant protein is internalized and processed by APCs. The process of antigen uptake, processing, and presentation continues during the co-culture with T cells.Optional: the antigen-loading incubation can be adjusted in a range from 2 h to 6 h depending on the experimental design and the antigen used. For instance, 6 h may be needed in the case you use microbial lysates as an antigen source and you want to wash away the unloaded antigen before the co-culture with T cells because it may be an undesired source of stimulatory pathogen-associated molecular patterns (PAMPs). On the contrary, 2 h is enough in the case of purified peptide antigens, which replace the peptides already exposed on the APCs’ major histocompatibility complex class II (MHC-II).
-
a.
Sorting of memory CD4+ T cells
This step enriches, first, CD4+ T cells by positive selection with magnetic CD4 MicroBeads. Then, it enables the isolation of memory CD4+ T lymphocytes by fluorescence-activated cell sorting (FACS).
FACS sorting time may vary depending on the FACS facility.
Note: perform this step during the 4 h incubation time needed for antigen loading on APCs. Use cold MACS buffer and perform centrifugations at 4°C throughout this step.
Optional: You can pass the CD14– cell fraction through a new MS column to remove residual CD14+ cells that were eventually not retained by the MACS separator magnetic field to avoid possible contamination of the CD4+ cell fraction by residual CD14+ cells. This step is not necessary if you are sorting T cell subsets by FACS later.
-
8.Label CD14– cells with CD4 MicroBeads:
-
a.Centrifuge the CD14– cell fraction at 300 × g for 10 min.
-
b.Resuspend 20 × 106 cells in 160 μL of MACS buffer.
-
c.Vortex CD4 MicroBeads for 20 s to resuspend them well.
-
d.Add 40 μL of CD4 MicroBeads and mix.
-
e.Incubate at 4°C–8°C for 20 min.
-
f.Add 4 mL of MACS buffer.
-
g.Centrifuge at 300 × g for 10 min.
-
h.Resuspend cells in 3 mL of MACS buffer.
-
a.
-
9.Isolate CD4+ cells (T helper lymphocytes):
-
a.Place an MS column on a MACS magnetic separator.
-
b.Prime the MS column with 1 mL of MACS buffer.
-
c.Apply the magnetically labeled cells on top of the MS Column.
-
d.Collect the unlabeled CD14– CD4– cells into a 15 mL conical tube. Discard or use for other experiments.
-
e.Wash the MS column twice with 2 mL of MACS buffer.
-
f.Collect the effluent as CD14– CD4– cell fraction into a 15 mL conical tube, as described in sub-step d.
-
g.Remove the MS column from the magnetic field of the MACS separator and place it on a suitable collection tube (e.g., a 15 mL conic tube).
-
h.Pipette 3 mL of MACS buffer and firmly flush out the CD4+ cell fraction using the plunger supplied with the column.
-
i.Remove the plunger, pipette an additional 3 mL of MACS buffer, and repeat the flushing.
-
i.Count the CD4+ cells and proceed to the next step.
-
i.
-
a.
Note: CD4+ T cells are expected to be 30%–50% of the total PBMCs.
Optional: You can store the CD14– CD4– cell fraction in a freezing buffer at –150°C and use it for other experimental purposes.
Optional: If you are performing cell magnetic sorting for the first time, you may verify the purity of the sorted cells by flow cytometry after staining the sorted populations with the relevant antibody (e.g., CD14 for the CD14+ cell fraction and CD4 for the CD4+ cell fraction). The expected purity is > 95%.
-
10.Isolate memory CD4+ T lymphocytes by FACS:
-
a.Prepare the staining solution by mixing the following antibodies in 250 μL of MACS buffer: CD8-FITC (diluted 1:20), CD14-AF488 (1:100), CD19-BB515 (1:100), CD56-BB515 (1:100), CD25-PE (1:200), CD45RA-Qdot655 (1:400), and CCR7-BV711 (1:100) or other suitable antibodies.
-
b.Centrifuge the CD4+ cell fraction at 300 × g for 10 min.
-
c.Resuspend cells in the staining solution.
-
d.Incubate at 20°C–25°C for 25 min, then move the tube on ice for 5 min.Note: some antibodies, such as CCR7, stain better at 37°C or 20°C–25°C than at 4°C. We found 20°C–25°C to be the best compromise for the indicated staining mix. The 5-minute incubation on ice is optional and may stabilize the antibody-antigen interaction.
