

Abstract
Laser capture microdissection and mass spectrometry (LCM/MS) is a technique that involves dissection of glomeruli from paraffin-embedded biopsy tissue, followed by digestion of the dissected glomerular proteins by trypsin, and subsequently mass spectrometry to identify and semiquantitate the glomerular proteins. LCM/MS has played a crucial role in the identification of novel types of amyloidosis, biomarker discovery in fibrillary GN, and more recently discovery of novel target antigens in membranous nephropathy (MN). In addition, LCM/MS has also confirmed the role for complement proteins in glomerular diseases, including C3 glomerulopathy. LCM/MS is now widely used as a clinical test and considered the gold standard for diagnosis and typing amyloidosis. For the remaining glomerular diseases, LCM/MS has remained a research tool. In this review, we discuss the usefulness of LCM/MS in other glomerular diseases, particularly MN, deposition diseases, and diseases of complement pathways, and advocate more routine use of LCM/MS at the present time in at least certain diseases, such as MN, for target antigen detection. We also discuss the limitations of LCM/MS, particularly the difficulties faced from moving from a research-based technique to a clinical test. Nonetheless, the role of LCM/MS in glomerular diseases is expanding. Currently, LCM/MS may be used to identify the etiology in certain glomerular diseases, but in the future, LCM/MS can play a valuable role in determining pathways of complement activation, inflammation, and fibrosis.
Keywords: complement, fibrosis, glomerular disease, GN, immunology and pathology
The study of glomerular proteomic profiles using laser capture microdissection and mass spectrometry (LCM/MS) is a relatively new technique used mostly in the research setting. The basic technique involves dissection of glomeruli, trypsinization of the dissected tissue containing glomerular proteins, followed by mass spectrometry (MS) and analysis of the proteins identified. Importantly, paraffin-embedded archival tissue can be used for LCM/MS1 (LCM/MS details are given in the supplement to ref. 1). To study the accumulation of proteins in different glomerular diseases, knowledge of the baseline proteins in normal glomeruli is important because it allows comparison of the proteins in the diseased glomeruli with the normal glomeruli. Using this basic premise, LCM/MS has played a crucial role in the identification of novel markers and etiology in specific glomerular diseases. At the same time, LCM/MS has also helped in understanding the pathogenesis of many glomerular diseases. For example, LCM/MS was first used to detect new types of amyloidosis.2 Subsequently, a novel marker, dna J homolog subfamily B member 9 (DNAJB9), for fibrillary GN was discovered using LCM/MS.3 Recently, LCM/MS was the essential technology used to detect novel antigens in membranous nephropathy (MN).4 Finally, the pathogenesis of C3 glomerulopathy was confirmed by identification of complement factors and complement-regulating proteins in the C3G glomeruli by LCM/MS.5
LCM/MS is valuable not just for accurate diagnosis of the glomerular disease but can also contribute to identification of the disease burden and in identifying the predominant disease pathways (Figure 1). In this report, the role of LCM/MS in the diagnosis of glomerular diseases and in identification of major pathways of disease is briefly discussed. Perhaps it is not too early to suggest that incorporation of LCM/MS findings of at least certain glomerular diseases for both the diagnosis and disease pathways involved could be included in the kidney biopsy report. This would be a tremendous step forward from just a pure histopathologic report.
Figure 1.
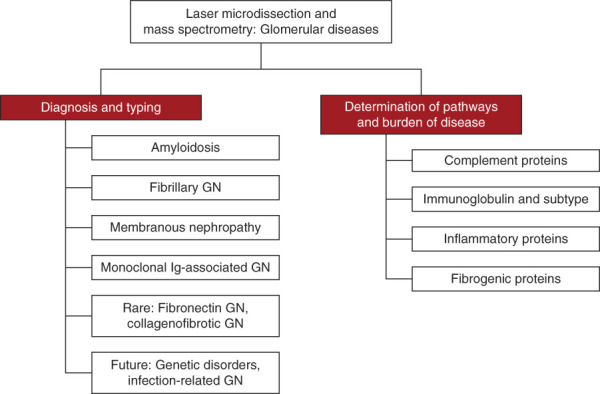
Role of laser microdissection and MS in glomerular diseases. MS, mass spectrometry.
It should be pointed out that not long ago, glomerular diseases, in particular GN, was classified on the basis of patterns of injury, such as mesangial proliferative GN, diffuse endocapillary GN, exudative GN, necrotizing and crescentic GN, membranoproliferative GN, or sclerosing GN. Although the pattern of injury is useful in identifying the severity and chronicity of the GN, it does not accurately confirm the underlying etiology, and dissimilar etiologies can give rise to the same pattern of injury. To provide a more etiology-based diagnosis, the emphasis shifted to the findings on immunofluorescence microscopy. Thus, GN is broadly classified as immune-complex GN, antiglomerular basement membrane (GBM) GN, pauci-immune (anti-ANCA–associated) GN, C3 glomerulopathy, and monoclonal Ig GN on the basis of the findings on immunofluorescence studies.6 Recently, a classification of GN on the basis of immunopathogenesis has also been proposed.7 Immunofluorescence microscopy is extremely helpful in identifying the etiology in many diseases. However, it still leaves a void in the accurate identification of the etiology and pathogenesis of many diseases because immunofluorescence microscopy can identify only a limited number of proteins, namely Igs and complement proteins, such as C3 and C1q. By contrast, LCM/MS identifies hundreds of proteins, including proteins specific to the disease compared with the typical 9–10 proteins usually stained by immunofluorescence (IF).
Recent advancements in LCM/MS include gentle infrared capture along with ultraviolet capture for more delicate handling and preserving morphologic details, improved technology for protein extraction, protein digestion and processing of multiple samples, plus new state-of-the-art mass spectrometers that achieve significant increases in protein identification and quantification of thousands of proteins per glomerular protein extraction. These advancements have made LCM/MS an attractive methodology for routine laboratory use rather than just for research purposes. LCM/MS is a technique that can now be used not just for confirmation of the diagnosis on routine kidney biopsy but also for providing additional data from the kidney biopsy sample to understand the pathophysiology and aid in the management of the kidney disease.
