

Summary
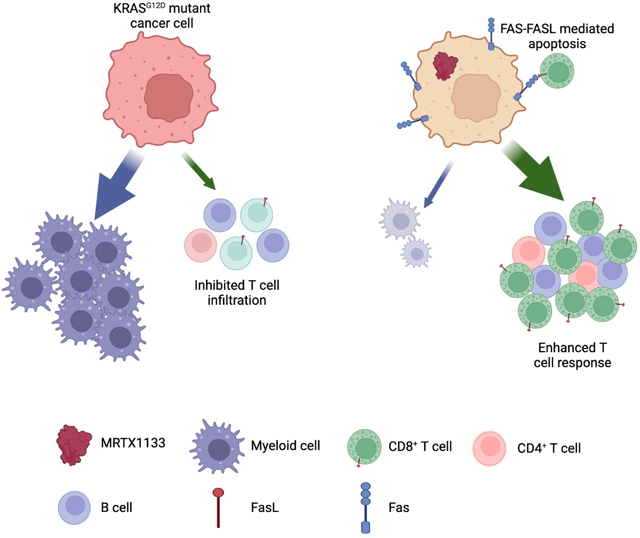
The KRASG12D mutation is present in nearly half of pancreatic adenocarcinomas (PDAC). We investigated the effects of inhibiting the KRASG12D mutant protein with MRTX1133, a non-covalent small molecule inhibitor of KRASG12D, on early and advanced PDAC and its influence on the tumor microenvironment. Employing 16 different models of KRASG12D-driven PDAC, we demonstrate that MRTX1133 reverses early PDAC growth, increases intratumoral CD8+ effector T cells, decreases myeloid infiltration, and reprograms cancer associated fibroblasts. MRTX1133 leads to regression of both established PanINs and advanced PDAC. Regression of advanced PDAC requires CD8+ T cells and immune checkpoint blockade (ICB) synergizes with MRTX1133 to eradicate PDAC and prolong overall survival. Mechanistically, inhibition of KRASG12D in advanced PDAC and human patient derived organoids induces FAS expression in cancer cells and facilitates CD8+ T cell-mediated death. Collectively, this study provides a rationale for a synergistic combination of MRTX1133 with ICB in clinical trials.
eTOC blurb
Mahadevan et al. demonstrate that the oncogenic KRASG12D inhibitor MRTX1133 reverses early and advanced PDAC growth and remodels the tumor microenvironment. MRTX1133 induces FAS expression in cancer cells and promotes CD8+ T cell mediated death.
Graphical Abstract
Introduction
KRAS mutations are a dominant genetic event in pancreatic adenocarcinoma (PDAC), with approximately 90% of patients presenting with such mutations, and 45% of patients having KRASG12D mutations 1. Genetically engineered mouse models revealed that KRASG12D is critical for the initiation and maintenance of PDAC 2–5, suggesting that mutant KRAS is a viable therapeutic target for the control of PDAC progression. Recently, inhibitors targeting the switch II pocket of mutant KRASG12C and KRASG12D have been developed, enabling suppression of mutant KRAS and downstream RAF-MEK-ERK (MAPK) signaling 6–10; however, the precise underlying mechanisms for the efficacy of such inhibitors are not fully understood. Mutant KRAS promotes tumor initiation and progression through a number of cell intrinsic mechanisms, including promoting cancer cell proliferation and rewiring cancer cell metabolic dependencies to promote growth 4,11–13. In addition, mutant KRAS can act through cancer cell extrinsic mechanisms to remodel the tumor microenvironment and impact cancer initiation and progression 14.
The complex desmoplastic response associated with PDAC is characterized by infiltration of immune cells and accumulation of fibroblasts and extracellular matrix. Studies with defined mutational drivers in cancer cells support a role of mutant KRAS in remodeling the tumor immune microenvironment (TIME) and, reciprocally, indicate that the TIME regulates the initiation and progression of cancer. Specifically, CD11b+ myeloid cells promote the initiation and maintenance of KRASG12D driven pancreatic cancer 15. KRASG12D is also associated with increased infiltration of CD4+ T cells 16 and mutant KRASG12V promotes the induction of Tregs 17, potentially contributing to the increase in Treg abundance during PDAC progression 18. Lung cancer and preinvasive pancreatic neoplasia driven by KRASG12D are associated with infiltration of IL17+CD4+ T cells (Th17) cells, which functionally promote tumor initiation and progression 19,20. Studies with genetic suppression of KrasG12D in PDAC revealed that mutant KRASG12D drives the accumulation of immunosuppressive CD4+Gata3+ Th2 cells through IL-33 secretion by cancer cells 11,21. Moreover, pharmacological KRASG12C inhibition with AMG510 increased T cell infiltration and was synergistic with immune checkpoint blockade (ICB) therapy 22, further supporting that mutant KRAS-associated remodeling of the TIME may be leveraged therapeutically to limit cancer progression. PDAC cancer associated fibroblasts (CAFs) are also implicated in cancer progression, with emerging findings on their functional heterogeneity 23–26, though their specific phenotypic distribution in the context of KRASG12D inhibition is unknown.
Here, we evaluate the efficacy of KRASG12D (KRAS*) inhibition with the small molecule inhibitor MRTX1133 in orthotopic and spontaneous mouse models of PDAC. KRAS* inhibition was associated with TIME remodeling and reprogramming of CAFs. We demonstrate that cancer cell clearance upon treatment of advanced PDAC with MRTX1133 is mediated by FAS-FASL interactions between cancer cells and CD8+ T cells, and that ICB synergizes with MRTX1133 to eradicate advanced PDAC, providing a rationale for clinical testing of such combination therapy.
Results
KRAS* inhibition suppresses the growth of human PDAC cells
To evaluate the impact of KRAS* inhibition on pancreatic cancer cells, a panel of cell lines were treated with MRTX1133. Treatment of a human PDAC KRASG12D mutant cell line (HPAC) and a murine PDAC KrasG12D mutant cell line (KPC689) with MRTX1133 resulted in effective downregulation of pERK (Fig. 1A), a downstream readout of KRAS* activity. In contrast, pERK was not impacted in KPC689 cells treated with the KRASG12C inhibitor MRTX849 (Fig. S1A). MRTX1133 inhibited the proliferation of cell lines with KRAS* mutation but had reduced impact on cell lines with other KRAS mutations (Fig. 1B, Fig. S1B). Collectively these results support the specific efficacy of MRTX1133 for PDAC cells with KRAS*.
Figure 1: KRAS* inhibition controls the growth of human cell lines and PDXs.

(A) Capillary immunoassay for pERK and vinculin abundance in HPAC and KPC689 cells in response to MRTX1133. Vehicle: DMSO. (B) Cell proliferation in response to MRTX1133. Cell lines and tissue of origin, and mutational status of KRAS/Kras are listed in parentheses (n=3 biological replicates/group). (C) Schematic of orthotopic experiments in NSG mice with HPAC and Panc1. Treatment started at day 21, and mice were euthanized at day 42. (D-E) Tumor/pancreas weights at endpoint for orthotopic HPAC (Healthy: n=5, VehicleMRTX1133: n=5, MRTX1133: n=9, VehicleMRTX849: n=5, MRTX849: n=9) (D) and Panc1 (Healthy: n=5, VehicleMRTX1133: n=5, MRTX1133: n=9, VehicleMRTX849: n=5, MRTX849: n=9) (E). The same pancreas weights for healthy are presented in both (D) and (E). (F-I) Histological analyses of orthotopic HPAC (VehicleMRTX1133: n=4, MRTX1133: n=5, VehicleMRTX849: n=4, MRTX849: n=6) and Panc1 (VehicleMRTX1133: n=5, MRTX1133: n=4, VehicleMRTX849: n=4, MRTX849: n=5) tumors. (F) Representative H&E images of orthotopic HPAC tumors with the indicated treatments. (G) Histological scoring of orthotopic HPAC tumors. (H) Representative H&E images of orthotopic Panc1 tumors with the indicated treatments. (I) Histological scoring of orthotopic Panc1 tumors. (J) Tumor growth curves of PDXs implanted subcutaneously into athymic nude mice. PATX148: n=5 mice per group; PATX140: VehicleMRTX1133 n=6 mice per group, MRTX1133 n=5 mice per group; PATX124: n=6 mice per group; PATX155: n=6 mice per group; PATX110: n=6 mice per group; PATX53: VehicleMRTX1133 n=5 mice per group, MRTX1133 n=6 mice per group; PATX66: n=6 mice per group; PATX69: n=6 mice per group. In D, E, G, I and J data are presented as mean ± SD. Significance was determined by unpaired t-test in D, E and by two-way ANOVA with Tukey’s multiple comparisons test in G and I. **** P<0.0001, ns: not significant. Scale bars: 100 μm.
See also figures S1–S3.
The anti-tumor efficacy of MRTX1133 on tumor growth kinetics was confirmed using orthotopic implantation of human PDAC cells in NSG mice (Fig. 1C). HPAC and Panc1 orthotopic xenografts treated with MRTX1133 (30 mg/kg BID i.p.) showed significantly reduced tumor/pancreas weights compared to vehicle treated mice (BID i.p. administered, VehicleMRTX1133), whereas MRTX849 treatment (30 mg/kg QD p.o.) had no impact on tumor growth compared to vehicle treatment (QD p.o. administered, VehicleMRTX849) (Fig. 1D–E, Fig. S1C–D). The observed reduction in tumor weight was consistent with histological findings, with mostly normal parenchyma and significantly reduced PDAC with MRTX1133 treatment compared to controls (Fig. 1F–I). MRTX1133 treatment was not associated with specific changes in bodyweight in either tumor models (Fig. S1E–F). Next, a panel of eight different KRAS* PDXs implanted subcutaneously in athymic nude mice were also evaluated for response to MRTX1133. All eight PDXs demonstrated tumor control or slight regression in response to MRTX1133 (Fig. 1J). Despite inhibition of tumor growth, hisopathological analysis at end point showed persistence of cancer cells in the MRTX1133 treated PDX bearing mice (Fig. S2A). Analysis of target engagement and downstream pathways demonstrated effective downregulation of pERK, DUSP6, and SPRY4 (Fig. S2B–G) and reduced proliferative (Ki67+) cancer cells (Fig. S3A).
Established PDAC regresses with KRAS* inhibition by MRTX1133 in immunocompetent mice
To evaluate the efficacy of MRTX1133 in an immunocompetent background, KPC689 cells were orthotopically implanted in C57BL/6J mice (Fig. 2A). MRTX1133 treatment significantly regressed PDAC and improved tumor histology without specific impact on body weight (Fig. 2B–E, Fig. S3B, S3C). MRTX1133 treated tumors also displayed reduced pERK abundance (Fig. 2D, F).
Figure 2: The efficacy of KRAS* inhibition in immunocompetent and immunodeficient backgrounds.