-
e.Wash the stained cells by adding 5 mL of MACS buffer.
-
f.Centrifuge at 300 × g for 10 min.
-
g.Carefully resuspend cells in 500 μL of MACS buffer and filter them through a 40 μm cell strainer to remove cell aggregates.Note: MACS buffer contains EDTA which reduces cation-dependent cell adhesion and helps to obtain the single-cell suspension necessary for efficient cell sorting.
-
h.Sort resting total memory CD4+ T lymphocytes by FACS based on the expression of CD45RA and CCR7, gating on lineage-negative (CD8– CD14– CD19– CD56–) cells and removing TREG and recently-activated T cells (CD25–/int) (see the sorting strategy in Figure 1A).Note: if possible, perform the cell sorting at 4°C.CRITICAL: add 500 μL of MACS buffer into the 5 mL sorting collection tube to avoid adhesion of cells to the plastic of the tube and the consequent cell loss or death.Optional: if necessary, any other T cell subset of interest may be sorted based on the expression of established cell surface markers, as described by Cossarizza et al.6 in section III.1 on human conventional αβ CD4+ T cells.Optional: if a FACS cell sorter is not available at your institution, you can perform the experiment using magnetically sorted total CD4+ T cells or use an appropriate kit for the magnetic enrichment of memory T cells. In this case, we recommend depleting CD25+ (Treg) cells by negative magnetic selection before isolating CD4+ cells by positive selection.
-
a.
Figure 1.

Gating strategies for T cell sorting
(A) Gating strategy for the identification and isolation of CD4+ total memory cells starting from CD4+ cells enriched by magnetic sorting.
(B) Gating strategy for the identification and isolation of in-vitro stimulated antigen-specific (CTV– CD25hi ICOShi), bystander-activated (CTV– CD25– ICOS–), and non-specific (CTV+ CD25– ICOS–) CD4+ T cells.
CD4+ T cells and antigen-loaded monocytes co-culture
This step is meant to label T cells to track their proliferation and stimulate antigen-specific T cells.
Note: use cold MACS buffer and DPBS 1× and perform centrifugations at 4°C until step 11 h; then, use warm DPBS 1× and T cell medium and perform centrifugations at 20°C–23°C.
-
11.CellTrace Violet (CTV) staining of CD4+ T cells:
-
a.Pre-warm T cell medium and DPBS 1× at 37°C.
-
b.Centrifuge the sorted CD4+ memory T cells at 300 × g for 10 min.
-
c.Resuspend cells in 5 mL of MACS buffer.
-
d.Centrifuge again CD4+ T cells at 300 × g for 10 min.
-
e.Resuspend cells in a suitable amount of MACS buffer and count them.
-
f.Centrifuge CD4+ T cells at 300 × g for 10 min.
-
g.Resuspend cells in 5 mL of DPBS 1×.
-
h.Centrifuge again CD4+ T cells at 300 × g for 10 min.
-
i.Prepare a 10 μM CTV solution (2× concentrated) in warm DPBS 1× (500 μL per 1 × 106 cells needed).
-
j.Carefully resuspend cells in warm DPBS 1× at a concentration of 2 × 106 cells/mL.Note: it is important to have a single-cell suspension without cell aggregates to ensure good CTV staining.
-
k.Add an equal volume of 2× concentrated CTV solution to the 2 × 106 cells/mL cell suspension to have, in the end, a 1 × 106 cells/mL cell suspension stained with a 5 μM CTV solution.
-
l.Incubate cells at 37°C for 20 min under slow rotation in a CO2 incubator.
-
m.Add five volumes of warm T cell medium and incubate cells at 37°C for 5 min under slow rotation in a CO2 incubator.
-
n.Centrifuge stained CD4+ T cells at 300 × g for 10 min.
-
o.Wash cells with 5 mL of T cell medium and centrifuge again as before.
-
p.Resuspend cells in a suitable amount of T cell medium (e.g., at a concentration of 2 × 106 cells/mL based on the previous count) and count them.Note: a 20%–50% reduction in the number of cells from the previous count can be expected and may vary depending on cell fitness and the expertise of the operator.
-
q.Adjust the cell suspension volume with T cell medium to have CTV-stained T cells at a final concentration of 1 × 106 cells/mL.
-
a.