The role of LCM/MS can be broadly divided into identification of (1) diagnosis of glomerular diseases and (2) pathways of glomerular inflammation, complement activation, and fibrosis. These will change as the etiology and more mechanistic pathways are identified in different diseases.
LCM/MS in the Diagnosis of Glomerular Diseases
Amyloidosis
Amyloidosis results from acellular accumulation of Congo red–positive amyloid fibrils. On the basis of the precursor protein, various types of amyloidosis are described, including Ig-light chain (AL) and Ig-heavy chain (AH) amyloidosis; serum amyloid A (AA) amyloidosis; leukocyte chemotactic factor 2 amyloidosis; fibrinogen α-chain (AFib) amyloidosis; gelsolin amyloidosis; transthyretin amyloidosis; lysozyme amyloidosis; and apolipoproteins I, II, IV and C-II amyloidosis. LCM/MS played an important role in not just the diagnosis but in the identification of many of these novel types of amyloidosis.8–14 The diagnosis and identification of novel types of amyloidosis by LCM/MS is based on the finding of the biochemical amyloid signature. The amyloid signature includes serum amyloid P component, apolipoprotein E (APOE), and the amyloidogenic protein. The amyloidogenic protein includes detection of any of the known amyloidogenic proteins, such as Ig-light chain, leukocyte chemotactic factor 2 protein, and fibrinogen α-chain, which then dictates the amyloid type.
The accurate diagnosis and classification of amyloidosis is of paramount importance because treatment and prognosis is dependent on the amyloid type. In fact, LCM/MS is now routinely used in clinical practice and considered the gold standard methodology for the diagnosis and typing of amyloidosis. Furthermore, LCM/MS can be used to detect known amino acid substitutions in patients with hereditary amyloidosis, thus expanding the discovery capability of LCM/MS.15 A recent study of 16,175 cases of amyloid typing in all organs by LCM/MS identified 22 different types of amyloids. The kidney was the second most common organ studied after the heart. AL-, ALECT2-, AA-, AH-, AFIB-, and AApoAIV amyloidosis were among the most common amyloid types detected in the kidney.15 Figure 2A shows the detection of the various amyloid types in the kidney. Note the presence of the amyloid signature proteins APOE and serum amyloid P component in all cases along with the individual amyloidogenic protein. Furthermore, as shown in Figure 2A, in patients with fibrinogen α-chain, transthyretin, and gelsolin amyloidosis, LCM/MS detected R573L mutation peptide, V122I mutation peptide, and N211K mutation peptide, respectively. Figure 2, B and C shows MS detection of the mutation in fibrinogen α-chain, and Figure 2, D and E shows MS detection of the mutation in transthyretin amyloidosis.
Figure 2.
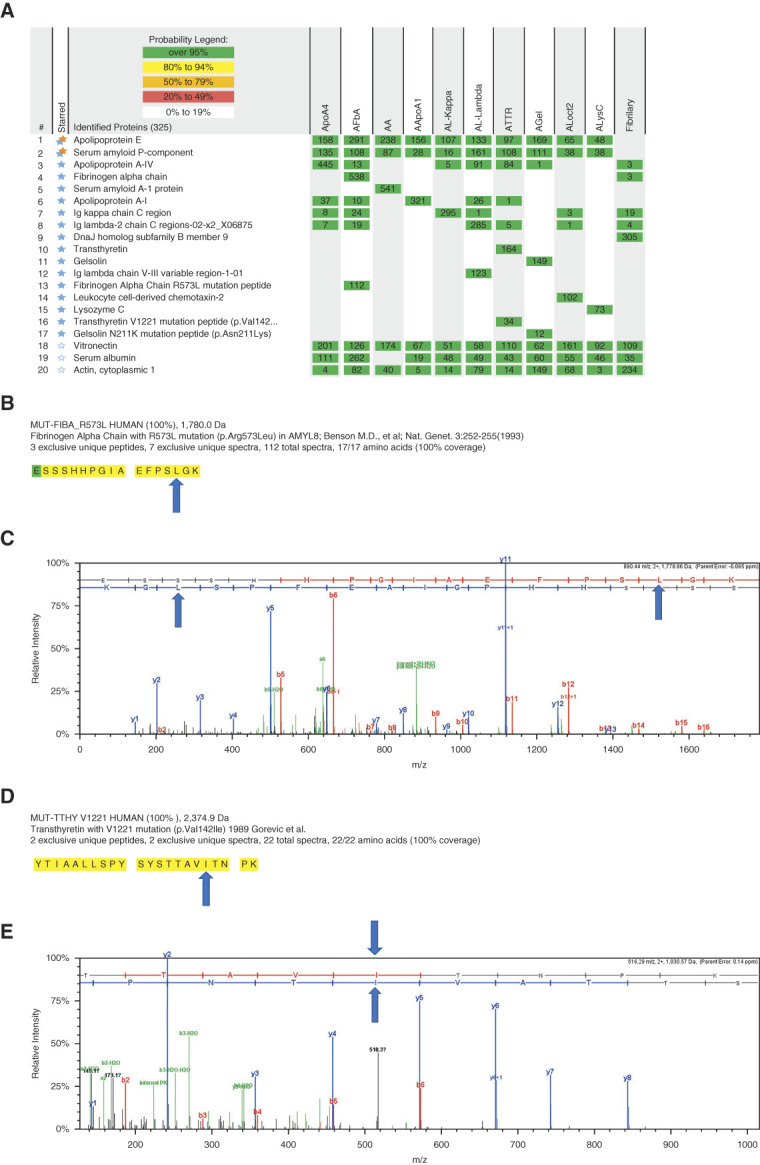
MS for amyloidosis and fibrillary GN. (A) Proteins indicated by double stars (blue and orange stars) are the amyloid signature proteins. Disease-specific proteins are indicated by blue stars. Numbers in the boxes represent the total number of spectra matched to the protein in a sample. Mascot and XTandem search engines performed the database searches against Swissprot human fasta files, and Scaffold (version Scaffold_4.8.3, Proteome Software Inc., Portland, OR) is used to compile the MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability by the Scaffold algorithm and contained at least two identified peptides. The spectra value indicates the total number of mass spectrum collected on the mass spectrometer and matched to the protein using the proteomics software. A higher number of mass spectra is indicative of greater abundance and will typically yield greater amino acid sequence coverage. A higher mass spectra value also indicates a higher confidence in the protein identification. (B) Portions of the fibrinogen alpha mutation sequence detected in case 2 are highlighted in bold black letters on a yellow background. Residues in green signify amino acids with artifactual chemical modifications (such as oxidation of methionine) that are induced by MS sample processing. (C) Tandem mass spectra of the fibrinogen alpha p.Arg573Leu mutant peptide detected in case 2 is shown. Blue arrows indicate the amino acid substitution. (D) Portions of the transthyretin mutation sequence detected in case 7 are highlighted in bold black letters on a yellow background, indicating complete coverage of the mutation containing peptide. (E) Tandem mass spectra of the transthyretin p.Val142Ile mutant peptide detected in case 7 is shown. Blue arrows indicate the amino acid substitution. Eleven individual cases are shown, ApoA4 amyloidosis is case 1, fibrinogen-a amyloidosis (AFbA) is case 2, AA amyloidosis is case 3, ApoA1 amyloidosis is case 4, AL-kappa amyloidosis is case 5, AL-lambda amyloidosis is case 6, transthyretin (ATTR) amyloidosis is case 7, gelsolin (AGel) amyloidosis is case 8, leukoycte chemotactic factor 2 (Alect2) amyloidosis is case 9, lysozyme (AlysC) amyloidosis is case 10, and fibrillary glomerulnephritis is case 11.
