

Abstract
In non–small cell lung cancer (NSCLC), response to immune checkpoint blockade (ICB) is associated with programmed cell death ligand 1 expression that is induced by interferon-γ–producing, tumor-infiltrating CD8+ T cells. However, not all tumors with a CD8+ T cell infiltrate respond to ICB, and little is known about the mechanisms governing ICB resistance in T cell–infiltrated NSCLC. We used an orthotopic NSCLC mouse model to study ICB-refractory CD8+ T cell responses. Single-cell RNA sequencing of the NSCLC mouse tumors revealed that lung cancer–specific tumor-infiltrating CD8+ T cells exhibited clonal expansion but lacked expression of genes associated with effector and exhausted T cell responses, indicating that they underwent a differentiation program distinct from conventional T cell exhaustion. This lung cancer–specific T cell dysfunction program was established early during priming in the mediastinal lymph node and was characterized by robust proliferation but a failed up-regulation of effector and exhausted T cell characteristics. Intriguingly, CD8+ T cells from patients with NSCLC expressed an analogous gene expression program, which appeared distinct from conventional T cell exhaustion. Administration of recombinant interleukin-2 (IL-2) and IL-12 was sufficient to restore effector T cell differentiation and induce control of KP lung tumors. These findings imply that a CD8+ T cell differentiation trajectory, activated during T cell priming in the mediastinal lymph node, limits the response of CD8+T cells to ICB and thereby may contribute to failure of ICB in a subset T cell–infiltrated NSCLC.
INTRODUCTION
Immune checkpoint blockade (ICB) provides long-term tumor control and survival for a fraction of patients with non–small cell lung cancer (NSCLC) (1–5). A preexisting, tumor-reactive T cell infiltrate is prognostic for ICB response in most cancers (6–8). Nonsquamous NSCLC is a heterogeneous disease whose treatment is determined by driver mutations (9, 10). Whereas EGFR-, ALK-, or ROS1-mutated tumors receive targeted small-molecule therapies that inhibit the activity of the corresponding oncogenic proteins, the remaining patients with NSCLC, dominated by KRAS mutations, receive ICB (9–11). Mutations that co-occur with KRAS play a role in shaping the T cell infiltrate (11, 12). Tumors with mutations in KRAS and LKB1/STK11 frequently lack T cell infiltration and are resistant to ICB (11, 12). Tumors with mutations in both KRAS and TP53 are frequently infiltrated with T cells (11, 12). Yet, in T cell–infiltrated KRAS/TP53-mutant tumors, the ICB response rate is only ~35% (11, 12). This discrepancy suggests that in KRAS/TP53 mutant NSCLC, not all tumor-infiltrating T cells respond to ICB, and a better understanding of the functional states of tumor-reactive T cells is needed.
That the functional state of antitumor T cells determines ICB response is supported by clinical reports correlating programmed cell death ligand 1 (PD-L1) expression to ICB outcome in NSCLC (3). PD-L1 expression within the tumor microenvironment (TME) is indicative of T cell function because PD-L1 is induced by the effector cytokine interferon-γ (IFN-γ) (13). Patients with PD-L1–positive, T cell–infiltrated tumors comprise about 40% of the ICB-treated patients with NSCLC and are the group most likely to respond to ICB (3, 9, 14). In contrast, patients with NSCLC with PD-L1–negative, T cell–infiltrated tumors respond poorly to ICB. Clinically, these T cell responses were described as “nonfunctional,” referring to their inability to produce IFN-γ (3). However, our understanding of the resistance mechanisms in T cell–infiltrated, ICB-refractory patients is limited.
To better understand ICB resistance in T cell–infiltrated NSCLC, we used an orthotopic model for NSCLC derived from an autochthonous genetically engineered mouse model driven by KrasG12D expression and deletion of Tp53 (KP cell line) (15). We found that antitumor T cell responses in this model entered into a T cell dysfunctional state that was distinct from T cell exhaustion (Tex). This dysfunctional state shared aspects of T cell responses found in ICB-refractory, T cell–infiltrated patients with NSCLC, including a lack of up-regulation of effector molecules after ICB (3). We found that the functional state of the tumor-infiltrating T cell response was determined during priming in the tumor-draining lymph node (TdLN) in our model system. ICB-refractory T cell responses were characterized by a lack of effector and exhaustion molecule expression despite robust T cell activation and migration into the KP tumors. Understanding how ICB-refractory T cell responses differ from ICB-sensitive, conventionally exhausted T cell responses is of high clinical interest. An improved understanding of the impact of T cell states on antitumor immunity will enable the development of targeted therapies to engage all T cell subsets in NSCLC.
RESULTS
ICB resistance in KP lung tumors despite a brisk T cell infiltrate
We inoculated KP tumor cells intravenously to establish lung tumors or subcutaneously to establish flank tumors and subsequently administered anti–cytotoxic T lymphocyte–associated protein 4 (anti–CTLA-4) and anti–PD-L1 combination ICB beginning on day 7 after tumor inoculation (Fig. 1A). KP lung tumors were unaffected by ICB (Fig. 1, B and C), but KP flank tumors had significantly delayed tumor outgrowth (Fig. 1D). Calculation of the fold change in tumor size, as a ratio of the treated tumor areas to the average of the control tumor areas, confirmed that flank tumors responded significantly better to ICB than lung tumors (Fig. 1E). Quantification of tumor-infiltrating lymphocyte (TIL) density in tumor areas using immunofluorescence (IF) staining, identified by keratin staining, confirmed that KP lung tumors had TIL (280.6 ± 29.66 T cells/ mm2, means ± SEM; Fig. 1F and fig. S1). The density of TIL in lung tumors was 29.6-fold higher compared with ICB-sensitive flank tumors (Fig. 1, F and G), indicating that ICB resistance was not driven by a lack of T cell infiltration. However, TIL in lung tumors expanded only 1.4-fold, whereas the density of TIL in flank tumors increased 11-fold after ICB (Fig. 1, F to H). This model system allowed us to gain insights into ICB resistance by contrasting ICB-resistant and ICB-sensitive T cell responses in T cell–infiltrated NSCLC driven by the same oncogenic mutations.
Fig. 1. Orthotopic KP tumors in the lung are resistant to ICB.
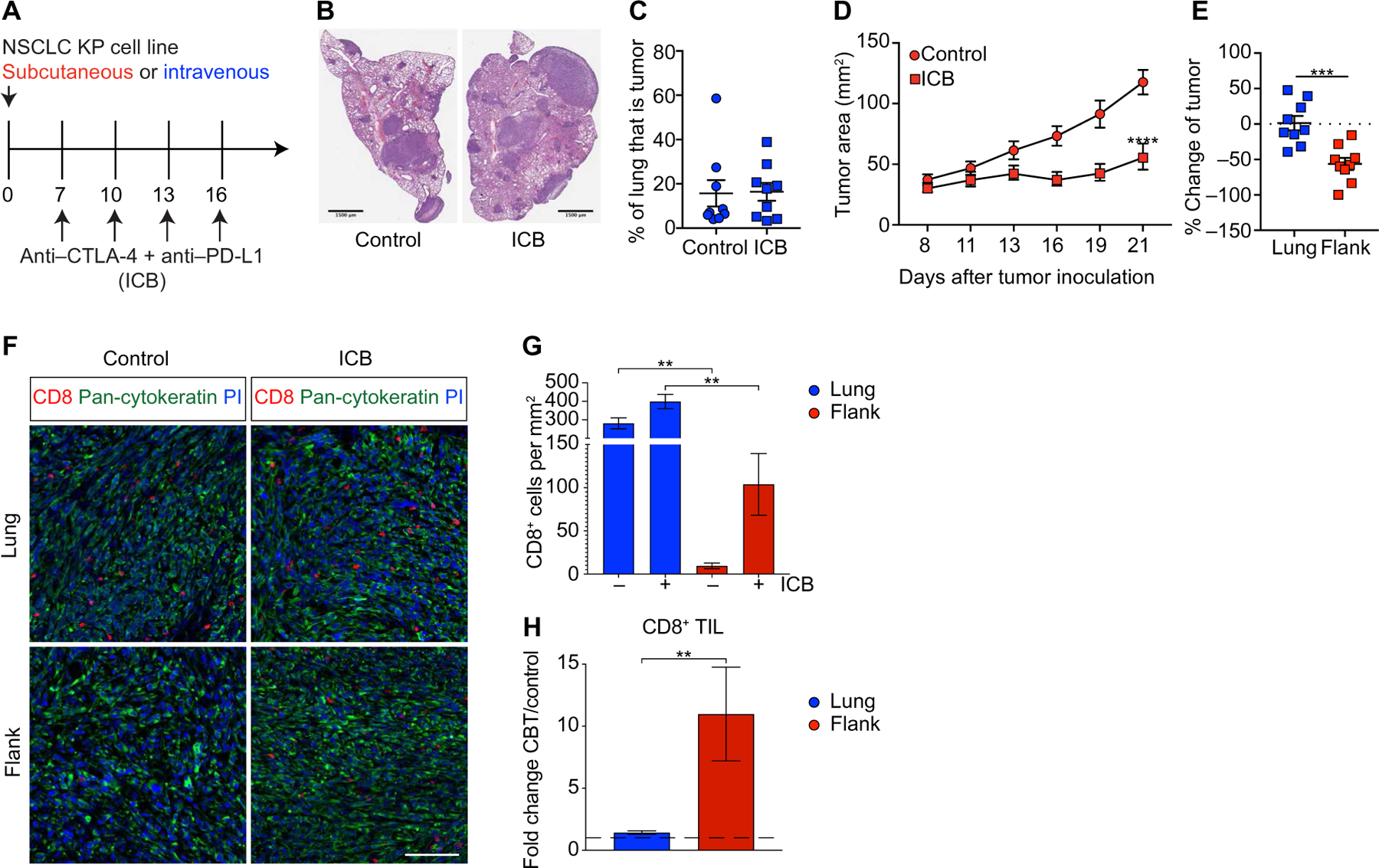
(A) Scheme of tumor inoculation and ICB. (B) Representative example and (C) quantification of lung tumor burden on day 21 in control and ICB-treated mice assessed by H&E stain (control n = 9, ICB n = 9, combined data from three experiments). (D) Outgrowth of flank KP tumors treated with ICB or control (control n = 9, ICB n = 9, combined data from three experiments). (E) Comparison between ICB efficacy in lung and flank tumors shown as percent change of tumor size over control treatment (lung n = 9, flank n = 9, combined data from three experiments). Dotted line represents no change from the untreated controls. (F) Representative IF images of lung and flank tumors on day 21 from control mice or mice treated with ICB. For uncropped images, see fig. S1. PI, propidium iodide. Scale bar, 100 mm. (G) Density of CD8+ T cells in control and ICB-treated lung and flank KP tumors on day 21 determined by IF. (H) Fold change of tumor-infiltrating CD8+ T cells between ICB-treated and control mice. Dashed line is at 1, which is no change from the control. Regions analyzed in (G) and (H): flank, n = 12 from three mice; flank + ICB, n = 14 from four mice; lung, n = 58 from three mice; and lung + ICB, n = 73 from four mice. Data are shown as means ± SEM, and statistical analysis was conducted using a two-way ANOVA (D), MWU test (C, E, and H), or one-way ANOVA (G) with **P < 0.01, ***P < 0.001, and ****P < 0.0001.
TIL from KP lung tumors do not exhibit conventional Tex
We performed single-cell RNA sequencing (scRNA-seq) on TIL (16). All analyses excluded circulating CD45+ cells using intravenous injection of a phycoerythrin (PE)–labeled anti-CD45 antibody before euthanasia, followed by magnetic depletion of PE-labeled cells. We sequenced 122,554 cells from eight flank and eight lung tumor–bearing mice on day 14 with and without ICB, including 10,774 T cells (Fig. 2A) and identified nine distinct T cell phenotypes (Fig. 2, B and C). Cluster proportions were not significantly different between control and ICB (fig. S2A) but were present at different proportions between sites of tumor growth (Fig. 2D). We focused our subsequent analysis on the two non-naïve CD8+ T cell clusters (CD8+ c1 and CD8+ c2) (6, 17, 18). CD8+ c1 were dominant in flank TIL, whereas CD8+ c2 were dominant in lung TIL (Fig. 2, A, B, and D). CD8+ c1 had increased Pdcd1, Lag3, Tnfrsf9, Tnfrsf4, Havcr2, Cd160, and Nrgn gene expression, all associated with conventional T cell activation and exhaustion (Fig. 2E and table S1) (19–22). CD8+ c2 had higher expression of transcripts associated with CD8+ T cell activation, including Klrg1 and Ccl5, and T cell survival and tissue homing, including Klf2, Klf3, S1pr1, S1pr4, Itga4, and Itgb1 (Fig. 2E) (23–31). Pathway analysis suggested interleukin-2 (IL-2)/signal transducer and activator of transcription 5 signaling to be enriched in CD8+ c1 (Fig. 2F, fig. S2B, and table S2). These data suggested that two distinct CD8+ T cell responses were induced against the KP tumor cell line depending on the location of the tumor growth; CD8+ T cell responses against flank tumors were characterized by effector function and conventional Tex, whereas CD8+ TIL in lung tumors lacked exhaustion molecules but expressed a distinct set of effector molecules.
Fig. 2. Lung tumor–infiltrating CD8+ T cells do not acquire effector or exhausted phenotypes observed in flank TIL.