-
12.Co-culture of CTV-stained CD4+ T cells with antigen-loaded monocytes:
-
a.Plate 1 × 105 cells CTV-stained CD4+ T cells with 5 × 104 antigen-loaded monocytes (2:1 ratio) in a flat-bottom 96 well plate in 200 μL of T cell medium supplemented with IL-2 (20 U/mL). Make as many technical replicates as possible depending on the cell number.Note: plate 1 × 105 cells CTV-stained CD4+ T cells alone and 1 × 105 cells CTV-stained CD4+ T cells with 5 × 104 unloaded monocytes as negative controls.
-
b.Quickly spin the plate (120 × g for 20 s) to let the cells settle down in the bottom of the well and incubate them at 37°C in a CO2 incubator for 6 days.Note: the incubation time is flexible in the 5–7 days range as the expression of the activation-induced markers described later is stable in this time window.
-
c.After 48 h resuspend the cells by carefully pipetting and transfer them to a U-bottom 96 well plate.
-
d.Incubate them at 37°C in a CO2 incubator for the remaining 4 days.
-
a.
Isolation of antigen-specific CD4+ T cells
This step is aimed at isolating antigen-specific CD4+ T cells.
Note: use cold MACS buffer and perform centrifugations at 4°C throughout this step.
-
13.Activation-induced markers staining and CD4+ T cell sorting:
-
a.Resuspend co-cultured cells and pool all the technical replicates by collecting them in a 15 mL centrifuge tube.
-
b.Centrifuge cells at 300 × g for 10 min.
-
c.Resuspend cells in 5 mL of MACS buffer and centrifuge them as in b.
-
d.Repeat sub-step c once more.
-
e.Prepare an antibody mix of CD4-BUV395 (diluted 1:100), CD14-AF488 (1:100), CD25-PE (1:200), and ICOS-PE-Cy7 (1:100) in MACS buffer.Note: the staining volume depends on the number of cells to stain; the antibody concentration depends on the antibody (e.g., clone and fluorochrome) used.
-
f.Stain cells with the antibody mix for 20 min at 20°C–25°C, followed by 5 min on ice.Note: CD4 and CD14 discriminate T cells from any residual monocytes while CD25 and ICOS are activation-induced markers.
-
g.Add 5 mL of MACS buffer and centrifuge cells at 300 × g for 10 min.
-
h.Resuspend cells in 5 mL of MACS buffer and centrifuge them as in g.
-
i.Carefully resuspend cells in a suitable amount of MACS buffer and filter them through a 40 μm cell strainer to remove cell aggregates.
-
j.Sort CD4+ CD14– CTV– CD25hi ICOShi cells by FACS (see the sorting strategy in Figure 1B). These will be CD4+ T cells that proliferated and upregulated the expression of activation-induced markers in response to antigen stimulation and are, thus, antigen-specific cells.7 Also, sort non-antigen-specific cells (CD4+ CD14– CTV+ CD25– ICOS–) and keep a small aliquot of unsorted T cells as controls to evaluate the presence of the TCR(s) of interest by qPCR (see the quantification and statistical analysis section).Note: if possible, perform the cell sorting at 4°C. The frequency of antigen-specific CD4+ T cells may vary depending on the antigen tested and the donor. For instance, in our experience, upon stimulation with SARS-CoV-2 Spike protein, CTV– CD25hi ICOShi cells were, on average, about 6.85% of the T cells harvested on Day 7, varying in a range between 1.83% and 12.65%.CRITICAL: add 500 μL of MACS buffer into the 5 mL sorting collection tube to avoid cell adhesion to the plastic of the tube and the consequent cell loss or death.
-
k.FACS-sorted antigen-specific CD4+ T cells and control samples can be directly subjected to the TCR-SMART protocol.Pause point: if necessary, sorted cells can be plated in a T cell medium supplemented with IL-2 (50 U/mL) and kept for 12–16 h at 37°C in a CO2 incubator before proceeding to the next step.Note: we recommend to start the TCR-SMART protocol with an input of 5,000–10,000 T cells. A lower number of T cells can be sufficient to obtain the result, but the experimental conditions haven’t been thoroughly tested.Optional: if needed, antigen-specific CD4+ T cells and control samples can be polyclonally expanded in a T cell medium supplemented with IL-2 (500 U/mL) for 5–6 days to increase the cell number.
-
a.
Identification of defined CD4+ T cell clones by TCR-SMART
This step is aimed at identifying pre-defined T cell clones by detecting their TCR sequences by RT-qPCR. The experimental approach is schematized in Figure 2.
