

Abstract
In this study, (E)-2-phenyl-N-(thiophen-2-ylmethylene)imidazo[1,2-a]pyrimidin-3-amine (3) is synthesized, and detailed spectral characterizations using 1H NMR, 13C NMR, mass, and Fourier transform infrared (FT-IR) spectroscopy were performed. The optimized geometry was computed using the density functional theory method at the B3LYP/6-311++G(d,p) basis set. The theoretical FT-IR and NMR (1H and 13C) analysis are agreed to validate the structural assignment made for (3). Frontier molecular orbitals, molecular electrostatic potential, Mulliken atomic charge, electron localization function, localized orbital locator, natural bond orbital, nonlinear optical, Fukui functions, and quantum theory of atoms in molecules analyses are undertaken and meticulously interpreted, providing profound insights into the molecular nature and behaviors. In addition, ADMET and drug-likeness studies were carried out and investigated. Furthermore, molecular docking and molecular dynamics simulations have been studied, indicating that this is an ideal molecule to develop as a potential vascular endothelial growth factor receptor-2 inhibitor.
1. Introduction
Nitrogen-based heterocycles have garnered significant attention in recent decades due to their significant contribution to medicinal chemistry.1 With their diverse pharmacological properties and structural versatility, these compounds have become invaluable building blocks in developing novel therapeutics.2 The incorporation of nitrogen atoms within heterocyclic frameworks imparts unique characteristics and biological activities, offering exciting opportunities for the design of efficacious and selective therapeutic agents.3,4 According to the U.S. Food and Drug Administration databases,5 more than 75% of approved drugs feature nitrogen-based heterocyclic moieties, highlighting their structural importance in drug design.
Among nitrogen-containing heterocycles, those featuring compounds that combine different heterocyclic scaffolds have garnered particular attention due to their broad applicability in synthetic chemistry.3 Imidazo[1,2-a]pyrimidine is a fused heterocyclic motif that combines imidazole and the pyrimidine moiety as its core structure; this fusion creates unique characteristics within the molecule that significantly influence its interactions with various biological targets, including enzymes, receptors, and nucleic acids. This makes it an interesting and valuable combination for pharmaceutical research.6 Imidazo[1,2-a]pyrimidine derivatives exhibit diverse biological activities, including antimicrobial,7,8 antifungal,9,10 antiviral,11,12 and anticancer13,14 activities.
Recently, Imidazo[1,2-a]pyrimidine derivatives have gained attention in drug discovery and medicinal chemistry due to their potential therapeutic applications. They serve as structural scaffolds for the development of novel drugs targeting various diseases. Structural modifications can be made to the imidazo[1,2-a]pyrimidine core to optimize drug-like properties, enhance potency, and improve selectivity toward specific biological targets.
Schiff bases, recognized as versatile pharmacophores, hold a prominent place among organic compounds and are extensively employed in designing and developing diverse bioactive lead compounds.15−17 Demonstrating a diverse range of biological activities, including antifungal,18,19 antibacterial,20 antimalarial,21 anticancer,22,23 antiviral,24 antitubercular,25 and antidepressant26 properties. The nitrogen atom within the imine group, which plays a pivotal role in forming hydrogen bonds with the active sites of receptors and enzyme constituents, lends these compounds a unique capability to intervene in cellular processes.27,28
Overall, combining Imidazo[1,2-a]pyrimidine and Schiff base in a single active pharmacophore could exhibit even more enhanced and innovative properties, thus opening new perspectives for their use in various fields. To the best of our knowledge, the literature survey reveals there have been no complete quantum chemical studies conducted on this selected moiety. Hence, this report presents the synthesis of a novel Imidazo[1,2-a]pyrimidine-Schiff base derivative and detailed experimental characterization using 1H NMR, 13C NMR, mass, and Fourier transform infrared (FT-IR) spectral methods. The theoretical spectroscopic investigation using the density functional theory (DFT) method at the B3LYP/6-311++G(d,p) basis set agreed to validate the structural assignment made for the newly synthesized compound. Furthermore, the highest occupied molecular orbital (HOMO) and least unoccupied molecular orbital (LUMO) molecular orbital energy values were studied, as well as key reactivity descriptors, including ΔEgap, chemical hardness (η), chemical softness (σ), global electrophilicity index (χ), molecular dipole moment (μ), and global electrophilicity index (ω), which have been computed. The molecular electrostatic potential (MEP) surface analysis is computed to identify potential binding sites on the molecule’s surface. Expanding our analysis, Mulliken atomic charge, electron localization function (ELF), localized orbital locator (LOL), natural bond orbital (NBO), Fukui functions, and quantum theory of atoms in molecules (QTAIM) were also analyzed and provided insights into the charge delocalization and interactions within the molecular system to understand and predict the compound’s chemical behavior.
Moreover, drug-likeness and ADMET studies were assessed and investigated, yielding properties conducive to the successful development of pharmaceutical agents. In addition, molecular docking studies targeting the vascular endothelial growth factor receptor-2 (VEGFR-2) receptor are conducted to predict the biological activity of the compound.
2. Results and Discussion
2.1. Chemistry
In this study, 2-phenylimidazo[1,2-a]pyrimidin-3-amine (1) as the basic precursor was synthesized according to our previous publication.29 Then, the synthesis of (E)-2-phenyl-N-(thiophen-2-ylmethylene)imidazo[1,2-a]pyrimidin-3-amine (3) was accomplished in ethanol at room temperature by the condensation reaction of (1) and thiophene-2-carbaldehyde (2) (Scheme 1). The structure of the new compound (3) described in this study was confirmed by 1H, 13C NMR, FT-IR, and mass spectrometry (Figures S1–S6, S8).
Scheme 1. Synthesis of (E)-2-Phenyl-N-(thiophen-2-ylmethylene)imidazo[1,2-a]pyrimidin-3-amine (3).

2.1.1. (E)-2-Phenyl-N-(thiophen-2-ylmethylene)imidazo[1,2-a]pyrimidin-3-amine (3)
Obtained as brown powder; yield 85%, mp; 178–180 °C. FT-IR (υmax/cm–1): υ(C–H) = 3108, 3067; υ(C=N) = 1642; υ(C=C) = 1573. 1H NMR (500 MHz, CHLOROFORM-D): δ 8.88 (d, J = 0.9 Hz, 1H), 8.63 (dd, J = 6.8, 2.1 Hz, 1H), 8.54 (dd, J = 4.1, 2.1 Hz, 1H), 7.87 (dd, J = 7.0, 1.4 Hz, 2H), 7.53 (ddd, J = 5.0, 1.1, 0.9 Hz, 1H), 7.48–7.39 (m, 2H), 7.36 (tt, J = 7.3, 1.4 Hz, 1H), 7.29 (dd, J = 3.7, 1.1 Hz, 1H), 7.10 (dd, J = 5.0, 3.7 Hz, 1H), 6.92 (dd, J = 6.8, 4.1 Hz, 1H). 13C NMR (126 MHz, CHLOROFORM-D): δ 151.90, 150.18, 146.03, 143.05, 135.24, 134.13, 132.75, 131.08, 131.03, 129.01, 128.59, 128.52, 128.28, 126.96, 108.88. LC–MS (ESI): m/z = 305.085 (M + 1).
2.2. Geometry and Quantum Chemical Investigations
2.2.1. Molecular Geometry
The molecular geometry of (3) was optimized using the DFT method with the B3LYP/6-311++G(d,p) basis set, incorporating diffuse functions to accurately capture the influence of lone pair electrons within the molecule. Through comprehensive frequency calculations, it was established that no negative or imaginary frequencies were present, thus affirming the validity of the optimized geometry (Figure 1) as an energy minimum configuration.30
Figure 1.
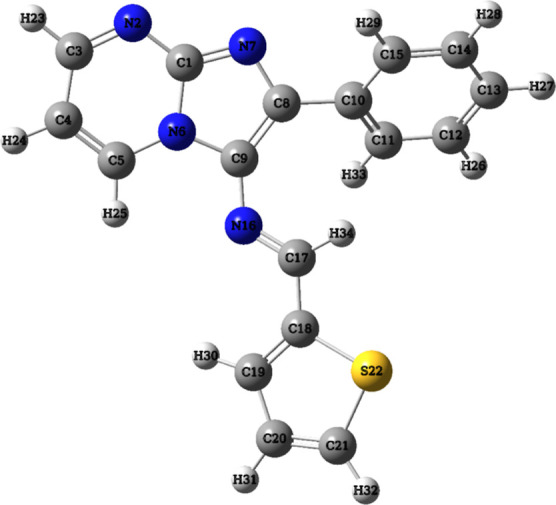
Optimized geometric structure with atom numbering of (3).
2.2.2. Vibrational Analysis
The synthesized compound (3), which consists of 34 atoms and thereby possesses 96 normal modes of vibration, was subjected to IR spectral analysis. Both the experimental (Figure S6) obtained and theoretical FT-IR spectra (Figure S7) are depicted in Figure 2. Vibrational frequencies were calculated using optimized structure parameters, and the Gauss view 6.0 visualization program was utilized to visualize the theoretical IR frequencies. Notably, after scaling the wavenumbers with a 0.961 scaling factor, the theoretical vibrational frequencies were in good agreement with the observed vibrational frequencies.
Figure 2.

Theoretical FT-IR (a) and experimental FT-IR (b) spectra of (3).
2.2.2.1. C–H Vibrations
According to the literature, the typical range for (C–H) stretching vibrations is between 3100 and 3000 cm–1.31 In this study, the aromatic (C–H) modes were observed between 3119 and 3031 cm–1, while the calculated (C–H) stretching vibration in the imine band (CH=N) was determined at 2955 cm–1. In the experimental spectrum, the vibrations detected between 3108 and 3067 cm–1 have been assigned to the stretching bands of the (C–H) bonds.
2.2.2.2. C=N Vibrations
Literature32 reports that the (C=N) stretching is commonly observed between 1700 and 1500 cm–1. The computed modes for the studied compound were determined at 1567, 1460, and 1394 cm–1. However, in the experimental spectrum, the (C=N) stretching band is recognized at 1642 cm–1.
2.2.2.3. C=C Vibrations
The literature reports a range of 1650–1200 cm–1 for the (C=C) stretching vibrations.33 In this molecule, the stretching vibration bands were calculated between 1580 and 1269 cm–1. In the experimental spectrum, 1573, 1490, and 1422 cm–1 vibrations were assigned to the C=C stretching bands.
2.2.2.4. C–N and C–C Vibrations
Assigning the (C–N) and (C–C) vibrations can be challenging due to their presence in a composite region with multiple potential bands. The literature suggests that (C–N) stretching vibrations typically occur between 1360 and 1000 cm–1,34 while (C–C) is found between 1590 and 1000 cm–1.35 In our investigated compound, the (C–N) modes were calculated at 1293, 1188, and 1117 cm–1, while the (C–C) modes were determined at 1406, 1269, and 1174 cm–1. In the experimental spectrum, vibrations observed at 1274 and 1144 cm–1 can be assigned to the (C–N) and (C–C) stretching bands, respectively.
2.2.3. NMR Spectral Studies
NMR spectroscopy is one of the most essential techniques for determining the structure of organic compounds. Theoretical approaches like DFT are valuable for predicting NMR spectra and investigating the relationship between molecular structure and chemical shifts. Combining experimental and theoretical methods makes it possible to evaluate and predict the structure of molecules more effectively. In this study, the 1H NMR (Figure 4) and 13C NMR (Figure 5) spectra of compound (3) were experimentally recorded in CDCl3, and the theoretical chemical shifts were calculated based on the optimized geometry of the molecule using the B3LYP method and the 6-311G + + (d,p) basis set with the standard Gauge Invariant Atomic Orbital (GIAO).35 The GIAO method is widely used for calculating isotropic nuclear magnetic shielding tensors, which are directly related to the observed chemical shifts in the NMR spectra. The experimental and theoretical NMR chemical shifts are given in Table 1.
Figure 4.

Experimental 1H NMR spectra of compound (3).
Figure 5.

