

Significance
The emergence of naturally drug-resistant opportunistic pathogens, such as Mycobacterium abscessus (Mab), presents a growing and deeply concerning public health threat due to few effective therapeutic options and a limited drug development pipeline. This research addresses this critical gap by introducing a unique series of N-ethylene aminomethyl spectinomycin (eAmSPC) analogs that show impressive activity against Mab. We have successfully unraveled the intricate mechanism by which the eAmSPC analogs overcome TetV-mediated efflux, thus reactivating the activity of spectinomycin against Mab. These mechanistic insights are enhanced by extensive developmental endeavors, showcasing the promising potential of this series as treatments against Mab infections.
Keywords: efflux, nontuberculous mycobacteria, antimicrobial resistance, antibiotic, protein synthesis inhibitor
Abstract
Mycobacterium abscessus (Mab), a nontuberculous mycobacterial (NTM) species, is an emerging pathogen with high intrinsic drug resistance. Current standard-of-care therapy results in poor outcomes, demonstrating the urgent need to develop effective antimycobacterial regimens. Through synthetic modification of spectinomycin (SPC), we have identified a distinct structural subclass of N-ethylene linked aminomethyl SPCs (eAmSPCs) that are up to 64-fold more potent against Mab over the parent SPC. Mechanism of action and crystallography studies demonstrate that the eAmSPCs display a mode of ribosomal inhibition consistent with SPC. However, they exert their increased antimicrobial activity through enhanced accumulation, largely by circumventing efflux mechanisms. The N-ethylene linkage within this series plays a critical role in avoiding TetV-mediated efflux, as lead eAmSPC 2593 displays a mere fourfold susceptibility improvement against Mab ΔtetV, in contrast to the 64-fold increase for SPC. Even a minor shortening of the linkage by a single carbon, akin to 1st generation AmSPC 1950, results in a substantial increase in MICs and a 16-fold rise in susceptibility against Mab ΔtetV. These shifts suggest that longer linkages might modify the kinetics of drug expulsion by TetV, ultimately shifting the equilibrium towards heightened intracellular concentrations and enhanced antimicrobial efficacy. Furthermore, lead eAmSPCs were also shown to synergize with various classes of anti-Mab antibiotics and retain activity against clinical isolates and other mycobacterial strains. Encouraging pharmacokinetic profiles coupled with robust efficacy in Mab murine infection models suggest that eAmSPCs hold the potential to be developed into treatments for Mab and other NTM infections.
The widespread use of broad-spectrum antibiotics in vulnerable populations, such as those with cystic fibrosis (CF) or chronic obstructive pulmonary disease (COPD), is driving the rise of naturally drug-resistant opportunistic pathogens such as nontuberculosis mycobacteria (NTM) (1, 2). The incidence of NTM pulmonary infection has surpassed that of new tuberculosis diagnoses in high-level industrialized countries, a statistic exacerbated by limited therapeutic options, prolonged treatment courses, and poor clinical outcomes (3). Among NTM species, Mycobacterium abscessus (Mab) is one of the most difficult to treat. Clinical practice guidelines recommend a drug regimen consisting of at least 3 antibiotics for upwards of 1 y to combat Mab infections (4). Despite aggressive therapy, cure rates remain poor, comparable to that of extensively drug-resistant tuberculosis (2, 5). Therefore, there is an urgent need for novel therapeutic strategies that improve the treatment options for Mab infections.
The development of successful anti-Mab therapies are complicated by the pathogen’s intrinsic resistance to many commonly prescribed antibiotics. Intrinsic drug resistance observed in Mab and other mycobacteria is linked to their unique cell wall structures, native efflux, and inducible resistance mechanisms (6). Because translational inhibitors (e.g., macrolides, aminoglycosides, and tetracyclines) serve as the cornerstone of Mab treatment, the WhiB7 “resistome” is among the most important determinants of antibiotic susceptibility. WhiB7 (Mab3508c) is the master transcriptional regulator associated with ribosomal stress and controls the expression of at least 127 response genes, including several involved in antimicrobial resistance, such as erm(41), eis1/2, N-acetyltransferases, and the multifacilitator pump (MFS) efflux pumps Tap and TetV (7–9). Developing effective therapeutic solutions that circumvent these inducible and innate drug resistance processes may lead to improved patient outcomes.
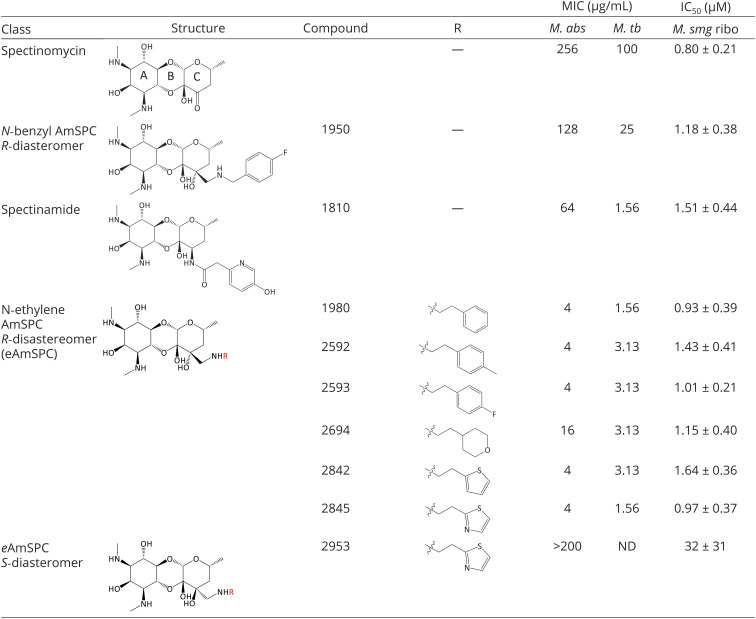
Our program has a long-standing interest in developing novel spectinomycin (SPC) analogs as antibacterial and antitubercular agents (10, 11). SPC is an aminocyclitol antibiotic that exhibits potent protein synthesis inhibition by binding to a unique ribosomal binding site, helix 34, within the head domain of the 16S rRNA of the bacterial 30S ribosomal subunit (12, 13). However, SPC has limited efficacy against mycobacterial species. Previous work from our group identified the spectinamides, a class of narrow-spectrum anti-TB drugs (see example spectinamide 1810 in Table 1) (14) with a strict requirement of a 2-N-heteroaryl acetamide side chain associated with avoidance of Tap (Rv1258c)-mediated efflux in Mycobacterium tuberculosis (10, 15, 16). Recent reports show that SPC resistance in Mab is similarly mediated by the expression of Mab2780c, a TetV-like efflux pump, and its avoidance may provide an avenue to exploit SPCs unique antimicrobial mechanism against this refractory pathogen (17).
Table 1.
Chemical structures and in vitro activity of SPCs
|
Structures of SPC and analogs with whole cell MIC activity and translation inhibitory activity against M. smegmatis ribosomes in cell-free translation assays. N-ethylene AmSPC (eAmSPC) series molecules are second-generation SPC analogues with enhanced antimicrobial properties over the parent compound, SPC. eAmSPCs improve upon the previously disclosed N-ethyl AmSPC analog, 1950. The eAmSPCs retain SPC ribosomal inhibition profiles while ameliorating whole cell antimicrobial activity. MICs, performed in triplicate for M. abs. and M. tb., were read on day 5 and day 7, respectively. Abbreviations: AmSPC, aminomethyl spectinomycins; M. abs, M. abscessus ATCC 19977; M. tb, M. tuberculosis H37Rv; M. smg ribo, purified M. smegmatis ATCC 19420 ribosomes; MIC, minimum inhibitory concentration; ND, not determined.
Herein, we report the identification and preclinical characterization of a subclass of N-ethylene linked aminomethyl spectinomycins (eAmSPCs), which are up to 64-fold more potent against Mab compared to the parent compound, SPC. These 2nd generation eAmSPCs are largely resilient toward the whiB7 resistance regulon and their increase in potency is attributed to avoidance of high-level TetV-mediated efflux, resulting in higher levels of accumulation. Importantly, the eAmSPCs synergize with several classes of anti-Mab antibiotics, retain activity against other NTM species and clinical Mab isolates, have favorable pharmacokinetic and safety profiles, and demonstrate efficacy in acute and chronic murine models of Mab infection. These results suggest that eAmSPCs have the potential to be developed into treatments for Mab and other NTM infections.
Results
Discovery and Structure–Activity Relationship of SPC Analogs that Target Mab.
Over the course of our studies on SPC, we have synthesized a library of >300 SPC analogs. These compounds were profiled for protein synthesis inhibition against Mycobacterium smegmatis ribosomes and antimicrobial activity against M. smegmatis, as this rapidly growing model organism is widely used to study antimycobacterial drug action (18, 19). The analogs responsive to these assays were then tested for their minimum inhibitory concentration (MIC) against Mab. This analysis led to the identification of the 2nd generation eAmSPCs subclass, which displayed promising activity against Mab not found in the 1st generation N-benzyl-substituted AmSPCs like 1950 (Table 1) (20).
The unsubstituted phenyl analog 1980 demonstrated a considerable increase in MIC potency compared to 1950, lead spectinamide 1810, and SPC (Table 1). Substitutions to the phenyl were explored to block potential sites of metabolism and to reduce the amphipathic character of analog 1980. The para-position (analogs 2592 and 2593, Table 1) was found to be optimal for maintaining MIC activity against Mab. The phenyl moiety was replaced with aliphatic and aromatic heterocycles to increase side chain polarity. This led to the identification of tetrahydropyran analog 2694, which showed a fourfold reduction in potency over 2593 and 1980, but maintained similar ribosomal inhibition activity (M. smegmatis ribosomal IC50 of 1.15 ± 0.40 µM; Table 1). Thiophene analog 2842 also showed promise with excellent anti-Mab activity, demonstrating a consistent structure–activity relationship. To further confirm that the anti-Mab activity was dependent on ribosome binding and not due to nonspecific interactions, such as membrane disruption, a pair of 3′ isomeric thiazoles, R-2845 and S-2953, were synthesized and compared. The R-2845 isomer was active in both microbiological and ribosome inhibition assays, whereas the S-2953 isomer abrogated activity in both assays (Table 1), confirming that the 3′ N-ethylene side-chain played an important stereospecific role in the activity of this new series.
eAmSPCs Display a Mode of Ribosomal Inhibition Consistent with SPC.
