

Abstract
Systemic autoinflammatory diseases are a heterogeneous group of rare genetic disorders caused by dysregulation of the innate immune system. Understanding the complex mechanisms underlying these conditions is critical for developing effective treatments. Cellular models are essential for identifying new conditions and studying their pathogenesis. Traditionally, these studies have used primary cells and cell lines of disease‐relevant cell types, although newer induced pluripotent stem cell (iPSC)‐based models might have unique advantages. In this review, we discuss the three cellular models used in autoinflammatory disease research, their strengths and weaknesses, and their applications to inform future research in the field.
Keywords: autoinflammation, disease modelling, induced pluripotent stem cell, systemic autoinflammatory diseases
Understanding the complex mechanisms underlying systemic autoinflammatory diseases is critical for developing effective treatments. Cellular models are essential for identifying new conditions and studying their pathogenesis. In this review, we discuss the different cellular models used in autoinflammatory disease research, their strengths and weaknesses, and their applications.
Introduction
Systemic autoinflammatory diseases (SAIDs) are a diverse group of chronic disorders characterised by antibody‐independent recurrent or persistent inflammation that affects multiple organ systems. 1 The term encompasses various clinical entities caused by the dysregulation of any pathway involved in the inflammatory response; as such a mechanistic classification system is useful in categorising SAIDs. 2 Major categories of SAIDs are (1) inflammasomopathies and other IL‐1 family conditions, (2) Type I interferonopathies and (3) diseases of NF‐𝛋B and/or aberrant TNF activity, although autoinflammatory conditions that cannot be classified into these categories have also been defined. Autoinflammation can also coexist with autoimmunity and immune deficiencies as a manifestation of immune dysregulation. 3 Many autoinflammatory diseases are rare hereditary monogenic disorders that mainly affect the paediatric age group.
Systemic autoinflammatory diseases are caused by the activation of the innate branch of the immune system, a rapid‐acting host defence mechanism operating through pattern recognition receptors in cellular components such as neutrophils, macrophages and natural killer cells. This system produces a fast but nonspecific response to a wide variety of pathogenic insults, in contrast to adaptive immunity where antigen‐specific activation results in the production of specific antibodies via B‐lymphocytes or T‐lymphocyte mediated cytotoxicity. The humoral and cellular components of innate immunity mediate inflammation, the key pathogenetic process in SAID. Inflammation is a complexly regulated defensive response that can involve multiple cell/tissue types and organ systems (Figure 1). 4 As a result of this complexity, the number of diseases categorised as SAIDs has steadily increased since the term was coined. 5 Identifying the molecular mechanisms of autoinflammatory diseases helps to uncover the various intricacies of inflammation regulation, aiding in our understanding of innate immune mechanisms in both health and disease.
Figure 1.
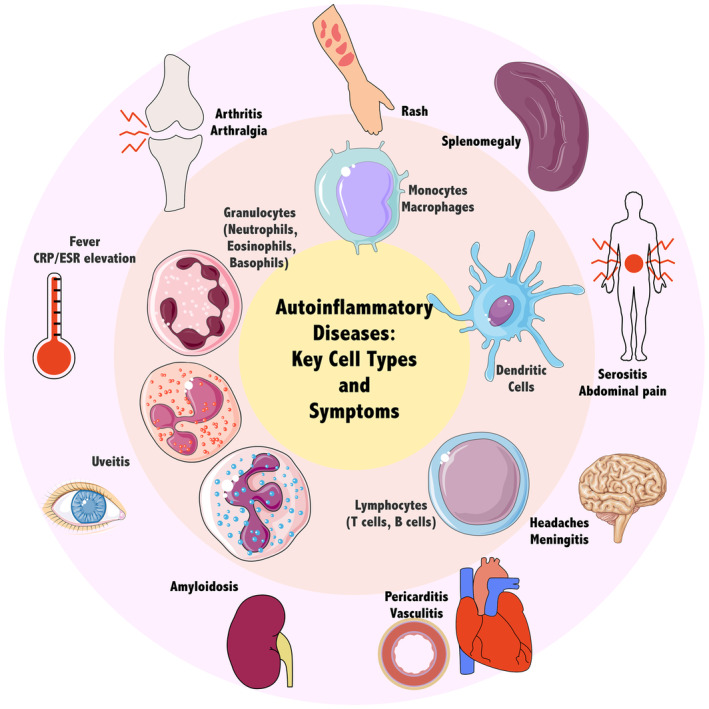
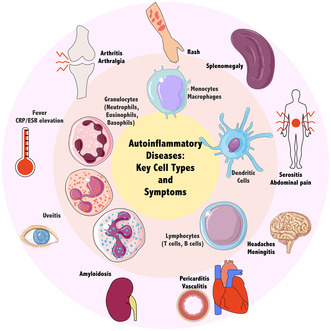
Hallmarks of autoinflammation and major effector cell types. These cell types may be modelled with primary cells isolated from patients, immortalised cell lines or patient‐derived induced pluripotent stem cells. These cellular models facilitate the identification of molecular events leading to the pathogenesis of autoinflammatory diseases. These events ultimately result in multisystem manifestations, including fever, inflammatory marker elevation and organ‐specific inflammation. Parts of the figure were drawn by using pictures from Servier Medical Art licensed under a Creative Commons Attribution 3.0 Unported Licence.
Numerous in vitro modelling approaches have been employed in the field of autoinflammatory disease research (Table 1). Traditionally, primary cells acquired from patients, cell lines that model immune cells, or cells transfected with disease‐related genes have been successfully used to investigate SAIDs. Such established cellular models can be used in various experimental setups to evaluate the function and activities of disease‐related gene products. Comparison of healthy cells with disease‐modelling cells yields valuable clues to the activities of different signalling pathways involved in inflammation and how they are dysregulated in the setting of autoinflammation. However, each of these options require compromises in terms of accessibility, ease of handling and physiological relevance. Recently, induced pluripotent stem cells derived from patient cells emerged as alternatives to these models that attempt to address some of the shortcomings of these models. To inform future research designs, this paper reviews established and emerging cellular models used in autoinflammatory disease research, comparing their strengths and disadvantages as well as their applications.
Table 1.