Experimental 1C NMR spectra of compound (3).
Table 1. Experimental and Theoretical 1H and 13C NMR Chemical Shifts in ppm.
| H-atom | experimental | B3LYP | C-atom | experimental | B3LYP |
|---|---|---|---|---|---|
| H-34 | 8.88 | 9.06 | C-17 | 151.90 | 151.15 |
| H-25 | 8.63 | 8.91 | C-3 | 150.18 | 153.52 |
| H-23 | 8.54 | 8.54 | C-1 | 146.03 | 152.14 |
| H-29 | 7.87 | 8.22 | C-8 | 143.05 | 143.41 |
| H-33 | 7.87 | 8.08 | C-9 | 135.24 | 132.88 |
| H-30 | 7.53 | 8.38 | C-10 | 134.13 | 142.39 |
| H-26 | 7.44 | 7.51 | C-19 | 132.76 | 129.49 |
| H-28 | 7.44 | 7.83 | C-5 | 131.09 | 136.01 |
| H-27 | 7.36 | 7.51 | C-20 | 131.03 | 132.64 |
| H-32 | 7.29 | 7.69 | C-12 | 129.01 | 133.01 |
| H-31 | 7.10 | 7.39 | C-14 | 129.01 | 134.53 |
| H-24 | 6.92 | 7.06 | C-11 | 128.59 | 131.86 |
| C-15 | 128.59 | 135.59 | |||
| C-13 | 128.52 | 132.90 | |||
| C-21 | 128.28 | 142.70 | |||
| C-18 | 126.96 | 154.37 | |||
| C-4 | 108.88 | 113.28 |
The experimental 1H NMR spectrum (Figures 4, 5) displayed an intense doublet at 8.88 ppm, which can be attributed to the proton of the imine group (N=CH) and computed at 9.06 ppm. Three doublets of doublets were experimentally observed at 8.63, 8.54, and 6.92 ppm, originating from the protons of the pyrimidine nucleus, and computed at 8.91, 8.54, and 7.06 ppm theoretically. The resonance signals assigned to the benzene protons appeared as doublets of doublets at 7.87 ppm, a multiplet in the range of 7.48–7.39 ppm integrating two hydrogens, and a triplet of triplets at 7.36 ppm. Theoretically, these protons were attained at 8.22, 8.08, 7.83, 7.51, and 7.51 ppm, respectively. Finally, the thiophene ring displays doublet at 7.53 ppm and two doublets o doublets at 7.29 and 7.10 ppm experimentally and computed at 8.38, 7.69, and 7.39 ppm, respectively. In 13C NMR spectra (Figures 5, S3–S5), the resonance signals of the imine group (N=CH) were obtained at 155.89 ppm experimentally and theoretically at 151.15 ppm. The chemical shifts of the carbons present in the pyrimidine ring are found experimentally at 150.18, 131.09, and 108.88 ppm and computed at 153.52, 136.01, and 113.28 ppm, respectively. The signal at 146.03 ppm associated with the carbon at the junction (N–C=N) was theoretically attained at 152.14 ppm. The signals at 134.13, 129.01, 128.59, and 128.52 ppm in the experimental spectrum are assigned to the carbons of the benzene ring, while they are calculated at 142.39, 134.53–133.01, 135.59–132.90, and 132.90 ppm. Additionally, the thiophene ring carbons appeared at 132.76, 131.03, 128.28, and 126.96 ppm and were calculated at 129.49, 132.64, 142.70, and 154.37 ppm, respectively. In summary, a reasonable agreement between the experimental and theoretical 1H and 13C NMR chemical shifts was achieved, validating the structural assignment for the compound (3) (Figure 3).
Figure 3.

Experimental and theoretical 1H and 13C NMR chemical shifts of (3).
2.2.4. ESI-MS Study
The ESI+-MS spectrum (Figure S8) exhibits a peak at m/z = 305.085, representing the molecular ion, [M + H]+, which aligns with the expected composition for the title molecule (C17H12N4S).
2.2.5. Frontier Molecular Orbital Studies
Frontier molecular orbitals (FMOs) refer to the HOMO and the LUMO of a molecule.36 The HOMO represents the electron-donating ability of the molecule, while the LUMO represents the electron-accepting ability. Understanding FMOs and the HOMO–LUMO energy gap (ΔEgap) is vital in predicting the behavior of molecules in chemical reactions and assessing their stability, reactivity, and electronic properties. The LUMO and HOMO energies and the global reactive descriptors of (3) have been calculated and tabulated in Table 2.37
Table 2. Energy and Quantum Chemical Parameters of (3).
| quantum chemical parameters | values | quantum chemical parameters | values |
|---|---|---|---|
| EHOMO (eV) | –5.6227 | η (eV) | 1.6115 |
| EHOMO–1 (eV) | –6.7491 | σ (eV–1) | 0.6205 |
| EHOMO–2 (eV) | –7.0731 | χ (eV) | 4.0112 |
| ELUMO (eV) | –2.3997 | ω (eV) | 4.9921 |
| ELUMO+1 (eV) | –1.8806 | μ (debye) | 5.306 |
| ELUMO+2 (eV) | –0.7115 | ||
| ΔEgap (eV) | 3.2230 |
The FMOs analysis of the molecular system under investigation reveals crucial insights into its electronic structure and reactivity (Figure 6, Table 2). The energy levels of the HOMO (EHOMO) and its adjacent orbitals (EHOMO–1 and EHOMO–2) are significant in determining the electron-donating capabilities and chemical stability. In this context, the calculated EHOMO values of −5.6227, −6.7491, and −7.0731 eV showcase the successive energy levels of these orbitals.38 On the other hand, the energy levels of the LUMO (ELUMO) and its higher energy counterparts (ELUMO+1 and ELUMO+2) indicate the electron-accepting potential and reactivity of the molecule. The ELUMO values of −2.3997, −1.8806, and −0.7115 eV depict these respective energy states. The ΔEgap of 3.2230 eV is indicative of the molecule’s kinetic stability, with a small gap suggesting promising reactivity in comparison to 4.55 eV for Sorafenib, 4.19 eV for Erlotinib, and 7.22 eV for Lenvatinib.39−41 Moreover, the chemical hardness (η) of 1.6115 eV highlights the molecule’s resistance to electron exchange and its overall stability. Specifically, it quantifies the energy required to add or remove an electron from the system. The values found are lower than Sorafenib 2.27, Erlotinib 2.09, and Lenvatinib 3.61 eV, suggesting that this system is relatively more reactive and responsive to changes in electron density (ED). The electrophilicity index (ω) of 4.9921 eV underscores the molecule’s ability to engage in electrophilic reactions. In this context, the relatively high ω value indicates that the molecule has a strong tendency to accept electrons from other species. This suggests that it is highly reactive in electrophilic reactions, where it would act as an electron acceptor. Consequently, the value found is higher than that of Sorafenib 3.44 eV, Erlotinib 3.27 eV, and Lenvatinib 2.11 eV, indicating that this molecule exhibits greater electrophilicity. Furthermore, the global electronegativity (χ) of 4.0112 eV emphasizes a relatively stronger tendency to attract electrons in a chemical system. This higher global electronegativity (χ) is indicative of the molecule’s affinity for electrons, suggesting that it tends to pull ED toward its atoms in a molecular environment. Comparatively, the higher χ value, when contrasted with Sorafenib (3.95 eV), Erlotinib (3.70 eV), and Lenvatinib (3.90 eV), implies a more pronounced electronegative character. This characteristic aligns with the molecule’s elevated electrophilicity index, indicating that it not only attracts electrons strongly but also actively engages in electrophilic reactions, portraying a dynamic reactivity profile. The combination of a higher global electronegativity, electrophilicity index, and lower chemical hardness collectively suggests that the molecule is predisposed to engaging in chemical reactions, particularly those involving electron transfer, while exhibiting stability in its electronic structure.40−47
Figure 6.

FMO diagram of (3).
In summary, the FMOs analysis provides insights into the electronic structure, with the calculated ΔEgap indicating both kinetic stability and reactivity. The lower chemical hardness (η) suggests heightened reactivity and responsiveness to ED changes. Additionally, the high electrophilicity index (ω) reflects a strong tendency for electrophilic reactions, while the global electronegativity (χ) signifies a pronounced affinity for electrons, presenting a dynamic reactivity profile. To further enhance the practical understanding of our findings, we have conducted a comparative analysis against established pharmaceutical drugs. This comparative approach aims to bridge the gap between the theoretical insights derived from the FMOs analysis and their real-world applications. By aligning the quantitative analysis with these known drugs, we intend to provide a more comprehensive understanding of the molecule’s characteristics and potential as a scaffold for further drug development. This comparison solidifies the link between theoretical concepts and practical utility, reinforcing the relevance and applicability of our study.48
2.2.6. MEP Surface Analysis
MEP analysis is a highly valuable technique within computational chemistry, offering profound insights into the electrostatic properties of molecules in comprehending molecular interactions, reactivity, and binding phenomena with other molecules or surfaces. MEP is particularly significant in drug design, enabling the identification of specific binding sites on a molecule’s surface, thereby facilitating a deeper understanding of interactions with receptor and enzyme targets.49 The MEP is represented as a three-dimensional surface used to depict different electrostatic potentials. The positive values represent regions with electron-deficient or electrophilic characteristics depicted in blue, while the negative values indicate regions with electron-rich or nucleophilic characteristics shown in red. Neutral regions are depicted in green.50 The MEP surface of (3) is shown in Figure 7.
Figure 7.
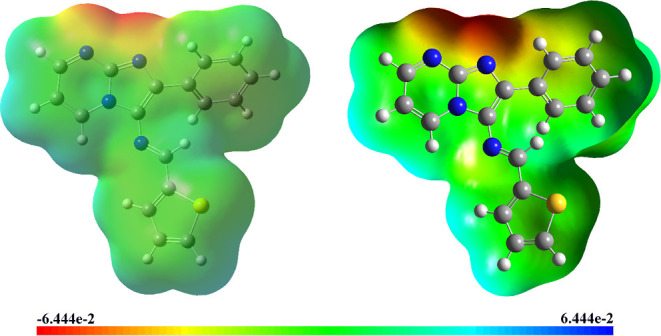
MEP surface of (3).
In the title molecule, the MEP values were predicted in the ranges of −6.444 × 10–2 and 6.444 × 10–2. The negative electrostatic potential region is prominently situated around the atoms N-2 and N-7. This region demonstrates the presence of nucleophilic reactive centers, indicating areas with a high tendency to attract electrophiles during chemical reactions. Conversely, the positive electrostatic potential sites are specifically observed around the hydrogen atoms H-24, H-25, H-30, and H-31. These sites offer valuable information about the regions where the molecule can potentially engage in intermolecular interactions. These MEP insights provide evidence of the molecule’s biological activity, suggesting potential sites for molecular recognition and binding with other molecules or biological targets.
2.2.7. Mulliken Analysis
Mulliken atomic charge analysis provides critical insights into the ED distribution within the molecular system (Figure 8), influencing various molecular properties such as dipole moment, polarizability, and reactivity.51 By interpreting the charge distribution, chemists can gain a deeper understanding of the electronic structure, bonding, and chemical behavior of the compound. It is essential to carefully consider the charge distribution when studying molecular interactions, as it guides the interpretation of chemical reactivity and the design of molecules with desired properties for specific applications.52
Figure 8.
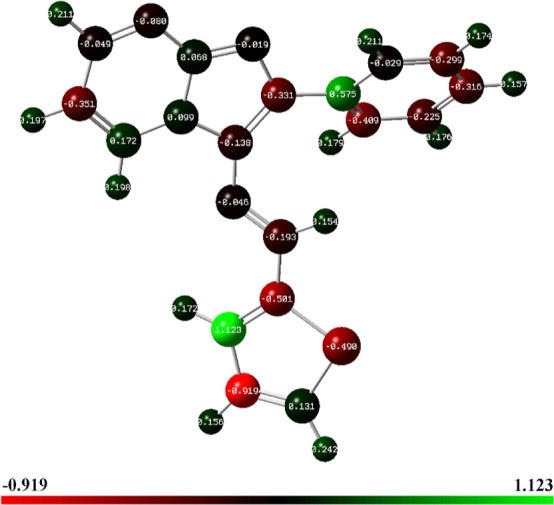
Optimized structure with Mulliken atomic charges of (3) designated by a color change on atoms.
The results presented in Table 3 reveal intriguing patterns of charge distribution, shedding light on the nature of the different atoms within the compound. All carbon atoms display both positive and negative charges. Carbon atoms C-19 and C-20 have the highest positive (1.12318 a.u.) and negative charges (−0.9195 a.u.), respectively, indicating electron-rich and electron-deficient regions. C-1, C-5, and C-10 exhibit positive charges, suggesting that these atoms have lost electrons and are electron-deficient.53 On the other hand, C-8, C-11, and C-18 display relatively higher negative charges, indicating an excess of electrons localized around these atoms. The positive charges on some carbon atoms are likely due to electron transfer to other more electronegative atoms or functional groups in the molecule.54 Conversely, negative charges on certain carbon atoms could arise from the electron-donating nature of the neighboring atoms or substituent groups. Nitrogen atoms in the compound are denoted by numbers 2, 6, 7, and 16. Among these, N-2, N-7, and N-16 display negative charges, indicating a higher ED around these atoms. This behavior is consistent with nitrogen’s electronegative nature, which allows it to attract electrons and carry a negative charge.47 Notably, nitrogen N-6 exhibits a positive charge (0.09902 a.u.), indicating its electron deficiency, primarily attributed to the mesmeric effect. This effect arises from sharing nonbonding electrons with adjacent atoms, leading to an altered ED around N-6. Furthermore, the involvement in resonance structures contributes to the redistribution of electrons, resulting in the observed positive charge. The presence of heteroatoms, such as nitrogen, often results in localized regions of ED due to their higher electronegativity and electron-withdrawing effects. Hydrogen atoms generally demonstrate positive charges, signifying that they have lost electrons and are electron-deficient.55 This is a common trend in organic molecules, where hydrogen atoms are less electronegative compared with carbon and nitrogen. Hydrogen atoms H-23 and H-32 stand out with the highest positive charges, indicating a pronounced electron deficiency, which might be attributed to their proximity to more electronegative or electron-withdrawing groups. Moreover, the sulfur atom (S-22) in the molecule exhibits a negative charge (−0.4902 a.u.), suggesting an excess of electrons in its vicinity.39 This negative charge on sulfur may be a consequence of its participation in various resonance structures or interactions with neighboring atoms, leading to the localization of ED.56
Table 3. Mulliken Atomic Charges of (3).
| atom | charge (a.u.) | atom | charge (a.u.) | atom | charge (a.u.) | atom | charge (a.u.) | atom | charge (a.u.) |
|---|---|---|---|---|---|---|---|---|---|
| C-1 | 0.06797 | C-9 | –0.1382 | C-17 | –0.1931 | H-25 | 0.19833 | H-33 | 0.17944 |
| N-2 | –0.0801 | C-10 | 0.57487 | C-18 | –0.5012 | H-26 | 0.17607 | H-34 | 0.15437 |
| C-3 | –0.0486 | C-11 | –0.4091 | C-19 | 1.12318 | H-27 | 0.15731 | ||
| C-4 | –0.3512 | C-12 | –0.2252 | C-20 | –0.9195 | H-28 | 0.17369 | ||
| C-5 | 0.17173 | C-13 | –0.3161 | C-21 | 0.13132 | H-29 | 0.21071 | ||
| N-6 | 0.09902 | C-14 | –0.299 | S-22 | –0.4902 | H-30 | 0.17159 | ||
| N-7 | –0.0185 | C-15 | –0.0289 | H-23 | 0.21136 | H-31 | 0.15623 | ||
| C-8 | –0.331 | N-16 | –0.0465 | H-24 | 0.19684 | H-32 | 0.24217 |
In summary, Mulliken charges play a crucial role in identifying electron accumulation and depletion regions. For instance, atoms with a net positive charge, such as C-10, C-19, and C-5, may act as electron acceptors, while those with a net negative charge, such as C-18, C-20, and S-22, can function as electron donors. This knowledge is instrumental in predicting and explaining the chemical reactivity. For example, understanding which atoms are prone to gaining or losing electrons guides the strategic placement of functional groups.
In the design of new molecules or materials, the nuanced information provided by Mulliken charges is indispensable. It allows for informed decisions about atom selection, enabling the creation of compounds with tailored properties. This approach is pivotal for the rational design of modifications, ensuring that the changes made align with desired outcomes.
2.2.8. ELF and LOL Analyses
The ELF and LOL analyses have been exhaustively investigated to elucidate the electron distribution and chemical bonding within various molecular systems.57 In the ELF image (Figure 9), regions highlighted in a distinct red hue conspicuously manifest around hydrogen atoms and within C–C and C–N bonds, denoting the prevalence of pronounced electron localization attributed to covalent bonding or the presence of lone electron pairs or nuclear shells in these regions. Furthermore, certain carbon atoms exhibit a discernible blue coloration, indicative of the existence of delocalized electron clouds around them.58 In contrast, the hydrogen atom in the LOL images (Figure 9) is depicted in a pristine white hue, suggesting that the ED surpasses the upper threshold of the color scale in these regions. Moreover, persistent blue hues are consistently discerned around sulfur atoms, which implies the existence of electron depletion regions between the valence shell and inner shell in the LOL maps. This comprehensive analysis furnishes invaluable insights into the nature of chemical bonding, electron localization, and electron pair distribution in diverse molecular complexes, thereby augmenting our comprehension of their structural and electronic attributes.59,60
Figure 9.