(A) Schematic of experiments with C57BL6/J mice orthotopically implanted with KPC689. Treatment started at day 15, and mice were euthanized at day 28. (B) Tumor/pancreas weights of orthotopic KPC689 tumors in C57BL6/J mice at endpoint (Healthy: n=5, VehicleMRTX1133: n=5, MRTX1133: n=8, VehicleMRTX849: n=5, MRTX849: n=9). (C) Bioluminescence of orthotopic KPC689 tumors in C57BL6/J mice over time. Treatment was initiated at day 15 (VehicleMRTX1133: n=5, MRTX1133: n=9 , VehicleMRTX849: n=5, MRTX849: n=9). (D-F) Representative H&E (VehicleMRTX1133: n=4, MRTX1133: n=6, VehicleMRTX849: n=5, MRTX849: n=8) and pERK (VehicleMRTX1133: n=4, MRTX1133: n=5 , VehicleMRTX849: n=3, MRTX849: n=8) immunostaining images of the pancreata of C57BL6/J mice orthotopically implanted with KPC689 (D) and quantification (E-F). Scale bar, 50 μm. (G) Schematic of experiments with NSG mice orthotopically implanted with KPC689. Treatment started at day 9, and mice were euthanized at day 21. (H) Bioluminescence of orthotopic KPC689 tumors in NSG mice over time. Treatment was initiated at day 9 (VehicleMRTX1133: n=5, MRTX1133: n=9 , VehicleMRTX849: n=5, MRTX849: n=10) (I) Tumor/pancreas weights of orthotopic KPC689 tumors in NSG mice at endpoint (VehicleMRTX1133: n=5, MRTX1133: n=9, VehicleMRTX849: n=5, MRTX849: n=9). Healthy pancreas weights are presented in Fig. 1D and 1E. (J-K) Representative H&E images of the pancreata of NSG mice orthotopically implanted with KPC689 (J) and quantification (VehicleMRTX1133: n=5, MRTX1133: n=6 , VehicleMRTX849: n=4, MRTX849: n=9) (K). Scale bar: 100 μm. (L) Histological scoring of the pancreata of MRTX1133 treated NSG and C57BL6/J mice orthotopically implanted with KPC689 at endpoint. Data included in this plot are also presented in panel (K) and (E). In B, C, E, F, H, I, K and L data are presented as mean ± SD. In B and I, unpaired t-test was used for comparison of VehicleMRTX849 and MRTX849 and Welch’s correction was used for VehicleMRTX1133 and MRTX1133 comparisons. In E and K, two-way ANOVA with Tukey’s multiple comparisons test was used. In L, two-way ANOVA with Sidak’s multiple comparison test was used. In C and H, significance was determined by one-way ANOVA with Sidak’s multiple comparison test and Kruskall-Wallis with Dunn’s multiple comparison test, respectively (VehicleMRTX1133 vs. MRTX1133 total flux comparisons). Significance was determined by unpaired t-test in F. * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001, ns: not significant.
See also figure S3.
In the context of an immunodeficient background lacking T cells, B cells, and NK cells in the NSG mice (Fig. 2G), MRTX1133 controlled orthotopic KPC689 tumor growth without specific impact on body weight (Fig. 2H–K, Fig. S3D, S3E). Histopathological assessment of MRTX1133 treated tumors showed greater PDAC involvement in NSG mice compared to C57BL/6J mice at end point suggesting that adaptive immune cells may contribute the therapeutic efficacy of MRTX1133 (Fig. 2L).
To evaluate the impact of KRAS* targeting in the context of tumor initiation and pancreatic intraepithelial neoplasia (PanIN) emergence, genetically engineered mouse models (GEMMs) driven by KrasG12D expression in the pancreas (KC mice; see STAR Methods) were employed. KC mice were treated with MRTX1133 starting at 7 weeks of age, when on average 5% of the pancreas histology involves PanIN lesions (Fig. 3A–C). KRAS* inhibition marginally decreased pancreas weight (Fig. S4A) and reduced the percentage of tissue with PanIN from 5% to 0.05% upon 13 weeks of treatment, whereas 35% pancreata of vehicle treated mice exhibited PanIN lesions after 13 weeks (Fig. 3B–D). CK19 staining confirmed regressed PanIN lesions in MRTX1133 treated mice (Fig. 3B, D). In addition, no specific differences in bodyweight or urine protein levels were observed between vehicle and MRTX1133 treated mice over the course of 13 weeks of treatment (Fig. S4B–C). Blood biochemistry and histological analyses revealed minimal changes upon 13 weeks of treatment with MRTX1133 compared to controls (Fig. S4D–E).
Figure 3: KRAS* inhibition reverses precancerous and established cancerous lesions.

(A) Schematic of spontaneous KC treatment experiments. Treatment was initiated at 7 weeks and mice euthanized at 20 weeks post-birth. (B-D) Representative H&E and CK19 immunostaining images of the pancreata of KC mice (B) and histological (Treatment initiation: n=4, VehicleMRTX1133: n=4 and MRTX1133: n=4) and CK19 immunostaining quantification (n=4 per group) (C-D). ADM: acinar to ductal metaplasia. (E) Schematic of spontaneous KPPC treatment experiments. Treatment was initiated at 6 weeks and mice euthanized at 9 weeks post-birth. (F-G) Representative H&E images of the pancreata of KPPC mice (F) and quantification (Treatment initiation: n=4, VehicleMRTX1133: n=1 and MRTX1133: n=1) (G). In C and D, data are presented as mean ± SD. Significance was determined by two-way ANOVA with Sidak’s multiple comparisons test in C and by Welch’s t-test in D. Scale bars: 100 μm. ** P<0.01, **** P<0.0001.
See also Figure S4.
Next, autochthonous genetic models of invasive PDAC with expression of KrasG12D and loss of Trp53 (KPPC) and advanced disease were treated with MRTX1133 or vehicle control (Fig. 3E). Treatment for 3 weeks demonstrated a reversion of PDAC to predominantly uninvolved pancreatic tissue and PanIN histology (Fig. 3F–G), suggesting that inhibition of KRAS* is sufficient to reprogram cancer cells to a premalignant state and potentially enable the regeneration of normal pancreatic parenchyma.
KRAS* inhibition is associated with reprogramming of stromal fibroblasts and immune cells in the PDAC microenvironment
scRNA-seq analysis of KPC689 tumors in C57BL6/J mice was conducted to evaluate the impact of KRAS* targeting with MRTX1133 on the tumor microenvironment (Fig. 4A, Fig. S5A–B). scRNA-seq analysis revealed a proportional increase in fibroblasts upon treatment with MRTX1133 (Fig. 4B). MRTX1133 treated KPC689 tumors in C57BL6/J mice demonstrated an increased proportion of podoplanin+ fibroblasts (Fig. 4D), a reported pan-fibroblast marker in PDAC 27. Evaluation of stromal fibroblast clusters in KPC689 tumors implanted in NSG and C57BL6/J mice revealed transcriptionally distinct subsets of fibroblasts (Fig. 4E–F, Fig. S5C–E, S6A–D) as previously reported 26. The diversity of CAFs subclusters were more pronounced in immune deficient (NSG) mice compared to immunocompetent mice with PDAC (C57BL/6J and KPPC mice), though in all groups MRTX1133 yield a shift in relative proportion toward aCAFs and bCAFs 26, which in part overlap with the iCAF gene signature 27 (Fig. 4E–H, Fig. S6E). Clustering of fibroblasts based on the previously reported myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen presenting CAFs (apCAFs) 27 revealed a shift to predominantly iCAFs with MRTX1133 treatment (Fig. S6E–H). Notably, a/bCAFs and iCAFs are associated with early stage of disease 26. Consistently with this observation, stromal fibroblasts in C57BL6/J KPC689 and KPPC tumors treated with MRTX1133 displayed a proportional distribution similar to healthy pancreas, whereas fibroblasts in NSG KPC689 tumors acquired an intermediate state between fibroblasts of healthy and tumor bearing tissue (Fig. 4H), suggesting that CAFs accumulation and heterogeneity in PDAC progression and therapy response may be regulated by tumor infiltrating immune cells.
Figure 4: Fibroblast composition is altered in an immune-dependent manner in response to KRAS* inhibition.

Fib (A-B) UMAP (A) and relative proportions of cell types (B) of orthotopic KPC689 tumors in C57BL6/J mice determined by scRNA-seq. (C-D) Representative immunofluorescent images of podoplanin stained orthotopic KPC689 tumors in C57BL6/J mice (C) and quantification of podoplanin+ area relative to nuclear area (D). Scale bar, 25 μm. (E-G) UMAP of fibroblasts in orthotopic KPC689 tumors in NSG mice (E) and C57BL6/J mice (F) and spontaneous KPPC tumors (G) determined by scRNA-seq. (H) Relative proportions of fibroblast subsets in orthotopic KPC689 tumors in NSG mice and C57BL6/J mice, and spontaneous KPPC tumors. Data are presented as mean in B, H and as mean ± SD in D. Significance was determined by unpaired t-test in D. ** P<0.01, ns: not significant.
See also Figures S5 and S6.
MRTX1133 treatment resulted in a significant increase in the frequencies of CD3+ T cells, CD8+ T cells, and CD19+ cells, as ascertained by immunotyping analyses (Fig. 5A). Analysis of T cell subsets by scRNA-seq revealed that MRTX1133 reduced the relative proportion of NK cells and increased the relative proportions of naïve CD4+,CD8+, and CD8+ effector T cells in both the KPPC mice and the orthotopic KPC689 model (Fig. 5B–D, Fig. S7A–D). Further analysis of effector CD8+ T cell population revealed increased expression of Ifng (encoding IFNγ), Prf1 (encoding perforin) Tbx21 (encoding T-bet), and Gzmb (encoding granzyme B) in the MRTX1133 treated tumors (Fig. 5E, Fig. S7E), suggestive of accumulation of activated T cells. Immunostaining confirmed an increase in Ki67+CD8+ T cells upon MRTX1133 treatment (Fig. 5F–G). MRTX1133 reduced frequencies of CD11b+ myeloid infiltration as evaluated by flow cytometry, immunohistochemistry and scRNA-seq analysis (Fig. 4B, 5A, Fig. S7A–B, S7F–G). Further analysis of other myeloid cell subsets or macrophage infiltration showed that although MRTX1133 suppressed Arg1+ and enriched Mrc1+ macrophage clusters (seen in scRNA-seq analysis), such shifts did not impact the overall frequency of CD11b+F4/80+ macrophage infiltration (Fig. 5A, Fig. S7H). Insignificant changes in CD11c+, CD11c+F4/80−, and CD3−NK1.1+ cell infiltration were observed upon treatment with MRTX1133 (Fig. S7I–K).
Figure 5: The efficacy of KRAS* inhibition is dependent on CD8+ T cells.

(A) Immunotyping by flow cytometry of orthotopic KPC689 tumors in C57BL6/J mice (VehicleMRTX1133: n=5, MRTX1133: n=8, VehicleMRTX849: n=5, MRTX849: n=9). (B-C) UMAPs and relative abundance of T cell subsets (C) of spontaneous KPPC tumors determined by scRNA-seq. (D) Relative abundance of T cell subsets of orthotopic KPC689 tumors in C57BL6/J mice determined by scRNA-seq. (E) Violin plots for Ifng, Prf1, and Txb21 expression in tumor infiltrating CD8+ effector T cells in orthotopic KPC689 C57BL6/J mice determined by scRNA-seq. (F-G) Representative immunofluorescent images (F) and quantification (G) of CD8+ and Ki67+ T cells in orthotopic KPC689 tumors in C57BL6/J mice. Scale bar: 25 μm. (VehicleMRTX1133: n=4, MRTX1133: n=5, VehicleMRTX849: n=3, MRTX849: n=3). (H) Schematic of experiments with C57BL6/J mice orthotopically implanted with PKC-HY19636. Treatment started when tumors were 685 mm3 by MRI. (I) Survival of mice with PKC-HY19636 orthotopic tumors with the indicated treatment groups (Iso/veh (n=7), MRTX1133 (n=3), MRTX1133+αCD8 (n=6), MRTX1133+αCTLA-4 (n=5), MRTX1133+αCTLA-4+αPD-1 (n=9), MRTX1133+αCD19 (n=6)) (J) Tumor volumes at 1 week post treatment initiation in the indicated treatment groups by MRI (Iso/veh (n=5), MRTX1133 (n=6), MRTX1133+αCD8 (n=5), MRTX1133+αCTLA-4 (n=8), MRTX1133+αCTLA-4+αPD-1 (n=10)). (K) Change in tumor volumes of indicated groups compared to baseline by MRI (Iso/veh (n=5), MRTX1133 (n=6), MRTX1133+αCD8 (n=5), MRTX1133+αCTLA-4 (n=8), MRTX1133+αCTLA-4+αPD-1 (n=10)). (L) Representative MRI of tumor burden 1 week post treatment initiation. (M) Representative H&E of the pancreas in the indicated group and (N) associated histopathological scoring (Iso/veh (n=5), MRTX1133 (n=3), MRTX1133 + αCD8 (n=3), MRTX1133 + αCTLA-4 (n=3), MRTX1133+ αCTLA-4 + αPD-1(n=3)). Scale bar: 100 μm. Data are presented as mean + SD in A, G, J, K and N; as mean in C and D, and as violin plots in E. In A, Mann-Whitney test was used for CD4+ T cells comparisons, comparisons between VehicleMRTX1133 and MRTX1133 for CD3+ T cells, and comparisons between VehicleMRTX849 and MRTX849 for CD19+ cells. Unpaired t-test was used for all other comparisons in A. In G, Mann-Whitney test was used for the comparisons between VehicleMRTX1133 and MRTX1133 and unpaired t-test was used for all other comparisons. In J and K, Brown-Forsythe ANOVA with Dunnett’s T3 multiple comparisons test was used to determine significance. In N, two-way ANOVA with Dunnett’s multiple comparisons test was used to determine significance. In I, log rank test was used to determine significance. * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001, ns: not significant.