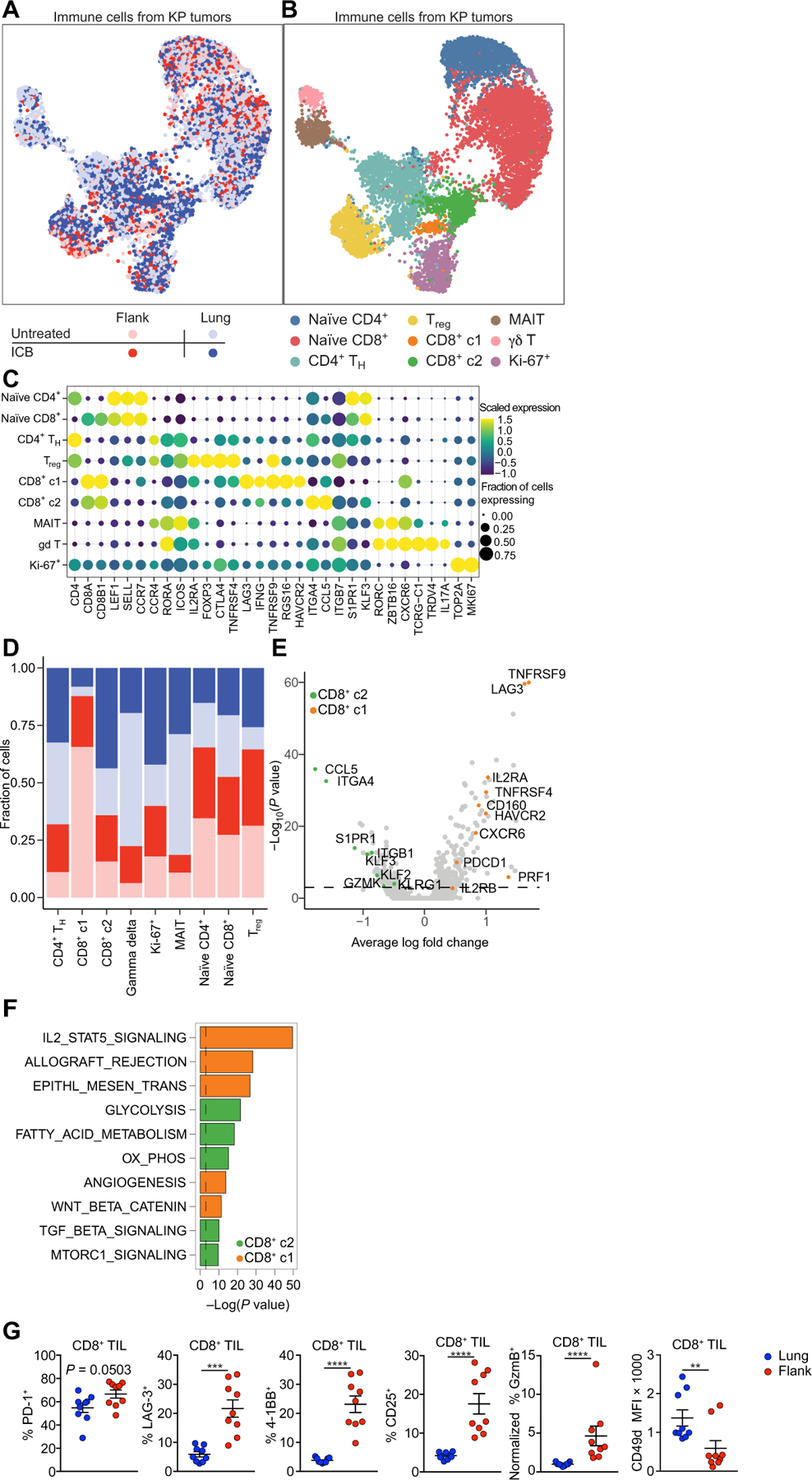
(A) UMAP plots of scRNA-seq indicating treatment status and tumor location and (B) curated clusters. TH, T helper. MAIT, mucosal associated invariant T cell. (C) Bubble plot of gene expression analysis of curated clusters. (D) Stack charts of contributions to each curated cluster based on treatment condition and tumor location. (E) Volcano plot of DEGs between CD8+ c1 and CD8+ c2 CD8+ T cells. Selected DEGs in CD8+ c2 (green) or CD8+ c1 (orange) TIL are highlighted. (F) Pathway analysis of DEGs showing enriched MSigDb hallmarks pathways for CD8+ c2 (green) and CD8+ c1 (orange) TIL. (G) Validation of differentially expressed key effector/exhaustion markers on lung and flank CD8+ TIL (lung, n = 9 and flank, n = 9, combined data from three experiments). Data are shown as means ± SEM, and statistical analysis was conducted using an MWU test (G) with **P < 0.01, ***P < 0.001, and ****P < 0.0001, or as indicated in Materials and Methods (A to F). MFI, mean fluorescence intensity.
Flow cytometry on TIL from lung or flank tumors was performed to validate differentially expressed genes (DEGs). Lung and flank TIL expressed programmed cell death 1 (PD-1), indicative of antigen recognition occurring at both tumor sites (Fig. 2G) (32). Flank TIL had higher expression of molecules associated with Tex: LAG-3 (Lag3) and 4–1BB (Tnfrsf9), effector T cell differentiation, CD25 (Il2ra), and granzyme B (Gzmb) (Fig. 2G), whereas CD49d (Itga4) was higher on lung TIL (Fig. 2H). A second model of lung cancer (LL/2) provided similar results (fig. S2C). See fig. S2D for example flow cytometry plots. Thus, in our model, lung tumor TIL exhibited signs of T cell activation yet lacked hallmarks of effector T cell differentiation and Tex. We therefore concluded that lung tumor–reactive TIL in our models were in a dysfunctional state distinct from Tex. For ease, we refer to this T cell state as lung cancer–specific T cell dysfunction, abbreviated TLdys, throughout the manuscript.
TIL with TLdys are tumor antigen specific
Differences in TIL between lung and flank KP tumors could be driven by differences in antigen specificities or T cell receptor (TCR) usage. We recovered Tcra and Tcrb sequences from single-cell libraries (33). CD8+ c1 and CD8+ c2 both showed clonal expansion in lung and flank KP tumors, suggesting that both sites of tumor growth induced a tumor-reactive T cell response (fig. S2E). Using grouping of lymphocyte interactions by paratope hotspots 2 (GLIPH2) (34) to analyze similarities in the TCR repertoire, we identified seven specificity groups that contained CDR3 sequences from three or more mice and that were shared across flank and lung tumors and both CD8+ phenotypes (fig. S2F). Identification of public clonotypes suggests a shared antigen present in both lung and flank KP tumors (table S3) capable of inducing clonal expansion.
To directly determine whether CD8+ T cell responses against the same antigen could enter the CD8+ c1 and CD8+ c2 states, we engineered KP cells to express the model antigen SIYRYYGL (SIY, KP.SIY cell line), which is presented on H-2Kb. Using SIY-loaded pentamers, we sorted SIY-reactive CD8+ TIL from day 14 lung and flank KP.SIY tumors. To elucidate whether SIY-reactive CD8+ T cells were transcriptionally similar to CD8+ T cell populations from KP parental lung and flank tumors, we conducted an integrated analysis of both datasets. We determined a high degree of overlap in the clustering of lung and flank SIY-reactive CD8+ TIL with their respective counterparts from KP parental tumors (Fig. 3A). Unsupervised analysis of the integrated data yielded two clusters consistent with the CD8+ c1 and CD8+ c2 in the KP parental cell line (Fig. 3B). The proportion of CD8+ c1 and CD8+ c2 within TIL from KP.SIY and KP parental tumors was similar (Fig. 3C), with CD8+ c1 being 47 and 59% of KP and KP.SIY flank TIL, respectively. Lung TIL consisted of 90 and 89% CD8+ c2 from KP and KP.SIY tumors, respectively, demonstrating that the TLdys phenotype is dominant in lung TIL (Fig. 3C). Next, we determined the DEGs between CD8+ c1 and CD8+ c2 from the KP.SIY TIL (Fig. 3D and table S4). Consistently, CD8+ c1 from KP.SIY tumors had higher expression of exhaustion markers, including Tnfrsf9, Tnfrsf4, Lag3, Cd160, and Havcr2, whereas CD8+ c2 had higher expression of lung-homing factors, including Ccl5 and Itga4. A comparison of DEGs between CD8+ c1 and CD8+ c2 from both the KP parental and KP.SIY CD8+ TIL datasets showed a highly significant degree of similarity (R = 0.55, P < 0.001; Fig. 3E), indicating that the transcriptional differences between CD8+ c1 and CD8+ c2 were conserved in TIL isolated from KP parental and KP.SIY tumors. This suggested that TLdys (CD8+ c2) was not a result of weak or absent reactivity to tumor-associated antigens because T cell responses against SIY induced differentiation to this T cell state.
Fig. 3. TLdys T cells in lung tumors and Tex T cells in flank tumor respond to the same antigen.
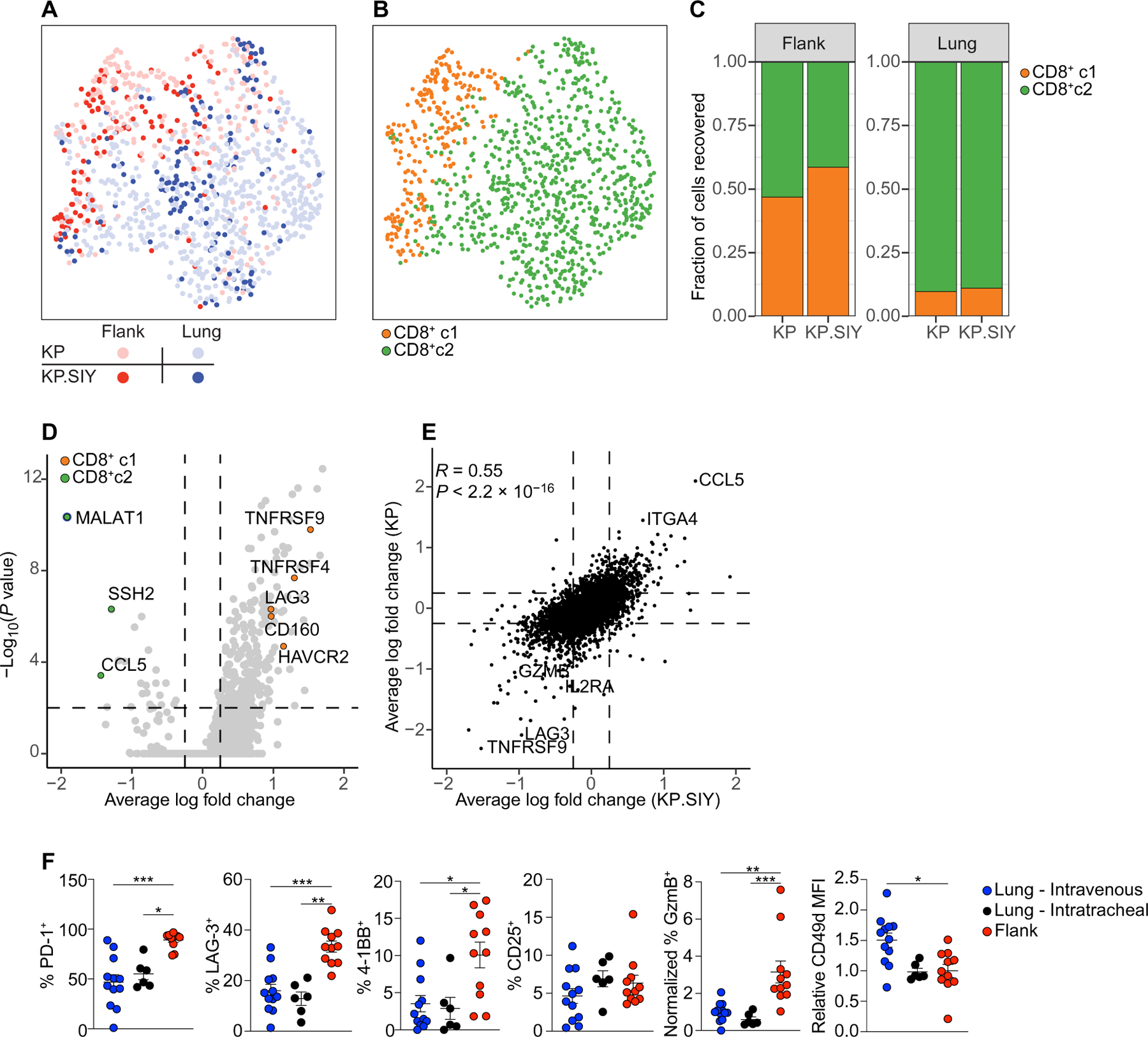
(A) Combined UMAP plot of scRNA-seq of polyclonal CD8+ T cells (lung and flank KP tumors) and SIY-pentamer+ CD8+ T cells (lung and flank KP.SIY tumors) indicating tumor cell line and location. (B) UMAP plot showing clusters CD8+ c1 and CD8+ c2 of combined datasets from (A). (C) Proportions of cluster CD8+ c1 or CD8+ c2 within flank and lung TIL. (D) Volcano plot of DEGs between CD8+ c1 and CD8+ c2 SIY-reactive CD8+ T cells. Genes of interest are highlighted. (E) The log fold change values of DEGs between CD8+ c1 and CD8+ c2 were compared between SIY-reactive CD8+ T cells from KP.SIY tumors and polyclonal CD8+ T cells from KP tumors. (F) Flow cytometry of SIY-reactive TIL from KP.SIY lung and flank tumors (lung intravenous, n = 12; lung intratracheal, n = 6; and flank n = 11; data pooled from four independent experiments, two with intratracheal tumor administration). Data are shown as means ± SEM, and statistical analysis was conducted using an MWU test (F) with *P < 0.05, **P < 0.01, and ***P < 0.001, or as indicated in Materials and Methods (A to E).
Flow cytometric analysis of SIY-reactive TIL (Fig. 3F) confirmed that the phenotypes of lung and flank TIL were similar to those observed in KP parental tumors (Figs. 2G and 3F). We also performed intratracheal administration of KP.SIY cells as a second method of tumor inoculation into the lung. We detected a difference in only CD49d between intravenously and intratracheally induced lung tumors, suggesting that intravenous tumor inoculation was not driving TLdys (Fig. 3F). In summary, differences between lung and flank TIL were preserved in the presence of a strong neoantigen and were consistent between intravenous and intratracheal lung tumor induction.
TLdys is not driven by the lung TME
Because separation of tumors from normal lung tissue before analysis was not feasible, we used IF staining to determine whether TLdys T cells were in the TME or within the adjacent lung. Using keratin to identify tumor areas, we found that CD8+ T cells were significantly enriched in lung tumors compared with the adjacent normal tissue (fig. S3, A and B). This was true for lung tumors inoculated intravenously or intratracheally (fig. S3, A and B), indicating that most CD8+ T cells from lung tumor–bearing mice resided in the tumor (see also fig. S1). We next quantified PD-1 expression on CD8+ T cells infiltrating lung tumors or the adjacent normal tissue because PD-1 is expressed upon antigen encounter (32). PD-1+CD8+ T cells were significantly enriched in lung tumors compared with adjacent tissue (fig. S3, A and C). PD-1 was expressed on 74.5, 78.3, and 73.7% of CD8+ T cells from intravenous lung, intratracheal lung, and flank tumors, respectively (fig. S3, A and D). These data indicate that most PD-1+ CD8+ T cells are within tumors and suggest that the differences between Tex and TLdys TIL are not due to failed infiltration of TIL with TLdys into the tumor.
