

Abstract
We present an unusual case of primary bilateral macronodular adrenal hyperplasia (PBMAH) in a 72-year-old African American man. The patient was found to harbor massively enlarged bilateral adrenal glands on imaging along with mild autonomous cortisol secretion. His workup for PBMAH included leukocyte analysis for the armadillo repeat-containing protein 5 (ARMC5) gene. The test revealed a novel heterozygous somatic ARMC5 mutation. The patient was initially managed conservatively. He subsequently presented with unprovoked bilateral pulmonary emboli. This was followed by the discovery of a nonsecreting pituitary macroadenoma, a hitherto unreported but putative association.
Keywords: pituitary macroadenoma, macronodular adrenal hyperplasia, armadillo repeat-containing protein 5 (ARMC5) mutation, Cushing syndrome
Introduction
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is a rare cause of Cushing syndrome (CS), and accounts for less than 2% of all cases of endogenous CS and approximately 10% to 15% of adrenal CS [1, 2]. PBMAH is characterized by multiple nonpigmented macronodules that are greater than 10 mm in size. Nodules as large as 30 to 40 mm have been described [3]. Overt CS is less common in PBMAH. It is more common for patients to present with mild autonomous cortisol secretion (MACS), in which hypercortisolism is mild and progresses insidiously over several years. Our understanding regarding the pathophysiology of PBMAH has improved over the last several years. PBMAH can occur due to an aberrant expression of G protein–coupled receptors [2]. Recent data show that a germline mutation in the armadillo repeat-containing protein 5 (ARMC5) gene is a more common etiology [3]. It appears that ARMC5 has a role in inhibiting cell proliferation and steroid synthesis. Inactivating mutations of ARMC5 lead to bilateral adrenal hyperplasia with consequent increased steroid synthesis [1]. Interestingly, PBMAH has been associated with multiple tumor syndromes including multiple endocrine neoplasia 1 (MEN-1) syndrome as well as meningiomas [1, 4].
Case Presentation
A 72-year-old African American man, with past medical history of dyslipidemia and well-controlled hypertension on a single agent, was referred to endocrinology for increased abdominal girth. A computed tomography scan (CT) of the abdomen showed massively enlarged bilateral adrenals concerning for macronodular hyperplasia vs metastatic disease (Fig. 1). This CT was compared with a CT performed 4 years prior, and adrenal size was similar in both scans, making a malignant process unlikely. Physical examination was significant only for generalized obesity (body mass index of 32) without any cushingoid features. Family history was negative for adrenal disorders.
Figure 1.
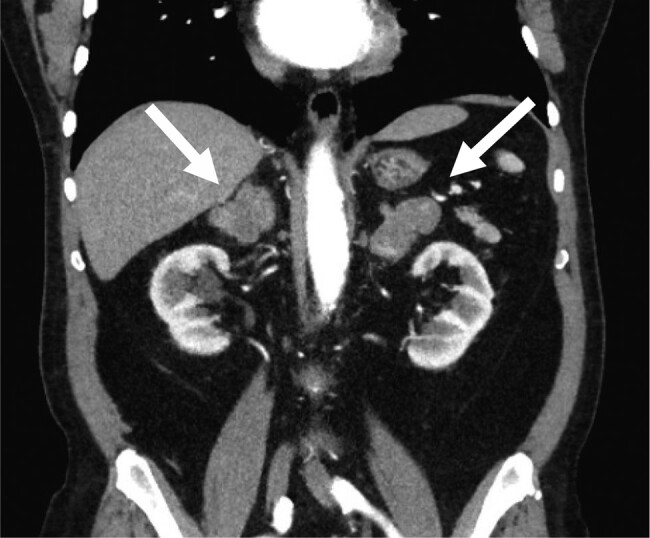
Computed tomography of the abdomen showing the massively enlarged bilateral adrenal glands (white arrows).