See also Figures S7–S9.
CD8+ T cells contribute to clearance of cancer cells in response to KRAS* inhibition
The increased infiltration of B cells and activated T cells suggested that such cell types may impact the efficacy of MRTX1133 and presented a rationale for the therapeutic combination of MRTX1133 with immune checkpoint blockade (ICB). To evaluate the functional contribution of immune cells, the PKC-HY19636 cell line was orthotopically implanted and treated with MRTX1133 at a starting tumor volume of 685 mm3 measured by MRI (Fig. 5H, Supplemental Fig. S8A–C). CD8 and CD19 depletion were performed to evaluate the functional contribution of CD8+ T cells and CD19+ B cells, respectively, and in another experimental group MRTX1133 combined with αCTLA-4 and/or αPD-1. After one week of treatment, all control mice (Veh/Iso) and MRTX1133 plus αCD8 treated mice succumbed to PDAC (Fig. 5I–L). All MRTX1133 alone treated mice displayed increased survival with significantly reduced tumor volumes compared to control mice and αCD8 treated mice (Fig. 5I–L, Fig. S8A–C). Further analysis of CD8+ T cells and CD3−CD11b−CD19+ B cells demonstrated systemic and intratumoral depletion in the mice treated with αCD8 or αCD19 antibodies and MRTX1133, respectively (Figure S9A–F). Analysis of pERK in tumors at 1 week post treatment demonstrated target engagement in all groups that received MRTX1133 (Fig. S9G–H). Although there was a modest increase in intra-tumoral CD8+ T cells in the MRTX1133 treated mice, addition of αCTLA-4 and αCTLA4+αPD-1 demonstrated a robust influx of CD8+ T cell infiltrates and enhanced tumor regression in these mice compared to MRTX1133 monotherapy (Fig. 5I, 5M, 5N and Fig. S9E–I). Depletion of CD8+ T cells with MRTX1133 abrogates the anti-tumor efficacy of the KRAS* inhibitor, whereas depletion of CD19+ B cells had no impact on MRTX1133 efficacy (Fig. 5I–N, Fig. S8A–C). Taken together, the data support that eradication of established, large tumors with KRAS* inhibition requires CD8+ T cells. MRTX1133 with αCTLA-4 and αPD-1 further increased the survival of mice and significantly reduced tumor volumes compared to MRTX1133 treated mice (Fig. 5I–L, Fig. S8A–C). At experimental endpoint, PDAC-related death was not observed in MRTX1133 with αCTLA-4 treated mice (Fig. 5I). Serial MRI measurements show a regression of tumor volume when MRTX1133 is given in combination with ICB compared to MRTX1133 as single agent or other controls (Fig. 5J–L, Fig. S8A–C). While MRTX1133 treated tumors eventually escape tumor stasis and succumb to PDAC, αCTLA-4 ICB enables sustained tumor inhibition and immune clearance of MRTX1133 treated tumors.
KRAS* inhibition upregulates FAS in cancer cells to facilitate apoptosis by CD8+ T cell
The efficacy of MRTX1133 in advanced PDAC is dependent on CD8+ T cell mediated clearance of cancer cells (Fig. 5I). To evaluate the mechanism(s) by which cancer cells become sensitive to adaptive immune responses, scRNA-seq analysis of cultured KPC689 cells treated with MRTX1133 was performed. A shift in transcriptional clusters was observed with MRTX1133 treatment, with cluster 0 and 7 becoming the most prevalent clusters in MRTX1133 treated cells (Fig. 6A–B). Pathway analysis revealed that KRAS signaling, MYC targets, and mTORC1 signaling were suppressed in these clusters (Fig. S10A–B), supporting effective targeting of KRAS* 4,28. In addition, several immune modulatory pathways were suppressed in MRTX1133 treated clusters, including TNFα signaling via NFκB, inflammatory response, and allograft rejection (Fig. S10A–B). Further analysis of death receptors in KPC689 cells revealed that Fas expression was increased upon MRTX1133 treatment (Fig. 6C). Moreover, MRTX1133 treated orthotopic KPC689 tumors revealed an increase relative proportions of CD8+ cells expressing Fasl (Fig. 6D, Fig. S10C), supporting the possibility of CD8+ T cell mediated cancer cell clearance through FAS-FASL interaction. MRTX849 and MRTX1133 induced FAS expression in KRASG12C and KRAS* mutant cell lines, respectively (Fig. 6E–H, Fig. S10D–E). Similar induction of FAS expression in response to MRTX1133 was observed in a PDAC patient derived organoid (PDO, line DH-50) at concentrations that specifically target KRAS* (Fig. 6I–K, Fig. S10F). Moreover, immunostaining of PDXs treated with MRTX1133 revealed an increase in FAS expression (Fig. S10G).
Figure 6: KRAS* inhibition induces FAS expression in cancer cells.

(A) UMAP projections and relative proportions of each cluster (B) of KPC689 cells evaluated by scRNA-seq analysis. (C) Expression of Fas in KPC689 cells evaluated by scRNA-seq analysis. (D) Number of CD8+ effector cells with Fasl (Fasl+) or without Fasl (Fasl−) expression in KPC689 tumors in C57BL6/J mice evaluated by scRNA-seq analysis. (E) Western blot analysis of pERK expression level in MIA PaCa-2 cells treated with KRASG12C inhibitor (MRTX849). (F) qPCR analysis of relative FAS expression in MIA PaCa-2 cells treated with KRASG12D inhibitor (MRTX1133) (n=3 biological replicates per group). (G) Representative flow plots and quantification of FAS expression at 24 hours post-treatment with vehicle (DMSO) or the indicated concentrations of MRTX1133 in KPC689 cells. n=3 biological replicates per group. (H) pERK and total ERK in KPC689 at 24 hours post-treatment with vehicle (DMSO) or the indicated concentrations of MRTX1133. (I) DH-50 organoids were treated with KRASG12D inhibitor (MRTX1133) (n=3 biological replicates per group). Representative phase-contrast microscope image. Scale bar, 50 μm. (J) Western blot analysis of pERK expression in DH-50 organoids treated with KRASG12D inhibitor (MTX1133). (K) qPCR analysis of relative FAS expression in DH-50 organoids treated with KRASG12D inhibitor (MRTX1133) (n=3 biological replicates per group). Data are presented as mean + SD in F, G, and K. Significance was determined by one-way ANOVA with Dunnett’s multiple comparisons test in F and G and by unpaired t-test in K. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, ns: not significant.
See also Figure S10.
Analysis of MRTX1133 treated tumors revealed that MRTX1133 effectively reduces cancer cell proliferation, independent of the presence of CD8+ T cells (Fig. 7A–B). In contrast, CD8+ T cell depletion inhibited induction of cleaved caspase 3 (CC3+) expression in cancer cells (Fig. 7C–D), suggesting that MRTX1133 functions to inhibit proliferation but CD8+ T cells are required to induce cancer cell killing. In order to functionally evaluate the contribution of FAS-FASL engagement to cancer cell clearance following MRTX1133 treatment, AK14837 shScramble (shScr) or shFas cells were orthotopically implanted and treated with MRTX1133 (Fig. 7E). MRI revealed that shFas tumors had significantly increased tumor growth compared to shScr tumors treated with MRTX1133 and the increased growth was associated with decreased survival of mice with shFas tumors (Fig. 7F–H). In contrast, similar growth kinetics and survival of mice bearing shScr and shFas tumors are observed (Mahadevan et al. 2023 in review). Histological analysis further confirmed increased tumor burden in shFas tumors (Fig. 7I–J, Fig. S11A). Proliferative cancer cells were similar between shScr and shFas tumors treated with MRTX1133 suggesting effective targeting of KRAS*; however, induction of cancer cell apoptosis was reduced in shFas tumors (Fig. 7K–N). A separate strategy to inhibit FASL with αFASL was employed wherein αFASL reduced the survival of mice (Fig. 7O–R, Fig. S11B–C). Similar histology was observed between isotype and αFASL treated tumors at endpoint (Fig. S11D–E). MRTX1133 treatment was associated with reduced Ki67, whereas αFASL treated tumors had reduced cancer cell apoptosis (Fig. S11H–I). Together these data indicate that clearance of cancer cells following pharmacological inhibition of KRAS* requires CD8+ T cell mediated killing through engagement of FAS on cancer cells with FASL on CD8+ T cells.
Figure 7: FAS-FASL engagement contributes to the clearance of cancer cells following MRTX1133 treatment.