To further determine whether TLdys were independent of the lung TME, we adoptively transferred 20,000 Thy1.2+ fluorescence-activated cell sorting (FACS)–sorted TIL from flank or lung tumors into Rag2−/− mice. Eight to 16 weeks later, we inoculated the reconstituted Rag2−/− mice with KP flank tumors. Adoptively transferred TIL from KP flank tumors led to slower tumor growth compared with those from KP lung tumors (fig. S3E), suggesting that TIL from KP lung and flank tumors are functionally different and that TLdys is a persistent T cell state that cannot be reversed by a flank tumor or homeostatic proliferation.
To probe for earlier differences in T cell responses against lung and flank tumors, we performed a concomitant immunity assay. Mice bearing flank or lung KP tumors were challenged with a second KP tumor implanted on the contralateral flank 7 days after the first tumor inoculation. Mice bearing an initial flank tumor experienced significantly improved tumor control of the second tumor compared with naïve mice (fig. S3F), but mice with an initial lung tumor showed no protection against a second flank tumor (fig. S3F). Similar results were obtained with the KP.SIY cell line (fig. S3G). Antibody-mediated depletion confirmed that this protection was due to CD8+ T cells (fig. S3H). To determine whether the lung TME was sufficient to induce TLdys after initial T cell activation, we assessed the effect of an initial flank tumor on the growth of subsequent lung tumors and observed protection against a second lung tumor challenge (fig. S3I), suggesting that the lung TME was not directly imposing TLdys. These data suggest that the TLdys induced by KP and LL/2 lung tumors is a persistent state of T cell dysfunction established early during the anti–lung tumor immune response.
TLdys is established during T cell priming in the mediastinal LN
We next postulated that the differences in CD8+ T cell responses were imposed during T cell priming in the lung tumor–draining mediastinal LN (mLN) or flank tumor–draining inguinal LN (iLN). To determine whether differences in antitumor T cell responses could be detected in the periphery, we performed an IFN-γ enzyme-linked immune absorbent spot (ELISpot) assay. KP lung tumors had significantly fewer IFN-γ–producing, SIY-reactive splenocytes compared with KP flank tumors (fig. S4A). To determine whether this was due to a lack of T cell proliferation during priming, we used an adoptive T cell transfer approach using TCR-transgenic 2C T cells (2C T cells) specific for SIY (35). Naïve, carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled 2C T cells were transferred into mice with KP.SIY lung or flank tumors on day 7, and the degree of T cell activation was determined in TdLN on day 10. The 2C T cells proliferated robustly in both the mLN and the iLN (fig. S4, B and C), suggesting that differences in antitumor CD8+ T cell responses were not due to a lack of T cell expansion during priming.
Next, we adoptively transferred 2C T cells to mice with KP.SIY lung or flank tumors on day 7. On day 10, 2C T cells from the mLN of lung tumor–bearing mice and the iLN of flank tumor–bearing mice were isolated using FACS, and scRNA-seq was performed using Smart-Seq to determine transcriptional differences between 2C T cells primed in the mLN or iLN (Fig. 4A). A total of 1744 genes were differentially expressed between 2C T cells priming in iLN or mLN (table S5). Consistent with the scRNA-seq data of endogenous flank TIL, 2C T cells primed in iLN showed high levels of genes associated with effector function (Gzma, Gzmb, Il2ra, Il12rb1, Il12rb2, Prdm1, Bhlhe40, and Id2) (36–39) and exhaustion (Pdcd1, Havcr2, Tnfrsf4, and Cd160) (19–22) (Fig. 4B). In contrast, 2C T cells primed in the mLN showed high levels of genes associated with inhibition of effector T cell differentiation (Sell, Pecam1, Bcl6, Id3, Lef1, Ctla4, Bach2, Tox, and Tox2) (36–44) and genes associated with lung T cell responses (Ccl5, Itga4, and Itgb1) (23–25) (Fig. 4B). Direct comparison of 2C T cells and endogenous CD8+ TIL revealed that 42 genes were consistently regulated between these early and late CD8+ T cell responses (fig. S4D and table S6), suggesting that differences in T cell responses between lung and flank KP tumors originated from differences during T cell priming. Flow cytometry affirmed that CD25 and GzmB were significantly reduced on T cells activated in mLN (Fig. 4, C and D), whereas mLN-primed 2C T cells showed higher levels of CD49d (Fig. 4E).
Fig. 4. CD8+ T cells primed in the mLN fail to acquire an effector phenotype.
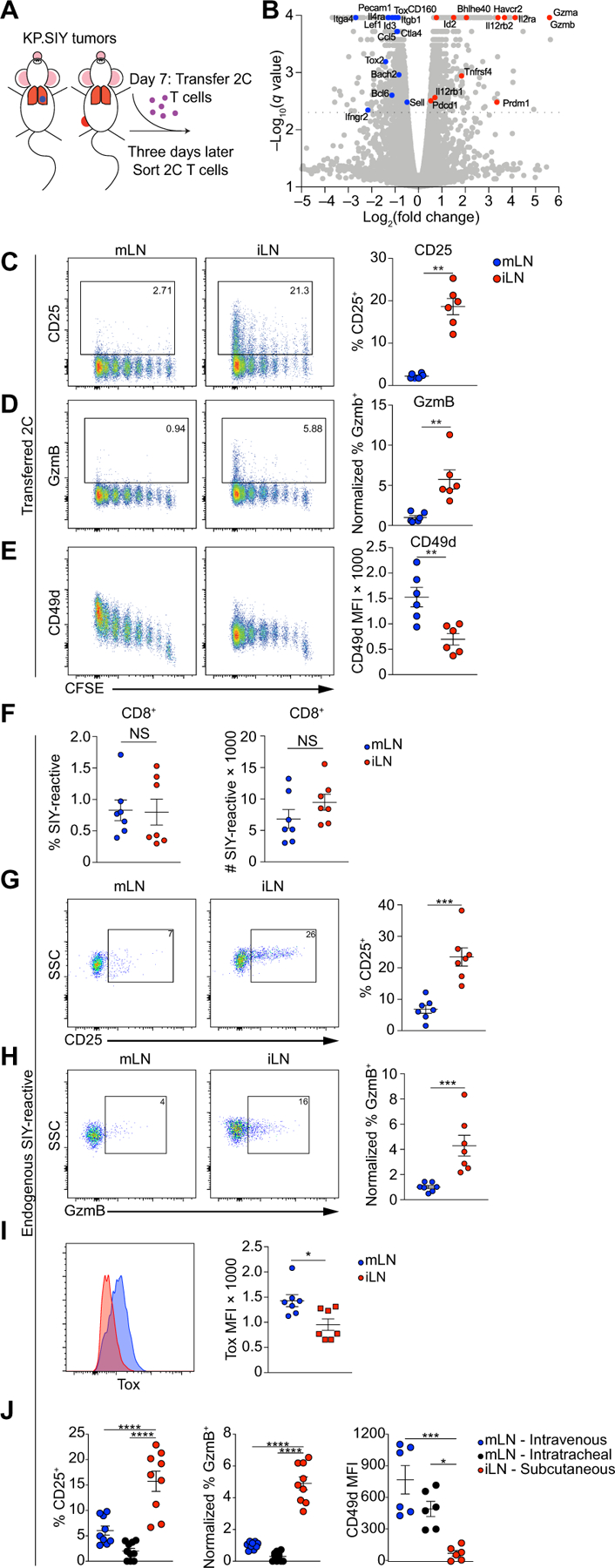
(A) Schematic of experimental setup. Mice were inoculated with KP.SIY lung or flank tumors, and on day 7 of tumor growth, 1 × 106 naïve, congenically marked 2C T cells were transferred to tumor-bearing mice. On day 10 of tumor growth, the 2C T cells were isolated from the mLN of lung tumor–bearing mice and iLN of flank tumor–bearing mice using FACS. (B) Volcano plot of DEGs between 2C T cells primed in the mLN and iLN of tumor-bearing mice. mLN, n = 3 and iLN, n = 4 biological replicates. Genes of interest are highlighted. (C to E) Representative example and quantification (means ± SEM) of CD25 (C), GzmB (D), and CD49d (E) expression levels on CFSE-labeled 2C T cells primed in the mLN and iLN of tumor-bearing mice at 72 hours after adoptive transfer, day 10 of tumor growth (mLN, n = 6 and iLN, n = 6; data pooled from two experiments). CFSE dilution is shown on the x axis. (F to I) Representative example and quantification (means ± SEM) of the percent and absolute number of SIY+-reactive T cells (F) as well as CD25 (G), GzmB (H), and Tox (I) expression levels on endogenous SIY-reactive T cells in the mLN and iLN of tumor-bearing mice 7 days after tumor inoculation. NS, not significant; SSC, side scatter. (J) Mice were inoculated with KP.SIY cells either intravenously, intratracheally, or subcutaneously. On day 7 of tumor growth, mLNs and iLNs were isolated, and endogenous SIY-reactive CD8+ T cells were analyzed with flow cytometry for the expression of CD25, GzmB, and CD49d (n = 9 for CD25 and GzmB and n = 6 for CD49d comparisons, data pooled from three experiments). Data are shown as means ± SEM, and statistical analysis was conducted using an MWU test (C to J) with *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 or as indicated in Materials and Methods (B).
To determine whether differences in antigen-presenting cells (APCs) in the TdLN were responsible for the differences in T cell priming, we examined tumor antigen–containing APC using KP tumor cells engineered to express ZsGreen, allowing extended detection of APC populations carrying tumor cell debris (45, 46). Flow cytometry of ZsGreen+ cells in the mLN of lung tumor–bearing mice and the iLN of flank tumor–bearing mice on day 7 after tumor inoculation revealed that conventional dendritic cells (DCs) were the dominant APC that acquired tumor antigen and migrated to the TdLN (fig. S4E). Furthermore, we detected equal numbers of ZsGreen+ DCs in both mLN and iLN (fig. S4F). To assess functional abilities of DCs, KP cells expressing ZsGreen and the SIIN peptide were inoculated into mice, and 7 days later ZsGreen+ DCs were sorted from TdLNs. ZsGreen+ DCs were cocultured with naïve TCR-transgenic CD8+ T cells specific for SIIN peptide (OT-I T cells). We detected identical T cell activation using ZsGreen+ DCs from mLN or iLN (fig. S4G), indicating that both DC populations have similar stimulatory capacity ex vivo, highlighting that differences between CD8+ T cell responses against lung and flank tumors were not due to differences in DC-mediated antigen presentation.
We next evaluated the priming of endogenous SIY-reactive T cells in response to KP.SIY lung and flank tumors. We found no significant differences in either the percentage or number of SIY-reactive CD8+ T cells on day 7 after tumor inoculation (Fig. 4F). However, SIY-reactive CD8+ T cells in the iLN of flank tumor–bearing mice expressed higher CD25 and GzmB (Fig. 4, G and H). SIY-reactive CD8+ T cells in the mLN of lung tumor–bearing mice expressed higher Tox (Fig. 4I), consistent with higher Tox transcript levels in 2C T cells primed in mLN (Fig. 4B). We next assessed the T cell phenotype of SIY-reactive CD8+ T cells in the mLN after intravenous or intratracheal KP tumor administration. SIY-reactive CD8+ T cells in the mLN had reduced CD25 and GzmB and increased CD49d compared with SIY-reactive CD8+ T cells in the iLN, indicating that these differences in priming occurred with both intravenous and intratracheal inoculation (Fig. 4J). To determine whether these differences in T cell activation were due to the orthotopic nature of KP lung tumor, we inoculated mice with the melanoma cell line B16.SIY. Responses in the mLN were similar to responses in the iLN (fig. S5, A and B), indicating that CD8+ T cell priming in the mLN in response to orthotopic tumors in the lung led to TLdys.
T cells with TLdys retain T cell factor 1 but are functionally distinct from precursor-exhausted T cells
Studies in chronic lymphocytic choriomeningitis virus (LCMV) have described two subsets of exhausted CD8+ T cells: terminally exhausted T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3+) CD8+ T cells, which gradually lose their functional properties and eventually undergo apoptosis, and precursor-exhausted T cell factor 1–positive (TCF-1+) CD8+ T cells that lack cytotoxicity but retain a stem-like capacity to differentiate into TIM-3+ T cells (36, 47). Because Havcr2, which encodes TIM-3, was differentially expressed on tumor-reactive CD8+ T cells in mLN and iLN and the corresponding TIL populations, we determined the expression of TIM-3 and TCF-1 during CD8+ T cell responses to lung and flank KP.SIY tumors. TIM-3 was up-regulated on CD8+ T cells in the iLN, both on transferred 2C T cells (Fig. 5A) and on endogenous SIY-specific CD8+ T cells (Fig. 5B). In contrast, CD8+ T cells activated in the mLN did not up-regulate TIM-3 and remained TCF1+ (Fig. 5, A and B). These differences were specific for orthotopic lung KP tumors because no differences were detected using B16.SIY tumors (fig. S5C).
Fig. 5. TLdys T cell state is functionally distinct from conventional Tex T cell state.