Selected studies showcasing the use of various cellular models in autoinflammatory disorders
| Disease | Important findings | Cellular model | References |
|---|---|---|---|
| Familial mediterranean fever (FMF) | iPSC‐derived macrophages from FMF patients exhibit inflammatory phenotype | Patient‐derived iPSCs | 90 |
| Neutrophil migration is increased in FMF | Primary neutrophils, HL‐60 | 21 | |
| Investigation of inflammatory features of iPSC‐derived macrophages containing various MEFV variants | Patient‐derived iPSCs | 91 | |
| Neutrophils from active FMF patients show increased migration and activation | Primary neutrophils | 22 | |
| FMF neutrophils transcriptionally and functionally resemble immature activated cells | Primary neutrophils | 23 | |
| miR‐197 is an anti‐inflammatory miRNA that decreases the expression of IL1R1 | THP‐1, SW982 | 61 | |
| Activation of the pyrin inflammasome is related to increased cell migration in monocytes | Primary PBMCs, THP‐1 | 40 | |
| CDC42 is required during pyrin activation | U‐937, THP‐1, HEK293T | 45 | |
| Pyrin activation is less dependent on transcriptional priming in FMF | Primary monocytes, THP‐1, BlaER1 | 46 | |
| TcdB is an activator of the pyrin inflammasome | THP‐1, U‐937, HEK293T | 44 | |
| Steroid hormone catabolites are pyrin inflammasome activators | U‐937 | 56 | |
| PKC inhibitors activate the pyrin inflammasome containing FMF‐associated variants | Primary PBMCs, U‐937 | 57 | |
| Periodic fever, Immunodeficiency and thrombocytopenia syndrome (PFIT) | WDR1 mutations cause an IL‐18–dependent autoinflammatory phenotype | Primary neutrophils, monocyte‐derived dendritic cells, PBMCs; patient‐derived lymphoblastoid cell line (LCL), HEK293T | 20 |
| WDR1 mutations effect immunological synapse formation and B‐cell development | Primary PBMCs, neutrophils, T‐cells, patient‐derived LCL | 12 | |
| Pyrin‐associated autoinflammatory disease (PAAND) | PAAND neutrophils display an activated phenotype and increased phagocytosis | Primary neutrophils | 23 |
| Cryopyrin‐associated periodic syndrome (CAPS) | Non‐mutant cells from a somatic mosaic CINCA syndrome patient may contribute to inflammatory phenotype | Patient‐derived iPSCs | 95 |
| NLRP3 mutation disrupts chondrogenesis via SOX9 activation independent of inflammatory effect | Patient‐derived iPSCs | 97 | |
| Phenotypic characterisation of iPSC‐derived cells from a NOMID patient led to the identification of the disease‐causing mutation | Patient‐derived iPSCs | 96 | |
| A novel TRAPS mutation increases NF‐KB activity, cytokine production and mtROS production | Primary PBMCs, HEK293 | 63 | |
| iPSC‐derived macrophages from a NOMID patient used to test compounds able to inhibit IL‐1β | Patient‐derived iPSCs | 104 | |
| Cold exposure promotes NLRP3 aggregation and activation | THP‐1, HeLa | 42 | |
| Functional investigation of different NLRP3 variants | Primary PBMCs, U‐937 | 59 | |
| Proteasome‐associated autoinflammatory syndrome (PRAAS) | Proteasome assembly dysfunction and resulting IFN signature are defining characteristics of CANDLE/PRAAS | Primary fibroblasts, keratinocytes, B‐cells, HeLa | 25 |
| POMP mutation leads to autoinflammation via dysfunction of the inflammasome and activation of the ER stress pathway | Primary fibroblasts, patient‐derived LCL, HEK293T | 13 | |
| POMP8 mutations lead to ubiquitinated protein accumulation, ROS production and type I IFN signature | Patient‐derived iPSCs | 103 | |
| PAC2 mutation leads to decreased proteasome activity | Primary fibroblasts, HeLa | 26 | |
| iPSC‐derived macrophages from a PRAAS patient used to test potential therapeutic compounds | Patient‐derived iPSCs | 105 | |
| Blau syndrome | Blau syndrome macrophages demonstrate an IFN‐ɣ dependent exaggerated inflammatory response to NOD2 ligands | Patient‐derived iPSCs | 101 |
| Evaluation of NF‐KB activation caused by various NOD2 mutations | HEK293 | 65 | |
| Anti‐inflammatory effect of anti‐TNF treatment on patient‐derived macrophages in terms of gene expression profiles is eliminated in iPSC‐derived macrophages | Patient‐derived iPSCs | 100 | |
| Autoinflammation, antibody deficiency, and immune dysregulation (APLAID) | Enhanced activity of Phospholipase Cɣ2 underlies the pathogenesis of PLAID | Primary PBMCs, COS‐7 | 66 |
| Coatomer protein complex, subunit alpha gene (COPA) syndrome | COPA syndrome is caused by defective retrograde ER‐Golgi transport | HEK293T, patient‐derived LCL | 14 |
| Autoinflammation, panniculitis and dermatosis syndrome | Mutations of OTULIN gene lead to aberrant ubiquitination of proteins in the NF‐KB signalling pathway | Primary fibroblasts, primary PBMCs, HEK293 | 29 |
| Early‐onset macrophage activation syndrome (MAS) | NLRC4 mutations lead to inflammatory cytokine production and cell death | THP‐1, HEK293T | 43 |
| Haploinsufficiency of A20 (HA20) | Identification of novel mutations of TNFAIP3 as the causative factor of autoinflammation in suspected cases of HA20 | HEK293 | 64 |
| Primary PBMCs, THP‐1 | 41 | ||
| NEMO deleted exon 5 autoinflammatory syndrome (NDAS) | Detailed characterisation of the molecular pathogenesis of three patients with autoinflammatory disease harbouring a NEMO‐Δex5 mutation | Primary fibroblasts, primary PBMCs, HEK293T, THP‐1, Jurkat, patient‐derived iPSCs | 27 |
| Cleavage‐resistant RIPK1‐induced autoinflammatory syndrome (CRIA syndrome) | Caspase‐8 resistant RIPK1 variants lead to increased necroptosis and autoinflammation | HEK293T, HeLa | 67 |
| Deficiency of adenosine deaminase 2 (DADA2) | ADA2 KO U‐937 cells recapitulate disease phenotype in terms of IL‐6 and TNF secretion, lentiviral correction of ADA2 alleviates inflammation | Primary PBMCs, primary CD34+ cells, U‐937 | 54 |
| Transcriptomic analysis of the pathogenicity of different ADA2 variants | Primary PBMCs, HEK293, U‐937 | 55 | |
| VEXAS syndrome | Functional analysis of UBA1 mutations in VEXAS syndrome | THP‐1, U‐937 | 58 |
| Neonatal onset of pancytopenia, autoinflammation, rash and episodes of haemophagocytic lymphohistiocytosis (NOCARH) syndrome | Mutant CDC42 accumulated in the Golgi apparatus leads to pyrin inflammasome activation | Patient‐derived iPSCs, primary PBMCs, THP‐1, HEK293T, COS‐1 | 92 |
| Multiple self‐healing palmoplantar carcinoma (MSPC), Familial keratosis lichenoides chronica (FKLC) | Gain‐of‐function mutations of NLRP1 lead to skin inflammatory phenotypes | Primary keratinocytes, primary fibroblasts, THP‐1, HEK293T | 34 |
| Juvenile recurrent respiratory papillomatosis | Activation of mutant NLRP1 underlies juvenile recurrent respiratory papillomatosis | Primary keratinocytes, HEK293T | 35 |
| DPP9 deficiency | DPP9 deficiency leads to spontaneous activation of the NLRP1 inflammasome | Primary keratinocytes, primary fibroblasts, HEK293T | 36 |
Human primary cells
Cells isolated from patient samples and cultured in vitro are an essential resource for studying disease. Primary cells provide a more physiologically relevant model than cell lines. Thus, these models are especially useful when dealing with previously undefined or rare diseases for which other models have not been established. Indeed, several autoinflammatory diseases and their causal mutations have been identified and studied using patient‐derived primary cells. However, human primary cells have a limited lifespan in cell culture conditions. Most mammalian somatic cells, such as fibroblasts, are subject to replicative senescence primarily because of telomere shortening, known as the Hayflick limit. 6 Cell types commonly studied in autoinflammatory disease research may have an even shorter lifespan, such as 5–7 days for PBMCs and less than 24 h for neutrophils. 7
