

Abstract
Classical histone deacetylases (HDACs) are enzymes that can hydrolytically cleave acetyl-Lys in histones and other properties and serve as established drug targets in some forms of cancer. Class I HDACs 1–3 typically exist in a range of multiprotein complexes inside cells and show distinct biological functions in modulating gene expression. In recent years it has become possible to purify and analyze the structure and enzymatic properties of several of these HDAC complexes including CoREST, MiDAC, NuRD, Sin3, SMRT, MIER, and RERE. Here we summarize what is experimentally established and or computationally predicted about the structure of these complexes and also describe their particular catalytic activities and site-specificities with modified nucleosome substrates.
Reversible Lys acetylation on histones and other proteins has emerged as a major post-translational modification (PTM) in epigenetics, gene regulation, and control of cell growth [1,2]. The enzymes that attach the acetyl group to proteins, Lys acetyltransferases (KATs), include the important small families: p300/CBP, PCAF/GCN5, and the MYST group [3]. The Lys deacetylases (KDACs) erase the acetyl-Lys PTMs and include 18 known enzymes in humans [3]. Eleven of these KDACs are Zn-dependent metallohydrolases, and have been called the histone deacetylases, HDAC1-HDAC11 [3]. These enzymes are sometimes known as classical HDACs and were molecularly identified in 1996 [4]. The other seven KDACs are known as the sirtuins and include Sirt1-Sirt7 were discovered several years later [4]. Sirtuins utilize an unusual chemical mechanism in which an NAD co-substrate, often involved in redox transformations, undergoes an attack by the acetamide carbonyl oxygen of acetyl-Lys [3]. The net result of the sirtuin reactions is the formation of O-acetyl-ADP-ribose and the unacetylated Lys residue [3].
Of the 11 classical HDACs, these have been subdivided into several classes [3,5]. The Class I HDACs are comprised of HDAC1, HDAC2, HDAC3, and HDAC8. HDAC1-HDAC3 are almost exclusively found in multiprotein complexes and are localized principally to the cell nucleus [3]. Class II HDACs are subdivided by apparent catalytic activity. HDAC6 is a well-established enzyme, and principally cytoplasmic, while the catalytic activities of HDAC4, HDAC5, HDAC7, and HDAC9 are uncertain [3]. HDAC4, HDAC5, HDAC7, and HDAC9 appear to be lacking key catalytic residues and their in vitro deacetylase activities are difficult to detect, although they in some cases they can process unnatural highly reactive trifluoroacetyl-Lys substrates [6]. HDAC10 is annotated as a polyamine deacetylase located in the cytosol [7] and HDAC11 seems to preferentially be involved in fatty deacylation of Lys residues [8]. Over the last several years, a number of small molecule metabolites have been shown to form amide linkages with Lys residues, giving rise to β-hydroxybutyrylation and lactylation. Removal of these unusual acylations by HDAC enzymes is just beginning to be characterized [9–11].
HDAC1, HDAC2, and HDAC3 share a number of similarities in structure and function. HDAC1 and HDAC2 show approximately 80% sequence identity and are believed to be interchangeable in a number of macromolecular complexes [3]. HDAC3 is somewhat more distantly related to HDAC1 and HDAC2 and is found in a distinct multi-protein complex as discussed below. Isolated HDAC1–3 proteins are considered to be unstable, and appear unable to deacetylate chromatin unless part of their cognate complexes.
In general, HDAC1–3 complexes contain corepressor proteins that help recruit DNA binding transcription factors and target them to specific chromatin loci. In some cases, the complexes can bind chromatin with high affinity in the absence of transcription factors. Several of the corepressor proteins possess a tandem domain pair known as ELM2-SANT which directly engages the HDAC catalytic subunit and is critical for complex stability. There are at least seven distinct core HDAC complexes that have been identified including CoREST, NuRD, Sin3, MiDAC, RERE, MIER, and SMRT (Figures 1 and 2) and a number of these will be discussed in more detail below [12–17].
Figure 1. Reported structures of class I HDAC complexes.