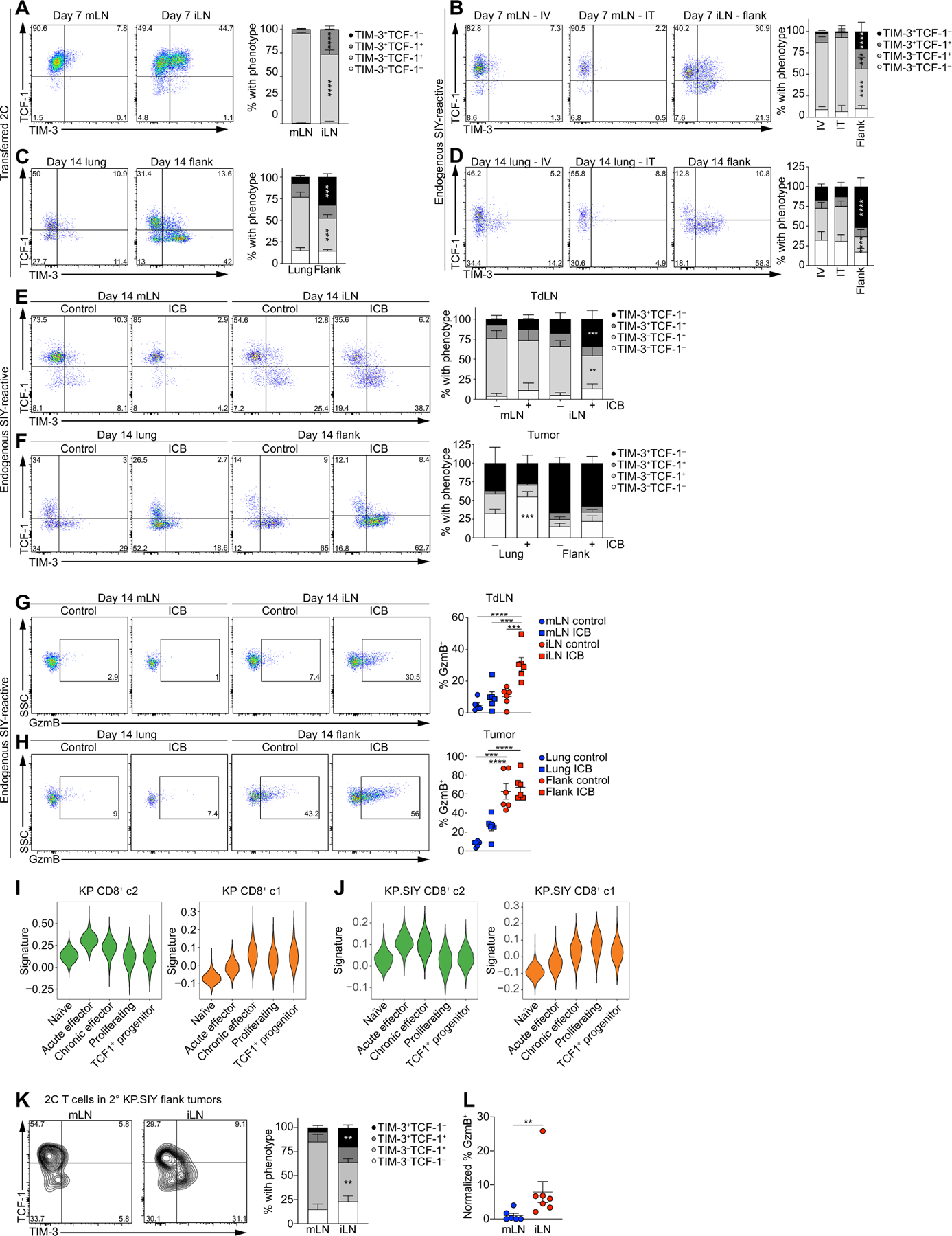
(A to D) Flow cytometry analysis of TCF-1 and TIM-3 expression on adoptively transferred 2C T cells in the mLN and iLN of KP.SIY tumor–bearing mice 72 hours after adoptive transfer (10 days after tumor inoculation; n = 14 for mLN and n = 10 for iLN, data pooled from two experiments). (B) Endogenous, SIY-reactive CD8+ T cells in KP.SIY tumor–bearing mice 7 days after intravenous, intratracheal, or subcutaneous inoculation (n = 9 for IV mLN, n = 10 for IT mLN, and n = 9 for iLN, data pooled from three experiments). (C) Adoptively transferred 2C T cells in lung or flank KP.SIY tumors 14 days after tumor inoculation and 7 days after adoptive transfer (n = 6 for lung and n = 5 for flank, data pooled from two experiments). (D) Endogenous, SIY-reactive CD8+ TIL in KP.SIY tumor–bearing mice 14 days after tumor inoculation (n = 6 for all conditions, data pooled from two experiments). (E to H) Analysis of SIY-reactive CD8+ T cells in KP.SIY tumor untreated or treated with ICB given on days 7 and 10 after tumor inoculation. TCF-1 and TIM-3 expression in mLN and iLN (E) and lung and flank tumors (F) 14 days after tumor inoculation (n = 6, data pooled from two experiments). GzmB expression on SIY-reactive CD8+ T cells in (G) mLN and iLN and lung and flank tumors (H) 14 days after tumor inoculation (n = 6, data pooled from two experiments). (I and J) Comparison of CD8+ c2 (green) or CD8+ c1 (orange) signatures from KP parental (I) and KP. SIY (J) datasets with previously published CD8+ T cell signatures from acute and chronic LCMV infection. (K and L) Flow cytometric analysis of TCF1–1, TIM-3 (K), and GzmB (L) expression in serially transferred 2C T cells on day 5 after transfer to RAG2−/− (n = 6 for mLN and n = 7 for iLN, data pooled from two experiments) Data are shown as means ± SEM, and statistical analysis was conducted using an MWU test with **P < 0.01, ***P < 0.001, and ****P < 0.0001 (A to H and K to L).
We next analyzed TIL on day 14 after tumor inoculation. Both adoptively transferred 2C T cells and endogenous T cells acquired a terminally exhausted TIM-3+ TCF1− phenotype in flank tumors (Fig. 5, C and D). However, neither 2C T cells nor SIY-reactive TIL in lung tumors acquired significant TIM-3+ expression and remained either TCF-1+ or became TIM-3− TCF-1− (Fig. 5, C and D) despite up-regulation of PD-1 (Figs. 2G and 3F and fig. S3, A, C, and D), suggesting that T cells were responding to their cognate antigen without acquiring T cell effector function. TCF-1 expression by SIY-reactive CD8+ T cells in mLN and lung tumors raised the possibility that KP lung tumors had an increased proportion of precursor-exhausted CD8+ T cells. Because ICB targets TCF-1+ precursor-exhausted CD8+ T cells and induces their differentiation into TIM-3+ TCF-1− effector T cells (37, 40, 48–51), we determined the potential of TCF-1+ T cells in mLN to differentiate to TIM3+ T cells. We treated mice on days 7 and 10 of KP.SIY tumor growth with ICB and evaluated the phenotypes of SIY-reactive CD8+ T cells. The proportion of TIM3+ TCF1− SIY-reactive CD8+ T cells in the iLN of flank tumor–bearing mice increased after ICB, with no differences in T cell phenotype detected in the mLN of lung tumor–bearing mice (Fig. 5E). Consistent with this observation, we detected no accumulation of TIM3+ TCF1− TIL in KP lung tumors; instead, ICB resulted in increased TIM-3− TCF-1− CD8+ TIL (Fig. 5F). SIY-specific CD8+ T cells in mLN and KP lung tumors also failed to up-regulate GzmB after ICB, whereas ICB enhanced GzmB expression in SIY-reactive CD8+ T cells in the iLN (Fig. 5, G and H). Lack of TIM-3 and GzmB up-regulation in SIY-reactive CD8+ T cells from mLN and lung tumors after ICB was consistent with the lack of ICB efficacy in KP lung tumors (Fig. 1, A to E) and suggested that the TCF-1+ CD8+ T cells in mLN and lung tumors are functionally distinct from precursor-exhausted CD8+ T cells.
We compared the transcriptional profiles of TIL with a published dataset of T cell exhaustion (36). CD8+ c2 from both the KP and KP. SIY scRNA-seq datasets, which constitutes TLdys, was most enriched for naïve and acute effector gene signatures despite low expression of effector molecules such as GzmB and CD25 (Fig. 5, I and J). CD8+ c1 from both the KP and KP.SIY scRNA-seq datasets were most similar to signatures from exhausted effectors and TCF-1+ precursor-exhausted T cells (Fig. 5, I and J). These data suggest that despite TCF-1 expression, lung tumor–reactive TLdys CD8+ T cells are transcriptionally and functionally distinct from precursor-exhausted T cells. Thus, T cells with TLdys are activated and gain tissue-homing capabilities but minimally differentiate into TIM-3+ CD8+ T cells and fail to acquire appreciable effector T cell functions even when exposed to ICB.
TLdys is associated with decreased T cell functionality
To measure the functionality of the TLdys state, we assessed cytokine production from SIY-reactive TIL from KP.SIY tumors. Although neither lung nor flank TIL produced high levels of IL-2, we detected significantly lower production of tumor necrosis factor–α and IFN-γ in lung TIL compared with flank TIL (fig. S6A). To evaluate cytotoxicity, we performed an in vivo cytotoxicity assay (52). Flank tumor–bearing mice showed increased eradication of SIY-pulsed cells, whereas no significant cytotoxicity was detected in lung tumor–bearing animals (fig. S6B). These results demonstrated that T cells with TLdys fail to differentiate into cytotoxic effector T cells despite robust T cell activation in the mLN (Fig. 4, C to E) and successful infiltration into lung tumors (fig. S3, A to C).
To determine the persistence of the TLdys state after T cell priming, naïve 2C T cells were transferred to lung or flank KP.SIY–bearing mice on day 7, isolated from mLN or iLN on day 10, and transferred to RAG2−/− mice bearing flank KP.SIY tumors. The 2C T cells primed in the mLN retained a TCF-1+ TIM-3− phenotype with decreased GzmB after infiltration into flank tumors, whereas the 2C T cells primed in iLN retained increased TIM-3 and GzmB expression (Fig. 5, K and L). This further affirmed that TLdys is a persistent state induced during T cell priming in the mLN rather than a consequence of differential stimulation in the TME.
TLdys is detectable in patients with NSCLC
To determine whether a TLdys T cell gene signature identifies subsets of human NSCLC TIL, we generated a uniform manifold approximation and projection (UMAP) plot from a scRNA-seq study of human NSCLC (Fig. 6A) (53) and mapped signatures corresponding to Tex and TLdys phenotypes onto this UMAP (Fig. 6B and table S7). The Tex signature enriched among the terminally differentiated CD8-LAYN cluster, whereas the TLdys signature was enriched among the less differentiated CD8-KLF2 (Kruppel like factor 2), CD8-GZMK, and CD8-XCL1 clusters (Fig. 6B). Pseudotemporal ordering revealed a trajectory from the CD8-KLF2, CD8-GZMK, and CD8-XCL1 populations to the terminally differentiated CD8-LAYN phenotype (Fig. 6C). Along this trajectory, the TLdys signature was down-regulated as a function of pseudotime, whereas the Tex signature was up-regulated (Fig. 6D). Thus, less differentiated CD8+ T cell populations in human NSCLC demonstrated transcriptomic similarity to the TLdys T cell state found in KP lung tumors, whereas terminally exhausted CD8+ T cell were closely related to the Tex T cells that dominate KP flank tumors. These observations suggest that both Tex and TLdys T cells are abundant, transcriptionally distinct, and mutually exclusive in the TIL of human NSCLC samples. Gueguen et al. (53) found minimal TCR sequence overlap between the KLF2 and GZMK clusters with the LAYN cluster, suggesting that, similar to our observations made in the mouse model, human CD8+ T cells enriched for the TLdys signature inefficiently differentiated to terminally exhausted TIL.
Fig. 6. TLdys T cell state is detectable in human NSCLC TIL populations.
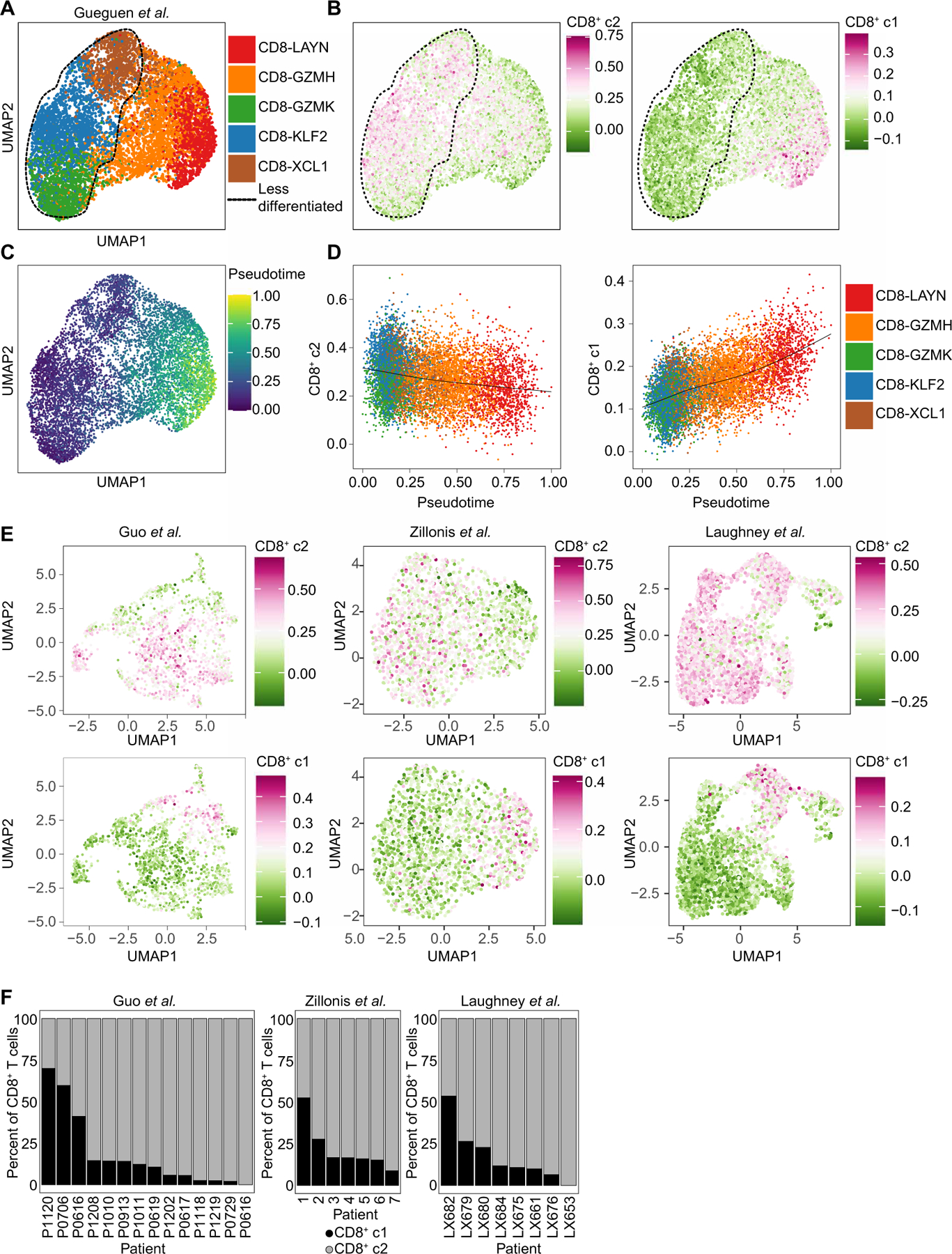
(A) UMAP plot of CD8+ T cell clusters from Gueguen et al. (53). (B) CD8+ c2 or CD8+ c1 signatures mapped onto the CD8+ T cell clusters from Gueguen et al. in (A). (C) Pseudotemporal analysis of CD8+ T cell clusters from Gueguen et al. in (A). (D) Pseudo-temporal analysis of CD8+ c2 or CD8+ c1 signatures and CD8+ T cell clusters from Gueguen et al. in (A). (E) UMAP feature plots from three human datasets analyzed for expression of CD8+ c2 (top) and CD8+ c1 (bottom) signatures. (F) Proportions of CD8+ T cells from individual patients that have CD8+ c1 (black) or CD8+ c2 (gray) signatures.