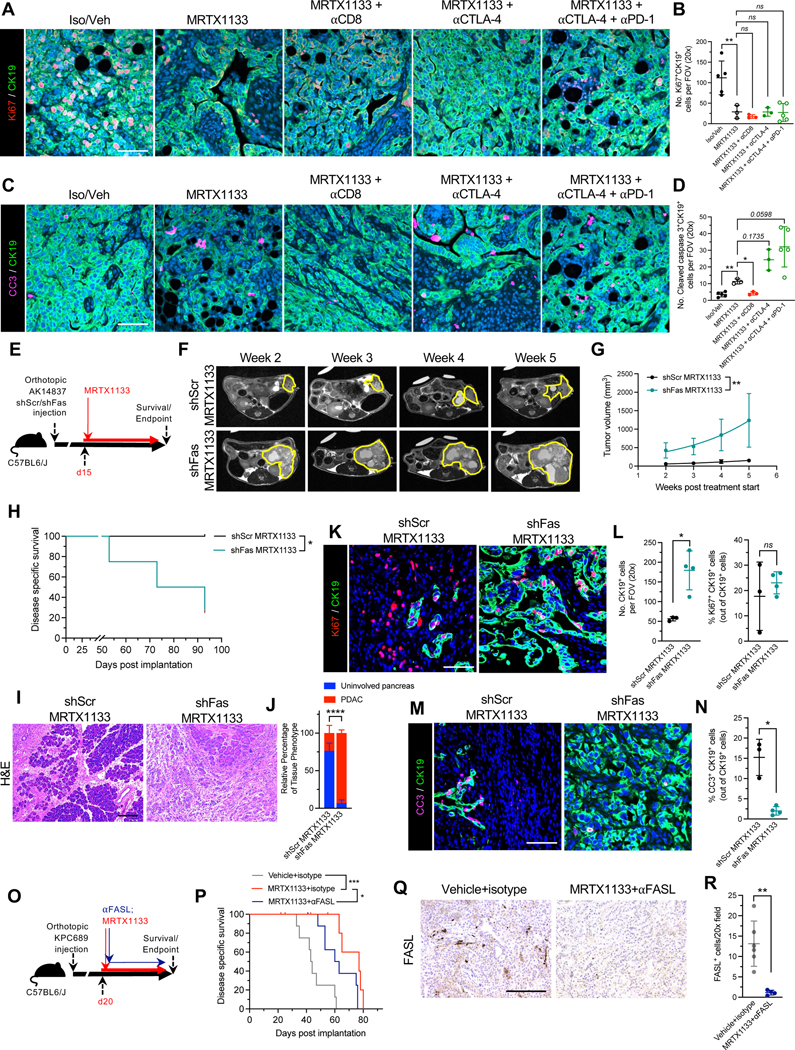
(A-B) Representative images of Ki67 (red) and CK19 (green) immunostained PKC-HY19636 tumors (A) and quantification of Ki67+CK19+ cells (B) (Iso/Veh, n=5; MRTX1133, n=3; MRTX1133 + αCD8, n=3; MRTX1133 + αCTLA-4, n=3; MRTX1133 + αCTLA-4 + αPD-1, n=5). (C-D) Representative images of cleaved caspase 3 (CC3, pink) and CK19 (green) immunostained PKC-HY19636 tumors (C) and quantification of CC3+CK19+ cells (D) (Iso/Veh, n=5; MRTX1133, n=3; MRTX1133 + αCD8, n=3; MRTX1133 + αCTLA-4, n=3; MRTX1133 + αCTLA-4 + αPD-1, n=5). (E) Schematic of treatment of AK14837 shCtrl and shFas with MRTX1133. (F-G) Representative MRI images (F) and quantification of tumor volume (G) (shCtrl, n=4; shFas, n=4). (H) Survival curve of mice with orthotopic KPC689 tumors treated with the indicated groups (shScr MRTX1133, n=4; shFas MRTX1133, n=4). (I-J) Representative H&E images (I) and associated histological scoring (J) (shScr MRTX1133, n=4; shFas MRTX1133, n=4). (K-L) Representative images of Ki67 (red) and CK19 (green) immunostained AK14837 tumors (K) and quantification (L) (shScr MRTX1133, n=3; shFas MRTX1133, n=4). (M-N) Representative images of cleaved caspase 3 (CC3, pink) and CK19 (green) immunostained AK14837 tumors (M) and quantification (N) (shScr MRTX1133, n=3; shFas MRTX1133, n=4). (O) Schematic of treatment of KPC689 with MRTX1133 and αFASL. (P) Survival curve of mice with orthotopic KPC689 tumors treated with the indicated groups (Vehicle+isotype, n=8; MRTX1133+isotype, n=9; MRTX1133+αFASL, n=9). (Q-R) Representative FASL immunostaining images (Q) and quantification (R) of KPC689 tumors (Vehicle+isotype, n=6, MRTX+αFASL, n=4). Scale bars, 100 μm. One-way ANOVA with Bonferroni’s multiple comparison test performed for (B). Brown-Forsythe ANOVA with Dunnett’s multiple comparison test performed for (D). The fits of exponential growth equations were compared for (G). Log-rank test performed for (H) and (P). Two-way ANOVA with Sidak’s multiple comparison test performed for (J). Unpaired t-test performed for right panel of (L) and unpaired Welch’s t-test performed for left panel of (L), (N), and (R). * P<0.05, ** P<0.01, *** P<0.001, ****, P<0.0001, ns: not significant.
See also Figure S11.
Discussion
Recent identification of a non-covalent small molecule inhibitor (MRTX1133) with specificity to the KRASG12D mutant protein has offered an opportunity to evaluate its efficacy and mechanism of action directly on KRAS* cancer cells and the entire PDAC tumor 6,29. Here, we study MRTX1133 in 16 different mouse models of pancreatic cancer, which include orthotopic xenografts, orthotopic syngeneic models, patient derived xenografts, the KC genetic model, and the KPPC genetic model. The goal was to focus on the efficacy of MRTX1133 against early and late stage PDAC and determine the impact of this inhibitor on KRAS* PDAC cells, along with its impact on the tumor microenvironment, with a specific focus on immune cells.
We show that MRTX1133 has significant impact on the growth of early stage tumors with tumor stasis observed in human xenograft models, as also shown in other recent studies 6,29. We demonstrate that MRTX1133 inhibits proliferation of PDAC cells and suppresses pERK, and such activity has a dominant impact on inducing inhibition of tumor growth; however, for sustained suppression of tumor growth and tumor clearance, CD8+ T cells are required. The critical function of CD8+ T cells in tumor clearance became apparent when treatment with MRTX1133 was initiated in mice with large tumor burden associated with advanced PDAC. In this regard, we show that genetic extinction of KRAS* results in upregulation of FAS via demethylation of FAS promoter on PDAC cells, rendering them vulnerable to FASL mediated apoptosis by CD8+ T cells (Mahadevan et al 2023 in review). Here, we demonstrate that FAS-FASL engagement is required for cancer cell clearance and efficacy of MRTX1133.
While other studies have suggested that T cells are not required for suppression of tumor growth in response to MRTX1133 in subcutaneous and orthotopic PDAC tumors, they involve short term experiments with low tumor burden (approximately 50 mm3) that is not reflective of typical clinical presentation and treatment of disease and such experiments depleted both CD4+ and CD8+ T cells 29, which includes both cytotoxic CD8+ T cells and tumor suppressive CD4+ T regulatory cells. Previous studies have demonstrated that the relative abundance of T cell subsets and overall T cell abundance changes during PDAC progression 18, suggesting that the contribution of T cells to MRTX1133 efficacy is dependent on the stage of disease. In the context of small tumors, it is likely that tumor stasis due to suspended proliferation can appear effective but in the absence of complete abrogation of proliferation and effective induction of cancer cell clearance, tumor reemergence driven by residual cancer cells or drug resistance is possible. Our study demonstrates that MRTX1133 is effective in regression of established tumors associated with advanced PDAC. Such activity depends on the activity of CD8+ cells and their suppression leads to rapid tumor growth despite treatment with MRTX1133.
These experiments suggest that evaluation of the efficacy of MRTX1133 in pre-clinical studies must involve mouse models with different tumor growth kinetics that include both early and late stage PDAC. Using this rationale, our study uniquely identifies a dominant impact of MRTX1133 in regressing pre-established, advanced and aggressive PDAC with large tumor burden. Guided by the data that demonstrates the emergence of T cells upon treatment with MRTX1133, we evaluated the combination of MRTX1133 with αCTLA-4 and αCTLA-4+αPD-1 ICB. These results show that in mice treated with MRTX1133 monotherapy, the PDAC tumors eventually escape tumor inhibition, whereas combination with αCTLA-4 therapy conferred sustained tumor inhibition and long term survival.
Collectively, our studies demonstrate that MRTX1133 is specific and efficient inhibitor of KRAS* and with the ability to regress PanINs and advanced PDAC in autochthonous and orthotopic models of PDAC. MRTX1133 treatment reprograms tumor immunity and stromal fibroblasts and synergizes with ICB to further regress tumors with increase in overall survival of treated mice. Such combination treatment opportunity informs current clinical development of MRTX1133.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Raghu Kalluri (rkalluri@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
scRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been included in the Supplemental Material and source data is deposited at Mendeley as listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact, upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse-FASL (CD178) | Bio X Cell | Catalog #: BE0319 RRID:AB_2819046 |
| InVivoPlus polyclonal Armenian hamster IgG | Bio X Cell | Catalog #: BE0091 RRID:AB_1107773 |
| InVivoMAb anti-mouse CD8α | Bio X Cell | Catalog #: BE0004–1 RRID:AB_1107671 |
| InVivoMab anti-mouse CD19 | Bio X Cell | Catalog #: BE0150 RRID:AB_10949187 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | Bio X Cell | Catalog #: BE0131 RRID:AB_10950184 |
| InVivoPlus anti-mouse PD-1 (CD279) | Bio X Cell | Catalog #: BE0273 RRID:AB_2687796 |
| InVivoPlus rat IgG2a isotype control | Bio X Cell | Catalog #: BE0089 RRID:AB_1107769 |
| InVivoPlus polyclonal Syrian hamster IgG | Bio X Cell | Catalog #: BE0087 RRID:AB_1107782 |
| Cytokeratin 19 | Abcam | Catalog #: ab52625 RRID:AB_2281020 |
| CD19 | Cell Signaling | Catalog #: 3574 RRID:AB_2275523 |
| CD8 | Cell Signaling | Catalog #: 85336 RRID:AB_2800052 |
| CD11b | Abcam | Catalog #: ab133357 RRID:AB_2650514 |
| pERK | Cell Signaling | Catalog #: 4376 RRID:AB_331772 |
| Goat Anti-Rabbit IgG Antibody (H+L), Biotinylated | Vector Laboratories | Catalog #: BA-1000 RRID:AB_2313606 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) | Cell Signaling | Catalog #: 4370 RRID:AB_2315112 |
| Alexa Fluor 647 anti-HLA A | Abcam | Catalog #:199837 RRID:AB_2728798 |
| Ki67 | Thermo Fisher Scientific | Catalog #: RM-9106-S RRID:AB_2341197 |
| FAS | Abcam | Catalog #: ab133619 N/A |
| Podoplanin | Abcam | Catalog #: ab11936 RRID:AB_298718 |
| Peroxidase-AffiniPure F(ab’)2 Fragment Goat Anti-Syrian Hamster IgG (H+L) | Jackson Immunoresearch Laboratories | Catalog #: 107–036-142 RRID:AB_2337455 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling | Catalog #: 9661 RRID:AB_2341188 |
| FASL | Santa Cruz Biotechnology | Catalog #: sc-834 RRID:AB_2100640 |
| CD16/32 | TONBO Biosciences | Catalog #: 70–0161 RRID:AB_2621487 |
| TruStain FcX (anti-mouse CD16/32) | BioLegend | Catalog #:101320 RRID:AB_1574975 |
| p44/42 MAPK (Erk1/2) | Cell Signaling | Catalog #: 9102 RRID:AB_330744 |
| Vinculin | Abcam | Catalog #: ab129002 RRID:AB_11144129 |
| Goat Anti-Rabbit Secondary HRP Conjugate | Protein Simple | Catalog #: 042–206 RRID:AB_2860577 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Catalog #: 4370 RRID:AB_2315112 |
| Donkey polyclonal Secondary Antibody to Rabbit IgG - H&L (HRP) | Abcam | Catalog #: ab16284 RRID:AB_955387 |
| Pacific Blue anti-mouse CD45 | BioLegend | Catalog #: 103126 RRID:AB_493535 |
| PE-Cyanine7 anti-mouse CD3e | Thermo Fisher Scientific | Catalog #: 25–0031-82 RRID:AB_469572 |
| BV605 anti-mouse CD4 | BioLegend | Catalog #: 100548 RRID:AB_2563054 |
| BV650 anti-mouse CD8 | BioLegend | Catalog #: 100742 RRID:AB_2563056 |
| BV711 anti-CD11b | BD Biosciences | Catalog #: 563168 RRID:AB_2716860 |
| RPE anti-mouse F4/80 | BioRad | Catalog #: MCA497PE RRID:AB_322048 |
| PerCP-Cy5.5 anti-mouse CD19 | BD Biosciences | Catalog #: 561113 RRID:AB_10563071 |
| PE-CF594 anti-mouse CD11c | BD Biosciences | Catalog #: 562454 RRID:AB_2737617 |
| PE anti-mouse NK1.1 | Thermo Fisher Scientific | Catalog #: 12–5941-83 RRID:AB_466051 |
| BV510 anti-mouse CD45 | BioLegend | Catalog #: 103137 RRID:AB_2561392 |
| BV786 anti-mouse CD11b | BD Biosciences | Catalog #: 740861 RRID:AB_2740514 |
| APC anti-mouse CD178 (FasL) | BioLegend | Catalog #: 106609 RRID:AB_2813951 |
| BV421 anti-mouse CD95/FAS | BD Biosciences | Catalog #: 562633 RRID:AB_2737690 |