Fibrillary GN
Fibrillary GN is a rare disease characterized by accumulation of Congo red–negative fibrillary deposits. Until recently, the diagnosis of fibrillary GN was primarily made on the detection of fibrillary deposits that were Congo red–negative. The use of LCM/MS was instrumental in discovering a novel biomarker DNAJB9 in fibrillary GN.3 Studies show that DNAJB9 is a specific biomarker for fibrillary GN, and detection of DNAJB9 by LCM/MS (or immunohistochemistry [IHC]) is extremely helpful not just in the diagnosis but also in distinguishing fibrillary GN from amyloidosis or other biopsies with fibrillar deposits. Figure 2A shows a case of fibrillary GN (fibrillary) showing high total spectral counts of DNAJB9. Note the absence of APOE and serum amyloid P component, thus clearly distinguishing fibrillary GN from all types of amyloidosis.
MN
MN is a common cause of nephrotic syndrome resulting from the deposition of antigen–antibody complexes along the GBMs. M-type phospholipase A2 receptor (PLA2R) in 2009 and thrombospondin type-1 domain-containing 7A (THSD7A) in 2014 were identified by MS as target antigens in a subset of MN.16,17 Both PLA2R and THSD7A account for approximately 50%–60% of all MNs, including primary and secondary MN, while the antigen(s) in remaining MNs was largely unknown.18,19 LCM/MS has since revolutionized the methodology for identification of the remaining antigens. The basic premise of discovery of a new antigen is similar to the detection of novel types of amyloidosis and biomarker DNAJB9 discovery in fibrillary GN, i.e., identification of novel/unique protein in a subset of MN that is absent in other MNs and controls. Confirmation of the antigen is then obtained by localization of the antigen along the GBM by IHC and confocal microscopy, detection of a bound antibody to the antigen in the tissue, and identification of the antibody in serum.
Using LCM/MS, over the past 4 years, at least eight new antigens/biomarkers have been detected with distinctive clinicopathologic findings.4 The first putative antigen detected by LCM/MS in MN was exostosin 1 and 2 (EXT1/EXT-2).1 Most importantly, EXT1/EXT-2 were present in MN patients with an autoimmune disease, such as lupus, mixed connective tissue disease, and Sjogren syndrome. Furthermore, it was recently shown that EXT1/EXT2-positive membranous lupus nephritis has better chronicity histologic indexes and kidney function outcomes compared with EXT1/EXT2-negative membranous lupus nephritis, regardless of whether proliferative features are present.20 Thus, these findings suggest that EXT1/EXT2-positive MN represents a distinct type of MN. Using LCM/MS, other new antigens have been recently discovered. These include neural epidermal growth factor like-1 protein, semaphorin 3B, protocadherin 7, neural cell adhesion molecule 1, serine protease HTRA1, protocadherin FAT1, neuron-derived neurotrophic factor, and recently proprotein convertase subtilisin/kexin type 6.21–28 The detection of these new antigens and distinctive clinicopathologic findings argue that each one represents a separate disease entity and suggest that MN may not be a specific disease but rather a pattern of injury of distinct diseases.29
Currently, at least 14 antigens have been identified in MN, and as the list of MN antigens expands, it will be difficult to test for individual antigens using IHC, especially because some of the antigens are rare and only specialized laboratories will be able to offer the IHC tests.30,31 Thus, LCM/MS is the most logical and alternative test to correctly diagnose and classify MN. LCM/MS offers advantages in that it is a one-stop test to identify the MN antigen, even if extremely rare, rather than performing multiple IHC tests for individual antigens (Figure 3A). It is also less prone to interpretation error and is a likely more sensitive test than IHC. In fact, we have identified PLA2R-positive MN on the basis of LCM/MS while they were negative on routine IF for PLA2R (data not shown).
Figure 3.

Antigens in MN. (A) IHC for individual antigens shows positive granular staining along the GBMs for (a) EXT1, (b) EXT2, (c) NELL1, (d) SEMA3B, (e) PCDH7, and (f) FAT1. (B) MS for detection of the antigen in MN. Each antigen is mutually exclusive and shows high spectral counts in the specific MN. Case 1 represents PLA2R-associated MN, case 2 represents THSD7A-associated MN, case 3 represents EXT1/EXT2-associated MN, case 4 represents NELL1-associated MN, case 5 represents SEMA3B-associated MN, case 6 represents PCDH7-associated MN, case 7 represents FAT1-associated MN, and case 8 represents NCAM1-associated MN. Criteria for identification and description of spectral counts similar to Figure 2A. EXT, exostosin; GBM, glomerular basement membrane; IHC, immunohistochemistry; MN, membranous nephropathy; NCAM1, neural cell adhesion molecule 1; NELL1, neural epidermal growth factor like-1 protein; PCDH7, protocadherin 7; PLA2R, phospholipase A2 receptor; SEMA3B, semaphorin 3B; THSD7A, thrombospondin type-1 domain-containing 7A.