ELF (a) and LOL (b) maps of (3).
2.2.9. NBO Analysis
NBO analysis is a powerful computational technique used in quantum chemistry to understand the nature of the chemical bonds within a molecule. It provides valuable insights into the interactions between atoms and the ED distribution, allowing researchers to characterize and interpret the bonding and electronic structure of the molecule.61 NBO analysis provides valuable information about charge transfers, intra- and intermolecular bonding, and stabilization energies, contributing significantly to understanding chemical compounds’ behavior and reactivity. This analysis enables scrutiny of interactions between donor and acceptor orbitals, quantified by stabilization energy E(2) values, indicating electron donation strength and molecular system conjugation degree.61
Throughout the analysis of Table 4, prominent hyperconjugative interactions were observed, particularly involving various π and π* bonds. For instance, the π bond between C-1 and N-6 exhibited interactions with π* bonds in C-4_C-5, C-8_C-9, and N-2_C-3, thus signifying significant electron donation. Similarly, the π bond between C-10_C-15 displayed hyperconjugative interactions with π* bonds in C-11_C-12 and C-13_C-14. Notably, the lone pair LP2 on atom N-7 proved to be an efficient electron donor, participating in a remarkable hyperconjugative interaction with the π* bond in C-1_N-6.62 Additionally, LP2 on atom S-22 also acted as an electron donor, engaging in hyperconjugative interactions with π* bonds in C-18_C-19 and C-20_C-21.63 Furthermore, we observed that the delocalization of ED between occupied Lewis-type and previously unoccupied non-Lewis-type orbitals led to the stabilization of donor–acceptor interactions. The molecule exhibited strong intramolecular hyperconjugative interactions, resulting in the stabilization of the ring systems. Moreover, the π bond ED was found to be weaker compared to the σ bond ED, indicating a higher contribution of σ bonding interactions to the overall stability of the molecule.51
Table 4. Selected Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis (E(2) > 15 kcal/mol) of (3).
| type | donor (i) | ED/e | type | acceptor (j) | ED/e | E(2) (kcal/mol) | Ej – Ei (a.u.) | Fi,j (a.u.) |
|---|---|---|---|---|---|---|---|---|
| Π | C-1_N-6 | 1.71534 | LP2 | N-7 | 1.25718 | 15.47 | 0.18 | 0.066 |
| Π | C-1_N-6 | 1.71534 | π* | C-4_C-5 | 0.00046 | 23.94 | 0.36 | 0.083 |
| Π | C-1_N-6 | 1.71534 | π* | C-8_C-9 | 0.00003 | 15.76 | 0.37 | 0.07 |
| Π | N-2_C-3 | 1.79368 | π* | C-1_N-6 | 0.00001 | 31.99 | 0.25 | 0.094 |
| Π | C-4_C-5 | 1.7694 | π* | N-2_C-3 | 0.00155 | 22.32 | 0.29 | 0.074 |
| Π | C-8_C-9 | 1.69399 | LP2 | N-7 | 1.25718 | 51.72 | 0.1 | 0.087 |
| Π | C-10_C-15 | 1.62906 | π* | C-11_C-12 | 0.00069 | 20.83 | 0.28 | 0.068 |
| Π | C-10_C-15 | 1.62906 | π* | C-13_C-14 | 0.00012 | 20.47 | 0.28 | 0.068 |
| Π | C-11_C-12 | 1.67453 | π* | C-10_C-15 | 0.00189 | 19.74 | 0.29 | 0.068 |
| Π | C-11_C-12 | 1.67453 | π* | C-13_C-14 | 0.00012 | 19.89 | 0.28 | 0.067 |
| Π | C-13_C-14 | 1.65701 | π* | C-10_C-15 | 0.00189 | 20.74 | 0.29 | 0.069 |
| Π | C-13_C-14 | 1.65701 | π* | C-11_C-12 | 0.00069 | 20.33 | 0.28 | 0.068 |
| Π | C-18_C-19 | 1.78074 | π* | N-16_C-17 | 0.00001 | 19.19 | 0.29 | 0.066 |
| Π | C-18_C-19 | 1.78074 | π* | C-20_C-21 | 0.00012 | 16.88 | 0.28 | 0.063 |
| Π | C-20_C-21 | 1.83494 | π* | C-18_C-19 | 0.00224 | 16.14 | 0.3 | 0.065 |
| LP2 | N-7 | 1.25718 | π* | C-1_N-6 | 0.00001 | 205.91 | 0.11 | 0.133 |
| LP2 | N-7 | 1.25718 | π* | C-8_C-9 | 0.00003 | 64.62 | 0.19 | 0.108 |
| LP2 | S-22 | 1.61174 | π* | C-18_C-19 | 0.00224 | 20.96 | 0.27 | 0.067 |
| LP2 | S-22 | 1.61174 | π* | C-20_C-21 | 0.00012 | 22.62 | 0.26 | 0.07 |
| π* | C-1_N-6 | 0.74149 | π* | N-2_C-3 | 0.00155 | 79.99 | 0.06 | 0.088 |
| π* | C-1_N-6 | 0.74149 | π* | C-4_C-5 | 0.00046 | 30.41 | 0.07 | 0.058 |
| π* | C-1_N-6 | 0.74149 | π* | C-8_C-9 | 0.00003 | 19.02 | 0.08 | 0.046 |
| π* | C-8_C-9 | 0.41509 | π* | C-10_C-15 | 0.00189 | 64.95 | 0.02 | 0.054 |
| π* | N-16_C-17 | 0.23307 | π* | C-8_C-9 | 0.00003 | 147.1 | 0.01 | 0.065 |
2.2.10. NLO Studies
Nonlinear optical (NLO) materials have garnered significant attention in the fields of physics, chemistry, and engineering because they possess potential applications in telecommunication, optical signal processing, data storage, biological imaging, and medical therapies. This phenomenon unveils a wide array of fascinating optical behaviors, offering unique insights and potential applications.64,65
As per the literature data, no NLO studies have been conducted on this selected moiety. Hence, this study presents a comprehensive analysis of the hyperpolarizability (β), dipole moment (μ), and polarizability (α), which serve as important parameters in pharmacology, drug designing, and industrial fields, offering a quantitative lens into the structure–activity relationships.66 Therefore, to investigate the NLO properties of the title compound, we concentrated on calculating the key parameters utilizing the x, y, and z components extracted from the Gaussian 09 output employing the DFT/B3LYP/6-311++G(d,p) basis set, using the following equations.67 The determined parameters are listed in Table 5.
with
Table 5. NLO Properties of (3)a.
| dipole moment μ (Debye) | polarizability (α) a.u. | hyperpolarizability (β) a.u. | |||
|---|---|---|---|---|---|
| μx | –4.3218 | αxx | 134.926 | βxxx | –153.69 |
| μy | –3.0768 | αxy | 8.8495 | βxxy | –77.22 |
| μz | 0.0953 | αyy | 113.56 | βxyy | 20.9204 |
| μ0 | 5.306 | αxz | 2.3696 | βyyy | –23.663 |
| αyz | 3.6526 | βxxz | –2.5495 | ||
| αzz | 136.389 | βxyz | –4.3247 | ||
| α0 | 128.291 | βyyz | 1.3138 | ||
| 190 127 × 10–24 esu | βxzz | 1.5831 | |||
| βyzz | 3.9899 | ||||
| βzzz | –0.948 | ||||
| β0 | 163.102 | ||||
| 140 909 × 10–32 esu | |||||
Standard value for urea (μ = 1.3732 D, β0 = 0.3728 × 10–30 esu).
For the exploration of NLO properties, urea serves as a fundamental reference, often employed for comparative analysis.68 Dipole moment and hyperpolarizability values have been compiled and are presented in Table 2. The nonzero characteristics of the dipole moment values provide a good description of the inherent dipolar nature of the optimized molecular structures. Notably, a calculated dipole moment (μ) of 5.306 D is observed, which is an approximation three times higher than the dipole moment of urea (μ = 1.768 D).69 The total polarization (α0) is recorded to be 19.0127 × 10–24 esu. Importantly, the first-order hyperpolarizability reaches a value of 140.909 × 10–32 esu, approximately four times greater than the value of urea’s hyperpolarizability (0.3728 × 10–30 esu). This substantial hyperpolarizability enhancement is attributed to charge transfer phenomena within the molecular structure, indicative of strong NLO attributes. Such findings underscore the potential utility of our molecule for NLO applications.70
2.2.11. Fukui Functions
Fukui functions are essential concepts in theoretical chemistry that provide insights into the reactivity of molecules, enabling the prediction of potential reactive sites and identifying regions susceptible to electrophilic and nucleophilic attacks, providing information on whether an atom is electrophilic or nucleophilic.71 Regarding our specific compound, the Fukui function calculations have yielded valuable insights into its reactivity characteristics (Table 6).
Table 6. Fukui Function of (3).
| atom | N | N – 1 | N + 1 | f+ | f– | f0 |
|---|---|---|---|---|---|---|
| C-1 | 0.0679 | 0.0532 | 0.0674 | –0.0005 | 0.0147 | –0.0071 |
| N-2 | –0.0801 | –0.0265 | –0.1109 | –0.0308 | –0.0537 | 0.0422 |
| C-3 | –0.0486 | 0.0039 | –0.0928 | –0.0442 | –0.0525 | 0.0483 |
| C-4 | –0.3512 | –0.4643 | –0.4411 | –0.0899 | 0.1132 | –0.0116 |
| C-5 | 0.1717 | 0.3426 | 0.21277 | 0.0411 | –0.1709 | 0.0649 |
| N-6 | 0.0990 | 0.1784 | 0.10126 | 0.0023 | –0.0794 | 0.0386 |
| N-7 | –0.0185 | 0.0396 | –0.0483 | –0.0298 | –0.0581 | 0.0439 |
| C-8 | –0.3310 | –0.1790 | –0.3297 | 0.0014 | –0.1520 | 0.0753 |
| C-9 | –0.1382 | –0.4060 | –0.1609 | –0.0227 | 0.2678 | –0.1226 |
| C-10 | 0.5749 | 0.7352 | 0.6347 | 0.0600 | –0.1603 | 0.0502 |
| C-11 | –0.4091 | –0.3697 | –0.5266 | –0.1175 | –0.0394 | 0.0785 |
| C-12 | –0.2252 | –0.2265 | –0.2918 | –0.0667 | 0.0014 | 0.0326 |
| C-13 | –0.3161 | –0.2796 | –0.2825 | 0.0336 | –0.0365 | 0.0015 |
| C-14 | –0.299 | –0.3564 | –0.3639 | –0.0650 | 0.0574 | 0.0038 |
| C-15 | –0.0289 | –0.0853 | 0.0221 | 0.0509 | 0.0564 | –0.0537 |
| N-16 | –0.0465 | –0.0160 | –0.1337 | –0.0872 | –0.0305 | 0.0588 |
| C-17 | –0.1931 | –0.7848 | –0.3077 | –0.1147 | 0.5917 | –0.2385 |
| C-18 | –0.5012 | –0.2963 | –0.3509 | 0.1503 | –0.2049 | 0.0273 |
| C-19 | 1.1232 | 1.7436 | 1.0098 | –0.1134 | –0.6204 | 0.3669 |
| C-20 | –0.9195 | –1.1579 | –0.9539 | –0.0344 | 0.2385 | –0.1020 |
| C-21 | 0.1313 | 0.3617 | 0.1031 | –0.0282 | –0.2304 | 0.1293 |
| S-22 | –0.4902 | –0.5191 | –0.6296 | –0.1394 | 0.0289 | 0.0552 |
| H-23 | 0.2113 | 0.2622 | 0.1710 | –0.0403 | –0.0509 | 0.0456 |
| H-24 | 0.1968 | 0.2418 | 0.1587 | –0.0381 | –0.0450 | 0.0415 |
| H-25 | 0.1983 | 0.2395 | 0.2031 | 0.0048 | –0.0412 | 0.0182 |
| H-26 | 0.1761 | 0.2047 | 0.1499 | –0.0262 | –0.0286 | 0.0274 |
| H-27 | 0.1573 | 0.1981 | 0.1229 | –0.0344 | –0.0407 | 0.0376 |
| H-28 | 0.1737 | 0.2089 | 0.1343 | –0.0394 | –0.0352 | 0.0373 |
| H-29 | 0.2107 | 0.2350 | 0.1929 | –0.0178 | –0.0243 | 0.0210 |
| H-30 | 0.1716 | 0.2351 | 0.1450 | –0.0266 | –0.0635 | 0.0450 |
| H-31 | 0.1562 | 0.1923 | 0.1012 | –0.0550 | –0.0361 | 0.0455 |
| H-32 | 0.2422 | 0.3011 | 0.1882 | –0.0540 | –0.0589 | 0.0564 |
| H-33 | 0.1794 | 0.1786 | 0.1918 | 0.0124 | 0.0008 | –0.0066 |
| H-34 | 0.1544 | 0.2121 | 0.1139 | –0.0405 | –0.0577 | 0.0491 |
Further analysis reveals that the compound exhibits distinct reactivity patterns. The Fukui functions have identified specific atoms as potential favorable sites for electrophilic and nucleophilic attacks. Notably, atoms C-4, C-9, C-17, C-20, and S-22 demonstrate a propensity for electrophilic attacks, indicating their ability to attract electrons.72 Conversely, atoms C-5, C-10, C-15, and C-18 are highlighted as favorable sites for nucleophilic attacks, suggesting their readiness to donate electrons.73 The presence of both electrophilic and nucleophilic sites underscores the compound’s capacity for engaging in chemical reactions with other species. This comprehensive investigation into the compound’s reactivity not only sheds light on its potential chemical interactions but also opens avenues for exploring its reactivity in diverse environments. The insights gleaned from these Fukui parameter analyses will undoubtedly contribute significantly to the broader understanding of the compound’s chemical behavior and its implications in various applications, including biological processes and catalytic reactions.74
2.2.12. QTAIM Analysis
The QTAIM analysis is a powerful and versatile tool used in computational chemistry to study noncovalent interactions in molecular systems. RDG analysis relies on the ED and its derivatives to generate informative 3D isosurfaces and scatter plots, revealing the distribution and strength of weak interactions such as hydrogen bonding and van der Waals forces. The color-coded isosurfaces display different regions representing distinct interaction types: blue for strong interactions (often associated with hydrogen bonding), green for weak interactions (like van der Waals forces), and red for steric hindrance effects.29,50
The AIM-RDG analysis (Figure 10) has provided valuable insights into the molecular structure, shedding light on its reactivity and behavior. One notable finding is the presence of weak noncovalent interactions within the compound. Specifically, the identification of feeble hydrogen interactions and van der Waals forces between specific atom pairs, such as N-7—H-29, N-16—H-25, N-16—H-30, and C-17—H-34, is noteworthy. These interactions exhibit relatively low energy levels yet play a crucial role in influencing the stability and physical properties of the compound.
Figure 10.

