

Summary
SARS-CoV-2 infection and vaccination elicit potent immune responses. Our study presents a comprehensive multimodal single-cell analysis of blood from COVID-19 patients and healthy volunteers receiving the SARS-CoV-2 vaccine and booster. We profiled immune responses via transcriptional analysis and lymphocyte repertoire reconstruction. COVID-19 patients displayed an enhanced interferon signature and cytotoxic gene upregulation, absent in vaccine recipients. B and T cell repertoire analysis revealed clonal expansion among effector cells in COVID-19 patients and memory cells in vaccine recipients. Furthermore, while clonal αβ T cell responses were observed in both COVID-19 patients and vaccine recipients, expansion of clonal γδ T cells was found only in infected individuals. Our dataset enables side-by-side comparison of immune responses to infection versus vaccination, including clonal B and T cell responses. Our comparative analysis shows that vaccination induces a robust, durable clonal B and T cell responses, without the severe inflammation associated with infection.
Subject areas: Immunology, Immune response, Transcriptomics
Graphical abstract
Highlights
-
•
Both COVID-19 infection and immunization elicited robust adaptive immune responses
-
•
SARS-CoV-2 infection results in profound upregulation of type I IFN signaling
-
•
Immune cells of COVID-19 patients have elevated expression of cytotoxic genes
-
•
Both infection and vaccine lead to clonal expansion of T cells and SHM in B cells
Immunology; Immune response; Transcriptomics
Introduction
In 2019, the novel coronavirus SARS-CoV-2 emerged, and the resulting pandemic has had unprecedented impact on the heath, economy, and social fabric of the global community. The clinical presentation of coronavirus disease 2019 (COVID-19), the disease caused by SARS-CoV-2, has been highly heterogeneous, with manifestations ranging from asymptomatic or mild illness to acute respiratory distress syndrome (ARDS), multiorgan failure, and death.
To date, a number of comprehensive studies have described immune responses to SARS-CoV-2 infection.1,2,3,4,5 These research efforts identified lymphopenia with concomitant innate cell expansion, while specific alterations in a number of immune subsets, including activated CD8 T cells, plasma cells, monocytes, and NK cells, are thought to shape patients’ clinical outcomes. Immune responses in individuals who survive COVID-19 eventually return to baseline, with establishment of memory T and B cell responses,6,7,8,9,10,11,12 and corresponding development of a neutralizing antibody repertoire.13,14
Infection with SARS-CoV-2 and vaccination against the virus have both been shown to stimulate immune responses and to protect against subsequent infection.8,11,15,16,17,18 Both infection and vaccination generate protective anti-Spike memory immune responses.15,16,17 A detailed comparison of immune responses to the virus versus vaccination and subsequent boosters provides a unique opportunity to contrast immune reactions linked to infection and immunization with well-defined antigens.
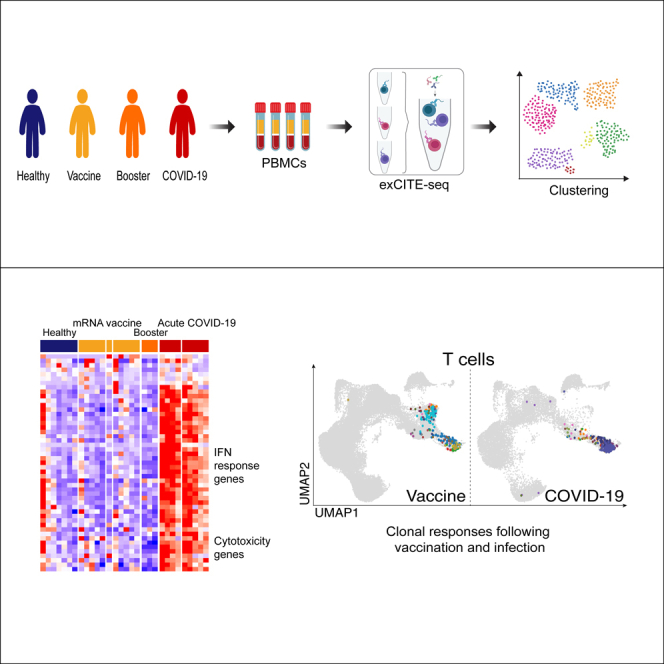
In our study, we took advantage of ExCITE-seq (Expanded Cellular Indexing of Transcriptomes and Epitopes by sequencing), a multimodal single-cell sequencing technique, to simultaneously characterize the surface phenotype and transcriptome of immune cells.19,20 This platform also enables reconstruction of B cell and T cell antigen receptor rearrangement of individual lymphocytes. We used the ExCITE-seq platform to characterize cellular and transcriptional responses to SARS-CoV-2 infection and vaccination in peripheral immune cells to better understand the host response to the pathogen and to immunization against defined viral antigens. We also quantified virus-specific antibody titers in the serum of COVID-19 patients and vaccine recipients using a recently developed multiplex bead-binding assay21 and antibody ELISA.22
Our multimodal analysis revealed dramatic alterations in the frequencies and transcriptional programs of several immune subsets in response to infection and highlighted differences in the breadth of immune responses observed upon infection and vaccination. In COVID-19 patients, transcriptional profiles of many immune cell populations were characterized by augmented IFN signaling, upregulation of genes associated with cytotoxicity, and changes in metabolic pathways. These transcriptional changes were also readily observed in an independent scRNA-seq dataset of 38 acute and 38 convalescent patients, with the cytotoxic signature persisting in circulating cells of convalescent patients.3 Analysis of peripheral immune cells following vaccination with the BNT162b2 mRNA vaccine also revealed alterations of transcriptional programs of several immune cell populations consistent with immune activation, but the highly augmented interferon (IFN) signaling and cytotoxic signature observed in COVID-19 patients were largely absent. We observed robust antibody response in both COVID-19 patients and immunized individuals, with vaccination inducing a remarkably consistent IgG response to S protein. Interestingly, B cell and T cell clonal responses differed dramatically between infected and vaccinated individuals, suggesting that infection-driven inflammation influences the trajectory of the adaptive immune response; this may have important implications for our understanding of the durability of protective immune responses.
Results
Overview of immune responses to COVID-19 infection and immunization
To improve our understanding of immune responses to SARS-CoV-2 antigens in different inflammatory contexts, we profiled circulating immune cells from five adults with acute COVID-19 and nine healthy adults, seven of whom received the BNT162b2 vaccine, and two who were SARS-CoV-2 naive. For three of the vaccine recipients, samples were also collected before and after receiving a booster. Samples were taken at multiple time points, resulting in a total of 42 post-vaccination and 9 post-infection samples (Scheme 1). COVID-19 samples were collected during the acute phase of infection. For four of the five COVID-19 patients, we obtained longitudinal samples. COVID-19 sample time points were recorded as days post-onset of symptoms, and clinical metadata were evaluated for clinical severity based on the WHO clinical progression scale, in which 1–3 represents mild, 4–5 moderate, and 6–9 severe disease.23 All subjects in the vaccinated group received two doses approximately three weeks apart, in accordance with its FDA Emergency Use Authorization. For vaccine responses, samples were collected at baseline, and then at approximately 1, 3, and 4 weeks after the first vaccine dose. For three of the vaccine recipients, we collected samples at additional time points: at 5 weeks post-vaccine as well as pre-booster, and 1 and 4 weeks post-booster. For all participants, demographic characteristics, clinical features, and outcomes are listed in Table S1. The age distribution of study participants was similar in vaccine and COVID-19 groups (Figure S1).
Scheme 1.

Overview of sample collection and analysis PBMCs and serum were collected from five COVID-19 patients, nine healthy volunteers, seven of whom received the Pfizer BNT162b2 SARS-CoV-2 mRNA vaccine, and two who were SARS-CoV-2 naïve
Time points for COVID-19 samples were recorded as days post-onset (DPO) of symptoms and samples were split into acute (≤10 DPO) and convalescent (>10 DPO). For vaccine responses, samples were collected at baseline, and then at approximately 1, 3, and 4 weeks after the first vaccine dose. For three of the vaccine recipients additional time points were collected at 5 weeks post-vaccine as well as pre-booster and 1 and 4 weeks after booster.
To assess the impact of SARS-CoV-2 infection and vaccination on each individual’s global immune landscape, we used a multimodal ExCITE-seq approach19 to identify discrete clusters based on the transcriptional profile and surface epitopes of circulating cells. To do this, peripheral blood mononuclear cells (PBMCs) were multiplexed and processed using 5′ droplet-based scRNA-seq technology (10x Genomics). Surface marker phenotypes were detected using an optimized 60-antibody ExCITE-seq panel,24 generating matching transcriptional and surface protein data. In addition, for each sample, we sequenced single-cell T cell receptors (TCR) αβ and γδ, as well as B cell receptors (BCRs), to evaluate antigen receptor repertoires. Samples from healthy volunteers prior to vaccination were grouped together with samples from unvaccinated COVID-19-naïve healthy donors as healthy controls (HC).
In total, we obtained 195,634 PBMCs from 51 individual samples, with an average of 3,600 cells/sample. Among these, 36,906 cells (∼19%) were from COVID-19 patients; 37,680 cells (∼19%) were from HCs and pre-vaccine samples; 90,753 cells (∼46%) were from post-vaccine samples; and 30,295 cells (∼15%) came from booster samples. All high-quality single cells were integrated across the RNA, antibody-derived tags (ADTs), TCR and BCR modalities for all subsequent analyses. Dimension reduction was performed using the combined RNA and ADT modalities to generate a uniform manifold approximation and projection (UMAP)25 representation of all 195,634 cells from HC, and from immunized volunteer and COVID-19 patient samples (Figures 1A and 1B). Using a combination of Louvain-based clustering,26 SingleR27 reference-based annotation, and literature markers, we identified 10 major lineages (Figures 1A and 1B) and 24 individual subpopulations of myeloid cells, B cells, conventional and innate-like T cells, and NK cells (Figures 1C–1F). Gene expression and canonical ADT markers further confirmed these lineages and sub-populations (Figures S2A and S2B; Table S2).
Figure 1.

Single-cell landscape of immunological responses to COVID-19 and SARS-CoV-2 BNT162b2 mRNA vaccine
(A) UMAP representation of over 195,000 PBMCs by scRNA-seq, clustered and colored by. indicated cell type. Clusters identified based on gene expression and surface epitopes.
(B) UMAP visualization of PBMCs from and COVID-19-naïve healthy donors (blue), healthy volunteers before receiving the BNT162b2 mRNA vaccine (blue), healthy volunteers after receiving the BNT162b2 mRNA vaccine (yellow) and booster (orange), and COVID-19 patients (red).
