

Abstract
There has recently been an explosion of studies implicating the human microbiome in playing a critical role in many disease and wellness states. The etiology of abnormal semen analysis (SA) parameters is not identified in 30% of cases; investigations involving the semen microbiome may bridge this gap. Here, we explore the relationship between the semen microbiome and alterations of sperm parameters. We recruited men presenting for fertility evaluation or vasectomy consultation with proven biological paternity. SA and next generation sequencing was performed. Differential abundance testing using Analysis of composition of Microbiota with Bias Correction (ANCOM-BC) was performed along with canonical correlational analysis for microbial community profiling. Men with abnormal (N = 27) sperm motility showed a higher abundance of Lactobacillus iners compared to those with normal (N = 46) sperm motility (mean proportion 9.4% versus 2.6%, p = 0.046). This relationship persisted on canonical correlational analysis (r = 0.392, p = 0.011). Men with abnormal sperm concentration (N = 20) showed a higher abundance of Pseudomonas stutzeri (2.1% versus 1.0%, p = 0.024) and Pseudomonas fluorescens (0.9% versus 0.7%, p = 0.010), but a lower abundance of Pseudomonas putida (0.5% versus 0.8%, p = 0.020), compared to those with normal sperm concentration (N = 53). Major limitations are related to study design (cross-sectional, observational). Our results suggest that a small group of microorganisms may play a critical role in observed perturbations of SA parameters. Some of these microbes, most notably Lactobacillus iners, have been described extensively within other, fertility-related, contexts, whereas for others, this is the first report where they have potentially been implicated. Advances in our understanding of the semen microbiome may contribute to potentially new therapeutic avenues for correcting impairments in sperm parameters and improving male fertility.
Subject terms: Microbiology, Urology, Gonads
Introduction
Male factor infertility is common and, despite advances in semen and genetic testing, still not fully understood1. The etiology of abnormal semen analysis (SA) parameters is not identified in 30% of cases2. Recent studies have implicated a role for the microbiome in health and human disease3,4. These investigations have recently expanded to an exploration of the semen microbiome and its potential role in male factor infertility.
Advances in next-generation sequencing (NGS) technologies have augmented our understanding of human health and disease by allowing for accurate, efficient, and relatively inexpensive characterization of the human microbiome. There have been a small handful of studies that have explored the semen microbiome, and an even smaller handful to explore the semen microbiome within the context of fertility. These studies have largely been limited by their superficial, retrospective nature and small sample sizes3. More recently, higher quality studies though still with relatively limited sample sizes have emerged. Lundy et al.5 explored the taxonomic and functional profile of the semen microbiome in 32 well-phenotyped men. Their findings underscore differences in alpha and beta diversity among infertile men compared with healthy controls, in addition to a direct association between genus Pseudomonas and total motile sperm count. Another study with a similar sample size found that men with non-obstructive azoospermia compared to healthy controls, demonstrate changes in semen microbiome beta diversity and associated taxonomic differences6.
Here, we expand on our understanding of the semen microbiome and its role in male infertility and alterations in SA parameters. Although results from a cross-sectional study do not allow us to make inferences related to causality, we hypothesize that a small, but critical group of microorganisms may play an important role in contributing to subfertility in men. Our findings will serve as the foundation for future, mechanistic, and potentially longitudinal studies. This is the largest study to explore this line of inquiry.
Methods
Study population and biospecimen collection
Men aged ≥ 18 years who presented either for an initial fertility evaluation (both primary and secondary infertility were included) or men with proven biological paternity prior to their vasectomy consultation were recruited for this study. The recruitment period was from 08/2021 through 06/2022, with an initial aim to recruit approximately 100 participants. All participants provided written informed consent and denied any acute illness. Information regarding participant clinical information was also collected, including age, body mass index, foreskin status, tobacco usage, and alcohol use. Social alcohol use was defined by ≤ 14 drinks per week; heavy alcohol use was > 14 drinks per week. Due to a lack of detailed information related to paternity timeline, some individuals who presented for vasectomy consultation may have fathered children many years ago so their current fertility cannot be guaranteed. Given this, we ultimately stratified our cohorts of interest by SA parameters, reflecting more objective, contemporary data. Semen samples were collected following 2–7 days of abstinence. All specimens were obtained prior to any surgical (including vasectomy) or pharmacologic intervention for fertility. Specimens for microbiome analysis were stored at − 80 °C until they were processed.