-
14.Cell lysis, mRNA retrotranscription (RT), and enrichment of TCR cDNA:
-
a.Wash the sorted cells in 1 mL of FACS buffer and centrifuge at 300 × g for 10 min at 4°C.
-
b.Resuspend cells in a suitable amount of FACS buffer and count them.Note: the resuspension volume can be calculated based on the cell count provided by the sorting cytometer to get a cell concentration compatible with a reliable count (e.g., in the range of 2.5 × 105 to 2.5 × 106 cells/mL in a Neubauer chamber). Troubleshooting 2.
-
c.Transfer the desired number of cells (in the range of 5,000–50,000) to a 0.5 mL PCR tube, wash with 200 μL of FACS buffer, and centrifuge at 300 × g for 10 min at 4°C.Note: if you do have not a centrifuge adapter for 0.5 mL PCR tubes, you can make your own by using a 1.5 mL PCR tube with the lid cut.
-
d.Carefully aspirate the supernatant and proceed with the cell lysis.CRITICAL: it is fundamental to remove all the supernatant after this centrifugation so as not to dilute the lysis buffer in the next step thus impairing the cell membrane lysis efficiency. To avoid disturbing the cell pellet, mark the orientation of the tube in the centrifuge and aspirate slowly moving along the tube wall opposite to the cell pellet. Keep in mind that using a fixed-angle rotor centrifuge the cell pellet will sit on the outer side of the tube bottom. First, aspirate the supernatant with a vacuum or with a 200 μL pipette (depending on your confidence level), leaving about 10 μL of residual supernatant. Then, aspirate the remaining volume with a 10 μL pipette using thin 0.1–10 μL filter tips. An eventual residual of 0.5–1 μL of supernatant will not impinge on the cell lysis. Troubleshooting 3.
-
e.Lyse the sorted cells (5,000–10,000) in 8.5 μL of cold lysis buffer, gently pipetting 5 times, and incubating 5 min on ice.Note: this protocol uses a relatively mild lysis buffer that lyses the cells without interfering with the RT reaction.CRITICAL: add the RNase inhibitor to the lysis buffer just before performing the cell lysis step to ensure its activity.
-
f.Centrifuge at 1350 × g for 10 min 4°C to pellet nuclei, recover 7.5 μL of supernatant and move it into a 0.2 mL PCR tube.Optional: depending on the starting number of cells, it is possible to isolate and purify RNA first and then proceed with the next steps of the protocol. For instance, you can isolate RNA using a silica membrane-based commercial kit, elute the purified RNA in nuclease-free H2O, and keep it at –80°C for long-term storage. When starting from purified RNA, take 10 ng of purified RNA (or 1 ng of rRNA-depleted purified RNA), resuspend it in 7.5 μL of nuclease-free H2O, and continue with the next steps.
-
g.Add 2.5 μL of RT Mix1.Note: Mix1 contains primers specific for the TRAC (SMART-TRAC), TRBC1, and TRBC2 genes (SMART-TRBC), which code for the constant region of the TCR α and β chains, linked to an adapter oligo that is exploited later for the unbiased cDNA amplification by PCR (primer sequences are listed in the key resources table). Mix1 also contains an oligo(dT)20 primer to retrotranscribe all the other polyadenylated mRNA in addition to the TCR genes.
-
h.Heat to 72°C for 3 min in a PCR thermocycler, then move directly on ice. Keep on ice for 2–3 min.
RT Mix 1 (1 sample) Reagent Final concentration Amount dNTPs (10 mM) 0.5 mM 1 μL Oligo(dT)20 (10 μM) 0.25 μM 0.5 μL SMART-TRAC oligo (10 μM) 0.25 μM 0.5 μL SMART-TRBC oligo (10 μM) 0.25 μM 0.5 μL Total volume 2.5 μL -
i.Add 10 μL of RT Mix2.Note: Mix2 contains the template-switch oligo (TSO) that has the same adapter sequence present in the SMART-TRAC and SMART-TRBC primers.
