

Abstract
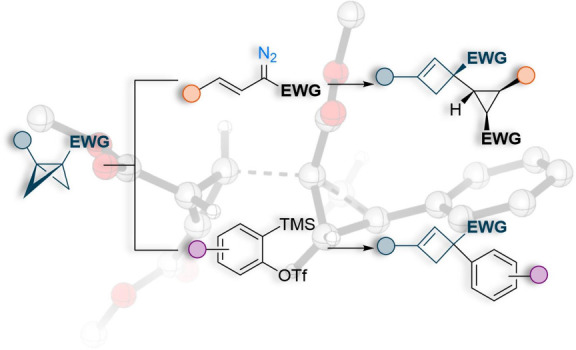
Bicyclo[1.1.0]butanes (BCBs), strained carbocycles comprising two fused cyclopropane rings, have become well-established building blocks in organic synthesis, medicinal chemistry, and chemical biology due to their diverse reactivity profile with radicals, nucleophiles, cations, and carbenes. The constraints of the bicyclic ring system confer high p-character on the interbridgehead C–C bond, leading to this broad reaction profile; however, the use of BCBs in pericyclic processes has to date been largely overlooked in favor of such stepwise, non-concerted additions. Here, we describe the use of BCBs as substrates for ene-like reactions with strained alkenes and alkynes, which give rise to cyclobutenes decorated with highly substituted cyclopropanes and arenes. The former products are obtained from highly stereoselective reactions with cyclopropenes, generated in situ from vinyl diazoacetates under blue light irradiation (440 nm). Cyclobutenes featuring a quaternary aryl-bearing carbon atom are prepared from equivalent reactions with arynes, which proceed in high yields under mild conditions. Mechanistic studies highlight the importance of electronic effects in this chemistry, while computational investigations support a concerted pathway and rationalize the excellent stereoselectivity of reactions with cyclopropenes.
Introduction
Bicyclo[1.1.0]butanes (BCBs, 1, Figure 1a) have emerged as versatile building blocks in organic synthesis due to the broad reactivity profile of their interbridgehead C1–C3 bond.1 With almost entirely p-character,2 ring-opening chemistry of this high-energy bond has been achieved using nucleophiles3 (including applications as bioconjugation agents),3b,4 radicals,5 electrophiles,6 and transition metals catalysts.7 In addition to such transformations, the ring expansion of BCBs to bicyclo[n.1.1]alkanes by formal one-,8 two-,9 or three-atom10 stepwise insertion processes has recently become a particularly valuable process due to the importance of these scaffolds in drug discovery.11 Finally, metalation of the acidic C–H bonds of BCBs provides further opportunities for scaffold diversification.3f,12
Figure 1.
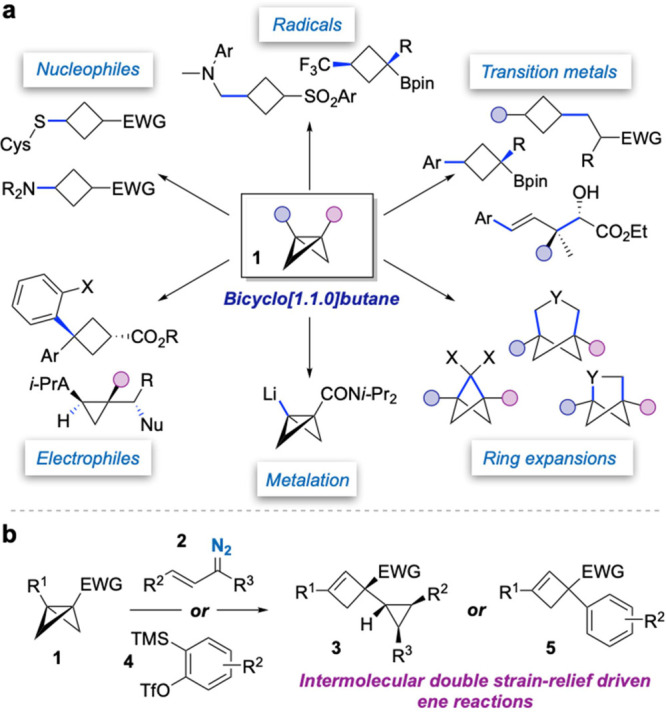
(a) Establishment of the reactivity of BCBs. (b) This work: stereoselective ene reactions of BCBs with cyclopropenes and arynes afford cyclopropyl- and aryl-substituted cyclobutenes.
In contrast to this rich chemistry, the exploration of BCBs as components in pericyclic reactions is limited, which may be due to the ease with which the aforementioned chemistries can occur. Reports of such processes have to date been limited to [3 + 2] cycloadditions and aza-ene reactions of diimides4b,13 and other activated alkenes,14 intramolecular ene-type reactions of N-allylated BCBs to give spirocyclic cyclobutanes,15 and isolated examples of reactions between electron-rich BCBs and benzyne.16 In some cases, there is debate as to whether these reactions are truly concerted or proceed via stepwise diradical, or ionic, pathways. We questioned whether other strained π-bonds might also engage with BCBs, thus affording unique small-ring product architectures from these readily accessible building blocks. Here, we describe the strain-relief-driven Alder-ene reaction of polyfunctionalized BCBs with cyclopropenes, generated in situ from visible light-promoted decomposition of vinyl diazo compounds (2, Figure 1b),17 to give cyclopropyl-substituted cyclobutenes (3); and also, the development of highly efficient ene reactions of BCBs with a selection of arynes18 generated under mild reaction conditions from arylsilane triflates194 to give arylated cyclobutenes 5. These reactions display a broad scope and high stereoselectivity; mechanistic studies support the involvement of an asynchronous concerted reaction pathway and highlight the importance of electronic effects in these transformations.