Figure 3A shows individual IHC for antigens, and Figure 3B shows representative LCM/MS findings in eight patients with MN with detection of different individual antigens. High total spectral counts of individual antigens are present in each case. On the basis of LCM/MS, the antigens are mutually exclusive, and we have not found any case of double positives. Baseline spectral counts of PLA2R and THSD7A may be present.
Monoclonal Ig–Associated Diseases
Detection of heavy and light chains, typically specific constant regions by LCM/MS, can be used to confirm the monoclonality of the immune deposits. Monoclonal Ig–associated diseases include AL and AH amyloidosis (discussed above); proliferative GN with monoclonal Ig deposits; monoclonal Ig–deposition disease; and less common diseases, such as crystal-storing histiocytosis, light chain proximal tubulopathy, and light chain crystalglobulinemia.32 Routine immunofluorescence studies are geared toward identification of monoclonal Ig by either light chain or, less commonly, heavy chain restriction. In addition, IgG subtyping can confirm the monoclonal IgG, and immunofluorescence studies after protease (pronase) digestion can help unmask hidden monoclonal Ig deposits.33–35 Yet, in certain instances, LCM/MS can be extremely useful to confirm the monoclonal Ig when routine IF is negative or is equivocal. (1) Routine IF does not detect monoclonal IgD and IgE deposits. Thus, rare cases of monoclonal Ig–deposition disease or amyloidosis resulting from deposition of monotypic IgD or IgE can be confirmed by LCM/MS.36 (2) In cases with truncated light or heavy chains, routine IF may not detect light chains or heavy chains; LCM/MS can accurately detect the truncated chain; and (3) IF may be negative in some cases of light chain deposition disease because of nonrecognition of the light chain epitopes or binding/masking of the light chain epitopes to matrix proteins. In such cases, identification of specific light chain Vk subgroups, such as Vk4, Vk2–28, or Vk3–20 subgroup, may help in confirmation of monotypic light chains.32
Rare Deposition Diseases: Fibronectin and Type III Collagen Glomerulopathy
Type III collagen glomerulopathy, also known as collagenofibrotic glomerulopathy (CG), results from glomerular accumulation of collagen type III. Type III collagen is not a normal constituent of glomerular matrix proteins, and its accumulation results in progressive sclerosing glomerulopathy and end stage kidney disease. Kidney biopsy shows mesangial expansion and glomerular wall thickening, including double-contour formation. The diagnosis is based on electron microscopy findings of subendothelial and mesangial accumulation of collagen fibrils; IHC with anti-collagen type III antibodies can be used to confirm the diagnosis.37,38 However, the diagnosis can be difficult during the early phase when deposition of collagen fibrils is not that abundant and may be confused with thrombotic microangiopathy.
Fibronectin glomerulopathy is an autosomal dominant disease and has been shown to be associated with mutations in FN1.39 Fibronectin glomerulopathy is characterized by deposition of fibronectin in the glomeruli, resulting in a light microscopy image similar to CG with mesangial expansion and variable thickening of the GBMs. Electron microscopy shows mesangial and subendothelial deposits with a focal fibrillar substructure, the diagnosis may be missed, and IHC is required using anti-fibronectin antibodies to confirm the diagnosis.
Both CG and fibronectin glomerulopathy are difficult diagnoses and need specialized IHC techniques for interpretation and confirmation of the diagnosis. IHC staining for both collagen III and fibronectin can be equivocal and difficult to interpret. On the other hand, LCM/MS can easily detect high spectral counts of either collagen type III or fibronectin in CG and fibronectin glomerulopathy, respectively (Figure 4). Interestingly, LCM/MS also shows accumulation of fibulin-1 in fibronectin glomerulopathy and periostin in CG, suggesting a proteomic signature for the two entities (unpublished data). Further studies are required to determine the role of fibulin-1 and periostin in these diseases. Both are markers of fibrosis (discussed below).
Figure 4.

Fibronectin and type III collagen glomerulopathy. Top panel shows very high total spectral counts of fibronectin in a case of fibronectin glomerulopathy and collagen alpha type III (samples run in duplicate). The bottom panel shows the EM findings of (A) fibrillar deposits in fibronectin glomerulopathy and (B) collagen fibrils in type III collagen glomerulopathy. Note the presence of fibulin-1 in fibronectin glomerulopathy and periostin in type III collagen glomerulopathy.
LCM/MS to Determine Pathways and Burden of Disease
Complement Pathways
Complement plays an important role in the pathogenesis of GN. Although the underlying etiology of GN might be different, complement activation with subsequent glomerular deposition of complement proteins results in glomerular injury and progression of the lesions. Routine immunofluorescence microscopy includes staining for only complement factors C3c and C1q. Therefore, for evaluation of the complement pathways, routine kidney biopsy provides only limited information.
LCM/MS can provide valuable information regarding both pathways of complement involved and the burden (semiquantitative) of complement proteins in the glomeruli. Using LCM/MS, studies have shown accumulation of complement proteins and pathways involved in C3 glomerulopathy, MN, and ANCA vasculitis.5,40–44 Preliminary data of glomerular diseases using LCM/MS show distinct complement pathways and burden of complement proteins, including complement-regulating proteins.45 LCM/MS studies show that C3 followed by C9 are the most abundant complement proteins in GN, indicating activation of classical/lectin/alternative and terminal pathways, either dominant or a combination of pathways. Furthermore, depending on the type of GN, C4A and/or C4B-dependent pathways were identified. Thus, MN, fibrillary GN, and infection-related GN showed C4A-dominant pathways while lupus nephritis, proliferative GN with monoclonal Ig deposits, monoclonal Ig–deposition disease, and immunotactoid glomerulopathy show C4B-dominant pathways. Significant deposition of complement regulatory proteins FHR1 and FHR5 was also present depending on the type of GN. To give an example of the data that LCM/MS can provide, Figure 5 shows the heat map of key complement proteins in C3GN and dense deposit disease (DDD) compared with controls. C3, C5, C6, C7, C8A, C8B, C9, CHR1, and complement factor H–related protein 5 are most significant proteins in C3GN and DDD compared with controls (Figure 5, A and B). Despite the abundance of these proteins and overlap of proteins (Figure 5C), there are significant differences in the amount of complement proteins that accumulate in each case, thus pointing to the heterogeneity in the complement profile of each case. Furthermore, accumulation of active versus inactive (or entrapped) complement proteins can be inferred by careful evaluation of amino acids belonging to corresponding complement fragments. Thus, the absence of amino acids belonging to C3a in C3 indicates activation of C3 into C3b, and the absence of amino acids belonging to C3f indicates further cleavage of C3b into iC3b.42
Figure 5.