| Biological samples | ||
| Patient-derived xenografts, see table S2 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MRTX849 (adagrasib) | MedChem Express | Catalog #: HY-130149 |
| MRTX1133 | WuXi AppTech | N/A |
| TrypLE Express | Gibco | Catalog #: 12605028https://www.thermofisher.com/order/catalog/product/12605028 |
| Doxycycline | West-Ward Pharmaceuticals | NDC#: 0143–2122-50 |
| D-Luciferin | GoldBio | Catalog #: LUCK-1G |
| Captisol | Selleck | Catalog #: S4592 |
| Sodium citrate pH 5 | Teknova | Catalog #: S8450 |
| Collagenase, type IV | Gibco | Catalog #: 17104019 |
| Dispase II | Gibco | Catalog #: 17105041 |
| Collagenase, type I | Gibco | Catalog #: 17018029 |
| cOmplete EDTA free-protease inhibitor | Roche | Catalog #: 4693159001 |
| PhosSTOP | Roche | Catalog #: 4906845001 |
| TRIzol reagent | Invitrogen | Catalog #: 15596026 |
| Critical commercial assays | ||
| CellTiter-Glo | Promega | Catalog #: G7570 |
| DUSP6 RNAscope probe | Advanced Cell Diagnostics | Catalog #: 405361 |
| SPRY4 RNAscope probe | Advanced Cell Diagnostics | Catalog #: 546711-C2 |
| URiSCAN test strips | BioSys Laboratories | Catalog #: U39 |
| Chromium Next GEM Single Cell 3ʹ Kit v3.1 | 10x Genomics | Catalog #: 1000268 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Catalog #: 20024907 |
| 12–230 kDa Separation Module | ProteinSimple | Catalog #: SM-W001 |
| Pierce BCA Protein Assay Kit | Thermo Fischer | Catalog #: 23225 |
| PureLink RNA Mini Kit | Invitrogen | Catalog #: 12183018A |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Applied Biosystems | Catalog #: 4374966 |
| Fast SYBR Green Master Mix | Applied Biosystems | Catalog #: 4385612 |
| Deposited data | ||
| scRNA-seq data | This paper | GEO: GSE228502 |
| Source data | This paper | https://data.mendeley.com/v1/datasets/h2mvr28n3k/draft?preview=1 |
| Experimental models: Cell lines | ||
| Cell lines, see Table S1 | This paper | N/A |
| Experimental models: Organisms/strains | ||
| B6.FVB-Tg(Pdx1-cre)6Tuv/J | Hingorani et al.2 | RRID:IMSR_JAX:014647 |
| B6.129S4-Krastm4Tyj/J | Hingorani et al.2 | RRID:IMSR_JAX:008179 |
| Trp53loxP/loxP | Chen et al.30 | N/A |
| C57BL/6J | Jackson laboratories | RRID:IMSR_JAX:000664 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson laboratories | RRID:IMSR_JAX:005557 |
| Crl:NU(NCr)-Foxn1nu | Charles River Laboratories | RRID:IMSR_CRL:490 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S5 | This paper | N/A |
| Recombinant DNA | ||
| pLKO.1 shFas: 5’-CCGGGTGTTCTCTTTGCCAGCAAATCTCGAGATTTGCTGGCAAAGAGAACACTTTTT-3’ | Sigma-Aldrich | Catalog #: TRCN0000012328 |
| pLKO.1 sh-Scr: 5’-CCGGGTCGGTATTCGTATCCTAACTCTCGAGATTTGCTGGCAAAGAGAACACTTTTT-3’ | Sigma-Aldrich | Catalog #: TRCN0000012328 |
| F-Luc-GFP Lentivirus | Capital Biosciences | Catalog #: VSL-0088P |
| Software and algorithms | ||
| Cell Ranger 7.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| R (version 4.0.0) package Seurat (version 4.0.3) | Butler et al.40 | https://github.com/satijalab/seurat/releases/tag/v4.0.3 |
| GSEA 4.2.2 | Mootha et al. and Subramanian et al.41,42 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| Other | ||
| VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Standard) | Vector Laboratories | Catalog #: PK-6100 |
| Rabbit-on-rodent HRP polymer | Biocare Medical | Catalog #: RMR622G |
| Opal 690 reagent | Akoya Biosciences | Catalog #: FP1497001KT |
| Opal 520 reagent | Akoya Biosciences | Catalog #: FP1487001KT |
| Opal 570 reagent | Akoya Biosciences | Catalog #: FP1488001KT |
| Hoechst 33342 | Invitrogen | Catalog #: 62249 |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories | Catalog #: H-1000–10 |
| Fluoroshield with DAPI | Sigma-Aldrich | Catalog #: F6057 |
| Fixable Viability Dye eFluor 780 | Invitrogen | Catalog #: 65–0865-14 |
| BD Horizon Brilliant Stain buffer | BD Biosciences | Catalog #: 563794 |
| BD Cytofix Fixation Buffer | BD Biosciences | Catalog #: 554655 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mouse models
All procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at MD Anderson Cancer Center (MDACC). Male and female, 6 to 16 weeks old C57BL/6J (KPC689 GFP Luc and PKC-HY19636 cells) and NSG (for KPC689 GFP Luc, Panc-1 GFP Luc, or HPAC cells) mice were purchased from Jackson Laboratories. Mice were randomly assigned to groups and treatment was initiated at the indicated days post-surgery. Genetically engineered mice includiing LSL-KrasG12D/+; Pdx1-Cre (KC) 2 and LSL-KrasG12D/+; Trp53L/L 30; Pdx1-Cre (KPPC) mice were maintained on mixed backgrounds and started on treatment at the indicated times post-birth. PDXs (Table S2) were derived as previously described 31 and implanted subcutaneously into 11 weeks old athymic nude mice (Charles River Laboratories). All animals received LabDiet 5053 chow and sterile water ad libitum and were group housed with a 12-hour-light/dark cycle at 21°C −23°C and 40%−60% humidity.
Cell lines and small molecule inhibitors
Cell lines employed in these studies, their origin, and culture conditions are listed in Table S1. All cells were cultured at 37°C and 5% CO2. Cells were routinely tested for mycoplasma with LookOut Mycoplasma PCR Detection Kit (Sigma Aldrich) and confirmed to be mycoplasma negative. HPAC, Panc1, A549, HCT116, MIA PaCa-2, and PSN-1 were STR verified by the MDACC Cytogenetics and Cell Authentication Core. MRTX849 (adagrasib) was purchased from MedChem Express. MRTX1133 was synthesized by WuXi AppTech. MRTX849 and MRTX1133 compounds were structurally verified by NMR and LC/MS. For cell culture experiments, MRTX849 and MRTX1133 were diluted in dimethyl sulfoxide (DMSO).
METHOD DETAILS
Cell culture
KPC689 cells were isolated from a spontaneous LSL-KrasG12D/+ 32; LSL-Trp53R172H/+ 33; Pdx1-Cre 2 (KPC) tumor as previously described 34,35 and serially passaged in C57BL6/J mice. PKC-HY19636 cells were isolated from a spontaneous P48-Cre; LSL-KrasG12D/+, Trp53L/+ tumor as previously described 36. AK14837 were isolated from a P48-Cre; tetO/CMV-LSL-KrasG12D/+, Trp53L/L, LSL-rtTA-EGFP tumor as previously described 4. shScr and shFas cells were generated by transducing AK14837 with shRNA lentiviral particles (Sigma Aldrich, TRCN0000012328) with 4 μg/mL polybrene (EMD Millipore, TR-1003-G) and selected with 10 μg/mL puromycin (Fischer scientific, AAJ672368EQ) 24 hours after infection. The shRNA oligo sequence (target sequence in bold) for is as follows: shFas: 5’-CCGGGTGTTCTCTTTGCCAGCAAATCTCGAGATTTGCTGGCAAAGAGAACACTTTTT-3’. The control (sh-Scr target 5’ GTCGGTATTCGTATCCTAACT 3’) was prepared by generating scrambled sequence from the sh-Fas#28 target sequence in the same pLKO.1 backbone. Panc1 and KPC689 were engineered to stably express GFP and firefly luciferase by lentiviral infection (Capital Biosciences) and maintained in 2 μg/mL puromycin (Sigma Aldrich).
Patient derived organoids
The DH-50 PDO was generated from a liver metastasis lesion of a PDAC patient and expanded as a patient derived xenograft. PDO cultures and expansion was performed as described previously 37. Briefly, PDO was cultured in Nunclon™ Delta Surface (Thermoscientific) on Geltrex dome (Reduced growth factor basement membrane, hESV qualified, Gibco) suspended in PaTOM growth media 37. NCI-65R was suspended in Tuveson growth media 38,39. TrypLE Express (Gibco) was used for passage of organoids and DMEM, 10% FBS and 1% P/S media was used to neutralize TrypLE.
Cell proliferation assays
Cells were plated at the density of approximately 2,500 cells/well in clear-bottom, white-wall 96-well plates (Corning) in DMEM (Gibco) supplemented with sodium pyruvate (Sigma Aldrich), PS and 2.5% FBS. After 24 hours, old media was removed and replaced with a serial dilution of MRTX1133 starting from 10 μM for three days. The CellTiter-Glo (Promega) assay was used to assess the viability of cells. CellTiter-Glo reagents and plates were allowed to equilibrate at room temperature, and 50 μl of reagent was added to 50 μl of media per well. The luminescent signal was read using a FLUOstar Omega plate reader (BMG LABTECH). Data were analyzed by normalizing the cell densities to the control wells. Constraints for IC50 calculations were set for the bottom to be less than 0.5 and top less than 1.0. Cell lines that did not have a calculable IC50 in the range of MRTX1133 evaluated are listed as ‘n.d.’ or not determined.
Animal studies
Mice were implanted orthotopically with KPC689 GFP Luc, PKC-HY19636, AK14837 shScr, or AK14837 shFas (0.5×106 cells in 20 μL of PBS), Panc-1 GFP Luc or HPAC (1×106 cells in 20 μL of PBS) by injection into the tail of the pancreas using a 27-gauge Hamilton syringe. Mice injected with AK14837 were given water with 2 g/L doxycycline (West-Ward, NDC 0143–2122-50) and sucrose (20 g/L) starting at one day prior to orthotopic injection and continued throughout the experiment. For detection of tumor burden, luciferase expression was measured by injecting the mice i.p. with D-Luciferin (Goldbio) per the manufacturer’s instructions and imaged via IVIS (Xenogen Spectrum). Alternatively, MRI imaging was performed on day 12 and followed up with serial measurements to assess tumor volumes using 7T Bruker MRI at the MDACC Small Animal Imaging Facility (SAIF). Genetically engineered mice includiing LSL-KrasG12D/+; Pdx1-Cre (KC) and LSL-KrasG12D/+; Trp53L/L 30; Pdx1-Cre (KPPC) mice were maintained on mixed backgrounds and started on treatment at the indicated times post-birth. PDXs (Table 2) were derived as previously described 31 and implanted subcutaneously into 11 weeks old athymic nude mice. Tumor volumes were captured by serial caliper measurements (twice weekly) and treatment was initiated when tumors reached 150 to 250 mm3. Tumor volume (TV) was calculated as TV = (D × d2/2), where “D” is the larger and “d” is the smaller superficial visible diameter of the tumor mass. All measurements were documented as mm3. Treatment groups included 6 mice per group. For acute pharmacodynamic (PD) biomarker studies (n=4 mice per group), tumors were allowed to grow to an average volume of 250–350 mm3 and collected 4 hours after the last dose of treatment and fixed in 10% neutral buffered formalin overnight and then processed and embedded in paraffin.