(C–F) UMAP representation of subclustered myeloid (C), B cell (D), T cell (E), and innate and unconventional T cell (F) populations colored and labeled by cell type.
(G and H) Graphs highlight specific populations that exhibited significant differences between healthy volunteers and COVID-19 patients. Vaccine samples are shown as weeks after first vaccine dose. Booster samples are shown as weeks after booster. COVID-19 patient samples are split by days post-onset (DPO) of symptoms into acute (≤10 DPO) and convalescent (>10 DPO). Line graphs show percentages for given cell population per subject, per time point. Connected lines indicate repeated measurements for the same subjects. P-values for these plots are determined by an ANOVA of linear mixed models and post-hoc pairwise comparison of estimated marginal means are listed on each plot. The bars represent the upper and lower confidence intervals for the estimated marginal means for the linear model.
A number of studies have demonstrated a highly heterogeneous anti-viral inflammatory responses in COVID-19 patients, likely due to variability of disease severity, stage of disease, and diversity of preexisting conditions.2,3,4,8,12 Similarly, our analysis revealed striking differences in the frequency of key immune cell populations between COVID-19 patients and healthy volunteers prior to and following vaccination (Figures 1G and 1H).
Dramatic difference in maturation of B cell responses triggered by SARS-CoV-2 infection and vaccination
Current COVID-19 vaccine efforts have focused on the generation of humoral immune responses against SARS-CoV-2, which has been demonstrated to be a correlate of protection against infection.28,29 To better understand the humoral responses following infection and vaccination, we examined B cell responses in the ExCITE-seq dataset. Single-cell analysis identified four distinct B cell populations based on gene expression and surface epitopes (Figure 1D). Relative to healthy volunteers, we observed striking expansion of circulating plasmablasts in COVID-19 patients at acute time points (Figure 1G). In contrast, we observed no apparent expansion of plasmablasts in circulation following vaccination, despite a successful humoral response in all subjects (Figure S3).
As plasmablasts are likely recent emigrants from lymphoid tissue, we hypothesized that they may carry a transcriptional imprint of the inflammatory milieu in tissue. We performed Gene Set Variation Analysis (GSVA) of plasmablasts from COVID-19 patients and healthy volunteers to find out whether signaling pathways were similarly expressed in both cohorts (Figures 2A, 2B, and S4). This analysis revealed that, relative to plasmablasts in healthy volunteers across all time points, plasmablasts from COVID-19 patients across all time points were highly enriched for genes involved in oxidative phosphorylation, type I and type II IFN responses (IFN-I, IFN-II), fatty acid metabolism, and mTORC1 signaling. The extent of upregulation of IFN response genes in COVID-19 patients correlated with severity of disease, as judged by fraction of inspired oxygen (Figure 2C). The elevated IFN-I signature is evident in our dataset and was validated using 76 COVID-19 patient samples stratified by disease severity in a large publicly available dataset (Figures 2B and S5).3 Plasmablasts from both COVID-19 patients and healthy volunteers after vaccination had elevated transcription of genes linked to IL-6 receptor signaling (JAK/STAT) and PI3K/AKT signaling pathways (Figures 2A and S5), two pathways associated with promoting plasmablast differentiation.30,31 In contrast to the transcriptional profile of plasmablasts from vaccinated individuals, which resembled that of healthy controls, the increased IFN signaling observed in plasmablasts from acute COVID-19 samples likely reflects the profound inflammation in infected patients. These transcriptional changes in response to IFN and other pro-inflammatory cytokines are likely to have broader implications for B cell differentiation and persistence.
Figure 2.

Maturation of B cell responses
(A) GSVA analysis of plasmablasts in cells from HC, Vaccine and COVID-19 (top color bar) patients from the Hallmark gene set32 and colored by oxygen requirement (bottom color bar) which is a clinical parameter defined as the fraction of inspired oxygen, where 21% represents oxygen content of room air without supplementation. HC, Vaccine, and Booster cells are colored at 21%. COVID-19 patient samples are split into acute (≤10 DPO) and convalescent (>10 DPO). See also Figure S4.
(B) IFN-I gene pathway module score in plasmablasts. Module scores were calculated using the AddModuleScore function from the Seurat package, which calculates the mean expression for a set of genes and adjusts for the collective expression of control features. Box and whisker plots were used to portray the distribution of the data, outliers, and the median. P-values were determined by the Wilcoxon test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). See also Figures S4 and S5.
(C) Oxygen requirement (shown as Fraction of inspired oxygen, FiO2) against GSVA enrichment score for interferon alpha response gene set as shown in A. P and tau values are determined by a Kendall rank correlation test.
(D) Clonal populations based on individual B cell IGH chain CDR3 sequence. CDR3 sequences occurring in at least 2 cells are colored blue, while any CDR3 sequences in at least 3 cells are colored uniquely in cells from Vaccinated (left) and COVID-19 patient samples (right).
(E) Number of mismatched bases according to IgBlast results of recovered VH gene sequences in memory (left), plasmablasts (middle), and resting (right) B cell subsets in cells from HC (blue), Vaccinated (yellow), Boosted (orange), and COVID-19 (red) patient samples. Box and whisker plots were used to portray the distribution of the data, outliers, and the median. P-values were determined by Wilcoxon test (ns p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
We next evaluated the B cell repertoire across these cohorts. Expansion of B cell clones, as well as convergent antibody repertoires, have been reported for a number of viral infections, including SARS-CoV-2.33,34,35 Analysis of the B cell receptor (BCR) repertoire revealed that a majority of clonal B-lineage cells were captured in the plasmablast cluster in COVID-19 patients. In vaccinated individuals, clonally expanded cells were primarily in resting and memory compartments (Figure 2D). This observation suggests that the IFN response associated with SARS-CoV-2 infection may promote rapid plasma cell differentiation in COVID-19 patients, while the BNT162b2 mRNA vaccine appears to favor clonal expansion of memory B cells.
To evaluate the extent of somatic hypermutation (SHM), we performed IgBlast-based36 alignment of the V(D)J repertoire for the sequenced samples and then evaluated single base-pair mismatches within the VH alignments for the previously defined B cell clusters. Increased SHM was apparent in memory B cells from all samples, with frequency of mutations notably higher when compared to resting B cells (Figure 2E). Although few plasmablasts were captured in PBMCs from vaccinated individuals, the rate of SHM in plasmablasts in these samples was comparable to that observed from COVID-19 patients at the peak of disease. Frequency of SHMs was significantly reduced in plasmablasts from convalescent patients’ COVID-19 samples compared to peak disease (Figure 2E), possibly as a consequence of long-lived plasma cells’ migrating out of circulation.37 It is also possible that some of the antibody-producing plasma cells are either retained at the site of infection or in the draining lymph nodes.38 The observation that Ig rearrangements in plasma cells from healthy individuals carry substantial somatic hyper mutation (SHM) burden is likely a consequence of affinity maturation in response to ongoing immune surveillance, as resting B cells from all individuals had very low background of mutations in their repertoire. There was an increase in SHM within the pool of memory cells one week after a person received the second dose of the vaccine compared to SHM one week after receiving the first dose (Figure 2E). Despite the inherent limitation of surveying only circulating memory B cells and plasma cells, these analyses highlight ongoing affinity maturation of B cell responses in both COVID-19 patients and vaccinated individuals.
NK and clonal T cell responses differ in infection and vaccination
Cell-mediated immune responses are carried out by NK cells, CD4, CD8 T cells, and unconventional T lymphocytes like gamma delta (γδ) T cells. Consistent with prior reports, we observed an increase in the frequency of proliferating T cells and NK cells in COVID-19 patients compared to healthy volunteers and vaccinated individuals (Figures 3A and 3B).39,40 In COVID-19 patients, we measured a dramatically elevated cytotoxic signature in NK cells, CD4 and CD8 T cells, and γδ T cells (Figures 3C and S6A). Both CD8 effector T cells and NK cells in COVID-19 patients showed significantly elevated expression of genes associated with cytotoxicity, such as GZMA, GZMB, GZMH, GNLY, NKG7, and PRF1 (Figures 3C and S6A). Because the number of COVID-19 patients in our study was limited, we also verified the elevation of cytotoxic gene signatures in a published dataset of PBMC biospecimens from COVID-19 patients (Figure S6B).3 This finding is consistent with previously published results that described a dysregulation of immune responses in COVID-19.41,42 Strikingly, while clonal expansion was evident only in CD8 effector T cells of COVID-19 patients, the BNT162b2 vaccine elicited robust clonal responses in both CD8 effector T cells and CD8 TEM cells, suggesting that the vaccine may elicit a more potent memory CD8 response than does infection (Figure 3D).
Figure 3.

Cytotoxic responses and clonality of conventional and innate-like T cells in COVID-19 and SARS-CoV-2 vaccine recipients
(A and B) Graphs of cell percentages of select cytotoxic cell populations that exhibited significant differences between COVID-19-naïve donors and healthy volunteers before receiving the BNT162b2 mRNA vaccine (blue), healthy volunteers after receiving the BNT162b2 mRNA vaccine (orange) and booster (dark orange), and COVID-19 patients (red). Line graphs show percentages for given cell population per subject, per time point. Connected lines indicate repeated measurements for the same subjects. P-values for these plots are determined by an ANOVA of linear mixed models and post-hoc pairwise comparison of estimated marginal means are listed on each plot. The bars represent the upper and lower confidence intervals for the estimated marginal means for the linear model.
(C) Average per-sample scaled expression of genes associated with cytotoxic effector function from the gene set T cell mediated cytotoxicity (GO:0001913) in proliferating NK and conventional T cells. See also Figure S6.
(D) UMAP visualization of clonal T cells from healthy volunteers after receiving the BNT162b2 mRNA vaccine (left) and COVID-19 patients (right). Clonality is determined by the CDR3 sequence in TCRβ chain. Identical CDR3 sequences in at least 5 cells are colored uniquely. See also Figure S6.
(E) UMAP visualization of CD8 T cell clones (>3 cells with identical CDR3) that match reported TCRβ sequences from natural and synthetic exposure to SARS-CoV-2. While CD8 T Effector Memory and CD4 T Activated clusters overlap in UMAP space, we confirmed that all mapped clonal cells were from the CD8 T Effector Memory population. 100% amino acid sequence identity threshold was used in mapping of CDR3 region to Spike-specific reported sequences.43 See also Figure S7.