To validate eAmSPCs retain the mode of action of the parent compound SPC and determine whether they form any additional ribosomal contacts that may enhance antimicrobial activity, X-ray crystallography studies were performed, and spontaneous mutants were generated to evaluate interactions at the helix 34 in the SPC binding site. To date, all our structural models defining binding of SPC analogs to the ribosome result from computer modeling experiments (10–12). Therefore, to understand the functional relevance of the side-chain moieties of eAmSPCs in the context of functional ribosome complex, we have determined the crystal structure of eAmSPC 2694 bound to the Thermus thermophilus 70S ribosome containing mRNA, and deacylated A-, P- and E-site tRNAs at 2.75 Å resolution (SI Appendix, Table S2). While two structures of ribosome-bound SPC have been reported previously, they were obtained either in complex with a vacant ligand-free 70S ribosome from Escherichia coli (PDB entry 4V56 in ref. 13) or 30S ribosomal subunit from T. thermophilus (PDB entry 1FJG in ref. 21). Since SPC binds the ribosome close to the decoding center, there is a possibility that its binding might be affected by the presence of mRNA and tRNA ligands. Therefore, to check whether mRNA and/or tRNAs affect SPC binding and to accurately evaluate the effect of SPC derivatization on its placement in the ribosome, we also determined a reference structure of the parent SPC bound to the same T. thermophilus 70S ribosome complex containing mRNA and all three tRNAs. An unbiased difference Fourier map revealed positive electron density peaks resembling characteristic features of SPC (SI Appendix, Fig. S1 A and B) and 2694 chemical structures (SI Appendix, Fig. S1 C and D). Our structures reveal binding of SPC and 2694 in the canonical SPC site located in the head domain of the small ribosomal subunit behind the A and P sites (Fig. 1 A and B). We found that the presence of mRNA and tRNAs does not affect the overall binding pose of SPC on the ribosome (Fig. 1C, blue vs. green). Moreover, the position of the aminocyclitol part of the ribosome-bound 2694 structure is identical to that of a parent SPC (Fig. 1C, green vs. yellow). This is not surprising because the aminocyclitol moiety, which interacts with the 16S rRNA, is critical for drug binding, and is the same between SPC and its eAmSPC 2694 analog. Similar to SPC, 2694 forms multiple hydrogen bonds (H-bonds) with the nucleotides G1064, C1066, G1068, A1191, C1192, and G1193 within helix 34 of the 16S rRNA of the 30S ribosomal subunit (Fig. 1 D and E). Additionally, the extended tetrahydropyran moiety of the 2694 analog encroaches upon the loop of the small ribosomal protein S5, where it establishes unique hydrophobic stacking interactions with the side chain of residue Arg25 (Fig. 1D). Interestingly, superposition of the ribosome structures determined in the absence and presence of 2694 revealed a prominent steric clash between the extended tetrahydropyran moiety of 2694 and residues Gln20 and Arg25 in the loop of protein S5 (Fig. 1F). This sterical hindrance is resolved via repositioning of the side chains of several nearby residues in the S5 loop (Arg18, Gln20, Arg25, and Arg27) not causing any significant structural rearrangements of the loop (Fig. 1F). Although SPC does not have the extended arm of 2694, its shared aminocyclitol moiety clashes with the side chain of Gln20, causing side chain rearrangements similar to those observed for 2694 (Fig. 1F).
Fig. 1.

Structure of SPC and eAmSPC 2694 (2694) in complex with the 70S ribosome, mRNA, and tRNAs. (A and B) Overview of the SPC/2694 binding site (yellow) in the T. thermophilus 70S ribosome viewed as a cross-cut sections from two different perspectives. The 30S subunit is shown in light gray, the 50S subunit is dark gray, the mRNA is magenta and the A-, P-, and E-site tRNAs are colored green, dark blue, and orange, respectively. Small ribosomal protein S5 is highlighted in blue. The view in (A) is a transverse section of the 70S ribosome. The view in (B) is from the Top after removing the head of the 30S subunit and protuberances of the 50S subunit, as indicated by the Inset. (C) Superposition of the ribosome-bound SPC (green) and 2694 (yellow) in the presence of tRNAs with the previous structure of SPC bound to 30S ribosomal subunit from T. thermophilus (blue, PDB entry 1FJG in ref. 21). The structures were aligned based on helix 34 of the 16S rRNA. (D and E) Close-up views of the 2694 interactions with helix 34 of the decoding center on the 30S ribosomal subunit. The E. coli numbering of the nucleotides in the 16S rRNA is used. Potential H-bond interactions are indicated with dashed lines. Note that the extended tetrahydropyranyl moiety of 2694 establishes van der Waals interactions with the Arg25 residue of the ribosomal protein S5. (F) Superposition of the ribosomal protein S5 structures in the absence (teal) and presence of the ribosome-bound SPC or 2694 (blue). While the binding of SPC/2694 to the bacterial ribosome does not cause significant rearrangements of the protein S5 loop, the side chains of several residues in the loop (Arg18, Gln20, Arg25, Arg27) are moved (red dashed arrows) to avoid steric hindrance with the ribosome-bound drugs.
To confirm that the ribosome remains the primary target of the eAmSPC analogs, we generated 2593-resistant spontaneous Mab mutants, which arose at a frequency of 2 to 4 x 10−7. Whole-genome sequencing of 17 mutants identified single polymorphisms at four nucleotides (G1064, C1066, A1191, C1192) within helix 34 (Fig. 1 D and E and SI Appendix, Table S3). Importantly, any of these individual mutations can confer resistance to 2593 (MIC > 200 µg/mL; SI Appendix, Table S3). These data are consistent with prior structural studies showing these nucleotides to be critical for SPC and its analogs binding to the bacterial ribosome (13, 21). Although the structural data show that eAmSPC compounds form additional contacts with the loop of ribosomal protein S5 (Fig. 1 D and E), no mutations conferring reduced susceptibility were observed in this protein. This finding together with similar translation inhibitory properties of lead compounds shown in Table 1 suggest that additional interactions with the loop of ribosomal protein S5 are unlikely to contribute to but do not detract from the enhanced Mab activity of the eAmSPCs.
eAmSPCs with Lipophilic R-Groups Accumulate in Mab at Higher Concentrations than SPC.
To investigate whether differences in drug accumulation may contribute to increased eAmSPC activity, we used a modified liquid chromatography-mass spectrometry LC/MS method to quantify compound accumulation in Mab (Fig. 2A) (22, 23). When concentrated Mab cells were treated with 100 µM of compound for 30 min, we found that the most potent eAmSPC compounds in whole-cell MIC assays (2593, 2842, 1980, 2592) also displayed the highest whole-cell accumulation, significantly higher than that of parent SPC (P < 0.0001, Fig. 2B). Of these leads, 2592 accumulated highest at 13.18-fold higher than SPC (503.7 ± 25.4 nM/1010 CFU vs. 38.2 ± 9.0 nM/1010 CFU), followed by 1980 (12.0-fold; 459.0 ± 52.7 nM/1010 CFU), 2593 (8.4-fold; 322.4 ± 64.1 nM/1010 CFU), and 2842 (8.3-fold; 315.7 ± 18.9 nM/1010 CFU). The 1st generation AmSPC 1950 also displayed an increase in accumulation at 4.3-fold higher than that of SPC (166.1 ± 7.3 nM/1010 CFU). Analog 2694, which is less potent than the other eAmSPCs, had the lowest whole-cell accumulation (2.5-fold; 96.8 ± 5.9 nM/1010 CFU), which was not significantly different when compared to SPC. Analysis of physicochemical properties of substituents at the 3′ position on the C-ring of the SPC scaffold revealed a high correlation between R-group lipophilicity and whole-cell accumulation (correlation coefficient r = 0.872; P = 0.0048; SI Appendix, Fig. S2 A and B). These data suggest enhanced intracellular accumulation of the eAmSPCs does contribute to their improved potency and is driven by R-group lipophilicity. This observation aligns with an analysis conducted by Davis et al., describing lipophilicity characteristics, such as LogP and LogD, being among the most positively correlated properties for compound accumulation in M. smegmatis (22).
Fig. 2.

eAmSPCs accumulate intracellularly and induce similar transcriptomic response as SPC. (A) Workflow of whole-cell accumulation assay in Mab. (B) Intracellular accumulation of compounds against Mab upon 30-min exposure. Data shown are representative of three independent replicates (mean ± SD). (C) Heat map of whiB7 genes differentially expressed (Log2FC > |3|; FDR < 0.01) in at least one treatment group. (D–F) Volcano plots depicting differential gene expression analysis [Log2 Fold-Change vs. –Log10 (FDR)] of (D) 2593 (1 µg/mL) vs. dimethyl sulfoxide (DMSO), (E) 2593 (1 µg/mL) vs. SPC (1 µg/mL), and (F) 2593 (1 µg/mL) vs. SPC (10 µg/mL), where the circles on to the right side of the respective plots indicate an upregulation of genes for 2593. Red circles indicate whiB7 response genes identified in ref 7. Statistical significance for (B) using ANOVA determined groups were of equal variance by Brown–Forsythe test and Dunnet’s post test was then used compare results of experimental compounds to that of parent molecule, SPC. (C–F) Transcriptomic data shown are representative of three independent replicates. All concentrations listed are per mL. Abbreviations: AMK, amikacin; CFU, colony forming units; FC, fold-change; FDR, false discovery rate; SPC, spectinomycin.
eAmSPC Induce Similar Transcriptional Response to SPC Despite Difference in Potency.