Diagnostic Assessment
Biochemical workup was consistent with adrenocorticotropin (ACTH)-independent hypercortisolism: 24-hour urine free cortisol (UFC) was 227.98 nmol/day or 82.6 mcg/24-hour. On repeat testing UFC was 375.64 nmol/day or 136.1 mcg/24-hour (normal: 11.04-138 nmol/day or 4-50 mcg/24-hour); midnight salivary cortisol was 17.9310 nmol/L or 0.65 mcg/dL (normal: 2.48 nmol/L or <0.09 mcg/dL); on a 1-mg dexamethasone suppression test cortisol remained unsuppressed at 463 nmol/L or 16.8 mcg/dL (normal: <49.68 nmol/L or <1.8 mcg/dL). Plasma ACTH remained undetectable at less than 1.1 pmol/L or less than 5 pg/mL (normal: 1.32-11 pmol/L or 6-50 pg/mL) on repeated tests. The remainder of the biochemical workup was unremarkable with normal plasma aldosterone concentration, renin activity, catecholamines and metanephrines, DHEAS (dehydroepiandrosterone sulfate), and 17-hydroxyprogesterone. Glycated hemoglobin A1c was 6.1%. Bone dual-energy x-ray absorptiometry scan was normal. Based on these metabolic abnormalities, the patient was diagnosed with MACS. Gene analysis showed a novel heterozygous variant c.2485T > C (p.Cys 829 Arg) in the ARMC5 gene (NM_001288767.1). We tested for MEN-1 gene mutation as it can on rare occasions be associated with adrenal nodules, but the test was negative in our patient.
Treatment
Conservative management ensued but a few months later, the patient was admitted to an outside hospital with shortness of breath. He was diagnosed with bilateral pulmonary emboli (PEs) and was started on anticoagulation. His repeat 24-hour UFC increased to 549.24 nmol/day or 199 mcg/24-hour and salivary cortisol increased to 24.29 nmol/L or 0.88 mcg/dL at that time. Given his hypercoagulability with consequent PEs and worsening hypercortisolism, the patient was started on oral ketoconazole 200 mg once daily in 2018. He remained on ketoconazole only for 2 weeks and then the medication was discontinued. Several weeks after stopping ketoconazole, the patient was admitted to the hospital with syncope. Magnetic resonance imaging of the brain revealed a 2.5 × 1.7 × 1.8 cm pituitary macroadenoma with mass effect on the optic chiasm and cavernous sinus extension (Fig. 2). Goldman visual field testing showed mild superior temporal depression bilaterally. His pituitary function confirmed a nonfunctional pituitary macroadenoma with central hypothyroidism due to mass effect. The patient was started on low-dose levothyroxine. The neurosurgery team, in light of his anticoagulation and recent bilateral PEs, deemed the patient high risk for transsphenoidal surgery.
Figure 2.
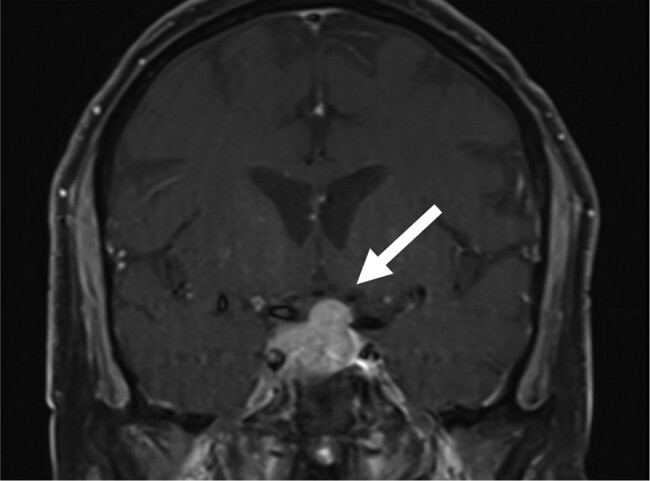
Pituitary magnetic resonance imaging showing the pituitary macroadenoma invading the cavernous sinus and compressing the overlying optic chiasm.