Semen analysis
Semen was evaluated according to World Health Organization 5th Edition criteria using a calibrated SQA-Vision Automated Semen Analyzer (Medical Electronic Systems, Encino, CA). Samples demonstrating oligozoospermia or azoospermia were independently evaluated using high-powered microscopy. Semen volume, pH, concentration, motility, and strict morphology (Kruger) are reported. Total motile sperm count was calculated by multiplying total concentration, total motility, and volume.
Microbiome community profiling
DNA extraction, PCR amplification, library preparation, and sequencing of 16S rRNA regions V1-V2 on Illumina Miseq platform was conducted as previously performed by MicroGenDX (Lubbock, TX)7,8. Briefly, DNA extraction was performed with a Qiagen TissueLyser and Zymo MagBead 96 DNA/RNA kit (Zymo Research, Tustin, CA, USA). Samples were mechanically lysed using Zirconium oxide beads (0.5 mm) and the Qiagen TissueLyser. The lysate was extracted for total DNA following the Zymo MagBead 96 DNA/RNA kit’s protocol on the KingFisher FLEX (ThermoFisher, Grand Island, NY, USA). The samples were amplified for sequencing using a 25uL reaction containing the 28F primer (GAGTTTGATCNTGGCTCAG) with the Illumina adapter and a unique barcode and 388R primer (GCTGCCTCCCGTAGGAGT) with the Illumina adapter and unique barcode along with Quanta AccuStart II Tough Mix (Quanta bio, Beverly, MA, USA). PCR reactions were conducted on ABI Veriti thermocyclers (Applied Biosystems, Carlsbad, CA, USA) with a thermal profile consisting of 5-min denaturation step at 95 °C, 35 cycles of 94 °C for 30 s, 52 °C for 40 s, and 72 °C for 60 s, and a final extension step of 72 °C for 10 min. PCR products were combined based on qualitative band strength to form the pooled amplicon libraries and size selection was performed using Agencourt AMPure XP beads (Beckman Coulter, Indianapolis, Indiana, USA) and Qiagen Minelute Kits (Qiagen). Pooled libraries were quantified using a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Paired read sequencing (2 × 250) was conducted on an Illumina MiSeq (Illumina, San Diego, CA, USA) and using a Reagent Kit v2 Nano, targeting an average ~ 2 k classified reads per sample. Positive controls, negative extraction controls, and no template PCR controls were also included for sequencing.
Bioinformatic curation, quality control filtering, and analysis of data was subsequently performed by RTL Genomics, a division of MicroGenDX. Denoising of sequence reads, chimera detection, and paired read assembly were conducted using Usearch79, UCHIME10, and PEAR11, respectively. Quality filtered and assembled reads were clustered into operational taxonomic units (OTUs) at 97% sequence similarity threshold using the UPARSE algorithm12. OTU assignment then performed using the MicroGenDX reference database. Where possible, taxonomic assignments are reported to the species level as used previously7. Multiple sequence alignment and phylogenetic tree construction for downstream analysis performed using MUSCLE12, and FastTree13. Prior to statistical analysis, additional quality control filtering performed in R to remove OTUs which failed to map to bacterial 16S database and OTUs suspected of being contaminants based on inspection of sequencing controls (i.e., Ralstonia, Pelomonas, and Marinobacter). Total abundance filtering was also used to remove any OTUs found at less than 0.001% of total read counts, which may reflect low level contamination or sequencing artifacts14. This methodology has previously been described8.
Statistical analysis
For statistical analysis of baseline participant characteristics, categorical variables are reported as counts and percentages, and continuous variables as means and standard deviations. Chi-squared and Fisher exact tests were used to compare categorical variables, and Student’s t-tests were used for continuous variables. Missing data is reported, where applicable; no imputation techniques or sensitivity analyses were performed.
Coverage of bacterial communities was estimated using Good’s coverage formula15. Bacterial alpha diversity was summarized using three indices (OTU richness, Hill1 diversity, and phylogenetic hill1 diversity)16. Beta diversity was summarized by Weighted UniFrac17 and qualitatively clustered using Principal Coordinate Analysis (PCoA). Alpha and beta diversity assist in interpreting higher level changes in microbial community structure. Alpha diversity measures the diversity in a single sample “within-sample,” whereas beta diversity measures the similarity or dissimilarity, or similarity, of two communities “between-sample”18. Samples were characterized as normal or abnormal according to sperm analysis results, with ANOVA or PERMANOVA used to compare each grouping for differences in alpha or beta diversity, respectively. Analysis of composition of Microbiota with Bias Correction (ANCOM-BC) was used to screen bacterial taxa for nonrandom distributions according to sperm analysis findings and reduce false positive rate19. Only taxa prevalent in more than 20% of samples were considered (zero_cut = 0.80) and less stringent parameters were used to maximize detection of possible discriminatory taxa (no p-value correction)8. Canonical correlation analysis (CCA) was used to measure the associations between bacterial relative abundance of species-level clusters more than 10% prevalent and participant metadata (age, SA volume, SA motility, and SA concentration).
