

Abstract
Fialuridine (FIAU) is a nucleoside-based drug that caused liver failure and deaths in a human clinical trial that were not predicted by nonclinical safety studies. A recent report concluded that a TK-NOG humanized liver (hu-liver) mouse model detected human-specific FIAU liver toxicity, and broader use of that model could improve drug safety testing. We further evaluated this model at similar dose levels to assess FIAU sensitivity and potential mechanistic biomarkers. Although we were unable to reproduce the marked acute liver toxicity with two separate studies (including one with a “sensitized” donor), we identified molecular biomarkers reflecting the early stages of FIAU mitochondrial toxicity, which were not seen with its stereoisomer (FIRU). Dose dependent FIAU-induced changes in hu-liver mice included more pronounced reductions in mitochondrial to nuclear DNA (mtDNA/nucDNA) ratios in human hepatocytes compared to mouse hepatocytes and kidneys of the same animals. FIAU treatment also triggered a p53 transcriptional response and opposing changes in transcripts of nuclear- and mitochondrial-encoded mitochondrial proteins. The time dependent accumulation of FIAU into mtDNA is consistent with the ≥9-week latency of liver toxicity observed for FIAU in the clinic. Similar changes were observed in an in vitro micro-patterned hepatocyte coculture system. In addition, FIAU-dependent mtDNA/nucDNA ratio and transcriptional alterations, especially reductions in mitochondrially encoded transcripts, were seen in livers of non-engrafted TK-NOG and CD-1 mice dosed for a shorter period.
Conclusion: These mechanistic biomarker findings can be leveraged in an in vitro model and in a more routine preclinical model (CD-1 mice) to identify nucleosides with such a FIAU-like mitochondrial toxicity mechanistic liability potential. Further optimization of the TK-NOG hu-liver mouse model is necessary before broader adoption for drug safety testing.
Keywords: fialuridine, mitochondrial DNA, biomarker, humanized liver mouse, liver, p53
Graphical Abstract
Graphical Abstract.

FIAU is a nucleoside drug that caused liver failure and deaths in a human clinical trial. FIRU is a diastereomer of FIAU, that has not been shown to induce in vitro toxicity. We tested both compounds in vivo in a TK-NOG humanized liver mouse model with a herpes simplex virus (HSV) thymidine kinase (TK) transgene, in mice without the TK transgene, and in an in vitro human liver model (HepatoPac®). We observed FIAU, but not FIRU-induced decreases in mtDNA/nucDNA ratios in all three models. We also observed a p53 transcriptional response and opposing changes in transcripts of nuclear- and mitochondrial-encoded mitochondrial proteins in mice with the TK transgene and in the in vitro HepatoPac® model. Toxicities of FIAU have been linked to the triphosphate form of this drug. The TK transgene (HSV-TK) may induce cytosolic, but not mitochondrial entrapment of FIAU triphosphate, while mitochondrial thymidine kinase 2 (TK-2) produces FIAU triphosphate that incorporates in mtDNA.
Introduction
Fialuridine (FIAU, Fig. 1a) is a nucleoside analog-based drug that was discontinued after 7 of 15 hepatitis B infected patients developed liver failure (5 deaths; 2 liver transplants) in a phase 2 clinical trial after approximately 3 months of treatment. The nonclinical safety studies supporting this trial did not identify liver toxicity in mice (6 months), rats (1 month), dogs (3 months), or monkeys (1 month).1–3 The observations in humans, including lactic acidosis, laboratory abnormalities, microscopic histologic liver findings, and ultrastructural mitochondrial alterations,2–6 were indicative of liver mitochondrial toxicity.
Fig. 1.

(a) FIAU (arabinose) and (b) FIRU (ribose) are stereoisomers differing only in the orientation of the 2′ fluorine atom.
While the precise mechanism of the severe liver injury seen in the human clinical trial is yet to be fully elucidated, subsequent in vitro and in vivo evaluations provide further evidence supporting mitochondrial toxicity as the cause of the severe liver toxicity. In vitro evidence indicates that FIAU can incorporate into mitochondrial DNA and inhibit mitochondrial DNA polymerase (Pol) γ.7–9
Additional evidence suggests that the mitochondrially-targeted equilibrative nucleoside transporter (ENT) 1 that transports FIAU is present in human mitochondria, but not in mouse mitochondria (rat, dog, or monkey not assessed), potentially providing a species-specific mechanistic explanation for the observed increased human sensitivity.10,11 It is also possible that species-specific differences in the active FIAU-triphosphate (TP) half-lives and/or in the amount of FIAU-TP generated in the mitochondria could lead to greater sensitivity in human, as has been reported for another nucleoside.12
In vivo experiments with FIAU offer additional clues to the potential mechanism leading to liver toxicity. The first set of experiments was conducted in woodchucks13,14 where dosing with FIAU resulted in adverse clinical signs, increases in plasma levels of alanine aminotransferase (ALT) and lactate, and incorporation of FIAU into DNA in both hepatitis positive and negative woodchucks after 12 weeks of dosing at a dose that was 4.5-fold greater than the adverse dose in the clinical studies [(0.25 mg/kg/day (mkd)]. Histopathologically, woodchucks showed ultrastructural changes in liver, heart, skeletal muscle, and kidney mitochondria and decreases in mitochondrial DNA content ranging from 55%–87% without declines in nuclear DNA.13,14 Another set of in vivo experiments was conducted in TK-NOG hu-liver mice15,16 where dosing with FIAU at a high dose of 400 mkd resulted in acute liver toxicity, mortality, increases in serum ALT and plasma lactate after 4 days in engrafted mice, but not in non-engrafted controls. After a longer dosing duration (2 weeks), similar effects were seen at lower dose levels (2.5–100 mkd). The liver toxicity observed by Xu et al. 2014 in TK-NOG hu-liver mice was observed 4 days to 2 weeks after initiation of dosing, which is a much shorter time than the human clinical trials (or woodchuck studies) where it took 9–12 weeks before liver toxicity was observed. Based on body weight, dose multiples for the TK-NOG hu-liver mice ranged from 10–1,600-fold above those used in the human clinical trial.15,16
The work we describe here summarizes our success in identifying early molecular mechanistic in vivo tissue biomarkers of FIAU toxicity across several models, and in developing an in vitro translational model that recapitulates many of the same mechanistic features, even though our attempts to reproduce the reported FIAU-mediated acute liver toxicity in TK-NOG hu-liver mice were unsuccessful. We also observed that FIAU can impact mitochondrial DNA/nuclear DNA (mtDNA/nucDNA) ratio not just in the liver, but also in the kidney. Finally, this mitochondrial toxicity mechanism observed in vivo can be recapitulated in vitro and in a more routine preclinical model (CD-1 mice); and it is highly specific to FIAU, given that it was not observed with its stereoisomer FIRU, also known as 5-IFDU (Fig. 1b).
Materials and methods
Chemicals
Fialuridine (1-(2-deoxy-2-fluoro-1-D-arabinofuranosyl)-5-iodouracil; FIAU), and 1-(2-fluoro-2-deoxyribofuranosyl)-5-iodouracil (FIRU, also known as 5-IFDU) were synthesized at Pharmaron (Louisville, KY). The structures were verified by 1H-NMR, 19F-NMR, and Chiral Analysis using six chiral columns. Both compounds were 99% pure. Mono and triphosphate standards of FIAU and FIRU were synthesized from the products provided by Pharmaron at NuBlocks LLC (Oceanside, CA). L-FMAU (Clevudine) was purchased from Ark Pharm.
Generation of humanized liver mice
For hu-liver mice studies, TK-NOG mice were generated and humanized at the Central Institute for Experimental Animals (CIEA), Kawasaki, Japan and shipped to Merck & Co., Inc., West Point, PA, USA. Two studies were conducted using aseptic conditions. For the first chimeric humanized liver mouse study, mice were provided CA-1 rodent diet (CLEA, Japan) and provided access to a MD-7 primate diet milled to the size of rodent pellets prior to shipment. For that study, cryopreserved human hepatocytes (Donor 2: Caucasian male, 5 years old) were obtained from BioPredic International, San Grègoire, France. Male TK-NOG mice received an intraperitoneal dose (4–10 mg/kg) ganciclovir at 8 weeks of age and were transplanted with the cryopreserved human hepatocytes at nine weeks of age. Some male TK-NOG mice received a second dose of 10 mg/kg Ganciclovir at 9 weeks of age and were transplanted at 10 weeks of age. Non-transplanted TK-NOG mice that did not receive Ganciclovir were used as controls, as this treatment is not tolerated in the absence of engraftment with human hepatocytes. Animals were placed on study at ~20 weeks of age, which was approximately one week after shipment from Japan. Table S1 summarizes human albumin (baseline), the baseline replacement index (reflective of engraftment), and plasma ALT levels.
For the second chimeric humanized liver mouse study, mice were provided CA-1 rodent diet (CLEA, Japan) prior to shipment. For that study, cryopreserved human hepatocytes (Donor 4: African American female, 30 years old) were obtained from Triangle Research Labs, Research Triangle Park, NC, USA. Male TK-NOG mice received 0.04–0.08 mg/mL val-ganciclovir in the drinking water for 3 days at 8 weeks of age and were engrafted with the cryopreserved human hepatocytes at 9 weeks of age. Animals were placed on study at 20 weeks of age, which was approximately one week after shipment from Japan. Table S1 summarizes human albumin (baseline), the baseline replacement index (reflective of engraftment), and plasma ALT levels.
Animal studies
Table S2 is a high-level summary of the in vivo mouse studies. More details are provided in the subsequent section.
Formulation and dosing
See supplementary methods for additional details.