RT Mix 2 (1 sample) Reagent Final concentration Amount Maxima RT buffer 5× 1× 4 μL RiboLock RNase Inhibitor (40 U/μL) 1 U 0.5 μL TSO (10 μM) 0.5 μM 1 μL Maxima RT RNase H– (200 U/μL) 100 U 0.5 μL Nuclease-free H2O 4 μL Total volume 10 μL Note: the final concentration indicated in the RT Mix1 and Mix2 tables refers to the 20 μL final volume of the RT reaction. -
j.Proceed with the RT protocol using the following cycling conditions.Note: the 5 cycles after the initial step at 42°C are not critical, but they may increase the final yield. The 50°C step helps in unfolding the RNA secondary structures that may cause the termination of cDNA elongation due to steric hindrance. The 25°C step favors the annealing of the TSO to the untemplated cytosines and the consequent template switch. The annealing of the TSO is further facilitated by the presence of a locked nucleic acid (LNA)-modified guanosine present at its 3′ end.3 The two steps at 42°C are aimed at completing the cDNA synthesis and the switched template extension.
RT cycling conditions Steps Temperature Time Cycles Retrotranscription and template-switch 42°C 45 min 1 Unfolding of RNA secondary structures 50°C 3 min 5 cycles Completing retrotranscription 42°C 7 min Facilitating TSO annealing and template-switch 25°C 15 s Completing switched template extension 42°C 90 s Retrotranscriptase inactivation 85°C 5 min 1 Hold 4°C Forever
-
a.
-
15.Unbiased PCR amplification of TCR cDNA and purification of the amplified DNA:
-
a.Prepare the PCR master mix as described in the table below. Assemble the PCR mix during the last 10 min of the RT reaction and keep it on ice.
PCR reaction master mix (1 sample) Reagent Final concentration Amount KAPA Buffer HiFi 5× 1× 10 μL dNTPs (10 mM) 0.3 mM 1.5 μL ISPCR oligo (10 μM) 0.3 μM 1.5 μL KAPA HiFi HotStart DNA Polymerase (1 U/μL) 0.5 U 0.5 μL Nuclease-free H2O 16.5 μL Total volume 30 μL -
b.Add 30 μL of the PCR master mix to the retrotranscribed cDNA. Amplify the cDNA by PCR using the cycling described in the table below.Note: the ISPCR primer anneals to the adapter sequence present both in the SMART-TCR oligos and the TSO, making it possible to run the PCR using a single primer.
PCR cycling conditions Steps Temperature Time Cycles Initial Denaturation 95°C 3 min 1 Denaturation 98°C 20 s 13–15 cycles Annealing 65°C 15 s Extension 72°C 1 min Final extension 72°C 5 min 1 Hold 4°C forever Note: the number of amplification cycles should be adjusted depending on the starting number of cells: 14 cycles are a good compromise in the range of 5,000–10,000 cells.Pause point: if necessary, you can keep the amplified DNA at 4°C up to 24 h. -
c.Purify the PCR product with SPRI beads with size selection properties using the appropriate volume ratio to get rid of primers. For instance, add 60 μL of SPRIselect beads to 50 μL of PCR product (1:1.2 ratio).CRITICAL: vortex the SPRI beads for 20 sec to resuspend them before use.
-
d.Mix the total reaction volume by pipetting 10 times, transfer the mix to a 1.5 mL DNA low-binding tube, and incubate at 20°C–25°C for 1 min.CRITICAL: insufficient mixing of sample and SPRIselect beads will lead to inconsistent size selection results.
-
e.Place the reaction tube on an appropriate magnetic stand and wait 4 min to allow the SPRI beads to settle to the magnet. Remove and discard the clear supernatant.CRITICAL: take care not to aspirate beads during this step, as the PCR product is associated with the beads. Bead loss will result in reduced yield.
-
f.Keeping the reaction tube on the magnet, add 180 μL of freshly prepared 85% ethanol solution and incubate at 20°C–25°C for 30 s. Aspirate and discard the ethanol supernatant.Note: ethanol is hygroscopic; prepare it fresh for optimal results.
-
g.Repeat the ethanol washing once more.
-
h.Remove the reaction tube from the magnet and let the beads air-dry for 5 min or until the bead pellet shows little cracks.CRITICAL: residual ethanol may impinge on the elution efficiency and downstream enzymatic reactions; on the opposite, beads over-drying may increase the elution time and reduce the yield. Troubleshooting 4.
-
i.Add 44 μL of Elution Buffer, mix the total elution volume by pipetting 10 times to resuspend the beads, and incubate at 20°C–25°C for 2 min.
-
j.Place the reaction tube back on the magnetic stand and wait 4 min to allow the SPRI beads to settle into the magnet. Recover 42 μL of eluate and transfer it to a new tube.Note: leaving 2 μL of eluate in the original reaction tube will avoid any carry-out of SPRI beads into the purified PCR product.