Ethics
This study was approved by the institutional review board of the University of California, Los Angeles, IRB#21–000,714. All methods were performed in accordance with the relevant guidelines and regulations.
Results
A total of 73 individuals were included in our study. Participants were then stratified into a priori defined groups: Group 1) normal sperm concentration and motility on SA versus at least one abnormality in sperm concentration or motility on SA; for brevity, we will refer to this comparison as “sperm result” in the figures (Table 1); Group 2) normal versus abnormal sperm motility (Supp Table 1); and Group 3) normal versus abnormal sperm concentration (Supp Table 2). To further contextualize this data, Supp Table 3 includes baseline participant characteristics stratified by recruitment groups (infertility versus vasectomy). Study participant characteristics are reported in the above-mentioned tables in detail. The average participant age was 37.94 with a standard deviation of 5.62 and the average BMI was 26.73 with a standard deviation of 6.15. Seventy-eight percent of participants were circumcised. There were no significant differences in age, BMI, circumcision status, smoking, or alcohol intake history among any of the three comparison groups. All participants recruited from the infertility evaluation cohort are engaging in regular sexual activity, whereas the sexual activity of those recruited from the vasectomy consultation cohort was not recorded.
Table 1.
Baseline Participant Characteristics.
| Cohort characteristics | Normal semen analysis group (n = 42) | Abnormal semen analysis group (n = 31) | p |
|---|---|---|---|
| Age (yr) | 37.25 ± 7.5 | 37.64 ± 6.7 | 0.704 |
| Body mass index (kg/m2) | 26.84 ± 7.1 | 26.55 ± 4.5 | 0.862 |
| Missing Data | 8 | 9 | |
| Circumcised | 32 (80%) | 21 (75%) | 0.625 |
| Missing Data | 2 | 3 | |