Heat map showing most abundant proteins in C3GN and DDD compared with controls. (A) CFHR1, C7, C6, C8B, CFHR5, C8A, SPP2, C9, and C3 are the most significant proteins in C3GN compared with controls, although the profile in each case is variable. (B) C6, C5, C8B, C7, C9, C8A, CFHR1, APOE, VTN, C3, CLU, and CFH are the most significant proteins in DDD compared with controls, although the profile in individual case is variable. (C) PCA shows overlap of overall proteins detected in C3GN and DDD. APOE, apolipoprotein E, CFHR1, complement factor H–related protein 1; CFHR5, complement factor H–related protein 5; CFH, complement factor H; CLU, clusterin; DDD, dense deposit disease; PCA, principal component analysis; SPP2, secreted phosphoprotein 2; VTN, vitronectin.
Recently, drugs that inhibit C5 have been used to treat a variety of glomerular diseases, in particular atypical hemolytic uremic syndrome. Kidney biopsy staining for C5b-9 has been used as a surrogate marker for use of C5-blocking drugs in atypical hemolytic uremic syndrome.46,47 The staining for C5b-9 has inherent difficulties in interpretation, and the technique is difficult to duplicate in laboratories. LCM/MS is another methodology that can provide information about accumulation of not just C5b-9 but also other proteins, including C3, and complement-regulating factors, such as complement factor H, CFB, CHFR1-5, and CD46. Finally, many complement-inhibiting drugs are in the pipeline for GN, including lupus nephritis, IgA nephropathy, ANCA vasculitis, and C3 glomerulopathy. LCM/MS on kidney biopsy specimens will be able provide important adjunct data on the glomerular complement profile in each case and possibly even predict the complement pathway that needs to be targeted and the probability of successful treatment by anti-complement drugs.
Ig Profile
Many types of GNs are mediated by immune complexes or Igs. These include IgA nephropathy, lupus nephritis, infection-related GN, monoclonal Ig–associated GN, cryoglobulinemic GN, and even ANCA-associated GN and anti-GBM GN. Noninflammatory diseases, including MN, are also mediated by autoantibodies (IgG) to target MN antigens. The IgG profile, including IgG subtypes, and semiquantitative estimation of Ig can done by LCM/MS. The IgG profile using LCM/MS is well-studied in different types of antigen-associated MNs and can be replicated in other forms of Ig-associated glomerular diseases. Furthermore, LCM/MS can also estimate the burden of immune deposits. Thus, it will be possible to estimate the amount of glomerular IgA in IgA nephropathy or amount of glomerular IgG (or other Igs) in a patient with lupus nephritis.
Inflammatory Markers
Recently, LCM/MS was used to compare inflamed (crescentic) versus normal-appearing glomeruli in ANCA-myeloperoxidase vasculitis. The authors found that in addition to complement proteins, such as C3, C5, C7, and C9 in crescentic glomeruli, there was also a significant increase in expression of actinin 4, laminin subunit 2, fibrinogen a, fibrillin 1, agrin, nidogen-2, and heat shock protein 90 in crescentic glomeruli compared with normal glomeruli. What was intriguing about the study was that there was an increase in expression of desmoplakin, protein S100, serpin B3, and thioredoxin in normal glomeruli compared with crescentic glomeruli, which may protect the glomeruli from developing crescents/necrosis.44 Recently, our group performed proteomic analysis in models of AKI, including coronavirus infection and sepsis, and showed proteins of different signaling pathways, including thesirtuin, ceramide, necroptosis, synaptic, oxidative, mammalian target of rapamycin, and NFRF2 signaling pathways.48 These types of studies portend a role of LCM/MS in identifying both inflammatory pathways and also protective biomarkers in GN.
Chronicity Markers
Similar to inflammatory markers, increased expression of structural proteins, such as collagens, vimentin, laminins, elastins, and fibronectin, indicate fibrogenic pathways. Furthermore, accumulation of heparan sulfates, such as agrin and nidogen-2, indicate GBM remodeling. Accumulation of matrix proteins, including tenascin C, fibrillin 1, periostin, and others, indicate fibrogenic niche.49 Indeed, we have found high spectral counts of tenascin C, fibrillin 1, and periostin in sclerosed glomeruli (unpublished data, on the basis of LCM/MS of MN). Although not yet studied by LCM/MS, proteins, such as PDGF, TGFβ, IL-6, plasminogen activator receptor, and others, can help determine the key pathways of glomerulosclerosis in different types of GN.49,50
Limitations of LCM/MS
There are two categories of limitation. The first one is related to the LCM/MS technology, and the second one is related to the kidney disease entities.
Limitations on Bringing a Research Discovery Test to a Clinical Test
There are a multitude of differences between biomarker discovery and clinical proteomics platforms. Owing to the unknown nature of the biomarker discovery experiments, LCM/MS for discovery is typically focused on maximizing sensitivity and accentuating sample composition differences using techniques that are not clinically viable. This begins at the microtomy. Thicker tissue sections (10 µM) may be used to increase the protein amount used for discovery experiments. By contrast, clinical proteomics methods are configured to detect known biomarkers, and hence, they start with 4–6-µm thick tissue sections to minimize the tissue usage of limited patient biopsies, which is a significant concern for renal biopsies. Likewise, the laser microdissection area is typically larger for discovery experiments, with some samples containing 500,000–1 million µm2 of glomerular area. This large tissue area enhances the ability to detect unknown biomarkers in a discovery setting. However, obtaining such large volumes of glomeruli is unrealistic for clinical assays because cases will require numerous tissue sections to obtain large amounts of tissue, which depletes the biopsies. Many of the renal clinical biopsies will fail to yield such large amounts, thereby making them ineligible for the assay. Thus, the first step of clinical proteomics translation is optimization of the tissue material and protein digestion methods that aim to extract the most signal from a minimal amount of the required tissue.