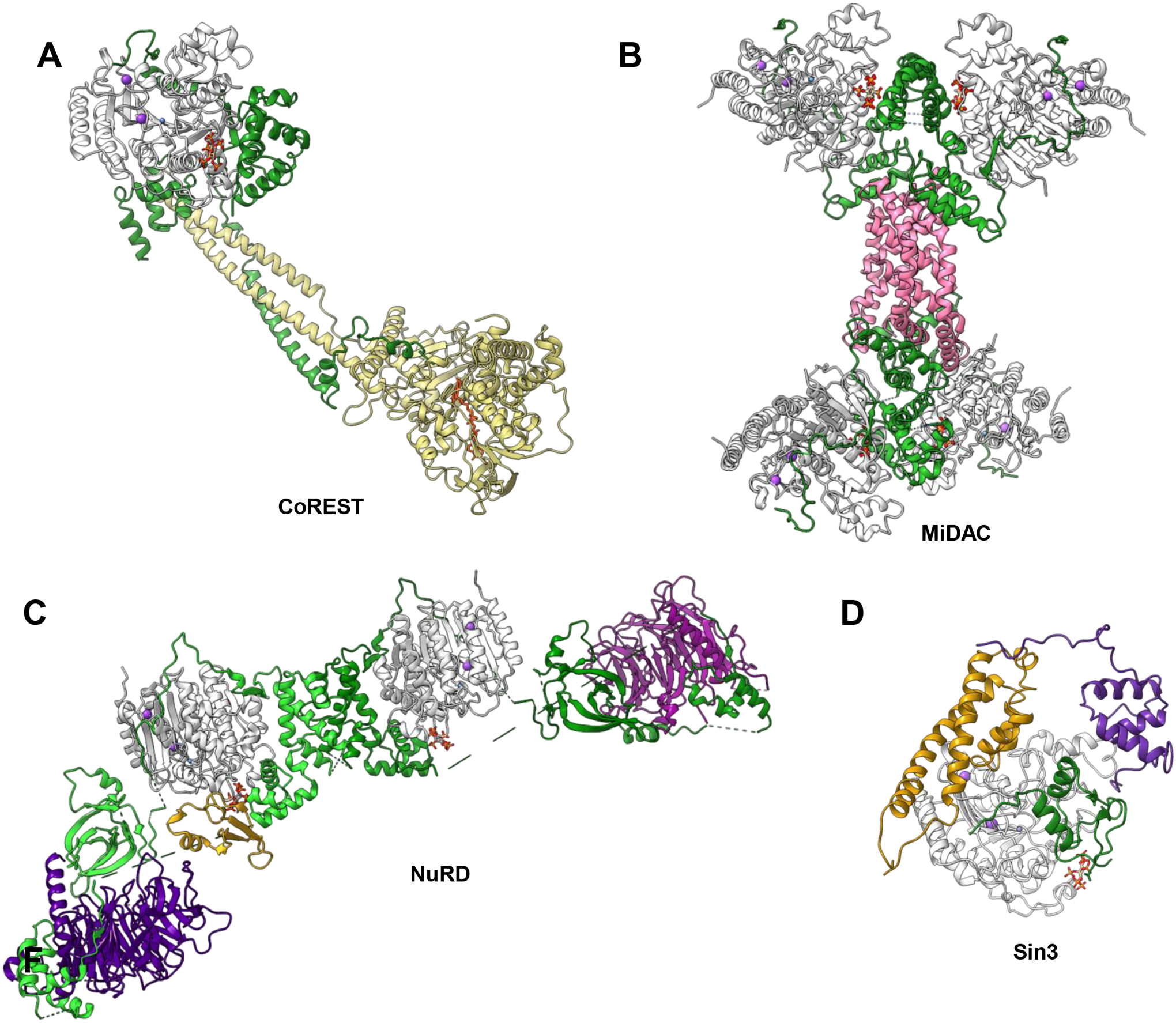
(A) LSD1-CoREST1-HDAC (gold-green-yellow) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). Alphafold-generated HDAC1-RCOR1elm2sant and LSD1-RCOR2linker-sant2 structures were fitted into the EM density map (EMD-10629) and were further refined with Rosetta Relax. (B) MiDEAS-DNTTIP1-HDAC (green-pink-grey) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). (C) NuRD deacetylase module with two each of MTA1 (green), HDAC1 (grey), and RBBP4 (indigo), as well as one MBD2 (gold), and IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue) (PDBID: 7AOA). (D) SIN3-HDAC1-SAP30L (gold-grey-green/purple) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue) (derived from PDBDEV_00000043).
Figure 2. Simulated class I HDAC complexes.