(F) UMAP visualization of clonal γδ T cells from healthy volunteers after receiving the BNT162b2 mRNA vaccine (left) and COVID-19 patients (right). Clonality is determined by the CDR3 sequence in TCRδ chain. Identical CDR3 sequences in at least 5 cells are colored uniquely. See also Figure S9.
To evaluate whether clonal CD8 cells are SARS-CoV-2 reactive, we mapped CDR3s from a large-scale database of SARS-CoV-2-specific TCRs.43 We found that many clonally expanded CD8 T cells within the CD8 effector T cell and CD8 TEM clusters have CDR3s that perfect match reported SARS-CoV-2-specific TCR sequences (Figures 3E and S7).
CD4 T helper cells orchestrate much of the adaptive anti-viral immune response. Strikingly, we observed that the majority of activated CD4 T cells from COVID-19 patients expressed genes associated with cytotoxic effector function, such as GZMH, GZMA, and PRF1 (Figures S6A and S6C). Cytotoxic CD4 T cells have been previously observed in COVID-19 and other viral infections44,45,46 and have also been noted in patients with autoimmune disease.47 Clonally expanded activated CD4 T cells were observed in COVID-19 patients and vaccinated individuals (Figure 3D), but only COVID-19 patients were characterized by elevated expression of cytotoxic genes (Figures S6A and S6C). Whether these cells contribute to virus clearance or to inflammation-associated pathology in COVID-19 is unclear.
To evaluate whether clonally expanded T cells in vaccinated individuals and COVID-19 patients in our single-cell sequencing dataset were reactive to SARS-CoV-2, we mapped CDR3s identified via scRNA-seq following in vitro activation with recombinant SARS-CoV-2 spike proteins.48 A notable number of clonally expanded T cells identified in COVID-19 patients and immunized individuals had matching CDR3s with in vitro activated spike-specific T lymphocytes (Figure S8).
Our single-cell analysis revealed that non-conventional lymphocytes were also engaged by both the viral infection in COVID-19 patients and the mRNA vaccine. γδ T cells are a subset of unconventional, non-MHC-restricted innate-like T cells with cytotoxic effector functions and the ability to regulate other immune cells.49,50 Transcriptional analysis revealed dramatic upregulation of genes associated with cytotoxic effector functions in the γδ T cells from COVID-19 patients, a feature not observed in γδ T cells from HCs or from vaccinated individuals (Figure S6). Repertoire analysis of γδ T cells revealed oligoclonal expansion in a majority of COVID-19 patients and a moderate dynamic response in vaccinated individuals (Figures 3F and S9). Overall, our single-cell analysis of lymphocyte responses demonstrated a persistent upregulation of genes associated with cytotoxic effector functions among NK cells, CD4 T, CD8 T, and γδ T cells in COVID-19 patients. The increased cytotoxicity of these cells likely contributes both to pathogen clearance and to immune-mediated pathology.
Discussion
In this study, we performed a multimodal analysis of samples from COVID-19 patients and from healthy volunteers before and after they received the SARS-CoV-2 BNT162b2 mRNA vaccine and booster. While both infection and immunization elicited robust humoral responses, our analysis revealed dramatic differences in cell composition and transcriptional profiles of circulating immune cells in response to the two different immune challenges.
Type I IFN mediates antiviral immunity, drives expression of a number of genes involved in viral clearance, and plays a critical role in initiating innate and adaptive immune responses during a viral infection.51 IFN signaling induced by viral infection orchestrates antigen presentation, cellular trafficking, and terminal differentiation of lymphocytes.52,53 However, prolonged IFN-I signaling also promotes immunopathology through induction of aberrant inflammatory responses during acute viral infection and can lead to immune dysfunction.54 Although the role of IFN-I signaling in COVID-19 awaits full elucidation, recent studies show that systemic production of type I IFN is negatively correlated with disease severity,55,56 while excessive local production exacerbates lung tissue damage and correlates with increased morbidity and mortality.57 Furthermore, individuals who died of severe COVID-19, which is associated with very high IFN-I, had poor germinal center (GC) responses.58 While SARS-CoV-2 infection led to a discernible IFN response, we did not find evidence of IFN induction by the BNT162b2 vaccine. This observation suggests that robust affinity maturation in response to viral antigens can occur in the absence of high levels of systemic IFN signaling. It is possible that IFN induction by the vaccine is transient and occurs early and therefore was not captured by our sample collection schedule.59,60 In addition, current mRNA vaccines incorporate chemically-modified nucleosides to prevent activation of TLR7 and other innate sensors, reducing IFN-I production.61 Our study lacks the timepoints to evaluate IFN-I signaling immediately after vaccination, but analysis of our ExCITE-seq data and of previously published single-cell dataset suggest that infection with SARS-CoV-2 results in profound upregulation of type I IFN signaling.
COVID-19 patients had a striking expansion of antibody-producing plasmablasts, with evidence of clonal cells in this cluster. Surprisingly, we did not detect plasmablast expansion in the blood of immunized individuals, despite a robust antibody response. This suggest that antibody-producing cells either migrate to their bone marrow niche at a time not captured by our weekly sampling, or remain in the tissues where they were generated. Recent studies have demonstrated that SARS-CoV-2 infection generates long-lived bone marrow plasma cells however, it remains to be elucidated whether mRNA vaccines drive a similar response.10,62 Further studies evaluating the presence of SARS-CoV-2-specific, long-lived plasma cells in the bone marrow following immunization would shed light on the durability of protective immunity and aid in vaccine development.
Recent studies have demonstrated that SARS-CoV-2 infection and mRNA vaccines elicit potent antigen-specific GC responses.11,18 Consistent with the idea of long-lived plasma cell trafficking to the bone marrow, convalescent patients have reduced plasmablasts in circulation relative to those with acute illness, and the SHM footprint in the repertoire of the remaining cells was significantly diminished from what we had observed at the peak of the disease.
In our study, plasmablasts in COVID-19 patients were characterized by a strong IFN-I signature relative to those in healthy volunteers. While it has been shown that an overzealous IFN response favors extrafollicular plasma cell differentiation at the expense of affinity maturation during an anti-viral response,63,64 we did observe an accumulation of SHMs in the repertoire of plasmablasts and memory cells from COVID-19 patients, as well as in vaccinated individuals. We ought to consider that our observation of SHMs in plasmablasts may reflect the fact that we are studying patients with less severe disease; an earlier study of postmortem thoracic lymph nodes that described muted GC response was conducted in patients with severe COVID-19.58 In our COVID-19 patients, clonal responses were most evident among plasmablasts. On the other hand, clonal cells were found within memory and resting B cells at multiple time points in vaccinated individuals. In future studies, stratifying patients by disease severity and a more detailed time course following vaccination should allow us to truly discern the impact of IFN on GC maturation in the context of SARS-CoV-2 infection and vaccination.
A number of studies have highlighted the shared IFN-induced gene signature in lymphocytes from patients with autoimmune disease and in subjects following viral infections.65,66 Our observation that B lymphocyte transcriptional programs in COVID-19 patients are dominated by a marked upregulation of IFN-response genes may be important for understanding the immunopathology of COVID-19. Dysregulation of IFN-I signaling is a common factor in multiple autoimmune diseases, and there is growing evidence that autoantibodies could be driving severe disease and long-term sequelae in some COVID-19 patients.67,68,69
Based on the role of antigen-specific T cells in protective immunity against SARS-CoV-2 infection, it is becoming increasingly clear that successful vaccines need to engage the cellular adaptive immune response.70,71,72 Indeed, humoral immune responses may be less effective against SARS-CoV-2 variants.73,74 Conversely, SARS-CoV-2-specific CD8 T cell responses, which target a broad range of epitopes, remain largely intact against variants.8,75 Our analysis revealed that both SARS-CoV-2 infection and, to a lesser degree, vaccination, elicit clonal CD8 effector T cell responses. We also observed a strong clonal response in CD8 TEM cells in all volunteers following immunization – a feature of adaptive response that was notably absent in COVID-19 patients. This finding corroborates recent studies that have demonstrated that vaccination induces durable SARS-CoV-2-specific T cell responses.76,77,78
Peripheral immune cells of COVID-19 patients were enriched in activated T cells, NK cells, and γδ T cells, with elevated expression of genes associated with cytotoxic effector functions (GZMA, GZMB, GZMH, PRF1, GNLY, NKG7, and IL-32). Clonal cytotoxic CD4 T cells in COVID-19 patients were largely absent in healthy volunteers following immunization. While hyperactivation of inflammatory responses and cytotoxic cells may contribute to immunopathology in severe illness, these features indicate protective immune responses and resolution of infection in mild and moderate disease.79 Furthermore, to our knowledge, our study is the first to highlight clonal expansion of γδ T cells in response to SARS-CoV-2 infection. Because few studies include analysis of γδ T cell repertoire, it remains to be elucidated whether these cells contribute to viral clearance or to the pathology associated with COVID-19.
This study underscores that SARS-CoV-2 infection and vaccination both lead to the development and maturation of antiviral adaptive immune responses. While the limited number of participants and absence of COVID-19 samples spanning a range of disease severities are important limitations of this study, we took steps to mitigate these limitations by validating our key findings with an analysis of a large publicly available dataset comprising 143 samples from healthy volunteers and COVID-19 patients. This dataset spans disease severity from asymptomatic to critical and supports the robustness of our findings.3 Future studies that include a granular analysis of a similarly high number of post-vaccination samples, preferably with time points immediately following the vaccine, will shed light on the differences in cytokine responses following these different types of immune challenges.
This study underscores the fine balance in COVID-19 between antiviral immune responses that achieve clearance of the infection and durable protective immunity, and those that lead to inflammation and immunopathology. Our highly granular dataset enables direct comparison of immune responses in both infected individuals and those that received the mRNA SARS-CoV-2 vaccine and booster. Better understanding of the immunological features associated with protective immunity, immunopathology, and durability of protective immunological memory will aid not only in better viral-disease therapeutics, but also facilitate the development of effective vaccines for new and re-emerging viral diseases that threaten public health.
Limitations of the study
Our study has some inherent limitations. The restricted number of participants and the lack of COVID-19 samples across a spectrum of disease severities are notable constraints. However, we have done our best to corroborate our key findings with analysis of a large publicly available dataset comprising 143 samples from healthy volunteers and COVID-19 patients. The vaccine samples, despite the relatively small sample size, provide detailed longitudinal sampling of circulating cells post-vaccination.