Results and Discussion
Our studies began with photochemical carbene generation from the vinyl diazo compound 2a, which is known to undergo spontaneous 2π-electrocyclization to the corresponding cyclopropene 6a upon carbene formation (Table 1).20,21 UV–vis spectra first confirmed that 2a is suitable for selective excitation in the presence of BCB 1a, which does not absorb at 440 nm, and that cyclopropene 6a was formed under these conditions.22 Pleasingly, irradiation of an equimolar quantity of 1a and 2a afforded the ene product 3a in 67% yield as a single diastereomer (entry 1).23 Incomplete conversion was observed, which we ascribed to decomposition of 2a over the 4 h reaction time. Increasing the equivalents of 2a, and employing a slow addition protocol (2 h of addition time, 0.28 M solution of 2a), improved the yield of 3a to 88% (entry 2). Decreasing the reaction temperature resulted in poor conversion (along with significant recovery of 1a), while no reaction was observed in the absence of light (entries 3, 4). Other solvents proved inferior to acetonitrile (entries 5–9).
Table 1. Optimization of Ene Reactionsa.

| entry | substrate | conditions | time (h) | 2a/4a (equiv) | yield 3a/5a (%) |
|---|---|---|---|---|---|
| 1 | 2a | MeCN | 4 | 1 | 67 |
| 2 | 2a | MeCN | 4 | 2 | 88 |
| 3 | 2a | MeCNb | 8 | 2 | 21 |
| 4 | 2a | MeCNc | 4 | 2 | n.r. |
| 5 | 2a | toluene | 4 | 2 | 20 |
| 6 | 2a | Et2O | 4 | 2 | 38 |
| 7 | 2a | CH2Cl2 | 4 | 2 | 38 |
| 8 | 2a | THF | 3 | 2 | 31 |
| 9 | 2a | MeOH | 3 | 2 | 44 |
| 10 | 4a | KF, THFd | 12 | 1.2 | 85 |
| 11 | 4a | CsF, MeCN | 12 | 1.2 | 82 |
| 12 | 4a | KF, THFd | 12 | 1.5 | 96 |
| 13 | 4a | KF, THFd | 4 | 1.5 | 98 |
| 14e | 4a | KF, THFd | 4 | 1.5 | 98 |
Reactions were carried out using 0.1 mmol of 1a (for reaction with 2a) or 0.2 mmol scale of 1a (for reaction with 4a). Isolated yields are reported.
Reaction conducted at 0 °C.
Reaction carried out in the dark.
Reaction conducted with 1 equiv 18-C-6 per equiv. KF.
Performed on 1.0 mmol scale. n.r. = no reaction.
Encouraged by the success of the ene reaction using cyclopropene, we questioned whether other enophiles could engage with 1a under similarly mild conditions. To this end, we were pleased to find that benzyne, produced in situ from 2-(trimethylsilyl)aryl triflate 4a using KF and 18-crown-6, underwent a smooth ene reaction at room temperature, delivering phenyl-substituted cyclobutene 5a in 85% yield (entry 10). Changing the fluoride source to CsF in CH3CN (to retard the rate of benzyne generation) did not improve the yield, but increasing the amount of benzyne precursor furnished 5a in 96% yield (entries 11 and 12). Finally, reducing the reaction time to 4 h led to a slight improvement, with 5a isolated in near quantitative yield on 0.2 and 1.0 mmol scale (98%, entries 13 and 14).
With optimized reaction conditions in hand for the reaction with both enophiles, we next explored the reaction scope (Figure 2). A variety of BCBs featuring ester, amide, and ketone groups were synthesized according to our previous studies22 and were combined with a selection of vinyl diazo esters (2a–2e). Pleasingly, good to excellent yields of cyclopropyl-substituted cyclobutenes were obtained (3b–3v, 41–93%), most as single diastereo- and regioisomers. We first found that reaction of C3-aryl substituted BCB esters 1b–1e with diester or aryl/ester-substituted diazos 2a–2d gave high yields of the product cyclobutenes 3b–3l (61–83%). Use of a monosubstituted vinyl diazo ester 2e gave a reduced yield of cyclobutene 3m, the lower yield of which likely reflects the instability of the intermediate monosubstituted cyclopropene.22
Figure 2.

Substrate scope for the cyclopropene–BCB and aryne–BCB ene reactions. Thermal ellipsoids are shown at 50%. H atoms have been omitted for clarity. All reactions of vinyldiazo compounds were carried out on a 0.1 mmol scale and of aryne precursors on 0.2 mmol scale. Yields are isolated yields. a3z was obtained in a 1:0.2 ratio with the regioisomeric ene product. bReaction of 1-naphthalyne. cReaction of 2-naphthalyne.
The effect of α-substitution of the diazo compound was investigated using trifluoromethyl-substituted vinyldiazo ester 2h.24 Interestingly, regioisomers 3ab and 3ab′ were obtained in 51 and 35% yield, respectively, the structures of which were unequivocally assigned by X-ray crystallographic analysis. The formation of these isomers arises from ene reaction at either end of the intermediate cyclopropene, illustrating the importance of steric effects in controlling regioselectivity using other (α-unsubstituted) vinyldiazo compounds.
Variation of the BCB electron-withdrawing group and arene was well-tolerated: Aryl-substituted BCB amides afforded the corresponding cyclobutenes 3n–3u in good to excellent yields (59–93%), with nitro, chloro, and aryl silyl ether groups all accommodated. Generally, electron-rich arenes were observed to give higher yields of ene products (e.g., 3u, 94%). Bicyclic substituents were also successful, such as naphthyl (3v, 79%) and indole (3w, 84%) groups. A BCB ketone proved to be equally effective (3x, 73%).
Modification of the BCB bridgehead substituent led to interesting results: a monosubstituted BCB amide (R1, R2 = H) exclusively afforded the opposite regioisomer of ene product, with C–C bond formation now distal from the amide electron-withdrawing group (3y, 92%). In contrast to aryl-substituted BCBs, a bridgehead methyl-substituted BCB afforded a mixture of regioisomers in favor of C–C bond formation at the amide-bearing carbon (3z, 1:0.2 rr); the major product 3z was isolated in 52% yield. Methyl substitution on the BCB bridge afforded a regioisomeric mixture of cyclobutenes, from which the major component 3aa, featuring the more-substituted alkene, was isolated in 41% yield.