| Infertility group | 17 (40%) | 28 (90%) | < 0.001 |
| Smoking status | |||
| Current smoker | 0 (0%) | 1 (5%) | 0.373 |
| Ex-smoker | 9 (24%) | 6 (27%) | |
| Never smoker | 28 (76%) | 15 (68%) | |
| Missing Data | 5 | 9 | |
| Alcohol use | |||
| None | 7 (18%) | 4 (20%) | 0.999 |
| Social | 28 (74%) | 16 (80%) | |
| Heavy | 3 (8%) | 0 (0%) | |
| Missing Data | 4 | 11 | |
| Semen analysis | |||
| Semen volume (mL) | 2.84 ± 1.4 | 2.14 ± 1.1 | 0.024 |
| Semen pH | 8.17 ± 0.2 | 8.12 ± 0.2 | 0.355 |
| Sperm concentration (million/mL) | 80.66 ± 49.0 | 29.79 ± 49.0 | < 0.001 |
| % Motile sperm | 59.21 ± 11.9 | 19.35 ± 17.6 | < 0.001 |
| Total sperm count (million) | 201.44 ± 122.7 | 62.74 ± 94.8 | < 0.001 |
| Total motile sperm count (million) | 116.67 ± 69.0 | 18.25 ± 30.2 | < 0.001 |
| % Normal morphology (Kruger strict criteria) | 26.17 ± 18.4 | 2.60 ± 4.7 | < 0.001 |
| Normozoospermia (> 15 million/mL) | 42 (100%) | 11 (35%) | < 0.001 |
| Oligospermia (< 15 million/mL) | 0 (0%) | 12 (39%) | |
| Azoospermia (0 million/mL) | 0 (0%) | 8 (26%) |
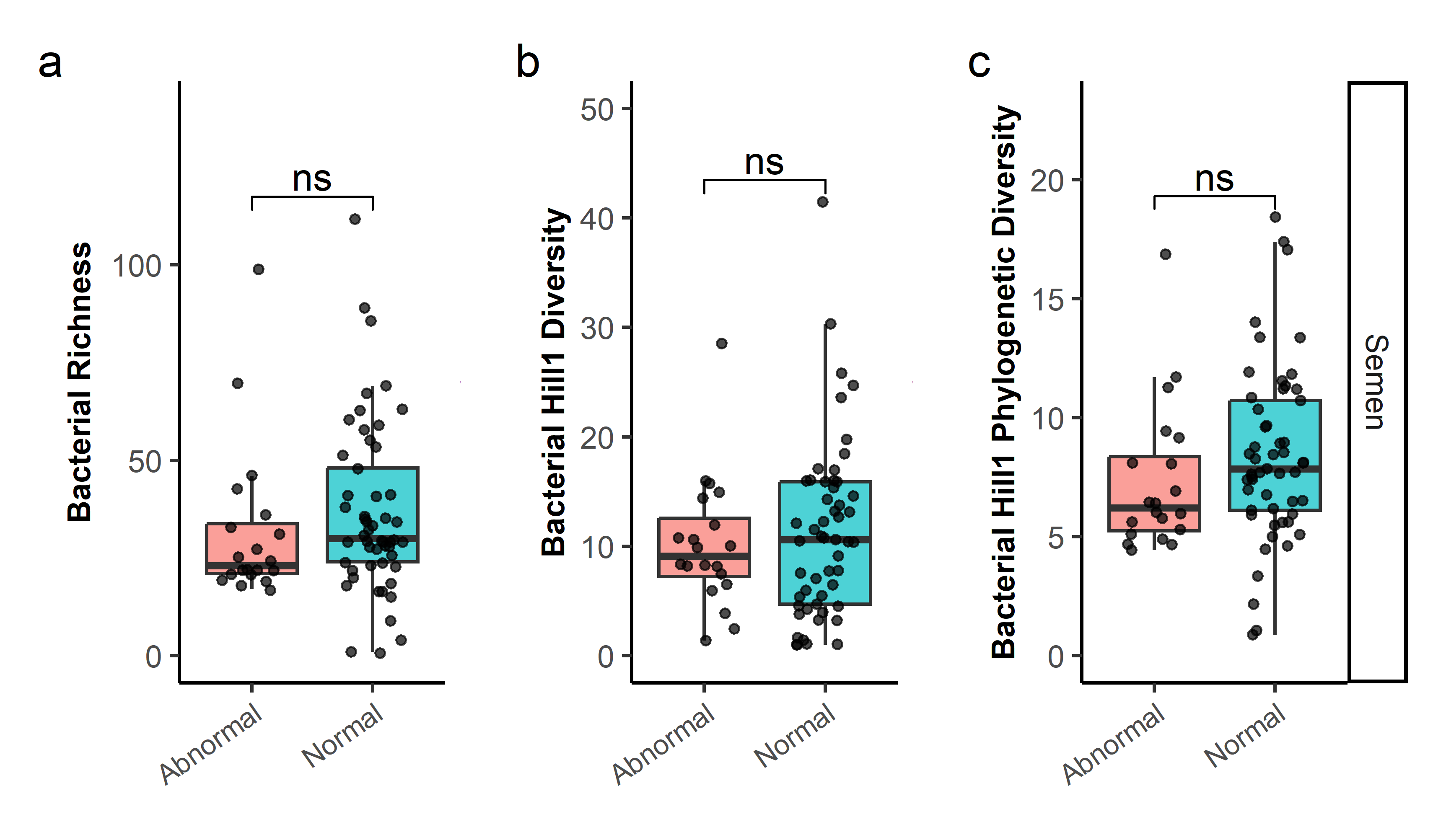
Samples were sequenced to a median depth of 2080 classified reads (Q1 = 1144, Q3 = 5531), providing a median 99.98% Good’s coverage estimate (Q1 = 99.91, Q3 = 100%) and suggesting that samples had been sequenced to sufficient depth (Supp Table 4). There were no significant differences in alpha or beta diversity among any groups investigated in this study. For OTU richness, Hill1 diversity, and phylogenetic hill1 diversity: p > 0.10 in all groups examined. This is shown in Fig. 1A–C for the Group 1 comparisons, Supp Figure 1A-C for the Group 2 comparisons, and Supp Figure 2A-C for the Group 3 comparisons. Bacterial communities looked similar when evaluated between group differences using Principal Co-ordinate Analysis with weighted UniFrac distances (Fig. 1D).
Figure 1.