WhiB7 (Mab3508c) is implicated in upregulating several key virulence and drug resistance elements for sustaining host colonization and antibiotic resistance (7, 24, 25). SPC resistance in Mab is linked to WhiB7 activation of MAB_2780c (tetV), as Mab Δ2780c deletion mutants have >100-fold increase in SPC susceptibility (7, 17). To determine whether SPC and eAmSPCs have differential abilities to induce whiB7, full transcriptomic responses of Mab exposed to SPC, 2593, and 2694 were analyzed. Mab cells were exposed to identical sub-MIC concentrations of SPC (1 and 10 µg/mL), 2593 (1 µg/mL), and 2694 (1 and 10 µg/mL) for 3 h. SPC and eAmSPC-treated cells were found to have similar transcriptomic profiles and similar abilities to induce genes within the whiB7 regulon (Fig. 2C and SI Appendix, Fig. S3A), with the strength of the transcriptomic response correlated with the ability of the compound to accumulate intracellularly. All treatment groups displayed at least 1,300 differentially expressed genes (DEGs; Log2FC > |1|, FDR < 0.01) compared to no treatment (SI Appendix, Fig. S3 B–D and G and Table S4). Treatment with 2593 resulted in 1876 DEGs, including 92 whiB7 regulon genes, when compared to no drug (Fig. 2D and SI Appendix, Table S4). When 2593 treatment was compared to SPC at 1 µg/mL, 242 (39 whiB7 genes) DEGs were observed (Fig. 2E); however, when compared to SPC at 10 µg/mL, only 3 (0 whiB7 genes) DEGs were observed (Fig. 2F). This is in line with the accumulation data, for which 2593 accumulates 8.4 times the amount of SPC. Accumulation of 2694 is not statistically different than SPC, and as expected, the number of DEGs is similar between cells treated with equivalent concentrations of 2694 and SPC (SI Appendix, Fig. S3 E, H, and I and Table S4). These data support that eAmSPCs are able to induce whiB7 resistance mechanisms but maintain potency despite induction of tetV along with other resistance genes.
eAmSPCs Avoid High-Level Efflux by TetV, the Main whiB7-Induced SPC Resistance Mechanism.
Since the potency and transcriptomic profiles of eAmSPCs correlated with intracellular accumulation, and the TetV efflux pump is responsible for resistance to parent SPC, we sought to determine how the potency of the eAmSPCs is impacted by TetV along with other WhiB7-mediated drug transporters, ribosomal protection proteins, and drug-modifying enzymes. MICs were determined for 2593 and 2694, the 1st generation AmSPC 1950 and SPC against strains having deletions of MAB_1846c (Δ1846c), MAB_2355c (Δ2355c), MAB_1409c (Δtap), MAB_4532c (Δeis2), MAB_2780c (ΔtetV), and MAB_3508c (ΔwhiB7). These genes were selected for deletion due to their substantial upregulation upon eAmSPC exposure and importance in drug resistance against Mab (genes labeled in Fig. 2D; see SI Appendix, Table S1 for genetically engineered strains) (17, 26, 27). Both 2593 and 2694 had modest fourfold increases in susceptibility in the ΔtetV strain, compared with 64-fold increase for SPC and 16-fold for 1950. Similarly, the susceptibility to 2593 and 2694 only increased twofold against the ΔwhiB7 strain, while susceptibility of SPC and 1950 increased eightfold (Table 2). Aside from a slight decrease in susceptibility of 1950, 2593, and 2694 against the Δtap strain, inactivation of all other genes had no effect on the susceptibility of SPC or the eAmSPCs. This indicates that SPC and 1950 are subject to a high-level of efflux by TetV, but the eAmSPCs are subject to a much lower level of efflux. The MIC of 2593 in ΔtetV strain is fourfold lower than 2694, while the MIC of 2694 in the ΔtetV strain is the same as SPC (MIC = 4 µg/mL; Table 2). This supports the notion that greater accumulation results in greater potency.
Table 2.
eAmSPCs are minimally impacted by TetV mediated resistance
| Isogenic deletion mutant MIC (µg/mL) | ∆3508c double deletion mutant MIC (µg/mL) | |||||||||||||
| Compound | WT | ∆3508c | ∆2355c | ∆1846c | ∆4532c | ∆1409c | ∆2780c | WT/∆2780c ratio | ∆2355c | ∆1846c | ∆4532c | ∆1409c | ∆2780c | ∆2780c/∆3508c ∆2780c ratio |
| 1950 | 128 | 16 to 32 | 128 | 64 | 64 | >128 | 8 | 16 | 16 | 16 | 16 | 16 | 4 | 2 |
| 2593 | 4 | 1 to 2 | 4 | 4 | 4 | 16 | 1 | 4 | 1 to 2 | 1 | 1 | 2 | 0.5 | 2 |
| 2694 | 16 | 8 | 16 | 16 | 16 | 32 | 4 | 4 | 8 | 8 | 8 | 8 | 4 | 1 |
| SPC | 256 | 32 | 256 to 512 | 256 to 512 | 256 to 512 | 256 | 4 | 64 | 32 to 64 | 32 to 64 | 32 to 64 | 32 to 64 | 4 | 1 |
| AMK | 2 | 0.25 | 2 | 1 to 2 | 0.25 | 2 | 2 | 1 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 8 |
Whole cell antimicrobial activity of 1950, 2593, 2694, SPC and AMK against M. abscessus ATCC 19977 (WT) strain and mutant strains. Single-deletion mutants constructed for this study consisted of ∆whiB7 (∆3508c), ∆2355c, ∆1846c, ∆eis2 (∆4532c), ∆tap (∆1409c) and ∆tetV (∆2780c). ∆whiB7 double-deletion mutants constructed for this study consisted of ∆whiB7 ∆2355, ∆whiB7 ∆1846c, ∆whiB7 ∆eis2, ∆whiB7 ∆tap and ∆whiB7 ∆tetV. The contribution of Mab2780c-mediated resistance mechanisms was calculated by dividing WT MIC by ∆tetV MICs. The contribution of secondary whiB7-regulated resistance mechanisms was calculated by dividing ∆tetV MIC by ∆whiB7 ∆tetV MIC. AMK and the ∆eis2 and ∆whiB7 ∆eis2 double-deletion mutants served as a control. Data shown are the range of MICs at day 5 from at least two independent experiments. Abbreviations: AMK, amikacin, SPC, spectinomycin; WT, wild-type.
To address possible synthetic gene relationships within the whiB7 resistance regulon, we combined the ∆whiB7 mutant with targeted deletions of the other genes—resulting in double-deletion mutants. The MICs of all tested compounds (SPC, AmSPC, and eAmSPC) against the double mutants were similar to the MICs obtained from the ∆whiB7 isogenic mutant with the exception of the ∆whiB7 and ∆tetV double mutant (Table 2). The SPC/AmSPC susceptibility pattern of the latter mutant corresponded to the phenotype of the ∆tetV isogenic mutant, indicating that the TetV transporter comprises the dominant whiB7-regulated SPC resistance mechanism (Table 2). These results show that the aminomethyl substitution, specifically the N-ethylene linkage of the aminomethyl group, has a critical role in diminishing TetV-mediated resistance in Mab and is responsible for the increased antimicrobial activity of the 2nd generation AmSPCs not seen in 1st generation analogs like 1950.
eAmSPCs Synergize with Other Anti-Mab Compounds.
Since combination therapy is critical for successful treatment of Mab infections, we determined the therapeutic potential of pairing eAmSPCs (2593 and 2694) with front-line Mab antimicrobials using a checkerboard approach. Mechanistic synergy and antagonism were determined using the web-based response surface method, bivariate response to additive interaction doses (BRAID) (28). Both 2593 and 2694 displayed similar degrees of synergy with clarithromycin (CLR), linezolid (LZD), bedaquiline (BDQ), and clofazimine (CLO), whereas both were found to be antagonistic with amikacin (AMK) and cefoxitin (CEFOX) (Fig. 3 A–G and SI Appendix, Fig. S4 A–F). Synergism between SPCs and macrolides aligns with previously reported results describing this combination in M. tuberculosis (29, 30). Encouragingly, this study also found that 2593/2694 synergized with LZD, indicating that the eAmSPC class performed favorably with two first-line, orally bioavailable protein synthesis inhibitors, both of which target the 50s ribosomal subunit (4). The antagonism observed with AMK could be mechanistic, as the eAmSPCs may inhibit the mistranslation key to AMK bactericidal potency (31), or they may compete for another unknown import mechanism. The combination with CEFOX was found to be the most antagonistic, which reflects the well-known antagonism between translational inhibitors and β-lactams, as the latter requires cell division for maximal activity. These results indicate the eAmSPC series has potential as part of therapeutic regimen.
Fig. 3.

Drug combination screening identifies synergistic/antagonistic combinations with antimycobacterial antibiotics. (A) Individual κ values and confidence intervals were calculated using the BRAID response surface model and plotted. κ > 0 are considered synergistic and κ < 0 are considered antagonistic with values further from 0 representing stronger interaction profiles (increase for synergy, decrease for antagonism) for that combination. Error bars crossing κ = 0 indicate a nonstatistically significant interaction. (B–G) Heatmap of individual checkerboard assay results where percent viability was determined by resazurin conversion over the final 24 h of a 72-h incubation at 37 °C and normalized to no drug (100% viability) and cell death controls (CLR 6 µg/mL; 0% viability) with darker blue indicating higher reduction potential (surrogate for cell viability). Checkerboard plates of 2593 in combinations with (B) AMK, (C) CLR, (D) LZD, (E) CEFOX, (F) BDQ, and (G) CLO. Median % viability values from three independent replicates were plotted. Abbreviations: AMK, amikacin; BDQ, bedaquiline; CEFOX, cefoxitin; CLO, clofazimine; CLR, clarithromycin; LZD, linezolid.
eAmSPCs Maintain Activity against Other NTMs, Clinical Isolates, and Gram-Positive Pathogens.
To explore the spectrum of activity of the eAmSPCs, lead compounds were tested against other NTMs, M. tuberculosis, and other clinically relevant microbial organisms (e.g., ESKAPE pathogens). Lead eAmSPCs performed favorably against other mycobacterial species, including M. tuberculosis and M. avium (Table 1 and SI Appendix, Table S5). Given the well-documented different responses to therapy between Mab sub-species, in particular macrolide antibiotics, lead eAmSPCs were then validated against two independent panels of recent mycobacterial clinical isolates from St. Jude Children’s Research Hospital and the University of Zürich (SI Appendix, Tables S5 and S6) (32–36). In vitro results indicate that all eAmSPCs maintain MIC activity between 1.6 and 16 µg/mL against the clinical isolates including those with significant loss of susceptibility to AMK and CLR (SI Appendix, Tables S5 and S6). Consistent with this observation, spontaneously generated AMK mutants of the WT strain remained susceptible against eAmSPCs, and spontaneously generated 2593-resistant mutants remained susceptible to AMK (SI Appendix, Table S5). In addition to their NTM activity, eAmSPCs were shown to be active against gram-positive bacteria with MICs as low as 1.6 µg/mL against Streptococcus species and as high as 25 µg/mL against methicillin-resistant Staphylococcus aureus (MRSA) for 2593 (SI Appendix, Table S7). The series lacked effectiveness against most gram-negative bacteria (MIC >200 µg/mL against Pseudomonas aeruginosa and Acinetobacter baumannii) outside E. coli K12, which was inhibited by 1980 and 2593 at 12.5 µg/mL (SI Appendix, Table S7). A shift in MIC between E. coli K12 and the isogenic ΔtolC knockout mutant indicate that SPC and its analogs can accumulate within gram-negative organisms, but these compounds are subject to efflux by resistance-nodulation-cell division pumps (SI Appendix, Table S7). These results suggest that the eAmSPCs are capable of potent activity against various Mycobacterium species and their use would not lead to cross-resistance with current treatment modalities.
eAmSPCs Demonstrate Favorable Pharmacokinetic and Safety Profiles.