Next, the mass spectometry instruments used in the biomarker discovery setting are also different, in types and configuration, than instruments that are typically used in most clinical environments. HPLC configurations in the discovery setting typically operate with longer columns (50 cm+), which subsequently require longer HPLC gradients to maximize signal response. By contrast, clinical platforms use shorter columns (15–25 cm) to balance sensitivity and throughput, which in turn reduces clinical costs. Clinical mass spectrometer method settings are also optimized for each assay. To emphasize this, typically, a biomarker discovery MS method is tailored toward detecting low-abundance proteins at the expense of higher abundance peptides by using lower triggering thresholds, larger fill times, and longer dynamic exclusion windows. Conversely, for a clinical assay, the MS instrument methods are optimized to accentuate the intensity differences for the target biomarkers by using more moderate fill time limits, slightly higher triggering thresholds, and shorter dynamic exclusion windows. All of these clinical optimizations are geared toward extracting the most sensitivity from the MS method while spending the least amount of analytical time per patient sample.
In addition, the transition from biomarker discovery to clinical production requires the creation of standardized sample processing controls, MS processes, quality assurance/quality control programs, and standardized reporting thresholds. Biomarker discovery platforms often are not reproducible beyond a batch of samples or certain time period. The coefficient of variability for biomarker discovery platforms is also very high (approximately 25%). Both issues are fundamentally incompatible in a clinical platform. Hence, extensive standardization and quality assurance/quality control programs are developed and instituted for each clinical assay to ensure a low coefficient of variability (approximately 5%) with high reproducibility over years on its end (i.e., <5% variation over ≥3 years).
Finally, a major factor is the cost and expertise required for setting up LCM/MS. Most institutions do not receive a high volume of kidney biopsies, and for specific indications, such as amyloidosis, LCM/MS remains a send-out referral test. However, as the indications for its use increase, there is a greater likelihood that LCM/MS will become available in larger institutions. Data sharing will be extremely important so that laboratories can not only share expertise but also standardize the LCM/MS methodology.
Limitations Based on Disease Entities
LCM/MS is a snapshot of the disease entity at the time of kidney biopsy. Diseases vary not only in that both active lesions and healing/resolved lesions may be present but also that lesions that may be focal and segmental. Thus, certain proteins may be absent or only low spectral counts may be present depending on the phase and extent of the disease process.
LCM/MS can only make semiquantitative assessment of proteins, and the abundance of proteins in question will, therefore, not be the same during all times of the disease process. Even so, studies are required whether the abundance of certain proteins (burden of disease), such as Igs and complement pathway proteins, correlate with outcomes.
LCM/MS does not answer whether the proteins detected are tissue phase-derived (i.e., local production) or are a consequence of the fluid phase/systemic disease process. In addition, small amounts of nonpathogenic proteins may be present in circulation/glomerular capillaries that will be detected by LCM/MS. This is particularly true for monoclonal Ig. In situ MS techniques and a potential correlation with spatial transcriptomics methods may be helpful in resolving this question.
Certain proteins may be common to a variety of glomerular diseases, particularly for inflammation or scarring. There may be a common theme for proteins related to active versus resolving/healed glomerular diseases. Some of these proteins may also represent an epiphenomenon rather than representing the specific pathways involved.
Certain proteins, such as MN target antigens protocadherin 7 and FAT1, are heavily glycosylated and can interfere with trypsin's access and inhibit its binding and cleavage of the arginine and lysine residues in the glycosylated region, resulting in false low spectral counts. False low counts can be inferred by examination of peptides detected in the amino acid sequence of the protein. Thus, examination of the coverage will reveal detection of peptides from nonglycosylated regions of proteins and absence of peptides from the heavily glycosylated regions.25,26
Biopsy Report
We propose that an additional section should be devoted to LCM/MS findings in specific disease entities in which LCM/MS was used for the diagnosis. These include amyloidosis, fibrillary GN, MN, monoclonal Ig–associated kidney diseases, and collagenofibrotic glomerulopathies. The findings should be presented in a logical sequence in the pathology report under additional tests performed. We anticipate, in the future, additional MS data in the biopsy report would include
Proteins detected that point to the etiology (such as detection of the monoclonal Ig or specific amyloid type or MN antigen)
Complement proteins and suggest the pathways involved
Ig profile
Inflammatory pathway proteins
Fibrogenic/chronicity pathway proteins
Future Directions
Infection-related GN: In most cases, the diagnosis is assumed on the basis of the history of infection and presence of GN. Yet, the infection or antigen is typically not demonstrated on the biopsy. Using LCM/MS, it may be possible to detect viral, bacterial, or fungal antigens in the glomeruli of infection-related GN. Clinical information regarding which microorganism is likely involved is critical so that database searches can then be set on the basis of sequences of individual microorganisms.
Glomerular diseases associated with mutations: Similar to various types of amyloidosis where mutations can be detected in some type's of amyloid, it should also be possible to detect mutations in various genetic forms in glomerular diseases, including Fabry and other storage diseases, basement membrane abnormalities (hereditary nephritis), and genetic causes of focal segmental glomerulosclerosis.
Artificial intelligence using MS: On the basis of the protein signatures, artificial intelligence should be able to identify proteins that result in disease and predict the pathways of both inflammation and scarring.
Summary
LCM/MS is a new technique that is mostly used in the research setting. However, its role has expanded beyond research to being an important clinical tool in the diagnosis of a variety of diseases, particularly those associated with unknown glomerular protein accumulation, such as amyloidosis and MN. LCM/MS in the future may also be used to reveal pathways of complement activation, inflammation, and fibrosis.
Acknowledgments
This work was supported in part by funding to SS from the Department of Laboratory Medicine and Pathology at the Mayo Clinic (Scholarly Clinician Award). We acknowledge assistance of Benjamin Madden in the Mayo Clinic Proteomics Core, which is a shared resource of the Mayo Clinic Cancer Center (NCI P30 CA15083).