Mice in control groups were administered 30 mg/kg MRTX1133 BID i.p. in 100 μL of 10% Captisol (Selleck) in 50mM sodium citrate pH 5 (Teknova, vehicle) or 30 mg/kg MRTX849 QD p.o. in 100 μL of vehicle. VehicleMRTX1133 consisted of vehicle administered BID i.p. and VehicleMRTX849 consisted of vehicle administered QD p.o. For FASL depletion experiments, mice were administered 250 μg of anti-FASL (BioXcell, BE0319) or isotype antibody (BioXcell, BE0091) diluted with PBS to a final volume of 100 μL three times per week. Concomitant with MRTX1133 (30 mg/kg, BID 5 days per week i.p.) treatment from day 13, depletion of CD8+ cells and CD19+ cells was performed using 200 μg of anti-mouse CD8 (BioXcell, clone 53–6.7) and anti-CD19 (BioXcell, clone 1D3) respectively twice per week (diluted to a final volume of 100 μL in PBS). For checkpoint immunotherapy, anti-mouse CTLA-4 (BioXcell, clone 9H10) and/or anti-mouse PD-1 (BioXcell, clone 29F.1A12) were injected three times per week i.p., starting dose 200 μg followed by 2 doses of 100 μg each (diluted to a final volume of 100 μL in PBS). The control (Isotype/vehicle) mice received a cocktail of Rat IgG2a (BioXcell, clone 2A3), Syrian hamster IgG (BioXcell, BE0087) (200 μg of each antibody diluted in a final volume of 100 μL in PBS) and 100 μL of vehicle i.p. The mice were euthanized at the indicated times post treatment.
Histological analyses
3 to 5 μm thick sections were made from formalin fixed paraffin embedded (FFPE) blocks and were deparaffinized and hydrated. Antigen retrieval was performed in Tris-EDTA buffer (pH 9.0) for 20 minutes at 95°C in a steamer. Subsequently, a hydrophobic barrier was created around the tissue sections using Pap pen, and sections were incubated with 3% H2O2 in PBS for 15 minutes. For blocking, the slides were incubated in 1.5% bovine serum albumin in PBST (0.1% Tween 20) for 30 minutes. The sections were incubated with primary antibodies, CK19 (Abcam ab52625, 1:250), CD19 (Cell Signaling 3574s; 1:100), CD8 (Cell Signaling 85336S, 1:300), CD11b (Abcam ab133357, 1:1000), pERK (Cell Signaling 4376S, 1:250), or FASL (Santa Cruz, sc-834; 1:100), diluted in blocking buffer overnight at 4 °C or for 3 hours at room temperature. For CD11b, FASL, pERK and CD8 immunohistochemistry, the slides were incubated in biotinylated anti-rabbit secondary (Vector Laboratories BA-1000, 1:250) for 30 minutes in blocking buffer followed by incubation in ABC reagent (Vector Laboratories PK-6100, prepared as per manufacturer’s instructions) for 30 minutes. Subsequently, slides were stained with DAB (Life Technologies), counterstained with haematoxylin, cover slipped and imaged with a Leica DM1000 LED microscope mounted with a DFC295 microscope camera (Leica) with LAS version 4.4 software (Leica) or a Zeiss AxioScan 7. Multiple representative images were taken from each tissue section and quantified by counting the number of positive cells in each visual field or using Image J (NIH) for quantification of area.
For both acute PD studies and efficacy studies on PDAC PDX models, sections were subjected to an initial heat-induced epitope retrieval (HIER) in citrate buffer, pH 6, at 95°C for 15 minutes. Anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (1:2000, Cell Signaling Technology #4370) was developed using Opal tyramide signal amplification (TSA) followed by direct immunofluorescence of HLA conjugated to Alexa 647 (1:250, Abcam #199837). RNAscope in situ hybridization assay was performed following manufacturer’s protocol (Advanced Cell Diagnostics, Inc) using DUSP6 (cat# 405361) and SPRY4 (cat# 546711-C2) probes. Appropriate positive and negative controls were included with the study sections. Digital images of whole-tissue sections were acquired using Vectra Polaris Automated Quantitative Pathology Imaging System (Akoya Biosciences) and representative regions were selected for each whole slide and processed using inForm Software v2.4 (Akoya Biosciences). Processed images were then analyzed using HALO Software v3.2 (Indica Labs Inc.). For immunostaining of PDXs for CK19, Ki67 and FAS, antigen retrieval was performed in citrate buffer (pH 6.0) at 95°C for 15 minutes in a BioGenex EZ – Retriever microwave, blocked with 1% BSA for 15 minutes, incubated in FAS antibody (Abcam ab133619, 1:200), washed with TBS followed by incubation in rabbit-on-rodent HRP polymer (Biocare Medical) for 10 minutes, and Opal 690 reagent (Akoya Biosciences, 1:100) for 10 minutes. The process was repeated with anti-CK19 (Abcam ab52625, 1:400) with Opal 520 (Akoya Biosciences, 1:100) and Ki67 (Thermo Fisher Scientific RM-9106-S, 1:400) with Opal 570 (Akoya Biosciences, 1:100). Tissues were stained with Hoechst 33342 (Invitrogen, 1:10,000) for 10 minutes, washed with TBS, mounted with Vectashield (Vector Laboratories), and imaged with a Zeiss Axio Observer Z1. Image analysis was performed in ImageJ.
For CK19 and CD8 immunofluorescence staining, the slides were incubated in rabbit-on rodent polymer (Biocare Medical) for 15 minutes following the primary antibody incubation described above. Subsequently, the sections were incubated in Opal 520 diluted in blocking buffer (for CK19 and CD8 primary) (Akoya Biosciences, 1:100) for 15 minutes. For CD8/Ki67 costaining, another round of antigen retrieval was performed as described above and the sections were incubated in blocking buffer for 30 minutes and in Ki67 primary antibody (Thermo Fischer Scientific RM-9106-S, 1:100) diluted in blocking buffer overnight at 4°C. The slides were then incubated in rabbit-on rodent polymer for 15 minutes, followed by incubation in Opal 690 reagent for 15 minutes (Akoya Biosciences, 1:100) in blocking buffer. Following another round of antigen retrieval, the slides were coverslipped with Fluoroshield with DAPI (Sigma Aldrich). PBST was used for washes between steps (3 mins each wash for 3 times). Imaging was performed with a Zeiss LSM800 confocal microscope with a 20x objective and quantified by counting the number of positive cells in each visual field.
For podoplanin immunofluorescence staining, antigen retrieval in TE buffer (pH 9.0) was performed for 15 minutes, followed by blocking in 1% BSA in TBS for 15 minutes. Slides were incubated in podoplanin primary antibody (Abcam ab11936, 1:200) for 1 hour at room temperature, washed with TBS for 2 minutes 3 times, then incubated with HRP goat anti-Syrian hamster IgG (Jackson Immunoresearch Laboratories 107–036-142, 1:250), washed with TBS for 2 minutes 3 times, and incubated in Opal 690 for 10 minutes. Antigen retrieval was repeated, followed by incubation in Hoechst 33342 (Invitrogen, 1:10,000) for 10 minutes and mounting with Vectashield mounting media (Vector Labs). Imaging was performed with a Zeiss LSM800 confocal microscope with a 20x objective and quantification of positive area performed in ImageJ (NIH). Cleaved caspase 3 (CC3), CK19, and Ki67 costaining was performed similar to CK19, Ki67, and FAS staining (described above), but with antigen retrieval for CC3 performed in TE buffer (pH 9.0) and incubated in CC3 antibody (Cell signaling 9661S, 1:400) and Opal 690 reagent (Akoya Biosciences, 1:100). Slides were imaged with a Zeiss Axio Observer Z1 or Zeiss LSM800 confocal microscope and image analysis was performed in ImageJ.
For histopathological analysis of H&E stained tissues, pancreas/tumors were analysed under bright field microscopy with multiple low power (10x magnification) representative images to define the relative percentages of uninvolved/normal looking pancreas, acinar-ductal metaplasia (ADM) or pancreatic intra-epithelial neoplasia (PanIN) leisons, and invasive PDAC lesions (classified further into well, moderate and poorly differentiated lesions). Alternatively, H&E stained pancreas sections were imaged with a Zeiss Axioscan 7 and the PDAC-containing regions measured in ImageJ (NIH). Liver necrosis was defined as areas of approximately than 5–10 hepatocytes or greater, and spot necrosis was defined as areas of 5 or less hepatocytes. Scores were set as 0 for no necrosis, 1 for 1–2 areas of necrosis detected, and 2 for 3 or more areas of necrosis detected per liver section. For binucleated cells, 5 periportal 400X field of view per mouse were selected at random and composite socres were defined as 0 for 1–2 binucleated cells, 1 for 3–5 binucleated cells, and 2 for greater than 6 binucleated cells per field of view. Kidney tubular damage was ascertained on 8 to 10 random 400X cortical fields of view and scored as 0 for no damaged tubules noted, 1 for 1 damaged tubule, and 2 for 2 or more damaged tubules. Kidney immune infiltration was ascertained on 5 to 8 random 400X cortical and papillary fields of view and scored as 0 for no significant (above healthy control) immune infiltrate, 1 for one or more fields of view with peritubular inflammation greater than observed in control (10–20 immune cells).
Blood chemistry and urine analyses
Blood was collected without anti-coagulant via retro-orbital bleeding or cardiac puncture and incubated on ice for 20 minutes. The samples were then centrifuged at 6,000 rpm for 5 minutes at 4°C and the serum samples were submitted to the MDACC DVMS Veterinary Pathology Services for assessment of total bilirubin, albumin, alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood-urea-nitrogen (BUN), lactate dehydrogenase (LDH), calcium, chloride, globulin, creatinine, potassium, sodium, phosphorus, glucose and total protein. Urine was collected and protein levels measured using URiSCAN test strips (BioSys Laboratories).
Pancreas digestion and scRNA-seq
Pancreata/tumors were collected and combined from 3 mice for vehicle groups and MRTX849 treated mice and 6 mice for MRTX1133 treated mice. Pancreata/tumors were minced and digested at 37°C for 30 minutes at 150 rpm in a 10 mL mixture of 4 mg/mL collagenase IV (Gibco) and 4 mg/mL dispase (Gibco). Cells were filtered once with a 70 μm filter and once more with a 40 μm filter. To stop the digestion, 25 mL of DMEM containing 10% FBS was added. Cells were then centrifuged at 500 g for 5 minutes, and the supernatant was removed, washed with 10 mL of FACS buffer (2% FBS in PBS), and centrifuged at 500 g for 5 minutes at 4°C. Cells were resuspended in 1 mL of FACS buffer and stained with 1 μL of Fixable Viability Dye eFluor™ 780 (eBioscience, 1:1,000) on ice for 10 minutes. Cells were washed with 3.5 mL of FACS buffer, centrifuged at 300 g for 5 minutes at 4°C, washed again, and resuspended in 1 mL of FACS buffer. Live cells were subsequently sorted via the BD FACSMelody cell sorter, followed by counting with Trypan Blue (Sigma Aldrich) exclusion with a Countess 3 (Invitrogen). For in vitro scRNA-seq, cells were treated with vehicle (DMSO), 50 nM MRTX1133, or 50 nM MRTX849 for 3 hours then trypsinized, washed, and counted. 5,000 cells were loaded into the 10x Chromium Controller to generate Single Cell 3’ Gene Expression dual indexed libraries (10x Genomics). The creation of Gel Bead-In-Emulsions (GEM) and subsequent cell barcoding, GEM-RT cleanup, cDNA amplification, and library assembly were performed with the Chromium Next GEM Single Cell 3’ Reagents Kits v3.1 (10x Genomics) according to manufacturer’s instructions. Paired-end, dual indexing sequencing was carried out using Illumina High Output Kits v2.5 (150 cycles) and the Illumina NextSeq500. The manufacturer’s specified parameters, combined with library loading and pooling, were used to determine the sequencing read cycles.
scRNA-seq data processing and analysis
Raw FASTQ files of scRNA sequencing data were processed using Cell Ranger 7.0.1 to generate the count matrices. Pre-built mm10, GRCh38, and barnyard reference genomes of both mouse (mm10) and human (GRCh38) downloaded from the 10X genomics website (https://support.10xgenomics.com/single-cell-gene expression/software/downloads/latest) were used for the alignment of single-cell data from different mouse models and cell lines. scRNA-seq analysis was conducted using the R (version 4.0.0) package Seurat (version 4.0.3)40 to process the expression matrices and perform downstream analysis. Multiple functions implemented in Seurat were used. To avoid the analysis driven by noise and low-quality cells, cells with a limited number of genes were discarded using the quantile function from R (version 4.0.0) to determine the range of genes to filter low-quality cells. The quantile range of genes was set from 2.5% to 97.5% and we discarded the cells with genes over 97.5% and less than 2.5%. Cells with more than 5% of mitochondrial counts were filtered for downstream analysis. The “Sctransform” function was used to normalize and stabilize the variance of expression matrices. The expression matrices were dimension reduced with principal component analysis (PCA). ‘FindNeighbors’ was used to define the nearest neighbors among cells in the PCA space, number of top principal components was determined by checking the plots generated by ‘JackStrawPlot’ and ‘ElbowPlot’ functions, and then ‘FindClusters’ was used to group cells with the Louvain algorithm based on the selected resolution. ‘RunUMAP’ function was used for visualizing the UMAP dimension reduction clusters. ‘DoHeatmap’ function was employed to display the top 10 genes for each meta cluster. ‘VinPlot’ function was used to show the expression probability distribution of genes across the defined cell clusters. GSEA was performed with GSEA 4.2.2 41,42 based on differentially expressed genes for each cluster and pathways with a FDR q-value < 0.1 reported.