In vitro cytotoxicity testing of the lead compounds against two mammalian cell lines showed the compounds are noncytotoxic (SI Appendix, Table S8). To alleviate potential concern for side effects, we then performed off-target profiling experiments using Eurofins safety screen and determined that 2593 does not interact with a wide range of mammalian receptors and avoids inhibition of common drug-metabolizing enzymes within the cytochrome P450 family (SI Appendix, Table S9). To further investigate eAmSPCs drug-like properties, we determined the in vivo pharmacokinetic (PK) properties of the 2592, 2593, 2694, and 2845. Mice that received a 10 mg/kg intravenous injection dose of 2592, 2593, and 2694 exhibited similar pharmacokinetic behavior with high peak concentrations (C0) of 38 to 74 mg/L and systemic exposure (AUCinf) between 15 and 19 h*mg/L (SI Appendix, Table S8). In contrast, mice that received thiazole-based 2845 exhibited substantially lower peak concentrations (19.3 mg/L), a twofold higher clearance, and a correspondingly reduced systemic exposure (6.95 h*mg/L). All leads exhibited a relatively short half-life (0.21 to 0.33 h) at therapeutically relevant plasma concentrations as previously observed for AmSPCs and other aminocyclitols (11, 37, 38). To exclude the possibility of metabolic instability, 2593 and 2694 were incubated with mouse, rat, and human liver microsomal preparations. Both compounds remained stable in the preparations of all three species without detectable degradation (SI Appendix, Table S10). Given these positive results, 2593 and 2694 were advanced to in vivo toxicity determination. These results demonstrated that mice can support subcutaneous (SC) bolus dosages of 2593 and 2694 at 50 mg/kg and 100 mg/kg, respectively (SI Appendix, Table S8).
eAmSPC Demonstrate Efficacy against Mab in Acute and Chronic Murine Infection Models.
The promising in vitro antimicrobial and in vivo pharmacokinetic data generated for our compounds supported moving to in vivo, proof-of-concept efficacy testing of lead compounds 2593 and 2694. Using an acute Mab infection model in immunocompromised mice (Fig. 4A) both 2593 (50 mg/kg SC twice daily) and 2694 (100 mg/kg SC twice daily) were found to significantly reduce the bacterial burden in the lung and spleen (P < 0.0001; Fig. 4 B and C). Strikingly, 2593 completely cleared infection below the limit of detection in pulmonary tissue with no burden observed in six infected mice, an improvement over standard-of-care control AMK (150 mg/kg SC once daily) (P < 0.0001; Fig. 4B). Additionally, 2593 and 2694 significantly decreased bacterial load in the spleen by 2-log colony-forming units (CFU), comparable to that of AMK (P < 0.001; Fig. 4C).
Fig. 4.

eAmSPC are efficacious in murine Mabs infection models. (A) Schematic of proof-of-concept studies investigating in vivo efficacy of lead eAmSPCs and AMK in an acute (Top; B and C) and chronic (Bottom; D and E) models of infection. (B and C) Bacterial counts in the (B) lungs and (C) spleens of SCID mice in the acute infection model. (D and E) Bacteria counts in the (D) lungs and (E) spleens of SCID mice in the chronic infection model. Results shown as mean ± SEM, n = 3 biological replicates at day 1 (and day 27 for chronic model) or six biological replicates at endpoint for all treatment conditions with the limit of detection being Log10 CFU of 1.7. Statistical significance using ANOVA and Dunnett multiple comparison test with calculated P-values shown. Abbreviations: AMK, amikacin; LOD, limit of detection; SCID, severe combined immune deficiency.
While the response in the acute model is a promising finding, Mab usually forms persistent infections requiring extended treatment periods in the clinic; therefore, 2593 was advanced for evaluation in a chronic infection model (Fig. 4A). When dosed at 30 mg/kg SC twice daily, 2593 was shown to significantly decrease Mab burden in the lungs (2.57 log CFU reduction, P < 0.0001) and spleens (2.69 log CFU reduction, P < 0.001) of chronically infected immunocompromised mice, similar to that of AMK (Fig. 4 D and E). Collectively, the demonstrated efficacy as a standalone monotherapy against Mab, coupled with the favorable interaction profile observed with commonly utilized anti-Mab therapeutics, signifies the promising capacity of the eAmSPCs in combating Mab infections.
Discussion
Infections with NTMs, particularly Mab, have poor prognosis due to high intrinsic drug resistance that leads to long treatment times, thus increasing the potential for side effects such as ototoxicity. Synthetic modification of existing antibiotics to avoid intrinsic resistance mechanisms can be used as a strategy to exploit mechanistically unique therapeutics for new disease indications. Previously, we used structure-guided drug design to modify SPC to generate the narrow spectrum antitubercular spectinamides, which avoid the intrinsic efflux mechanisms of M. tuberculosis (10). Herein, we report on the identification and development of the eAmSPCs, which have significantly improved therapeutic properties over SPC against Mab. We demonstrate that the eAmSPCs i) bind to the same site within helix 34 of the 16S rRNA of the bacterial ribosome as SPC, ii) have improved potency through increased accumulation, largely through avoiding high-level TetV-mediated efflux, iii) act synergistically with several classes of anti-Mab antimicrobials, iv) are active against drug-resistant clinical Mab isolates and lack cross-resistance to common therapeutics, v) have favorable pharmacokinetic and safety profiles, and vi) demonstrate efficacy in acute and chronic murine models for Mab.
Consistent with previous SAR studies involving SPC analogs, substitutions of the ketone from the C-ring of SPC are tolerated provided the 3′-(R) aminomethyl stereochemistry is maintained (10, 11, 34). Contrary to Tap (Rv1258c)-mediated SPC efflux in M. tuberculosis, an isogenic knockout of the closest homolog in Mab, MAB_1409c (also known as Tap; 55% identical to Rv1258c), had no impact on SPC susceptibility and a paradoxical effect on the eAmSPCs. Instead, the WhiB7-dependent Mab2780c efflux pump, TetV (17% identical to the closest homolog in M. tuberculosis, Rv1258c), was responsible for SPC resistance in Mab (17). The results in this study indicate that the enhanced antimicrobial action of the eAmSPC series over SPC in Mab is extraribosomal—through avoidance of high-level, TetV-mediated efflux. This model is supported by observations in mechanistic studies, which demonstrated ribosomal binding activity and transcriptomic induction of WhiB7 resistance mechanisms by eAmSPCs are consistent with SPC and confirmed that TetV is the dominant WhiB7 resistance mechanism for both SPC and eAmSPCs. However, the eAmSPCs were found to be subject to a much lower level of efflux by TetV, resulting in only a fourfold increase in susceptibility against ΔtetV, in contrast to the 64-fold increase for SPC. Accumulation studies demonstrated the ability of eAmSPCs to accumulate in whole cells correlated with the strength of the transcriptional response, MIC, and lipophilicity of the R-group. Collectively, these results imply that N-ethylene linkage to cyclic substituents of the 2nd generation eAmSPCs plays a crucial role in their activity against Mab. Even a minor shortening of the linkage by a single carbon, as observed in the comparison between 1st generation AmSPC 1950 and 2nd generation AmSPC 2593, leads to elevated MICs and TetV efflux ratios (16-fold) against Mab. This shift suggests that longer linkages might modify the kinetics of drug expulsion by TetV, ultimately tilting the equilibrium towards heightened intracellular concentrations and enhanced antimicrobial efficacy. The low-level efflux by TetV observed for eAmSPCs leaves room for future improvement through structure-guided design to fully abolish this remaining efflux. Additional studies regarding accumulation, expression of genes in the whiB7 resistance regulon, and their relationship to potency of the eAmSPCs are ongoing and will provide the basis for further development of more potent compounds.
The in vitro and in vivo studies were encouraging, demonstrating that eAmSPCs have many attributes that make them attractive preclinical candidates for treating NTM infections. Lead compounds exhibited broad-spectrum activity across a panel of Mycobacterium species, including M. avium and M. tuberculosis, as well as a collection of drug-resistant Mab clinical isolates derived from patients in North America and Europe. eAmSPCs avoid resistance by inactivation as they are not substrates for the mycobacterial aminoglycoside modifying enzyme Eis. The mechanism of protein synthesis inhibition of the eAmSPCs is advantageous, as it does not overlap with other ribosomal inhibitors used against NTMs and their mechanisms of resistance. We have also demonstrated that eAmSPCs synergize with several classes of anti-Mab agents, including a representative macrolide and oxazolidinone, providing further rationale for their position within a therapeutic regimen. In vitro studies demonstrate that lead eAmSPCs display minimal cytotoxicity in mammalian cell lines (>100 µM) and have little potential for off-target effects that result in adverse or drug–drug interactions. Hepatic microsomal stability studies in mice, rats, and humans indicated no detectable degradation for either 2593 or 2694. Pharmacokinetic profiling of leads identified 2593 and 2694 as having the most favorable exposures, lowest clearance, and in vivo disposition behavior similar to other aminocyclitols. These properties, combined with the high safety index of SPC and the spectinamides, suggest that eAmSPC also have a favorable pharmacologic profile. Most importantly, the eAmSPCs were found to have similar or greater efficacy to standard-of-care control AMK in acute and chronic infection models in mice. In an acute Mab infection model in immunocompromised mice, both 2593 and 2694 were found to significantly reduce the bacterial burden in the lung and spleen. Impressively, 2593 completely cleared infection in pulmonary tissue with no burden observed in all six infected mice. In a chronic infection model, which better represents the persistent infections found in the clinic, 2593 was shown to decrease Mab burden greater than 2.5 log CFU in both the lungs and spleens of chronically infected immunocompromised mice, similar to that of AMK. The primary limitations of the eAmSPCs are low oral bioavailability which is favored for NTM therapies, and the maximum tolerated doses are lower than other semisynthetic SPC analogs. To enhance bioavailability at the site of infection, our spectinamide program evaluated intrapulmonary delivery of lead 1599, where it was shown to be well tolerated and effective in combination with oral pyranzinamide (38). Given spectinamide’s similar physicochemical properties, the eAmSPCs could also benefit from similar delivery strategies.