The Xu, et al.15 manuscript used dose levels ranging from 2.5–400 mg/kg/day with toxicities observed at all dose levels in a dose and time-dependent manner. A dose of 90 mg/kg/day (mkd) was initially targeted (Cohort A, 1st hu-liver study) as a top dose to try to bracket the middle dose where Xu, et al report a death and poor tolerability in all remining 14 mice within 4 days. After no liver toxicity or deaths were observed in our study at this dose level after 29 days, we decided to evaluate a higher dose level. However, after many unsuccessful efforts were made to consistently formulate a 400 mkd dose. Subsequently, 250 mkd was used as the high dose since this dose formulation was consistent.
First TK-NOG Hu-liver mouse study
All studies were approved by the Merck & Co., Inc., Rahway, NJ, USA Institutional Animal Care and Use Committee and conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility in compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. Animals were provided autoclaved tap water throughout the study and were housed and handled in sterile conditions. FIAU, FIRU, or vehicle (10% DMSO) were orally administered to male TK-NOG mice (non, low, or high-engrafted) once daily at either 90 mkd (Cohort A) for 60 days or 250 mkd (Cohort B) for 57 days (planned for 60 days but terminated early due to clinical signs of toxicity). In addition to the test article, all animals received approximately 1,500 ppm of vitamin C supplemented PMI irradiated rodent diet 5,002 to mitigate the potential risk of vitamin C deficiency, as human hepatocytes lack a critical enzyme needed for vitamin C synthesis.17 Due to concerns that the vitamin C diet could potentially protect against the intended drug-induced liver toxicity, vitamin C was removed from the feed for approximately half of the humanized mice in Cohort B on Study Day (SD) 29 throughout the remainder of the study (rodent autoclavable diet #5010 only). On SDs 6, 20, 34, and 48, non-terminal bleeds (~160 μL) were taken from fasted animals by orbital sinus or by tail artery and samples were processed to plasma (EDTA). Samples of serum and plasma were also obtained via vena cava at study termination, and both liver and kidney were collected for postmortem analysis. Animals were weighed at pretest (one day prior to study) and weekly throughout the study. There were 3 animals per group in the FIRU and vehicle control cohorts, and 4 animals per group in the FIAU cohort.
2nd TK-NOG Hu-liver mouse study with screened and selected “sensitive donor”
Animals were administered a PMI vitamin C-free rodent autoclavable diet (#5010) and autoclaved tap water throughout the study and were housed and handled in sterile conditions. FIAU, FIRU, or vehicle (10% DMSO) were administered orally to male TK-NOG mice (non, low, or high-engrafted) once daily at 250 mkd for 32 days (planned for 60 days but terminated early due to clinical signs of toxicity). Both non-terminal and terminal bleeds followed the same procedure as the first study. There were three animals per group for the non-engrafted vehicle control and FIRU conditions, 6–7 animals for the low and high-engrafted FIRU conditions, and 11–13 animals for the low and high-engrafted FIAU conditions.
Fifteen-day non-engrafted TK-NOG and CD-1 mouse study
FIAU (250 mkd), FIRU (250 mkd), or vehicle (10% DMSO) were administered orally to male non-engrafted TK-NOG mice (14–18 weeks old, from Taconic Farms; original stock from CIEA) or male CD-1 mice (8 weeks old, from Charles River Laboratories) for 15 days. Animals were sacrificed on the last day of dosing approximately 4 (+/− 30 min) hours post dose. Animals were administered a PMI vitamin C-free rodent autoclavable diet (#5010) and autoclaved tap water throughout the study. TK-NOG mice were handled/housed in sterile conditions. Serum and plasma were sampled by vena cava at study termination and both liver and kidney were evaluated for postmortem analysis. There were 4 animals per group.
Clinical chemistry analyses
Serum parameters, including ALT, aspartate aminotransferase (AST), alkaline phosphatase (Alk Phos), glucose, and bilirubin were determined by routine clinical pathology procedures using a Beckman AU5800 Chemistry Analyzer. Plasma ALT was also measured on a Beckman AU5800 Chemistry Analyzer.
Post-mortem evaluations
Microscopic histomorphologic examinations were limited to liver and kidney. Hematoxylin and eosin (H&E)-stained tissue sections were examined, and grades were assigned in a five-point grading scale with 0 being no observable pathology, 1 being minimal, 2 mild, 3 moderate, 4 marked, and 5 severe. Non-engrafted TK-NOG mouse liver samples from the first humanized liver mouse study were also evaluated using transmission electron microscopy (TEM) (see supplementary methods).
Pharmacokinetics and analysis of parent (FIAU or FIRU) and their mono- and triphosphate anabolites
Ten μL of whole blood were collected into 0.1 M sodium citrate tubes with microsampling methods from the tail vein from control and treated animals at 1, 2, 4, 7, and 24 h post dose after 14 oral doses, except for Cohort B in the first TK-NOG hu-liver mouse study where blood sampling was not conducted at 2 and 7 h post dose. For the 15-day non-engrafted TK-NOG and CD-1 mouse study and the second TK-NOG hu-liver mouse study, liver was collected at terminal sacrifice approximately 4 or 24 h post dose. Liver samples were analyzed to determine levels of FIAU, FIRU, and their corresponding anabolites (mono or triphosphate) by liquid chromatography-mass spectrometry (LC-MS/MS) analysis. Details are in the supplementary methods section. The whole blood concentration curves were analyzed with Phoenix for AUC0–24 h and Cmax. Data are expressed as mean ± the standard deviation.
Liver samples (~150–500 mg) were excised and placed immediately in liquid nitrogen. The samples were subsequently stored in a − 70 °C freezer. For processing, the samples were homogenized in 3 v/w ice cold 70/30 methanol/20 mM EDTA+20 mM EGTA, pH 8.0 in RNAase/DNAase free microvials (Cat No 19-628S-3: 2 mL with 2.8 mm ceramic beads) at ~− 20 °C using an Omni Bead Ruptor 24 cryomill. The samples were cryomilled to yield homogenous liver homogenates using the following conditions: speed = 3.1 m/s; cycle time = 0.15 min; #cycles = 10, dwell time = 1 min.
A dimethylhexylamine (DMHA) based ion-pairing HPLC-MS/MS method was used to measure drug-related FIAU or FIRU monophosphate (NMP) and FIAU or FIRU triphosphate (NTP) metabolite concentrations in liver samples from each cohort of the 1–15 day study. The electrospray MS/MS method was run on a SCIEX API 4,000 mass spectrometer with turboV ionspray source in negative ion mode monitoring the following transitions (NMP metabolite: [M-H]- = 451 > 79; NTP: [M-H]- = 611 > 159). As FIAU and FIRU are isomers, their NMP and NTP metabolites are also isomeric and thus the same MS/MS transitions were used for FIAU and FIRU NMP and likewise FIAU and FIRU NTP. Absence of endogenous interference(s) was verified for NTP analysis by monitoring of the same MRM transitions in unspiked control cell lysates.
Liver nucleosides were measured by LC-MS/MS analysis after protein precipitation of the liver homogenates prepared for NTP analysis. Concentration (μM) in undiluted liver was reported after correction by the factor of 4 dilution used in homogenate preparation.
Liver concentrations (nmol/g liver) of nucleoside triphosphate (NTP) anabolites of parent FIAU and FIRU were quantitated against calibration standards of the same nucleoside standards (see Chemicals Section from Materials and Methods) prepared from pooled control liver homogenates.
Mitochondrial DNA/nuclear DNA (mtDNA/nucDNA) ratio assessment
Approximately 50 mg of liver and kidney (first hu-liver mice study only) were collected at the final necropsy in tubes, and samples were subsequently frozen on dry ice before being stored at −70 °C until processing. The mtDNA/nucDNA ratios for liver (mouse and human) and kidney (mouse only) were assessed by quantitative real time PCR (qPCR). qPCR specific for mouse or human mtDNA and nucDNA targets was performed using TaqMan Universal PCR master mix no AmpErase UNG (Applied Biosytems Cat# 4324020). Mouse clusterin (nuclear DNA target) and MT-Co2 (mitochondrial DNA target) copy number assays were amplified with primer/probes custom designed by ABI (see Table 1 below). The human nuclear target was amplified using a custom designed ABI copy number assay and for the human mitochondrial target, MT-CO1 an assay from the ABI inventory was used (see Table 1 below). Two tailed heteroscedastic T-tests were performed in Microsoft Excel with two tails using < 0.05 as a cutoff to compare group levels of significance. The strain (i.e. TK-NOG or CD-1) and engraftment level (i.e. Non, Low, and High) were matched when considering comparisons on each study.
Table 1.
Primer and genes assayed for the mitochondrial to nuclear DNA ratio assay.
| Target Species | Genome | assay ID | Gene symbol | Gene name |
|---|---|---|---|---|
| mouse | nuclear | Mm00397583_cn | Clu | Clusterin |
| mouse | mitochondrial | CC1RUG7 | MT-CO2 | cytochrome c oxidase 2 |
| human | nuclear | Hs06509577_cn | – | located in Chr.1:205664468 |
| human | mitochondrial | Hs02596864_g1 | MT-CO1 | cytochrome c oxidase I |
Calculation of mtDNA/nucDNA ratios was performed with the following equation 2^-(mitoCT-nuclearCT) and expressed as percent of mean of controls.
Assessment of human albumin levels
Plasma samples were assayed for human albumin using a human albumin ELISA kit from Bethyl Laboratories, Incorporated at a 1:125000 dilution.
RNA Seq for 1 st chimeric Hu-liver mouse study and for 15-day non-engrafted TK-NOG and CD-1 mouse study
Approximately 50 mg of liver was collected from each mouse in tubes at the final necropsy, frozen on dry ice, and samples stored at −70 °C prior to processing to RNA. See supplementary methods for details on sample preparation and sequencing.
For the p53 response evaluation (Table S3), 10 genes commonly associated with p53-mediated DNA damage response18,19 were selected, similar to what was reported by Podtelezhnikov and colleagues.20 All mitochondrially encoded gene transcripts were used except for short mt-tRNA (Table S3). Ten representative nuclear encoded mitochondrial proteins for electron transport chain complexes were selected since these transcripts form a co-regulated network (Table S3).21
In vitro HepatoPac® studies with different donors
Human HepatoPac® micropatterned hepatocyte co-cultures were leveraged for in vitro studies. See supplementary methods for details, including handling, donor information (Table S4), and RNA Seq.