As blood is an easily accessible source of primary cells, peripheral blood mononuclear cells (PBMCs) are the cells of choice when investigating immune diseases, including SAIDs. PBMCs consist of a diverse population of immune cells, including lymphocytes, monocytes, dendritic cells and natural killer cells, 8 so they are widely used in autoinflammatory disease research as they represent the general state of the immune system. As many immune cells can contribute to a disease phenotype, studying PBMCs as a whole can be especially valuable when studying undefined conditions. PBMCs also allow for innate immune cells to be examined separately. Monocytes, precursors of macrophages and major players in inflammation, can be isolated/enriched from PBMCs using methods such as Percoll density gradient separation, 9 plastic adhesion, 10 or magnetic separation then differentiated into monocyte‐derived macrophages (MDMs) with M‐CSF or GM‐CSF. 11 PBMCs can further be used to generate B‐lymphoblastoid cell lines (LCLs) via infection with EBV in vitro. These LCLs provide a continuous alternative to finite primary cells and have been utilised in researching autoinflammatory diseases with autoimmune features where adaptive immune cells are also affected. 12 , 13 , 14 It is important to note that different isolation methods can produce differing cell populations in terms of viability, purity and gene expression. 15 , 16 PBMCs, primary monocytes and MDMs are valuable tools in autoinflammatory disease research that provide an easily accessible, physiologically relevant platform for modelling.
Neutrophils, another major player in innate immunity, can also be obtained from patients' blood; however, they are more challenging to isolate, are viable for a shorter period than PBMCs, and are prone to activation and apoptosis under culture conditions. 17 , 18 , 19 These properties necessitate researchers to work quickly with neutrophils, making them a less attractive choice for modelling. Despite this, primary neutrophils have been used to demonstrate increased IL‐18 production in periodic fever, immunodeficiency and thrombocytopenia (PFIT), 20 increased polarisation, cell migration, 21 and ensuing activation 22 in familial Mediterranean fever (FMF); and an immature activated phenotype with increased capacity for phagocytosis, oxidative burst and spontaneous migration in patients with pyrin‐associated autoinflammation with neutrophilic dermatosis (PAAND) and FMF. 23
Fibroblasts derived from skin biopsies of patients have also been used to investigate autoinflammatory diseases. 13 , 24 , 25 , 26 , 27 , 28 , 29 Although relatively invasive to acquire, primary fibroblast cultures are easier to set up and maintain compared to other cell types, such as immune cells. Fibroblasts may be an appropriate initial model to investigate changes in gene expression in SAIDs since many inflammation‐related genes are expressed in this cell type. Fibroblasts can contribute to inflammatory phenotypes, recognise DAMPs and PAMPs, and participate in cytokine signalling. 30 As autoinflammatory syndromes can also result from defects that affect multiple cell types and commonly have skin manifestations, fibroblasts can be a cost‐effective and relevant model in autoinflammatory diseases. However, it is important to note that even if disease‐relevant genes are expressed, fibroblast phenotypes may differ from those of innate immune cells 31 , 32 and they do not provide an opportunity to perform functional assays to evaluate immune‐relevant functions such as inflammasome formation.
Patients' skin biopsies can alternatively be used to culture primary keratinocytes. These models can be especially helpful when investigating diseases with skin manifestations. The NLRP1 inflammasome is highly expressed in keratinocytes, therefore these cells have been used to model NLRP1‐related autoinflammatory diseases. 33 , 34 , 35 , 36 Keratinocytes have also been derived from skin biopsy taken from active lesions to model proteasome‐associated autoinflammatory syndromes (PRAAS). 25 In a similar manner to fibroblasts, primary keratinocyte cultures offer an easily accessible and valuable tool for studying autoinflammatory diseases when disease‐related genes are expressed.
While primary cells are arguably the most relevant disease model, logistical and technical difficulties necessitate the use of alternative models. SAIDs are rare diseases that mainly affect the paediatric population which limits the number of primary cells that can be acquired from patients. In addition, primary cells are viable for a limited time under culture conditions. The resulting scarcity of primary cells makes it challenging to expand study groups, and replicate results or experimental conditions to eliminate patient‐to‐patient variability.
Cell lines
Immortalised cell lines are ubiquitous models for monocytes/macrophages used in studies of innate immune mechanisms, host‐pathogen interactions and roles in disease. Compared to primary cells, cell lines are easier to handle and have a homogeneous genetic background. While primary cells require suitable donors and have a short lifespan in culture, cell lines are widely available, can be cultured indefinitely, cryopreserved, and are more amenable to transfection to generate disease models. However, these cells usually have malignant heritage and genetic changes because of adaptation to culture conditions, making them of uncertain biological relevance. 37 Another prevalent problem is cross‐contamination with other cell lines; 535 cell lines have no known authentic stock, and another 47 have some stocks that are known to be contaminated according to the ICLAC Register of Misidentified Cell Lines. 38 To alleviate this problem, researchers must be aware of misidentified cell lines, follow good cell culture practice, and cell lines should be authenticated via STR or SNP profiling. 39
One widely used cell line in SAIDs research is THP‐1, 40 , 41 , 42 , 43 , 44 , 45 , 46 a monocyte‐like cell line isolated from an acute myeloid leukaemia patient. 47 This line is morphologically and functionally similar to monocytes and can be differentiated in vitro into macrophage‐like cells. 48 However, it is important to keep in mind that THP‐1 cells functionally differ from primary blood cells. One important difference is response to LPS. For example, while LPS‐induced (NF‐KB dependent) gene expression response in THP‐1 cells has been shown to be highly similar to primary cells, 49 these cells express variable levels of CD14 depending on culture conditions 50 ; therefore, they may demonstrate variable sensitivity to LPS. As a result, decreased release of cytokines IL‐8, IL‐6 and IL‐10 upon LPS stimulation has been reported in THP‐1 cells. 51 In addition, a recent analysis of the chromosomal conformation of THP‐1 cells and primary monocytes using high‐throughput chromosome conformation capture revealed significant differences in chromosomal arrangement that were correlated with changes in gene expression. Genes involved in cell proliferation, differentiation and adhesion were predictably upregulated in THP‐1 cells; whereas genes downregulated in THP‐1 cells were found to be linked to immune functions including type I IFN signalling, host defence and chemokine response. 52 Overall, these structural, transcriptomic and functional differences suggest that THP‐1 cells are less sensitive to activating stimuli and care needs to be exercised when interpreting functional data obtained using these cell lines.