An inherent limitation of this study is the focus on circulating leukocytes, which limits the conclusions we can derive. Exploring lymphocytes in lymph nodes and bone marrow would provide insights into B cell response maturation and germinal center dynamics. B cell analyses from lymph node biopsies would also allow for a more in-depth study of SHMs. Lastly, the absence of early time points, notably in the initial days after infection or vaccination, hampers our capacity to evaluate early innate responses. Furthermore, while we have an accurate timeline for our vaccinated and boosted individuals’ samples, we rely on self-reported symptoms to establish the timeline of infection for COVID-19 samples.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq™-C0159 anti-human HLA-DR | BioLegend | 307663; RRID: AB_2800795 |
| TotalSeq™-C0080 anti-human CD8 | BioLegend | 301071; RRID: AB_2800730 |
| TotalSeq™-C0391 anti-human CD45 | BioLegend | 304068; RRID: AB_2800762 |
| TotalSeq™-C0064 anti-human CD123 | BioLegend | 306045; RRID: AB_2800789 |
| TotalSeq™-C0367 anti-human CD2 | BioLegend | 309231; RRID: AB_2810464 |
| TotalSeq™-C0034 anti-human CD3 | BioLegend | 300479; RRID: AB_2800723 |
| TotalSeq™-C0124 anti-human CD31 | BioLegend | 303139; RRID: AB_2800757 |
| TotalSeq™-C0072 anti-human CD4 | BioLegend | 300567; RRID: AB_2800725 |
| TotalSeq™-C0050 anti-human CD19 | BioLegend | 302265; RRID: AB_2800741 |
| TotalSeq™-C0139 anti-human TCRgd | BioLegend | 353747; RRID: AB_2800949 |
| TotalSeq™-C0140 anti-human CD183 (CXCR3) | BioLegend | 331231; RRID: AB_2814199 |
| TotalSeq™-C0125 anti-human CD44 | BioLegend | 338827; RRID: AB_2800900 |
| TotalSeq™-C0176 anti-human CD39 | BioLegend | 328237; RRID: AB_2800853 |
| TotalSeq™-C0063 anti-human CD45RA | BioLegend | 304163; RRID: AB_2800764 |
| TotalSeq™-C0224 anti-human TCRab | BioLegend | 306743; RRID: AB_2800793 |
| TotalSeq™-C0061 anti-human CD117 | BioLegend | 313243; RRID: AB_2810474 |
| TotalSeq™-C0161 anti-human CD11b | BioLegend | 301359; RRID: AB_2800732 |
| TotalSeq™-C0145 anti-human CD103 | BioLegend | 350231; RRID: AB_2749996 |
| TotalSeq™-C0147 anti-human CD62L | BioLegend | 304851; RRID: AB_2800770 |
| TotalSeq™-C0123 anti-human EpCAM | BioLegend | 324247; RRID: AB_2814181 |
| TotalSeq™-C0058 anti-human HLA-ABC | BioLegend | 311449; RRID: AB_2800816 |
| TotalSeq™-C0154 anti-human CD27 | BioLegend | 302853; RRID: AB_2800747 |
| TotalSeq™-C0006 anti-human CD86 | BioLegend | 305447; RRID: AB_2800786 |
| TotalSeq™-C0138 anti-human CD5 | BioLegend | 300637; RRID: AB_2800726 |
| TotalSeq™-C0146 anti-human CD69 | BioLegend | 310951; RRID: AB_2800810 |
| TotalSeq™-C0160 anti-human CD1c | BioLegend | 331547; RRID: AB_2800871 |
| TotalSeq™-C0158 anti-human CD134 | BioLegend | 350035; RRID: AB_2800932 |
| TotalSeq™-C0402 anti-human CD1a | BioLegend | 300135; RRID: AB_2814107 |
| TotalSeq™-C0088 anti-human CD279 | BioLegend | 329963; RRID: AB_2800862 |
| TotalSeq™-C0028 anti-human CD30 | BioLegend | 333919; RRID: AB_2800883 |
| TotalSeq™-C0151 anti-human CD152 | BioLegend | 369621; RRID: AB_2801015 |
| TotalSeq™-C0148 anti-human CD197 (CCR7) | BioLegend | 353251; RRID: AB_2800943 |
| TotalSeq™-C0171 anti-human ICOS | BioLegend | 313553; RRID: AB_2800823 |
| TotalSeq™-C0410 anti-human CD38 | BioLegend | 356637; RRID: AB_2820007 |
| TotalSeq™-C0144 anti-human CD185 (CXCR5) | BioLegend | 356939; RRID: AB_2800968 |
| TotalSeq™-C0156 anti-human CD95 | BioLegend | 305651; RRID: AB_2800787 |
| TotalSeq™-C0053 anti-human CD11c | BioLegend | 371521; RRID: AB_2801018 |
| TotalSeq™-C0366 anti-human CD184 (CXCR4) | BioLegend | 306533; RRID: AB_2800791 |
| TotalSeq™-C0181 anti-human CD21 | BioLegend | 354923; RRID: AB_2800953 |
| TotalSeq™-C0090 anti-human IsoIgG1 | BioLegend | 400187; RRID: AB_2888921 |
| TotalSeq™-C0091 anti-human IsoIgG2A | BioLegend | 400293; RRID: AB_2888922 |
| TotalSeq™-C0027 anti-human CD70 | BioLegend | 355119; RRID: AB_2800955 |
| TotalSeq™-C0071 anti-human CD194 (CCR4) | BioLegend | 359425; RRID: AB_2800988 |
| TotalSeq™-C0396 anti-human CD26 | BioLegend | 302722; RRID: AB_2810435 |
| TotalSeq™-C0007 anti-human CD274 | BioLegend | 329751; RRID: AB_2800860 |
| TotalSeq™-C0386 anti-human CD28 | BioLegend | 302963; RRID: AB_2800751 |
| TotalSeq™-C0166 anti-human CD66b | BioLegend | 392909; RRID: AB_2801027 |
| TotalSeq™-C0081 anti-human CD14 | BioLegend | 301859; RRID: AB_2800736 |
| TotalSeq™-C0390 anti-human CD127 | BioLegend | 351356; RRID: AB_2800937 |
| TotalSeq™-C0163 anti-human CD141 | BioLegend | 344125; RRID: AB_2810541 |
| TotalSeq™-C0085 anti-human CD25 | BioLegend | 302649; RRID: AB_2800745 |
| TotalSeq™-C0087 anti-human CD45RO | BioLegend | 304259; RRID: AB_2800766 |
| TotalSeq™-C0155 anti-human CD107a | BioLegend | 328649; RRID: AB_2800854 |
| TotalSeq™-C0169 anti-human CD366 | BioLegend | 345049; RRID: AB_2800925 |
| TotalSeq™-C0005 anti-human CD80 | BioLegend | 305243; RRID: AB_2800783 |
| TotalSeq™-C0831 anti-human CD138 | BioLegend | 352327; RRID: AB_2814282 |
| TotalSeq™-C0143 anti-human CD196 (CCR6) | BioLegend | 353440; RRID: AB_2810563 |
| TotalSeq™-C0180 anti-human CD24 | BioLegend | 311143; RRID: AB_2800813 |
| TotalSeq™-C0152 anti-human CD223 | BioLegend | 369335; RRID: AB_2814327 |
| TotalSeq™-C0804 anti-human CD186 (CXCR6) | BioLegend | 362559; RRID: AB_2801002 |
| TotalSeq™-C0047 anti-human CD56 | BioLegend | 369621; RRID: AB_2801015 |
| TotalSeq™-C0251 anti-human Hashtag 1 | BioLegend | 394661; RRID: AB_2801031 |
| TotalSeq™-C0252 anti-human Hashtag 2 | BioLegend | 394663; RRID: AB_2801032 |
| TotalSeq™-C0253 anti-human Hashtag 3 | BioLegend | 394665; RRID: AB_2801033 |
| TotalSeq™-C0254 anti-human Hashtag 4 | BioLegend | 394667; RRID: AB_2801034 |
| TotalSeq™-C0255 anti-human Hashtag 5 | BioLegend | 394669; RRID: AB_2801035 |
| TotalSeq™-C0256 anti-human Hashtag 6 | BioLegend | 394671; RRID: AB_2820042 |
| TotalSeq™-C0257 anti-human Hashtag 7 | BioLegend | 394673; RRID: AB_2820043 |
| TotalSeq™-C0258 anti-human Hashtag 8 | BioLegend | 394675; RRID: AB_2820044 |
| TotalSeq™-C0259 anti-human Hashtag 9 | BioLegend | 394677; RRID: AB_2820045 |
| TotalSeq™-C0260 anti-human Hashtag 10 | BioLegend | 394679; RRID: AB_2820046 |
| TotalSeq™-C Human Universal Cocktail, V1.0 | Biolegend | 399905; RRID: AB_2876728 |
| HTO21 anti-human hashing antibody | Stoekus et al.79 | New York Genome Center |
| HTO22 anti-human hashing antibody | Stoekus et al.79 | New York Genome Center |
| HTO23 anti-human hashing antibody | Stoekus et al.79 | New York Genome Center |
| Alexa Fluor® 488 AffiniPure Goat Anti-Human IgG, Fcγ fragment specific | Jackson ImmunoResearch | 109-545-098; RRID:AB_2337840 |
| R-Phycoerythrin AffiniPure Goat Anti-Human Serum IgA, α chain specific | Jackson ImmunoResearch | 109-115-011 |
| DyLight™ 405 AffiniPure Donkey Anti-Human IgM, Fc5μ fragment specific | Jackson ImmunoResearch | 709-475-073; RRID:AB_2340551 |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 (2019-nCoV) Nucleocapsid-AVI & His recombinant Protein, Biotinylated | Sino Biological | 40588-V27B-B |
| SARS-CoV-2 (2019-nCoV) Nucleocapsid-His recombinant Protein | Sino Biological | 40588-V08B |
| SARS-CoV-2 (2019-nCoV) Spike S1-His Recombinant Protein | Sino Biological | 40591-V08H |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 5’ Kit v1.1 | 10x Genomics | PN-1000165 |
| Chromium Next GEM Single Cell 5' Kit v2 | 10x Genomics | PN-1000263 |
| Single Index Kit T Set A | 10x Genomics | PN-1000213 |
| Dual Index Kit TT Set A | 10x Genomics | PN-1000215 |
| Dual Index Kit TN Set A | 10x Genomics | PN-1000250 |
| Chromium Single Cell V(D)J Enrichment Kit, Human T Cell | 10x Genomics | PN-1000005 |
| Chromium Single Cell V(D)J Enrichment Kit, Human B Cell | 10x Genomics | PN-1000016 |