NCI and RDG surface map of (3).
The AIM-RDG analysis highlights the occurrence of steric effects within the molecular cycle, elucidating how spatial hindrance due to bulky groups influences the three-dimensional shape of the molecule. These steric effects, resulting from the interaction of atoms with neighboring bulky groups, impact the stability of different spatial arrangements and, consequently, the overall reactivity and behavior of the compound. This spatial arrangement, while primarily governing the molecule’s shape, extends its influence to affect intramolecular associations and can alter reactivity in chemical reactions, emphasizing the pivotal role of steric effects in shaping the compound’s behavior and impacting properties such as reactivity, stability, and interactions with other molecules. Despite their weakness, these noncovalent interactions significantly shape the molecular structure and govern intramolecular associations.44,75
2.2.13. Pharmacokinetics and Drug Likeliness Prediction
The comprehensive in silico ADMET analysis conducted on the novel molecule (3) using SwissADME provided valuable insights into its drug-like properties.76 The radar chart in Figure 13 illustrates the bioavailability assessment of the selected ligand. The bioavailability radar, employed by SwissADME, delineates an optimal region represented by a pink-colored area, signifying the favorable range for six physicochemical parameters crucial for oral bioavailability, namely, flexibility (FLEX), insolubility (INSOLU), unsaturation (INSATU), lipophilicity (LIPO), polarity (POLAR), and SIZE. Upon closer examination of the radar charts, it is evident that the compound aligns closely with the desired range for all parameters except for unsaturation (INSATU), where it deviates significantly. However, despite this total deviation, it is important to note that the oral bioavailability of the compound is unlikely to be affected. The remaining physicochemical parameters within the optimal range suggest that the compound still possesses favorable characteristics for efficient absorption and distribution (Figure 11).
Figure 13.

(a) 2D diagram showing the interaction between (3) and the VEGFR-2 active site. (b) 3D protein–ligand interactions between (3) and the VEGFR-2 active site.
Figure 11.
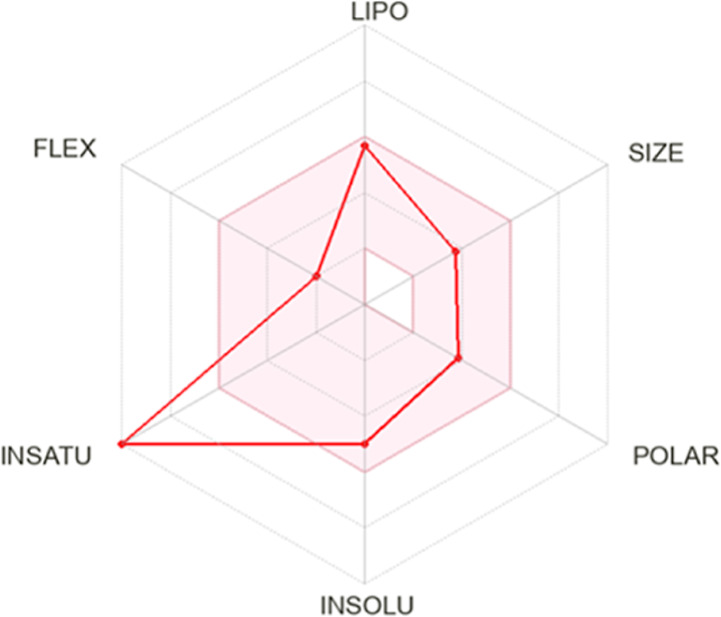
Bioavailability radar plot of (3) provided by SwissADME.
The assessed properties using the SwissADME server demonstrated desirable characteristics (Figure S9), including excellent solubility (log S = −4.99) and a high potential for gastrointestinal absorption, indicating favorable oral bioavailability. Furthermore, this scaffold was predicted to have permeability across the blood–brain barrier.77 These attributes are crucial for efficient drug delivery and therapeutic efficacy. Moreover, the molecule was found to be noninhibitory to both CYP2D6 and CYP3A4 enzymes, which are key players in drug metabolism. Inhibition of these enzymes can lead to altered pharmacokinetics, affecting the efficacy and safety of coadministered drugs.78 Thus, the absence of inhibition suggests a reduced likelihood of potential drug–drug interactions, enhances its safety profile, and highlights the molecule’s compatibility with a wide range of medications. The physicochemical properties of the molecule further support its drug-like characteristics. With a moderate topological polar surface area (TPSA = 70.79 Å2) and lipophilicity (XLOGP3 = 4.42), it exhibits an appropriate balance between hydrophilicity and lipophilicity, which can influence factors such as permeability, distribution, and interaction with biological targets. One notable observation is that the molecule adheres to the Lipinski Rule of Five and other essential drug-likeness rules, including Ghose,79 Veber,80 Egan,81 and Muegge,82 without any violations. This rule serves as a guide to assess the drug-likeness of a compound, considering factors such as molecular weight, lipophilicity, hydrogen bond donors, and acceptors.83 In the case of the molecule, it has a molecular weight of 304.37 g/mol, which falls within the acceptable range for drug-like compounds. Additionally, it has zero hydrogen bond donors (less than 5) and three hydrogen bond acceptors (less than 10), suggesting a favorable interaction potential with biological targets. And finally, an MLOGP (octanol–water partition coefficient) value of 2.65 (less than 5) indicates balanced hydrophilic and lipophilic properties, making it suitable for targeting intracellular or membrane-bound proteins.84 The absence of violations indicates favorable drug-like properties, suggesting the potential utility of the molecule as a promising scaffold for further drug development. However, it is important to note that these predictions are based on the current scaffold, and modifications in future analogs may impact these properties.
The pan-assay interference compounds (PAINS) and Brenk technique are used to identify potentially toxic and unstable fragments in a compound. They help in the evaluation and recognition of misleading compounds that may exhibit undesirable characteristics.85 Despite the absence of PAINS alerts, a notable observation was the presence of a Brenk alert, which is the presence of an imine_1 group. The imine_1 group, consisting of a carbon–nitrogen double bond, can be reactive and may lead to hazardous interactions with other biological molecules that could result in various adverse effects or unpredictable biological responses.86 Therefore, the presence of this functionality highlights the necessity of evaluating the toxicity profile of the molecule thoroughly.
2.2.14. Prediction Via ProTox-II Server
The prediction of chemical toxicity serves as a powerful tool to mitigate late-stage failures in drug design, and computational-based methodologies provide valuable tools for this work. By analyzing, simulating, visualizing, and predicting the toxicity of chemicals, in silico toxicology plays a significant role in reducing reliance on animal testing and expediting toxicity assessments.87 To evaluate the toxicity properties of the novel molecule (3), an analysis was conducted using the ProTox-II server. ProTox-II is a virtual laboratory that employs machine learning and molecular similarity to predict the toxicities of small molecules. It utilizes 33 models to predict various toxicity endpoints, adverse outcome pathways, and toxicity targets based on fragment propensities and similarity.88 The toxicity was estimated by recognizing the ligand fragments and structure. The various toxicity profiles estimated through the ProTox-II server are given in Table 7.
Table 7. Toxicity Profiles of (3) Predicted Using ProTox-II.
| classification | Target | prediction | probability |
|---|---|---|---|
| organ toxicity | hepatotoxicity | active | 0.50 |
| toxicity endpoints | carcinogenicity | active | 0.52 |
| toxicity endpoints | immunotoxicity | inactive | 0.99 |
| toxicity endpoints | mutagenicity | active | 0.65 |
| toxicity end points | cytotoxicity | inactive | 0.73 |
| Tox21-nuclear receptor signaling pathways | AhR | active | 0.61 |
| Tox21-nuclear receptor signaling pathways | androgen receptor | inactive | 0.98 |
| Tox21-nuclear receptor signaling pathways | androgen receptor ligand binding domain (AR-LBD) | inactive | 0.96 |
| Tox21-nuclear receptor signaling pathways | aromatase | inactive | 0.76 |
| Tox21-nuclear receptor signaling pathways | estrogen receptor alpha | inactive | 0.78 |
| Tox21-nuclear receptor signaling pathways | estrogen receptor ligand binding domain | inactive | 0.99 |
| Tox21-nuclear receptor signaling pathways | peroxisome proliferator activated-receptor gamma (PPAR-Gamma) | inactive | 0.94 |
| Tox21-stress response pathways | nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ARE) | inactive | 0.86 |
| Tox21-stress response pathways | heat shock factor response element | inactive | 0.86 |
| Tox21-stress response pathways | mitochondrial membrane potential | inactive | 0.70 |
| Tox21-stress response pathways | phosphoprotein (tumor suppressor) p53 | inactive | 0.76 |
| Tox21-stress response pathways | ATPase family AAA domain-containing protein 5 (ATAD5) | inactive | 0.69 |
The results revealed promising findings concerning its safety profile. The predicted LD50 was estimated to be 1800 mg/kg (Figure 12), indicating a relatively high dose required to cause lethality in experimental animals. In terms of specific toxicity pathways, the molecule was found to be inactive in both the Tox21-Nuclear receptor signaling pathways and the Tox21-Stress response pathways, which suggests a lack of significant interactions with these key cellular pathways, further supporting its favorable safety profile. However, it is noteworthy that there is a very low probability of activation of the aryl hydrocarbon receptor (AhR), which could potentially lead to hazardous effects.89 Additionally, the analysis indicated a mild to low hepatotoxicity potential, indicating a reduced likelihood of adverse effects on liver function. Furthermore, the molecule exhibits a low probability of cytotoxicity and mutagenicity, indicating a reduced likelihood of inducing genetic mutations. These findings contribute to the comprehensive interpretation of the molecule’s toxicity profile and emphasize its potential as a promising candidate with relatively low toxicity risks. However, it is important to note that experimental validation and further investigations are necessary to fully understand the molecule’s toxicity profile and ensure its safety for potential therapeutic applications.
Figure 12.