This report is the first to demonstrate that intrinsic efflux in Mab can be effectively eluded through the semisynthetic modification of natural products like SPC. Currently, no evidence-based regimen is available to predict the successful resolution of Mab infection. Given that Mab most common infectious niche is pulmonary, 2593’s effectiveness in reducing Mab burden in the lung highlights the promising potential eAmSPCs exhibit in treating pulmonary-associated infections. The vulnerable nature of CF, pulmonary transplant recipients, and COPD populations’ lungs provides an inviting environment for drug-refractory mycobacterial infections. Our results show that eAmSPCs are bioactive in pulmonary tissue as monotherapy, and as such, the advancement of eAmSPCs as novel therapeutics as part of a combinatorial antimicrobial regimen warrants further exploration for pulmonary infections caused by Mab and other NTM pathogens.
Materials and Methods
Bacterial Cell Culture.
A. baumannii (ATCC 19606), Bacillus anthracis (Strerne 34F2), Bacillus subtilis (ATCC 23857), Burkholderia cepacia (ATCC 25416), E. coli (K12), Klebsiella pneumoniae (ATCC 33495), Proteus mirabilis (Hauser ATCC 25933), Proteus vulgaris (ATCC 33420), P. aeruginosa (PA01), S. aureus (ATCC 29213; MSSA), S. aureus (NRS70; MRSA), Staphylococcus epidermidis (ATCC 14990), and Stenotrophomonos maltophilia (ATCC 13637) were maintained in Mueller Hinton (MH) (Fisher Scientific) broth or agar plates. E. coli ΔtolC (K12 ΔtolC:Kan) was maintained on MH broth or agar containing 25 µg/mL kanamycin. Streptococcus pneumoniae (R6) and Streptococcus pyogenes (ATCC 700294) were cultured in MH in the presence of 10% (v/v, final) defibrinated, lysed horse blood. Mycobacterium chelonae ATCC 35752, Mab ATCC 19977, clinical Mab isolates and genetically engineered mutants, Mycobacterium peregrinum ATCC 14467, Mycobacterium fortuitum ATCC 6841, and M. smegmatis ATCC 19420 were maintained in MH2 (cation adjusted) (Fisher Scientific) broth and agar. Mycobacterium marinum ATCC 927, Mycobacterium ulcerans ATCC 19423, Mycobacterium kansasii ATCC 12478, and Mycobacterium avium ATCC 25291 were maintained and cultured in MH2 broth and agar supplementary in 5% (v/v) oleic albumin dextrose catalase (Sigma Aldrich). M. tuberculosis (H37Rv) was cultured in Middlebrook 7H9 broth (Difco Laboratories) supplemented with 10% albumin-dextrose complex and 0.05% (v/v) Tween 80 (pH 7.4).
Genetically Engineered Mab Deletion Mutants.
Unmarked Mab deletion mutants (SI Appendix, Table S1A) deficient in open reading frames MAB_1409c (tap), MAB_2355c, MAB_1846, and MAB_2780c (tetV) were constructed by transformation of electrocompetent Mab ATCC 19977 using suicide vectors derived from pSE-apr-katG (27) or pKH_aac(3)IVDsRed2_KatG (39), respectively. Briefly, upstream and downstream fragments of the target genes (each approx. 1.5 kbp) were amplified by PCR with primers shown in SI Appendix, Table S1B. Subsequently, fragments were cloned into backbone vectors adjacent to an aac(3)IV resistance cassette by restriction enzyme digestion or Gibson assembly. Resulting plasmids pSE-Δ1409c, pKH-Δ2355c, pKH-Δ1846, and pKH-Δ2780c were verified by sequencing and subsequently electroporated into competent Mab. Mab transformants were selected on LB agar plates containing apramycin (50 mg/L). In case of pKH-derived vectors, red fluorescence was exploited as an additional marker. Transformants, grown after 5 to 7 d of incubation at 37 °C, were confirmed by PCR amplification of the aac(3)IV resistance cassette. Subsequently, putative single cross-over transformants were subjected to counter-selection on agar plates containing isoniazid (32 mg/L). Putative deletion mutants grown after 5 to 7 d of incubation at 37 °C were screened by PCR amplification of the target locus and confirmed by Southern blot analysis. Deletion of open reading frames MAB_2355c, MAB_1846, MAB_1409c (tap), and MAB_2780c (tetV) was demonstrated by digestion of genomic DNA (gDNA) with Eco47III, BfuI, PvuII, and NsbI and hybridization with DNA probes generated by PCR amplification or restriction enzyme digestion (SI Appendix, Table S1C). Double-deletion mutants were generated by transforming electrocompetent single-deletion mutants with the target vectors of choice according to the procedure described for generation of single-deletion mutants. Subsequently, double-deletion mutants were confirmed as described for single-deletion mutants. The absence of the aac(3)IV cassette in deletion mutants was further confirmed by phenotypic apramycin susceptibility testing.
Antibacterial Susceptibility Testing.
MICs were determined using broth microdilution in appropriate media (indicated above) according to Clinical Laboratory Standards Institute (CLSI; M24 for Mycobacterium, M100 for all other species tested), using twofold serial dilutions in 96-well plates (ThermoFisher Scientific, cat. 163320) starting at a drug concentration of at least 64 µg/mL and a final volume of 0.2 mL. Plates were incubated in 5% CO2 at 37 °C except for M. chelonae, M. marinum, and M. ulcerans (30 °C). MICs were recorded after 16 to 20 h of incubation, except for mycobacterial MICs, which were recorded after 3 to 14 d of incubation, dependent on CLSI recommendation for specific bacterium and compound. Mab MICs in Table 2 were determined after 5 d of incubation. Median or range of MIC values are reported. All experiments were performed in biologic triplicate unless otherwise stated.
Ribosomal Inhibition Assays.
Mycobacterial cell-free translation inhibition assays were performed as described previously (40). In brief, M. smegmatis strain SZ380 bacterial cells were disrupted and emulsified with a microfluidizer processor (Microfluidics, Westwood, MA, USA) at 25,000 lb/in2. S30 extracts were prepared by addition of dithiothreitol (DTT) to 1 mM and centrifugation at 30,000 × g at 4 °C. Translation reaction mixtures containing either test article or dimethyl sulfoxide (DMSO) vehicle control and 4 µL of the S30 extract, 0.2 mM amino acid mix, 6 µg tRNA (Sigma), 0.4 µg hFluc mRNA, 0.3 µL protease inhibitor (complete, EDTA-free, Roche), 12 U RNAse inhibitor (Ribolock, Thermo Scientific), 6 µL S30 premix without amino acids (Promega), plus water to a final reaction volume of 15 µL were incubated for 1 h at 37 °C. The reaction was stopped on ice before adding 75 µL of luciferase assay reagent (Promega) and recording of luminescence. Regression analysis and IC50 calculation were performed using GraphPad Prism version 9.3.1 by using the equation [log(inhibitor) vs. response–Variable slope (four parameters)] with the fitting method: least squares (ordinary) fit. Y = Bottom + (Top-Bottom)/(1+10^((X-LogIC50))).
X-Ray Crystallographic Structure Determination.
Wild-type 70S ribosomes from T. thermophilus (strain HB8) were prepared as described previously (41–44). Synthetic mRNA with the sequence 5′-GGC-AAG-GAG-GUA-AAA-AUG-UUC-UAA-3′ containing Shine-Dalgarno sequence followed by the P-site methionine (AUG) and A-site phenylalanine (UUC) codons was obtained from Integrated DNA Technologies (Coralville, IA, USA). Ribosome complexes with mRNA and tRNAs were formed by mixing 5 µM T. thermophilus 70S ribosomes with 10 µM mRNA and incubation at 55 °C for 10 min, followed by addition of 20 µM P-site tRNAifMet and 20 µM A-site tRNAPhe substrates (43–47). For cocrystallization of SPC or 2694 with the ribosome, the antibiotic was added to a final concentration of 100 µM, and the complex was left at room temperature for an additional 15 min prior to crystallization. Ribosome complexes were prepared in the buffer containing 5 mM HEPES-KOH (pH 7.6), 50 mM KCl, 10 mM NH4Cl, and 10 mM Mg(CH3COO)2 and then crystallized in the buffer containing 100 mM Tris-HCl (pH 7.6), 2.9% (w/v) PEG-20K, 9 to 10% (v/v) MPD, 175 mM arginine, 0.5 mM β-mercaptoethanol. Crystals were grown by the vapor diffusion method in sitting drops at 19 °C, stabilized and cryo-protected stepwise using a series of buffers with increasing MPD concentrations (25%, 30%, 35%) until reaching the final concentration of 40% (v/v) MPD as described previously.
Collection and processing of the X-ray diffraction data, model building, and structure refinement were performed as described in our previous reports (43–52). Diffraction data were collected at beamlines 24ID-C and 24ID-E at the Advanced Photon Source (Argonne National Laboratory). A complete dataset was collected using 0.979 Å X-ray irradiation at 100K from multiple regions of the same crystal, using 0.3-degree oscillations. Raw data were integrated and scaled using the XDS software package. Molecular replacement was performed using PHASER from the CCP4 program suite (version 7.0). The search model was generated from the previously published structures of the T. thermophilus 70S ribosome with bound mRNA and aminoacylated tRNAs [PDB entry 6XHW (47)]. Initial molecular replacement solutions were refined by rigid-body refinement with the ribosome split into multiple domains, followed by positional and individual B-factor refinement using the PHENIX software (version 1.17). Noncrystallographic symmetry restraints were applied to four parts of the 30S ribosomal subunit (head, body, spur, and helix 44) and four parts of the 50S subunit (body, L1-stalk, L10-stalk, and C-terminus of the L9 protein). Structural models were built in Coot (version 0.8.2). The statistics of data collection and refinement are compiled in SI Appendix, Table S2. All figures showing atomic models were rendered using PyMOL Molecular Graphics System software (version 1.8.6, Schrödinger, www.pymol.org).