Verification that FIAU-triphosphate (TP) and FIRU-TP had no effects on the mtDNA/nucDNA PCR analyses
To confirm that the reduced mtDNA/nucDNA ratio was indeed due to decreases in mtDNA and not due to a lack of PCR amplification of the mtDNA when FIAU or FIRU are incorporated, we conducted an investigation demonstrating that DNA incorporation of FIAU or FIRU does not impact the mtDNA/nucDNA ratio assay. See supplementary methods.
Results
In the first Hu-liver mouse study, FIAU (but not FIRU) resulted in clinical signs of toxicity without transaminase increases in Hu-liver mice, while transaminase increases were seen for FIAU (but not FIRU) in the non-engrafted TK-NOG mice controls
In our first FIAU TK-NOG hu-liver mouse study, animals treated with FIAU showed clinical signs of toxicity that were more severe in the human hepatocyte-engrafted mice toward the end of the approximately 2-month treatment period. FIAU-related clinical signs of toxicity consisted of decreased activity in animals dosed with 90 mkd (Cohort A) and decreased activity, thin appearance, decreased skin turgor, and pale appearance in animals dosed with 250 mkd (Cohort B). FIAU treatment also resulted in 16.4% body weight loss in animals dosed with 250 mkd (compared to pretest, see Fig. S1). Based on these effects, animals receiving 250 mkd were terminated early on SD 58 for humane reasons instead of the planned SD 61 study termination.
Despite the clinical signs of toxicity seen toward the end of the study, there were no sustained increases in ALT levels (as compared to their respective vehicle control groups) in either the low- or the high-engrafted groups dosed with 90 or 250 mkd of FIAU. Interestingly, ALT increases of 189 and 549% were observed after approximately one-month of dosing in the non-engrafted TK-NOG control animals receiving 90 or 250 mkd of FIAU, respectively (Fig. 2, Fig. S2, and Table S7). In contrast, and as expected, FIRU treatment resulted in minimal or no ALT changes in any of the dose groups. Importantly, it is worth noting that the average ALT baseline levels for the low- and high-engrafted animals were approximately 5-fold greater than those for non-engrafted groups (i.e. ~100 vs. ~500 U/L for non-engrafted vs. engrafted animals, respectively). These elevated and variable baseline differences may have confounded the detection of potential treatment-related changes.
Fig. 2.

Alanine aminotransferase (ALT) responses (average ± standard deviation) over time (study day) for vehicle control (blue), FIAU (red), and FIRU (green) in non-engrafted, low-engrafted, and high-engrafted TK-NOG mice with donor 2. The top trellis is the 90 mkd dose or vehicle from cohort a, and the bottom trellis is the 250 mkd dose or vehicle from cohort B.
There was no difference in the ALT levels in animals in Cohort B that had Vitamin C removed from their diets as compared to those that received supplementation for the entire study.
Changes observed in other serum clinical chemistry parameters while inconsistent in engrafted animals, were more consistent in non-engrafted animals and more pronounced in response to FIAU as compared to FIRU treatment (See Table S7, supplementary results). This included changes in AST, in the non-engrafted group, with greater responses to FIAU (+227–321%) compared to no changes with 90 mkd FIRU and only modest changes with 250 mkd FIRU treatment. Also, Alk Phos increased, with the greatest responses to FIAU in the non-engrafted group (+308%–529%) and increases in bilirubin were also seen with FIAU, but not FIRU treatment in non-engrafted animals. Serum glucose levels generally decreased with FIAU treatment but not FIRU treatment in non-engrafted animals.
Humanization of TK-NOG engrafted mice was maintained through the duration of the study
Human albumin levels in serum provide a metric of the degree of engraftment of human hepatocytes in chimeric hu-liver mice. A decrease in human albumin levels over time can represent a loss of human hepatocytes and a repopulation with mouse hepatocytes. Human albumin was not detected for non-engrafted mice (Fig. S3), confirming the species specificity of the assay. The human serum albumin levels in engrafted mice were stable throughout both the Cohort A and Cohort B phases of the study apart from “high” engrafted mice treated with FIRU. Those mice, which had been randomized before the study independent of their pre-study human albumin levels, had 1.7-fold higher human albumin levels to begin with, and over the study their levels dropped to the ranges observed for the other high engrafted groups (Fig. S4).
Histomorphology and TEM evaluations reveal minimal to mild alterations in liver and no alteration in kidney
Histopathologically, the human hepatocytes exhibited cytoplasmic vacuolation, irrespective of FIAU or FIRU treatment. Such vacuolation has been reported for the TK-NOG humanized liver mouse model and is consistent with glycogen storage in the cytoplasm,22 which is hypothesized to be an adaptation to the higher serum glucose in mice vs. humans. For the mouse hepatocytes, FIAU-related minimal to mild hepatocellular hypertrophy and minimal single cell necrosis were observed in both non-engrafted and engrafted animals, perhaps leading to the high ALT baseline. The hepatocellular hypertrophy was characterized by enlarged cells with an eosinophilic granular cytoplasm. An occasional necrotic hepatocyte was observed predominantly in non-engrafted animals. No FIAU or FIRU treatment-related effects were noted in the kidneys.
Transmission electron microscopy (TEM)
Due to the complexity and lack of hepatic histopathological response in TK-NOG humanized mice, we focused the TEM analysis on the liver of non-engrafted TK-NOG mice, as they also showed significant decreases in mtDNA/nucDNA. There were no apparent FIAU-treatment related differences in mitochondrial morphology by TEM (Fig. S4).
FIAU, but not FIRU, induces reductions in mtDNA/nucDNA ratio and transcriptional changes after 2 months of dosing in non-engrafted and engrafted TK-NOG mice
Consistent with the previous Woodchuck study,14 we observed significant FIAU-related decreases in mtDNA/nucDNA in human and mouse hepatocytes. The mean liver mtDNA/nucDNA ratios for the human hepatocytes from both low- and high-engrafted hu-liver mice dosed with FIAU or FIRU compared to their respective engraftment controls are shown in Fig. 3. For the 90 mkd (Cohort A) FIAU-treated group, the mtDNA/nucDNA levels decreased by 72%–82% (mean ratios from 0.18–0.28), while the mtDNA/nucDNA levels for the 250 mkd (Cohort B) FIAU-treated group decreased by 89% for both cohorts (mean ratio 0.11). For FIRU, there was no difference between FIRU-treated animals for either Cohort or for the different engraftment levels. No human mitochondrial or human nuclear DNA was detected in non-engrafted mice, confirming the specificity of the assay.
Fig. 3.

Human liver mtDNA/nucDNA responses (average ± standard deviation) for vehicle control (blue), FIAU (red), and FIRU (green) in non-, low-, and high-engrafted TK-NOG mice with donor 2. Each response is ratioed to the respective engraftment vehicle control. A dashed line is depicted at a ratio of 0.5, and a dotted line is depicted at a ratio of 0.2. The top trellis is the 90 mkd dose or vehicle, and the bottom trellis is the 250 mkd dose or vehicle following 2 months of dosing. When P < 0.05 for a treated group compared to the respective vehicle control, an asterisk (*) was added above it. When P < 0.05 for a FIRU group compared to the respective FIAU group, a dagger (†) was added above it.
Decreases in mtDNA/nucDNA ratios were also observed in the mouse remnant livers in hu-liver mice and in non-engrafted mouse livers. Figure 4 summarizes FIAU-related decreases in mouse mtDNA/nucDNA mean ratios from non-, low-, and high-engrafted hu-liver mice with respect to their engraftment groups. For the 90 mkd (Cohort A) FIAU-treated group, there was a 16% decrease (mean ratio 0.84) for non-engrafted mice, and it ranged from a 42%–54% decrease (mean ratio 0.46–0.58) for the low- and high-engrafted mice. For the 250 mkd (Cohort B) FIAU-treated group, there was a 77%–82% decrease (mean ratio 0.18–0.23). There was no difference between FIRU treated animals in Cohort B for the different engraftment levels compared to their respective engraftment controls. For Cohort A (90 mkd), while the high-engrafted FIRU treated mice had a 37% decrease (mean ratio 0.63), this effect was not significant given the variability seen in the mouse mtDNA/nucDNA data for this Cohort.
Fig. 4.
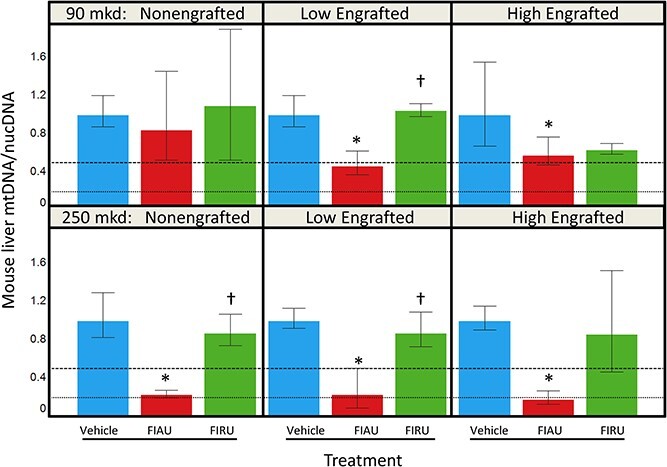
Mouse liver mtDNA/nucDNA (average ± standard deviation) for vehicle control (blue), FIAU (red), and FIRU (green) in non-engrafted, low-engrafted, and high-engrafted TK-NOG mice with donor 2. Each response is ratioed to the respective engraftment vehicle control. A dashed line is depicted at a ratio of 0.5, and a dotted line is depicted at a ratio of 0.2. The top trellis is the 90 mkd dose or vehicle, and the bottom trellis is the 250 mkd dose or vehicle. When P < 0.05 for a treated group compared to the respective vehicle control, an asterisk (*) was added above it. When P < 0.05 for a FIRU group compared to the respective FIAU group, a dagger (†) was added above it.