Graphical representation of the molecular weight and dose value distribution of (3).
2.2.15. Vascular Endothelial Growth Factor Receptor-2
VEGFR-2, belonging to the receptor tyrosine kinase (RTK) family, is primarily expressed on the surface of endothelial cells that form the inner lining of blood vessels, facilitating their response to extracellular cues.90 VEGFR-2 plays a crucial role in regulating mitogenesis (cell division) and angiogenesis (the formation of new blood vessels) in endothelial cells.91 In the context of cancer, tumor cells exhibit aberrant production of vascular endothelial growth factor (VEGF), a key signaling molecule. VEGF specifically binds to VEGFR-2, triggering a cascade of events that promote tumor vascularization and leading to the development of a robust blood supply. This vascular network supplies essential nutrients for tumor growth, proliferation, and migration.92 Given its pivotal role in tumor angiogenesis, the VEGFR-2 pathway has garnered significant interest as a promising target for the discovery of inhibitors aimed at obstructing pathological angiogenesis and impeding cancer progression, metastasis, and uncontrolled growth.93−95
2.2.16. Molecular Docking Analysis
Molecular docking studies represent a premier approach within the pharmaceutical design sector for comprehending how small molecules (ligands) interact with larger molecules (proteins). Molecular docking is adeptly employed to estimate drug–target interactions through the precise calculation of energy minimization and binding energy. It aids in understanding binding interactions and plays a key role in designing new drugs by identifying potential compounds that could effectively target specific proteins involved in diseases.96−98
The molecular docking study of compound (3) with the VEGFR-2 receptor using AutoDock Vina revealed a docking score of −8.4 kcal/mol (Table 8). This score suggests a strong affinity of compound (3) toward the VEGFR-2 receptor, indicating its potential as a VEGFR-2 inhibitor. When compared to established reference compounds,41,98−103 (3) demonstrated a competitive binding affinity, closely aligning with Axitinib (−9.3 kcal/mol) and Sunitinib (−9.3 kcal/mol). Although its score was lower than that of Sorafenib (−11.0 kcal/mol), it outperformed that of Tamoxifen (−7.1 kcal/mol). This comparative analysis highlights the acceptable binding affinity of (3), supporting its potential for further development as an effective VEGFR-2 inhibitor. The 2D interaction diagram of (3) (Figure 13) displayed a common binding pattern with the key residues ASP1046 and GLU885 in the VEGFR-2 active site, comparable to the reference compounds.41,98−103 (Figures 14–17). The analysis revealed eight types of bonds formed between compound (3) and the receptor, including two conventional hydrogen bonds with ASP1046, pi–anion bonds with GLU885 and LYS868, alkyl bonds with ILE1044, LEU1019 and VAL898, pi–alkyl bonds with VAL899 and VAL916, a pi–sulfur bond with CYS1045, a pi–σ bond with LE889, and van der Waals bonds with VAL848, ILE888, and ILE892. However, to validate these findings, it is essential to perform further investigations, such as molecular dynamics (MD) simulations, to assess the dynamic behavior and robustness of the compound–receptor interaction.
Table 8. Docking Energy Scores in Kcal/mol for the Synthesized Compound (3) and Reference Compounds in the VEGFR-2 Active Site.
| compound | docking score (kcal/mol) | compound | docking score (kcal/mol) |
|---|---|---|---|
| 3 | –8.4 | Tamoxifen | –7.1 |
| Axitinib | –9.3 | Sunitinib | –9.3 |
| Sorafenib | –11.0 |
Figure 14.

(a) 2D diagram showing the interaction between Axitinib and the VEGFR-2 active site. (b) 3D protein–ligand interactions between Axitinib and the VEGFR-2 active site.
Figure 17.

(a) Two-dimensional diagram showing the interaction between Tamoxifen and the VEGFR-2 active site. (b) 3D protein–ligand interactions between Tamoxifen and the VEGFR-2 active site.
Figure 15.

(a) Two-dimensional diagram showing the interaction between Sorafenib and the VEGFR-2 active site. (b) 3D protein–ligand interactions between Sorafenib and the VEGFR-2 active site.
Figure 16.

(a) 2D diagram showing the interaction between Sunitinib and the VEGFR-2 active site. (b) 3D protein–ligand interactions between Sunitinib and the VEGFR-2 active site.
2.2.17. MD Analysis
In order to investigate the interactions between the VEGFR-2 and (3), as well as to examine the stability of the bound conformation and the dynamic behavior of the receptor and ligand, we conducted MD studies based on the protein’s rigid crystal structure. To further elucidate the protein–ligand interactions in a biologically relevant context, we employed MD simulations. These simulations, which replicate physiological conditions, provide valuable insights into the dynamic nature of protein–ligand complexes. The obtained results were analyzed by using the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) parameters. The analysis of the RMSD provides insights into the equilibration and stability of the protein–ligand complex. A low RMSD value indicates a higher degree of stability in the complex, whereas a high RMSD value suggests greater fluctuations and reduced stability. In this study, we examined the RMSD graph of the (3)-VEGFR-2 complex, as depicted in Figure 18. Upon the initiation of the simulation, we observed an initial fluctuation in the RMSD values, which can be attributed to the equilibration process. However, subsequent to this initial phase, the complex displayed a remarkable level of stability throughout the 30 ns (ns) simulation.104−109 The mean RMSD value indicates a consistently low deviation from that of the initial conformation. These findings suggest that the (3)-VEGFR-2 complex achieved a well-equilibrated state and maintained a highly stable structure over the course of the simulation. The low RMSD values imply that the interactions between the compound (3) and the VEGFR-2 target remained intact, highlighting the robustness of the protein–ligand binding.110
Figure 18.

RMSD of VEGFR-2 alone and in complex with (3) over the simulation time.
The analysis of the RMSF plot provides insights into the flexibility and rigidity of individual amino acids within the protein during the simulation. Lower RMSF values indicate a higher level of stability, while higher values suggest increased flexibility. In this study, we investigated the RMSF plot of the (3)-VEGFR-2 complex, depicted in Figure 19. The RMSF plot exhibited fluctuations primarily observed in the N-terminal and C-terminal residues of the complex. However, these fluctuations were minimized, and the protein displayed enhanced stability within the residues of the active site that interacted with the compound (3). This finding suggests that the presence of the compound contributes to the stabilization of the protein.
Figure 19.