We next evaluated the scope of the reaction with arynes as enophiles. Under the optimized conditions, a wide variety of arylated BCB esters with substitution at the C4-position and C3-position of the arene gave excellent yields of the corresponding phenyl-substituted arylcyclobutenes 5b–j (93–99%) on reaction with benzyne. Moreover, substitution at the C2-position of the BCB arene did not affect the reactivity (5k, 95%). Increasing the steric bulk of the BCB ester substituent and reaction with a BCB morpholine amide also proceeded with high efficiency (5l–5o, 91–98%). Interestingly, as observed with the cyclopropene ene reaction, an inversion of regioselectivity was observed in the reaction of benzyne with monosubstituted BCBs to give ester 5p (87%) and Weinreb amide 5q (69%).
The ene reaction also proceeded smoothly using an array of symmetrical and unsymmetrical-substituted arynes to give the corresponding arylated phenylcyclobutenes. The formation of 5r–5v from symmetric arynes proceeded in excellent yields (89–99%), which were not compromised by ortho substituents (5w, 97%). Pleasingly, 3-methoxybenzyne delivered a single regioisomer 5x in 97% yield;12 in contrast, 4-methyl- and 4-chlorobenzyne afforded mixtures of regioisomers with respect to the aryne (5y and 5z), albeit in high yields. Interestingly, the symmetric and unsymmetric naphthalynes generated from the corresponding regioisomeric silyl triflate precursors produced the same naphthyl cyclobutene 5aa in 95 and 97% yield, respectively. Finally, phenanthryne furnished the desired cyclobutene 5ab in 97% yield. Arynes bearing electron-withdrawing groups such as NO2, and heterocyclic arynes such as pyridyne, did not afford the desired cyclobutene product under the optimized conditions.
To study the influence of electronic effects on the cyclopropene–BCB ene reaction, competition experiments were carried out between vinyl diazo 2a and pairs of BCB amide substrates differing in the nature of the aryl group at the C3 position (using 2.5 equiv of each BCB relative to 2a, Figure 3a). A plot of relative rate constants (log(kX/kH)) against the corresponding Hammett substituent constants revealed a surprising insensitivity to electron-donating groups (X = Me, OMe) but a dramatic retardation of reaction rate for electron-withdrawing substituents (Cl, CF3). We also subjected the bridge exo-deuterated BCB D-1p to reaction with 2a (Figure 3b), which afforded a 1:1 mixture of ene products D-3n and D-3n′ with the deuterium atom entirely retained on the cyclobutene ring. This confirms that the cyclopropene reacts exclusively with a C–H bond on the endo face of the BCB. We then explored the possibility that the aryne ene reaction could operate via stepwise radical or ionic pathways (Figure 3c). When the reaction was carried out in the presence of the radical scavengers TEMPO and BHT, 5a was formed without significant detriment to the yield, suggesting the reaction is not radical-mediated.25 Moreover, treatment of BCB 1a with aryne precursor 4a under the optimized conditions in the presence of 2.0 equiv of D2O resulted in no incorporation of deuterium at the ortho-position (Figure 3c). This supports a concerted mechanism, rather than stepwise nucleophilic attack of the BCB on the aryne intermediate followed by protonation.
Figure 3.
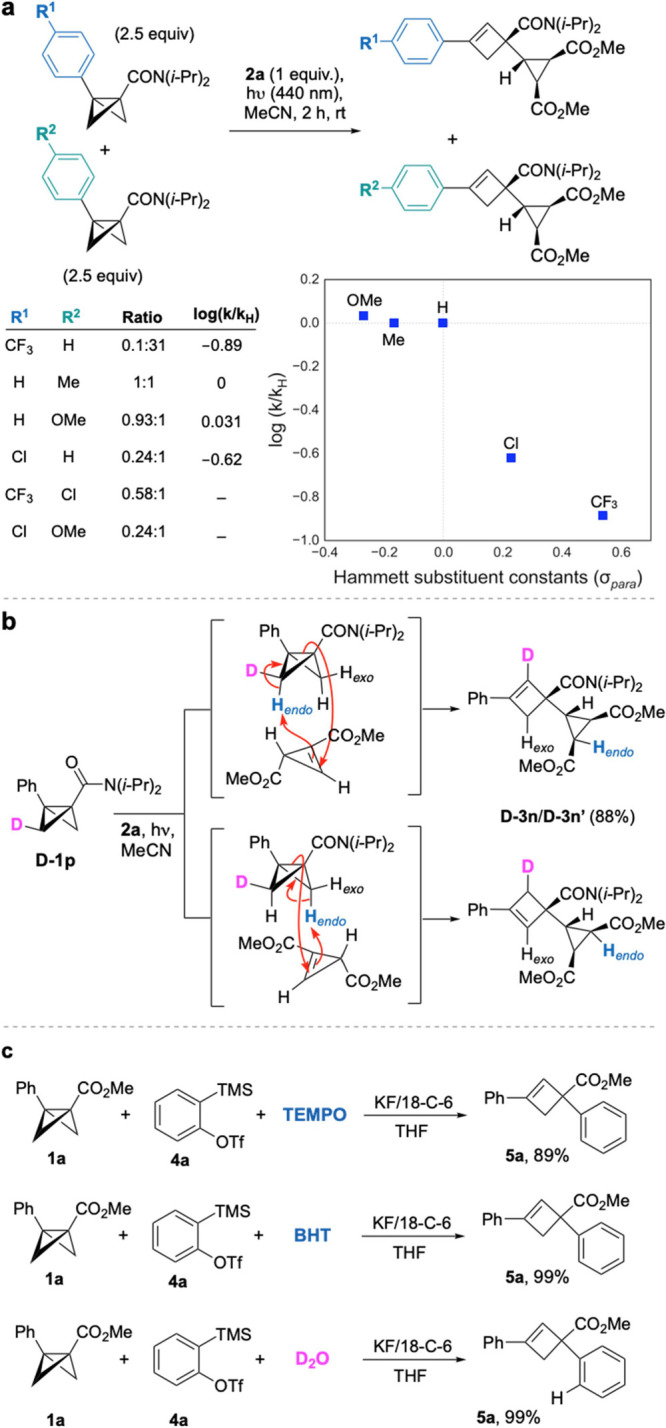
(a) Hammett study of the electronic effect of the bridgehead arene on the ene reaction; see the Supporting Information for details. (b) Deuterium labeling of the BCB bridge demonstrates exclusive endo C–H transfer to the cyclopropene. (c) Exploration of stepwise radical and ionic pathways for the aryne ene reaction.