-
k.Quantify the amplified DNA with the dsDNA High Sensitivity Assay on a Qubit fluorometer, using 2 μL of the eluate.
-
l.Dilute the amplified DNA with the Elution Buffer to reach a concentration of 2 ng/mL.CRITICAL: different SPRI beads kits may have different properties in terms of size selection capacity (e.g., they use different sample-to-SPRI beads ratios to select for the same DNA fragment size) and complementary reagents (e.g., ethanol solution for the washing steps may vary in the range of 70%–85%). We recommend always following the manufacturer’s indications.Pause point: the purified amplified DNA can be frozen at –20°C for long-term storage.
-
a.
-
16.Detection of pre-defined T cell clones by qPCR:
-
a.Perform the qPCR using the TCR-specific qPCR primers (Table 1) designed in advance and loading 4 ng of purified amplified DNA per reaction.
-
b.Prepare a 10 μM stock solution of forward and reverse TCR-specific primers by mixing 10 μL of 100 μM forward primer and 10 μL of 100 μM reverse primer with 80 μL of nuclease-free H2O.
-
c.Assemble the qPCR reaction master mix as described in the table below. Prepare a master mix per each primer pair, including two mixes for the TRAC-specific and TRBC-specific primers (Table 1).Note: these primers are designed to be nested compared to the ones used in the RT reaction and work as housekeeping controls providing a relative quantification of the TCR of interest.
qPCR reaction master mix Reagent Final concentration/Amount Amount Fast SYBR Green Master Mix 2× 1× 10 μL Fwd + Rev primer mix (10 μM) 0.1 μM 0.2 μL Nuclease-free H2O 7.8 μL Total volume 18 μL -
d.Dispense 18 μL of the qPCR master mix per well in a 96-well qPCR microplate.
-
e.Add 4 ng of the amplified DNA (2 μL of a 2 ng/μL sample) in each well and pipette 2–3 times to mix.Note: despite the SYBR Green master mix contains a hot start polymerase, we recommend assembling the reaction on ice.
-
f.Seal the microplate with an optical clear sealing film and quickly spin it down (300 × g for 30 s).
-
g.Run the qPCR choosing the “Fast” protocol.Note: run at least two technical replicates per each qPCR primer pair; three technical replicates may be better if you are approaching qPCR experiments for the first time or if you frequently experience delta-Cts between replicates higher than 0.5.
-
h.Analyze the data as described in the quantification and statistical analysis section.
-
a.
Figure 2.

Schematic representation of the TCR-SMART core protocol
The cartoon visualizes the steps leading to the synthesis of amplified TCRα and TCRβ DNA. These steps include cell lysis, cDNA synthesis, template switch, the addition of an adapter sequence at the cDNA 5′ and 3′ ends, and the TCR-targeted amplification of cDNA.
Expected outcomes
The TCR-SMART protocol describes how to detect the presence of T cells with a known TCR in a heterogeneous population of any kind. Here, we combined the protocol with a workflow leading to the identification of antigen-specific T cells, since in many immunological studies it is relevant to identify the T cells specific for a defined antigen of interest. Using the same workflow, it is also possible to isolate bystander-activated T cells, which are lymphocytes that get activated by the inflammatory microenvironment and proliferate independently of their antigen specificity. Representative gating strategies of flow cytometry data are shown in Figure 1. Usually, the frequency of T cells specific for a specific antigen or for the immunodominant antigens of a certain microbe is relatively low. Therefore, the number of sorted antigen-specific T cells can be limiting for the isolation and purification of RNA by commercial kits. Despite the T cells can be polyclonally expanded with allogeneic irradiated feeder cells and phytohemagglutinin in an IL-2-containing medium, the expansion may create clonal biases due to the differential proliferative potential of more or less differentiated T cell populations. For instance, central memory T cells have a higher capacity for proliferating compared with effector memory T cells.8 By performing the RT reaction directly on the cell lysate, the TCR-SMART protocol allows the identification of TCR sequences starting from a few thousand cells, thus bypassing the need for clonal expansion. Non-antigen-specific T cells from the same donor can be tested in parallel to verify the relative enrichment of the TCR of interest in the antigen-specific T cell population compared to the non-specific one (Figure 3).
Figure 3.