Disclosures
L.M.P. Palma reports the following: Employer: State University of Campinas (UNICAMP); Consultancy: OrphanDC/Apellis Brazil; Honoraria: Speaker for Alexion Pharma Brazil; Speaker for Apellis; Advisory or Leadership Role: Committee of Pediatric Nephrology of the Brazilian Society of Nephrology and Committee of Pediatric Transplantation of the Brazilian Association of Organ Transplantation; and Speakers Bureau: Speaker for Alexion Pharma Brazil; Speaker for Apellis. S. Sethi reports Consultancy: Novartis; Honoraria: Honorarium for Teaching, Grand Rounds, Lectures, Up to date, reviewing slides for a study for Novartis; Patents or Royalties: Patents for membranous nephropathy antigens; and Speakers Bureau: Reach MD, MJH videos. All remaining authors have nothing to disclose.
Funding
None.
Author Contributions
Conceptualization: Lilian M.P. Palma, Sanjeev Sethi.
Data curation: Benjamin Madden, Sanjeev Sethi, Jason D. Theis.
Formal analysis: Benjamin Madden, Jason D. Theis.
Funding acquisition: Sanjeev Sethi.
Investigation: Sanjeev Sethi.
Methodology: Benjamin Madden, Sanjeev Sethi, Jason D. Theis.
Project administration: Sanjeev Sethi.
Resources: Sanjeev Sethi.
Writing – original draft: Sanjeev Sethi.
Writing – review & editing: Benjamin Madden, Lilian M.P. Palma, Sanjeev Sethi.
References
- 1.Sethi S Madden BJ Debiec H, et al. Exostosin 1/exostosin 2–associated membranous nephropathy. J Am Soc Nephrol. 2019;30(6):1123–1136. doi: 10.1681/ASN.2018080852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957–4959. doi: 10.1182/blood-2009-07-230722 [DOI] [PubMed] [Google Scholar]
- 3.Dasari S Alexander MP Vrana JA, et al. DnaJ heat shock protein family B member 9 is a novel biomarker for fibrillary GN. J Am Soc Nephrol. 2018;29(1):51–56. doi: 10.1681/ASN.2017030306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sethi S. New ‘antigens’ in membranous nephropathy. J Am Soc Nephrol. 2021;32(2):268–278. doi: 10.1681/ASN.2020071082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sethi S Gamez JD Vrana JA, et al. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int. 2009;75(9):952–960. doi: 10.1038/ki.2008.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sethi S Haas M Markowitz GS, et al. Mayo clinic/renal pathology society consensus report on pathologic classification, diagnosis, and reporting of GN. J Am Soc Nephrol. 2016;27(5):1278–1287. doi: 10.1681/ASN.2015060612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders HJ, Kitching AR, Leung N, Romagnani P. Glomerulonephritis: immunopathogenesis and immunotherapy. Nat Rev Immunol. 2023;23(7):453–471. doi: 10.1038/s41577-022-00816-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sethi S Theis JD Leung N, et al. Mass spectrometry based proteomic diagnosis of renal immunoglobulin heavy chain amyloidosis. Clin J Am Soc Nephrol. 2010;5(12):2180–2187. doi: 10.2215/cjn.02890310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sethi S Theis JD Shiller SM, et al. Medullary amyloidosis associated with apolipoprotein A-IV deposition. Kidney Int. 2012;81(2):201–206. doi: 10.1038/ki.2011.316 [DOI] [PubMed] [Google Scholar]
- 10.Nasr SH Dasari S Hasadsri L, et al. Novel type of renal amyloidosis derived from apolipoprotein-CII. J Am Soc Nephrol. 2017;28(2):439–445. doi: 10.1681/ASN.2015111228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sethi S Dasari S Amin MS, et al. Clinical, biopsy, and mass spectrometry findings of renal gelsolin amyloidosis. Kidney Int. 2017;91(4):964–971. doi: 10.1016/j.kint.2016.11.017 [DOI] [PubMed] [Google Scholar]
- 12.Sethi S Theis JD Quint P, et al. Renal amyloidosis associated with a novel sequence variant of gelsolin. Am J Kidney Dis. 2013;61(1):161–166. doi: 10.1053/j.ajkd.2012.07.016 [DOI] [PubMed] [Google Scholar]
- 13.Sethi S, Theis JD. Pathology and diagnosis of renal non-AL amyloidosis. J Nephrol. 2018;31(3):343–350. doi: 10.1007/s40620-017-0426-6 [DOI] [PubMed] [Google Scholar]
- 14.Nasr SH Dasari S Mills JR, et al. Hereditary lysozyme amyloidosis variant p.Leu102Ser associates with unique phenotype. J Am Soc Nephrol. 2017;28(2):431–438. doi: 10.1681/ASN.2016090951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dasari S Theis JD Vrana JA, et al. Amyloid typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. Mayo Clinic Proc. 2020;95(9):1852–1864. doi: 10.1016/j.mayocp.2020.06.029 [DOI] [PubMed] [Google Scholar]
- 16.Beck LH Bonegio RGB Lambeau G, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. New Engl J Med. 2009;361(1):11–21. doi: 10.1056/nejmoa0810457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomas NM Beck LH Meyer-Schwesinger C, et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. New Engl J Med. 2014;371(24):2277–2287. doi: 10.1056/nejmoa1409354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol. 2017;12(6):983–997. doi: 10.2215/cjn.11761116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronco P Beck L Debiec H, et al. Membranous nephropathy. Nat Rev Dis Primers 2021;7(1):69. doi: 10.1038/s41572-021-00303-z [DOI] [PubMed] [Google Scholar]
- 20.Ravindran A Casal Moura M Fervenza FC, et al. In patients with membranous lupus nephritis, exostosin-positivity and exostosin-negativity represent two different phenotypes. J Am Soc Nephrol. 2021;32(3):695–706. doi: 10.1681/ASN.2020081181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sethi S Debiec H Madden B, et al. Semaphorin 3B–associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int. 2020;98(5):1253–1264. doi: 10.1016/j.kint.2020.05.030 [DOI] [PubMed] [Google Scholar]
- 22.Sethi S Debiec H Madden B, et al. Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int. 2020;97(1):163–174. doi: 10.1016/j.kint.2019.09.014 [DOI] [PubMed] [Google Scholar]