| Chromium Single Cell Human TCR Amplification Kit | 10x Genomics | PN-1000252 |
| Chromium Single Cell Human BCR Amplification Kit | 10x Genomics | PN-1000253 |
| Dead Cell Removal Kit | Miltenyi Biotec | 130-090-101 |
| KAPA HiFi HotStart ReadyMix | Roche | KK2601 |
| MultiCyt® QBeads® Streptavidin Coated panel QSAv1,2,3 and 5 | Sartorius | 90792 |
| Deposited data | ||
| SARS-CoV-2 infection and vaccination ECCITE-seq data | This paper | https://cellxgene.cziscience.com/collections/ecb739c5-fe0d-4b48-81c6-217c4d64eec4 |
| Raw and processed data | This paper | GEO: GSE247917 |
| Haniffa COVID-19 single-cell RNA-seq dataset | Stephenson et al.3 | https://www.covid19cellatlas.org/ |
| Experimental models: Cell lines | ||
| Human: Expi293 cells | Thermo Fisher | A14527 |
| Oligonucleotides | ||
| ADT additive: CCTTGGCACCCGAGAATTCC | Stoekus et al.79 | IDT, Inc. |
| HTO additive: GTGACTGGAGTTCAGACGTGTGCTC | Stoekus et al.79 | IDT, Inc. |
| Index 1 (i7) Adapter: CAAGCAGAAGACGGC ATACGAGATxxxxxxxxGTCTCGTGGGCTCGG x: Barcode or index sequence |
Illumina | IDT, Inc. |
| D7xx_s (HTO indexing primer): CAAGCAGA AGACGGCATACGAGATxxxxxxxxGTGACTG GAGTTCAGACGTGTGC x: Barcode or index sequence |
Stoekus et al.79 | IDT, Inc. |
| Target enrichment 1 hTRDC primer (v1.1): AGCTTGACAGCA TTGTACTTCC |
Mimitou et al.19 | IDT, Inc. |
| Target enrichment 1 hTRGC primer (v1.1): TGTGTCGTTAGTCTTCATGGTGTTCC | Mimitou et al.19 | IDT, Inc. |
| Target enrichment 2 hTRDC primer (v1.1): TCCTTCACCAGACAAGCGAC |
Mimitou et al.19 | IDT, Inc. |
| Target enrichment 2 hTRGC primer (v1.1): GATCCCAGAATCGTGTTGCTC |
Mimitou et al.19 | IDT, Inc. |
| Target enrichment forward primer (v2): GATCTACACTCTTTCCCTACACGACGC |
10x Genomics | IDT, Inc. |
| Software and algorithms | ||
| Cell Ranger | 10x Genomics | https://www.10xgenomics.com/support/software/cell-ranger; RRID:SCR_017344 |
| Seurat | Stuart et al.80 | https://satijalab.org/seurat; RRID:SCR_016341 |
| kallisto kb-count v0.24.1 | Bray et al.81 | https://github.com/pachterlab/kallisto_paper_analysis/; RRID:SCR_018213 |
| totalVI | Gayoso et al.82 | https://github.com/YosefLab/scvi-tools |
| IgBLAST | Ye et al.36 | https://www.ncbi.nlm.nih.gov/igblast/; RRID:SCR_002873 |
| GSVA package v1.38 | Hänzelmann et al.32 | https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-7; RRID:SCR_021058 |
| scDblFinder | Germaine et al.83 | https://github.com/plger/scDblFinder; RRID:SCR_022700 |
Resource availability
Lead contact
-
•
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Sergei Koralov (sergei.koralov@nyulangone.org).
Materials availability
-
•
This study did not generate new unique reagents.
Data and code availability
-
•
Raw and processed single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
Single-cell RNA-seq data have also been deposited at CELLxGENE and are publicly available as of the date of publication. Data can be explored interactively through the web at https://cellxgene.cziscience.com/. The download link to the data object, as an h5ad file, is listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. The link to the dataset is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Participant/sample details
-
•
Peripheral blood samples were drawn from both outpatients and hospitalized patients with confirmed COVID-19 as well as healthy volunteers at NYU Langone Health. Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood. Peripheral blood was collected in accordance with a NYU Institutional Review Board protocols (IRB 18-02035, 18-02037 and 20-00463). Informed consent was obtained from all participants. Samples were de-identified and assigned coded identification numbers prior to analysis.
-
•
Samples were collected from five subjects with confirmed COVID-19 and nine healthy participants, seven of which received the BNT162b2 mRNA vaccine. For three of the vaccinated subjects, samples were also collected after receiving the booster.
-
•
Demographic characteristics, clinical features, and outcomes for study participants are reported in the table below as well as in Table S1.
Patients and sample collection
Peripheral blood samples were drawn from both outpatients and hospitalized patients with confirmed COVID-19 at NYU Langone Health. SARS-CoV-2 was detected in patients’ nasopharyngeal swab using the cobas® SARS-CoV-2 real time PCR under EUA. Samples were also collected from healthy volunteers before and after receiving the BNT162b2 mRNA vaccine and booster. Peripheral blood was collected in accordance with a NYU Institutional Review Board protocols (IRB 18-02035, 18-02037 and 20-00463). Samples were de-identified and assigned coded identification numbers prior to analysis.
Whole blood was collected in commercially available heparin-coated tubes (BD). Plasma was collected from whole blood by centrifugation at 2000 x g at 4°C, aliquoted, and stored at -20°C. For serum collection, whole blood was collected in serum separator tubes (SST). The blood was allowed to clot undisturbed at room temperature for 30-45 minutes and the clot was removed by centrifugation at 2000 x g at 4°C, the serum aliquoted and stored at -20°C.
PBMCs were isolated from peripheral blood by gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare) and SepMate™ PBMC Isolation Tubes (Stemcell) according to the manufacturer’s instructions. Buffy coat PBMCs were cryopreserved in FBS (Corning) supplemented with 10% DMSO (Sigma-Aldrich) and stored in liquid nitrogen.
| Participant | Cohort | Age | Sex | Race | BMI |
|---|---|---|---|---|---|
| SK-010 | Acute COVID-19 | 36-40 | Female | Asian | NA |
| SK-011 | Acute COVID-19 | 56-60 | Female | Caucasian | 35.7 |
| SK-012 | Acute COVID-19 | 41-45 | Male | Caucasian | 24.7 |
| SK-013 | Acute COVID-19 | 66-65 | Male | NA | NA |
| SK-014 | Acute COVID-19 | 51-55 | Female | Asian | NA |
| CV-00 1 | mRNA vaccine | 36-40 | Male | Asian | 28.4 |
| CV-003 | mRNA vaccine | 31-35 | Male | Asian | 27.9 |
| CV-011 | mRNA vaccine | 36-40 | Male | Caucasian | 28.6 |
| CV-012 | mRNA vaccine | NA | NA | Asian | 28.7 |
| CV-022 | mRNA vaccine | 41-45 | Male | Caucasian | 22.4 |
| CV-053 | mRNA vaccine | 46-50 | Female | African-American | NA |
| CV-056 | mRNA vaccine | 51-55 | Female | Caucasian | 25.4 |
| SK-007 | HC | 36-40 | Female | Caucasian | 20.4 |
| SK-008 | HC | 36-40 | Female | Caucasian | 28.6 |
Method details
Single-cell RNA-seq sample processing
PMBCs were thawed at 37°C and diluted slowly with warm RPMI supplemented with 20% heat-inactivated FBS. The cells were washed twice and resuspended in warm media then rested on ice for 30-60 minutes. After resting, cells were counted and viability determined. Live cells were enriched using Dead Cell Removal Kit (Miltenyi Biotec) to ensure viability of all samples is at least 95% prior to staining. Cells were stained with barcoded antibodies for CITE-seq and cell hashing (BioLegend) in DNA LoBind tubes (Eppendorf, 22431021).19,20,24,81 Approximately 200,000 cells per sample were resuspended in staining buffer containing PBS, 2% BSA, 0.01% Tween-20. Cells were centrifuged at 300-350 x g for 5 minutes at 4°C and the supernatant carefully removed. The cell pellet was resuspended in 12.5 μl staining buffer with Fc blocking reagent (TruStain FcX, BioLegend, 422302; FcR blocking reagent, Miltenyi Biotec, 130-059-901) and cells were incubated for 10 minutes on ice. Following incubation 12.5 μl 2x barcoded antibody cocktail was added to the cells/Fc block solution and mixed gently. Each sample was hashed with a unique hashing antibody. Cells were incubated for 30 minutes on ice. After staining, cells were washed 3 times in staining buffer and one more time in staining buffer without Tween-20. Cells were filtered using a 70 μm filter and counted. Equal number of cells were pooled in PBS supplemented with 0.04% BSA for loading on 10x controller.