U‐937 is another monocytic cell line, isolated from a patient with histiocytic lymphoma, 53 that is also used widely in autoinflammatory disease research. 45 , 54 , 55 , 56 , 57 , 58 , 59 These cells display a similar phenotype to THP‐1 cells, can be cultured in similar conditions, and can also be differentiated into macrophage‐like cells. However, LPS‐induced gene expression response in these cells has been found to be weak in comparison to both THP‐1 cells and PBMCs. 49 Interestingly, one study showed that under similar differentiation conditions, THP‐1 cells are more responsive to the pro‐inflammatory M1 activation pathway, while U‐937 cells are more responsive to the anti‐inflammatory M2 activation. 60 These findings suggest that using both THP‐1 cells and U‐937 to validate results may be advisable when it comes to SAIDs research.
Cell line models of non‐immune cells can also be used to demonstrate their contribution to disease pathogenesis. The SW982 cell line (synovial fibroblast) has been used to demonstrate the anti‐inflammatory effect of miR‐197‐3p 61 in familial Mediterranean fever, where synovial inflammation (arthritis) is a characteristic feature. Furthermore, cell lines receptive to transfection such as HEK293, 62 , 63 , 64 , 65 COS‐7 66 and HeLa 25 , 26 , 42 , 67 have been employed in autoinflammatory disease research as a less technically demanding option to study the molecular effects of novel disease‐causing mutations.
Cell line based models are widely utilised in SAIDs research, because of their convenience and availability. However, functional differences of immortalised cell lines from primary cells necessitate caution when generalising findings. Researchers must keep these differences in mind when selecting cell lines and interpreting results to enhance the reliability of their findings.
Patient‐derived iPS cells
The discovery of induced pluripotent stem cells (iPSCs), somatic cells that have been reprogrammed into an embryonic stem cell‐like state, 68 has opened new avenues of research in disease modelling, regenerative medicine and drug discovery. 69 One and a half decades after their discovery, the introduction of various cell types as a source for iPSCs (e.g. fibroblasts, 68 blood cells, 70 urinary epithelial cells 71 ), the discovery of non‐integrative reprogramming methods (e.g. Sendai virus, 72 episomal, 73 mRNA transfection 74 ), and new developments that significantly increased reprogramming efficiency 75 have expanded the accessibility of the platform. iPSCs derived from patient samples provide an inexhaustible resource of disease‐specific cells, making them an attractive choice for disease modelling. iPSC‐based models have been established for numerous diseases including, but not limited to, neurodegenerative, 76 hereditary cardiac 77 and immune 78 disorders.
In order to be useful in modelling autoinflammatory disorders, iPSCs need to be differentiated into the relevant cell types; indeed many different protocols for the differentiation of iPSCs towards monocytes/macrophages have been developed, with varying techniques and efficiencies. 79 These differentiation protocols generally last 3–4 weeks and rely on sequential incubation with various combinations of cytokines and growth factors that mimic primitive haematopoiesis. iPSC‐derived macrophages (iPSDMs) have been studied more extensively than monocytes, and their morphological and functional phenotypes have been compared with those of primary monocyte‐derived macrophages (MDMs). iPS‐derived macrophages have been reported to be highly similar to donor‐derived cells 80 , 81 , 82 , 83 and THP‐1 cells 84 in terms of morphological features and surface marker expression. They have also been shown to be functionally comparable to primary MDMs in terms of phagocytosis, 80 , 82 cell migration 81 and ability to be polarised. 83 , 85 Secretome analyses of iPS‐derived cells in response to LPS have also been reported to be largely similar to primary cells; however, variations depending on the cytokine measured and differentiation protocols exist. 80 , 81 , 86
Transcriptomic analyses have also shown that iPSDMs and human macrophages are largely similar with vast overlap in expressed genes; however, small differences enable discrimination between iPSDMs and their primary counterparts. 81 , 83 , 86 , 87 Ontogenically, iPSDMs show more resemblance to tissue‐resident macrophages (TRMs) than monocyte‐derived macrophages. 80 , 81 , 88 Significant changes in gene expression were observed in antigen binding, phagosome, lysosome, immune response pathways (downregulated in iPSDMs) and cell cycle, adhesion, ECM and developmental processes (upregulated in iPSDMs). 83 , 86 , 87 Based on cell surface marker expression 80 and transcriptome changes, 83 , 89 unpolarized (M0) macrophages derived from iPSCs also show a phenotype closer to alternatively activated (M2) macrophages; but these differences seem to be minimised upon polarisation.
Altogether, findings from many studies demonstrate that iPSDMs share morphological and transcriptomic features with primary cells and retain many important properties, such as cell marker expression, cytokine release, migration and plasticity with remarkable similarity. The differences that have been reported can be difficult to correlate because of varying differentiation protocols, small sample numbers, and donor‐to‐donor variability between primary cells used as benchmarks. Regardless, iPSDMs have been shown to be valid alternatives to model cellular functions compared to primary cells.
The demonstration of iPS‐derived monocytes and macrophages as adequate alternatives to their primary counterparts has paved the way for the use of these cells to model autoinflammatory diseases. Patient‐derived iPSDMs have been used to recapitulate the autoinflammatory phenotypes of their primary counterparts, such as inflammasome formation and cytokine secretion in FMF, 90 , 91 and the molecular events leading to pyrin inflammasome activation in the presence of CDC42 mutations. 92 Studies utilising this model system showcase not only the potential of iPS‐based models as an alternative to primary cells, but also some of their unique advantages over primary cell‐based models.