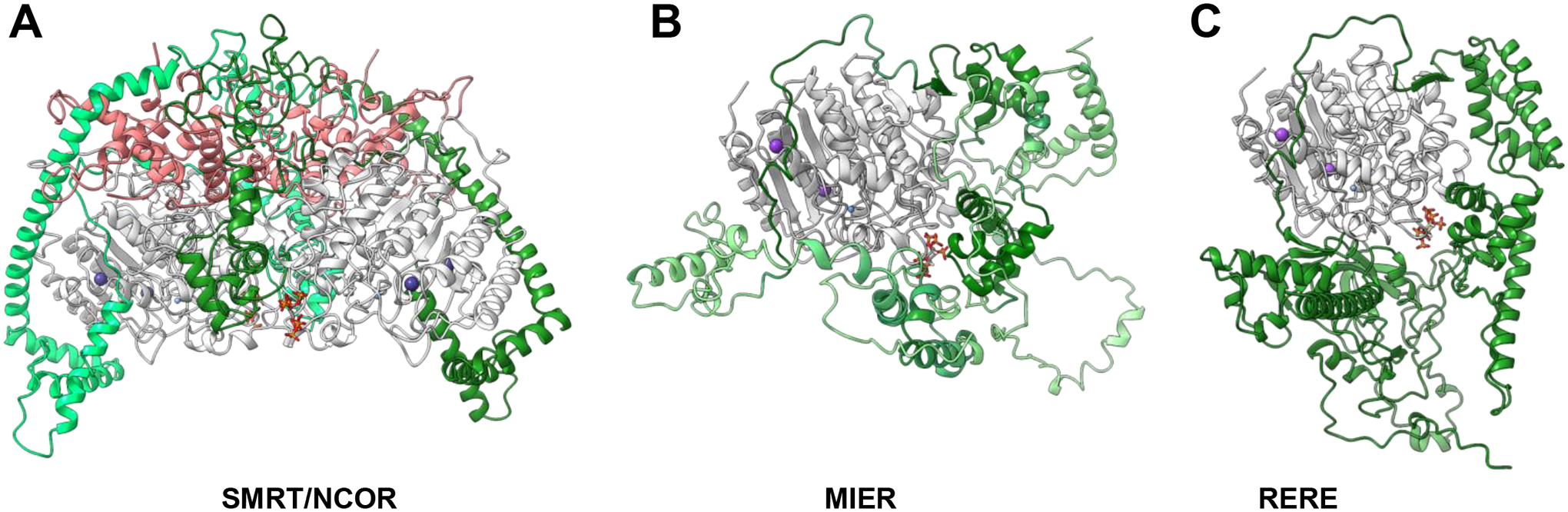
(A) Two NCOR1-GPS2 (green) fusion complexed with two HDAC3 (grey), four equivalents of TBL1(pink) tetramerization domain, IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). (B) MIER-HDAC1 (green-grey) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). (C) RERE-HDAC1 (green-grey) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). (A-C) Complexes simulated with AlphaFold multimer; pLDDT quintiles depicted by green gradient with darker color corresponding to higher pLDDT. Top-scoring models were selected and potassium, zinc, and inositol hexaphosphate were added based on an existing model (PDBID: 5ICN), then subjected to Rosetta relax for further refinement. Lowest energy structures were selected for depiction. All structures rendered with ChimeraX.
HDAC enzymes have attracted because HDAC dysregulation has been linked to cancer and other diseases. There are now six clinically used HDAC inhibitors for the treatment of malignancies [1]. They are principally used for the treatment of cutaneous T cell lymphoma. Two well-known subtypes of cutaneous T cell lymphoma are mycosis fungoides and Sezary syndrome. These diseases involve mutant, neoplastic T cells that appear to attack the skin, causing a combination of skin rash and skin tumors. Overall, cutaneous T cell lymphoma is relatively rare with about 1000 cases diagnosed annually in the United States (https://rarediseases.org/rare-diseases/cutaneous-t-cell-lymphomas/). Beyond cutaneous T cell lymphoma, HDAC inhibitors have been approved to a limited extent for the related malignancy peripheral T cell lymphoma as well as refractory multiple myeloma and acute myeloid leukemia. The clinically employed HDAC inhibitors are primarily hydroxamic acids, that act through coordination of the active site Zn [1]. The macrocycle peptide based agent romidepsin lacks a hydroxamic acid but possesses a disulfide that can be reduced in vivo, liberating an extended alkyl thiol that mimics a Lys and also engages the catalytic Zn [1].
A number of studies have investigated the substrate specificity of HDAC enzymes, primarily as isolated enzymes in the absence of their binding partners and with substrates consisting of acetyl-Lys peptide libraries [94,95]. In this setting, HDAC enzymes appear to show little sequence selectivity. HDAC selectivity has also been studied in cellular experiments using genetic knockouts or different HDAC inhibitors with mass spec based acetylomics as a readout [18]. Some selectivity has been observed in this setting, although the directness of deacetylation events in a cell-based assay is uncertain. Recently, HDAC complexes have been analyzed with nucleosome substrates and revealing some interesting complex-specific deacetylase site-selectivity [11,19,20]. Below we discuss several HDAC complexes and summarize some of their structural, enzymatic, and biological properties.