Microbial Community Profiling, Group 1. Global diversity measures comparing participants with normal sperm concentration and motility on SA versus participants with at least one abnormality in sperm concentration or motility on SA are outlined in 1A–C (alpha-diversity), and 1D (beta-diversity).
Regardless of our SA-stratified groupings, there was significant overlap between the most abundant species identified; the top five species always included: Enterococcus faecalis, Corynebacterium tuberculostearicum, Lactobacillus iners, Staphylococcus epidermidis, and Finegoldia magna. Figure 2, Supp Figure 3, and Supp Figure 4 outline the relative abundance of the top 30 species in the Group 1, 2, and 3 comparisons, respectively.
Figure 2.

Relative Abundance, Group 1. Relative abundance of the top 30 species of participants with normal sperm concentration and motility on SA versus participants with at least one abnormality in sperm concentration or motility on SA.
Using the ANCOM-BC procedure, we found: Group 1) participants with normal SA parameters showed a lower abundance of Peptoniphilus coxii (p = 0.0469) but a higher abundance of Staphylococcus hominis (p = 0.00335) compared to participants with any abnormality in sperm concentration or motility; Group 2) participants with normal sperm motility showed a lower abundance of Lactobacillus iners (p = 0.0464) compared to participants with abnormal sperm motility; Group 3) participants with normal sperm concentration showed a lower abundance of Paraburkholderia phenazinium (p = 0.0247), Pseudomonas fluorescens (p = 0.0101), and Pseudomonas stutzeri (p = 0.0241), but a higher abundance of Pseudomonas putida (p = 0.00478) compared to participants with anormal sperm concentration (Fig. 3, Supp Table 5).
Figure 3.

Differential Relative Abundance. Only statistically significant differences are shown. Figure 1A–C, represent the comparisons of groups 1, 2 and 3, respectively.
To query beyond our binary analysis of abnormal versus normal SA parameters, we also performed a CCA, which interprets participant data as a continuous variable. All constraining variables (participant age, SA volume, SA concentration, SA motility) were significantly associated with bacterial composition (Supp Table 6). Table 2 outlines the top 20 species with the highest correlation to the two CCA axes that explain the greatest amount of variance in the data. The top five species that account for the greatest degree of variance across the entire data set were: Lactobacillus iners (Rcumulative = 0.392), Negativicoccus massiliensis (Rcumulative = 0.379), Corynebacterium simulans (Rcumulative = 0.374), Peptinophilus grossensis (Rcumulative = 0.363), and Dermabacter vaginalis (Rcumulative = 0.280), as visualized in Fig. 4.
Table 2.
Canonical correlation analysis.
| Species | CCA1 P | CCA1 R | CCA2 P | CCA2 R | Cumulative R |
|---|---|---|---|---|---|
| Lactobacillus iners | 0.011 | 0.297 | 0.429 | − 0.094 | 0.392 |
| Negativicoccus massiliensis | 0.044 | − 0.237 | 0.231 | 0.142 | 0.379 |
| Corynebacterium simulans | 0.118 | − 0.184 | 0.109 | 0.189 | 0.374 |
| Peptoniphilus grossensis | 0.602 | 0.062 | 0.010 | − 0.300 | 0.363 |
| Brevibacterium mcbrellneri | 0.134 | − 0.177 | 0.132 | 0.178 | 0.355 |
| Lactobacillus crispatus | 0.231 | − 0.142 | 0.117 | 0.185 | 0.327 |
| Corynebacterium pyruviciproducens | 0.434 | − 0.093 | 0.057 | 0.224 | 0.317 |
| Mobiluncus curtisii | 0.348 | 0.111 | 0.091 | − 0.199 | 0.310 |
| Agrobacterium rhizogenes | 0.468 | 0.086 | 0.061 | 0.220 | 0.307 |
| Paraburkholderia phenazinium | 0.231 | 0.142 | 0.181 | − 0.158 | 0.300 |
| Fusobacterium nucleatum | 0.290 | 0.125 | 0.159 | − 0.167 | 0.292 |
| Actinotignum schaalii | 0.146 | 0.172 | 0.325 | 0.117 | 0.289 |
| Phyllobacterium myrsinacearum | 0.690 | 0.047 | 0.045 | 0.235 | 0.283 |
| Dermabacter vaginalis | 0.025 | − 0.262 | 0.875 | 0.019 | 0.280 |
| Actinomyces radingae | 0.733 | − 0.041 | 0.042 | 0.239 | 0.280 |
| Anaerococcus octavius | 0.135 | − 0.176 | 0.408 | 0.098 | 0.275 |
| Corynebacterium tuberculostearicum | 0.263 | − 0.133 | 0.281 | 0.128 | 0.261 |
| Streptococcus mitis | 0.385 | 0.103 | 0.194 | − 0.154 | 0.257 |
| Helcobacillus massiliensis | 0.312 | − 0.120 | 0.277 | 0.129 | 0.249 |
| Peptoniphilus coxii | 0.083 | 0.204 | 0.786 | − 0.032 | 0.237 |
Figure 4.