Cell capture and library preparation
Sample processing of COVID-19 patient samples for scRNA-seq was performed in the ABSL3 facility of NYU Grossman School of Medicine (New York, NY), in accordance with its Biosafety Manual and Standard Operating Procedures. The concentration of single cell suspension was determined using Countess II Automated Cell Counter (Invitrogen) or NucleoCounter NC-3000 (Chemometec) and adjusted to ∼1450 cells/μl. Using single cell Chromium Next GEM Single Cell 5' Library & Gel Bead Kit (v1.1 chemistry) (10x Genomics, PN-1000165) and Chromium Next GEM Chip G (10x Genomics, PN-1000120) or Chromium Next GEM Single Cell 5' Kit (v2 chemistry) (10x Genomics, PN-1000263) and Chromium Next GEM Chip K (10x Genomics, PN-1000286), the cell suspension was loaded onto the Chromium single cell controller (10x Genomics) to generate gel beads-in-emulsion (GEM) according to the manufacturer’s protocol. Approximately 56,000 cells were loaded into each lane targeting recovery of 25,000 cells. Captured cells were lysed and barcoded cDNA generated from released mRNA through reverse transcription in individual GEMs. The reverse transcription reaction was performed in 200 μl PCR tubes (Scientific American, 1402-4700) in Mastercycler Nexus thermal cycler (Eppendorf) at 53°C for 45 minutes, followed by 85°C for 5 minutes, and hold at 4°C. The generated cDNA and antibody-derived tags (ADTs) were amplified with 10x reagents from the Next GEM kit according to the manufacturer’s protocol. For experiments using v1.1 chemistry, ADT and hashtag oligo (HTO) additive primers were spiked into the cDNA amplification mix at 2 nM concentration. The additive primers can be found in the key resources table. Gene Expression (GEX) libraries were prepared from amplified cDNA using indexing primers from Single Index Kit T Set A (10x Genomics, PN-1000213) while ADT and HTO libraries were prepared from amplified ADT/HTO fragments using indexing primers found in key resources table. For experiments using v2 chemistry, GEX libraries were prepared using indexing primers from Dual Index Kit TT Set A (10x Genomics) while ADT libraries were prepared using indexing primers from Dual Index Kit TN Set A (10x Genomics). To prepare V(D)J libraries, target enrichment was performed. For TCRαβ, target enrichment was performed using Chromium Single Cell V(D)J Enrichment Kit, Human T Cell (v1.1) (10x Genomics, PN-1000005) or Chromium Single Cell Human TCR Amplification Kit (v2) (10x Genomics, PN-1000252). For BCR, target enrichment was performed using Chromium Single Cell V(D)J Enrichment Kit, Human B Cell (v1.1) (10x Genomics, PN-1000016) or Chromium Single Cell Human BCR Amplification Kit (v2) (10x Genomics, PN-1000253). Target enrichment for TCRγ/δ was done using KAPA HiFi ReadyMix (Roche) and custom reverse primers which can be found in the key resources table. The enrichment reaction was supplemented with 5% DMSO, the final concentration of forward primer was 0.4 μM and reverse primers 0.2 μM each, and the amplification was carried out using the following program: 45 seconds at 98°C followed by 20 seconds at 98°C, 30 seconds at 62°C, 1 minute at 72°C for 12 cycles. V(D)J libraries from experiments using v1.1 chemistry were constructed using indexing primers from Single Index Kit T Set A (10x Genomics, PN-1000213) according to the 10x Genomics protocol and experiments using v2 chemistry were constructed using indexing primers from Dual Index Kit TT Set A (10x Genomics). The quality of the libraries was assessed using the TapeStation 4150 (Agilent Technologies). All libraries were sequenced using an Illumina NovaSeq 6000 system with sequencing depth as follows: GEX 30,000 reads/cell, V(D)J 5,000 reads/cell (each library), ADT (v1.1) 6,000 reads/cell, HTO (v1.1) 2,400 reads/cell, ADT (v2) 8,000 reads/cell with paired-end 100 bp reads. Libraries were pooled to desired quantities and sequenced on a NovaSeq 6000 100 cycle flow cell (R1: 26 cycles i5/i7: 10 cycles, R2: 74 cycles) (Illumina). Reads were trimmed as required for downstream processing.
scRNA-seq data processing, quality control, and analysis
Cell Ranger software suite from 10x Genomics was used to demultiplex cellular barcodes, align reads to the human genome (GRCh38 ensemble, http://useast.ensembl.org/Homo_sapiens), and perform UMI counting. ADT and HTO count matrices were generated using kallisto kb-count v0.24.184,80 and demultiplexed HTOs using HTODemux from Seurat v4.0.0.83 Low quality cells were filtered out (< 200 genes/cell, > 15% mitochondrial genes). Seurat was used to process the single-cell data, generate uniform manifold approximation and projection (UMAP) representation based on totalVI82 dimension reduction of RNA and ADT modalities. R package scDblFinder82 was used to identify and filter out doublets. RNA was normalized and batch-corrected by totalVI, while ADT values were corrected by the built-in integration function FindIntegrationAnchors in Seurat. Clustering was performed by the Louvain algorithm26 and cell type identification was determined by clustering, SingleR annotations, corrected ADT levels and canonical markers for various immune cell subsets. Further, we performed sub-clustering on Myeloid, B, conventional T and innate-like T and NK cells to identify 32 individual populations. BCR/TCR sequences were processed by Cell Ranger VDJ and added to the metadata of the combined Seurat object for each sample. Gene set variation analysis was performed using the R package GSVA v1.3885 with various named gene sets from MSigDB (http://www.gsea-msigdb.org/gsea/index.jsp)32 in order to determine pathway enrichment. The enrichment scores from GSVA for the interferon-alpha pathway were plotted against the oxygen requirement for each sample. Module scores were calculated using Seurat’s AddModuleScore function for interferon-alpha pathway genes. For memory, plasmablasts, and resting B cells, the IgBlast command ‘igblastn’ was used to query sample VH gene sequences against the IMGT human reference sequences to determine the number of mutated bases.36,86 All clonal analysis was performed by plotting only high confidence (as determined by Cellranger VDJ) clones.
Validation dataset
We used a public scRNA-seq dataset to validate some of the key findings of the study.3 The dataset includes 143 samples from healthy volunteers and COVID-19 patients, stratified by disease severity. In our analysis of data from Stephenson et al., we included COVID-19 samples with mild, moderate, severe, and critical disease.
Multiplex bead-binding assay for antibody profiling
We obstained the amino acid sequence of the Spike protein ectodomain (residues 16-1213) of SARS-CoV-2 (GenBank: MN908947.3). A synthetic gene encoding a stabilized Spike protein,87 including the removal of the furin cleavage site, K986P and V987P mutations, the addition of a T4 foldon trimerization domain, a His6-tag and a biotinylation tag (Avi-tag) at the C-terminus was synthesized (Integrated DNA technologies) and cloned into the pBCAG mammalian expression vector. The codon-optimized genes encoding the receptor-binding domain of SARS-CoV-2 (RBD, residues 328–531; GenBank: MN908947.3) and the RBD of SARS-CoV (residues 315–517; GenBank: AFR58742.1) with the His-tag and the Avi-tag at the C-terminus were synthesized (Integrated DNA Technologies) and cloned into the pBCAG vector. The G476S mutation, common in SARS-CoV-2 samples in North America,88 was introduced in the expression vector for SARS-CoV-2 RBD using PCR. Expi293 cells (Thermo Fisher, A14527) were transiently transfected with the vectors according to the manufacturer’s protocol, and the transfected cells were grown at 37°C with 8% CO2 for 7 days. The recombinant proteins were purified from filtered cell culture supernatants by immobilized metal ion affinity chromatography (IMAC) using HiSTrap excel column (GE Healthcare). The purified proteins were biotinylated using the BirA enzyme produced in-house in presence of 500 μM biotin and 10 mM ADT. After biotinylating, the BirA enzyme was removed by IMAC and recombinant proteins were dialyzed against PBS and stored at -80°C. Biotinylated nucleocapsid protein was purchased from Sino Biological (40588-V27B-B). We used MultiCyt® QBeads® Streptavidin Coated panel QSAv1,2,3 and 5 (Sartorius, 90792) to immobilize SARS-CoV-2 antigens; spike to QSAv1, Nucleocapsid to QSAv2, the receptor-binding-domain (RBD) of spike to QSAv3, and biotin only to QSAv5. The antigens were diluted to 25 nM in PBS with 0.5 % BSA and mixed with the same volume of the twice-washed QBeads. We detected antigen-specific antibodies in heat-inactivated serum or plasma using anti-human IgG-Alexa 488 (Jackson ImmunoResearch, 109-545-098; 1:800 in PBS 0.1 % Tween 20 and with 1 % BSA), anti-human IgA-PE (Jackson ImmunoResearch, 109-115-011; 1:100) and anti-human IgM-DyLight405 (Jackson ImmunoResearch, 709-475-073; 1:200). We measured the samples on a Yeti ZE5 Cell Analyzer (Bio-Rad) and analyzed the data using FlowJo (BD, version 10.7.1).
ELISA
We used direct ELISA to quantify Spike-specific antibody titers in healthy volunteers before and after receiving BNT162b2 mRNA vaccine and booster as described.22 Briefly, 96-well plates were coated with 1 μg/ml S1 protein diluted in PBS and incubated overnight at 4°C (40591-V08H and 40588-V08B, Sino Biological). Plates were washed with PBS containing 0.05% Tween-20 (PBS-T) and blocked with 5% non-fat milk in PBS-T. Sera were heat-inactivated at 56°C for one hour, diluted in blocking solution, and added to the coated plate. After two hours’ incubation, the plate was washed with PBS-T. Horseradish peroxidase (HRP)-conjugated anti-human IgG antibody was diluted in blocking buffer, added to each well, incubated for one hour at room temperature, and washed with PBS-T. After developing with TMB Peroxidase Substrate 3,3′,5,5′-Tetramethylbenzidine (Thermo Fisher Scientific) for five minutes, we stopped the reaction with 1N hydrochloric acid and determined the optical density by measuring absorbance at 450 nm on a Synergy 4 (BioTek) plate reader.
Quantification and statistical analysis
All statistical analysis was done in base R. In Figures 1G, 1H, 3A, and 3B, line graphs show percentages for given cell population per subject, per time point. P-values for these plots are determined by an ANOVA of linear mixed models and post-hoc pairwise comparison of estimated marginal means are listed on each plot. The bars represent the upper and lower confidence intervals for the estimated marginal means for the linear model. In Figures 2B and 2E, box and whisker plots were used to portray the distribution of the data, outliers, and the median. P-values were determined by the Wilcoxon test and represented as asterisks on the graph (ns p > 0.05, ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001). In Figure 2C, Kendal rank correlation coefficient was used to establish the statistical dependence between oxygen requirement (shown as fraction of inspired oxygen) and GSVA enrichment score for interferon alpha response gene set. P and tau values are determined by a Kendall rank correlation test and are shown on the plot. The statistical details can be found in the figure legends.
Acknowledgments
We are grateful for support of this work from NYU Grossman School of Medicine. Work in Dr. Koralov’s laboratory was further supported by the NIH (HL125816, CA271245, 2R44AI136141), LEO Foundation Grant (LF-OC-20-000351), NYU Cancer Center Pilot Grant (P30CA016087),the Judith and Stewart Colton Center for Autoimmunity Pilot grant. Presented work was also supported by NIH grant R21 AI158997, R01 CA194864 and R01 CA212608 to S.K.; NIH grants AI114852 and AI082630 to R.S.H.; K08AI163457 to R.J.U.; and AI148574 to M.J.M. T.B.B. and NØ received support from the Danish Cancer Society (Kræftens Bekæmpelse), the Danish Council for Independent Research (Danmarks Frie Forskningsfond) and the LEO Foundation. This work was also supported by funding from the NCATS/NIH Centers for Translational Science Awards (CTSA) to New York University (UL1 TR001445). We thank all members of NYU Vaccine Center processing and clinical staff, including Michael Tuen, Jimmy Wilson, Abdonnie Holder, Shelby Goins, Meron Tasissa, Sara Wesley Hyman, and Farzana Antara. We are also grateful to the NYU Genome Technology Core, and Dr. Heguy in particular, for technical assistance and support. We sincerely thank Ms. Cathy Shufro for her valuable contributions to improving the manuscript. Finally, we would like to thank all the study participants who have contributed to our studies.