To validate that the TLdys gene signature could also be detected in other NSCLC datasets, we mapped gene signatures of Tex or TLdys T cell states onto three additional NSCLC datasets (54–56). We found that across NSCLC datasets, most of the CD8+ TIL appeared enriched for the TLdys signature, whereas only a fraction of TIL were enriched for the Tex signature (Fig. 6E). For individual patients, the contribution of the TLdys and Tex phenotype varied drastically, supporting the notion that a fraction of patients with NSCLC completely lack a population of T cells in the Tex state found in KP flank tumors and are dominated by the TLdys phenotype found in KP lung tumors (Fig. 6F). In summary, we were able to validate that the TLdys T cell state is detectable in human NSCLC TIL, and similar to our observation in the mouse model, they have limited differentiation to the Tex state despite signs of T cell activation.
IL-2 and IL-12 therapy prevents TLdys
Our data suggested that the TLdys state was induced during T cell priming in the mLN, where transcriptional profiling indicated significantly lower expression of Il2ra, Il12rb, and Il12rb2 (Fig. 4B). In addition, pathway analysis had found that IL-2 signaling was significantly enriched in flank TIL compared with lung TIL (Fig. 2F). Given these observations, we determined whether cytokine therapy with IL-2 or IL-12 could prevent TLdys induction in our model. To deliver IL-2 or IL-12 to tumor-bearing mice, we used fusion proteins of IL-2 or IL-12 to murine serum albumin (MSA) (MSA-IL2 and MSA-IL12, respectively), which lengthens the half-life of IL-2 or IL-12 in vivo and has potent antitumor effects in mouse flank tumor models (57, 58). We transferred 2C T cells to mice bearing KP.SIY lung tumors and, on the same day, administered MSA-IL2 or MSA-IL12. MSA-IL2 treatment slightly increased CD25 and GzmB expression, whereas MSA-IL12 had no effect (Fig. 7B). Combined administration of MSA-IL2 and MSA-IL12 led to a significant increase in both CD25 and GzmB (Fig. 7B). To determine whether differentiation of endogenous T cells to the TLdys state could be overcome, we treated mice bearing KP.SIY lung tumors with MSA on day 7 after tumor inoculation and analyzed the endogenous SIY-reactive CD8+ T cells in mLN and tumors on day 10 after tumor inoculation. SIY-reactive CD8+ T cells in the mLN down-regulated TCF-1 and up-regulated TIM-3 (Fig. 7C), CD25, GzmB, and 4–1BB, whereas CD49d was unchanged (Fig. 7D). We also found a notable up-regulation of TIM-3, CD25, GzmB, and 4–1BB (Fig. 7, E and F) and a modest down-regulation of CD49d expression (Fig. 7F) in SIY-reactive CD8+ TIL, indicating that IL-2 and IL-12 were sufficient to overcome TLdys in our model.
Fig. 7. TLdys can be overcome by combined IL-2 and IL-12 therapy.
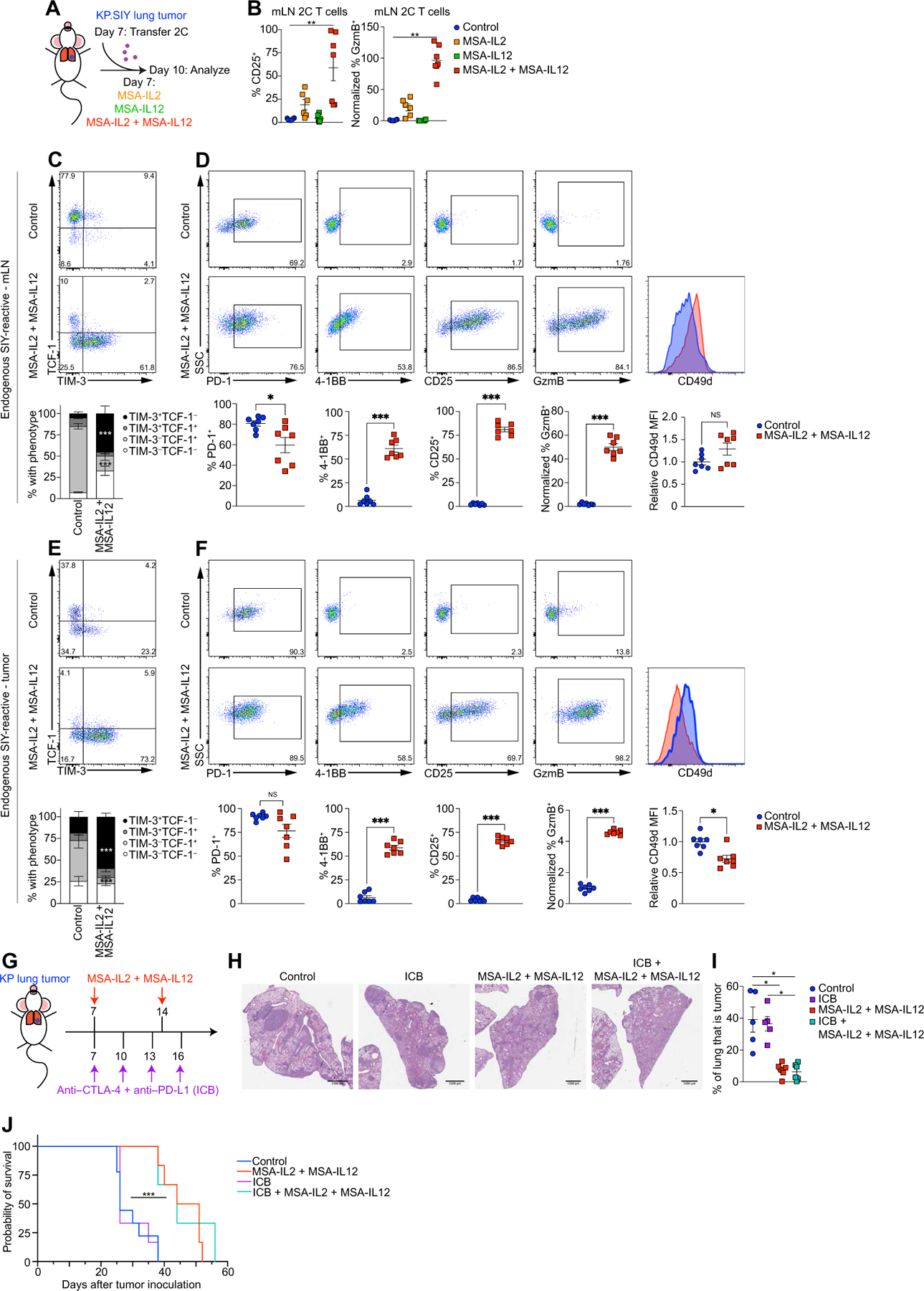
(A) Schematic of experimental design. Mice were inoculated with KP.SIY lung tumors. On day 7 of tumor growth, 2C T cells were adoptively transferred to tumor-bearing mice. Immediately after adoptive transfer, mice were given either IL-2 or IL-12 fusions to MSA or the combination. Seventy-two hours after adoptive 2C T cell transfer, the mLNs were analyzed with flow cytometry. (B) Percentage of CD25 (left) and Gzmb (right) on 2C T cells in mLNs for control or cytokine treatments (control, IL-2, and IL-12, n = 6 and IL-2 + IL-12, n = 7, data pooled from two experiments). (C and D) Endogenous SIY-reactive CD8+ T cells in the mLNs were analyzed for (C) TCF-1 and TIM-3 expression or (D) for PD-1, 4–1BB, CD25, GzmB, and CD49d expression. (E and F) Analysis of endogenous SIY-reactive CD8+ TIL from the same mice as (C) and (D). (E) TCF-1 and TIM-3 expression. (F) PD-1, 4–1BB, CD25, GzmB, and CD49D expression. [(C to F) n = 7 each condition, data pooled from two experiments] (G) Schematic of experiment combining ICB with MSA-IL2 + MSA-IL12 therapy. Mice were inoculated with KP.SIY lung tumors. Mice received either control treatments, ICB on days 7, 10, 13, and 16, MSA-IL2 + MSA-IL12 on days 7 and 10, or the combination of ICB and MSA-IL2 and MSA-12. (H and I) Lung tumor burden assessed on day 21 with (H) showing a representative H&E example and (I) showing the quantification of tumor area per lung lobe (control and ICB, n = 5 and MSA-IL2 + MSA-IL12 and ICB + MSA-IL2 + MSA-IL12, n = 6, data pooled from two experiments). (J) Mice were inoculated with lung KP tumors, treated as in (G), and monitored for survival (control, n = 9; ICB, n = 6; MSA-IL2 + MSA-IL12, n = 6; and ICB + MSA-IL2 + MSA-IL12, n = 3; data pooled from three experiments). Data are shown as means ± SEM, and statistical analysis was conducted using an MWU test with *P < 0.05, **P < 0.01, and ***P < 0.001 (B to I), or with a Kaplan-Meier analysis (J).
On the basis of the significant change in T cell differentiation induced by MSA-IL2 and MSA-IL12, we next assessed whether this therapy could induce control of KP lung tumors. Mice were inoculated with KP parental lung tumors and treated with ICB, MSA-IL2, and MSA-IL12 or the combination on day 7 (Fig. 7G). ICB had no effect on tumor growth (Fig. 7, H and I), whereas MSA-IL2 and MSA-IL12 and the combination of ICB + MSA-IL2 and MSA-IL12 induced marked reductions in lung KP tumors on day 21 (Fig. 7, H and I). In addition, a longitudinal study using micro–computed tomography found that MSA-IL2 and MSA-IL12 treatment even reduced tumor size over time (fig. S7, A and B, and movies S1 to S4). Consistently, MSA-IL2– and MSA-IL12–treated mice had significantly longer survival (Fig. 7J). Consistent with previous reports (57), KP flank tumors also benefitted from MSA-IL2 and MSA-IL12 (fig. S8), suggesting that this therapy also enhances T cell responses in flank TIL. In summary, our data suggest that CD8+ T cells with TLdys fail to respond to ICB but respond to combined IL-2 and IL-12 treatment in the KP lung tumor model.
DISCUSSION
We identified a state of T cell dysfunction induced during T cell priming in the mLN of KP lung tumor–bearing mice, which we termed TLdys. TLdys was characterized by reduced expression of effector and exhaustion molecules on CD8+ T cells and ICB resistance (Fig. 8). TLdys resembles T cell responses in an ICB-refractory sub-set of human NSCLCs that have been referred to as nonfunctional. In this subset of patients, tumors are infiltrated by T cells, but these T cells fail to up-regulate effector molecules and PD-L1 in response to ICB (3). Therefore, our observations on the induction and characteristics of the TLdys T cell state may allow for the rational development of new treatment strategies for T cell–infiltrated but ICB-refractory NSCLC.
Fig. 8. Schematic summary.