To further explore the mechanism of the cyclopropene ene reaction26 and explain the stereoselectivity of the process, DFT calculations were carried out at the CPCM(acetonitrile)-DLPNO-CCSD(T)/def2-TZVPP//IEFPCM(acetonitrile)-B2PLYP(D3)/ def2-SVP level of theory at 298 K, using 1a and 6a as substrates (Figure 4a). Four possible arrangements of the cyclopropene and BCB were explored (A–D) in which the cyclopropene engages the BCB with the ester group on its sp3 carbon atom oriented exo (A, B) or endo (C, D). For each of these possibilities, the cyclopropene ring can then also orient exo (A, C) or endo (B, D) relative to the BCB. ‘Ester-exo’ conformations A and B would result in products 3a (observed) and 3ab, featuring a meso-cyclopropane group (i.e., ester groups syn) following transfer of the hydrogen atom from the BCB, and differ in the relative stereochemistry on the cyclobutene ring; ‘ester-endo’ conformations C and D lead to products 3ac and 3ad which also differ in the cyclobutene stereochemistry but now feature a chiral cyclopropane motif (ester groups anti).
Figure 4.

(a) Computation of conformations and ene transition states for the reaction of cyclopropene 3a and cyclopropene 6a. (b) Intrinsic reaction coordinate scan of the pathway via TSA reveals a delayed C–H transfer in the ene reaction, and a ‘one step–two-stage’ concerted pathway. (c) Electronic effects on transition state energies (TSA). E = CO2Me.
We first identified the transition state TSA which leads to the formation of the observed product 3a with an energy barrier of 22.5 kcal mol–1. This TS shows that the ene reaction proceeds via a concerted but highly asynchronous mechanism in which C–H bond breaking/formation is delayed compared to C–C bond formation between the cyclopropene and the C3 bridgehead carbon atom (see discussion below). Two other transition states were identified: TSC (25.5 kcal mol–1), and TSD (32.5 kcal mol–1) that would lead to the (unobserved) (R,R,S)- and (R,R,R)-diastereomers 3ac and 3ad, respectively. A transition state corresponding to the fourth (R,S,s,S)-diastereomer 3ab could not be located, with attempts to minimize conformation B instead converging on the presumably lower energy TSA (which is effectively a rotamer of TSB around the forming C–C bond).
The asynchronous nature of these transition states is consistent with previous calculations on cyclopropene ene reactions.26 To further explore this process, we performed an intrinsic reaction coordinate (IRC) scan (Figure 4b). This revealed an intriguing reaction profile that is characteristic of ‘one step–two stage’ asynchronous pericyclic processes:24TSA (point X) is characterized by a C1–C4 bond length of 1.96 Å with a Mayer bond order of 0.418, which contrasts with a breaking C2–H2 bond length of 1.11 Å (bond order 0.870) and forming C5–H2 bond length of 1.52 Å (bond order 0.023). C–H bond formation is thus largely delayed until the molecule distorts sufficiently to bring H2 into proximity to C5 (C5–H2 bond order 0.322, point Y). At this point, the IRC scan reveals significant C–H transfer, developing cyclobutene double bond character, and rapid, exergonic H transfer.
Finally, we compared the relative transition state energies of BCB 1a with those of equivalent BCB esters featuring electron-donating (OMe) and electron-withdrawing (CF3) substituents on the arene ring (Figure 4c). In comparison to TSA (unsubstituted Ph ring, 22.5 kcal mol–1), these displayed barriers of 21.2 kcal mol–1 for TSA(OMe) and 25.7 kcal mol–1 for TSA(CF3), which is in good agreement with the experimentally observed relative rates from the competition experiments (see Figure 3a).
The products of these ene reactions are rich in functionality and could be of use in other settings such as medicinal chemistry research, where highly substituted small ring systems are of importance. In terms of further chemistry, related cyclobutenes have recently been shown to be suitable substrates for (3 + 2) and (2 + 1) cycloaddition reactions to form rigid cyclobutane-fused ring systems.27 As an alternative to additional ring formation, we were able to demonstrate successful cyclobutene ring cleavage via ozonolysis of the double bond in 3e (Figure 5), which afforded the complex cyclopropane-containing product 7 in 74% yield. Featuring three different carbonyl environments, this ketoaldehyde would be expected to undergo a variety of other chemistries toward stereochemically rich carbon backbones.
Figure 5.

Product derivatization via the oxidative cleavage of cyclobutene 3e.
In conclusion, we have demonstrated highly regioselective and diastereoselective ene reactions of BCBs with cyclopropenes and arynes to afford cyclopropyl- and aryl-substituted cyclobutenes. The reactions proceed under mild conditions in good to near-quantitative yields. Experimental and DFT studies support an asynchronous concerted ‘one step–two-stage’ pathway, with exclusive reaction on the endo-face of the BCB. The cyclobutene products, which feature a quaternary carbon center to which the cyclopropane or arene is attached, are of potential use as small molecule building blocks in medicinal chemistry. Together, these methods further enhance the array of reactivities displayed by BCBs as valuable strain-relief building blocks in organic synthesis.