Author contributions
E.I., R.H., and S.B.K. conceived and planned the experiments. E.I. carried out the experiments. J.S., J.D., T.B., E.I., and K.R. planned and carried out the computational analysis. S.G., A.C., M.S., C.Z., and T.K. contributed to sample preparation. E.I. drafted the manuscript. E.I., J.S., and J.D. designed the figures. A.K. and S.K. performed the multiplex bead-binding assay. M.S. performed the ELISA to quantify Spike-specific Ab titers. L.D. helped adopt experimental methods for high containment BSL3 laboratories. E.M., P.S., N.O., S.G., and A.H. helped with experimental design. M.M., P.U., M.S., and R.H. established protocols and guided patient recruitment. All authors provided critical feedback and helped shape the research, analysis and text of the manuscript and approved the final version.
Declaration of interests
M.J.M. reported potential competing interests: laboratory research and clinical trials contracts with Lilly, Pfizer (exclusive of the current work), and Sanofi for vaccines or MAB vs. SARS-CoV-2; contract funding from USG/HHS/BARDA for research specimen characterization and repository; research grant funding from USG/HHS/NIH for SARS-CoV-2 vaccine and MAB clinical trials; personal fees from Meissa Vaccines, Inc. and Pfizer for Scientific Advisory Board service. RSH has received research support from CareDx for SARS-CoV-2 vaccine studies and has performed consulting work for Bristol-Myers-Squibb.
Published: November 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108572.
Contributor Information
Kelly V. Ruggles, Email: kelly.ruggles@nyulangone.org.
Ramin S. Herati, Email: ramin.herati@nyulangone.org.
Sergei B. Koralov, Email: sergei.koralov@nyulangone.org.
Supplemental information
References
- 1.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369 doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohammed R.N., Tamjidifar R., Rahman H.S., Adili A., Ghoreishizadeh S., Saeedi H., Thangavelu L., Shomali N., Aslaminabad R., Marofi F., et al. A comprehensive review about immune responses and exhaustion during coronavirus disease (COVID-19) Cell Commun. Signal. 2022;20:79. doi: 10.1186/s12964-022-00856-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carsetti R., Zaffina S., Piano Mortari E., Terreri S., Corrente F., Capponi C., Palomba P., Mirabella M., Cascioli S., Palange P., et al. Different Innate and Adaptive Immune Responses to SARS-CoV-2 Infection of Asymptomatic, Mild, and Severe Cases. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.610300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kared H., Redd A.D., Bloch E.M., Bonny T.S., Sumatoh H., Kairi F., Carbajo D., Abel B., Newell E.W., Bettinotti M.P., et al. SARS-CoV-2–specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest. 2021;131 doi: 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laidlaw B.J., Ellebedy A.H. The germinal centre B cell response to SARS-CoV-2. Nat. Rev. Immunol. 2022;22:7–18. doi: 10.1038/s41577-021-00657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moss P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022;23:186–193. doi: 10.1038/s41590-021-01122-w. [DOI] [PubMed] [Google Scholar]
- 9.Pape K.A., Dileepan T., Kabage A.J., Kozysa D., Batres R., Evert C., Matson M., Lopez S., Krueger P.D., Graiziger C., et al. High-affinity memory B cells induced by SARS-CoV-2 infection produce more plasmablasts and atypical memory B cells than those primed by mRNA vaccines. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner J.S., Kim W., Kalaidina E., Goss C.W., Rauseo A.M., Schmitz A.J., Hansen L., Haile A., Klebert M.K., Pusic I., et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature. 2021;595:421–425. doi: 10.1038/s41586-021-03647-4. [DOI] [PubMed] [Google Scholar]
- 11.Turner J.S., O'Halloran J.A., Kalaidina E., Kim W., Schmitz A.J., Zhou J.Q., Lei T., Thapa M., Chen R.E., Case J.B., et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature. 2021;596:109–113. doi: 10.1038/s41586-021-03738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J.-Y., Wang X.-M., Xing X., Xu Z., Zhang C., Song J.-W., Fan X., Xia P., Fu J.-L., Wang S.-Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- 13.Jeewandara C., Jayathilaka D., Gomes L., Wijewickrama A., Narangoda E., Idampitiya D., Guruge D., Wijayamuni R., Manilgama S., Ogg G.S., et al. SARS-CoV-2 neutralizing antibodies in patients with varying severity of acute COVID-19 illness. Sci. Rep. 2021;11:2062. doi: 10.1038/s41598-021-81629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Guo X., Xin Q., Pan Y., Hu Y., Li J., Chu Y., Feng Y., Wang Q. Neutralizing Antibody Responses to Severe Acute Respiratory Syndrome Coronavirus 2 in Coronavirus Disease 2019 Inpatients and Convalescent Patients. Clin. Infect. Dis. 2020;71:2688–2694. doi: 10.1093/cid/ciaa721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chodick G., Tene L., Patalon T., Gazit S., Tov A.B., Cohen D., Muhsen K. The effectiveness of the first dose of BNT162b2 vaccine in reducing SARS-CoV-2 infection 13-24 days after immunization: real-world evidence. medRxiv. 2021 doi: 10.1101/2021.01.27.21250612. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371 doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson M.G., Burgess J.L., Naleway A.L., Tyner H.L., Yoon S.K., Meece J., Olsho L.E.W., Caban-Martinez A.J., Fowlkes A., Lutrick K., et al. Interim Estimates of Vaccine Effectiveness of BNT162b2 and mRNA-1273 COVID-19 Vaccines in Preventing SARS-CoV-2 Infection Among Health Care Personnel, First Responders, and Other Essential and Frontline Workers - Eight U.S. Locations, December 2020-March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021;70:495–500. doi: 10.15585/mmwr.mm7013e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lederer K., Castaño D., Gómez Atria D., Oguin T.H., 3rd, Wang S., Manzoni T.B., Muramatsu H., Hogan M.J., Amanat F., Cherubin P., et al. SARS-CoV-2 mRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated with Neutralizing Antibody Generation. Immunity. 2020;53:1281–1295.e85. doi: 10.1016/j.immuni.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimitou E.P., Cheng A., Montalbano A., Hao S., Stoeckius M., Legut M., Roush T., Herrera A., Papalexi E., Ouyang Z., et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods. 2019;16:409–412. doi: 10.1038/s41592-019-0392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hattori T., Koide A., Panchenko T., Romero L.A., Teng K.W., Corrado A.D., Koide S. Multiplex bead binding assays using off-the-shelf components and common flow cytometers. J. Immunol. Methods. 2021;490 doi: 10.1016/j.jim.2020.112952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samanovic M.I., Oom A.L., Cornelius A.R., Gray-Gaillard S.L., Karmacharya T., Tuen M., Wilson J.P., Tasissa M.F., Goins S., Herati R.S., Mulligan M.J. Vaccine-Acquired SARS-CoV-2 Immunity versus Infection-Acquired Immunity: A Comparison of Three COVID-19 Vaccines. Vaccines (Basel) 2022;10:2152. doi: 10.3390/vaccines10122152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect. Dis. 2020;20:e192–e197. doi: 10.1016/S1473-3099(20)30483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buus T.B., Herrera A., Ivanova E., Mimitou E., Cheng A., Papagiannakopoulos T., Smibert P., Ødum N., Koralov S.B. Improving oligo-conjugated antibody signal in multimodal single-cell analysis. bioRxiv. 2020 doi: 10.1101/2020.06.15.153080. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McInnes L., Healy J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv. 2018 doi: 10.48550/arXiv.1802.03426. Preprint at. [DOI] [Google Scholar]
- 26.Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008;2008 [Google Scholar]
- 27.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finch E., Lowe R., Fischinger S., de St Aubin M., Siddiqui S.M., Dayal D., Loesche M.A., Rhee J., Beger S., Hu Y., et al. SARS-CoV-2 antibodies protect against reinfection for at least 6 months in a multicentre seroepidemiological workplace cohort. PLoS Biol. 2022;20 doi: 10.1371/journal.pbio.3001531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harvey R.A., Rassen J.A., Kabelac C.A., Turenne W., Leonard S., Klesh R., Meyer W.A., 3rd, Kaufman H.W., Anderson S., Cohen O., et al. Real-world data suggest antibody positivity to SARS-CoV-2 is associated with a decreased risk of future infection. medRxiv. 2020 doi: 10.1101/2020.12.18.20248336. Preprint at. [DOI] [Google Scholar]
- 30.Hirano T., Ishihara K., Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 31.Limon J.J., Fruman D.A. Akt and mTOR in B Cell Activation and Differentiation. Front. Immunol. 2012;3:228. doi: 10.3389/fimmu.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson K.J.L., Liu Y., Roskin K.M., Glanville J., Hoh R.A., Seo K., Marshall E.L., Gurley T.C., Moody M.A., Haynes B.F., et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe. 2014;16:105–114. doi: 10.1016/j.chom.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nielsen S.C.A., Yang F., Jackson K.J.L., Hoh R.A., Röltgen K., Jean G.H., Stevens B.A., Lee J.-Y., Rustagi A., Rogers A.J., et al. Human B Cell Clonal Expansion and Convergent Antibody Responses to SARS-CoV-2. Cell Host Microbe. 2020;28:516–525.e5. doi: 10.1016/j.chom.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robbiani D.F., Gaebler C., Muecksch F., Lorenzi J.C.C., Wang Z., Cho A., Agudelo M., Barnes C.O., Gazumyan A., Finkin S., et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–442. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye J., Ma N., Madden T.L., Ostell J.M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–W40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halliley J.L., Kyu S., Kobie J.J., Walsh E.E., Falsey A.R., Randall T.D., Treanor J., Feng C., Sanz I., Lee F.E.H. Peak frequencies of circulating human influenza-specific antibody secreting cells correlate with serum antibody response after immunization. Vaccine. 2010;28:3582–3587. doi: 10.1016/j.vaccine.2010.02.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacLean A.J., Richmond N., Koneva L., Attar M., Medina C.A.P., Thornton E.E., Gomes A.C., El-Turabi A., Bachmann M.F., Rijal P., et al. Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity. 2022;55:718–733.e8. doi: 10.1016/j.immuni.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Vito C., Calcaterra F., Coianiz N., Terzoli S., Voza A., Mikulak J., Della Bella S., Mavilio D. Natural Killer Cells in SARS-CoV-2 Infection: Pathophysiology and Therapeutic Implications. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.888248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanna S.J., Codd A.S., Gea-Mallorqui E., Scourfield D.O., Richter F.C., Ladell K., Borsa M., Compeer E.B., Moon O.R., Galloway S.A.E., et al. T cell phenotypes in COVID-19 - a living review. Oxf. Open Immunol. 2021;2:iqaa007. doi: 10.1093/oxfimm/iqaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georg P., Astaburuaga-García R., Bonaguro L., Brumhard S., Michalick L., Lippert L.J., Kostevc T., Gäbel C., Schneider M., Streitz M., et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell. 2022;185:493–512.e25. doi: 10.1016/j.cell.2021.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu K., He J., Wu Y., Xie B., Liu X., Wei B., Zhou H., Lin B., Zuo Z., Wen W., et al. Dysregulated adaptive immune response contributes to severe COVID-19. Cell Res. 2020;30:814–816. doi: 10.1038/s41422-020-0391-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nolan S., Vignali M., Klinger M., Dines J.N., Kaplan I.M., Svejnoha E., Craft T., Boland K., Pesesky M., Gittelman R.M., et al. A large-scale database of T-cell receptor beta (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res. Sq. 2020 [Google Scholar]