Flow cytometry
Tumors or pancreata were minced and digested in 1.5 mg/mL type I collagenase (Thermo Fisher Scientific) in 5 mL PBS at 37°C for 20 minutes. Subsequently, the cell pellet was resuspended in cRPMI (10% heat inactivated FBS (Atlanta Biologicals, Atlanta, Georgia, USA), 1% PS, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol, filtered using 70 μm strainer (Corning), and centrifuged at 600 g for 3 minutes at 4°C. Subsequently, cells were incubated for 5 minuutes in RBC lysis buffer (ThermoFischer). Each sample was incubated with 100 μL surface antibody cocktail diluted in FACS buffer, 20% Brilliant Stain buffer (BD Bioscience) and 50 μg/mL anti-mouse CD16/32 (TONBO Biosciences) for 30 minutes on ice. Fixable viability dye-eFluor780 (FVD) (eBioscience) was used at 1:1000 concentration in the buffer with the antibody cocktail. To confirm systemic depletion of CD19+ cells following treatment with anti-CD19 antibody, spleens of WT and MRTX1133+αCD19 treated mice were strained through 70 μm filter in FACS buffer. Subsequently, the isolated splenocytes were processed for RBC lysis, stained with antibody cocktail and fixed as described above. For analysis of FASL expression, spleens were filtered with a 100 μm strainer (Corning), washed with FACS buffer (2% FBS in PBS), resuspended in staining buffer (50% FACS buffer, 50% Brilliant Stain buffer, 50 μg/mL TruStain FcX (Biolegend), antibodies, and fixable viability dye-eFluor780 (1:1000), and incubated for 1 hour on ice. For in vitro flow cytometry analysis, cells were detached with TrypLE Express, washed with PBS, resuspended in 50% FACS buffer and 50% Brilliant Stain buffer, and incubated in anti-mouse CD95/FAS-BV421 antibody (BD 562633, 1:100) for 1 hour on ice. Subsequently, cells were fixed with fixation buffer (BD Bioscience) and data were acquired using a BD LSR Fortessa-X20 and analysed with FlowJo v10.7.1. Antibodies panels used for flow cytometry are listed in Table S3–4.
Capillary western immunoassay (WES/Simple Western)
400,000 to 500,000 cells were seeded and after 24 hours, treated with vehicle (DMSO), MRTX1133, or MRTX849 for 3 hours prior to protein isolation with RIPA buffer containing 1x cOmplete EDTA free protease inhibitor (Roche) and 1x phosSTOP (Roche). In other experiments, 40,000 cells were seeded and after 24 hours, treated with vehicle (DMSO), MRTX1133, or MRTX849 for 24 hours and protein isolated as described above. 0.4–0.5 μg/μL of protein was prepared for Simple Western (Bio-Techne) according to manufacturer’s instructions. Samples were immunoassayed for phospho-p44/42 MAPK (Erk1/2) (Cell Signaling 4376S, 1:10), p44/42 MAPK (Erk1/2) (Cell Signaling 9102S, 1:10) and vinculin (Abcam ab129002, 1:250–1:500) and anti-rabbit secondary antibody (Bio-Techne) with the 12–230 kDa separation module on a Wes instrument (30 min separation at 475 V, 5 min blocking, 30 min primary antibody, 30 min secondary antibody, 15 min chemiluminescence detection). Compass software (Bio-Techne) was used to generate gel images.
Western blot
To analyze protein expression in MIA PaCa-2 and KPC689 cell lines and DH-50 and NCI-65R patient derived organoids (PDOs), cell lysates were prepared in RIPA lysis buffer and protein concentrations were measured using Bicinchoninic Acid (BCA) assay (Pierce™ BCA Protein Assay Kit, Thermo Fischer). The same amount of protein was loaded for each sample (20 to 40 μg of protein lysates) and diluted with Laemmli Sample Buffer (Biorad, 1610747) and RIPA buffer in a final volume of 30 μL and incubated at 95ºC for 5 min. 10 μg of NCI-65R derived proteins were loaded for each sample and diluted with with NuPAGE™ LDS Sample Buffer (Invitrogen) and NuPAGE™ Sample Reducing Agent (Invitrogen) in a final volume of 4 μL and incubated at 95°C for 5 min. Subsequently, the protein lysates were loaded into Bolt™ 4–12% Bis-Tris Plus gel (Thermo Fischer) for electrophoretic separation and transferred onto nitrocellulose membranes (Amersham™, Protran™). 5% non-fat milk in TBST was used as the blocking buffer for MIA PaCa-2 and KPC cell lysates and DH50 PDO lysates, and 5% BSA in TBST for NCI-65R PDO lysates. Following blocking, membranes were incubated overnight at 4ºC in the following primary antibodies: phospho-p44/42 MAPK (Erk1/2) (Cell Signaling 4376S or 4370, 1:1000), p44/42 MAPK (Erk1/2) (Cell Signaling 9102, 1:1000) and vinculin (Abcam ab129002, 1:10,000). Membranes were incubated in secondary antibodies (Donkey Anti-rabbit IgG H&L: Abcam ab16284, 1:10,000) for 1 hour at room temperature. Membranes were developed with chemiluminescence reagents (Pierce™ ECL Western Blotting Substrate) as per manufacturer’s recommendation. Uncropped, full length images from the western blots are included in the Supplemental Material.
Quantitative real-time PCR analyses
TRIzol reagent (Invitrogen) was added to cell culture after indicated treatments (on the culture dish after removing the media and washing with PBS) and incubated on ice for 5 minutes. Subsequently, RNA was extracted using Invitrogen™ PureLink™ RNA Mini Kit (Invitrogen) and stored at −80°C. 0.5 – 1.5 μg of the RNA (measured by Nanodrop spectrophotometer) was used for cDNA synthesis using High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems) as per the manufacturer’s instructions. RNA from cells were tested for knockdown of Fas/FAS by qPCR. The primer sequences are listed in Table S5. qPCR was run using Fast SYBR Green Master Mix (Applied Biosystems) and the QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). The relative fold change in gene expression was determined using the 2-ΔΔCt method, with the control group was arbitrarily set to 1. Statistical analyses were computed on biological replicates values of ΔCt.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphical representation of the data and statistical tests were performed using GraphPad Prism 9 and reported in the respective figure legends. Shapiro-Wilk test was used to assess normality of distribution of samples. For comparison of two groups, unpaired t-test (for samples with normal distribution) and Mann-Whitney test (for samples with non-normal distribution) were used for comparison of means. For comparison of more than 2 groups, one-way analysis of variance (ANOVA) with Tukey’s, Sidak’s, or Dunnett’s multiple comparisons test (for samples with normal distribution) and Kruskal-Wallis with Dunn’s multiple comparisons test (for samples with non-normal distribution) was used. Two-way ANOVA with Tukey’s or Sidak’s multiple comparisons test (for samples with normal distribution) or Brown-Forsythe ANOVA with Dunnett’s T3 multiple comparisons test (for samples with non-normal distribution) was used for comparison of histopathological analysis of tissue phenotypes. Log-rank test was used to compare Kaplan-Meier survival curves. P-values are reported as *P<0.05, **P<0.01, ***P<0.001, **** P<0.0001, ns: not significant. Exact p-values are reported where indicated.
Supplementary Material
Highlights.
KRASG12D inhibition with MRTX1133 reverses early and advanced PDAC growth
MRTX1133 increases CD8+ T cells and reprograms cancer associated fibroblasts
Regression of advanced PDAC in response to MRTX1133 requires CD8+ T cells
KRASG12D inhibition induces FAS to facilitate CD8+ T cell mediated death
Acknowledgements
This work was supported in part by Break Through Cancer and the Lustgarten Foundation for Pancreatic Cancer Research. KKM and KMM were supported by Ergon Foundation Post-Doctoral Trainee Fellowships. RK is a Distinguished University Chair supported by Sid W. Richardson Foundation. The Kalluri laboratory PDAC related studies are supported by NCI P01 CA117969. AM is also supported by the Sheikh Khalifa bin Zayed Foundation and NCI U54CA274371. Other support includes the Small Animal Imaging Facility and veterinary pathology services core facility at MD Anderson Cancer Center supported by NCI P30CA16672. We thank the MDACC Cytogenetics and Cell Authentication Core for STR testing, Lori Wilson and Traver Hart for assistance with sequencing, Nicolas Ryujin and Shabnam Shalapour for assistance with FACS sorting, Charles Kingsley for assistance with animal imaging, Sarah Patel for assistance with tissue processing, and Xiaoyan Ma, Andy Zuniga, Briana Epps, Diego Torres, Robert Mullinax and Angela Harris for their assistance with PDX related studies.
Footnotes
Declaration of interests
A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology, and this financial relationship is managed and monitored by the UTMDACC Conflict of Interest Committee. A.M. is also listed as an inventor on a patent that has been licensed by Johns Hopkins University to Thrive Earlier Detection and serves as a consultant for Freenome and Tezcat Biosciences.