Identification of T cell clone-specific TCR sequences by qPCR
Bar plots show the expression of clone-specific TCRα and TCRβ variable regions relative to the TCRα and TCRβ constant regions in the same samples. Shown are the data from an N-specific T cell clone (upper panels) and from a non-specific T cell clone (lower panels). This figure partially uses some of the data shown in Figure 6a of Notarbartolo et al.1
The TCR-SMART protocol is scalable and versatile. As mentioned before, it can be performed starting from very limited amounts of cells, but also from purified RNA, when the cell number is sufficient for its isolation. It can be applied to any T cell-containing population and can be easily modified to detect simultaneously the other polyadenylated transcripts by adding a different adapter to the oligo-dT primer and increasing the extension time in the PCR amplification step.
Quantification and statistical analysis
Analyze qPCR data following the example reported in Table 2. Calculate the difference between the threshold cycle (Ct) of technical replicates to evaluate the reproducibility of measurements. Then, calculate the average Ct between technical replicates for each target region. Subtract the constant region-specific (TRAC or TRBC) Ct to the clone-specific Ct. This calculation will provide the ΔCt value. Elevate 2 to the power of –ΔCt to obtain the expression of the clone-specific TCR relative to the alpha or beta TCR constant region. Troubleshooting 5. Plot the obtained data in bar plots to visualize the presence of the T cell clone in the population of interest (Figure 3). We consider a T cell clone to be present in the sample when the qPCR reaction with clone-specific primers shows Ct values 30 in technical duplicates or 35 in at least three technical replicates. In addition, you should observe a reduction in the relative expression of the TCR in the non-specific T cell population compared with the same sample stimulated with an independent antigen (Figure 3) or stimulated with the same antigen but not sorted. The presence of the clone-specific TCR in the antigen-specific population and its depletion from the non-specific one ratify the specificity of the selected T cell clone.
Summary table with the time needed to perform each task of the TCR-SMART protocol
| Task | Timing | Day |
|---|---|---|
| Identification of TCR sequences in single-cell TCR-seq data | 3 h | Before you begin |
| qPCR primer design | 10 min (per primer pair) | Before you begin |
| Isolation of PMBCs | 1.5 h | Day 1 |
| Magnetic sorting of CD14+ monocytes | 70 min | Day 1 |
| Irradiation of CD14+ cells and antigen loading | 75 min hands-on + 4 h of incubation | Day 1 |
| Sorting of memory CD4+ T cells | 1.5 h + FACS sorting time | Day 1 |
| CD4+ T cells and antigen-loaded monocytes co-culture | 1 h + 6 days of incubation | Day 1 |
| Isolation of antigen-specific CD4+ T cells | 85 min + FACS sorting time | Day 7 |
| Identification of defined CD4+ T cell clones by TCR-SMART | 4 h + 2 h per qPCR plate | Day 7–8 |
Table 2.
Example of qPCR data analysis
| Target name | Sample name | Ct_1 | Ct_2 | Ct difference (Ct_1 – Ct_2) | Ct average | ΔCt (Ct_clone-specific – Ct_constant region) | Relative expression (2–ΔCt) |
|---|---|---|---|---|---|---|---|
| TRAC (TCR-α constant region) | Nucleoprotein-specific T cells | 23.211 | 23.402 | ‒0.192 | 23.306 | ||
| Clone_X TCR-α (variable region) | Nucleoprotein-specific T cells | 29.416 | 29.320 | 0.096 | 29.368 | 6.061 | 0.015 |
| TRBC (TCR-β constant region) | Nucleoprotein-specific T cells | 21.492 | 21.554 | ‒0.062 | 21.523 | ||
| Clone_X TCR-β (variable region) | Nucleoprotein-specific T cells | 28.428 | 28.531 | ‒0.103 | 28.479 | 6.956 | 0.008 |
Limitations
The TCR-SMART protocol detects the presence of α and β TCR sequences separately. When performed on polyclonal populations, it is not able to provide information on the matched α and β TCR sequences at the single-cell level. Nonetheless, since the β chain of the TCR is the main determinant of TCR diversity and specificity, the identification of a certain β TCR sequence can be considered a good approximation for the presence of a T cell clone, as usually done when performing bulk TCR-sequencing experiments. Currently, the TCR-SMART protocol remains a qualitative rather than a quantitative assay.
Troubleshooting
Problem 1
The isolated PBMCs are heavily contaminated by red blood cells and platelets (related to step 2).
Potential solution
You can remove the contaminating cells by performing a final washing step with 10 mL of wash buffer and centrifuging cells at 120 × g for 10 min. If this is not enough, you can lyse the red blood cells by osmotic shock, for instance using a commercial or home-made red blood cell lysis buffer.