Canonical Correlation Analysis Plot. Visual representation of the canonical correlational analysis, highlighting non-microbiome (3A) and microbiome (3B) variables differentiating participants with normal compared to abnormal SA parameters.
Discussion
In this study, we add to the still nascent body of research exploring the relationship between the semen microbiome and fertility. We understand that changes to SA parameters do not necessarily reflect or predict fertility or subfertility, which is an important distinction that should be taken into consideration in the interpretation of this data. Our results suggest that a small group of microorganisms play a critical role in observed alterations of SA parameters. Some of these microbes have been described extensively within other fertility-related contexts, whereas for others, this is the first report where they have been implicated in fertility and subfertility. The present findings do not indicate causality, but these results may inform future mechanistic studies to tease apart the complex relationship between human microbial populations and male fertility.
Aligned with previous studies, our results reveal that semen harbors a diverse, but largely consistent, microbiome. The most abundant members of this niche were Enterococcus faecalis, Staphylococcus epidermidis, Corynebacterium tuberculostearicum, and Lactobacillus iners. These findings match with near fidelity to the recently published seminal report by Suarez Arbelaez et al.,8 which explored taxonomic differences in the semen microbiome in men pre- and post-vasectomy, albeit in a somewhat smaller sample (58 men). Remarkably, we did not identify any significant differences in alpha or beta diversity regardless of our SA-based classifications, suggesting that it may not be a global dysbiosis that contributes to subfertility, but perhaps a more subtle change in certain cornerstone species. Somewhat in contrast to our findings, Lundy and colleagues5 recently published a high quality study, that did find differences in alpha and beta diversity in fertile (n = 12) versus infertile men (n = 25). This discordance may reflect different patient populations in addition to methodological differences in analyses.
When taking into consideration semen volume, concentration, and motility, Lactobacillus iners emerged as the strongest differentiator between men with normal versus abnormal SA parameters. This finding was then recapitulated in our subgroup analysis, which showed that men with abnormal motility have a semen microbiome particularly enriched in Lactobacillus iners. Previously published investigations implicate this species as playing a prominent negative role in fertility; however, much of this literature is related to the vaginal microbiome and female factors. In a study that included 25 couples undergoing the use of assisted reproductive technologies, researchers found that increased abundance of vaginal lavage Lactobacillus iners was associated with infertility.20 Looking beyond fertility, the relationship between the vaginal microbiome and Lactobacillus iners is highly nuanced; in some contexts, this microbe takes on the role of a vaginal symbiont, whereas in others it may worsen vaginal dysbiosis and predispose the host to developing bacterial vaginosis, sexually transmitted infections, and adverse pregnancy outcomes.21–23 Our study represents the first report of a negative association between Lactobacillus iners and male-factor fertility.
Lactobacillus iners has the smallest reported genome of any member of this genus24 while also having a subset of enzymes not available to other members of this genus.25 For example, Lactobacillus iners can produce the less common L-lactic acid (rather than D-lactic acid) isomer, which can induce a pro-inflammatory local milieu. This may be sufficient to impair sperm motility in a clinically meaningful way.26 The female literature has even shown associations between the presence of Lactobacillus iners and pro-inflammatory cytokines including tumor necrosis factor-alpha and macrophage migration inhibitory factor.25,27.
We also found that three species in the genus Pseudomonas were differentially abundant when groups were stratified by abnormal and normal sperm concentrations, with Pseudomonas fluorescens and Pseudomonas stutzeri being more abundant in patients with abnormal SA concentrations, whereas Pseudomonas putida less abundant in abnormal SA concentration samples. Previous investigations have reported on the relationship between Pseudomonas-predominant genera and low-quality semen.28 However, some studies suggest the opposite, with a positive association between genus Pseudomonas and total motile sperm counts.5 These seemingly conflicting findings in the literature likely reflect, in part, differences in patient populations and sample sizes but also suggest – as do our findings – that not every member of the same genus may function in the same way to impact, be it positively or negatively, fertility.