Decreases in mouse mtDNA/nucDNA ratios were also observed in kidneys from non, low, and high engrafted hu-liver mice dosed with 250 mkd of FIAU (Cohort B) (Fig. S5; only kidneys from Cohort B were assayed; treatment-related histomorphological changes not seen). FIAU decreased the mean mtDNA/nucDNA by 52% (mean ratio 0.48) as compared to the respective non-engraftment controls and by 69%–74% (mean ratio 0.26–0.31) for the low and high engrafted groups as compared to their respective engraftment controls. There were no FIRU-related mouse mtDNA/nucDNA changes for the different engraftment levels compared to their respective engraftment controls. The kidney does not have engrafted human cells, and no human mitochondrial or nuclear DNA signal was seen confirming the specificity of the assay.
Pharmacokinetics analysis shows similar plasma exposures between engrafted and non-engrafted TK-NOG mice
To put the findings into perspective, FIAU and FIRU plasma exposures after 14 days of dosing were assessed as part of this study. As shown in Table S8, the FIAU exposures were within 2-fold for both cohorts of engrafted mice compared to those in the non-engrafted mice, suggesting engraftment with human hepatocytes did not influence plasma exposure.
For FIRU, the AUC0–24 h exposures of engrafted mice dosed with 250 mkd FIRU were ~3-fold higher than the exposure in non-engrafted mice, suggesting a small potential engraftment effect. However, the Cmax parameter for FIRU treated animals were all within 2-fold, and not considered to be a noteworthy change (Table S8).
FIAU caused significant activation of a liver P53 transcriptional DNA damage response, reductions in mitochondrial encoded transcripts, and increases in nuclear encoded mitochondrial transcripts
Livers from the 250 mkd dosed non-engrafted TK-NOG and engrafted TK-NOG hu-liver mice were profiled to assess whether FIAU treatment results in any significant transcriptional responses (Fig. 5). Profiling was not assessed on kidney samples from this study. Similar to the mtDNA/nucDNA analyses, mouse and human-specific responses can be separated by species. In chimeric mice the average (log10 ratio) P53 driven gene signature transcriptional response was 0.35 (2.3-fold) for low and 0.39 (2.5-fold) for high engrafted mice for the human compartment. In the mouse compartment, the P53 transcriptional response was present but to a lesser degree [0.26 (1.8-fold), 0.19 (1.5-fold], and 0.15 (1.4-fold) seen for non-, low-, and high-engrafted animals, respectively. Consistent with FIAU treatment-related decreases in mtDNA/nucDNA, a clear reduction [−0.20 (0.63-fold), −0.20 (0.63-fold), and −0.15 (−0.71), log10 ratio] for non-, low-, and high-engrafted mice in the mouse compartment in mitochondrial encoded gene transcripts was seen. There was an unanticipated concurrent elevation in non-engrafted mice of nuclear encoded mitochondrial gene transcripts [0.15 (1.4-fold), log10 ratio] in livers following treatment with FIAU for 57 days but not with FIRU. For the livers from the 90 mkd dosed mice, the magnitude of the response for p53 and mitochondrially encoded transcripts was less than the response for the 250 mkd group, and responses for nuclear encoded transcripts for mitochondrial proteins were more variable (Fig. S6).
Fig. 5.

Individual P53-mediated transcriptional responses, responses of mitochondrial encoded transcripts, and responses of nuclear encoded transcripts of mitochondrial proteins in non, low, and high engrafted chimeric mice for cohort B (250 mkd FIAU, 250 mkd FIRU, or vehicle; dosed for 57 days in (a) mouse or (b) human hepatocytes (only engrafted mice).
In engrafted hu-liver mice treated with FIAU, the remnant mouse liver tissue maintained a robust P53 transcriptional response and marked reduction in mitochondrial encoded transcripts, while concurrent elevations of nuclear encoded transcripts for mitochondrial proteins were apparent but blunted compared to livers from non-engrafted mice. No decreases were seen, but interestingly, some increases in mitochondrial encoded transcripts were observed with FIRU treatment, especially in the human hepatocytes. Among the human hepatocytes from livers of engrafted mice, the elevated p53 transcriptional response and reduction in mitochondrial encoded transcripts were evident following FIAU but not FIRU treatment. Modest elevations of nuclear encoded transcripts for mitochondrial proteins were also evident following FIAU treatment in human hepatocytes.
FIAU did not result in ALT increases or histopathological changes in liver in a repeat (2nd) Hu-liver mouse study engrafted with a sensitized donor that was pre-screened in an in vitro hepatocyte model
To explore that the lack of phenotypic hepatotoxicity of the first FIAU study in hu-liver mouse was not limited by the human hepatocyte donor used in that study, we conducted a second study using hu-liver mice engrafted with hepatocytes from a different human donor pre-screened for sensitivity (Figs S7–S9) in an in vitro hepatocyte co-culture assay. The increased sensitivity seen in vitro with this second donor did not translate to ALT increases or liver histopathological changes (Supplement, Figs S10–S12). The results obtained in this hu-liver mouse study were generally similar to those obtained in the first study. Like in the first study, FIAU dosing was also not well-tolerated at the high dose (250 mkd). In fact, FIAU tolerability was even poorer in this study, especially for engrafted mice. Additionally, FIAU-dependent, and not FIRU-dependent, mtDNA/nucDNA decreases were again seen in liver (Fig. S13). There were no meaningful differences in exposures between the non-engrafted and the engrafted animals (Table S9). Nucleoside triphosphate liver levels at termination for FIAU were higher the lower the engraftment degree (e.g., Non>Low>High, Fig. S14). For FIRU, they were similar for non and low engrafted mice and lowest for high engrafted mice (Fig. S14).
FIAU, but not FIRU, induces mtDNA/nucDNA decreases and transcriptional changes in the TK-NOG (non-engrafted) and CD-1 mice in a 15-day study
Given that the non-engrafted TK-NOG mice showed FIAU treatment related mtDNA/nucDNA decreases in the initial hu-liver TK-NOG donor study, we conducted a 2-week study to compare the effects of FIAU and FIRU treatments in wildtype (CD-1) mice and non-engrafted TK-NOG mice to assess whether CD-1 mice would show similar mtDNA/nucDNA decreases in response to FIAU treatment, even in the absence of the herpes simplex virus (HSV) thymidine kinase (TK) present in the TK-NOG mice.
For TK-NOG (non-engrafted) mice orally administered 250 mkd FIAU, elevations were again observed in ALT and other clinical chemistry parameters, but not TK-NOG mice given FIRU. The percent difference in mean values from the respective controls were: +261% AST, +630% ALT (Fig. 6), and +250% total bilirubin. In contrast, there were no changes in serum chemistry parameters in the CD-1 mice administered 250 mkd FIAU, or 250 mkd FIRU.
Fig. 6.
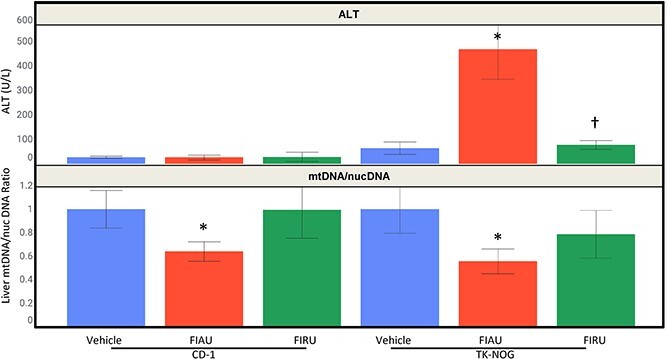
Top trellis: ALT responses (average ± standard deviation) for vehicle control (blue), 250 mkd FIAU (red), and 250 mkd FIRU (green) in CD-1 and non-engrafted TK-NOG mice. Bottom trellis: Liver mtDNA/nucDNA (average ± standard deviation) for vehicle control (blue), 250 mkd FIAU (red), and 250 mkd FIRU (green) in CD-1 and non-engrafted TK-NOG mice for 2 weeks. A dashed line is depicted at a ratio of 0.5. When P < 0.05 for a treated group compared to the respective vehicle control, an asterisk (*) was added above it. When P < 0.05 for a FIRU group compared to the respective FIAU group, a dagger (†) was added above it.
Although FIAU treatment-related ALT increases were only seen in TK-NOG mice, and not in CD-1 mice, liver mtDNA/nucDNA mean ratios decreased to similar levels in both TK-NOG (0.56 or 44% decrease) and CD-1 (0.64 or 36% decrease) mice treated with 250 mkd FIAU (Fig. 6). In contrast, these decreases were not seen in either strain treated with FIRU (250 mkd).
For this study, the average body-weight loss in TK-NOG mice (all non-engrafted) orally administered 250 mkd FIAU was −2.2 g compared to −1.3 g loss in TK-NOG mice receiving vehicle. In addition, decreased body weight gain of −65% compared to controls was observed in the CD-1 mice orally administered 250 mkd FIAU. There were no changes in body weight in mice administered 250 mkd FIRU in either strain.There were no meaningful differences in exposures between the non-engrafted and the engrafted animals (Table S11).
Histopathologically, there were no treatment-related liver findings in any of the CD-1 mice dose groups. For TK-NOG mice (all non-engrafted) orally administered 250 mkd FIAU, hepatocellular hypertrophy, single cell necrosis, and bile duct hyperplasia were observed (see Table S10). The livers from non-engrafted TK-NOG mice treated with 250 mkd FIRU were considered not remarkable.
Higher parent drug and triphosphate anabolite levels seen in liver for both FIAU and FIRU in the TK-NOG strain
As shown in Table S16, the FIAU and the FIRU exposures in the TK-NOG (non-engrafted) mice compared to the CD-1 mice were within 2-fold, which is not considered a meaningful difference.