Outcome and Follow-up
Ketoconazole was stopped after 2 weeks of therapy because of drug interactions with several of his medications. Laboratory tests repeated 1 month after discontinuing ketoconazole showed normalization of 24-hour UFC and midnight salivary cortisol, with 24-hour UFC of 103.78 nmol/day or 37.6 mcg/24-hour (normal: 11.04-138 nmol/day or 4-50 mcg/24-hour) and late-night salivary cortisol level of 14.08 nmol/L or 0.51 mcg/dL (normal: 2.48 nmol/L or <0.09 mcg/dL). But post 1-mg dexamethasone suppression test the cortisol remained high at 419.52 nmol/L or 15.2 mcg/dL (normal: <49.68 nmol/L or <1.8 mcg/dL).
Currently, in 2023, the patient is 5 years out from the initial diagnosis and ketoconazole therapy. The patient lost about 10 pounds in the year following the ketoconazole, but between late 2019 and 2023 has gained about 20 pounds. In the past 2 years, the patient has had uneventful hip replacements due to degenerative joint disease. While his dual-energy x-ray absorptiometry scan remains stable in the spine, the patient suffered 2 fractured ribs after a minor trauma. In the past year he has been diagnosed with nonischemic cardiomyopathy with an ejection fraction of 45%. The patient remains on lifelong anticoagulation because of persistently elevated D-dimer levels.
Between 2018 and 2023, his 24 hour UFC has ranged from 61.82 to 187.13 nmol/day or 22.4-67.8 mcg/24-hour, and the most recent level was 171.40 nmol/day or 62.1 mcg/24-hour (normal: 11.04-138 nmol/day or 4-50 mcg/24-hour) (Fig. 3). His late-night salivary cortisol, while lower than the peak levels of 2018, has mostly remained above 2.5 nmol/L or 0.10 mcg/dL (Fig. 4), and his ACTH has remained undetectable. His glycated hemoglobin A1c increased to 6.7% in June 2023, despite being on empagliflozin for his cardiomyopathy. His prolactin level is normal and the most recent level was 0.922 nmol/L or 21.2 ng/mL (reference range, 0.141-0.971 nmol/L or 3.24–22.34 ng/mL). His follicle-stimulating hormone, luteinizing hormone, and thyrotropin remain low, and insulin-like growth factor-1 remains low normal.
Figure 3.

Trends in 24-hour urine free cortisol (UFC) over time. The patient received 2 weeks of ketoconazole in 2018 when his UFC was 199 mcg/24 hours.
Figure 4.

The patient had a late-night salivary cortisol value of 0.88 mcg/dL in May 2018, prior to his 2-week course of ketoconazole.
A follow-up CT in 2023 shows no change in the size of the enlarged adrenal glands. Pituitary magnetic resonance imaging scan shows stable macroadenoma size. Neurosurgery has been postponed indefinitely due to both the location of the pituitary macroadenoma and being on anticoagulation posing high surgical risks for the patient.
Discussion
We report an unusual case of PBMAH with MACS but with subsequent development of bilateral PEs. This patient has unique findings of a pituitary macroadenoma as well as a novel heterozygous germline mutation in the ARMC5 gene, NM_001288767.1:c.2485T > C(p.Cys829Arg) (gene sequencing was performed at Fulgent Laboratory, Temple City, California). It appears PBMAH can result from various germline and somatic mutations [5]. Different mechanisms involving the cyclic adenosine monophosphate/protein kinase pathway A, including germline and/or somatic defects, have been reported as causing PBMAH, such as mutations in GNAS, PRKR1A, and PDE11A. Consequently, PBMAH is now recognized to be associated with tumor syndromes such as familial adenomatous polyposis coli, MEN-1, fumarate hydratase deficiency syndromes, etc. However, more recently ARMC5 has been recognized as a major genetic cause of PBMAH in 80% of familial and 30% of sporadic cases [1]. Another notable mutation is associated with germline mutation and inactivation of lysine demethylase type 1A (KDM1A), which can give rise to aberrant gastric inhibitory polypeptide receptors associated with dependent PBMAH with hypercortisolemia and adrenal myelolipoma, monoclonal gammopathy of undetermined significance, or multiple myeloma [4].