CoREST Complex
The REST Corepressor (CoREST) complex is unique among Class I HDAC complexes because it contains both the deacetylase HDAC1 and the histone demethylase LSD1 (Figure 1). The dual histone erasers are held together by the CoREST1 scaffold protein. The complex fine-tunes gene repression by removing PTMs associated with active transcription near the promoters and enhancers of genes involved in development. The developmental role of the complex makes its activity important in various cell types and implicates it in cancer and many other diseases [21–27].
The CoREST complex gets recruited to specific genomic loci with the help of transcription factors, such as REST, to remove mono- or di-methylation from histone H3 Lys 4 (H3K4me; hereon, 1-letter code is used to denote histone residues and 3-letter codes are used to denote residues on other proteins), and various acylations of the histone H3 and H2B tails. To study the CoREST complex’s physiological functions, targets, and spatiotemporal regulation, various chemical biology strategies have been employed, including small molecule, peptide, semi-protein synthesis, protein engineering, and gene editing-based tools (Table 1).
Table 1. Class I HDAC complexes.
Seven representative class I HDAC complexes’ core components, interaction partners, biological functions, structural information, and complex-specific molecular tools are outlined.
| # | Complex name | Core components | Interaction partners | Biological functions | Available structures | Complex specific molecular tools (with references if available) |
|---|---|---|---|---|---|---|
| 1 | CoREST | HDAC1/2 | REST | Represses neuronal differentiation genes | EMD 10626–10630: CoREST complex alone and nucleosome-bound [28] | Rodin-A: HDAC1/2 inhibitor [58] |
| CoREST1/3 (RCOR1/3) | SNAG family transcription factors (SNAI1/2 GFI-1/1Betc) | Regulates stem cell fate and development [21–23,27] | SASDH45: CoREST complex SAXS [28] | corin: HDAC1/2-LSD1 dual inhibitor [25] | ||
| LSD1 (KDM1A) | HMG20A/B | Regulates epithelial/endothelial to mesenchymal transition [26,59] | PDBID 6VYP: LSD1-RCOR1 complex bound to nucleosome [38] | SNAG peptide: noncovalent LSD1 inhibitor [26] | ||
| CTBP1 | Mediates cell plasticity in cancer & vascular diseases [60,61] | tranylcypromine-derivatives: LSD1 covalent inhibitor | ||||
| GSE1 | tetrahydrofolate: LSD1 formaldehyde scavenger [62] | |||||
| ZNF217 | UM171: complex degrader [63,64] | |||||
| PHF21A | TALE-LSD1: LSD1 gene repressor [65] | |||||
| TERRA lncRNA | SP2509: LSD1 noncovalent inhibitor [66] | |||||
| HOTAIR lncRNA | domatinostat (4SC202): LSD1 & HDAC1 dual inhibitor and microtubule polymerization inhibitor [66,67] | |||||
| GATA2/3 | ||||||
| *More targets identified using BioID [68] | ||||||
| 2 | NuRD | HDAC1/2 | FOG1 | ATP-dependent chromatin remodeling | PDBID 5ICN: HDAC/MTA1 [69] | BPK-25: NuRD complex degrader [70] |
| MTA1/2/3 | BCL11A | Modulates developmental gene expression | PDBID 5FXY, 6G16: RBBP4/MTA1 [71] | p66α peptide: disrupts p66α MBD2 interaction [72] | ||
| RBBP4/7 | SALL4 | Maintains genome integrity | PDBID 7AO8, 7AO9, 7AOA: HDAC1/MTA1 dimer with MDB2 and with or without RBBP4 [50] | Biphenyl Calix[4]arenes: prevents CHD4-H3K9me3 interaction [73,74] | ||
| MBD2/3 | ZMYND8 | Cell cycle progression | EMD 22895: Low resolution model of NuRD complex [54] | |||
| CHD3/4/5 | ZNF187 | EMD-21382: MTA/HDAC/MBD complex [54] | ||||
| GATAD2A/B (p66α/β) | ATR (through CHD4) | PXD017378: CHD4 and CDK2AP2 XL-MS [52] | ||||
| CDK2AP1 | PWWP2A (replaces CHD MBD GATA and CDK2AP1) | PXD017244: RBBP7, MBD3, HDAC1, ZNF687, CHD4, and MTA1 XL-MS [52] | ||||