- 23.Sethi S Madden B Casal Moura M, et al. Membranous nephropathy in syphilis is associated with neuron-derived neurotrophic factor. J Am Soc Nephrol. 2023;34(3):374–384. doi: 10.1681/ASN.0000000000000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sethi S Casal Moura M Madden B, et al. Proprotein convertase subtilisin/kexin type 6 (PCSK6) is a likely antigenic target in membranous nephropathy and nonsteroidal anti-inflammatory drug use. Kidney Int. 2023;104(2):343–352. doi: 10.1016/j.kint.2023.04.006 [DOI] [PubMed] [Google Scholar]
- 25.Sethi S Madden B Casal Moura M, et al. Hematopoietic stem cell transplant-membranous nephropathy is associated with protocadherin FAT1. J Am Soc Nephrol. 2022;33(5):1033–1044. doi: 10.1681/ASN.2021111488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sethi S Madden B Debiec H, et al. Protocadherin 7-associated membranous nephropathy. J Am Soc Nephrol. 2021;32(5):1249–1261. doi: 10.1681/ASN.2020081165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Rabadi LF Caza T Trivin-Avillach C, et al. Serine protease HTRA1 as a novel target antigen in primary membranous nephropathy. J Am Soc Nephrol. 2021;32(7):1666–1681. doi: 10.1681/ASN.2020101395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caza TN Hassen SI Kuperman M, et al. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney Int. 2021;100(1):171–181. doi: 10.1016/j.kint.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sethi S. Membranous nephropathy: a single disease or a pattern of injury resulting from different diseases. Clin Kidney J. 2021;14(10):2166–2169. doi: 10.1093/ckj/sfab069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sethi S, Madden B. Mapping antigens of membranous nephropathy: almost there. Kidney Int. 2023;103(3):469–472. doi: 10.1016/j.kint.2023.01.003 [DOI] [PubMed] [Google Scholar]
- 31.Caza T Storey A Hassen S, et al. Discovery of seven novel putative antigens in membranous nephropathy and membranous lupus nephritis identified by mass spectrometry. Kidney Int. 2023;103(3):593–606. doi: 10.1016/j.kint.2023.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sethi S, Rajkumar SV, D’Agati VD. The complexity and heterogeneity of monoclonal immunoglobulin–associated renal diseases. J Am Soc Nephrol. 2018;29(7):1810–1823. doi: 10.1681/ASN.2017121319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasr SH, Galgano SJ, Markowitz GS, Stokes MB, D'Agati VD. Immunofluorescence on pronase-digested paraffin sections: a valuable salvage technique for renal biopsies. Kidney Int. 2006;70(12):2148–2151. doi: 10.1038/sj.ki.5001990 [DOI] [PubMed] [Google Scholar]
- 34.Larsen CP Messias NC Walker PD, et al. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. 2015;88(4):867–873. doi: 10.1038/ki.2015.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen CP Ambuzs JM Bonsib SM, et al. Membranous-like glomerulopathy with masked IgG kappa deposits. Kidney Int. 2014;86(1):154–161. doi: 10.1038/ki.2013.548 [DOI] [PubMed] [Google Scholar]
- 36.Royal V Quint P Leblanc M, et al. IgD heavy-chain deposition disease: detection by laser microdissection and mass spectrometry. J Am Soc Nephrol. 2015;26(4):784–790. doi: 10.1681/ASN.2014050481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen AH. Collagen type III glomerulopathies. Adv Chronic Kidney Dis. 2012;19(2):101–106. doi: 10.1053/j.ackd.2012.02.017 [DOI] [PubMed] [Google Scholar]
- 38.Duggal R, Nada R, Rayat CS, Rane SU, Sakhuja V, Joshi K. Collagenofibrotic glomerulopathy-a review. Clin Kidney J. 2012;5(1):7–12. doi: 10.1093/ndtplus/sfr144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castelletti F Donadelli R Banterla F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A. 2008;105(7):2538–2543. doi: 10.1073/pnas.0707730105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sethi S Fervenza FC Zhang Y, et al. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82(4):465–473. doi: 10.1038/ki.2012.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ravindran A Madden B Charlesworth MC, et al. Proteomic analysis of complement proteins in membranous nephropathy. Kidney Int Rep. 2020;5(5):618–626. doi: 10.1016/j.ekir.2020.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sethi S Vrana JA Fervenza FC, et al. Characterization of C3 in C3 glomerulopathy. Nephrol Dial Transplant. 2017;32(3):459–465. doi: 10.1093/ndt/gfw290 [DOI] [PubMed] [Google Scholar]
- 43.Sethi S Zand L De Vriese AS, et al. Complement activation in pauci-immune necrotizing and crescentic glomerulonephritis: results of a proteomic analysis. Nephrol Dial Transplant. 2017;32(suppl 1):i139–i145. doi: 10.1093/ndt/gfw299 [DOI] [PubMed] [Google Scholar]
- 44.Sethi A, Grande J, Specks U, Fervenza FC. Proteomic profile of uninvolved versus crescentic glomeruli in MPO-ANCA-associated vasculitis. Clin Kidney J. 2023;16(7):1180–1182. doi: 10.1093/ckj/sfad030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sethi S, Palma LMP, Theis JD, Fervenza FC. Proteomic analysis of complement proteins in glomerular diseases. Kidney Int Rep. 2023;8(4):827–836. doi: 10.1016/j.ekir.2023.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galbusera M Noris M Gastoldi S, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis. 2019;74(1):56–72. doi: 10.1053/j.ajkd.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 47.Noris M, Remuzzi G. Terminal complement effectors in atypical hemolytic uremic syndrome: C5a, C5b-9, or a bit of both? Kidney Int. 2019;96(1):13–15. doi: 10.1016/j.kint.2019.02.038 [DOI] [PubMed] [Google Scholar]
- 48.Alexander MP Mangalaparthi KK Madugundu AK, et al. Acute kidney injury in severe COVID-19 has similarities to sepsis-associated kidney injury: a multi-omics study. Mayo Clinic Proc. 2021;96(10):2561–2575. doi: 10.1016/j.mayocp.2021.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. 2022;18(9):545–557. doi: 10.1038/s41581-022-00590-z [DOI] [PubMed] [Google Scholar]
- 50.Sarnak MJ Katz R Ix JH, et al. Plasma biomarkers as risk factors for incident CKD. Kidney Int Rep. 2022;7(7):1493–1501. doi: 10.1016/j.ekir.2022.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]