ARMC5 functions as a tumor suppressor gene, therefore inactivating mutations result in a tumorigenic state like the one seen in PBMAH [1]. Assié et al [3] genotyped blood and tumor DNA of 33 patients with PBMAH and found that in all 33 patients, both alleles of ARMC5 carried mutations, one somatic and the other germline, which is consistent with a 2-hit model. The most frequently detected mutation (in 18/33) involved the ARMC5 gene, located at chromosome 16p11.2. The most common somatic change was loss of heterozygosity at 16p (in 8/33). Alencar et al [5] analyzed 47 members of a Brazilian family, 29 additional patients, and 125 randomly selected healthy controls They identified a heterozygous germline variant in the ARMC5 gene (p.Leu365Pro) and also noted inactivation in both alleles of ARMC5 in affected individuals. Other authors have similarly reported a biallelic inactivation [4, 6]. Notably, a study looking at the expression of 4 ARMC5 isoforms, ARMC5-001, ARMC5-003, ARMC5-201/ARMC5-002, and ARMC5-002, in 46 human normal tissues revealed that all 4 isoforms are present in the adrenal and the brain. Interestingly, pituitary has one of the highest expressions of ARMC5-003 but lacks ARMC5-001 [6]. While pituitary adenomas have been reported with familial PBMAH in syndromes like McCune-Albright and MEN-1, this is the first report of PBMAH with pituitary macroadenoma with ARMC5 mutation [1].
The ARMC5 mutations have been noted to be associated with more severe disease with larger adrenal glands, more numerous nodules, higher cortisol levels, with associated clinical manifestations [1]. Even though CS is well recognized as a hypercoagulable state due to alterations in von Willebrand factor, fibrinogen, and factor VIII [7, 8], leading to increased risk of venous thromboembolism, there are relatively few reports of the development of PE. Our patient had the novel missense mutation variant c.2485T > C (p.Cys829Arg) in the ARMC5 gene affecting isoform ARMC5-001 (NM_001288767.1). It is unknown if this variant predisposed him to development of PEs and/or pituitary macroadenoma. Due to his high surgical risk, he cannot undergo surgery and we are unable to report adrenal and pituitary ARMC5 expression. Although it is recommended to consider further testing for gastric inhibitory polypeptide–dependent regulation of cortisol secretion after a mixed meal, the patient at this time has declined testing, including testing for other G protein–coupled receptor mutations like the angiotensin 1 receptor and the vasopressin V1 receptor. Worsening hypercortisolism was managed medically with ketoconazole in our patient. Lower than usual doses of ketoconazole were used and for 2 weeks only. Interestingly, we noted a sustained normalization in the 24-hour UFC and lower salivary cortisol levels post ketoconazole. The typical dose of ketoconazole used in the treatment of CS of any etiology is 400 to 1200 mg daily. In one case reported in the literature, there was rapid normalization in 24-hour UFC in a PBMAH patient when treated with 400 mg daily of ketoconazole within the first month and sustained for 10 years with continuous use of ketoconazole [9]. It is unclear if the improvement in our patient is related to ketoconazole in the setting of the low secretory capacity of PBMAH or part of a cyclical hypersecretory pattern by the autonomous adrenal nodules.