Acknowledgments
A.D.G. and E.A.A. thank the EPSRC (EP/S013172/1) for funding. A.T.B. thanks the Science and Engineering Research Board (SERB), Government of India for financial support (CRG/2021/001803). S.B. thanks CSIR (for a Senior Research Fellowship), and A.G. thanks MHRD (for a Research Fellowship). R.E.M. thanks the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB, and Vertex (EP/L015838/1). The authors gratefully acknowledge the EPSRC for a Strategic Equipment Grant (EP/V028995/1). The authors thank Nils Frank for helpful discussions concerning theoretical calculations.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c13080.
Author Contributions
§ A.D. and S.B. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Golfmann M.; Walker J. C. L. Bicyclobutanes as unusual building blocks for complexity generation in organic synthesis. Commun. Chem. 2023, 6, 9. 10.1038/s42004-022-00811-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kelly C. B.; Milligan J. A.; Tilley L. J.; Sodano T. M. Bicyclobutanes: from curiosities to versatile reagents and covalent warheads. Chem. Sci. 2022, 13, 11721–11737. 10.1039/D2SC03948F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fawcett A. Recent advances in the chemistry of bicyclo- and 1-azabicyclo[1.1.0]butanes. Pure Appl. Chem. 2020, 92, 751–765. 10.1515/pac-2019-1007. [DOI] [Google Scholar]
- a Schulman J. M.; Newton M. D. Contributions to the nuclear spin-spin coupling constants of directly bonded carbons. J. Am. Chem. Soc. 1974, 96, 6295–6297. 10.1021/ja00827a009. [DOI] [Google Scholar]; b Whitman D. R.; Chiang J. F. Electronic structures of bicyclo[1.1.0]butane and bicyclo[1.1.1]pentane. J. Am. Chem. Soc. 1972, 94, 1126–1129. 10.1021/ja00759a016. [DOI] [Google Scholar]; c Pomerantz M.; Abrahamson E. W. The Electronic Structure and Reactivity of Small Ring Compounds. I. Bicyclobutane. J. Am. Chem. Soc. 1966, 88, 3970–3972. 10.1021/ja00969a015. [DOI] [Google Scholar]
- a Guo L.; Noble A.; Aggarwal V. K. α-Selective Ring-Opening Reactions of Bicyclo[1.1.0]butyl Boronic Ester with Nucleophiles. Angew. Chem., Int. Ed. 2021, 60, 212–216. 10.1002/anie.202011739. [DOI] [PubMed] [Google Scholar]; b Lopchuk J. M.; Fjelbye K.; Kawamata Y.; Malins L. R.; Pan C.-M.; Gianatassio R.; Wang J.; Prieto L.; Bradow J.; Brandt T. A.; Collins M. R.; Elleraas J.; Ewanicki J.; Farrell W.; Fadeyi O. O.; Gallego G. M.; Mousseau J. J.; Oliver R.; Sach N. W.; Smith J. K.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-Release Heteroatom Functionalization: Development, Scope, and Stereospecificity. J. Am. Chem. Soc. 2017, 139, 3209–3226. 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gianatassio R.; Lopchuk J. M.; Wang J.; Pan C.; Malins L. R.; Prieto L.; Brandt T. A.; Collins M. R.; Gallego G. M.; Sach N. W.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-release amination. Science 2016, 351, 241. 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Panish R. A.; Chintala S. R.; Fox J. M. A Mixed-Ligand Chiral Rhodium(II) Catalyst Enables the Enantioselective Total Synthesis of Piperarborenine B. Angew. Chem., Int. Ed. 2016, 55, 4983–4987. 10.1002/anie.201600766. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Panish R.; Chintala S. R.; Boruta D. T.; Fang Y.; Taylor M. T.; Fox J. M. Enantioselective Synthesis of Cyclobutanes via Sequential Rh-catalyzed Bicyclobutanation/Cu-catalyzed Homoconjugate Addition. J. Am. Chem. Soc. 2013, 135, 9283–9286. 10.1021/ja403811t. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Gaoni Y. New bridgehead-substituted 1-(arylsulfonyl)bicyclo [1.1.0]butanes and some novel addition reactions of the bicyclic system. Tetrahedron 1989, 45, 2819–2840. 10.1016/S0040-4020(01)80112-5. [DOI] [Google Scholar]; g Gaoni Y.; Tomazic A. Bridgehead reactivity, nucleophilic and radical additions, and lithium aluminum hydride reduction of 1-(arylsulfonyl)bicyclobutanes: general access to substituted, functionalized cyclobutanes. Syntheses of (+)-citrilol acetate, (+)-junionone, and the tricyclo[3.3.0.01,4]octane and tricyclo[4.3.0.01,7]nonane ring systems. J. Org. Chem. 1985, 50, 2948–2957. 10.1021/jo00216a028. [DOI] [Google Scholar]