Established iPSC lines are clonal as each iPSC colony is derived from a single somatic cell. This property, has proved useful in the study of the autoinflammatory disease NOMID (Neonatal‐Onset Multisystem Inflammatory Disease), where around 30% of patients are found to exhibit somatic mosaicism. 93 , 94 iPSDMs derived from such a patient are automatically separated into mutant and WT populations and the comparison of these two populations helped to identify the effect of the disease‐causing NLRP3 mutation on the macrophage secretome. 95 In another study of NOMID, separate genetic analyses of iPSDMs exhibiting the cytokine release profile characteristic of the mutant cells enabled researchers to pinpoint the disease‐causing mutation when whole exome sequencing of the patient and his parents failed to provide a conclusive diagnosis. 96
Although most autoinflammatory disease research focuses on myeloid cells in pathogenesis, pluripotent iPSC lines enable different cell types to be studied. This property is especially useful when the cell type to be studied is hard to access or difficult to model in vitro. For example, chondroprogenitor cells derived from NOMID patients' iPS cells were used by Yokoyama et al. 97 to study the effects of NLRP3 mutations on chondrogenesis. The arthropathy in this disease is distinct from inflammatory arthritis seen in other autoinflammatory conditions and unresponsive to IL‐1 targeting therapies. Using patient‐derived iPS cells, this study constructed a new in vitro model for NOMID arthropathy, where primary cells are difficult to access and mouse models don't fully recapitulate the disease condition. 98 Another study used patient‐specific iPSC‐derived endothelial cells from a patient with gain‐of‐function mutation of Lyn kinase. 99 This study demonstrated the contribution of mutant Lyn in endothelial to the phenotype of leucocytoclastic vasculitis for the first time.
Furthermore, iPSCs are more amenable to genome editing compared to primary cells, making it possible to generate iPSC‐derived cells with desired or corrected mutations. Introducing or correcting pathogenic mutations in vitro is an indispensable tool in autoinflammatory disease research as patient samples are hard to come by and can be highly heterogeneous. The CRISPR‐Cas9 platform has been used to generate genetically edited iPS‐derived macrophage lines to study Blau 100 , 101 and CANDLE 102 , 103 syndromes as well as the Lyn kinase mutation mentioned above. 99 In all three cases, the disease‐causing mutation was corrected in patient‐derived iPSCs while the same mutation was introduced into iPSCs derived from healthy controls, creating two isogenic disease‐control pairs. This approach enables the use of more relevant controls than traditional options, such as cells from healthy family members.
As a result of their expandability and homogeneity, iPSC‐derived cells are also suitable for drug screening applications. For autoinflammatory disease research, the concept was demonstrated when multiple compounds known to inhibit the NLRP3 pathway were tested on iPSDMs of NOMID patients and healthy controls. 95 Although not aimed at identifying a new therapeutic compound, this study exhibited the value of an unlimited patient‐specific cell supply in future studies. Later, this investigation was expanded to include more than four thousand compounds using high‐throughput approaches. 104 This high‐throughput cytokine‐release‐based approach was also used to identify three inhibitors of the chemokines MCP‐1 and IP‐10 for the treatment of CANDLE syndrome using patient‐specific iPSDMs. 105
Pathogenetic mechanisms leading to autoinflammation are numerous, therefore a wide range of genetic defects can be identified in these patients; including newly identified/ultra‐rare variants causing undefined or unclassified autoinflammatory conditions. Studying the pathogenetic effects of these variants can illuminate the complex mechanisms that regulate innate immunity. However, patient scarcity and disease fatalities may limit available samples and time for research. Therefore, iPS cells derived from these patients' samples can provide a theoretically infinite source of cells that may be used in future applications. iPSC lines have been established and used in functional studies of patients presenting with autoinflammation not part of a previously identified SAID, carrying novel mutations in IKBKG, 27 OAS1 106 and NFKBIA. 107 iPSC lines have alsso been established from patients with STING‐associated vasculopathy with onset in infancy (SAVI), 108 otulipenia 109 and Aicardi‐Goutières syndrome. 110 , 111
Conclusions
Cellular models, mainly primary cells, cell lines and patient‐derived iPSCs, have been key in shaping our understanding of autoinflammatory diseases. The choice of models depends on various factors including the availability of clinical samples, experimental design and resources at hand. While primary cells are considered the ideal model of the in vivo environment, alternative models are required because of their scarcity and limited lifespan. Cell lines are easily accessible and expandable; however, concerns about the validity of cell line‐based models arise because of adaptation to culture conditions, genetic drift and resulting functional differences from primary cells. iPSC‐based models emerge as a newer option that combines the phenotypic similarity to primary cells with the ease of handling of cell lines, while eliminating the need for repeated sample collection from paediatric patients. Disease modelling of autoinflammatory disorders using patient‐derived iPSCs has been demonstrated successfully in multiple studies (Table 1). However, establishing an iPSC‐based model may require specialised training and resource availability, with research still ongoing to optimise these models. Overall, each research team should understand the advantages and limitations of each cellular model in investigating SAIDs to ensure a holistic and complete understanding of innate immune mechanisms in both health and disease.
To date, various methods have been established for the differentiation of iPSCs into haematopoietic‐lineage cells, including protocols that have been scaled to produce large numbers of cells. While it is still unclear whether the resulting cells are phenotypically and functionally consistent, further research into the differentiation process and characterisation of resulting cells will enable the widespread adoption of these models. Ongoing research in the field iPSC reprogramming and differentiation is on track to improve our understanding of barriers to these processes, in addition to providing ways to overcome them. These developments enable a future where numerous patient‐derived iPSC lines can be established more easily and efficiently for these rare diseases, biobanks can be built, and large‐scale experiments can be designed independent of the patient population. Genetically edited patient‐specific iPSCs may also provide an opportunity for autologous haematopoietic stem cell transplantation as a curative approach for some of the more severe phenotypes associated with autoinflammatory diseases.
Author contributions
Başak Şen: Conceptualization; writing – original draft. Banu Balcı‐Peynircioğlu: Conceptualization; writing – review and editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgments
The authors thank YZ Akkaya‐Ulum and TH Akbaba for their contributions to the conception of the manuscript, and S Çapan for reading the manuscript and offering comments and suggestions. This work was supported by Technical and Scientific Research Council of Turkey (TUBITAK), Grant number: 122S379.