Selection of Spontaneous Resistant Mutants.
First, 2593-resistant Mab mutants were generated by plating 100 µL of drug-free, supersaturated culture (OD600 > 1) of Mab ATCC 19977 onto MH agar plates with 4X (16 mg/L) and 10X (40 mg/L) MIC concentration of 2593. Colonies were checked for mutations by evaluating MIC value against 2593 and other eAmSPCs. Method was replicated to select for AMK-resistant mutants.
gDNA Extraction, Whole-Genome Sequencing, and Analysis of Clinical Isolates and Spontaneously Generated 2593 Mutants.
gDNA from spontaneously generated mutants and St. Jude clinical isolates were extracted using the DNeasy Blood and Tissue Kit (Qiagen, cat. 69504) according to the manufactures’ directions on gDNA extraction of gram-positive bacteria. Purified gDNA were then sequenced using MiSeq Illumina sequencing at a depth of 30X genome coverage at the Hartwell Center at St. Jude’s Children’s Research Hospital (SJCRH). Sequencing data were saved in FASTQ format and data were processed through the High-Performance Computing Facility at SJCRH. Adapter content and quality trimming were performed using Cutadapt (53). The quality of the raw and trimmed reads was assessed using FastQC and MultiQC (54, 55). The resulting processed reads were aligned to the reference Mab ATCC 19977 genome (Genome accession: NC_010397) using Bowtie2 (56). Variant calling was performed using VarScan (57) and annotated using SNPeff (58). Variants were confirmed through the integrative genomics viewer (Broad) (59). MAB_3869c (rpoB) and MAB_2297 (erm) were used to speciate St. Jude clinical isolates. All sequencing reads associated with this study have been deposited to National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number PRJNA865308.
Whole-Cell Accumulation Assay.
Treatment.
Mab ATCC 19977 was grown from single colonies in 200 mL of MHB2 containing 0.05% Tyloxapol at 37 °C with shaking to an optical density (OD600) of 0.5 to 0.6. The bacteria were harvested at 3,220 RCF for 10 min at 4 °C in 200-mL centrifuge tubes (ThermoFisher Scientific, cat. 376813), and the supernatant was discarded. The cells were then washed in 20 mL of buffer PBS+ (phosphate-buffered saline) buffer [PBS + 0.05% Tween-80 + 5% glucose (w/v)] and pelleted as before, and the supernatant was discarded. Glucose was added to energize active transport processes in the minimal buffer to account for efflux. Cells were washed again by resuspended pellets in 12 mL of PBS+ and transferring the cell suspension to a 50-mL sterile centrifuge tube (Corning, cat. 430290) and pelleted as before. The pellets were then resuspended in 6 mL of PBS+ buffer, and 1 mL of concentrated cell suspension was aliquoted into 5-mL screw-cap centrifuge tubes (Eppendorf, cat. 13864407) containing DMSO-solubilized compound (100 µM final volume; 1% DMSO). The concentrated cell suspension was incubated for 30 min at 37 °C in a shaking incubator. After incubation, the cell mixture was centrifuged at 3,220 RCF for 10 min at 4 °C. The cell pellet was then washed by resuspending in 3 mL of PBS+ buffer and centrifuged as before. After a total of three consecutive wash steps, the cell pellet was resuspending in the original 1 mL of PBS+ buffer and transferred to a 1.5-mL sterile screw top centrifuge tube (USA Scientific, cat. 14153500) where 10 µL was used for CFU enumeration by serial dilution and the remaining 990 µL centrifuged 14,500 RCF for 2 min at 4 °C. To lyse the samples, each pellet was resuspending in 300 µL of 2.5% TCA and 100 µL of 0.1 mm silicone spheres (BioSpec, 11079101Z) were added. Each sample was then subject to three cycles of bead beating using the Bead Mill four homogenizer (Fisher Scientific, Atlanta GA) at maximum speed for 45 s followed by 3 min on ice. Lysates were pelleted at 14,500 RCF for 2 min at 4 °C and 250 µL of the lysate-supernatant was collected and filtered using 1.5-mL centrifugal 0.22 µm filter tubes (Millipore; Cat. UFC30GVNB) before being analyzed by LC-MS/MS.
Analysis.
Samples were analyzed using an Acquity ultra performance liquid chromatography (UPLC, Waters Corporation) coupled with 6500 Triple Quad System (AB Sciex). Briefly, 10 µL of extract was separated on an Acquity UPLC HSS T3 1.8 mm, 2.1 × 50 mm column, which was maintained at 60 °C. Solvent A was 0.1% Heptafluorobutyric Acid (HFBA) in MilliQ Water and solvent B was 0.1% HFBA in acetonitrile. The inlet method for these samples utilized a flow rate of 0.9 mL min−1 with the following gradient: 0 to 0.5 min, 99.0% solvent A, and 1% solvent B; 0.5 to 1.4 min, gradient of 99 to 5% solvent A and 1 to 95% solvent B; 1.4 to 1.8 min, hold on 5% solvent A and 95% solvent B; 1.8 to 0.82 min gradient of 5 to 99.0% solvent A and 95 to 1% solvent B; and 1.82 to 2 min, hold on 99% solvent A and 1% solvent B, as previously described (17). The first 0.5 min of eluate was desalted to waste by an integrated Valco valve. Multiple reaction monitoring was used to quantify each compound metabolite based on linear calibration curve. Data were acquired using Analyst Software (AB Sciex, version 1.6.3) and analyzed using MultiQuant Software (AB Sciex, version 3.0.3). The mass spectrometer was operated in positive ion mode with electrospray ionization. Reaction monitoring parameters were as follows: the pressure of ion source gas 1 = 50 psi, the pressure of ion source gas 2 = 50 psi, the pressure of curtain gas = 30 psi, the pressure of collision gas = 10 psi, ion spray voltage = 5.5 kV, source temperature = 600 °C, and entrance potential = 10 V. Parent ion m/z, fragment m/z, declustering potential, collision energy, collision cell exit potential, retention time, and correlation coefficient for standard curve are described in SI Appendix, Table S11. Accumulation values (A) were calculated as previously described (60) using the following equation: , where C is compound concentration, V is extraction volume (0.3 mL), and CFU is number of CFU recorded normalized to 1010. All experiments were performed in biologic triplicate, and data was plotted as mean and SD in GraphPad Prism v9.5.0 (GraphPad Software, La Jolla California USA). Statistical significance using ANOVA determined groups were of equal variance by Brown–Forsythe test and Dunnet’s posttest was then used compare results of experimental compounds to that of parent molecule, SPC. Methods schematic in Fig. 2B was created with BioRender.com.
RNA Sequencing.
Treatment.
Transcriptomic analysis was performed using the type strain Mab ATCC 19977 grown in MHB II at 37 °C to an OD600 of 0.4 to 0.8. At mid-log, cultures were exposed to 1 µg/mL or 10 µg/mL of SPC or 2694, 1 µg/mL of 2593, or an equal volume of vehicle control (DMSO) for 3 h at 37 °C followed by addition of 2 volumes of RNA protect (Qiagen, Carol Stream IL), then centrifuged for 5 min at 10,000 RCF at 4 °C.
Extraction.
Supernatant was poured off and pellets stored at −80 °C. Next day samples were thawed out and resuspended in 500 µL RLT + 1% betamercaptoethanol and transferred to Lysing Matrix B (VWR, Pittsburgh PA). Samples were placed into a Bead Mill four homogenizer (Fisher Scientific, Atlanta GA) and homogenized 4 times for 40 s at 4.5 m/s with 1 min of ice cooling between each round of homogenization. Lysate was spun down at 8000 RCF for 2 min and supernatant removed to clean centrifuge tube. Subsequent extraction of RNA was performed according to the RNAeasy extraction kit (Qiagen, Carol Stream IL). RNA was eluted in 40 µL of elution buffer yielding a typical concentration of 400 to 500 ng/µL. RNA samples were then DNase treated to remove residual gDNA from samples. This was performed using the gDNA Clean and Concentrator Kit (Zymo Research, Irvine CA). Samples were eluted in 30 to 45 µL of RNase free water, yields ranging from 100 to 250 ng/µL.
Sequencing, data processing, and analysis.
Libraries were prepared from total RNA with the TruSeq Stranded mRNA Library Prep Kit according to the manufacturer’s instructions (Illumina, PN 20020595). Libraries were analyzed for insert size distribution using the 2100 BioAnalyzer High Sensitivity kit (Agilent). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (ThermoFisher) and by low-pass sequencing with a MiSeq nano kit (Illumina). Paired-end 100 cycle sequencing was performed on a NovaSeq 6000 (Illumina) or HiSeq 2500 (Illumina) by the St. Jude Children's Research Hospital sequencing core at a depth of 100M reads/sample. Reads were saved in FASTQ format and data were processed through the High-Performance Computing Facility at St. Jude. Adapter content and quality trimming was performed using Cutadapt (53). The quality of the raw and trimmed reads was assessed using FastQC and MultiQC (54, 55). The resulting processed reads were aligned to the reference M. abscessus ATCC 19977 genome (Genome accession: NC_010397) using STAR2.7.1 and aligned reads were counted using featureCounts (61, 62). Differential gene expression analysis was analyzed using DESeq2 in the R statistical computing environment (63). Volcano plots were constructed in GraphPad Prism v9.5.0 (GraphPad Software, La Jolla California USA) and heatmaps were made using heatmaps.2 command in the R package gplots (version 3.1.3). All RNA-seq data shown are representative of three independent experiments and the raw reads and processed counts table have been deposited to NCBI Gene Expression Omnibus (GEO) under accession number GSE222081.
Checkerboard Drug Combination Screening.