- a Kaur A.; Lin W.; Dovhalyuk V.; Driutti L.; Di Martino M. L.; Vujasinovic M.; Löhr J. M.; Sellin M. E.; Globisch D. Chemoselective bicyclobutane-based mass spectrometric detection of biological thiols uncovers human and bacterial metabolites. Chem. Sci. 2023, 14, 5291–5301. 10.1039/D3SC00224A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schwartz B. D.; Smyth A. P.; Nashar P. E.; Gardiner M. G.; Malins L. R. Investigating Bicyclobutane–Triazolinedione Cycloadditions as a Tool for Peptide Modification. Org. Lett. 2022, 24, 1268–1273. 10.1021/acs.orglett.1c04071. [DOI] [PubMed] [Google Scholar]; c Tokunaga K.; Sato M.; Kuwata K.; Miura C.; Fuchida H.; Matsunaga N.; Koyanagi S.; Ohdo S.; Shindo N.; Ojida A. Bicyclobutane carboxylic amide as a cysteine-directed strained electrophile for selective targeting of proteins. J. Am. Chem. Soc. 2020, 142, 18522–18531. 10.1021/jacs.0c07490. [DOI] [PubMed] [Google Scholar]; d Zhang P.; Zhuang R.; Wang X.; Liu H.; Li J.; Su X.; Chen X.; Zhang X. Highly Efficient and Stable Strain-Release Radioiodination for Thiol Chemoselective Bioconjugation. Bioconjugate Chem. 2018, 29, 467–472. 10.1021/acs.bioconjchem.7b00790. [DOI] [PubMed] [Google Scholar]; e Lopchuk J. M.; Fjelbye K.; Kawamata Y.; Malins L. R.; Pan C. M.; Gianatassio R.; Wang J.; Prieto L.; Bradow J.; Brandt T. A.; Collins M. R.; Elleraas J.; Ewanicki J.; Farrell W.; Fadeyi O. O.; Gallego G. M.; Mousseau J. J.; Oliver R.; Sach N. W.; Smith J. K.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-Release Heteroatom Functionalization: Development, Scope, and Stereospecificity. J. Am. Chem. Soc. 2017, 139, 3209–3226. 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pratt C. J.; Aycock R. A.; King M. D.; Jui N. T. Radical α-C–H Cyclobutylation of Aniline Derivatives. Synlett 2020, 31, 51–54. 10.1055/s-0039-1690197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Silvi M.; Aggarwal V. K. Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters. J. Am. Chem. Soc. 2019, 141, 9511–9515. 10.1021/jacs.9b03653. [DOI] [PubMed] [Google Scholar]
- a Guin A.; Bhattacharjee S.; Harariya M. S.; Biju A. T. Lewis acid-catalyzed diastereoselective carbofunctionalization of bicyclobutanes employing naphthols. Chem. Sci. 2023, 14, 6585–6591. 10.1039/D3SC01373A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kerner M. J.; Wipf P. Semipinacol-Type Rearrangements of [3-(Arylsulfonyl)bicyclo[1.1.0]butan-1-yl]alkanols. Org. Lett. 2021, 23, 3615–3619. 10.1021/acs.orglett.1c01004. [DOI] [PubMed] [Google Scholar]; c Bennett S. H.; Fawcett A.; Denton E. H.; Biberger T.; Fasano V.; Winter N.; Aggarwal V. K. Difunctionalization of C–C σ-Bonds Enabled by the Reaction of Bicyclo[1.1.0]butyl Boronate Complexes with Electrophiles: Reaction Development, Scope, and Stereochemical Origins. J. Am. Chem. Soc. 2020, 142, 16766–16775. 10.1021/jacs.0c07357. [DOI] [PubMed] [Google Scholar]
- a Wölfl B.; Winter N.; Li J.; Noble A.; Aggarwal V. K. Strain-Release Driven Epoxidation and Aziridination of Bicyclo[1.1.0]butanes via Palladium Catalyzed σ-Bond Nucleopalladation. Angew. Chem., Int. Ed. 2023, 62, e202217064 10.1002/anie.202217064. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang Z.; Gevorgyan V. Palladium Hydride-Enabled Hydroalkenylation of Strained Molecules. J. Am. Chem. Soc. 2022, 144, 20875–20883. 10.1021/jacs.2c09045. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pinkert T.; Das M.; Schrader M. L.; Glorius F. Use of Strain-Release for the Diastereoselective Construction of Quaternary Carbon Centers. J. Am. Chem. Soc. 2021, 143, 7648–7654. 10.1021/jacs.1c03492. [DOI] [PubMed] [Google Scholar]; d Ociepa M.; Wierzba A. J.; Turkowska J.; Gryko D. Polarity-Reversal Strategy for the Functionalization of Electrophilic Strained Molecules via Light-Driven Cobalt Catalysis. J. Am. Chem. Soc. 2020, 142, 5355–5361. 10.1021/jacs.0c00245. [DOI] [PubMed] [Google Scholar]; e Fawcett A.; Biberger T.; Aggarwal V. K. Carbopalladation of C–C σ-bonds enabled by strained boronate complexes. Nat. Chem. 2019, 11, 117–122. 10.1038/s41557-018-0181-x. [DOI] [PubMed] [Google Scholar]; f Walczak M. A. A.; Krainz T.; Wipf P. Ring-Strain-Enabled Reaction Discovery: New Heterocycles from Bicyclo[1.1.0]butanes. Acc. Chem. Res. 2015, 48, 1149–1158. 10.1021/ar500437h. [DOI] [PubMed] [Google Scholar]; g Walczak M. A. A.; Wipf P. Rhodium(I)-Catalyzed Cycloisomerizations of Bicyclobutanes. J. Am. Chem. Soc. 2008, 130, 6924–6925. 10.1021/ja802906k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bychek R.; Mykhailiuk P. K. A Practical and Scalable Approach to Fluoro-Substituted Bicyclo[1.1.1]pentanes. Angew. Chem., Int. Ed. 2022, 61, e202205103 10.1002/anie.202205103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ma X.; Pinto W.; Pham L. N.; Sloman D. L.; Han Y. Synthetic Studies of 2,2-Difluorobicyclo[1.1.1]pentanes (BCP-F2): The Scope and Limitation of Useful Building Blocks for Medicinal Chemists. Eur. J. Org. Chem. 2020, 2020, 4581–4605. 10.1002/ejoc.202000679. [DOI] [Google Scholar]; c Ma X.; Sloman D. L.; Han Y.; Bennett D. J. A Selective Synthesis of 2,2-Difluorobicyclo[1.1.1]pentane Analogues: “BCP-F2. Org. Lett. 2019, 21, 7199–7203. 10.1021/acs.orglett.9b02026. [DOI] [PubMed] [Google Scholar]; d Bychek R. M.; Hutskalova V.; Bas Y. P.; Zaporozhets O. A.; Zozulya S.; Levterov V. V.; Mykhailiuk P. K. Difluoro-Substituted Bicyclo[1.1.1]pentanes for Medicinal Chemistry: Design, Synthesis, and Characterization. J. Org. Chem. 2019, 84, 15106–15117. 10.1021/acs.joc.9b01947. [DOI] [PubMed] [Google Scholar]; e Applequist D. E.; Renken T. L.; Wheeler J. W. Polar substituent effects in 1,3-disubstituted bicyclo[1.1.1]pentanes. J. Org. Chem. 1982, 47, 4985–4995. 10.1021/jo00146a031. [DOI] [Google Scholar]