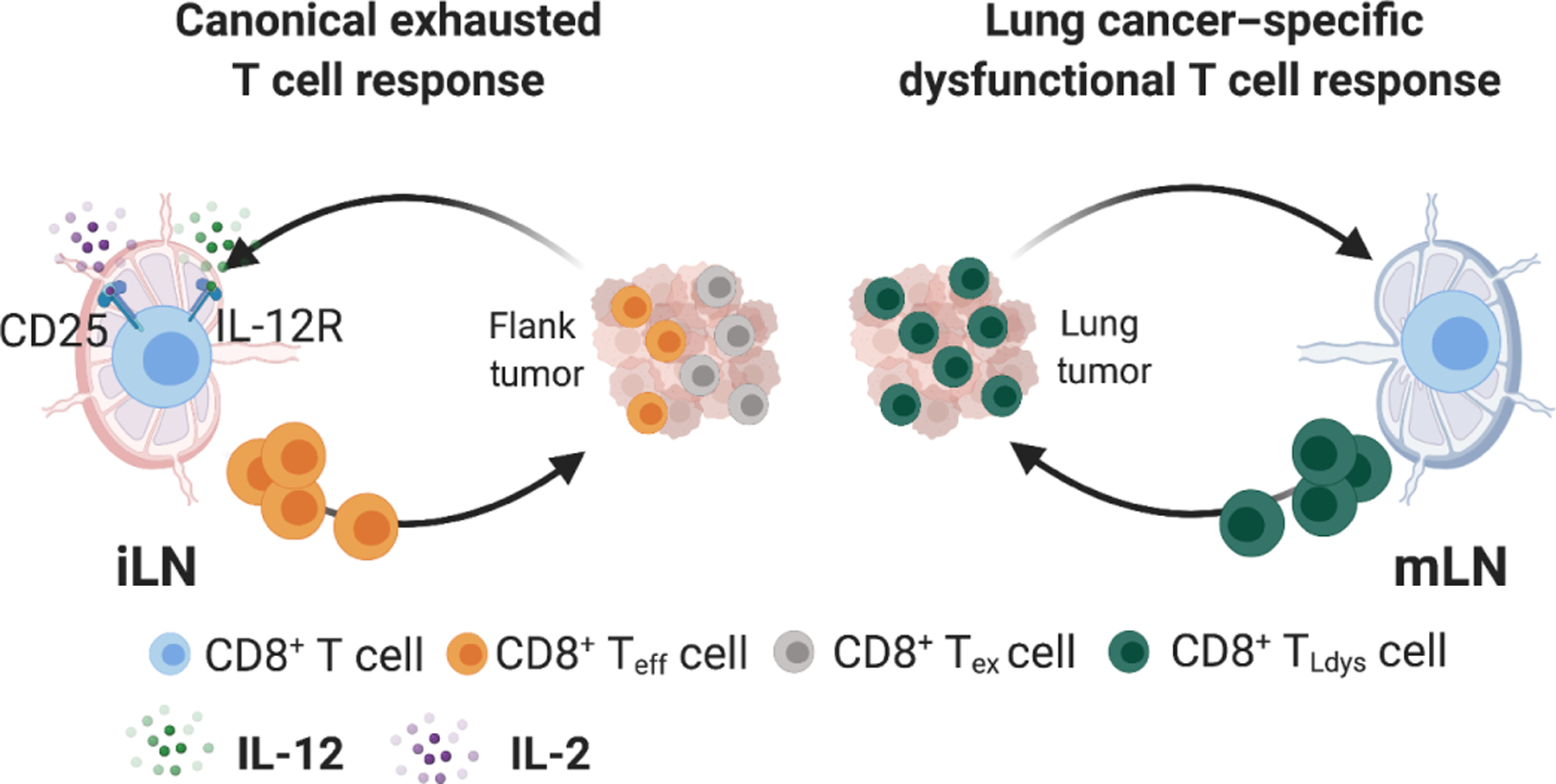
(Left) In response to flank tumors, tumor-reactive CD8+ T cells primed in the iLN up-regulate CD25 and IL-12R, express effector molecules, show signs of conventional exhaustion, and respond to CBT. (Right) In response to lung tumors, however, tumor-reactive CD8+ T cells primed in the mLN activate a lung-specific dysfunctional program (TLdys), do not up-regulate CD25 and IL-12R, fail to gain effector molecule expression, and do not acquire a conventional Tex phenotype. CD8+ TLdys cells do not respond to CBT. This suggests that differentiation of CD8+ T cells into dysfunctional states other than conventional Tex drives CBT resistance in a subset of T cell–infiltrated NSCLCs. Teff, effector T cell. Created with Biorender.com.
T cell infiltration into tumors is predictive for ICB response, but the quality of T cells is critical (40). We provide evidence that not all tumor-reactive T cells are functionally interchangeable and that not all tumor-specific CD8+ T cells will react to ICB. T cells with TLdys, induced in response to orthotopic lung cancer, failed to produce IFN-γ, a feature of antitumor immune responses that mediate PD-L1 up-regulation, a biomarker in NCSLC. ICB failed to expand TIL TLdys and did not affect the phenotype or function of lung tumor–reactive T cells. Furthermore, T cells with TLdys had low GzmB and poor cytotoxicity in vivo. Thus, TLdys closely resembles the nonfunctional T cell responses described in an ICB-refractory NSCLC patient population (3, 59–62). In contrast, flank TIL gained effector function and responded to ICB with T cell expansion and tumor control, suggesting that TLdys was specific to orthotopic lung tumors.
We determined that T cell fate decisions leading to Tex or TLdys occurred during priming in the TdLN. Transcriptional differences occurred within 72 hours after T cell activation. T cells primed in the mLN never gained robust CD25 expression, whereas priming in the iLN resulted in CD25 up-regulation. CD8+ T cells that express high levels of CD25 differentiate into effector T cells (38, 39), suggesting that CD25 expression is required for differentiation into effector and exhausted T cell states. Consistently, T cells primed in the mLN had low GzmB, a phenotype that was preserved in lung TIL. These data suggested that T cell fate decisions are made early during T cell priming, depend on the local cytokine milieu, and can predetermine the quality of the resulting TIL.
T cell activation is affected by antigen drainage to the TdLN and the functional capacity of APC. We found comparable antigen load and stimulatory capacity of DC between lung and flank tumors. TLdys was also observed with intratracheal administration of tumor cells, a condition used to control for potential systemic dissemination of tumor cells or different tumor loads induced between intravenous and subcutaneous settings. However, it remains a possibility that greater immunogenic cell death is induced in flank tumors, leading to a more inflammatory environment and thus effective CD8+ T cell activation. Alternatively, the lung could be a tolerogenic environment (63), requiring a greater degree of immunogenic cell death for innate immune activation. It should be noted, however, that only inoculation of lung cancers KP and LL/2 into the lung induced TLdys, whereas inoculation of B16 melanoma cells resulted in Tex, suggesting that only orthotopic lung tumors may induce TLdys. Additional tumor cell lines and modes of cancer induction will be needed to fully elucidate the details resulting in Tex or TLdys induction and whether induction of TLdys is restricted to the mLN. Our analyses indicate the presence of both phenotypes in flank TIL, suggesting that T cell differentiation may be complex.
Adoptive transfer studies suggested that TLdys persisted after T cell activation. Neither homeostatic proliferation in RAG2−/− mice nor ICB treatment induced effector function, supporting that epigenetic imprinting might mediate TLdys (64, 65). We found increased Tox levels in T cells primed in the mLN. Tox is an epigenetic modifier, previously found to be a critical factor for not only epigenetic remodeling of the chromatin during induction and maintenance of Tex but also long-lived memory T cell responses (42, 66). In addition, Tox expression was found in stem-like memory CD8+ T cells induced after intravenous nanoparticle vaccine administration (67). Therefore, we speculate that Tox up-regulation during induction of the TLdys suggests a context-specific effect of Tox. Further assessment of the epigenetic state of TLdys and Tex will be needed to fully elucidate whether Tox or additional epigenetic modifiers are important for the induction of TLdys and how the epigenetic state of TLdys compares with other T cell states. Consistent with decreased expression of CD25 and GzmB after priming in the mLN, we found decreased differentiation of CD8+ T cells into TIM-3+ effector cells. TCF-1 and TIM-3 define precursor- and terminally exhausted CD8+ T cells, respectively, in chronic LCMV and melanoma models (36, 37, 40, 47, 51). Expression of TCF1+ in SIY-reactive T cells in the mLN together with elevated levels of Tox, Bach2, and Bcl6 suggested a relation to precursor-exhausted CD8+ T cells (47). However, adoptive transfer studies of 2C T cells primed in mLN clearly demonstrated that T cells with TLdys failed to differentiate into effector or exhausted T cells. Direct comparison of CD8+ TIL from KP lung tumors to transcriptional signatures of precursor-exhausted CD8+ T cells suggested that T cells with TLdys do not transcriptionally resemble precursor-exhausted CD8+ T cells. These data suggest that TLdys is induced early and does not give rise to conventionally exhausted T cells.
Integration of our findings with clinical datasets suggested that a fraction of T cell–infiltrated patients lacked exhausted T cells and were dominated by TLdys T cells. A recent publication described five CD8+ TIL populations in NSCLC: blood-derived CD8-KLF2 and CD8-GZMK precursors, tumor-infiltrating CD8-XCL1 precursors, a transitory CD8-GZMH population, and terminally exhausted CD8-LAYN T cells (53). Mapping the TLdys signature onto this dataset suggested a close association of T cells with TLdys with KLF2 and GZMK precursor clusters, whereas the exhaustion signature derived from CD8+ c1 T cells mapped to the exhausted LAYN cluster. Gueguen et al. (53) found limited differentiation of KLF2 and GZMK precursor populations to exhausted LAYN T cells, consistent with our data that T cells with TLdys failed to acquire effector and exhaustion molecules. Multiple studies found that exhausted CD8+ T cells correlate with good responses to ICB in patients with NSCLC (60–62), and the presence of a T cell–derived IFN-γ signature and PD-L1 up-regulation is predictive for ICB response in NSCLC. This is supported by our studies in which TLdys T cells failed to respond to ICB or make IFN-γ; therefore we propose that the patient populations dominated by TLdys T cells may represent patients that would be ICB refractory. However, this requires clinical validation. We also showed that IL-2/IL-12 cytokine treatment could mediate differentiation of TLdys T cells into highly potent effector T cells. This observation demonstrates that whereas TLdys T cells fail to spontaneously differentiate into effector T cells, cytokine therapy could facilitate this differentiation. Further studies will be required to determine the phenotype of T cells responding to IL-2/IL-12 therapy in flank tumors because about 50% of flank TIL were of TLdys T cell state. How natural cytokine signals might affect the spontaneous differentiation into Tex and TLdys state will also require additional interrogations. However, immunosuppressive populations such as regulatory T cells or tumor-associated macrophages might affect T cell differentiation by cytokine production or consumption (68, 69).
Whereas Gueguen et al. (53) found limited evidence for transition of T cell clonotypes from the KLF2 or GZMK clusters to the LAYN cluster, our TCR clonotype analyses identified shared clonotypes between Tex and TLdys. Furthermore, we were able to demonstrate the induction of TLdys and exhausted T cell phenotypes among endogenous SIY-reactive T cells in KP.SIY tumors and in TCR-transgenic T cells. In addition, only subtle differences between the CD8+ TIL responses against KP and KP.SIY tumors were found. Together, these observations provide strong support that decisions between Tex and TLdys are independent of TCR usage or affinity. Whether TCR signal strength affects T cell states in the TME requires further investigation. In summary, analyses of both clinical samples and our mechanistic studies agree that T cell priming in LN is involved in inducing different T cell states and highlights the importance of further understanding the determinants of early T cell fate decisions.
To determine whether the TLdys phenotype could be therapeutically manipulated, we assessed whether cytokine therapy might be effective when administered during priming. Combined treatment with IL-2 and IL-12 induced differentiation of TLdys to an effector T cell state. This raises questions about IL-2 and IL-12 signals during priming. IL-2 availability can be significantly affected by immunosuppressive populations such as regulatory T cells (70, 71). Regulatory T cells, in turn, are frequently localized to tissue-draining LN based on tissue-specific antigen expression patterns (72, 73). Lung-specific regulatory T cells may therefore affect effector T cell activation in the mLN. IL-12 is produced by myeloid cells (74), but the exact cellular source of IL-12 is still unclear. It is conceivable that IL-12 is induced once CD8+ T cells gain the ability to produce IFN-γ (75). IL-12 has also been shown to induce down-regulation of Klf2 and S1pr1 expression in T cells (76). Down-regulation of KLF2 and S1PR1 is a common feature of CD8+ T cells that express high levels of PD-1, LAG-3, TIM-3, and 4–1BB both in human TIL (59–62) and TIL isolated from KP flank tumors. This together with the requirement of IL-2 might suggest that IL-2 and IL-12 signaling sensitizes T cells during priming for the optimal acquisition of effector function. Thus, IL-2– and IL-12–based immunotherapies may help differentiate TIL toward an ICB-responsive state. In a recent study, new derivatives of IL-12 were generated to reduce natural killer cell–mediated toxicity but retain the ability of IL-12 to induce T cell effector function, primarily IFN-γ and GzmB (77). This preclinical work, together with our observations, points to a potential use for cytokine therapy in patients with nonfunctional T cell responses who fail to respond to current treatments.
One shortcoming of our studies was that we could not separate lung tumors and normal lung tissue, and therefore, we cannot fully exclude that T cells from healthy lung affect our analysis; however, our IF experiments suggest that TIL were strongly enriched in the TME. Another shortcoming was that our study is based on a limited number of transplantable cell lines and tumor inoculation methods. In addition, there were insufficiently available human NSCLC data to determine whether the gene signature associated with CD8+ c2 is predictive of ICB resistance. Future longitudinal human studies comparing ICB outcome with pretreatment CD8+ TIL scRNA-seq will be required to address this question.
Together, our data suggest that responsiveness to ICB in NSCLC can be predetermined during T cell priming in the mLN. If T cells enter TLdys, a T cell response lacking cytotoxic effector functions becomes established in tumors. It is conceivable that this early decision point precludes T cells with TLdys from reinvigoration by ICB. This study provides mechanistic insight into the induction of ICB-refractory T cell responses and provides rationale for potential strategies to increase effector CD8+ T cell differentiation in NSCLC, potentially benefiting ICB-refractory patients with nonfunctional immune responses, improving outcomes for patients with NSCLC.
MATERIALS AND METHODS
A full description of the methods can be found in Supplementary Methods.
Study design
The purpose of this study was to determine how differences in T cell activation between lung and flank KP tumors led to ICB resistance in KP lung tumors. We characterized T cell differentiation in these two tumor settings and defined how this contributed to ICB resistance. We used transplantable syngeneic tumor mouse models and publicly available human datasets. Mice were randomized into study groups after tumor inoculation and before treatments. All experiments were performed at least twice independently with at least three mice per group. All data were included unless there were technical issues with experimental setup or data collection. Outliers were not removed. All tumor outgrowth studies were planned to end on day 21 after tumor injection. Early termination occurred if tumors grew past the allowed size or if mice were flagged by veterinarians for tumor ulceration, changes in body condition, or other health reasons that required euthanasia before day 21. Other tumor analysis time points were days 7 and 14 after tumor injection or 3 days after adoptive transfer of transgenic T cells.
Mice
All mice were housed and bred under specific pathogen–free conditions at the Koch Institute animal facility. All experimental animal procedures were approved by the Committee on Animal Care at the Massachusetts Institute of Technology (MIT).
Generation of expression vectors
The pLV-EF1a-IRES-puro vector (Addgene no. 85132) was used in conjunction with the cerulean-SIY insert generated using the Cerulean-N1 template (Addgene no. 54742) or the ZsGreen insert zsGreen_minOVA (a gift from M. Krummel).
Tumor cell lines and tumor outgrowth studies
KP, LL/2, KP.SIY, and B16.SIY were cultured under standard conditions. Tumor cells (2.5 × 105) were injected. Subcutaneous tumor area measurements were collected two to three times a week, whereas lung tumor areas of hematoxylin and eosin (H&E) sections were measured using QuPath software.
Immunohistochemistry
Flank tumors and tumor-bearing lungs were fixed in 10% formalin. Fixed tissues were processed, paraffin-embedded, sectioned, and stained by the Hope Babette Tang Histology Facility at the Koch Institute at MIT. For anti-CD8 staining, a 1:200 dilution of anti-CD8 (clone 4SM16, eBioscience 14–0195-82) was used.