Problem 2
The cell count is much lower than what is indicated by the FACS sorter or cells are lost (related to step 14 b).
Potential solution
To minimize post-sorting cell loss, pre-wet the collection tube by filling it with MACS buffer and incubating it at 4°C for 20 min. Then aspirate the buffer and add 500 μL of MACS buffer. If you have a few hundred cells you can expand them as indicated in the optional notes at the end of Step 13. Alternatively, you can blindly continue the protocol, even if you cannot see a cell pellet, and verify the amount of amplified DNA at the end of the procedure.
Problem 3
The cell pellet gets disturbed during the aspiration step and looks loose (related to step 14 d).
Potential solution
Stop aspirating and repeat the centrifugation increasing the speed up to 500 × g for 10 min. Slowly aspirate the supernatant with a 200 μL pipette.
Problem 4
The SPRI beads get overdried and are difficult to resuspend in the Elution Buffer (related to step 15 h).
Potential solution
Increase the incubation time with the Elution Buffer to 5 min and pipette several times until the SPRI beads aggregates are dissolved. Calculate the 2 min of incubation for the elution starting from the moment the aggregates are dissolved and the SPRI beads are well resuspended.
Problem 5
The reaction with constant region-specific primers (TRAC or TRBC) reaches the threshold before the linear exponential phase of the qPCR (e.g., it has a Ct value ) (related to quantification and statistical analysis).
Potential solution
Perform a serial dilution of the amplified cDNA (e.g., 8, 16, 32, and 64-fold dilutions) and repeat the qPCR reaction. Verify the linearity of the qPCR signal, namely that you observe an increase of one Ct for each 2-fold dilution, and choose a Ct value falling in the linear range. If the amplified cDNA sample was diluted only for the reaction with constant region-specific primers to avoid too high Ct values in the reaction with clone-specific primers, consider the sample dilution when calculating the relative expression of the clone-specific TCR.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Samuele Notarbartolo (samuele.notarbartolo@policlinico.mi.it).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Samuele Notarbartolo (samuele.notarbartolo@policlinico.mi.it).
Materials availability
The sequence of SMART and qPCR primers specific for the TCR alpha and beta chain constant genes generated in this study is readily available from the key resources table and Table 1.
Data and code availability
This study did not generate datasets or code.
Acknowledgments
We thank the INGM Flow Cytometry Facility for cell sorting and M. Bombaci for the production of recombinant SARS-CoV-2 RBD and N proteins. S.N. is supported by a Ricerca Finalizzata Giovani Ricercatori grant from the Italian Ministry of Health (RF GR-2021-12374097). The graphical abstract and Figure 2 were created with the help of BioRender.com.
Author contributions
Conceptualization, S.N.; methodology, S.N.; software, A.G.; investigation, S.N. and A.G.; resources, A.B.; writing – original draft, S.N.; writing – review and editing, S.N. and A.G.; visualization, S.N.; supervision, S.N., R.G., and S.A.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2023.102787.
Supplemental information
References
- 1.Notarbartolo S., Ranzani V., Bandera A., Gruarin P., Bevilacqua V., Putignano A.R., Gobbini A., Galeota E., Manara C., Bombaci M., et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abg5021. [DOI] [PubMed] [Google Scholar]
- 2.Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 4.Huang J., Wang Q.J., Yang S., Li Y.F., El-Gamil M., Rosenberg S.A., Robbins P.F. Irradiation enhances human T-cell function by upregulating CD70 expression on antigen-presenting cells in vitro. J. Immunother. 2011;34:327–335. doi: 10.1097/CJI.0b013e318216983d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teresa Pinto A., Laranjeiro Pinto M., Patrícia Cardoso A., Monteiro C., Teixeira Pinto M., Filipe Maia A., Castro P., Figueira R., Monteiro A., Marques M., et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 2016;6 doi: 10.1038/srep18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cossarizza A., Chang H.D., Radbruch A., Abrignani S., Addo R., Akdis M., Andrä I., Andreata F., Annunziato F., Arranz E., et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition) Eur. J. Immunol. 2021;51:2708–3145. doi: 10.1002/eji.202170126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu M., Notarbartolo S., Foglierini M., Jovic S., Mele F., Jarrossay D., Lanzavecchia A., Cassotta A., Sallusto F. Clonal composition and persistence of antigen-specific circulating T follicular helper cells. Eur. J. Immunol. 2023;53 doi: 10.1002/eji.202250190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.