Adjacent investigations from the female infertility literature found that targeted microbial therapy based off of NGS of the endometrial microbiome improved assisted reproductive technology outcomes in women with recurrent implantation failure.29 Though more in its infancy, our findings suggest that there may be similar microbial signatures in semen, which could be worth targeting to improve male factor infertility.
The present study has limitations. Although this work represents the largest semen microbiome sampling in any published work to date, the patient population at our institution is not socioeconomically diverse given the relative affluence of our geographical region. A multi-institutional, longitudinal study design may offer stronger reinforcement or contradiction to our local findings. Collection of semen specimens through masturbation invariably includes inhabitants of the urethral microbiome, which may confound our findings though this still remains the most reasonable method of specimen collection given the alternative of seminal vesicle aspiration is not a practical option. We did not collect information related to recent antibiotic use or genitourinary infections, which may confound our results. Furthermore, we did not collect data on sexual intercourse frequency and whether barrier methods of contraception were used so we are unable to query the impact of potential partner-influenced changes in the microbiome that may have emerged through intercourse. Only a single sample of semen was collected from each participant, which may limit generalizability as the inter-specimen stability of the semen microbiome has not been previously explored. Furthermore, incorporation of additional biomarkers believed to be associated with sperm health such as systemic markers for oxidative stress or DNA fragmentation index may provide important mechanistic context and should be explored in future investigations.
Conclusions
Our findings highlight a small but critical group of microorganisms that may play an important role in male fertility; namely, Lactobacillus iners and members of the genus Pseudomonas. This exploratory analysis supports data reported in previously published, smaller, studies while also revealing novel insights that will be critical in guiding future, mechanistic investigations that will help us understand the complex relationship between the semen microbiome and fertility.
Supplementary Information
{kind=link}
{kind=link}
Author contributions
V.O. contributed to study concept and design, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, and study supervision. A.B. contributed to study concept and design, participant recruitment, and critical revision of the manuscript for important intellectual content. R.K. contributed to participant recruitment and critical revision of the manuscript for important intellectual content. J.T.S. contributed to study concept and design, participant recruitment, and critical revision of the manuscript for important intellectual content. J.S.A. contributed to analysis and interpretation of data, drafting of the manuscript, and critical revision of the manuscript for important intellectual content. T.K. contributed to participant recruitment, administrative support, and critical revision of the manuscript for important intellectual content. S.F.M. contribute to administrative support and critical revision of the manuscript for important intellectual content. C.D.T. contributed to analysis and interpretation of data, drafting of the manuscript, administrative support, and critical revision of the manuscript for important intellectual content. T.M.H. contributed to participant recruitment, study supervision, and critical revision of the manuscript for important intellectual content. J.N.M. contributed to study concept and design, participant recruitment, critical revision of the manuscript for important intellectual content, and study supervision. S.V.E. contributed to study concept and design, participant recruitment, critical revision of the manuscript for important intellectual content, and study supervision. All authors read and approved the final manuscript.
Funding
This work was supported in part by the 2023 Urology Care Foundation Residency Research Award Program and the Russell Scott, Jr., MD, Urology Research Fund.
Data availability
Raw 16S rRNA sequence data has been uploaded to the SRA under BioProject Accession PRJNA1040881.
Competing interests
JNM is a consultant for Halozyme Pharma, Boston Scientific, and Endo Pharmaceuticals. All other authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-51686-4.