Learning Points
Patients with ARMC5 mutations should be monitored for other tumor entities to delineate the full spectrum of ARMC5-related neoplasia. While meningiomas have been reported in patients with ARMC5 mutations, pituitary adenomas have not. The ARMC5 gene is highly expressed in the pituitary and may contribute to the hyperplastic pituitary abnormalities seen in our patient.
Although PBMAH is a rare cause of CS, it is frequently associated with ARMC5 mutations.
ARMC5 mutations are frequently associated with more severe CS. Whereas CS is known to be a hypercoagulable state, PE is uncommon.
Contributors
All authors made individual contributions to authorship. S.K. and F.A. were involved in case presentation, diagnosis, and patient course and graphs. E.N. and S.K. were involved in discussion. All authors reviewed and approved the final draft.
Abbreviations
- ACTH
adrenocorticotropin
- ARMC5
armadillo repeat-containing protein 5 gene
- CS
Cushing syndrome
- CT
computed tomography
- MACS
mild autonomous cortisol secretion
- MEN-1
multiple endocrine neoplasia 1
- PBMAH
primary bilateral macronodular adrenal hyperplasia
- PEs
pulmonary emboli
- UFC
urine free cortisol
Contributor Information
Shikha Khosla, Division of Endocrinology, Veterans Affairs Medical Center, Washington, DC 20422, USA; Department of Endocrinology, George Washington University, Washington, DC 20037, USA.
Farah Alsarraf, Department of Endocrinology, Mubarak Alkabeer University Hospital, Kuwait City, Kuwait.
Eric S Nylen, Division of Endocrinology, Veterans Affairs Medical Center, Washington, DC 20422, USA; Department of Endocrinology, George Washington University, Washington, DC 20037, USA.
Funding
No public or commercial funding.
Disclosures
The authors have no conflicts of interest or other relevant disclosures.
Informed Patient Consent for Publication
Signed informed consent obtained directly from the patient.
Data Availability Statement
Some or all data sets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request. These data, like other pituitary indices and bone density results, are mentioned in the article.
References
- 1. Charchar HLS, Fragoso MCBV. An overview of the heterogeneous causes of Cushing syndrome resulting from primary macronodular adrenal hyperplasia (PMAH). J Endocr Soc. 2022;6(5):bvac041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Louiset E, Duparc C, Young J, et al. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med. 2013;369(22):2115‐2125. [DOI] [PubMed] [Google Scholar]
- 3. Assié G, Libé R, Espiard S, et al. ARMC5 Mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med. 2013;369(22):2105‐2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertherat J, Bourdeau I, Bouys L, et al. Clinical, Pathophysiologic, Genetic, and Therapeutic Progress in Primary Bilateral Macronodular Adrenal Hyperplasia. Endocr Rev. 2023. Jul 11;44(4):567‐628. [DOI] [PubMed] [Google Scholar]
- 5. Alencar GA, Lerario AM, Nishi MY, et al. ARMC5 Mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99(8):E1501‐E1509. [DOI] [PubMed] [Google Scholar]
- 6. Berthon A, Faucz F, Bertherat J, et al. Analysis of ARMC5 expression in human tissues. Mol Cell Endocinol. 2017;441:140‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. St-Jean M, Lim DST, Langlois F. Hypercoagulability in Cushing's syndrome: from arterial to venous disease. Best Pract Res Clin Endocrinol Metab. 2021;35(2):101496. [DOI] [PubMed] [Google Scholar]
- 8. Wagner J, Langlois F, Lim DST, et al. Hypercoagulability and risk of venous thromboembolic events in endogenous Cushing's syndrome: A systematic meta-analysis. Front Endocrinol (Lausanne). 2019;28(9):805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Comte-Perret S, Zanchi A, Gomez F. Long-term low-dose ketoconazole treatment in bilateral macronodular adrenal hyperplasia. Endocrinol Diabetes Metab Case Rep. 2014;2014(1):140083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Some or all data sets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request. These data, like other pituitary indices and bone density results, are mentioned in the article.