- a Agasti S.; Beltran F.; Pye E.; Kaltsoyannis N.; Crisenza G. E. M.; Procter D. J. A catalytic alkene insertion approach to bicyclo[2.1.1]hexane bioisosteres. Nat. Chem. 2023, 15, 535–541. 10.1038/s41557-023-01135-y. [DOI] [PubMed] [Google Scholar]; b Kleinmans R.; Dutta S.; Ozols K.; Shao H.; Schäfer F.; Thielemann R. E.; Chan H. T.; Daniliuc C. G.; Houk K. N.; Glorius F. ortho-Selective Dearomative [2π + 2σ] Photocycloadditions of Bicyclic Aza-Arenes. J. Am. Chem. Soc. 2023, 145, 12324–12332. 10.1021/jacs.3c02961. [DOI] [PubMed] [Google Scholar]; c Kleinmans R.; Pinkert T.; Dutta S.; Paulisch T. O.; Keum H.; Daniliuc C. G.; Glorius F. Intermolecular [2π+2σ]-photocycloaddition enabled by triplet energy transfer. Nature 2022, 605, 477–482. 10.1038/s41586-022-04636-x. [DOI] [PubMed] [Google Scholar]; d Guo R.; Chang Y.-C.; Herter L.; Salome C.; Braley S. E.; Fessard T. C.; Brown M. K. Strain-release [2π + 2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes Initiated by energy transfer. J. Am. Chem. Soc. 2022, 144, 7988–7994. 10.1021/jacs.2c02976. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Liang Y.; Kleinmans R.; Daniliuc C. G.; Glorius F. Synthesis of Polysubstituted 2-Oxabicyclo[2.1.1]hexanes via Visible-Light-Induced Energy Transfer. J. Am. Chem. Soc. 2022, 144, 20207–20213. 10.1021/jacs.2c09248. [DOI] [PubMed] [Google Scholar]; f Dhake K.; Woelk K. J.; Becica J.; Un A.; Jenny S. E.; Leitch D. C. Beyond Bioisosteres: Divergent Synthesis of Azabicyclohexanes and Cyclobutenyl Amines from Bicyclobutanes. Angew. Chem., Int. Ed. 2022, 61, e202204719 10.1002/anie.202204719. [DOI] [PubMed] [Google Scholar]
- a Nguyen T. V. T.; Bossonnet A.; Waser J. Photocatalyzed [3σ + 2σ] and [3σ + 2π] cycloadditions for the synthesis of bicyclo[3.1.1]heptanes and cyclopentenes. ChemRxiv 2023, 10.26434/chemrxiv-2023-s8j30. [DOI] [PubMed] [Google Scholar]; b Zheng Y.; Huang W.; Dhungana R. K.; Granados A.; Keess S.; Makvandi M.; Molander G. A. Photochemical Intermolecular [3σ + 2σ]-Cycloaddition for the Construction of Aminobicyclo[3.1.1]heptanes. J. Am. Chem. Soc. 2022, 144, 23685–23690. 10.1021/jacs.2c11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shire B. R.; Anderson E. A. Conquering the Synthesis and Functionalization of Bicyclo[1.1.1]pentanes. JACS Au 2023, 3, 1539–1553. 10.1021/jacsau.3c00014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mykhailiuk P. K. Saturated bioisosteres of benzene: Where to go next?. Org. Biomol. Chem. 2019, 17, 2839–2849. 10.1039/C8OB02812E. [DOI] [PubMed] [Google Scholar]; c Measom N. D.; Down K. D.; Hirst D. J.; Jamieson C.; Manas E. S.; Patel V. K.; Somers D. O. Investigation of a bicyclo[1.1.1]pentane as a phenyl replacement within an LpPLA2 inhibitor. ACS Med. Chem. Lett. 2017, 8, 43–48. 10.1021/acsmedchemlett.6b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McNamee R. E.; Haugland M. M.; Nugent J.; Chan R.; Christensen K. E.; Anderson E. A. Synthesis of 1,3-disubstituted bicyclo[1.1.0]butanes via directed bridgehead functionalization. Chem. Sci. 2021, 12, 7480–7485. 10.1039/D1SC01836A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McNamee R. E.; Thompson A. L.; Anderson E. A. Synthesis and Applications of Polysubstituted Bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 2021, 143, 21246–21251. 10.1021/jacs.1c11244. [DOI] [PubMed] [Google Scholar]; c Tyler J. L.; Aggarwal V. K. Synthesis and Applications of Bicyclo[1.1.0]butyl and Azabicyclo[1.1.0]butyl Organometallics. Chem. – Eur. J. 2023, 29, e202300008 10.1002/chem.202300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chang M. H.; Dougherty D. A. 2,3-Diazabicyclo[2.1.1]hex-2-ene. Synthesis and thermal decomposition. J. Org. Chem. 1981, 46, 4092–4093. 10.1021/jo00333a040. [DOI] [Google Scholar]; b Amey R. L.; Smart B. E. Bicyclo[1.1.0]butanes. Reactions with cyclic azo compounds. J. Org. Chem. 1981, 46, 4090–4092. 10.1021/jo00333a039. [DOI] [Google Scholar]
- Cairncross A.; Blanchard E. P. Jr. Bicyclo[1.1.0]butane Chemistry. II. Cycloaddition Reactions of 3-Methylbicyclo[1.1.0]butanecarbonitriles. The Formation of Bicyclo[2.1.1]hexanes. J. Am. Chem. Soc. 1966, 88, 496–504. 10.1021/ja00955a021. [DOI] [Google Scholar]