Liver levels of mono (NMP) and tri-phosphorylated nucleotides (NTP) for both FIAU or FIRU were assessed at both 4- and 24-h post-dose. However, only the 4 h post-dose time point is plotted in Fig. 7, as levels at the 24-h post dose time point were undetectable (data not shown). Compared to non-engrafted TK-NOG mice, CD-1 mice generated only negligible levels of NMP and NTP for both FIAU and FIRU, and the level of the respective parent molecules were also very low. This contrasts with the relatively high phosphorylated liver levels of both FIAU and FIRU in non-engrafted TK-NOG mice observed at the same time point. This likely results from the overexpression of the TK transgene,23,24 which is an enzyme capable of mono phosphorylating FIAU and FIRU.25,26 Levels of parent FIAU and FIRU nucleosides in non-engrafted TK-NOG mice were also considerably higher than for CD-1 mice (Fig. 7). The method used for liver sampling (flash frozen in liquid nitrogen shortly after animal sacrifice rather than in situ freeze clamping of liver prior to sacrifice) may have resulted in partial dephosphorylation of phosphorylated nucleotide.
Fig. 7.

Liver levels of nucleotide monophosphate (e.g. FIAU-MP or FIRU-MP; nmol/g, average ± standard deviation; left trellis), nucleotide triphosphate (e.g. FIAU-TP or FIRU-TP; nmol/g, average ± standard deviation; middle trellis), and parent nucleosides (e.g. FIAU or FIRU; μM, average ± standard deviation; right trellis) in CD-1 and non-engrafted TK-NOG mice dosed with 250 mkd FIAU (red) or 250 mkd FIRU(green). All levels assessed 4 h post-dose after 13 days of daily dosing.
Livers from CD-1 and TK-NOG non-engrafted mice treated with 250 mkd FIAU were also transcriptionally profiled. The average p53 response (log10 ratio) was elevated (0.14, 1.4-fold) in TK-NOG mice but not in CD-1 (−0.01) mice (Fig. 8). Consistent with FIAU treatment-related decreases in mtDNA/nucDNA observed in both strains, a similar reduction in the average mitochondrial encoded gene transcripts response (log10 ratio) was also seen for CD-1 (−0.13, 0.74-fold) and for TK-NOG mice (−0.12, 0.76-fold). No increases in nuclear encoded mitochondrial gene transcripts are apparent following FIAU treatment of either strain.
Fig. 8.
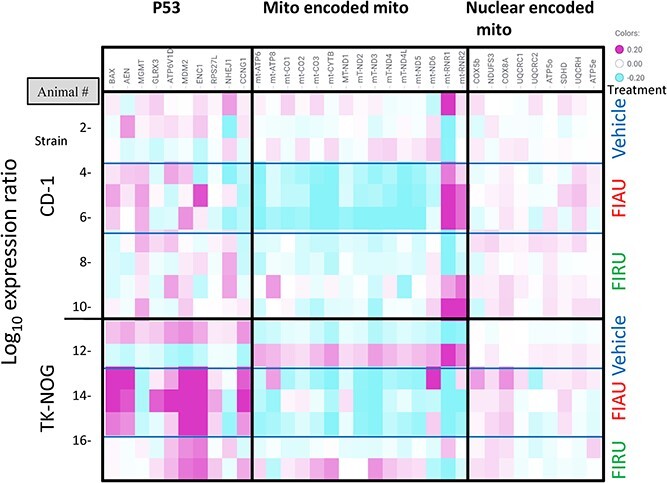
Individual P53, mitochondrially encoded and nuclear encoded mitochondrial transcriptional responses in CD-1 and non-engrafted TK-NOG mice (250 mkd FIAU, 250 mkd FIRU, or vehicle treated for 14 days).
Table 2 is a high-level summary of responses for in vivo mouse studies focused on clinical signs, ALT, liver histomorphology, liver mtDNA/nucDNA, and RNA profiling.
Table 2.
Summary of clinical signs, ALT, liver histomorphology, liver mtDNA/NucDNA and RNA profiling from in vivo mouse studies.
| Endpoint | 1st TK-NOG hu-liver mouse | 2nd TK-NOG hu-liver mouse | NE TK-NOG and CD-1 mouse | ||
|---|---|---|---|---|---|
| 60 days of dosing/ 61 total (Cohort A/ 90 mkd: 60 days) | 60 days/ 61 total (Cohort B/ 250 mkd: 57 days) | 60 days (250 mkd: 32 days) | 15 days (250 mkd: 15 days) | ||
| Clinical signs | Decreased activity (FIAU) | Decreased activity, thin appearance, decreased skin turgor, and pale appearance (FIAU) | Hunched posture & unkempt appearance in LE & HE FIAU groups. Incidence greatest in HE FIAU group, and increased with dosing. | None | |
| ALT (% increase) | +189% NE FIAU; +42% LE FIAU; other groups not changed | +549% (NE FIAU); +120% (NE FIRU) | Although a + 100% was seen for NE FIAU on day 20, a sustained increase seen for any group | +630% ALT (250 mkd FIAU NE TK-NOG) | |
| Liver Histo-morphology | Mouse remnant | FIAU-related minimal to mild hepatocellular hypertrophy & minimal single cell necrosis observed in both NE & LE & HE mice. Occasional necrotic hepatocyte seen predominantly in NE mice. | No findings, except background findingsa | Moderate hypertrophy, mild bile duct hyperplasia, & minimal to mild single cell necrosis of hepatocytes observed in all livers of NE TK-NOG mice dosed with 250 mkd FIAU | |
| Human | No findings, except backgrounda | No findings, except backgrounda | Not applicable | ||
| mtDNA /nucDNA (Average ratio) | Mouse Liver | 0.46 (LE FIAU); 0.58 (HE FIAU) | 0.26 (NE FIAU); 0.23 (LE FIAU); 0.18 (HE FIAU) | 0.51 (NE FIAU); 0.34 (LE FIAU); 0.50 (HE FIAU) | CD-1: 0.64 (FIAU) TK-NOG: 0.57 (FIAU) |
| Human Liver | 0.18 (LE FIAU); 0.28 (HE FIAU) | 0.11 for both LE and HE (FIAU) | 0.41 (LE FIAU); 0.55 (HE FIAU) | Not applicable | |
| Mouse Kidney | Not assessed | 0.48 NE FIAU; 0.31 (LE FIAU); 0.31 (HE FIAU) | Not assessed | Not assessed | |
| Liver Profiling (Range average Log10)b | Mouse Liver | p53: 0.14 (NE FIAU); 0.20 (LE FIAU); 0.19 (LE FIAU) Mito encoded: −0.11 (LE FIAU); −0.24 (HE FIAU); −0.14 (NE FIRU) -0.10 (HE FIRU) |
p53: 0.26 (NE FIAU); 0.19 (LE FIAU); 0.15 (HE FIAU) Mito encoded: −0.20 (NE FIAU); −0.20 (LE FIAU); −0.15 (HE FIAU) Nuclear encoded mito: 0.15 (NE FIAU); 0.15 (LE FIAU); 0.10 (HE FIAU) |
Not applicable | p53: 0.14 (FIAU) Mito encoded: −0.13 (FIAU) |
| Human Liver | p53: 0.13 (LE FIAU) Mito encoded: −0.19 (HE FIAU) |
p53: 0.35 (LE FIAU); 0.39 (HE FIAU) Mito encoded: −0.30 (LE FIAU); −0.21 (HE FIAU) |
Not applicable | ||
No effect if not mentioned.
Abbreviations: Non-engrafted (NE); Low-engrafted (LE); High engrafted (HE)
aModel-related background histomorphologic findings were present in human hepatocytes (rarefaction and vacuolation), and in the remaining mouse tissue, single cell necrosis of hepatocytes, bile duct ectasia and bile ductile hyperplasia were observed.
bNo effect considered if average Log10 value was between −0.1 and 0.1.
FIAU-TP or FIRU-TP incorporation does not interfere with qPCR for the mtDNA/nucDNA
Given the significant reduction in mtDNA/nucDNA ratio observed in the various studies, we initiated an effort to confirm that FIAU incorporation into mtDNA does not impact the mtDNA/nucDNA ratio assay, and that any reductions observed in mtDNA/nucDNA were real and not due to an artifact. As part of this effort, PCR was conducted using a proofreading deficient polymerase to amplify the NADH Dehydrogenase 6 gene (957 bases, from mitochondria, see Table S5 for primers) to ensure that the polymerase would amplify the DNA regardless of the potential incorporation of whether FIAU or FIRU were incorporated. Varying concentrations of FIAU-TP (100–175 μM, see Table S6) or FIRU-TP (100–188 μM, see Table S6) were added to the PCR reaction as the dT-TP (0.02–2 mM, see Table S6) concentrations were also varied. dT-TP concentrations were adjusted because FIAU-TP and FIRU-TP structures are similar to the dT-TP structure. A standard concentration (10 mM) was used for the other nucleotide-TP. The PCR product was run on a 1.2% agarose gel to confirm amplification and assayed with PicoGreen to determine the concentration of the PCR products. In addition, qPCR (with a proofreading deficient polymerase) was used to quantify the 957 bp product by targeting an 89 bp sequence within the 957 bp product (see Table S5 for primer, probes) to look at whether the PCR products from the different reactions could be amplified and if there was any interference from integrated FIAU-TP or FIRU-TP on the reaction. This was compared against the PicoGreen signal to determine whether FIAU-TP or FIRU-TP incorporation impacted the PCR reaction. We also demonstrated with cell culture studies in HepG2 cells with control, FIAU or FIRU treatment for 1 week that the Anti BrdU antibody binds only to the wells of cells treated with FIAU or FIRU (data not shown). BrdU is structurally similar to both FIAU and FIRU, and this supports the use of the antibody to probe for incorporation into DNA. As can be seen in and Fig. 9 and Table S12, an increase in FIAU-TP or FIRU-TP while decreasing the dT-TP in the reaction mixture decreased the production of transcript. The FIAU-TP loss of transcript was more pronounced. Additionally, the Anti BrdU antibody reacted with the product, supporting FIAU-TP and FIRU-TP incorporation; although, we do not know the differential reactivity between the two compounds. Both the DNA concentrations by qPCR or PicoGreen were comparable, and a ratio of the qPCR product to the PicoGreen reaction was ~1.0, supporting the conclusion that FIAU-TP or FIRU-TP incorporation does not interfere with qPCR for the mtDNA/nucDNA.