A screen of 2593 and 2694 in combination with Mab therapeutics was conducted. Each drug combination was assessed using a 10-point concentration curve checkerboard design performed in triplicate, resulting in 300 data points per combination (1:2 dilutions; concentration range for each compound constructed for MIC values to be at 3rd to 4th dilution if solubility allowed). All compounds were dissolved in ≥90% DMSO and serially diluted into a 384-well Echo PP plate (Labcyte, cat no. PP-0200) where 125 nL of the eAmSPCs and 250 nL for all other compounds were transferred to a 384-well black clear bottom plate (ThermoFisher Scientific, cat. 142761) using a Beckman Echo 650 Acoustic Liquid Handler. Mid-log Mab ATCC 19977 cultures were diluted to an OD600 of 0.005 in MHB2 and 50 µL was delivered to each well of 384-well assay plate containing compound using a Multidrop Combi (ThermoFisher Scientific). This resulted in a DMSO concentration of 0.75% and the following final concentration ranges: 2593 (25 to 0.05 µg/mL), 2694 (64 to 0.125 µg/mL), AMK (32 to 0.063 µg/mL), CLR (4 to 0.008 µg/mL), LZD (12.8 to 0.025 µg/mL), CEFOX (256 to 0.5 µg/mL), BDQ (1 to 0.002 µg/mL), and CLO (8 to 0.016 µg/mL). Additionally, 12 individual cell-death (CLR at 6 µg/mL) and no drug (DMSO) controls were transferred and included for each plate, as well as monotreatment for each compound. After media transfer, assay plates were then briefly centrifuged at 300 RCF and incubated for 48 h at 37 °C. After 48 h, plates were removed and 5 µL of 0.77 mM of resazurin (final concentration 70 µM) was added to each well via a MultiDrop Combi and incubated for an additional 24 h. After incubation, plates were sealed with a black light-absorbing film (AbsorbMax, RPI, cat. 202537) and the fluorescent intensity (540/590) was read using a PHERAstar FS Multilabel reader (BMG, Cary, NC). Raw fluorescent intensity values were normalized to no drug (100% viability) and cell death controls (0% viability) to estimate % viability. Synergy and antagonism were determined using a web-based response surface method, BRAID (https://braid.stjude.org/webapp/) after converting concentration units to molar (28). Kappa (κ) values and CIs were recorded and interpreted as κ > 0 are synergistic and κ < 0 are antagonistic with values further from 0 representing stronger interaction profiles (increase for synergy, decrease for antagonism). Error bars crossing κ = 0 indicate a nonstatistically significant interaction. All checkerboard experiments were performed in technical triplicate. The BRAID plot and heat maps (median % viability values plotted) were constructed in GraphPad Prism version 9.5.0 (GraphPad Software, La Jolla California USA).
Murine Model Studies.
All murine model studies were conducted in accordance with the Animal Welfare Act and the Public Health Service Policy on Humane Care and Use of Laboratory Animals and were prior to initiation approved by Institutional Animal Care and Use Committees of Colorado State University.
Acute model of Mab infection.
The acute model of efficacy against Mab was performed similarly to previously published methods (64, 65). Six- to eight-week-old SCID (severe combined immune deficiency) female mice were purchased from Charles River Laboratories and rested 1 wk prior to infection. Mice were divided into five groups containing 3 or 6 mice each and infected with a tail vein injection of 100 µL containing 1 × 106 CFU of Mab 103 (clinical isolate from CF patient). Three mice were killed 1-d post infection to determine bacteria uptake. Whole lungs and spleen were extracted and homogenized in 4.5 mL of PBS, plated on 7H11 agar plates, and incubated for 7 d in 37 C dry-air incubator (limit of detection: Log10 1.7 CFU/organ). To determine efficacy, therapy or no drug control was initiated on the other four groups at 2 d post infection. Treatment with 2593 (50 mg/kg/dose) and 2694 (100 mg/kg/dose) were delivered twice daily via 100 µL SC injections for 8 d. Treatment with AMK (150 mg/kg/dose; Sigma) was delivered once daily via 100 µL SC injections for 8 d. All compounds were prepared in Plasmalyte. Six mice from each group (untreated, 2593, 2694, AMK) were then killed 2 d after administration of the final dose and bacterial loads in the whole lungs and spleens were determined.
Chronic model of Mab infection.
The chronic model (also known as prolonged acute model) of efficacy against Mab was performed similarly as described above with several changes. Mice were divided into five groups containing 3 or 6 mice each and infected with a tail vein injection of 100 µL containing 1 × 106 CFU of Mab subsp. abscessus 1513, a virulent strain previously used in the chronic SCID treatment mouse model (66). Three mice were killed at day 1 and 27 post infection to determine bacteria uptake in whole lungs and livers (limit of detection: Log10 1.7 CFU/organ). Therapy or no drug control was initiated on the other three groups at 28 d postinfection. Treatment with 2593 (30 mg/kg/dose) was delivered twice daily via 100 µL SC injections and continued for 28 d. Treatment with AMK (150 mg/kg/dose) was delivered once daily via 100 µL SC injections for 28 d. Six mice from each group (untreated, 2593, AMK) were killed 5 d after administration of the final dose and bacterial loads in the whole lungs, and spleens were determined.
Statistical analysis.
Bacterial burdens in animal models in the untreated vs. compound-treated animal organs were analyzed using GraphPad Prism version 9.5.0 (GraphPad software, San Diego, CA), using ANOVA and Dunnett multiple comparison test. Data were presented using mean ± SEM (n = 6). Significance was considered below a P value of <0.05. Schematic in Fig. 4A was created with BioRender.com.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the NIH (R01-AI157312 to R.E.L.), NIH Ruth L. Kirschstein National Research Service Award (F31-AI169961 to G.A.P.), National Cancer Institute of the NIH (P30-CA021765 to SJCRH), National Institute of General Medical Sciences of the NIH (R01-GM132302 to Y.S.P.), the Illinois State startup funds (to Y.S.P.), the American Lebanese Syrian Associated Charities, and SJCRH. Research in the laboratory of P. Sander is supported by the Swiss NSF (310030_197699), the Federal Office of Public Health (3632001500), the Joint Program Initiative Antimicrobial Resistance (JPIAMR—ACOMa-2022-050), and University of Zurich. We thank the Hartwell Center Core Facility from SJCRH for whole genome and RNA sequencing services. We thank Dr. Diane Ordway and Dr. Anne Lenaerts from Colorado State University, (Fort Collins, CO, USA) for their assistance in the conduct of murine efficacy models supported by NIH and the National Institute of Allergy and Infectious Diseases (Contract No. HHSN272201100009I). We thank Victoria Loudon-Hossler from SJCRH for assistance with the mammalian cytotoxicity assays described. We thank Madison Sluter-Phelps of SJCRH for assistance with scientific editing of this manuscript. We thank Dimitri Shcherbakov, Tanja Valoria, and Klara Haldimann for help with the in vitro translation assays. We thank Kelley Hurst-Hess and Dr. Pallavi Ghosh of Wadsworth Center, New York Department of Health for their critical discussion of this study. We thank the staff at Northeastern Collaborative Access Team (NE-CAT) beamlines 24ID-C and 24ID-E for help with X-ray diffraction data collection, especially Drs. Malcolm Capel, Frank Murphy, Surajit Banerjee, Igor Kourinov, David Neau, Jonathan Schuermann, Narayanasami Sukumar, Anthony Lynch, James Withrow, Kay Perry, Ali Kaya, and Cyndi Salbego. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH (P30-GM124165 to NE-CAT). The Eiger 16M detector on 24-ID-E beamline is funded by an NIH-ORIP HEI grant (S10-OD021527 to NE-CAT). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Author contributions
G.A.P., E.C.B., P. Sander, and R.E.L. designed research; G.A.P., M.N.C., D.M.F., P. Selchow, C.J.M., S.L.W., S.D., J.L., M.M., M.D.M., B.Y.K., P.A.M., S.M.R., L.A.W., S.M.A., L.Y., R.B.L., Z.H.T., P.B.L., and S.N.H. performed research; S.L.W., S.D., S.M.R., and P. Sander contributed new reagents/analytic tools; G.A.P., M.N.C., L.A.W., R.B.L., B.M., Y.S.P., S.N.H., E.C.B., P. Sander, and R.E.L. analyzed data; and G.A.P., B.M., Y.S.P., S.N.H., E.C.B., P. Sander, and R.E.L. wrote the paper.
Competing interests
S.L.W., J.L., and R.E.L. disclose patent filings on this chemical series.
Footnotes
This article is a PNAS Direct Submission. T.D. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
Whole-genome sequencing data associated with this study have been deposited to NCBI SRA under accession number PRJNA865308 (67). Raw and processed transcriptomic sequencing data have been deposited to NCBI GEO under accession number GSE222081 (68). Coordinates and structure factors were deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank with accession codes: 8UVR (69) for the wild-type T. thermophilus 70S ribosome in complex with mRNA, deacylated A-site tRNAPhe, deacylated P-site tRNAiMet, deacylated E-site tRNAPhe, and SPC; 8UVS (70) for the wild-type T. thermophilus 70S ribosome in complex with mRNA, deacylated A-site tRNAPhe, deacylated P-site tRNAiMet, deacylated E-site tRNAPhe, and 2694.