Inclusion and Diversity Statement
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G, et al. (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203.e113. 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 3.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 4.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, and Pasca di Magliano M. (2012). Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122, 639–653. 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, Laguer J, Medwid J, Vanderpool D, Lifset E, et al. (2022). Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med 28, 2171–2182. 10.1038/s41591-022-02007-7. [DOI] [PubMed] [Google Scholar]
- 7.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, Dahlke JR, Fell JB, Fischer JP, Gunn RJ, et al. (2022). Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J Med Chem 65, 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- 9.Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, Chen JJ, Chen N, Frohn MJ, Goodman G, et al. (2020). Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J Med Chem 63, 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 10.Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, Liao WT, Cavallaro F, Denz P, Bernard V, et al. (2020). Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov 10, 608–625. 10.1158/2159-8290.CD-19-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao W, Rose JL, Wang W, Seth S, Jiang H, Taguchi A, Liu J, Yan L, Kapoor A, Hou P, et al. (2019). Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 568, 410–414. 10.1038/s41586-019-1062-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D. (2011). RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11, 761–774. 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, Rhim AD, Simeone DM, Beatty GL, and Pasca di Magliano M. (2017). Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 66, 124–136. 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, and Rustgi AK (2003). The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res 63, 2005–2009. [PubMed] [Google Scholar]
- 17.Zdanov S, Mandapathil M, Abu Eid R, Adamson-Fadeyi S, Wilson W, Qian J, Carnie A, Tarasova N, Mkrtichyan M, Berzofsky JA, et al. (2016). Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol Res 4, 354–365. 10.1158/2326-6066.CIR-15-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiraoka N, Onozato K, Kosuge T, and Hirohashi S. (2006). Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 12, 5423–5434. 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 19.McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H, et al. (2014). Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637. 10.1016/j.ccr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, Moghaddam SJ, and Dong C. (2014). T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci U S A 111, 5664–5669. 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, Zhang Y, Gomez EC, Morreale B, Senchanthisai S, et al. (2022). Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 40, 153–167 e111. 10.1016/j.ccell.2022.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 23.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, McAndrews KM, and Kalluri R. (2021). Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol 18, 792–804. 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, Li B, Wang H, Miyake T, Correa de Sampaio P, et al. (2022). Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov 12, 1580–1597. 10.1158/2159-8290.CD-20-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, and Evan GI (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J, Shoush J, Quinones L, Brown NV, Bassett JB, et al. (2023). Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov 13, 298–311. 10.1158/2159-8290.CD-22-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. (2005). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730. 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D, Cicalese A, Shah PK, Viale A, Pettazzoni PF, et al. (2016). In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep 16, 133–147. 10.1016/j.celrep.2016.05.063. [DOI] [PubMed] [Google Scholar]
- 32.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243–3248. 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, and Jacks T. (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, and Kalluri R. (2017). Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503. 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, and Kalluri R. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng Y, Xia X, Zhao Y, Zhao Z, Martinez C, Yin W, Yao J, Hang Q, Wu W, Zhang J, et al. (2021). Glucocorticoid receptor regulates PD-L1 and MHC-I in pancreatic cancer cells to promote immune evasion and immunotherapy resistance. Nat Commun 12, 7041. 10.1038/s41467-021-27349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, et al. (2015). Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 21, 1364–1371. 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, Sojoodi M, Li VS, Schuijers J, Gracanin A, et al. (2013). Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J 32, 2708–2721. 10.1038/emboj.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been included in the Supplemental Material and source data is deposited at Mendeley as listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact, upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMAb anti-mouse-FASL (CD178) | Bio X Cell | Catalog #: BE0319 RRID:AB_2819046 |
| InVivoPlus polyclonal Armenian hamster IgG | Bio X Cell | Catalog #: BE0091 RRID:AB_1107773 |
| InVivoMAb anti-mouse CD8α | Bio X Cell | Catalog #: BE0004–1 RRID:AB_1107671 |
| InVivoMab anti-mouse CD19 | Bio X Cell | Catalog #: BE0150 RRID:AB_10949187 |
| InVivoPlus anti-mouse CTLA-4 (CD152) | Bio X Cell | Catalog #: BE0131 RRID:AB_10950184 |
| InVivoPlus anti-mouse PD-1 (CD279) | Bio X Cell | Catalog #: BE0273 RRID:AB_2687796 |
| InVivoPlus rat IgG2a isotype control | Bio X Cell | Catalog #: BE0089 RRID:AB_1107769 |
| InVivoPlus polyclonal Syrian hamster IgG | Bio X Cell | Catalog #: BE0087 RRID:AB_1107782 |
| Cytokeratin 19 | Abcam | Catalog #: ab52625 RRID:AB_2281020 |
| CD19 | Cell Signaling | Catalog #: 3574 RRID:AB_2275523 |
| CD8 | Cell Signaling | Catalog #: 85336 RRID:AB_2800052 |
| CD11b | Abcam | Catalog #: ab133357 RRID:AB_2650514 |
| pERK | Cell Signaling | Catalog #: 4376 RRID:AB_331772 |
| Goat Anti-Rabbit IgG Antibody (H+L), Biotinylated | Vector Laboratories | Catalog #: BA-1000 RRID:AB_2313606 |
| Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) | Cell Signaling | Catalog #: 4370 RRID:AB_2315112 |
| Alexa Fluor 647 anti-HLA A | Abcam | Catalog #:199837 RRID:AB_2728798 |
| Ki67 | Thermo Fisher Scientific | Catalog #: RM-9106-S RRID:AB_2341197 |
| FAS | Abcam | Catalog #: ab133619 N/A |
| Podoplanin | Abcam | Catalog #: ab11936 RRID:AB_298718 |
| Peroxidase-AffiniPure F(ab’)2 Fragment Goat Anti-Syrian Hamster IgG (H+L) | Jackson Immunoresearch Laboratories | Catalog #: 107–036-142 RRID:AB_2337455 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling | Catalog #: 9661 RRID:AB_2341188 |
| FASL | Santa Cruz Biotechnology | Catalog #: sc-834 RRID:AB_2100640 |
| CD16/32 | TONBO Biosciences | Catalog #: 70–0161 RRID:AB_2621487 |
| TruStain FcX (anti-mouse CD16/32) | BioLegend | Catalog #:101320 RRID:AB_1574975 |
| p44/42 MAPK (Erk1/2) | Cell Signaling | Catalog #: 9102 RRID:AB_330744 |
| Vinculin | Abcam | Catalog #: ab129002 RRID:AB_11144129 |
| Goat Anti-Rabbit Secondary HRP Conjugate | Protein Simple | Catalog #: 042–206 RRID:AB_2860577 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Catalog #: 4370 RRID:AB_2315112 |
| Donkey polyclonal Secondary Antibody to Rabbit IgG - H&L (HRP) | Abcam | Catalog #: ab16284 RRID:AB_955387 |
| Pacific Blue anti-mouse CD45 | BioLegend | Catalog #: 103126 RRID:AB_493535 |
| PE-Cyanine7 anti-mouse CD3e | Thermo Fisher Scientific | Catalog #: 25–0031-82 RRID:AB_469572 |
| BV605 anti-mouse CD4 | BioLegend | Catalog #: 100548 RRID:AB_2563054 |
| BV650 anti-mouse CD8 | BioLegend | Catalog #: 100742 RRID:AB_2563056 |
| BV711 anti-CD11b | BD Biosciences | Catalog #: 563168 RRID:AB_2716860 |
| RPE anti-mouse F4/80 | BioRad | Catalog #: MCA497PE RRID:AB_322048 |
| PerCP-Cy5.5 anti-mouse CD19 | BD Biosciences | Catalog #: 561113 RRID:AB_10563071 |
| PE-CF594 anti-mouse CD11c | BD Biosciences | Catalog #: 562454 RRID:AB_2737617 |
| PE anti-mouse NK1.1 | Thermo Fisher Scientific | Catalog #: 12–5941-83 RRID:AB_466051 |
| BV510 anti-mouse CD45 | BioLegend | Catalog #: 103137 RRID:AB_2561392 |
| BV786 anti-mouse CD11b | BD Biosciences | Catalog #: 740861 RRID:AB_2740514 |
| APC anti-mouse CD178 (FasL) | BioLegend | Catalog #: 106609 RRID:AB_2813951 |
| BV421 anti-mouse CD95/FAS | BD Biosciences | Catalog #: 562633 RRID:AB_2737690 |
| Biological samples | ||
| Patient-derived xenografts, see table S2 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MRTX849 (adagrasib) | MedChem Express | Catalog #: HY-130149 |
| MRTX1133 | WuXi AppTech | N/A |
| TrypLE Express | Gibco | Catalog #: 12605028https://www.thermofisher.com/order/catalog/product/12605028 |
| Doxycycline | West-Ward Pharmaceuticals | NDC#: 0143–2122-50 |
| D-Luciferin | GoldBio | Catalog #: LUCK-1G |
| Captisol | Selleck | Catalog #: S4592 |
| Sodium citrate pH 5 | Teknova | Catalog #: S8450 |
| Collagenase, type IV | Gibco | Catalog #: 17104019 |
| Dispase II | Gibco | Catalog #: 17105041 |
| Collagenase, type I | Gibco | Catalog #: 17018029 |
| cOmplete EDTA free-protease inhibitor | Roche | Catalog #: 4693159001 |
| PhosSTOP | Roche | Catalog #: 4906845001 |
| TRIzol reagent | Invitrogen | Catalog #: 15596026 |
| Critical commercial assays | ||
| CellTiter-Glo | Promega | Catalog #: G7570 |
| DUSP6 RNAscope probe | Advanced Cell Diagnostics | Catalog #: 405361 |
| SPRY4 RNAscope probe | Advanced Cell Diagnostics | Catalog #: 546711-C2 |
| URiSCAN test strips | BioSys Laboratories | Catalog #: U39 |
| Chromium Next GEM Single Cell 3ʹ Kit v3.1 | 10x Genomics | Catalog #: 1000268 |
| NextSeq 500/550 High Output Kit v2.5 (150 Cycles) | Illumina | Catalog #: 20024907 |
| 12–230 kDa Separation Module | ProteinSimple | Catalog #: SM-W001 |
| Pierce BCA Protein Assay Kit | Thermo Fischer | Catalog #: 23225 |
| PureLink RNA Mini Kit | Invitrogen | Catalog #: 12183018A |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Applied Biosystems | Catalog #: 4374966 |
| Fast SYBR Green Master Mix | Applied Biosystems | Catalog #: 4385612 |
| Deposited data | ||
| scRNA-seq data | This paper | GEO: GSE228502 |
| Source data | This paper | https://data.mendeley.com/v1/datasets/h2mvr28n3k/draft?preview=1 |
| Experimental models: Cell lines | ||
| Cell lines, see Table S1 | This paper | N/A |
| Experimental models: Organisms/strains | ||
| B6.FVB-Tg(Pdx1-cre)6Tuv/J | Hingorani et al.2 | RRID:IMSR_JAX:014647 |
| B6.129S4-Krastm4Tyj/J | Hingorani et al.2 | RRID:IMSR_JAX:008179 |
| Trp53loxP/loxP | Chen et al.30 | N/A |
| C57BL/6J | Jackson laboratories | RRID:IMSR_JAX:000664 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson laboratories | RRID:IMSR_JAX:005557 |
| Crl:NU(NCr)-Foxn1nu | Charles River Laboratories | RRID:IMSR_CRL:490 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S5 | This paper | N/A |
| Recombinant DNA | ||
| pLKO.1 shFas: 5’-CCGGGTGTTCTCTTTGCCAGCAAATCTCGAGATTTGCTGGCAAAGAGAACACTTTTT-3’ | Sigma-Aldrich | Catalog #: TRCN0000012328 |
| pLKO.1 sh-Scr: 5’-CCGGGTCGGTATTCGTATCCTAACTCTCGAGATTTGCTGGCAAAGAGAACACTTTTT-3’ | Sigma-Aldrich | Catalog #: TRCN0000012328 |
| F-Luc-GFP Lentivirus | Capital Biosciences | Catalog #: VSL-0088P |
| Software and algorithms | ||
| Cell Ranger 7.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| R (version 4.0.0) package Seurat (version 4.0.3) | Butler et al.40 | https://github.com/satijalab/seurat/releases/tag/v4.0.3 |
| GSEA 4.2.2 | Mootha et al. and Subramanian et al.41,42 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| Other | ||
| VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Standard) | Vector Laboratories | Catalog #: PK-6100 |
| Rabbit-on-rodent HRP polymer | Biocare Medical | Catalog #: RMR622G |
| Opal 690 reagent | Akoya Biosciences | Catalog #: FP1497001KT |
| Opal 520 reagent | Akoya Biosciences | Catalog #: FP1487001KT |
| Opal 570 reagent | Akoya Biosciences | Catalog #: FP1488001KT |
| Hoechst 33342 | Invitrogen | Catalog #: 62249 |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories | Catalog #: H-1000–10 |
| Fluoroshield with DAPI | Sigma-Aldrich | Catalog #: F6057 |
| Fixable Viability Dye eFluor 780 | Invitrogen | Catalog #: 65–0865-14 |
| BD Horizon Brilliant Stain buffer | BD Biosciences | Catalog #: 563794 |
| BD Cytofix Fixation Buffer | BD Biosciences | Catalog #: 554655 |