- 44.Juno J.A., van Bockel D., Kent S.J., Kelleher A.D., Zaunders J.J., Munier C.M.L. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front. Immunol. 2017;8:19. doi: 10.3389/fimmu.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaneko N., Boucau J., Kuo H.H., Perugino C., Mahajan V.S., Farmer J.R., Liu H., Diefenbach T.J., Piechocka-Trocha A., Lefteri K., et al. Expansion of Cytotoxic CD4+ T Cells in the Lungs in Severe COVID-19. medRxiv. 2021 doi: 10.1101/2021.03.23.21253885. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meckiff B.J., Ramírez-Suástegui C., Fajardo V., Chee S.J., Kusnadi A., Simon H., Eschweiler S., Grifoni A., Pelosi E., Weiskopf D., et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4(+) T Cells in COVID-19. Cell. 2020;183:1340–1353.e16. doi: 10.1016/j.cell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi A., Saito T. CD4 CTL, a Cytotoxic Subset of CD4(+) T Cells, Their Differentiation and Function. Front. Immunol. 2017;8:194. doi: 10.3389/fimmu.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gray-Gaillard S.L., Solis S., Chen H.M., Monteiro C., Ciabattoni G., Samanovic M.I., Cornelius A.R., Williams T., Geesey E., Rodriguez M., et al. Molecularly distinct memory CD4+ T cells are induced by SARS-CoV-2 infection and mRNA vaccination. bioRxiv. 2023 doi: 10.1101/2022.11.15.516351. Preprint at. [DOI] [Google Scholar]
- 49.Sabbaghi A., Miri S.M., Keshavarz M., Mahooti M., Zebardast A., Ghaemi A. Role of γδ T cells in controlling viral infections with a focus on influenza virus: implications for designing novel therapeutic approaches. Virol. J. 2020;17:174. doi: 10.1186/s12985-020-01449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawand M., Déchanet-Merville J., Dieu-Nosjean M.-C. Key Features of Gamma-Delta T-Cell Subsets in Human Diseases and Their Immunotherapeutic Implications. Front. Immunol. 2017;8:761. doi: 10.3389/fimmu.2017.00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu W., Metcalf J.P. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J. Innate Immun. 2020;12:437–447. doi: 10.1159/000508379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gessani S., Conti L., Del Cornò M., Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins. 2014;6:1696–1723. doi: 10.3390/toxins6061696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adamo S., Michler J., Zurbuchen Y., Cervia C., Taeschler P., Raeber M.E., Baghai Sain S., Nilsson J., Moor A.E., Boyman O. Signature of long-lived memory CD8+ T cells in acute SARS-CoV-2 infection. Nature. 2022;602:148–155. doi: 10.1038/s41586-021-04280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McNab F., Mayer-Barber K., Sher A., Wack A., O'Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C., Martins A.J., Lau W.W., Rachmaninoff N., Chen J., Imberti L., Mostaghimi D., Fink D.L., Burbelo P.D., Dobbs K., et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell. 2021;184:1836–1857.e22. doi: 10.1016/j.cell.2021.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broggi A., Ghosh S., Sposito B., Spreafico R., Balzarini F., Lo Cascio A., Clementi N., De Santis M., Mancini N., Granucci F., Zanoni I. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369:706–712. doi: 10.1126/science.abc3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaneko N., Kuo H.H., Boucau J., Farmer J.R., Allard-Chamard H., Mahajan V.S., Piechocka-Trocha A., Lefteri K., Osborn M., Bals J., et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell. 2020;183:143–157.e13. doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li C., Lee A., Grigoryan L., Arunachalam P.S., Scott M.K.D., Trisal M., Wimmers F., Sanyal M., Weidenbacher P.A., Feng Y., et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 2022;23:543–555. doi: 10.1038/s41590-022-01163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bergamaschi C., Terpos E., Rosati M., Angel M., Bear J., Stellas D., Karaliota S., Apostolakou F., Bagratuni T., Patseas D., et al. Systemic IL-15, IFN-γ, and IP-10/CXCL10 signature associated with effective immune response to SARS-CoV-2 in BNT162b2 mRNA vaccine recipients. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Granados-Riveron J.T., Aquino-Jarquin G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed. Pharmacother. 2021;142 doi: 10.1016/j.biopha.2021.111953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen D.C., Lamothe P.A., Woodruff M.C., Saini A.S., Faliti C.E., Sanz I., Lee F.E.-H. COVID-19 and plasma cells: Is there long-lived protection? Immunol. Rev. 2022;309:40–63. doi: 10.1111/imr.13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Braun D., Caramalho I., Demengeot J. IFN-α/β enhances BCR-dependent B cell responses. Int. Immunol. 2002;14:411–419. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- 64.Soni C., Perez O.A., Voss W.N., Pucella J.N., Serpas L., Mehl J., Ching K.L., Goike J., Georgiou G., Ippolito G.C., et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity. 2020;52:1022–1038.e7. doi: 10.1016/j.immuni.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kyogoku C., Smiljanovic B., Grün J.R., Biesen R., Schulte-Wrede U., Häupl T., Hiepe F., Alexander T., Radbruch A., Grützkau A. Cell-specific type I IFN signatures in autoimmunity and viral infection: what makes the difference? PLoS One. 2013;8 doi: 10.1371/journal.pone.0083776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rönnblom L., Leonard D. Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci. Med. 2019;6 doi: 10.1136/lupus-2018-000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ehrenfeld M., Tincani A., Andreoli L., Cattalini M., Greenbaum A., Kanduc D., Alijotas-Reig J., Zinserling V., Semenova N., Amital H., Shoenfeld Y. Covid-19 and autoimmunity. Autoimmun. Rev. 2020;19 doi: 10.1016/j.autrev.2020.102597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Acosta-Ampudia Y., Monsalve D.M., Rojas M., Rodríguez Y., Zapata E., Ramírez-Santana C., Anaya J.-M. Persistent Autoimmune Activation and Proinflammatory State in Post-Coronavirus Disease 2019 Syndrome. J. Infect. Dis. 2022;225:2155–2162. doi: 10.1093/infdis/jiac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Psarras A., Emery P., Vital E.M. Type I interferon–mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology. 2017;56:1662–1675. doi: 10.1093/rheumatology/kew431. [DOI] [PubMed] [Google Scholar]
- 70.Hirai T., Yoshioka Y. Considerations of CD8(+) T Cells for Optimized Vaccine Strategies Against Respiratory Viruses. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.918611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeyanathan M., Afkhami S., Smaill F., Miller M.S., Lichty B.D., Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020;20:615–632. doi: 10.1038/s41577-020-00434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sauer K., Harris T. An Effective COVID-19 Vaccine Needs to Engage T Cells. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.581807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geers D., Shamier M.C., Bogers S., den Hartog G., Gommers L., Nieuwkoop N.N., Schmitz K.S., Rijsbergen L.C., van Osch J.A.T., Dijkhuizen E., et al. SARS-CoV-2 variants of concern partially escape humoral but not T cell responses in COVID-19 convalescent donors and vaccine recipients. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abj1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edara V.-V., Pinsky B.A., Suthar M.S., Lai L., Davis-Gardner M.E., Floyd K., Flowers M.W., Wrammert J., Hussaini L., Ciric C.R., et al. Infection and Vaccine-Induced Neutralizing-Antibody Responses to the SARS-CoV-2 B.1.617 Variants. N. Engl. J. Med. 2021;385:664–666. doi: 10.1056/NEJMc2107799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu J., Yu J., McMahan K., Jacob-Dolan C., He X., Giffin V., Wu C., Sciacca M., Powers O., Nampanya F., et al. CD8 T cells contribute to vaccine protection against SARS-CoV-2 in macaques. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abq7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hurme A., Jalkanen P., Heroum J., Liedes O., Vara S., Melin M., Teräsjärvi J., He Q., Pöysti S., Hänninen A., et al. Long-Lasting T Cell Responses in BNT162b2 COVID-19 mRNA Vaccinees and COVID-19 Convalescent Patients. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.869990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maringer Y., Nelde A., Schroeder S.M., Schuhmacher J., Hörber S., Peter A., Karbach J., Jäger E., Walz J.S. Durable spike-specific T cell responses after different COVID-19 vaccination regimens are not further enhanced by booster vaccination. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.add3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sureshchandra S., Lewis S.A., Doratt B.M., Jankeel A., Coimbra Ibraim I., Messaoudi I. Single-cell profiling of T and B cell repertoires following SARS-CoV-2 mRNA vaccine. JCI Insight. 2021;6 doi: 10.1172/jci.insight.153201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Melsted P., Booeshaghi A.S., Liu L., Gao F., Lu L., Min K.H.J., da Veiga Beltrame E., Hjörleifsson K.E., Gehring J., Pachter L. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. 2021;39:813–818. doi: 10.1038/s41587-021-00870-2. [DOI] [PubMed] [Google Scholar]
- 81.Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Germain P.L., Lun A., Garcia Meixide C., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2021;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 85.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giudicelli V., Chaume D., Lefranc M.-P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33:D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amanat F., Stadlbauer D., Strohmeier S., Nguyen T.H.O., Chromikova V., McMahon M., Jiang K., Arunkumar G.A., Jurczyszak D., Polanco J., et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Q., Wu J., Nie J., Zhang L., Hao H., Liu S., Zhao C., Zhang Q., Liu H., Nie L., et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell. 2020;182:1284–1294.e9. doi: 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw and processed single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
Single-cell RNA-seq data have also been deposited at CELLxGENE and are publicly available as of the date of publication. Data can be explored interactively through the web at https://cellxgene.cziscience.com/. The download link to the data object, as an h5ad file, is listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. The link to the dataset is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.