RMSF of VEGFR-2 upon compound (3) binding. The vertical green lines represent the amino acid residue of VEGFR-2 making contact with ligands.
Consistently, both the RMSD and the RMSF analyses demonstrated the stability of the complex throughout the entire simulation. Based on these results, compound (3) shows potential as an inhibitor of the VEGFR-2 receptor, making it a promising candidate for further exploration and assessment of its biological potential through in vitro and in vivo studies.
3. Conclusions
The integration of quantum computational calculations in several domains, particularly in pharmaceutical research, can significantly accelerate the discovery, design, and optimization of new small-molecule therapeutics. This approach has the potential to revolutionize drug development by providing a deeper understanding of molecular properties and interactions, leading to the creation of more effective and targeted therapeutics.
In this present study, a novel Imidazo[1,2-a]pyrimidine Schiff base derivative was synthesized and experimentally characterized using 1H NMR, 13C NMR, and mass and FT-IR spectroscopic methods. The DFT method at the B3LYP/6-311++G(d,p) basis set was used to optimize the molecular structures, and the theoretical parameters were identified. Theoretical FT-IR and NMR (1H and 13C) analyses were reported and compared, showing excellent agreement with experimental values. The FMOs demonstrate that the ΔEgap of the title molecule was 3.22 eV in comparison to 4.55 eV for Sorafenib, 4.19 eV for Erlotinib, and 7.22 eV for Lenvatinib.40,41 This suggests good chemical stability, indicating that the molecules are less likely to undergo spontaneous reactions. This characteristic is desirable for applications prioritizing stability. Simultaneously, the moderate ΔEgap indicates reasonable reactivity, establishing a well-balanced foundation for controlled chemical interactions when needed. The predicted MEP surface reveals potential nucleophilic and electrophilic sites for chemical interactions. Notably, the negative electrostatic potential region is prominently concentrated around N-2 and N-7, indicating the presence of nucleophilic centers in this region. In addition, Mulliken charges offer a qualitative perspective on the distribution of ED within a molecule, allowing for the identification of active interaction sites. For instance, atoms exhibiting a net positive charge, such as C-10, C-19, and C-5, are likely candidates for electron acceptance in chemical reactions. Conversely, atoms with a net negative charge, including C-18, C-20, and S-22, are poised to act as electron donors. This understanding of charge distribution enhances our ability to predict and interpret chemical reactivity, facilitating a more nuanced comprehension of the molecule’s interactive behavior.
The molecule’s stability as a result of hyperconjugative interaction and charge delocalization was analyzed using NBO orbital analysis. The results obtained by NBO analysis show that the ED in the antibonding π* and π* orbitals and the second-order delocalization energies E(2) prove the existence of intramolecular charge transfer (ICT) within the molecule and contribute to an increase in the stability of the molecule. The most important interaction energies of charge transfer are mainly generated by the orbital overlap between n (LP2_ N-7) π* (C-1_N-6) with a stabilization energy of 205.91 kcal/mol and ED of 1.26e, which results in the ICT that stabilizes the system. A strong interaction was observed due to ED transfer from the π* (N-16_C-17) orbital to the antibonding π* orbital (C-8_C-9) with a high stabilization energy of 147.1 kcal/mol. Meanwhile, NLO properties have been investigated theoretically, and promising results were obtained. The dipole moment μ (5.306 D) of the title compound is approximately three times larger than the urea value, and the first-order hyperpolarizability (β) was 140.909 × 10–32 esu, approximately four times greater than the reference material urea. Consequently, the title molecule could be an effective NLO material candidate.
The comprehensive in silico ADMET analysis and drug-likeness parameters conducted on the novel molecule reveal its adherence to the Lipinski Rule of Five and other essential drug-likeness criteria with relatively low toxicity risks. These findings underscore the scaffold’s potential as a liability-free framework for further development to incorporate into medicinal chemistry campaigns. The molecular docking results revealed a promising binding affinity of −8.4 kcal/mol, showing a similar binding pattern to the key amino acids in the VEGFR-2 active site in comparison to Sorafenib (−11.0 kcal/mol), Axitinib (−9.3 kcal/mol), Sunitinib (−9.3 kcal/mol), and Tamoxifen (−7.1 kcal/mol). Additionally, MD analysis of the synthesized ligand with the VEGFR-2 receptor demonstrated a strong affinity and stable interactions with key residues, indicating the potential of the synthesized ligand as a promising candidate for further development as a targeted therapy against VEGFR-2-driven diseases, such as cancers, angiogenesis, and disorders affecting the cardiovascular system.
4. Experimental Section
4.1. General Information and Materials
All chemicals are reagent grade and were used as received. Reactions were examined with TLC (silica gel, Merck 60 F254). 1H NMR, as well as 13C NMR spectra, were acquired in CDCl3 using a JNM-ECZ500R/S1 FT NMR SYSTEM (JEOL) functioning at 500 MHz for 1H and 126 MHz for 13C. The chemical shifts were expressed in parts per million (ppm), and the residual solvent peak was taken as an internal reference. The mass spectra were obtained using Thermo Scientific TSQ 8000 Evo Triple Quadrupole GC–MS/MS spectrometry. The FT-IR spectrum was measured utilizing JASCO FT/IR-4700 spectrophotometers over a range of 4000–400 cm–1. Melting points were measured using a Stuart melting point apparatus, SMP20.
4.2. Synthesis
The synthesis of intermediate 2-phenylimidazo[1,2-a]pyrimidin-3-amine (1) was reported in our previous publication.29 Target compound (3) was obtained by condensing equimolar quantities of (1) (20 mmol) with thiophene-2-carbaldehyde (2) (20 mmol) in ethanol (40 mL) in the presence of two drops of acetic acid. The stirring for 24 h at room temperature resulted in the formation of a brown precipitate that was filtered and purified by recrystallization in ethanol to give an analytically pure product.
4.3. Computational Details
Generally, the DFT method was applied to establish a connection between the investigated compound structure and its associated behaviors. The geometry optimization and all other calculations were done in the Gaussian 09 package.111 The quantum chemical calculations were performed utilizing the B3LYP method at the 6-311++G(d,p) basis set. Moreover, the Gauss View 6112 software was utilized to compute and visually represent vibrational frequencies, and the chemical NMR (1H and 13C) shift values were obtained using the GIAO method with the same basis set. Using the same theoretical framework, we conducted FMOs, Mulliken charges, MEP, NBO, and NLO calculations through the Gaussian program to gain insights into the chemical stability, reactivity, ED hyperconjugation, and the material’s electronic response.113 ELF and LOL analyses were obtained using the Multiwfn code.59 For generating QTAIM 3D and 2D graphs, we employed VMD and Gnuplot tools.114,115
4.4. Pharmacokinetics, Drug Likeliness, and Toxicity Prediction
In order to gain insights into the pharmacokinetic properties and toxicity profile of the highly promising VEGFR-2 inhibitor, a comprehensive analysis was performed using the SwissADME online tool.76 The ligand’s SMILES representation was used as input to evaluate various ADME (absorption, distribution, metabolism, and excretion) principles, pharmacokinetics, drug-likeness, and medicinal chemistry parameters. Additionally, to assess the potential toxicity and determine the toxicity dosage and class of the ligands, the ProTox-II server was used.88
4.5. Molecular Docking Studies
The three-dimensional crystal structure of VEGFR-2 (PDB ID: 4ASD) (Figure 20) was obtained from the Protein Data Bank (PDB) (https://www.rcsb.org/structure/4ASD). The protein was prepared using AutoDockTools, water molecules and heteroatoms were removed, polar hydrogens and Kollman atomic charges were added, and then saved in PDBQT format. The molecule (3) was converted to 3D PDB format by OpenBabel and then to 3D PDBQT format using AutoDockTools. Sorafenib (CID: 216239), Axitinib (CID: 6450551), Sunitinib (CID: 5329102), and Tamoxifen (CID: 2733526) were used as reference compounds.41,98−103 To facilitate the molecular docking study, a grid box was generated to cover all target active site residues described in the literature;116 details of the molecular docking active site residues and grid box parameters are shown in Table 9.
Figure 20.
3D structure of VEGFR-2 (PDB id: 4ASD).
Table 9. Active Residues, Size, and Coordinates of the Grid Box.
| grid box parameters |
|||||||
|---|---|---|---|---|---|---|---|
| center |
size |
||||||
| protein | X | Y | Z | X | Y | Z | residues |
| VEGFR-2 | –24 | –3 | –9 | 21 | 21 | 21 | ASP814 VAL848 LYS868 GLU885 ILE888 LEU889 ILE892 VAL898 VAL899 VAL914 VAL916 LEU1019 ILE1044 CYS1045 ASP1046 PHE1047 |
For the visualization of interactions between compound (3) and the active site of VEGFR-2, PyMOL was utilized for 3D visualization, while Discovery Studio was employed for 2D visualization. These software tools allowed for a comprehensive analysis and visualization of the specific interactions occurring between compound (3) and the active site residues of VEGFR-2.
4.6. Molecular Dynamics Simulations
MD simulations were conducted to assess the stability of interactions between compound (3) and the VEGFR-2 target. Schrodinger LLC Desmond software was used for MD for 30 ns.110 Before the simulations, the ligand–receptor complex was subjected to preprocessing using Maestro’s Protein Preparation Wizard. This involved optimization, minimization, and the filling of missing residues, if necessary. The system was then constructed by using the System Builder tool. For the simulations, the TIP3P solvent model was employed in an orthorhombic box, maintaining a temperature of 300 K, a pressure of 1 atm, and utilizing the OPLS_2005 force field.117 To simulate physiological conditions, counterions, and 0.15 M sodium chloride were added to neutralize the models. Before the actual simulation, the models were equilibrated, and the trajectories were saved at intervals of 100 ps for further analysis and inspection.
Acknowledgments
The authors thank their respective institutions for providing the necessary facilities to complete this work. The authors extend their appreciation to the Deanship of Scientific Research atKing Khalid University for funding this work through large group Research Projectunder grant number RGP2/390/44.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c06841
(PDF)
Author Contributions
⊥ M.A. and O.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Kalaria P. N.; Karad S. C.; Raval D. K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. 10.1016/j.ejmech.2018.08.040. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Studer A. Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. 10.1039/C5CS00083A. [DOI] [PubMed] [Google Scholar]
- Smith B. R.; Eastman C. M.; Njardarson J. T.; Beyond C.; H O. Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures: Miniperspective. J. Med. Chem. 2014, 57, 9764–9773. 10.1021/jm501105n. [DOI] [PubMed] [Google Scholar]
- Fang W.-Y.; Ravindar L.; Rakesh K. P.; Manukumar H. M.; Shantharam C. S.; Alharbi N. S.; Qin H.-L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerru N.; Gummidi L.; Maddila S.; Gangu K. K.; Jonnalagadda S. B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. 10.3390/molecules25081909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerru N.; Bhaskaruni S. V. H. S.; Gummidi L.; Maddila S. N.; Maddila S.; Jonnalagadda S. B. Recent advances in heterogeneous catalysts for the synthesis of imidazole derivatives. Synth. Commun. 2019, 49, 2437–2459. 10.1080/00397911.2019.1639755. [DOI] [Google Scholar]
- Mantipally M.; Gangireddy M. R.; Gundla R.; Badavath V. N.; Mandha S. R.; Maddipati V. C. Rational design, molecular docking and synthesis of novel homopiperazine linked imidazo[1, 2-a]pyrimidine derivatives as potent cytotoxic and antimicrobial agents. Bioorg. Med. Chem. Lett. 2019, 29, 2248–2253. 10.1016/j.bmcl.2019.06.031. [DOI] [PubMed] [Google Scholar]
- Ramírez-Trinidad Á.; Carrillo-Jaimes K.; Rivera-Chávez J. A.; Hernández-Vázquez E. Synthesis and cytotoxic/antimicrobial screening of 2-alkenylimidazo[1, 2-a]pyrimidines. Med. Chem. Res. 2023, 32, 144–157. 10.1007/s00044-022-02997-6. [DOI] [Google Scholar]
- Fisher M. H.; Lusi A. Imidazo[1, 2-a]pyridine anthelmintic and antifungal agents. J. Med. Chem. 1972, 15, 982–985. 10.1021/jm00279a026. [DOI] [PubMed] [Google Scholar]
- Rival Y.; Grassy G.; Michel G. Synthesis and antibacterial activity of some Imidazo [1, 2-α] pyrimidine derivatives. Chem. Pharm. Bull. 1992, 40, 1170–1176. 10.1248/cpb.40.1170. [DOI] [PubMed] [Google Scholar]
- Alqarni S.; Cooper L.; Galvan Achi J.; Bott R.; Sali V. K.; Brown A.; Santarsiero B. D.; Krunic A.; Manicassamy B.; Peet N. P.; Zhang P.; Thatcher G. R. J.; Gaisina I. N.; Rong L.; Moore T. W. Synthesis, Optimization, and Structure-Activity Relationships of Imidazo[1, 2-a]pyrimidines as Inhibitors of Group 2 Influenza A Viruses. J. Med. Chem. 2022, 65, 14104–14120. 10.1021/acs.jmedchem.2c01329. [DOI] [PubMed] [Google Scholar]