References
- 1. Krainer J, Siebenhandl S, Weinhausel A. Systemic autoinflammatory diseases. J Autoimmun 2020; 109: 102421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Masumoto J, Zhou W, Morikawa S et al. Molecular biology of autoinflammatory diseases. Inflamm Regen 2021; 41: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol 2020; 146: 925–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 2020; 6: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McDermott MF, Aksentijevich I, Galon J et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97: 133–144. [DOI] [PubMed] [Google Scholar]
- 6. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621. [DOI] [PubMed] [Google Scholar]
- 7. Pillay J, den Braber I, Vrisekoop N et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010; 116: 625–627. [DOI] [PubMed] [Google Scholar]
- 8. Autissier P, Soulas C, Burdo TH, Williams KC. Evaluation of a 12‐color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A 2010; 77: 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Almeida MC, Silva AC, Barral A, Barral NM. A simple method for human peripheral blood monocyte isolation. Mem Inst Oswaldo Cruz 2000; 95: 221–223. [DOI] [PubMed] [Google Scholar]
- 10. Davies JQ, Gordon S. Isolation and culture of human macrophages. Methods Mol Biol 2005; 290: 105–116. [DOI] [PubMed] [Google Scholar]
- 11. Kelly A, Grabiec AM, Travis MA. Culture of human monocyte‐derived macrophages. Methods Mol Biol 2018; 1784: 1–11. [DOI] [PubMed] [Google Scholar]
- 12. Pfajfer L, Mair NK, Jimenez‐Heredia R et al. Mutations affecting the Actin regulator WD repeat‐containing protein 1 lead to aberrant lymphoid immunity. J Allergy Clin Immunol 2018; 142: 1589–1604. [DOI] [PubMed] [Google Scholar]
- 13. Poli MC, Ebstein F, Nicholas SK et al. Heterozygous truncating variants in POMP escape nonsense‐mediated decay and cause a unique immune dysregulatory syndrome. Am J Hum Genet 2018; 102: 1126–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watkin LB, Jessen B, Wiszniewski W et al. COPA mutations impair ER‐Golgi transport and cause hereditary autoimmune‐mediated lung disease and arthritis. Nat Genet 2015; 47: 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chometon TQ, Siqueira MDS, Sant Anna JC et al. A protocol for rapid monocyte isolation and generation of singular human monocyte‐derived dendritic cells. PloS One 2020; 15: e0231132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nielsen MC, Andersen MN, Moller HJ. Monocyte isolation techniques significantly impact the phenotype of both isolated monocytes and derived macrophages in vitro . Immunology 2020; 159: 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Avci E, Akkaya‐Ulum YZ, Yoyen‐Ermis D, Esendagli G, Balci‐Peynircioglu B. A method for high‐purity isolation of neutrophil granulocytes for functional cell migration assays. Turk J Biochem 2019; 44: 810–821. [Google Scholar]
- 18. Blanter M, Gouwy M, Struyf S. Studying neutrophil function in vitro: cell models and environmental factors. J Inflamm Res 2021; 14: 141–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheel‐Toellner D, Wang K, Craddock R et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood 2004; 104: 2557–2564. [DOI] [PubMed] [Google Scholar]
- 20. Standing AS, Malinova D, Hong Y et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin‐regulatory gene WDR1. J Exp Med 2017; 214: 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balcı‐Peynircioğlu B, Akkaya‐Ulum YZ, Avcı E et al. Potential role of pyrin, the protein mutated in familial Mediterrenean fever, in inflammatory cell migration. Clin Exp Rheumatol 2018; 36(6 Suppl 115): 116–124. [PubMed] [Google Scholar]
- 22. Martirosyan A, Poghosyan D, Ghonyan S, Mkrtchyan N, Amaryan G, Manukyan G. Transmigration of neutrophils from patients with familial Mediterranean fever causes increased cell activation. Front Immunol 2021; 12: 672728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Malengier‐Devlies B, Metzemaekers M, Gouwy M et al. Phenotypical and functional characterization of neutrophils in two pyrin‐associated auto‐inflammatory diseases. J Clin Immunol 2021; 41: 1072–1084. [DOI] [PubMed] [Google Scholar]
- 24. Boisson B, Laplantine E, Dobbs K et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med 2015; 212: 939–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brehm A, Liu Y, Sheikh A et al. Additive loss‐of‐function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 2015; 125: 4196–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Jesus AA, Brehm A, VanTries R et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol 2019; 143: 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee Y, Wessel AW, Xu J et al. Genetically programmed alternative splicing of NEMO mediates an autoinflammatory disease phenotype. J Clin Invest 2022; 132: e128808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y, Jesus AA, Marrero B et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 371: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Q, Yu X, Demirkaya E et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early‐onset autoinflammatory disease. Proc Natl Acad Sci USA 2016; 113: 10127–10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davidson S, Coles M, Thomas T et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol 2021; 21: 704–717. [DOI] [PubMed] [Google Scholar]
- 31. Boisson B, Laplantine E, Prando C et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL‐1 and LUBAC deficiency. Nat Immunol 2012; 13: 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Damgaard RB, Walker JA, Marco‐Casanova P et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 2016; 166: 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhong FL, Robinson K, Teo DET et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem 2018; 293: 18864–18878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhong FL, Mamai O, Sborgi L et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via Inflammasome activation. Cell 2016; 167: 187–202. [DOI] [PubMed] [Google Scholar]
- 35. Drutman SB, Haerynck F, Zhong FL et al. Homozygous NLRP1 gain‐of‐function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci USA 2019; 116: 19055–19063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harapas CR, Robinson KS, Lay K et al. DPP9 deficiency: an inflammasomopathy that can be rescued by lowering NLRP1/IL‐1 signaling. Sci Immunol 2022; 7: eabi4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bosshart H, Heinzelmann M. THP‐1 cells as a model for human monocytes. Ann Transl Med 2016; 4: 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Capes‐Davis A, Theodosopoulos G, Atkin I et al. Check your cultures! A list of cross‐contaminated or misidentified cell lines. Int J Cancer 2010; 127: 1–8. [DOI] [PubMed] [Google Scholar]
- 39. Yu M, Selvaraj SK, Liang‐Chu MM et al. A resource for cell line authentication, annotation and quality control. Nature 2015; 520: 307–311. [DOI] [PubMed] [Google Scholar]
- 40. Akbaba TH, Akkaya‐Ulum YZ, Demir S, Ozen S, Balci‐Peynircioglu B. The pyrin inflammasome aggravates inflammatory cell migration in patients with familial Mediterranean fever. Pediatr Res 2022; 91: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 41. Jiang W, Deng M, Gan C, Wang L, Mao H, Li Q. A novel missense mutation in TNFAIP3 causes haploinsufficiency of A20. Cell Immunol 2022; 371: 104453. [DOI] [PubMed] [Google Scholar]