Fig. 9.

a) Effect of decreasing dT-TP(teal) while increasing either FIAU-TP (red) or FIRU-TP (bright green) in the PCR reaction mixture. FIAU-TP (red) has more effect than FIRU-TP (bright green) on qPCR for amplifying 957 bp human mitochondrial DNA. b) BrdU antibody cross-reactivity with DNA supports both FIAU (red) and FIRU (bright green) incorporation, but the FIAU incorporates at lower concentration ratios. Notes: dTP = deoxytriphosphate PCR mix; bp = base pairs; OD = optical density.
Discussion
We evaluated two hepatocyte donors for FIAU liver toxicity in hu-liver mice using the TK-NOG mouse strain, one randomly chosen and the other purposely selected based on in vitro sensitivity to FIAU toxicity following screening of 6 additional donor lots in human HepatoPac® co-cultures. The timing of the clinical signs of toxicity, the lack of ALT changes, and absence of histopathologic alterations observed in these hu-liver mice studies are in contrast to the results previously reported by Xu and colleagues15,16 where hu-liver mice dosed with FIAU showed signs of acute liver toxicity (mortality and/or morbidity) in a dose dependent manner after 4–14 days of treatment with histopathologic and ultrastructural alterations that reflected results reported from the FIAU clinical trial. This rapid manifestation of liver injury is also in contrast to the timing (≥9 weeks) of this toxicity observed in the clinical trial.2 The timing and response of biomarkers and tolerability for our study are more consistent with the delay in the clinical study findings and the literature on FIAU incorporation into mtDNA, which would be expected to take time to manifest as a toxicity. However, the lack of serum clinical chemistry changes and lack of histopathologic changes in chimeric mice with human hepatocytes after 2 months of dosing in our studies is puzzling.
There are several variables between our studies and the study conducted by Xu and colleagues.15,16 One such variable is that the human hepatocyte donors we used are different than those used by Xu and colleagues. Moreover, Xu et al. reported 2 donors having similar hepatotoxicity.15,16 In addition, the 50% incidence among 15 subjects in the clinical trial with FIAU was not anticipated to present difficulty with reproducibility among individual donors.2 Nevertheless, in an attempt to minimize the potential impact of this variable, we screened 7 human hepatocyte donors (including the donor from the 1st study, Donor 2) using the HepatoPac® system. One of the donors (Donor 4) was identified as having increased sensitivity to FIAU, as assessed by earlier and more pronounced reduction of urea synthesis for FIAU, more prominent drop in the mtDNA/nucDNA ratio for both FIAU and FMAU, and a more pronounced P53 gene signature response for FIAU compared to other donors. We then used this donor to engraft the mice for the second TK-NOG hu-liver mouse study. Nonetheless, the increased sensitivity of this donor was not sufficient to recapitulate the acute liver toxicity previously reported for this model from the Xu, et al. 2014 report.15,16 Other potential variables are summarized in Table 3. Note that while we assessed plasma and liver drug exposures on our studies, this was not reported for the Xu, et al. 2014 report,15,16 and thus a direct comparison cannot be established.
Table 3.
Variables between our studies and the Xu et al. 2014 study.
| Parameter | 1st Study (Donor 2) | 2nd Study (Donor 4) | Xu & colleagues’ study [14, 15] | ||
|---|---|---|---|---|---|
| Doses & Duration | 90 mpk: 60 days | 250 mpk: 57 days | 250 mkd: 32 days (planned for 60 days) | 4 days: 400 mpk | 14–17 days: 2.5, 25, and 100 mpk |
| Human Albumin for Highly Engrafted Mice, U/L (median) | 6.2–9.9 (9.7)a | 3.5–9.5 (6.6)a | 5.5–11.1 (8.1)b | 6.4–18.2 (11.6) | |
| Change in Human Albumin for Highly Engrafted Mice, U/L (median) | Not observed from SD6 to end of study | Not observed from SD6 to end of study | 31% decrease for vehicle control | Not mentioned | |
| Baseline ALT for Highly Engrafted Mice, U/L (median) | 330–355 (335)a | 465–540 (470)a | 313–522 (415)a 225–1,062 (509)b |
SD0-14: 80–152 | |
| Exposure, AUC 0–24 h (μM*hr) for high engrafted mice | 92.2 | 506 | 255 | Not assessed | |
| Vitamin C in Feed | - 1,500 PPM of Vitamin C supplemented PMI irradiated rodent diet 5,002 - Cohort B (250 mpk) switched on SD 29 to rodent autoclavable diet 5,010 with no Vitamin C |
No Vitamin C in rodent autoclavable diet 5,010 | Not mentioned | ||
| Donors | 5 YO Male Caucasian | 30 YO African American | 3 YO Female, 2 YO Female (no ethnicity provided) | ||
| Dosing Prior to Engraftment Procedure | 4–10 mpk Ganciclovir (ip), one or two doses | Val-ganciclovir in drinking water for 3 days | 25 mpk Ganciclovir (ip) | ||
Abbreviations: SD = Study Day; mpk = mg/kg; ppm = parts per million; ip = Intraperitoneal; YO = year old.
Comments:
aSampled on SD 6. Reflects control values only (n = 3 for first study and n = 7 for second MSD study) Due to a company-wide cybersecurity/data loss incident in 2017, we do not have a pretest of hu-albumin and ALT for the 1st hu-liver mouse study, and we used results from the study day 6 collection.
bSampled ~2–3 weeks prior to the study.
While we did not observe FIAU-induced acute liver toxicity, our assessments did identify important FIAU-treatment related effects that were independent of TK transgene effects, including decreases in mtDNA/nucDNA levels in liver and kidney, and certain specific and biologically relevant changes in RNA expression patterns, especially a consistently marked reduction in transcripts for mitochondrial encoded proteins. The FIAU-induced mtDNA/nucDNA decreases and reductions in mitochondrial encoded transcripts in liver were observed not only in the human hepatocytes in the TK-NOG hu-liver animals, but also in non-engrafted TK-NOG mice and in CD-1 mice and were not observed following FIRU treatment in any of these models. This finding suggests that monitoring mtDNA/nucDNA levels and mitochondrial encoded transcripts in wild-type rodents may be useful as biomarkers to screen for molecules with FIAU-like mechanistic mitochondrial toxicities that could be clinically relevant. Interestingly, the magnitude of mtDNA/nucDNA changes in the initial study did not translate to apparent changes in mitochondria quality or numbers in FIAU treated TK-NOG mice as assessed by electron microscopy. Changes in ultrastructure for liver mitochondria have been reported with FIAU treatment in severe clinical cases, woodchucks, and humanized-liver mice including lipid droplets, and mitochondria with an abnormal size, shape, and cristae loss.6,14,15 In addition, the less severe phenotype of enlarged mitochondria was observed in rats treated with FIAU.2 Given that the mtDNA/nucDNA assessments are PCR based, we conducted additional evaluations to ensure that our PCR assay would work when FIAU or FIRU is incorporated into DNA. To that end, we incorporated FIAU or FIRU into DNA using their TP anabolites and a proofreading deficient PCR enzyme and then verified their incorporation into DNA using a BrdU antibody that recognizes these incorporated nucleosides. We then amplified a smaller fragment using a conventional qPCR enzyme and compared that response to PicoGreen detection. Although addition of FIAU-TP or FIRU-TP decreased DNA amplification for the initial reaction, the levels assessed using PicoGreen and qPCR were comparable, supporting the suitability of the qPCR-based assay to assess mt/nucDNA levels.
Generally, the active form of nucleoside-based drugs is the TP anabolite. To assess whether the FIAU treatment-related effects observed in our mouse studies correlated with the level of FIAU-TP, we measured both the nucleotide mono and triphosphate anabolites for FIAU and FIRU in the TK-NOG (non-engrafted) and CD-1 mice study. The much higher levels observed in the TK-NOG (non-engrafted) mice as compared to the levels seen in the CD-1 mice are most likely explained by the presence of the HSV TK transgene in TK-NOG mice. In fact, FIAU has been shown to be a substrate for HSV-TK,23 which has cytoplasmic and nuclear localization,27,28 as well as human TK1 (cytoplasmic localization) and TK2 (mitochondrial localization),24 which would add the first phosphate group to FIAU. This rate limiting step of adding the initial phosphate group, which for HSV TK would occur in the cytoplasm, can drive subsequent production of the tri-phosphorylated nucleotide. The mtDNA/nucDNA ratios of TK-NOG and CD-1 mice were similar, perhaps because FIAU-TP is trapped in the cytoplasm of TK-NOG mice and inaccessible to mitochondria. FIAU levels, however, appear to remain equivalent and similarly accessible to the mitochondrial compartment of both TK-NOG and CD-1 mice. Compartmental trapping of phosphorylated nucleotide analogs has been reported to play a role in toxicity.29 In addition, a recent study30 performed using hepatocyte spheroids showed knockdown of mitochondrial TK-2 was protective of the FIAU-related mitochondria toxicity, further supporting that formation of FIAU-TP in mitochondria is an imperative trigger for mitochondrial toxicity.
Mitochondrial toxicity due to an impact on mtDNA/nucDNA has a cumulative effect on mitobiogenesis and may not manifest until an excessive mitochondrial capacity is sufficiently impaired. In addition, cellular or tissue biochemical toxicity is delayed from mtDNA reduction due to propagation to mitochondrial proteins and cellular biochemistry. It is unclear how much mtDNA depletion is needed for toxicity to manifest. It is possible that there may be species differences between humans and rodents in terms of responses in these parameters that make rodents less sensitive.