- Gudmundsson K. S.; Johns B. A. Imidazo[1, 2-a]pyridines with potent activity against herpesviruses. Bioorg. Med. Chem. Lett. 2007, 17, 2735–2739. 10.1016/j.bmcl.2007.02.079. [DOI] [PubMed] [Google Scholar]
- Reddy Gangireddy M.; Mantipally M.; Gundla R.; Nayak Badavath V.; Paidikondala K.; Yamala A. Design and Synthesis of Piperazine-Linked Imidazo[1, 2-a]pyridine Derivatives as Potent Anticancer Agents. ChemistrySelect 2019, 4, 13622–13629. 10.1002/slct.201902955. [DOI] [Google Scholar]
- Annareddygari S.; Kasireddy V.; Reddy J. Synthesis of Novel Amide-Functionalized Imidazo[1, 2-a]pyrimidin-5(1H)-ones and Their Biological Evaluation as Anticancer Agents. Russ. J. Org. Chem. 2022, 58, 412–418. 10.1134/S1070428022030216. [DOI] [Google Scholar]
- Berhanu A. L.; Gaurav; Mohiuddin I.; Malik A. K.; Aulakh J. S.; Kumar V.; Kim K.-H. A review of the applications of Schiff bases as optical chemical sensors. TrAC, Trends Anal. Chem. 2019, 116, 74–91. 10.1016/j.trac.2019.04.025. [DOI] [Google Scholar]
- Uddin M. N.; Ahmed S. S.; Alam S. M. R. REVIEW: Biomedical applications of Schiff base metal complexes. J. Coord. Chem. 2020, 73, 3109–3149. 10.1080/00958972.2020.1854745. [DOI] [Google Scholar]
- Verma C.; Quraishi M. A. Recent progresses in Schiff bases as aqueous phase corrosion inhibitors: Design and applications. Coord. Chem. Rev. 2021, 446, 214105. 10.1016/j.ccr.2021.214105. [DOI] [Google Scholar]
- Hamad A.; Chen Y.; Khan M. A.; Jamshidi S.; Saeed N.; Clifford M.; Hind C.; Sutton J. M.; Rahman K. M. Schiff bases of sulphonamides as a new class of antifungal agent against multidrug-resistant Candida auris. MicrobiologyOpen 2021, 10, 1218. 10.1002/mbo3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Zhang J.; Tan W.; Wang G.; Li Q.; Dong F.; Guo Z. Antifungal activity of double Schiff bases of chitosan derivatives bearing active halogeno-benzenes. Int. J. Biol. Macromol. 2021, 179, 292–298. 10.1016/j.ijbiomac.2021.02.184. [DOI] [PubMed] [Google Scholar]
- da Silva C. M.; da Silva D. L.; Modolo L. V.; Alves R. B.; de Resende M. A.; Martins C. V. B.; de Fátima Â. Schiff bases: A short review of their antimicrobial activities. J. Adv. Res. 2011, 2, 1–8. 10.1016/j.jare.2010.05.004. [DOI] [Google Scholar]
- Tople M. S.; Patel N. B.; Patel P. P.; Purohit A. C.; Ahmad I.; Patel H. An in silico-in vitro antimalarial and antimicrobial investigation of newer 7-chloroquinoline based Schiff-bases. J. Mol. Struct. 2023, 1271, 134016. 10.1016/j.molstruc.2022.134016. [DOI] [Google Scholar]
- Uddin N.; Rashid F.; Ali S.; Tirmizi S. A.; Ahmad I.; Zaib S.; Zubair M.; Diaconescu P. L.; Tahir M. N.; Iqbal J.; Haider A. Synthesis, characterization, and anticancer activity of Schiff bases. J. Biomol. Struct. Dyn. 2020, 38, 3246–3259. 10.1080/07391102.2019.1654924. [DOI] [PubMed] [Google Scholar]
- Shekhar S.; Khan A. M.; Sharma S.; Sharma B.; Sarkar A. Schiff base metallodrugs in antimicrobial and anticancer chemotherapy applications: a comprehensive review. Emergent Mater. 2022, 5, 279–293. 10.1007/s42247-021-00234-1. [DOI] [Google Scholar]
- Kaushik S.; Paliwal S. K.; Iyer M. R.; Patil V. M. Promising Schiff bases in antiviral drug design and discovery. Med. Chem. Res. 2023, 32, 1063–1076. 10.1007/s00044-023-03068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro R.; Kachroo M. Synthesis and biological evaluation of anti-tubercular activity of Schiff bases of 2-Amino thiazoles. Bioorg. Med. Chem. Lett. 2020, 30, 127655. 10.1016/j.bmcl.2020.127655. [DOI] [PubMed] [Google Scholar]
- Yuldasheva N.; Acikyildiz N.; Akyuz M.; Yabo-Dambagi L.; Aydin T.; Cakir A.; Kazaz C. The Synthesis of Schiff bases and new secondary amine derivatives of p-vanillin and evaluation of their neuroprotective, antidiabetic, antidepressant and antioxidant potentials. J. Mol. Struct. 2022, 1270, 133883. 10.1016/j.molstruc.2022.133883. [DOI] [Google Scholar]
- Nath B. D.; Islam M. M.; Karim M. R.; Rahman S.; Shaikh M. A. A.; Georghiou P. E.; Menelaou M. Recent Progress in Metal-Incorporated Acyclic Schiff-Base Derivatives: Biological Aspects. ChemistrySelect 2022, 7, 202104290. 10.1002/slct.202104290. [DOI] [Google Scholar]
- Kajal A.; Bala S.; Kamboj S.; Sharma N.; Saini V. Schiff Bases: A Versatile Pharmacophore. J. Catal. 2013, 2013, 1–14. 10.1155/2013/893512. [DOI] [Google Scholar]
- Azzouzi M.; Ouafi Z. E.; Azougagh O.; Daoudi W.; Ghazal H.; Barkany S. E.; Abderrazak R.; Mazières S.; Aatiaoui A. E.; Oussaid A. Design, synthesis, and computational studies of novel imidazo[1, 2-a]pyrimidine derivatives as potential dual inhibitors of hACE2 and spike protein for blocking SARS-CoV-2 cell entry. J. Mol. Struct. 2023, 1285, 135525. 10.1016/j.molstruc.2023.135525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayer K.; Haque T. Density Functional Theory Calculation on the Structural, Electronic, and Optical Properties of Fluorene-Based Azo Compounds. ACS Omega 2020, 5, 4507–4531. 10.1021/acsomega.9b03839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja M.; Muhamed R. R.; Muthu S.; Suresh M. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV-Visible), first order hyperpolarizability, NBO and molecular docking study of (E)-1-(4-bromobenzylidene)semicarbazide. J. Mol. Struct. 2017, 1128, 481–492. 10.1016/j.molstruc.2016.09.017. [DOI] [Google Scholar]
- Ermiş E.; Durmuş K.; Aygüzer Ö. U.; Berber H.; Güllü M. A new 2, 2′-oxydianiline derivative symmetrical azomethine compound containing thiophene units: Synthesis, spectroscopic characterization (UV-Vis, FTIR, 1H and 13C NMR) and DFT calculations. J. Mol. Struct. 2018, 1168, 115–126. 10.1016/j.molstruc.2018.05.021. [DOI] [Google Scholar]
- de Freitas L. V.; da Silva C. C. P.; Ellena J.; Costa L. A. S.; Rey N. A. Structural and vibrational study of 8-hydroxyquinoline-2-carboxaldehyde isonicotinoyl hydrazone - A potential metal-protein attenuating compound (MPAC) for the treatment of Alzheimer’s disease. Spectrochim. Acta, Part A 2013, 116, 41–48. 10.1016/j.saa.2013.06.105. [DOI] [PubMed] [Google Scholar]
- Asadi Z.; Esrafili M. D.; Vessally E.; Asnaashariisfahani M.; Yahyaei S.; Khani A. A structural study of fentanyl by DFT calculations, NMR and IR spectroscopy. J. Mol. Struct. 2017, 1128, 552–562. 10.1016/j.molstruc.2016.09.027. [DOI] [Google Scholar]
- Patil P. G.; Melavanki R.; Radder S. B.; Kusanur R.; Hiremath C. S.; Patil N. R.; Hiremath S. M. Synthesis, Structural Characterizations, and Quantum Chemical Investigations on 1-(3-Methoxy-phenyl)-3-naphthalen-1-yl-propenone. ACS Omega 2021, 6, 25982–25995. 10.1021/acsomega.1c02688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakumari G.; Iyandurai N.; Raja M.; Vetrivelan V.; Thamarai A.; Javed S.; Muthu S. Chemical reactivity, solvent effects, spectroscopic (FTIR, Raman, SERS, UV-Visible), Hirshfeld analyses and antimalarial investigation of 3-Acetylbenzoic acid. Chemical Physics Impact 2023, 6, 100190. 10.1016/j.chphi.2023.100190. [DOI] [Google Scholar]
- Yu J.; Su N. Q.; Yang W. Describing Chemical Reactivity with Frontier Molecular Orbitalets. JACS Au 2022, 2, 1383–1394. 10.1021/jacsau.2c00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerlings P.; De Proft F. Chemical Reactivity as Described by Quantum Chemical Methods. Int. J. Mol. Sci. 2002, 3, 276–309. 10.3390/i3040276. [DOI] [Google Scholar]
- Abd El-Lateef H. M.; Khalaf M. M.; Amer A. A.; Kandeel M.; Abdelhamid A. A.; Abdou A. Synthesis, Characterization, Antimicrobial, Density Functional Theory, and Molecular Docking Studies of Novel Mn(II), Fe(III), and Cr(III) Complexes Incorporating 4-(2-Hydroxyphenyl azo)-1-naphthol (Az). ACS Omega 2023, 8, 25877–25891. 10.1021/acsomega.3c01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Otaibi J. S.; Ullah Z.; Mary Y. S.; Mary Y. S.; Soman S.; Thirunavukkarasu M.; Kwon H. W. DFT investigations on conformational analysis, solvation effects, reactivity studies, chemical descriptors and docking of two anti-cancerous drugs, Lenvatinib and Regorafenib. Vietnam J. Chem. 2022, 60, 636–652. 10.1002/vjch.202200013. [DOI] [Google Scholar]
- Al-Muntaser S. M.; Al-Karmalawy A. A.; El-Naggar A. M.; Ali A. K.; Abd El-Sattar N. E. A.; Abbass E. M. Novel 4-thiophenyl-pyrazole, pyridine, and pyrimidine derivatives as potential antitumor candidates targeting both EGFR and VEGFR-2; design, synthesis, biological evaluations, and in silico studies. RSC Adv. 2023, 13, 12184–12203. 10.1039/D3RA00416C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizwana B. F.; Muthu S.; Prasana J. C.; Abraham C. S.; Raja M. Spectroscopic (FT-IR, FT-Raman) investigation, topology (ESP, ELF, LOL) analyses, charge transfer excitation and molecular docking (dengue, HCV) studies on ribavirin. Chem. Data Collect. 2018, 17–18, 236–250. 10.1016/j.cdc.2018.09.003. [DOI] [Google Scholar]
- Tarika J. D. D.; Dexlin X. D. D.; Madhankumar S.; Jayanthi D. D.; Beaula T. J. Tuning the Computational Evaluation of Spectroscopic, ELF, LOL, NCI analysis and Molecular Docking of Novel Anti COVID-19 Molecule 4-Dimethylamino Pyridinium 3, 5-Dichlorosalicylate. Spectrochim. Acta, Part A 2021, 259, 119907. 10.1016/j.saa.2021.119907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthanveedu V.; Muraleedharan K. Study on structural detailing of gossypetin and its medicinal application in UV filtering, radical scavenging, and metal chelation open up through NCI, TD-DFT, QTAIM, ELF, and LOL analysis. Comput. Theor. Chem. 2023, 1225, 114126. 10.1016/j.comptc.2023.114126. [DOI] [Google Scholar]
- Jeyanthi G.; Gnana Sambandam C. Spectral profiling, structural, molecular docking and ELF elucidation of bioactive molecule Benzoguanamine. J. Mol. Struct. 2021, 1243, 130879. 10.1016/j.molstruc.2021.130879. [DOI] [Google Scholar]
- Janani S.; Rajagopal H.; Sakthivel S.; Kadaikunnan S.; Abbas G.; Muthu S. Comparison of experimental and theoretical spectral groups, electronic properties, topological, and molecular docking investigations of 1-N-Cbz-piperidine-4-carboxylic acid- Potential Cancer drug. J. Mol. Struct. 2023, 1289, 135832. 10.1016/j.molstruc.2023.135832. [DOI] [Google Scholar]
- Kubota Y.; Tanaka S.; Funabiki K.; Matsui M. Synthesis and Fluorescence Properties of Thiazole-Boron Complexes Bearing a β-Ketoiminate Ligand. Org. Lett. 2012, 14, 4682–4685. 10.1021/ol302179r. [DOI] [PubMed] [Google Scholar]
- Al Sheikh Ali A.; Khan D.; Naqvi A.; Al-blewi F. F.; Rezki N.; Aouad M. R.; Hagar M. Design, Synthesis, Molecular Modeling, Anticancer Studies, and Density Functional Theory Calculations of 4-(1, 2, 4-Triazol-3-ylsulfanylmethyl)-1, 2, 3-triazole Derivatives. ACS Omega 2021, 6, 301–316. 10.1021/acsomega.0c04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sayed D. S.; Abdelrehim E.-s. M. Computational details of molecular structure, spectroscopic properties, topological studies and SARS-Cov-2 enzyme molecular docking simulation of substituted triazolo pyrimidine thione heterocycles. Spectrochim. Acta, Part A 2021, 261, 120006. 10.1016/j.saa.2021.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azougagh O.; Essayeh S.; Achalhi N.; El Idrissi A.; Amhamdi H.; Loutou M.; El Ouardi Y.; Salhi A.; Abou-Salama M.; El Barkany S. New benzyltriethylammonium/urea deep eutectic solvent: Quantum calculation and application to hyrdoxylethylcellulose modification. Carbohydr. Polym. 2022, 276, 118737. 10.1016/j.carbpol.2021.118737. [DOI] [PubMed] [Google Scholar]
- Prabakaran A.; Vijayakumar V.; Radhakrishnan N.; Chidambaram R.; Muthu S. Experimental and Quantum Chemical Computational Analysis of Novel N<sup>4</sup>,N<sup>4′</sup>-Dimethyl-[1,1′-Biphenyl]-3,3′,4,4′-Tetraamine. Polycyclic Aromat. Compd. 2022, 42, 925–941. 10.1080/10406638.2020.1756356. [DOI] [Google Scholar]
- Azad I.; Akhter Y.; Khan T.; Azad M. I.; Chandra S.; Singh P.; Kumar D.; Nasibullah M. Synthesis, quantum chemical study, AIM simulation, in silico ADMET profile analysis, molecular docking and antioxidant activity assessment of aminofuran derivatives. J. Mol. Struct. 2020, 1203, 127285. 10.1016/j.molstruc.2019.127285. [DOI] [Google Scholar]
- Al-Sehemi A. G.; Irfan A.; Asiri A. M.; Ammar Y. A. Molecular design of new hydrazone dyes for dye-sensitized solar cells: Synthesis, characterization and DFT study. J. Mol. Struct. 2012, 1019, 130–134. 10.1016/j.molstruc.2012.02.035. [DOI] [Google Scholar]
- Jeelani A.; Muthu S.; Raajaraman B. R.; Sevvanthi S. Spectroscopic, quantum chemical calculations, and molecular docking analysis of 3-Chlorophenyl boronic acid. Spectrosc. Lett. 2020, 53, 778–792. 10.1080/00387010.2020.1834410. [DOI] [Google Scholar]
- Al-Janabi A. S. M.; Elzupir A. O.; Yousef T. A. Synthesis, anti-bacterial evaluation, DFT study and molecular docking as a potential 3-chymotrypsin-like protease (3CLpro) of SARS-CoV-2 inhibitors of a novel Schiff bases. J. Mol. Struct. 2021, 1228, 129454. 10.1016/j.molstruc.2020.129454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geethapriya J.; Shanthidevi A.; Arivazhagan M.; Elangovan N.; Thomas R. Synthesis, structural, DFT, quantum chemical modeling and molecular docking studies of (E)-4-(((5-methylfuran-2-yl)methylene)amino) benzenesulfonamide from 5-methyl-2-furaldehyde and sulfanilamide. J. Indian Chem. Soc. 2022, 99, 100418. 10.1016/j.jics.2022.100418. [DOI] [Google Scholar]
- Sundaram S.; Vijayakumar V. N.; Balasubramanian V. Electronic and structure conformational analysis (HOMO-LUMO, MEP, NBO, ELF, LOL, AIM) of hydrogen bond binary liquid crystal mixture: DFT/TD-DFT approach. Comput. Theor. Chem. 2022, 1217, 113920. 10.1016/j.comptc.2022.113920. [DOI] [Google Scholar]