Immunofluorescence
Tumors were processed as previously described (78). Tissues were stained with antibodies (see the Supplementary Materials) and washed with phosphate-buffered saline (PBS), and nuclei were counterstained with propidium iodide. Tissue sections were imaged using the TissueFAXS whole-slide scanning system (TissueGnostics, USA) using a Zeiss 20x Plan-Neofluar air, 0.5 numerical aperture and analyzed using the TissueQuest image analysis software (TissueGnostics, USA). Images were processed with the image processing package FIJI (79).
Tumor dissociation
Spleens and LNs were dissected from mice and physically dissociated. Tumors were dissociated using the gentleMACS Octo Dissociator (Miltenyi).
Flow cytometry and cell sorting
All analyses excluded circulating CD45+ cells using intravenous injection of a PE-CF594–labeled anti-CD45 antibody before euthanasia, which allowed circulating cells to be excluded from analyses. Cells were resuspended in antibody-containing staining buffer plus eBioscience Fixable Viability Dye eFluor 780 or eFluor 506 to distinguish live and dead cells and with anti-CD16/CD32 to prevent nonspecific antibody binding. Cell surface proteins were stained for 20 min on ice with fluorophore-conjugated antibodies at a 1:200 dilution. Cells were washed and resuspended in fixation/permeabilization buffer followed by staining with antibodies against intracellular targets. To obtain absolute counts of cells, Precision Count Beads (BioLegend) were added. Flow cytometry sample acquisition was performed on an LSRFortessa cytometer (BD Biosciences), and the collected data were analyzed using FlowJo v10.5.3 software (TreeStar). For cell sorting, the surface staining was performed as described above under sterile conditions, and cells were acquired and sorted into TRIzol or buffer using a FACSAria III sorter (BD Biosciences). For TIL analysis, cells were pregated on live, CD45+, CD45-IV−, TCRb+, single cells, CD4−, and CD8+. See the Supplementary Materials for all antibodies. See fig. S9 for example gating strategy of CD8+ and SIY-reactive CD8+ cells.
Seq-Well sequencing and analysis
Single-cell suspensions were prepared as described above. Mice received anti–CD45-PE antibody retro-orbitally 3 min before euthanasia, followed by bead-mediated depletion of antibody-labeled circulating cells. SIY pentamer–reactive CD8+ T cells were isolated using FACS. Cells were then processed using the Seq-Well platform with second strand chemistry, as previously described (16, 33). Libraries were barcoded and amplified using the Nextera XT kit (Illumina) and were sequenced on a NovaSeq 6000 or a NextSeq 550 (Illumina).
Single-cell data processing and visualization
Raw read processing of scRNA-seq reads was performed as previously described (80). Principal components analysis was performed. The number of principal components used for visualization was determined by examination of the elbow plot, and two-dimensional embeddings were generated using UMAP. Clusters were determined using Louvain clustering, as implemented in the FindClusters function in Seurat, and clusters that contained T cells were selected for further analysis. These cells were reprocessed with the same processing and clustering steps described above. Differential gene expression was performed for each cluster, and clusters corresponding to similar phenotypes were merged.
Pathway enrichment analysis
Gene sets representing relevant pathways were obtained from MSigDB (81, 82). A score for the expression of genes in each pathway was calculated for each cell using the AddModuleScore function in Seurat (83). Pathways in the hallmarks gene collection were used to analyze CD8+ T cells and KP.zsGreen tumor cells.
Paired single-cell TCR sequencing
Paired TCR sequencing and read alignment was performed as previously described (84). Processing of reads was performed using the Immcantation software suite (85, 86). GLIPH2 analysis was performed as described previously (34) using the provided data from mouse CD8+ T cells as a reference. For the analysis of global motifs, sub-stitutions between any pair of amino acids were permitted. GLIPH specificity groups that were subsets of other specificity groups were not analyzed further.
Integrated analysis of KP parental and KP.SIY single-cell data
Single-cell sequencing data from KP parental and KP.SIY TIL were integrated using Seurat v3 (87). Genes that were detected in less than 3% of cells in either the KP TIL or KP.SIY TIL were excluded. Default parameter values and the top 30 canonical correlation analysis (CCA) components were used to find transfer anchors between datasets and generate an integrated matrix.
Adoptive TIL transfer
Flank and lung tumors were dissociated and prepared for FACS as described above. TIL were identified as Live, CD45+, noncirculating Thy1+ cells. Recipient RAG2−/− mice received equal numbers (20,000) of either lung or flank TIL intravenously. TIL were allowed more than 12 weeks to reconstitute RAG2−/− mice, after which the reconstituted mice were inoculated with flank tumors.
Concomitant immunity assay
Mice were injected with KP tumors subcutaneously or intravenously as outlined above. Seven days after tumor injection, mice were injected with a second KP tumor on the contralateral flank or intravenously.
CD8 antibody depletion
Mice were injected with 200 µg of anti-CD8 antibody (clone 53–6.7 Bio X Cell) weekly for the duration of the experiment, beginning 48 hours before tumor inoculation.
IFN-γ–ELISpot
Splenocytes (1 × 106) were plated on coated and washed ELISpot plates and stimulated with 160 nM SIY peptide or a mixture of phorbol 12-myristate 13-acetate (100 ng/ml; Sigma-Aldrich) and ionomycin (1 µg/ml; Sigma-Aldrich) as a positive control or medium only as negative control. Plates were developed the next day using the BD Biosciences mouse IFN-γ–ELISpot kit, following the manufacturer’s protocol.
2C T cell adoptive transfer
T cells were isolated from 2C RAG2−/−CD45.1+ mice (spleen and LN) and labeled with CFSE or CellTrace Violet (Life Technologies). T cells (106) were transferred to mice bearing KP.SIY tumor 7 days after tumor inoculation and isolated 72 hours later. For CFSE analysis, the percent proliferated was calculated as the percentage of cells that had undergone one or more rounds of division based on CFSE staining intensity using an unstimulated, CFSE-labeled sample as an undivided reference control. For transfers into secondary recipients, 2C T cells were transferred into primary recipients as described above, isolated from mLN or iLN, and enriched using the Miltenyi CD8+ T cell isolation kit before CD45.1+ 2C T cells were sorted using FACS. Sorted cells were transferred intravenously into secondary recipient RAG2−/− mice bearing day 7 flank KP.SIY tumors. Secondary recipients were analyzed 5 days after adoptive transfer.
DC analysis and ex vivo coculture assay
LN processing procedure was adapted from Ruhland et al. (45), and CD19+ and CD3+ cells were depleted using magnetic beads before flow analysis or sort of DC. For ex vivo cocultures, OT-I T cells were isolated and CFSE-labeled as described above. CD8+ OT-I cells (5 × 104) and FACS-sorted ZsGreen+ DCs (5 × 103) were cocultured and analyzed by flow cytometry at 72 hours.
Smart-seq bulk RNA-seq and analysis
2C T cells were adoptively transferred to tumor-bearing mice as described above. Seventy-two hours after transfer, TdLN cells were isolated and prepared for FACS. 2C T cells were FACS-sorted directly into TRIzol reagent and RNA-extracted. RNA library preparation and sequencing were performed at the KI Genomics Core / MIT BioMicro Center. Sequencing was performed using the HiSeq 2000 sequencing system (Illumina). Sequence alignment and .bam file generation were performed by the KI Genomics Core / MIT BioMicro Center. The resulting .bam files were sorted using Samtools. Differential gene expression analysis was performed on the sorted .bam files with Cufflinks.
Analysis of data from Chen et al.
Data from the scRNA-seq of P14 cells published by Chen et al. (36) were downloaded from the Gene Expression Omnibus (GEO) database under accession number GSE131535. The data were processed using the processing pipeline described above, and clusters were determined using the FindClusters function in Seurat. Gene signature scores were calculated using the AddModuleScore function in Seurat.
Cytokine and detection
Single-cell suspensions were isolated from tumors as described above. Round-bottom 96-well plates were coated overnight in PBS with anti-CD3 (0.2 mg/ml; clone 145–2C11, BD Biosciences), and anti-CD28 (0.5 µg/ml; clone 37.51, BD Biosciences). Cells were plated in coated round-bottom 96-well plates for 4 hours in the presence of BrefeldinA (BioLegend). After 4 hours, the cells were stained for extra-cellular and intracellular antigens.
In vivo cytotoxicity assay
The protocol was adapted from Kim et al. (52). Wild-type splenocytes were ammonium-chloride-potassium–lysed and pulsed for 1 hour at 37°C with 0.2 µM SIY peptide. SIY-pulsed splenocytes were then labeled with 0.5 µM CFSE, whereas control, unpulsed splenocytes were labeled with 5 µM CFSE for 10 min at 37°C. After CFSE labeling, SIY-pulsed and control splenocytes were mixed at a 1:1 ratio, and 10 × 106 of each were intravenously injected into mice bearing KP.SIY lung or flank tumors on day 7 of tumor growth or into naïve control mice. Four hours after injection of CFSE-labeled splenocytes, spleens from tumor-bearing mice were isolated, single-cell splenocyte suspensions were generated by physical dissociation, and splenocytes were stained with a live/ dead discriminator dye and analyzed for CFSE+ populations by flow cytometry.
Analysis of human NSCLC datasets
Human NSCLC single-cell datasets were downloaded from the GEO database, under accession numbers GSE99254 (54), GSE127465 (56), GSE123902 (55), and GSE162500 (53). Data from Gueguen et al. were processed using integration in Seurat 3, as described by Stuart et al. (87). Gene signature scores were computed using the AddModuleScore function in Seurat. Pseudotemporal ordering using diffusion pseudotime was performed using sc.tl.dpt function in SCANPY (88, 89). Data from Guo et al. (54), Zillonis et al. (56), and Laughney et al. (55) were processed using the processing pipeline described above to identify clusters corresponding to CD8+ T cells. Gene signature scores were computed using the AddModuleScore function in Seurat. Clusters were identified using the FindClusters function in Seurat and were classified as CD8+ c1 or CD8+ c2 according to their expression of the corresponding gene signatures.
ICB treatment
Mice were injected intraperitoneally with anti–CTLA-4 (clone UC10–4F10–11) and anti–PD-L1 (clone 10F.9G2) antibodies on days 7, 10, 13, and 16 after tumor inoculation. Each mouse received 100 µg of each antibody per treatment.
MSA-IL2 and MSA-IL12 generation
MSA-cytokine fusions were generated as previously described (57, 58). Human embryonic kidney (HEK) 293 cells were transfected with plasmid DNA using polyethylenimine in OptiPro serum-free medium (Thermo Fisher Scientific). His-tagged proteins were isolated from HEK293 supernatant using TALON Metal Affinity Resin (Takara Bio Inc.). Cytokine fusion proteins were then further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 pg column on an ÄKTA fast protein liquid chromatography system (GE Healthcare). All proteins were stored at 4°C, but before therapeutic injection, cytokine fusion proteins were warmed to room temperature.
MSA treatment
Each mouse was injected retro-orbitally with MSA-IL2 (5.94 × 10−10 mol per mouse) and/or MSA-IL12 (1.42 × 10−11 mol per mouse) per treatment. For experiments phenotyping 2C T cells or endogenous SIY-reactive CD8+ T cells, mice received one dose of MSA-cytokine fusions on day 7 after tumor inoculation and were analyzed on day 10 after tumor inoculation. For therapeutic experiments, mice were dosed with MSA-IL2 and/or MSA-IL12 on days 7 and 14 after tumor inoculation and analyzed on day 21 after tumor inoculation and monitored for survival after treatment.
Micro–computed tomography
A Bruker Skyscan 1276 was used to acquire a series of images with a rotation step of 0.65° over a 360° rotation. Images were acquired with x-ray tube settings of 100 kV, 200 µA, and an exposure time of 90 ms with a 0.5-mm aluminum beam filter. A 4 × 4 detector binning was used for an isotropic resolution of 40.16 µm. Anesthesia was induced at 3% isoflurane and maintained at 2.0 to 2.5% during imaging, which lasted 76 s. Image reconstruction was performed using the Bruker NRecon software.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (GraphPad) and R. All data are shown as means ± SEM. For flow cytometry, immunohistochemistry, and tumor outgrowth studies, statistical analyses were performed with Mann-Whitney U (MWU) test for comparisons of two groups or two-way analysis of variance (ANOVA) for multiple comparisons over time, with *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001. For differential gene expression between T cell clusters, P values were calculated using a two-sided Wilcoxon rank-sum test and were corrected with Bonferroni correction. For comparison of gene modules with previously published T cell states, false discovery rate q values were calculated using a one-tailed hypergeometric test and were corrected with Bonferroni correction. For pathway analysis, P values were calculated using a two-tailed Wilcoxon rank-sum test and were corrected with Bonferroni correction.
Supplementary Material
Acknowledgments:
We would like to thank M. Duquette for technical assistance and the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Flow Cytometry, Histology, and Preclinical Imaging and Testing facilities.
Funding:
S.S. was funded by the Pew-Stewart Scholars for Cancer Research. B.L.H. was funded by a Ludwig Center for Molecular Oncology Post-Doctoral Fellowship. S.S. and J.C.L. were funded by the Koch Institute Frontier Research Program through the Casey and Family Foundation Cancer Research Fund. This work was supported by the Koch Institute Support Grant P30-CA14051 from the National Cancer Institute.
Footnotes
Competing interests: S.S. is a SAB member for Arcus Biosciences and Venn Therapeutics. S.S. is a consultant for TAKEDA, Merck, Tango Therapeutics, and Ribon Therapeutics and received funding for unrelated projects from Exelicis and TAKEDA. J.C.L. has interests in Sunflower Therapeutics PBC, Pfizer, Honeycomb Biotechnologies, OneCyte Biotechnologies, SQZ Biotechnologies, Alloy Therapeutics, QuantumCyte, Amgen, and Repligen. J.C.L.’s interests are reviewed and managed under MIT’s policies for potential conflicts of interest. J.C.L. receives sponsored research support at MIT from Amgen, the Bill & Melinda Gates Foundation, Biogen, Pfizer, Roche, Takeda, and Sanofi. B.L.H., D.M.M., K.D.W., J.C.L., and S.S. are inventors on patent application 63/158,225 submitted by the MIT that covers lung cancer–specific T cell dysfunction.