While there is evidence that suggests mitochondrially targeted ENT1 transports FIAU more specifically into human mitochondria10,11 uptake for FIAU into mitochondria from other species is not well understood. One candidate gene besides ENT1 is ENT3 (SLC29A3), which has been localized to mitochondria.31 Our studies support that there is uptake in the mitochondria of mice as evidenced by the decline in mtDNA/nucDNA levels in both CD-1 and non-engrafted TK-NOG mice, as well as in hu-liver mice. We have also seen declines in mtDNA/nucDNA levels in rats (manuscript in preparation), and this may be a mechanism of uptake for the woodchuck. Besides uptake, which can impact accumulation, other aspects such as capacity, energy metabolism, and DNA damage response may impact species and tissue sensitivity.
Our hypothesis is that the expression of the TK transgene drives cytoplasmic FIAU-triphosphate production in cells containing this transgene (e.g. nonengrafted liver remnant). Given the higher degree of p53 damage and ALT excursion after 15 days of dosing in non-engrafted TK-NOG mice, we suggest the cytoplasmic transgene leads to generation and trap of FIAU phosphorylated anabolites in cytoplasm. On the other hand, FIAU-triphosphate is expected to be generated in mitochondria of cells irrespective of the TK transgene. This is supported by the fact that the mtDNA/nucDNA levels were similar in both non-engrafted TK-NOG and CD-1 mice.
RNA expression profile analyses performed on the initial hu-liver mouse study (~2 months), the 2-week non-engrafted TK-NOG and CD-1 mouse study, and the HepatoPac® donor study revealed decreases in mitochondrially-encoded transcripts that paralleled the mtDNA/nucDNA decreases. Interestingly, transcripts for nuclear-encoded mitochondrial proteins were increased for the high dose for the initial hu-liver mouse study and for the HepatoPac® donor study, as were transcription factors that regulate mitochondrial biogenesis, such as nuclear respiratory factor 1 and 2 which upregulate mitochondrial and nuclear encoded transcripts for mitochondrial proteins.32,33 The lack of an apparent decrease in mitochondria quality as concluded from resistance to hepatotoxicity, or reduction in numbers of mitochondria in FIAU treated TK-NOG hu-liver mice as assessed by electron microscopy may partly be due to the upregulation of nuclear mitochondrial transcripts or to a threshold effect for mtDNA decline which was not surpassed in the animals on this study. The increase in transcripts for nuclear encoded mitochondrial proteins was more variable in the low dose (90 mkd) for the initial hu-liver mouse studies, suggesting that group had a delay in this adaptive response. In addition, the decreases in mitochondrial encoded transcripts and the increases in transcripts for nuclear encoded mitochondrial proteins were dose-responsive in the in vitro HepatoPac® model and showed differences between donors.
RNA expression profiling analyses also revealed that FIAU induced a p53 DNA damage gene signature response in all the studies (in vitro and in vivo) that were profiled. Induction of p53 responsive genes is known to occur by nuclear DNA damage by genotoxicants, which in turn activates DNA repair, and in cases of irreparable DNA damage, by executing programmed cell death.34 In addition to the nuclear p53 response, recent data suggest the p53 can localize and elicit a response in mitochondria and may affect the accuracy of mtDNA synthesis by acting as a proofreader35–38 and that p53 plays a role in maintenance of mtDNA abundance.39 It is likely that both nuclear and mitochondrial DNA damage arms of the p53 response were seen in these studies. One interesting finding was that the p53 gene signature response for the 2-week non-engrafted TK-NOG and CD-1 mouse study was more pronounced for the TK-NOG than for the CD-1 mouse strain. This suggests the HSV TK transgene had an effect, perhaps increasing FIAU-TP in the cellular, but not mitochondrial compartment. This in turn would be expected to increase levels of the active FIAU-TP incorporated into nuclear DNA, which is consistent with the specific injury in TK-NOG mice, whereas both the mtDNA decline and also the decline in mitochondrial encoded transcripts were similar. Perhaps the mitochondrial p53 response would have been triggered later in time for the CD-1 mice. Similar to the TK-NOG and CD-1 mouse study, for the hu-liver study where profiling was done, we saw the mouse p53 response was greatest for nonengrafted mice followed by low engrafted mice which have a higher mouse remnant liver compared to high engrafted mice. Alternatively, since p53 is a stress-responsive gene, the lack of P53 activation in FIAU-treated.
CD-1 mice, which did not develop liver injury, could be interpreted as evidence that the p53 activation is indeed secondary to injury. For the human donors evaluated in HepatoPac® in vitro, the most sensitive donor (Donor 4) had the highest p53 response as well as the lowest mitochondrially encoded protein transcript levels; and the inverse was seen for donor 6, which had low sensitivity, potentially suggesting p53 response effects on mitochondria in vitro.
In conclusion, we had initially attempted to recapitulate a report of acute FIAU liver injury in hu-liver mice15,16 using conventional endpoints (ALT and liver degeneration/necrosis) as we sought to further qualify the model for improved assessment of liver safety of novel antiviral nucleosides and define additional translational molecular biomarkers useful for both in vitro/in vivo model systems. Even though the liver injury phenotype results were not recapitulated, we did identify additional mechanistic biomarkers in mouse tissues and in an in vitro liver model (HepatoPac®), including changes in mtDNA/nucDNA and transcriptional changes especially reductions in mitochondrial encoded transcripts, that can be used to assess FIAU-like sensitivity. These biomarkers might even be applicable to tissues beyond liver and kidney that may not reach thresholds sufficient for achieving a fulminant organ toxicity, and may apply to species beyond mice. Additionally, the timing and response for our study is more consistent with the clinical study and literature on FIAU incorporation into mtDNA, which would be expected to take time to manifest as a toxicity. We also showed that these biomarkers can differentiate in both in vitro and in vivo studies between toxic (FIAU) and non-toxic (FIRU) nucleosides only differing by the stereospecificity of 1 atom (2’fluoro ara or ribo, respectively).
The mtDNA/nucDNA biomarker is not “human specific.” Nevertheless, human cells are shown by our data to be consistently more responsive to the same concentrations of FIAU administered to rodent cells (manuscript in preparation). Taken together the data suggest that both human and rodent livers have excess mitochondrial functional capacity, but human hepatocytes are more sensitive to FIAU, which is supported by the reported clinical toxicity of FIAU.2 The mtDNA/nucDNA ratio responds quantitively to changes across multiple in vitro and in vivostudies in both human and animal models.1,8,13 Further, this mechanistic biomarker is concluded to be logically indicative of biochemical adversity, as it shows specificity for nucleosides that have adverse reports (manuscript in preparation). Nevertheless, tying responses to a quantitative clinical correlate such as clinical chemistry or histopathology still needs more consideration of the downstream impact of mtDNA on mitochondrial protein production, and ultimately cellular function. Additionally, a universal biomarker to link mtDNA/nucDNA decreases is complicated since mitochondrial effects from nucleosides occur across different tissue/organs with different mitochondrial capacities, available bioenergetic substrates, and accumulation patterns, and mitochondrial injury manifests in different ways. The molecular biomarkers identified in the study should be further evaluated as mechanistic tools for evaluating drugs for exposure relevant FIAU-like mitochondrial toxicity potential.
Author contributions
Amy G. Aslamkhan (Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Supervision, Visualization, Writing—Original Draft, Writing—Review & Editing), Laura Michna (Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Writing—Original Draft), Alexei Podtelezhnikov (Formal Analysis, Investigation, Methodology, Writing—Original Draft), Katerina Vlasakova (Formal Analysis, Investigation, Methodology, Writing—Original Draft, Writing—Review & Editing), Hiroshi Suemizu, Yasuyuki Ohnishi (Methodology, Investigation, Project Administration), Liping Liu (Data Curation, Formal Analysis, Investigation, Methodology, Writing—Original Draft, Writing—Review & Editing), Pamela Lane, Matthew C. Kuhls, Zhibin Wang, Stephen Pacchione, Zoltan Erdos, Rodger William Tracy, Kenneth Koeplinger, Nagaraja Muniappa, John Valentine (Formal Analysis, Investigation, Methodology, Writing—Original Draft), Qiuwei Xu, Alema Galijatovic-Idrizbegovic (Conceptualization, Writing—Review & Editing), Warren E Glaab (Conceptualization, Formal Analysis, Investigation, Methodology, Supervision), Frank D. Sistare (Conceptualization, Investigation, Methodology, Project Administration, Supervision, Writing—Original Draft, Writing—Review & Editing), Jose Lebron (Conceptualization, Investigation, Methodology, Project Administration, Supervision, Writing—Original Draft, Writing—Review & Editing).
Conflict of interest statement: None declared.
Supplementary Material
Contributor Information
Amy G Aslamkhan, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Laura Michna, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Alexei Podtelezhnikov, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Katerina Vlasakova, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Hiroshi Suemizu, Laboratory Animal Research, Central Institute for Experimental Animals, 210-0821 Kawasaki-ku, Kawasaki 3-25-12 Tonomachi, Japan.
Yasuyuki Ohnishi, Laboratory Animal Research, Central Institute for Experimental Animals, 210-0821 Kawasaki-ku, Kawasaki 3-25-12 Tonomachi, Japan.
Liping Liu, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Pamela Lane, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Qiuwei Xu, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Matthew C Kuhls, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Zhibin Wang, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Stephen Pacchione, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Zoltan Erdos, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Rodger William Tracy, Pharmacokinetics, Dynamics, Metabolism and Bioanalytics, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA, United States.
Kenneth Koeplinger, Pharmacokinetics, Dynamics, Metabolism and Bioanalytics, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA, United States.
Nagaraja Muniappa, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
John Valentine, Pharmacokinetics, Dynamics, Metabolism and Bioanalytics, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA, United States.