- 42. Karasawa T, Komada T, Yamada N et al. Cryo‐sensitive aggregation triggers NLRP3 inflammasome assembly in cryopyrin‐associated periodic syndrome. Elife 2022; 11: e75166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moghaddas F, Zeng P, Zhang Y et al. Autoinflammatory mutation in NLRC4 reveals a leucine‐rich repeat (LRR)‐LRR oligomerization interface. J Allergy Clin Immunol 2018; 142: 1956–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu H, Yang J, Gao W et al. Innate immune sensing of bacterial modifications of rho GTPases by the pyrin inflammasome. Nature 2014; 513: 237–241. [DOI] [PubMed] [Google Scholar]
- 45. Spel L, Zaffalon L, Hou C, Nganko N, Chapuis C, Martinon F. CDC42 regulates PYRIN inflammasome assembly. Cell Rep 2022; 41: 111636. [DOI] [PubMed] [Google Scholar]
- 46. Mangan MSJ, Gorki F, Krause K et al. Transcriptional licensing is required for pyrin inflammasome activation in human macrophages and bypassed by mutations causing familial Mediterranean fever. PLoS Biol 2022; 20: e3001351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP‐1). Int J Cancer 1980; 26: 171–176. [DOI] [PubMed] [Google Scholar]
- 48. Chanput W, Mes JJ, Wichers HJ. THP‐1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol 2014; 23: 37–45. [DOI] [PubMed] [Google Scholar]
- 49. Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND. Transcriptional profiling of the LPS induced NF‐kappaB response in macrophages. BMC Immunol 2007; 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aldo PB, Craveiro V, Guller S, Mor G. Effect of culture conditions on the phenotype of THP‐1 monocyte cell line. Am J Reprod Immunol 2013; 70: 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP‐1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm 2013; 2013: 697972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu Y, Li H, Czajkowsky DM, Shao Z. Monocytic THP‐1 cells diverge significantly from their primary counterparts: a comparative examination of the chromosomal conformations and transcriptomes. Hereditas 2021; 158: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U‐937). Int J Cancer 1976; 17: 565–577. [DOI] [PubMed] [Google Scholar]
- 54. Zoccolillo M, Brigida I, Barzaghi F et al. Lentiviral correction of enzymatic activity restrains macrophage inflammation in adenosine deaminase 2 deficiency. Blood Adv 2021; 5: 3174–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nihira H, Izawa K, Ito M et al. Detailed analysis of Japanese patients with adenosine deaminase 2 deficiency reveals characteristic elevation of type II interferon signature and STAT1 hyperactivation. J Allergy Clin Immunol 2021; 148: 550–562. [DOI] [PubMed] [Google Scholar]
- 56. Magnotti F, Chirita D, Dalmon S et al. Steroid hormone catabolites activate the pyrin inflammasome through a non‐canonical mechanism. Cell Rep 2022; 41: 111472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Magnotti F, Lefeuvre L, Benezech S et al. Pyrin dephosphorylation is sufficient to trigger inflammasome activation in familial Mediterranean fever patients. EMBO Mol Med 2019; 11: e10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu Z, Gao S, Gao Q et al. Early activation of inflammatory pathways in UBA1‐mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med 2023; 4: 101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cosson C, Riou R, Patoli D et al. Functional diversity of NLRP3 gain‐of‐function mutants associated with CAPS autoinflammation. Preprint. bioRxiv 2023: 2023-09. doi: 10.1101/2023.09.22.558949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nascimento CR, Rodrigues Fernandes NA, Gonzalez Maldonado LA, Rossa JC. Comparison of monocytic cell lines U937 and THP‐1 as macrophage models for in vitro studies. Biochem Biophys Rep 2022; 32: 101383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Akkaya‐Ulum YZ, Akbaba TH, Tavukcuoglu Z et al. Familial Mediterranean fever‐related miR‐197‐3p targets IL1R1 gene and modulates inflammation in monocytes and synovial fibroblasts. Sci Rep 2021; 11: 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masters SL, Lagou V, Jeru I et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med 2016; 8: 332ra345. [DOI] [PubMed] [Google Scholar]
- 63. Tsuji S, Matsuzaki H, Iseki M et al. Functional analysis of a novel G87V TNFRSF1A mutation in patients with TNF receptor‐associated periodic syndrome. Clin Exp Immunol 2019; 198: 416–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kadowaki S, Hashimoto K, Nishimura T et al. Functional analysis of novel A20 variants in patients with atypical inflammatory diseases. Arthritis Res Ther 2021; 23: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Matsuda T, Kambe N, Ueki Y et al. Clinical characteristics and treatment of 50 cases of Blau syndrome in Japan confirmed by genetic analysis of the NOD2 mutation. Ann Rheum Dis 2020; 79: 1492–1499. [DOI] [PubMed] [Google Scholar]
- 66. Zhou Q, Lee GS, Brady J et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet 2012; 91: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tapiz IRAJ, Cochino AV, Martins AL et al. Characterization of novel pathogenic variants leading to Caspase‐8 cleavage‐resistant RIPK1‐induced autoinflammatory syndrome. J Clin Immunol 2022; 42: 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872. [DOI] [PubMed] [Google Scholar]
- 69. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 2017; 16: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brown ME, Rondon E, Rajesh D et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PloS One 2010; 5: e11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moad M, Pal D, Hepburn AC et al. A novel model of urinary tract differentiation, tissue regeneration, and disease: reprogramming human prostate and bladder cells into induced pluripotent stem cells. Eur Urol 2013; 64: 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene‐free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009; 85: 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008; 322: 949–953. [DOI] [PubMed] [Google Scholar]
- 74. Warren L, Manos PD, Ahfeldt T et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010; 7: 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang Z, Zheng J, Pan R, Chen Y. Current status and future prospects of patient‐derived induced pluripotent stem cells. Hum Cell 2021; 34: 1601–1616. [DOI] [PubMed] [Google Scholar]
- 76. Chang CY, Ting HC, Liu CA et al. Induced pluripotent stem cells: a powerful neurodegenerative disease modeling tool for mechanism study and drug discovery. Cell Transplant 2018; 27: 1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Moretti A, Laugwitz KL, Dorn T, Sinnecker D, Mummery C. Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med 2013; 3: a014027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Saito MK. Disease modeling of immunological disorders using induced pluripotent stem cells. Immunol Med 2018; 41: 68–74. [DOI] [PubMed] [Google Scholar]