Data and materials availability:
All data and materials are available upon request from the corresponding author. Sequencing data can be found in the GEO database under accession number GSE184388. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rafei H, El-Bahesh E, Finianos A, Nassereddine S, Tabbara I, Immune-based therapies for non-small cell lung cancer. Anticancer Res 37, 377–387 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS, Predictive correlates of response to the anti–PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, Zou W, Chen DS, Yi J, Sandler A, Rittmeyer A; POPLAR Study Group, Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–1846 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, Ferrucci PF, Smylie M, Hogg D, Hill A, Marquez-Rodas I, Haanen J, Guidoboni M, Maio M, Schoffski P, Carlino MS, Lebbe C,McArthur G, Ascierto PA, Daniels GA, Long GV, Bastholt L, Rizzo JI, Balogh A, Moshyk A, Hodi FS, Wolchok JD, Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 381, 1535–1546 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski M. Spasic, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G, The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol 14, 717–734 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, Zhang C, Lunceford JK, Joe A, Cheng J, Webber AL, Ibrahim N, Plimack ER, Ott PA, Seiwert TY, Ribas A, McClanahan TK, Tomassini JE, Loboda A, Kaufman D, Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L, Melero I, Schalper KA, Herbst RS, Immunotherapy in non-small cell lung cancer: Facts and hopes. Clin. Cancer Res 25, 4592–4602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brahmer JR, Govindan R, Anders RA, Antonia SJ, Sagorsky S, Davies MJ, Dubinett SM, Ferris A, Gandhi L, Garon EB, Hellmann MD, Hirsch FR, Malik S, Neal JW, Papadimitrakopoulou VA, Rimm DL, Schwartz LH, Sepesi B, Yeap BY, Rizvi NA, Herbst RS, The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J. Immunother. Cancer 6, 75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, Zhang J, Giri U, Gudikote J, Cortez MA, Yang C, Fan Y, Peyton M, Girard L, Coombes KR, Toniatti C, Heffernan TP, Choi M, Frampton GM, Miller V, Weinstein JN, Herbst RS, Wong KK, Zhang J, Sharma P, Mills GB, Hong WK, Minna JD, Allison JP, Futreal A, Wang J, Wistuba II,Heymach JV, Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 5, 860–877 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF,Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, Ali SM, Elvin JA, Singal G, Ross JS, Fabrizio D, Szabo PM, Chang H, Sasson A, Srinivasan S, Kirov S, Szustakowski J, Vitazka P, Edwards R, Bufill JA, Sharma N, Ou SI, Peled N, Spigel DR, Rizvi H, Aguilar EJ, Carter BW, Erasmus J, Halpenny DF, Plodkowski AJ, Long NM, Nishino M, Denning WL, Galan-Cobo A, Hamdi H, Hirz T, Tong P, Wang J, Rodriguez-Canales J, Villalobos PA, Parra ER, Kalhor N, Sholl LM, Sauter JL, Jungbluth AA, Mino-Kenudson M, Azimi R, Elamin YY, Zhang J, Leonardi GC, Jiang F, Wong KK, Lee JJ, Papadimitrakopoulou VA, Wistuba II, Miller VA, Frampton GM, Wolchok JD, Shaw AT, Janne PA, Stephens PJ, Rudin CM, Geese WJ, Albacker LA, Heymach JV, STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF, Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med 5, 200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokito T, Azuma K, Kawahara A, Ishii H, Yamada K, Matsuo N, Kinoshita T, Mizukami N, Ono H, Kage M, Hoshino T, Predictive relevance of PD-L1 expression combined with CD8+ TIL density in stage III non-small cell lung cancer patients receiving concurrent chemoradiotherapy. Eur. J. Cancer 55, 7–14 (2016). [DOI] [PubMed] [Google Scholar]
- 15.LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, Naranjo S, Westcott PMK, Duarte FM, Sankar V, Chiang Z, Brack A, Law T, Hauck H, Okimoto A, Regev A, Buenrostro JD, Jacks T, Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell 38, 212–228.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gierahn TM, Wadsworth II MH, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK, Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD, Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, Wylie T, Allison JP, Smyth MJ, Old LJ, Mardis ER, Schreiber RD, Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF, The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med 214, 381–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chibueze CE, Yoshimitsu M, Arima N, CD160 expression defines a uniquely exhausted subset of T lymphocytes in HTLV-1 infection. Biochem. Biophys. Res. Commun 453, 379–384 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA, Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72, 917–927 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med 207, 2187–2194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galkina E, Thatte J, Dabak V, Williams MB, Ley K, Braciale TJ, Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J. Clin. Invest 115, 3473–3483 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grau M, Valsesia S, Mafille J, Djebali S, Tomkowiak M, Mathieu AL, Laubreton D, de Bernard S, Jouve PE, Ventre E, Buffat L, Walzer T, Leverrier Y, Marvel J, Antigen-induced but not innate memory CD8 T cells express NKG2D and are recruited to the lung parenchyma upon viral infection. J. Immunol 200, 3635–3646 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, Doherty PC, de Fougerolles AR, Topham DJ, The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity 20, 167–179 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Eberlein J, Davenport B, Nguyen TT, Victorino F, Jhun K, van der Heide V, Kuleshov M, Ma’ayan A, Kedl R, Homann D, Chemokine signatures of pathogen-specific T cells I: Effector T cells. J. Immunol 205, 2169–2187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorner BG, Scheffold A, Rolph MS, Huser MB, Kaufmann SH, Radbruch A, Flesch IE, Kroczek RA, MIP-1a, MIP-1b, RANTES, and ATAC/lymphotactin function together with IFN-γ as type 1 cytokines. Proc. Natl. Acad. Sci. U.S.A 99, 6181–6186 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R, Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol 4, 1191–1198 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, Wani MA, Lingrel JB, Hogquist KA, Jameson SC, Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442, 299–302 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Rivera J, Proia RL, Olivera A, The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol 8, 753–763 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hart GT, Hogquist KA, Jameson SC, Krüppel-like factors in lymphocyte biology. J. Immunol 188, 521–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN, Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 40, 235–247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes TK, Wadsworth II MH, Gierahn TM, Do T, Weiss D, Andrade PR, Ma F, de Andrade Silva BJ, Shao S, Tsoi LC, Ordovas-Montanes J, Gudjonsson JE, Modlin RL, Love JC, Shalek AK, Second-strand synthesis-based massively parallel scRNA-Seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity 53, 878–894.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM, Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genomewide antigen screening. Nat. Biotechnol 38, 1194–1202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stegall MD, Elices M, Shepard G, Gup C, Ninova D, Ferguson D, Gill RG, The 2C T-cell transgenic mouse: An in vivo model of allospecific cytotoxic T-cell activation and homing. Transplant. Proc 31, 779 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra J-C, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, Wherry EJ, TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, Leonard WJ, Kissick HT, Ahmed R, Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 51, 1043–1058.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R, Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A, Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, Schietinger A, TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D,Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, Zehn D, TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roychoudhuri R, Eil RL, Clever D, Klebanoff CA, Sukumar M, Grant FM, Yu Z, Mehta G, Liu H, Jin P, Ji Y, Palmer DC, Pan JH, Chichura A, Crompton JG, Patel SJ, Stroncek D, Wang E, Marincola FM, Okkenhaug K, Gattinoni L, Restifo NP, The transcription factor BACH2 promotes tumor immunosuppression. J. Clin. Invest 126, 599–604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruhland MK, Roberts EW, Cai E, Mujal AM, Marchuk K, Beppler C, Nam D, Serwas NK, Binnewies M, Krummel MF, Visualizing synaptic transfer of tumor antigens among dendritic cells. Cancer Cell 37, 786–799.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, Kallies A, Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol 21, 1256–1266 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hacohen N, Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC, Checkpoint blockade immunotherapy induces dynamic changes in PD-1−CD8+ tumor-infiltrating T cells. Immunity 50, 181–194.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, Cardenas M, Wilkinson S, Lake R, Sowalsky AG, Valanparambil RM, Hudson WH, McGuire D, Melnick K, Khan AI, Kim K, Chang YM, Kim A, Filson CP, Alemozaffar M, Osunkoya AO, Mullane P, Ellis C, Akondy R, Im SJ, Kamphorst AO, Reyes A, Liu Y, Kissick H, An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO, Murine in vivo CD8+ T cell killing assay. Bio Protoc 4, e1172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gueguen P, Metoikidou C, Dupic T, Lawand M, Goudot C, Baulande S, Lameiras S, Lantz O, Girard N, Seguin-Givelet A, Lefevre M, Mora T, Walczak AM, Waterfall JJ, Amigorena S, Contribution of resident and circulating precursors to tumor-infiltrating CD8+ T cell populations in lung cancer. Sci. Immunol 6, eabd5778 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, Gao R, Zhang L, Dong M, Hu X, Ren X, Kirchhoff D, Roider HG, Yan T, Zhang Z, Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallee VP, Xie Y, Masilionis I, Carr AJ, Kottapalli S, Allaj V, Mattar M, Rekhtman N, Xavier JB, Mazutis L, Poirier JT, Rudin CM, Pe’er D, Massague J, Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, Choi S, Richards WG, De Rienzo A, Tenen DG, Bueno R, Levantini E, Pittet MJ, Klein AM, Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, Wittrup KD, Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med 11, eaaw2614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, Kauke MJ, Moynihan KD, Angelini A, Williams RT, Stephan MT, Kim JS, Yaffe MB, Irvine DJ, Weiner LM, Dranoff G, Wittrup KD, Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 27, 489–501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, Chee SJ, Eschweiler S, King EV, Awad AS, Hanley CJ, McCann KJ, Bhattacharyya S, Woo E, Alzetani A, Seumois G, Thomas GJ, Ganesan AP, Friedmann PS, Sanchez-Elsner T, Ay F, Ottensmeier CH, Vijayanand P, Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med 216, 2128–2149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, Halse H, Grynszpan L, Signolle N, Dayris T, Leclerc M, Droin N, de Montpreville V, Mercier O, Validire P, Scoazec JY, Massard C, Chouaib S, Planchard D, Adam J, Besse B, Mami-Chouaib F, CD103(+)CD8(+) TRM cells accumulate in tumors of anti–PD-1-responder lung cancer patients and are tumor-reactive lymphocytes enriched with Tc17. Cell Rep Med 1, 100127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, Woo E, Friedmann PS, King EV, Thomas GJ, Sanchez-Elsner T, Vijayanand P, Ottensmeier CH, Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol 18, 940–950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, Savic Prince S, Wiese M, Lardinois D, Ho PC, Klein C, Karanikas V, Mertz KD, Schumacher TN, Zippelius A, A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med 24, 994–1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T, Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD, Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335, 723–727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin HM, Kim G, Kim S, Sim JH, Choi J, Kim M, Kwon M, Ye SK, Lee DS, Cho SW, Kim ST, Lee J, Kim HR, Chromatin accessibility of circulating CD8(+) T cells predicts treatment response to PD-1 blockade in patients with gastric cancer. Nat. Commun 12, 975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sekine T, Perez-Potti A, Nguyen S, Gorin JB, Wu VH, Gostick E, Llewellyn-Lacey S, Hammer Q, Falck-Jones S, Vangeti S, Yu M, Smed-Sorensen A, Gaballa A, Uhlin M, Sandberg JK, Brander C, Nowak P, Goepfert PA, Price DA, Betts MR, Buggert M, TOX is expressed by exhausted and polyfunctional human effector memory CD8+ T cells. Sci. Immunol 5, eaba7918 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mule MP, Martins AJ, Finnigan JP, Zhang XM, Hamerman JA, Bhardwaj N, Tsang JS, Hickman HD, Ginhoux F, Ishizuka AS, Seder RA, Intravenous nanoparticle vaccination generates stem-like TCF1(+) neoantigen-specific CD8(+) T cells. Nat. Immunol 22, 41–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li A, Herbst RH, Canner D, Schenkel JM, Smith OC, Kim JY, Hillman M, Bhutkar A, Cuoco MS, Rappazzo CG, Rogers P, Dang C, Jerby-Arnon L, Rozenblatt-Rosen O, Cong L, Birnbaum M, Regev A, Jacks T, IL-33 signaling alters regulatory T Cell diversity in support of tumor development. Cell Rep 29, 2998–3008.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L, Chen ST, Sastre-Perona A, Park MD, Tabachnikova A, Dhainaut M, Hamon P, Maier B, Sawai CM, Agullo-Pascual E, Schober M, Brown BD, Reizis B, Marron T, Kenigsberg E, Moussion C, Benaroch P, Aguirre-Ghiso JA, Merad M, Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O’Garra A, Stockinger B, CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int. Immunol 17, 279–288 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, Rudensky AY, An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol 17, 1322–1333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leonard JD, Gilmore DC, Dileepan T, Nawrocka WI, Chao JL, Schoenbach MH, Jenkins MK, Adams EJ, Savage PA, Identification of natural regulatory T cell epitopes reveals convergence on a dominant autoantigen. Immunity 47, 107–117.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND, Savage PA, Aire-dependent thymic development of tumor-associated regulatory T cells. Science 339, 1219–1224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trinchieri G, Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol 3, 133–146 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, Freeman GJ, Warren SE, Ong S, Browning E, Twitty CG, Pierce RH, Le MH, Algazi AP, Daud AI, Pai SI, Zippelius A, Weissleder R, Pittet MJ, Successful Anti–PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC, Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glassman CR, Mathiharan YK, Jude KM, Su L, Panova O, Lupardus PJ, Spangler JB, Ely LK, Thomas C, Skiniotis G, Garcia KC, Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 184, 983–999.e24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, Robbins R, Crowley DM, Bronson RT, Jacks T, Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity 43, 579–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA, Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tu AA, Gierahn TM, Monian B, Morgan DM, Mehta NK, Ruiter B, Shreffler WG, Shalek AK, Love JC, TCR sequencing paired with massively parallel 3’ RNA-seq reveals clonotypic T cell signatures. Nat. Immunol 20, 1692–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, Kleinstein SH, pRESTO: A toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH, Change-O: A toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck III WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haghverdi L, Buttner M, Wolf FA, Buettner F, Theis FJ, Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Wolf FA, Angerer P, Theis FJ, SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials are available upon request from the corresponding author. Sequencing data can be found in the GEO database under accession number GSE184388. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.