- Sevvanthi S.; Muthu S.; Aayisha S.; Ramesh P.; Raja M. Spectroscopic (FT-IR, FT-Raman and UV-Vis), computational (ELF, LOL, NBO, HOMO-LUMO, Fukui, MEP) studies and molecular docking on benzodiazepine derivatives- heterocyclic organic arenes. Chem. Data Collect. 2020, 30, 100574. 10.1016/j.cdc.2020.100574. [DOI] [Google Scholar]
- Arulaabaranam K.; Mani G.; Muthu S. Computational assessment on wave function (ELF, LOL) analysis, molecular confirmation and molecular docking explores on 2-(5-Amino-2- Methylanilino)-4-(3-pyridyl) pyrimidine. Chem. Data Collect. 2020, 29, 100525. 10.1016/j.cdc.2020.100525. [DOI] [Google Scholar]
- Janani S.; Rajagopal H.; Muthu S.; Javed S.; Irfan A. Structural, electronic properties (different solvents), chemical reactivity, ELF, LOL, spectroscopic insights, molecular docking and in vitro anticancer activity studies on methyl (4-nitro-1-imidazolyl)acetate. J. Indian Chem. Soc. 2022, 99, 100438. 10.1016/j.jics.2022.100438. [DOI] [Google Scholar]
- Jeba Reeda V. S.; Bena Jothy V. Vibrational spectroscopic, quantum computational (DFT), reactivity (ELF, LOL and Fukui), molecular docking studies and molecular dynamic simulation on (6-methoxy-2-oxo-2H-chromen-4-yl) methyl morpholine-4-carbodithioate. J. Mol. Liq. 2023, 371, 121147. 10.1016/j.molliq.2022.121147. [DOI] [Google Scholar]
- Atlam F. M.; Awad M. K.; Gaber M.; Fathalla S. New Zn (II) and Cd (II) complexes of 2, 4-dihydroxy-5-[(5-mercapto-1H-1, 2, 4-triazole-3-yl)diazenyl]benzaldehyde: Synthesis, structural characterization, molecular modeling and docking studies, DNA binding and biological activity. Appl. Organomet. Chem. 2020, 34, 5635. 10.1002/aoc.5635. [DOI] [Google Scholar]
- Akram N.; Mansha A.; Premkumar R.; Franklin Benial A. M.; Asim S.; Iqbal S. Z.; Ali H. S. Spectroscopic, quantum chemical and molecular docking studies on 2, 4-dimethoxy-1, 3, 5-triazine: a potent inhibitor of protein kinase CK2 for the development of breast cancer drug. Mol. Simul. 2020, 46, 1340–1353. 10.1080/08927022.2020.1822526. [DOI] [Google Scholar]
- Lokhande P. K. M.; Patil D. S.; Kadam M. M.; Sekar N. Theoretical Investigation of Optical and Nonlinear Optical (NLO) Properties of 3-Azabenzanthrone Analogues: DFT and TD-DFT Approach. ChemistrySelect 2019, 4, 10033–10045. 10.1002/slct.201901681. [DOI] [Google Scholar]
- Green K. A.; Cifuentes M. P.; Samoc M.; Humphrey M. G. Syntheses and NLO properties of metal alkynyl dendrimers. Coord. Chem. Rev. 2011, 255, 2025–2038. 10.1016/j.ccr.2011.05.017. [DOI] [Google Scholar]
- Sevvanthi S.; Muthu S.; Raja M.; Aayisha S.; Janani S. PES, molecular structure, spectroscopic (FT-IR, FT-Raman), electronic (UV-Vis, HOMO-LUMO), quantum chemical and biological (docking) studies on a potent membrane permeable inhibitor: dibenzoxepine derivative. Heliyon 2020, 6, 04724. 10.1016/j.heliyon.2020.e04724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. U.; Hussain S.; Asghar M. A.; Munawar K. S.; Khera R. A.; Imran M.; Ibrahim M. M.; Hessien M. M.; Mersal G. A. M. Exploration of Nonlinear Optical Properties for the First Theoretical Framework of Non-Fullerene DTS(FBTTh2)2-Based Derivatives. ACS Omega 2022, 7, 18027–18040. 10.1021/acsomega.2c01474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eşme A.; Sağdınç S. Spectroscopic (FT-IR, FT-Raman, UV-Vis) analysis, conformational, HOMO-LUMO, NBO and NLO calculations on monomeric and dimeric structures of 4-pyridazinecarboxylic acid by HF and DFT methods. J. Mol. Struct. 2017, 1147, 322–334. 10.1016/j.molstruc.2017.06.110. [DOI] [Google Scholar]
- Raja M.; Raj Muhamed R.; Muthu S.; Suresh M. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR; UV-Visible), NLO; NBO, HOMO-LUMO, Fukui function and molecular docking study of (E)-1-(5-bromo-2-hydroxybenzylidene)semicarbazide. J. Mol. Struct. 2017, 1141, 284–298. 10.1016/j.molstruc.2017.03.117. [DOI] [Google Scholar]
- Chérif I.; Raissi H.; Abiedh K.; Gassoumi B.; Caccamo M. T.; Magazu S.; Said A. H.; Hassen F.; Boubaker T.; Ayachi S. Computational studies on optoelectronic and nonlinear optical properties of para-substituted nitrobenzofurazan compound. Mater. Today Commun. 2023, 35, 106133. 10.1016/j.mtcomm.2023.106133. [DOI] [PubMed] [Google Scholar]
- Fukui K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. 10.1126/science.218.4574.747. [DOI] [PubMed] [Google Scholar]
- K V. A.; Rajagopal H.; Muthu S.; Jayanthi V.; Girija R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro-N-methylpyridine-2-carboxamide. J. Mol. Struct. 2020, 1210, 128053. 10.1016/j.molstruc.2020.128053. [DOI] [Google Scholar]
- Bendjeddou A.; Abbaz T.; Gouasmia A.; Villemin D. Molecular Structure, HOMO-LUMO, MEP and Fukui Function Analysis of Some TTF-donor Substituted Molecules Using DFT (B3LYP) Calculations. Int. Res. J. Pure Appl. Chem. 2016, 12, 1–9. 10.9734/IRJPAC/2016/27066. [DOI] [Google Scholar]
- Jeeva P.; Sudha S.; Rakić A.; Dimić D.; Ramarajan D.; Barathi D. Structural, spectroscopic, quantum chemical, and molecular docking study towards cartilage protein of (3E, 3′E)-3, 3′-(1, 4-phenylenebis(azanediyl))bis(cyclohex-2-en-1-one). J. Mol. Struct. 2023, 1274, 134429. 10.1016/j.molstruc.2022.134429. [DOI] [Google Scholar]
- Thirunavukkarasu M.; Prabakaran P.; Saral A.; Alharbi N. S.; Kadaikunnan S.; Kazachenko A. S.; Muthu S. Molecular level solvent interaction (microscopic), electronic, covalent assembly (RDG, AIM & ELF), ADMET prediction and anti-cancer activity of 1-(4-Fluorophenyl)-1-propanone): Cytotoxic agent. J. Mol. Liq. 2023, 380, 121714. 10.1016/j.molliq.2023.121714. [DOI] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Zheng F.; Zhan C.-G. Improved Prediction of Blood-Brain Barrier Permeability Through Machine Learning with Combined Use of Molecular Property-Based Descriptors and Fingerprints. AAPS J. 2018, 20, 54. 10.1208/s12248-018-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T.; Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [PubMed] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Egan W. J.; Merz K. M.; Baldwin J. J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- Muegge I.; Heald S. L.; Brittelli D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2012, 64, 4–17. 10.1016/j.addr.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Nissink J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic P.; Somanathan R. Mechanism of conjugated imine and iminium species, including marine alkaloids: electron transfer, reactive oxygen species, therapeutics and toxicity. Curr. Bioact. Compd. 2010, 6, 46–59. 10.2174/157340710790711782. [DOI] [Google Scholar]
- Raies A. B.; Bajic V. B. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2016, 6, 147–172. 10.1002/wcms.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Eckert A. O.; Schrey A. K.; Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, 257–263. 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V.; Quintana F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. 10.1038/s41577-019-0125-8. [DOI] [PubMed] [Google Scholar]
- da Rocha-Azevedo B.; Lee S.; Dasgupta A.; Vega A.; de Oliveira L.; Kim T.; Kittisopikul M.; Malik Z. A.; Jaqaman K. Heterogeneity in VEGF Receptor-2 Mobility and Organization on the Endothelial Cell Surface Leads to Diverse Models of Activation by VEGF. Cell Rep. 2020, 32, 108187. 10.1016/j.celrep.2020.108187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abhinand C. S.; Raju R.; Soumya S. J.; Arya P. S.; Sudhakaran P. R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signaling 2016, 10, 347–354. 10.1007/s12079-016-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouladzadeh A.; Dorraki M.; Min K. K. M.; Cockshell M. P.; Thompson E. J.; Verjans J. W.; Allison A.; Bonder C. S.; Abbott D. The development of tumour vascular networks. Commun. Biol. 2021, 4, 1111. 10.1038/s42003-021-02632-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzaneh Behelgardi M.; Zahri S.; Gholami Shahvir Z.; Mashayekhi F.; Mirzanejad L.; Asghari S. M. Targeting signaling pathways of VEGFR1 and VEGFR2 as a potential target in the treatment of breast cancer. Mol. Biol. Rep. 2020, 47, 2061–2071. 10.1007/s11033-020-05306-9. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Li Y.; Wang Y.; Lin C.; Zhang D.; Chen J.; Ouyang L.; Wu F.; Zhang J.; Chen L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 89. 10.1186/s13045-022-01310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervarrec T.; Gaboriaud P.; Tallet A.; Berthon P.; Houben R.; Schrama D.; Guyétant S.; Touzé A.; Samimi M. Le VEGF-A comme cible thérapeutique potentielle dans le carcinome à cellules de Merkel. Ann. Dermatol. Venereol. 2018, 145, 128. 10.1016/j.annder.2018.09.145.29248189 [DOI] [Google Scholar]
- El-Adl K.; Sakr H.; Nasser M.; Alswah M.; Shoman F. M. A. 5-(4-Methoxybenzylidene)thiazolidine-2, 4-dione-derived VEGFR-2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluations. Arch. Pharm. 2020, 353, 2000079. 10.1002/ardp.202000079. [DOI] [PubMed] [Google Scholar]
- Elkaeed E. B.; Yousef R. G.; Elkady H.; Gobaara I. M. M.; Alsfouk B. A.; Husein D. Z.; Ibrahim I. M.; Metwaly A. M.; Eissa I. H. Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects. Molecules 2022, 27, 4606. 10.3390/molecules27144606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullaziz M. A.; Abdel-Mohsen H. T.; El Kerdawy A. M.; Ragab F. A. F.; Ali M. M.; Abu-bakr S. M.; Girgis A. S.; El Diwani H. I. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur. J. Med. Chem. 2017, 136, 315–329. 10.1016/j.ejmech.2017.04.068. [DOI] [PubMed] [Google Scholar]
- Abd El-Meguid E. A.; Naglah A. M.; Moustafa G. O.; Awad H. M.; El Kerdawy A. M. Novel benzothiazole-based dual VEGFR-2/EGFR inhibitors targeting breast and liver cancers: Synthesis, cytotoxic activity, QSAR and molecular docking studies. Bioorg. Med. Chem. Lett. 2022, 58, 128529. 10.1016/j.bmcl.2022.128529. [DOI] [PubMed] [Google Scholar]
- Dawood D. H.; Nossier E. S.; Ali M. M.; Mahmoud A. E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. 10.1016/j.bioorg.2020.103916. [DOI] [PubMed] [Google Scholar]
- Alanazi M. M.; Elkady H.; Alsaif N. A.; Obaidullah A. J.; Alkahtani H. M.; Alanazi M. M.; Alharbi M. A.; Eissa I. H.; Dahab M. A. New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies. RSC Adv. 2021, 11, 30315–30328. 10.1039/D1RA05925D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar D. R.; Soni J. Y.; Guduru R.; Rayani R. H.; Kusurkar R. V.; Vala A. G.; Talukdar S. N.; Eissa I. H.; Metwaly A. M.; Khalil A.; Zunjar V.; Battula S. Discovery of new anticancer thiourea-azetidine hybrids: design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. 10.1016/j.bioorg.2021.105206. [DOI] [PubMed] [Google Scholar]
- Eissa I. H.; Ibrahim M. K.; Metwaly A. M.; Belal A.; Mehany A. B. M.; Abdelhady A. A.; Elhendawy M. A.; Radwan M. M.; ElSohly M. A.; Mahdy H. A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg. Chem. 2021, 107, 104532. 10.1016/j.bioorg.2020.104532. [DOI] [PubMed] [Google Scholar]
- Fayyazi N.; Fassihi A.; Esmaeili S.; Taheri S.; Ghasemi J. B.; Saghaie L. Molecular dynamics simulation and 3D-pharmacophore analysis of new quinoline-based analogues with dual potential against EGFR and VEGFR-2. Int. J. Biol. Macromol. 2020, 142, 94–113. 10.1016/j.ijbiomac.2019.09.077. [DOI] [PubMed] [Google Scholar]
- Unadkat V.; Rohit S.; Parikh P.; Sanna V.; Singh S. Rational design-aided discovery of novel 1, 2, 4-oxadiazole derivatives as potential EGFR inhibitors. Bioorg. Chem. 2021, 114, 105124. 10.1016/j.bioorg.2021.105124. [DOI] [PubMed] [Google Scholar]
- Rampogu S.; Baek A.; Zeb A.; Lee K. W. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer 2018, 18, 264. 10.1186/s12885-018-4050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C.-m.; Liu D.-q.; Wang X.-y.; Yu R.-l.; Lv Y.-t. The unbinding studies of vascular endothelial growth factor receptor-2 protein tyrosine kinase type II inhibitors. J. Mol. Graphics Modell. 2015, 59, 130–135. 10.1016/j.jmgm.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Elkaeed E. B.; Yousef R. G.; Khalifa M. M.; Ibrahim A.; Mehany A. B. M.; Gobaara I. M. M.; Alsfouk B. A.; Eldehna W. M.; Metwaly A. M.; Eissa I. H.; El-Zahabi M. A. Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies. Molecules 2022, 27, 6203. 10.3390/molecules27196203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D.; Mahanta S.; Tag H.; Das S. K.; Das Gupta D.; Tanti B.; Ananthan R.; Das R.; Jambhulkar S.; Hui P. K. Identification of tyrosine kinase inhibitors from Panax bipinnatifidus and Panax pseudoginseng for RTK—HER2 and VEGFR2 receptors, by in silico approach. Mol. Diversity 2022, 26, 1933–1955. 10.1007/s11030-021-10304-5. [DOI] [PubMed] [Google Scholar]
- Ferreira L. G.; Dos Santos R. N.; Oliva G.; Andricopulo A. D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M.Gaussian 09; Gaussian Inc., 2009.
- Frisch A.; Nielson A.; Holder A.. Gaussview user manual; Gaussian Inc.: Pittsburgh, PA, 2000; Vol. 556. [Google Scholar]
- Lu T.; Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Racine J. gnuplot 4.0: a portable interactive plotting utility. J. Appl. Econ. 2006, 21, 133–141. 10.1002/jae.885. [DOI] [Google Scholar]
- McTigue M.; Murray B. W.; Chen J. H.; Deng Y.-L.; Solowiej J.; Kania R. S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 18281–18289. 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D.; Williams J.; Wu Y.; Damm W.; Shelley J.; Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.