Supporting Information
References
- 1.Gannon A. D., Darch S. E., Same game, different players: Emerging pathogens of the CF lung. MBio 12, e01217-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansen M. D., Herrmann J.-L., Kremer L., Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 18, 392–407 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Winthrop K. L., et al. , Incidence and prevalence of nontuberculous mycobacterial lung disease in a large US managed care health plan, 2008–2015. Ann. Am. Thor. Soc. 17, 178–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daley C. L., et al. , Treatment of nontuberculous mycobacterial pulmonary disease: An official ATS/ERS/ESCMID/IDSA clinical practice guideline. Clin. Infect. Dis. 71, e1–e36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.CDC, Extensively drug-resistant tuberculosis (XDR TB) (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA, 2016). [Google Scholar]
- 6.van Ingen J., Boeree M. J., van Soolingen D., Mouton J. W., Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist. Updat. 15, 149–161 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Hurst-Hess K., Rudra P., Ghosh P., Mycobacterium abscessus WhiB7 regulates a species-specific repertoire of genes to confer extreme antibiotic resistance. Antimicrob. Agents Chemother. 61, e01347-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lilic M., Darst S. A., Campbell E. A., Structural basis of transcriptional activation by the Mycobacterium tuberculosis intrinsic antibiotic-resistance transcription factor WhiB7. Mol. Cell 81, 2875–2886.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pryjma M., Burian J., Kuchinski K., Thompson C. J., Antagonism between front-line antibiotics clarithromycin and amikacin in the treatment of Mycobacterium abscessus infections is mediated by the whiB7 gene. Antimicrob. Agents Chemother. 61, e01353-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee R. E., et al. , Spectinamides: A new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med. 20, 152–158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruhn D. F., et al. , Aminomethyl spectinomycins as therapeutics for drug-resistant respiratory tract and sexually transmitted bacterial infections. Sci. Transl. Med. 7, 288ra275 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dharuman S., et al. , Synthesis, antibacterial action, and ribosome inhibition of deoxyspectinomycins. J. Antibiot. (Tokyo) 74, 381–396 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borovinskaya M. A., Shoji S., Holton J. M., Fredrick K., Cate J. H. D., A steric block in translation caused by the antibiotic spectinomycin. ACS Chem. Biol. 2, 545–552 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson G. T., et al. , Spectinamides are effective partner agents for the treatment of tuberculosis in multiple mouse infection models. J. Antimicrob. Chemother. 72, 770–777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J., et al. , Structure-activity relationships of spectinamide antituberculosis agents: A dissection of ribosomal inhibition and native efflux avoidance contributions. ACS Infect. Dis. 3, 72–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balganesh M., et al. , Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob. Agents Chemother. 56, 2643–2651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurst-Hess K. R., Phelps G. A., Wilt L. A., Lee R. E., Ghosh P., Mab2780c, a TetV-like efflux pump, confers high-level spectinomycin resistance in Mycobacterium abscessus. Tuberculosis 138, 102295 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sander P., et al. , Ribosomal and non-ribosomal resistance to oxazolidinones: Species-specific idiosyncrasy of ribosomal alterations. Mol. Microbiol. 46, 1295–1304 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Mishra S., Ahmed T., Tyagi A., Shi J., Bhushan S., Structures of Mycobacterium smegmatis 70S ribosomes in complex with HPF, tmRNA, and P-tRNA. Sci. Rep. 8, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iverson A., et al. , Efficacy of aminomethyl spectinomycins against complex upper respiratory tract bacterial infections. Antimicrob. Agents Chemother. 63, e02096-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter A. P., et al. , Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407, 340–348 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Davis T. D., Gerry C. J., Tan D. S., General platform for systematic quantitative evaluation of small-molecule permeability in bacteria. ACS Chem. Biol. 9, 2535–2544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richter M. F., et al. , Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim K. H., et al. , Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. U.S.A. 109, 7729–7734 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldenburg M., et al. , TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Guo Q., et al. , Efflux pumps contribute to intrinsic clarithromycin resistance in clinical, Mycobacterium abscessus isolates. Infect. Drug Resist. 13, 447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rominski A., et al. , Elucidation of Mycobacterium abscessus aminoglycoside and capreomycin resistance by targeted deletion of three putative resistance genes. J. Antimicrob. Chemother. 72, 2191–2200 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Twarog N. R., Stewart E., Hammill C. V., Shelat A. A., BRAID: A unifying paradigm for the analysis of combined drug action. Sci. Rep. 6, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramón-García S., et al. , Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob. Agents Chemother. 55, 3861–3869 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruhn D. F., et al. , In vitro and in vivo evaluation of synergism between anti-tubercular spectinamides and non-classical tuberculosis antibiotics. Sci. Rep. 5, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wohlgemuth I., et al. , Translation error clusters induced by aminoglycoside antibiotics. Nat. Commun. 12, 1830 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffith D. E., Brown-Elliott B. A., Benwill J. L., Wallace R. J. Jr., Mycobacterium abscessus. “Pleased to meet you, hope you guess my name...”. Ann. Am. Thorac. Soc. 12, 436–439 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Kim S. Y., et al. , The drug susceptibility profile and inducible resistance to macrolides of Mycobacterium abscessus and Mycobacterium massiliense in Korea. Diagn. Microbiol. Infect. Dis. 81, 107–111 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Li G., et al. , Antimicrobial susceptibility and MIC distribution of 41 drugs against clinical isolates from China and reference strains of nontuberculous mycobacteria. Int. J. Antimicrob. Agents 49, 364–374 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Wallace R. J. Jr., et al. , Genetic basis for clarithromycin resistance among isolates of Mycobacterium chelonae and Mycobacterium abscessus. Antimicrob. Agents Chemother. 40, 1676–1681 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mougari F., et al. , Standardized interpretation of antibiotic susceptibility testing and resistance genotyping for Mycobacterium abscessus with regard to subspecies and erm41 sequevar. J. Antimicrob. Chemother. 71, 2208–2212 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Nichols D., et al. , Pharmacokinetics and fate of 3H-trospectomycin sulphate, a novel aminocyclitol antibiotic, in male and female rats. Xenobiotica 21, 827–837 (1991). [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Juarrero M., et al. , Preclinical evaluation of inhalational spectinamide-1599 therapy against tuberculosis. ACS Infect. Dis. 7, 2850–2863 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schäfle D., et al. , Rifabutin is inactivated by Mycobacterium abscessus Arr. Antimicrobial. Agents Chemother. 65, e02215-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tharra P. R., et al. , Short synthesis of (+)-actinobolin: Simple entry to complex small-molecule inhibitors of protein synthesis. Angew. Chem. Int. Ed. Engl. 61, e202116520 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Selmer M., et al. , Structure of the 70 S ribosome complexed with mRNA and tRNA. Science 313, 1935–1942 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Polikanov Y. S., Blaha G. M., Steitz T. A., How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science 336, 915–918 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polikanov Y. S., Steitz T. A., Innis C. A., A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 21, 787–793 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polikanov Y. S., Melnikov S. V., Söll D., Steitz T. A., Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 22, 342–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khabibullina N. F., et al. , Structure of dirithromycin bound to the bacterial ribosome suggests new ways for rational improvement of macrolides. Antimicrob. Agents Chemother. 63, e02266-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batool Z., Lomakin I. B., Polikanov Y. S., Bunick C. G., Sarecycline interferes with tRNA accommodation and tethers mRNA to the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 117, 20530–20537 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Svetlov M. S., et al. , Structure of Erm-modified 70S ribosome reveals the mechanism of macrolide resistance. Nat. Chem. Biol. 17, 412–420 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Syroegin E. A., Aleksandrova E. V., Polikanov Y. S., Insights into the ribosome function from the structures of non-arrested ribosome–nascent chain complexes. Nat. Chem. 15, 143–153 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Syroegin E. A., Aleksandrova E. V., Polikanov Y. S., Structural basis for the inability of chloramphenicol to inhibit peptide bond formation in the presence of A-site glycine. Nucleic Acids Res. 50, 7669–7679 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Syroegin E. A., et al. , Structural basis for the context-specific action of the classic peptidyl transferase inhibitor chloramphenicol. Nat. Struct. Mol. Biol. 29, 152–161 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitcheltree M. J., et al. , A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 599, 507–512 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paranjpe M. N., et al. , Insights into the molecular mechanism of translation inhibition by the ribosome-targeting antibiotic thermorubin. Nucleic Acids Res. 51, 449–462 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011). [Google Scholar]
- 54.Ewels P., Magnusson M., Lundin S., Kaller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrews S., FastQC: A Quality Control Tool for High Throughput Sequence Data (Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom, 2010). [Google Scholar]
- 56.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koboldt D. C., et al. , VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cingolani P., et al. , A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorvaldsdóttir H., Robinson J. T., Mesirov J. P., Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geddes E. J., Li Z., Hergenrother P. J., An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protocols 16, 4833–4854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y., Smyth G. K., Shi W., FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Obregón-Henao A., et al. , Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob. Agents Chemother. 59, 6904–6912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Selchow P., et al. , Apramycin overcomes the inherent lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob. Agents Chemother. 66, e0151021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Critchley I. A., et al. , In vitro activity and in vivo efficacy against non-tuberculous mycobacteria of SPR719, the active moiety of the novel oral benzimidazole prodrug SPR720. bioRxiv [Preprint] (2022). 10.1101/2022.12.08.519697. Accessed 12 February 2023. [DOI]
- 67.Cheramie M. N., Phelps G. A., Lee R. E., From “Spectinomycin Analogs for the Treatment of Mycobacterium abscessus Infections”. NCBI BSRA. https://www.ncbi.nlm.nih.gov/bioproject/865308. Deposited 2 August 2022.
- 68.Lee R. E., Phelps G. A., Fernando D. M., from Data. “Spectinomycin Analogs for the Treatment of Mycobacterium abscessus Infections”. NCBI GEO. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222081. Deposited 9 January 2023.
- 69.Killam B. Y., Phelps G. A., Lee R. E., Polikanov Y. S., from Data. “Crystal structure of the wild-type Thermus thermophilus 70S ribosome in complex with spectinomycin, mRNA, deacylated A- and E-site tRNAphe, and deacylated P-site tRNAmet at 2.60A resolution”. Protein Data Bank. https://www.rcsb.org/structure/unreleased/8UVR. Deposited 3 November 2023. [Google Scholar]
- 70.Killam B. Y., Phelps G. A., Lee R. E., Polikanov Y. S., from Data. “Crystal structure of the wild-type Thermus thermophilus 70S ribosome in complex with spectinomycin derivative 2694, mRNA, deacylated A- and E-site tRNAphe, and deacylated P-site tRNAmet at 2.75A resolution”. Protein Data Bank. https://www.rcsb.org/structure/unreleased/8UVS. Deposited 3 November 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Whole-genome sequencing data associated with this study have been deposited to NCBI SRA under accession number PRJNA865308 (67). Raw and processed transcriptomic sequencing data have been deposited to NCBI GEO under accession number GSE222081 (68). Coordinates and structure factors were deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank with accession codes: 8UVR (69) for the wild-type T. thermophilus 70S ribosome in complex with mRNA, deacylated A-site tRNAPhe, deacylated P-site tRNAiMet, deacylated E-site tRNAPhe, and SPC; 8UVS (70) for the wild-type T. thermophilus 70S ribosome in complex with mRNA, deacylated A-site tRNAPhe, deacylated P-site tRNAiMet, deacylated E-site tRNAPhe, and 2694.