- 79. Lyadova I, Gerasimova T, Nenasheva T. Macrophages derived from human induced pluripotent stem cells: the diversity of protocols, future prospects, and outstanding questions. Front Cell Dev Biol 2021; 9: 640703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cao X, Yakala GK, van den Hil FE, Cochrane A, Mummery CL, Orlova VV. Differentiation and functional comparison of monocytes and macrophages from hiPSCs with peripheral blood derivatives. Stem Cell Reports 2019; 12: 1282–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cui D, Franz A, Fillon SA et al. High‐yield human induced pluripotent stem cell‐derived monocytes and macrophages are functionally comparable with primary cells. Front Cell Dev Biol 2021; 9: 656867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hiramatsu N, Yamamoto N, Isogai S et al. An analysis of monocytes and dendritic cells differentiated from human peripheral blood monocyte‐derived induced pluripotent stem cells. Med Mol Morphol 2020; 53: 63–72. [DOI] [PubMed] [Google Scholar]
- 83. Zhang H, Xue C, Shah R et al. Functional analysis and transcriptomic profiling of iPSC‐derived macrophages and their application in modeling Mendelian disease. Circ Res 2015; 117: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hong D, Ding J, Li O et al. Human‐induced pluripotent stem cell‐derived macrophages and their immunological function in response to tuberculosis infection. Stem Cell Res Ther 2018; 9: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yanagimachi MD, Niwa A, Tanaka T et al. Robust and highly‐efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum‐ and feeder cell‐free conditions. PloS One 2013; 8: e59243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Monkley S, Krishnaswamy JK, Goransson M et al. Optimised generation of iPSC‐derived macrophages and dendritic cells that are functionally and transcriptionally similar to their primary counterparts. PloS One 2020; 15: e0243807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Alasoo K, Martinez FO, Hale C et al. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci Rep 2015; 5: 12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Buchrieser J, James W, Moore MD. Human induced pluripotent stem cell‐derived macrophages share ontogeny with MYB‐independent tissue‐resident macrophages. Stem Cell Reports 2017; 8: 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gutbier S, Wanke F, Dahm N et al. Large‐scale production of human iPSC‐derived macrophages for drug screening. Int J Mol Sci 2020; 21: e4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Takata K, Kozaki T, Lee CZW et al. Induced‐pluripotent‐stem‐cell‐derived primitive macrophages provide a platform for modeling tissue‐resident macrophage differentiation and function. Immunity 2017; 47: 183–198. [DOI] [PubMed] [Google Scholar]
- 91. Shiba T, Tanaka T, Ida H et al. Functional evaluation of the pathological significance of MEFV variants using induced pluripotent stem cell‐derived macrophages. J Allergy Clin Immunol 2019; 144: 1438–1441. [DOI] [PubMed] [Google Scholar]
- 92. Nishitani‐Isa M, Mukai K, Honda Y et al. Trapping of CDC42 C‐terminal variants in the Golgi drives pyrin inflammasome hyperactivation. J Exp Med 2022; 219: e20211889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lasiglie D, Mensa‐Vilaro A, Ferrera D et al. Cryopyrin‐associated periodic syndromes in Italian patients: evaluation of the rate of somatic NLRP3 mosaicism and phenotypic characterization. J Rheumatol 2017; 44: 1667–1673. [DOI] [PubMed] [Google Scholar]
- 94. Tanaka N, Izawa K, Saito MK et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an international multicenter collaborative study. Arthritis Rheum 2011; 63: 3625–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tanaka T, Takahashi K, Yamane M et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood 2012; 120: 1299–1308. [DOI] [PubMed] [Google Scholar]
- 96. Kawasaki Y, Oda H, Ito J et al. Identification of a high‐frequency somatic NLRC4 mutation as a cause of autoinflammation by pluripotent cell‐based phenotype dissection. Arthritis Rheumatol 2017; 69: 447–459. [DOI] [PubMed] [Google Scholar]
- 97. Yokoyama K, Ikeya M, Umeda K et al. Enhanced chondrogenesis of induced pluripotent stem cells from patients with neonatal‐onset multisystem inflammatory disease occurs via the caspase 1‐independent cAMP/protein kinase a/CREB pathway. Arthritis Rheumatol 2015; 67: 302–314. [DOI] [PubMed] [Google Scholar]
- 98. Bonar SL, Brydges SD, Mueller JL et al. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PloS One 2012; 7: e35979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. de Jesus AA, Chen G, Yang D et al. Constitutively active Lyn kinase causes a cutaneous small vessel vasculitis and liver fibrosis syndrome. Nat Commun 2023; 14: 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kitagawa Y, Kawasaki Y, Yamasaki Y, Kambe N, Takei S, Saito MK. Anti‐TNF treatment corrects IFN‐gamma‐dependent proinflammatory signatures in Blau syndrome patient‐derived macrophages. J Allergy Clin Immunol 2022; 149: 176–188. [DOI] [PubMed] [Google Scholar]
- 101. Takada S, Kambe N, Kawasaki Y et al. Pluripotent stem cell models of Blau syndrome reveal an IFN‐gamma‐dependent inflammatory response in macrophages. J Allergy Clin Immunol 2018; 141: 339–349. [DOI] [PubMed] [Google Scholar]
- 102. Yu Q, Mehta A, Zou J et al. Human induced pluripotent stem cells generated from chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome patients with a homozygous mutation in the PSMB8 gene (NIHTVBi016‐a, NIHTVBi017‐a, NIHTVBi018‐a). Stem Cell Res 2022; 62: 102820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Honda‐Ozaki F, Terashima M, Niwa A et al. Pluripotent stem cell model of Nakajo‐Nishimura syndrome untangles proinflammatory pathways mediated by oxidative stress. Stem Cell Reports 2018; 10: 1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seki R, Ohta A, Niwa A et al. Induced pluripotent stem cell‐derived monocytic cell lines from a NOMID patient serve as a screening platform for modulating NLRP3 inflammasome activity. PloS One 2020; 15: e0237030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kase N, Terashima M, Ohta A et al. Pluripotent stem cell‐based screening identifies CUDC‐907 as an effective compound for restoring the in vitro phenotype of Nakajo‐Nishimura syndrome. Stem Cells Transl Med 2021; 10: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Magg T, Okano T, Koenig LM et al. Heterozygous OAS1 gain‐of‐function variants cause an autoinflammatory immunodeficiency. Sci Immunol 2021; 6: eabf9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tan EE, Hopkins RA, Lim CK et al. Dominant‐negative NFKBIA mutation promotes IL‐1β production causing hepatic disease with severe immunodeficiency. J Clin Invest 2020; 130: 5817–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mehta A, Yu Q, Liu Y et al. Human induced pluripotent stem cells generated from STING‐associated vasculopathy with onset in infancy (SAVI) patients with a heterozygous mutation in the STING gene. Stem Cell Res 2022; 65: 102974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chen D, Li Z, Liu Y et al. Human induced pluripotent stem cells generated from a patient with a homozygous L272P mutation in the OTULIN gene (NIHTVBi014‐a). Stem Cell Res 2020; 47: 101921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hanchen V, Kretschmer S, Wolf C et al. Generation of induced pluripotent stem cell lines from two patients with Aicardi‐Goutieres syndrome type 1 due to biallelic TREX1 mutations. Stem Cell Res 2022; 64: 102895. [DOI] [PubMed] [Google Scholar]
- 111. Hanchen V, Kretschmer S, Wolf C et al. Generation of induced pluripotent stem cell lines from three patients with Aicardi‐Goutieres syndrome type 5 due to biallelic SAMDH1 mutations. Stem Cell Res 2022; 64: 102912. [DOI] [PubMed] [Google Scholar]