Alema Galijatovic-Idrizbegovic, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Warren E Glaab, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Frank D Sistare, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
Jose Lebron, Nonclinical Drug Safety, Merck & Co., Inc., 770 Sumneytown Pike, West Point, PA 19486, United States.
References
- 1. Richardson FC, Engelhardt JA, Bowsher RR. Fialuridine accumulates in DNA of dogs, monkeys, and rats following long-term oral administration. Proc Natl Acad Sci U S A. 1994:91(25):12003–12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manning FJ, Swartz S. Committee to review the fialuridine (FIAU/FIAC) clinical trials (Institute of Medicine): review of the fialuridine (FIAU) clinical trials. Washington, D.C.: National Academies Press; 1995. p. 280. [PubMed] [Google Scholar]
- 3. Straus SE. Unanticipated risk in clinical research. In: Gallin J, Ognibene FP, Johnson LL, editors. Principles and practice of clinical research. 4th ed. Academic Press, London, United Kingdom; 2018. pp. 141–159. [Google Scholar]
- 4. McKenzie R, Fried MW, Sallie R, Conjeevaram H, di Bisceglie AM, Park Y, Savarese B, Kleiner D, Tsokos M, Luciano C, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med. 1995:333(17):1099–1105. [DOI] [PubMed] [Google Scholar]
- 5. Marwick C. NIH panel report of `no flaws' in FIAU trial at variance with FDA report, new probe planned. JAMA. 1994:272(1):9–11. [PubMed] [Google Scholar]
- 6. Kleiner DE, Gaffey MJ, Sallie R, Tsokos M, Nichols L, McKenzie R, Straus SE, Hoofnagle JH. Histopathologic changes associated with fialuridine hepatotoxicity. Mod Pathol. 1997:10:192–199. [PubMed] [Google Scholar]
- 7. Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001:276(44):40847–40857. [DOI] [PubMed] [Google Scholar]
- 8. Lewis W, Levine ES, Griniuviene B, Tankersley KO, Colacino JM, Sommadossi JP, Watanabe KA, Perrino FW. Fialuridine and its metabolites inhibit DNA polymerase gamma at sites of multiple adjacent analog incorporation, decrease mtDNA abundance, and cause mitochondrial structural defects in cultured hepatoblasts. Proc Natl Acad Sci U S A. 1996:93(8):3592–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui L, Yoon S, Schinazi RF, Sommadossi JP. Cellular and molecular events leading to mitochondrial toxicity of 1-(2-deoxy-2-fluoro-1-beta-D-arabinofuranosyl)-5-iodouracil in human liver cells. J Clin Invest. 1995:95(2):555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee EW, Lai Y, Zhang H, Unadkat JD. Identification of the mitochondrial targeting signal of the human equilibrative nucleoside transporter 1 (hENT1): implications for interspecies differences in mitochondrial toxicity of fialuridine. J Biol Chem. 2006:281(24):16700–16706. [DOI] [PubMed] [Google Scholar]
- 11. Lai Y, Tse CM, Unadkat JD. Mitochondrial expression of the human equilibrative nucleoside transporter 1 (hENT1) results in enhanced mitochondrial toxicity of antiviral drugs. J Biol Chem. 2004:279(6):4490–4497. [DOI] [PubMed] [Google Scholar]
- 12. Sykes C, van Horne B, Jones J, Kashuba ADM, Gatto G, van der Straten A, Johnson L, Cottrell ML. Intracellular islatravir pharmacology differs between species in an in vitro model: implications for preclinical study design. J Antimicrob Chemother. 2022:77(4):1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tennant BC, Baldwin BH, Graham LA, Ascenzi MA, Hornbuckle WE, Rowland PH, Tochkov IA, Yeager AE, Erb HN, Colacino JM, et al. Antiviral activity and toxicity of fialuridine in the woodchuck model of hepatitis B virus infection. Hepatology. 1998:28(1):179–191. [DOI] [PubMed] [Google Scholar]
- 14. Lewis W, Griniuviene B, Tankersley KO, Levine ES, Montione R, Engelman L, de Courten-Myers G, Ascenzi MA, Hornbuckle WE, Gerin JL, et al. Depletion of mitochondrial DNA, destruction of mitochondria, and accumulation of lipid droplets result from fialuridine treatment in woodchucks (Marmota monax). Lab Investig. 1997:76(1):77–87. [PubMed] [Google Scholar]
- 15. Xu D, Nishimura T, Nishimura S, Zhang H, Zheng M, Guo YY, Masek M, Michie SA, Glenn J, Peltz G. Fialuridine induces acute liver failure in chimeric TK-NOG mice: a model for detecting hepatic drug toxicity prior to human testing. PLoS Med. 2014:11(4):e1001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen J. Toxicology. 'Humanized' mouse detects deadly drug side effects. Science. 2014:344(6181):244–245. [DOI] [PubMed] [Google Scholar]
- 17. Linster CL, van Schaftingen E, Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007:274(1):1–22. [DOI] [PubMed] [Google Scholar]
- 18. Auerbach SS. In vivo signatures of genotoxic and non-genotoxic chemicals. In: Waters MD, Thomas RS, editors. Toxicogenomics in predictive carcinogenicity. Cambridge: Royal Society of Chemistry; 2016. pp. 113–153. [Google Scholar]
- 19. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017:36(28):3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Podtelezhnikov AA, Monroe JJ, Aslamkhan AG, Pearson K, Qin C, Tamburino AM, Tanis KQ. Quantitative transcriptional biomarkers of xenobiotic receptor activation in rat liver for the early assessment of drug safety liabilities. Toxicol Sci. 2020:175(1):98–112. [DOI] [PubMed] [Google Scholar]
- 21. Isaac RS, McShane E, Churchman LS. The multiple levels of mitonuclear coregulation. Annu Rev Genet. 2018:52(1):511–533. [DOI] [PubMed] [Google Scholar]
- 22. Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, Ito M, Suematsu M, Peltz G, Nakamura M, et al. The reconstituted 'humanized liver' in TK-NOG mice is mature and functional. Biochem Biophys Res Commun. 2011:405(3):405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tjuvajev JG, Finn R, Watanabe K, Joshi R, Oku T, Kennedy J, Beattie B, Koutcher J, Larson S, Blasberg RG. Noninvasive imaging of herpes virus thymidine kinase gene transfer and expression: a potential method for monitoring clinical gene therapy. Cancer Res. 1996:56(18):4087–4095. [PubMed] [Google Scholar]
- 24. Wang J, Eriksson S. Phosphorylation of the anti-hepatitis B nucleoside analog 1-(2′-deoxy-2′-fluoro-1-beta-D-arabinofuranosyl)-5-iodouracil (FIAU) by human cytosolic and mitochondrial thymidine kinase and implications for cytotoxicity. Antimicrob Agents Chemother. 1996:40(6):1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morin KW, Duan W, Xu L, Zhou A, Moharram S, Knaus EE, McEwan AJB, Wiebe LI. Cytotoxicity and cellular uptake of pyrimidine nucleosides for imaging herpes simplex type-1 thymidine kinase (HSV-1 TK) expression in mammalian cells. Nucl Med Biol. 2004:31(5):623–630. [DOI] [PubMed] [Google Scholar]
- 26. Schinazi RF, Fox JJ, Watanabe KA, Nahmias AJ. Activities of 1-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-5-iodocytosine and its metabolites against herpes simplex virus types 1 and 2 in cell culture and in mice infected intracerebrally with herpes simplex virus type 2. Antimicrob Agents Chemother. 1986:29(1):77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Degrève B, Esnouf R, de Clercq E, Balzarini J. Characterization of multiple nuclear localization signals in herpes simplex virus type 1 thymidine kinase. Biochem Biophys Res Commun. 1999:264(2):338–342. [DOI] [PubMed] [Google Scholar]
- 28. Söling A, Simm A, Rainov N. Intracellular localization of herpes simplex virus type 1 thymidine kinase fused to different fluorescent proteins depends on choice of fluorescent tag. FEBS Lett. 2002:527(1-3):153–158. [DOI] [PubMed] [Google Scholar]
- 29. Zhu C, Johansson M, Karlsson A. Incorporation of nucleoside analogs into nuclear or mitochondrial DNA is determined by the intracellular phosphorylation site. J Biol Chem. 2000:275(35):26727–26731. [DOI] [PubMed] [Google Scholar]
- 30. Hendriks DFG, Hurrell T, Riede J, van der Horst M, Tuovinen S, Ingelman-Sundberg M. Mechanisms of chronic fialuridine hepatotoxicity as revealed in primary human hepatocyte spheroids. Toxicol Sci. 2019:171(2):385–395. [DOI] [PubMed] [Google Scholar]
- 31. Govindarajan R, Leung GP, Zhou M, Tse CM, Wang J, Unadkat JD. Facilitated mitochondrial import of antiviral and anticancer nucleoside drugs by human equilibrative nucleoside transporter-3. Am J Physiol Gastrointest Liver Physiol. 2009:296(4):G910–G922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cannino G, Di Liegro CM, Rinaldi AM. Nuclear-mitochondrial interaction. Mitochondrion. 2007:7(6):359–366. [DOI] [PubMed] [Google Scholar]
- 33. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008:88(2):611–638. [DOI] [PubMed] [Google Scholar]
- 34. Meek DW. The p53 response to DNA damage. DNA Repair (Amst). 2004:3(8-9):1049–1056. [DOI] [PubMed] [Google Scholar]
- 35. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000:275(21):16202–16212. [DOI] [PubMed] [Google Scholar]
- 36. Marchenko ND, Moll UM. Mitochondrial death functions of p53. Mol Cell Oncol. 2014:1(2):e955995:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bakhanashvili M, Grinberg S, Bonda E, Simon AJ, Moshitch-Moshkovitz S, Rahav G. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ. 2008:15(12):1865–1874. [DOI] [PubMed] [Google Scholar]
- 38. Park JH, Zhuang J, Li J, Hwang PM. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016:590(7):924–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lebedeva MA, Eaton JS, Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009:1787(5):328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.