References
- 1.Punab M, et al. Causes of male infertility: A 9-year prospective monocentre study on 1737 patients with reduced total sperm counts. Hum. Reprod. 2017;32:18–31. doi: 10.1093/humrep/dew284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minhas S, et al. European association of urology guidelines on male sexual and reproductive health: 2021 update on male infertility. Eur. Urol. 2021;80:603–620. doi: 10.1016/j.eururo.2021.08.014. [DOI] [PubMed] [Google Scholar]
- 3.Osadchiy V, Mills JN, Mayer EA, Eleswarapu SV. The seminal microbiome and male factor infertility. Curr. Sex Health Rep. 2020;12:202–207. doi: 10.1007/s11930-020-00273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osadchiy V, Martin CR, Mayer EA. The gut-brain axis and the microbiome: mechanisms and clinical implications. Clin. Gastroenterol. Hepatol. 2019;17:322–332. doi: 10.1016/j.cgh.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundy SD, et al. Functional and taxonomic Dysbiosis of the gut, urine, and semen microbiomes in male infertility. Eur. Urol. 2021;79:826–836. doi: 10.1016/j.eururo.2021.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Campbell K, et al. Next generation sequencing analysis of semen microbiome taxonomy in men with non-obstructive azoospermia vs fertile controls: A pilot study. F. S. Sci. 2023 doi: 10.1016/j.xfss.2023.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowman ME, Tipton CD, Labordere AL, Brown JA. Equine sinusitis aetiology is linked to sinus microbiome by amplicon sequencing. Equine Vet J. 2022 doi: 10.1111/evj.13884. [DOI] [PubMed] [Google Scholar]
- 8.Suarez Arbelaez MC, et al. Pilot study: Next-generation sequencing of the semen microbiome in vasectomized versus nonvasectomized men. Eur. Urol. Focus. 2023;9:75–82. doi: 10.1016/j.euf.2022.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 10.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake LE, et al. An assessment of minimum sequence copy thresholds for identifying and reducing the prevalence of artefacts in dietary metabarcoding data. Methods Ecol. Evolut. 2022;13:694–710. doi: 10.1111/2041-210X.13780. [DOI] [Google Scholar]
- 15.Gilbert JA, et al. The taxonomic and functional diversity of microbes at a temperate coastal site: A 'multi-omic' study of seasonal and diel temporal variation. PLoS One. 2010;5:e15545. doi: 10.1371/journal.pone.0015545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D. hillR: Taxonomic, functional, and phylogenetic diversity and similarity through Hill Numbers. J. Open Source Softw. 2018;3:1041. doi: 10.21105/joss.01041. [DOI] [Google Scholar]
- 17.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kers JG, Saccenti E. The power of microbiome studies: Some considerations on which alpha and beta metrics to use and how to report results. Front Microbiol. 2021;12:796025. doi: 10.3389/fmicb.2021.796025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020;11:3514. doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campisciano G, et al. Lactobacillus iners and gasseri, Prevotella bivia and HPV belong to the microbiological signature negatively affecting human reproduction. Microorganisms. 2020 doi: 10.3390/microorganisms9010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng N, Guo R, Wang J, Zhou W, Ling Z. Contribution of lactobacillus iners to vaginal health and diseases: a systematic review. Front Cell Infect. Microbiol. 2021;11:792787. doi: 10.3389/fcimb.2021.792787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabbatini S, et al. Lactobacillus iners Cell-Free Supernatant Enhances Biofilm Formation and Hyphal/Pseudohyphal Growth by Candida albicans Vaginal Isolates. Microorganisms. 2021 doi: 10.3390/microorganisms9122577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakobsson T, Forsum U. Lactobacillus iners: A marker of changes in the vaginal flora? J. Clin. Microbiol. 2007;45:3145. doi: 10.1128/JCM.00558-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macklaim JM, Gloor GB, Anukam KC, Cribby S, Reid G. At the crossroads of vaginal health and disease, the genome sequence of Lactobacillus iners AB-1. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl 1):4688–4695. doi: 10.1073/pnas.1000086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cocomazzi G, et al. The impact of the female genital microbiota on the outcome of assisted reproduction treatments. Microorganisms. 2023 doi: 10.3390/microorganisms11061443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witkin SS, et al. Influence of vaginal bacteria and D- and L-lactic acid isomers on vaginal extracellular matrix metalloproteinase inducer: Implications for protection against upper genital tract infections. mBio. 2013 doi: 10.1128/mBio.00460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Seta F, Campisciano G, Zanotta N, Ricci G, Comar M. The vaginal community state types microbiome-immune network as key factor for bacterial vaginosis and aerobic vaginitis. Front Microbiol. 2019;10:2451. doi: 10.3389/fmicb.2019.02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng SL, et al. Bacterial communities in semen from men of infertile couples: Metagenomic sequencing reveals relationships of seminal microbiota to semen quality. PLoS One. 2014;9:e110152. doi: 10.1371/journal.pone.0110152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwami N, et al. Therapeutic intervention based on gene sequencing analysis of microbial 16S ribosomal RNA of the intrauterine microbiome improves pregnancy outcomes in IVF patients: A prospective cohort study. J. Assist. Reprod. Genet. 2023;40:125–135. doi: 10.1007/s10815-022-02688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw 16S rRNA sequence data has been uploaded to the SRA under BioProject Accession PRJNA1040881.