- a Ueda M.; Walczak M. A. A.; Wipf P. Formal Alder-ene reaction of a bicyclo[1.1.0]butane in the synthesis of the tricyclic quaternary ammonium core of daphniglaucins. Tetrahedron Lett. 2008, 49, 5986–5989. 10.1016/j.tetlet.2008.07.179. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wipf P.; Walczak M. A. A. Pericyclic Cascade Reactions of (Bicyclo[1.1.0]butylmethyl)amines. Angew. Chem., Int. Ed. 2006, 45, 4172–4175. 10.1002/anie.200600723. [DOI] [PubMed] [Google Scholar]
- a Pomerantz M.; Wilke R. N.; Gruber G. W.; Roy U. Electronic structure and reactivity of small ring compounds. V. Reaction of some bicyclobutanes with various dienophiles. J. Am. Chem. Soc. 1972, 94, 2752–2758. 10.1021/ja00763a037. [DOI] [Google Scholar]; b Pomerantz M.; Gruber G. W.; Wilke R. N. Electronic structure and reactivity of small-ring compounds. III. Mechanistic studies of the bicyclobutane-benzyne reaction. J. Am. Chem. Soc. 1968, 90, 5040–5041. 10.1021/ja01020a057. [DOI] [Google Scholar]
- a Jurberg I. D.; Davies H. M. L. Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci. 2018, 9, 5112–5118. 10.1039/C8SC01165F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang Z.; Stivanin M. L.; Jurberg I. D.; Koenigs R. M. Visible light-promoted reactions with diazo compounds: a mild and practical strategy towards free carbene intermediates. Chem. Soc. Rev. 2020, 49, 6833–6847. 10.1039/D0CS00224K. [DOI] [PubMed] [Google Scholar]
- a Jayanth T. T.; Jeganmohan M.; Cheng M.-J.; Chu S.-Y.; Cheng C.-H. Ene Reaction of Arynes with Alkynes. J. Am. Chem. Soc. 2006, 128, 2232–2233. 10.1021/ja058418q. [DOI] [PubMed] [Google Scholar]; b Xu H.; He J.; Shi J.; Tan L.; Qiu D.; Luo X.; Li Y. Domino Aryne Annulation via a Nucleophilic–Ene Process. J. Am. Chem. Soc. 2018, 140, 3555–3559. 10.1021/jacs.8b01005. [DOI] [PubMed] [Google Scholar]; c Bhojgude S. S.; Bhunia A.; Gonnade R. G.; Biju A. T. Efficient Synthesis of 9-Aryldihydrophenanthrenes by a Cascade Reaction Involving Arynes and Styrenes. Org. Lett. 2014, 16, 676–679. 10.1021/ol4033094. [DOI] [PubMed] [Google Scholar]; d Chen Z.; Liang J.; Yin J.; Yu G.-A.; Liu S. H. Alder-ene reaction of aryne with olefins. Tetrahedron Lett. 2013, 54, 5785–5787. 10.1016/j.tetlet.2013.08.049. [DOI] [Google Scholar]; e Yang Y.; Jones C. R. The Aryne Ene Reaction. Synthesis 2022, 54, 5042–5054. 10.1055/a-1827-2987. [DOI] [Google Scholar]
- a Himeshima Y.; Sonoda T.; Kobayashi H. Fluoride-induced 1,2-Elimination of O-trimethylsilylphenyl Triflate to Benzyne Under Mild Conditions. Chem. Lett. 1983, 12, 1211–1214. 10.1246/cl.1983.1211. [DOI] [Google Scholar]; b Peña D.; Cobas A.; Pérez D.; Guitián E. An Efficient Procedure for the Synthesis of ortho-Trialkylsilylaryl Triflates: Easy Access to Precursors of Functionalized Arynes. Synthesis 2002, 34, 1454–1458. 10.1055/s-2002-33110. [DOI] [Google Scholar]; c Shi J.; Li L.; Li Y. o-Silylaryl Triflates: A Journey of Kobayashi Aryne Precursors. Chem. Rev. 2021, 121, 3892–4044. 10.1021/acs.chemrev.0c01011. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Clark D. M.; Alligood D. B.; Eiband G. R. Mechanistic aspects of formal [3 + 4] cycloadditions between vinylcarbenoids and furans. Tetrahedron 1987, 43, 4265–4270. 10.1016/S0040-4020(01)90301-1. [DOI] [Google Scholar]
- Hommelsheim R.; Guo Y.; Yang Z.; Empel C.; Koenigs R. M. Blue-Light-Induced Carbene-Transfer Reactions of Diazoalkanes. Angew. Chem., Int. Ed. 2019, 58, 1203–1207. 10.1002/anie.201811991. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- The structure of the cyclopropane products was determined by NMR analysis of coupling constants and 2D NMR experiments, as well as by analogy with X-ray crystal structures for products 3i and 3n.
- Supurgibekov M. B.; Prakash G. K. S.; Nikolaev V. A. Two-Stage Synthesis of 3-(Perfluoroalkyl)-Substituted Vinyldiazocarbonyl Compounds and Their Nonfluorinated Counterparts: A Comparative Study. Synthesis 2013, 45, 1215–1226. 10.1055/s-0032-1318309. [DOI] [Google Scholar]
- Scherübl M.; Daniliuc C. G.; Studer A. Arynes as Radical Acceptors: TEMPO-Mediated Cascades Comprising Addition, Cyclization, and Trapping. Angew. Chem., Int. Ed. 2021, 60, 711–715. 10.1002/anie.202012654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q.; Thomas B. E.; Houk K. N.; Dowd P. Transition Structures of the Ene Reactions of Cyclopropene. J. Am. Chem. Soc. 1997, 119, 6902–6908. 10.1021/ja963248q. [DOI] [PubMed] [Google Scholar]
- Lin S.-L.; Chen Y.-H.; Liu H.-H.; Xiang S.-H.; Tan B. Enantioselective Synthesis of Chiral Cyclobutenes Enabled by Bro̷nsted Acid-Catalyzed Isomerization of BCBs. J. Am. Chem. Soc. 2023, 145, 21152–21158. 10.1021/jacs.3c06525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.