| 3 | MiDAC | HDAC1/2 | CCNA2 | Modulates developmental gene expression during embryogenesis | PDBID 4D6K & 2MWI: domain structures & stoichiometry [14] | n/a |
| MIDEAS | CDKN1A (p21) | Orchestrates chromosome alignment during mitosis | BMRB 25326: domain structures & stoichiometry [14] | |||
| DNTTIP1 | CDKN1B (p27) | PDBID 6Z2J & 6Z2K: dimeric and tetrameric complex [16] | ||||
| DNTT (TdT) | EMD 11041–11042: dimeric and tetrameric complex [16] | |||||
| 4 | MIER | HDAC 1/2 | CDYL1 | Maintenance of stemness | AF-Q8N108-F1 (predicted) | XY-07–104: Mier1/3 scaffold degrader [75] |
| MIER1/2/3 | REST [76] | Represses neuronal differentiation genes by REST association [76] | ||||
| EHMT2 [77]) | endothelial to mesenchymal transition [78] | |||||
| H2B [57] | ||||||
| SP1 [79] | ||||||
| WIZ [80] | ||||||
| 5 | RERE | HDAC1/2 | EHMT2 (G9a) | Regulates developmental gene expression | PDB 2YQK: SANT domain solution structure (unpublished) | miR-200b: regulates expression of RERE [81] |
| RERE | FAT1 [82]) | Epithelial-to-mesenchymal transition and mesenchymal cell proliferation [83] | AF-Q9P2R6-F1 (predicted) | miR-429: regulates expression of RERE [81] | ||
| ATN1 [84] | Retinoic acid signaling coactivation [17] | |||||
| 6 | Sin3 (A/B) | HDAC1/2 | BRMS1 | Regulates tissue development | PDBDEV_00000043: In silico model with XL-MS distance restraints – SIN3A/SAP30L/HDAC1 complex [85] | SID peptide: blocks MAD-SID interaction [86] |
| SAP30/30L | MAD1 | Cell cycle | PDB 2N2H: Solution structure of Sds3 in complex with Sin3A [87] | entinostat [88] | ||
| SIN3A/B | NANOG | Embryogenesis/pluripotency | PDB 2N1U: Structure of SAP30L [89] | tacedinaline [88] | ||
| SAP18 | ARID4A/B | DNA replication | PDB 2LD7: Solution structure of the mSin3A PAH3-SAP30 SID complex [90] | BML-210 [88] | ||
| SDS3 (SUDS3) | ING1 | |||||
| RBBP4/7 | SAP25 | |||||
| MXD1 | ||||||
| REST | ||||||
| HBP1 | ||||||
| 7 | SMRT | HDAC3/4/7 | TBL1XR1 | Lipid metabolism | PDBID 4A69: SMRT/HDAC3/IP4 [91] | Cpd-60: HDAC3 inhibitor that allows association with SMRT [48] |
| NCOR2 | TBL1Y | Macrophage polarization and inflammation | PDBID 2L5G: GPS2-SMRT [44] | Arsenic trioxide: MAPK-dependent SMRT phosphorylation causes complex dissociation [92] | ||
| GPS2 | SCF E3 ligase complex | Fibroblast cell cycle progression | PDBID 2XTC, 2XTD, 2XTE: TBL1 [44] | miRNA-100: silencing SMRT/NCOR2 gene [93] | ||
| TBL1X | E2-UBC5HC | Development | XY-07–090: NCOR1/HDAC3-degrader [75] | |||
| SUMOylated-NR1H2/NR5A2 | Circadian rhythms | |||||
| NR1H3 | ||||||
| ANKRD26 | ||||||
| ZBTB7A | ||||||
| AR | ||||||
| p53 | ||||||
| unliganded nuclear receptors (TR, RAR, RXR, PPAR) | ||||||
| orphan nuclear receptors | ||||||
| COUP-TFs | ||||||
| DAX1 | ||||||
| Rev-Erb |
Relative to most of the HDAC complexes, the CoREST complex can deacetylate nucleosome substrates rather swiftly and with little site specificity (V/[E] ~0.03–0.1 min−1 for H3 tail acetylations and ~0.02 min−1 for H2B tail acetylations), except for H3K14ac (V/[E] < 0.005 min−1) [11,19,20]. Curiously, H3K14ac is not a disfavored substrate in assays with isolated histone H3 protein substrates. To understand whether H3K14ac is unusually inaccessible in nucleosome substrates hydroxamic acid analogues of acetyl-lysine analogs were incorporated at either H3K9 or H3K14 using sortase-mediated semisynthesis followed by nucleosome reconstitution. Such hydroxamic acids can tightly coordinate the active site Zn in HDACs and in principle provide insight into steric accessibility based on the position in the tail. However, inhibition assays of CoREST activity with these hydroxamic acid nucleosomes revealed that the hydroxamic acid-14 (Ki ~ 60 nM) and hydroxamic acid-9 (Ki ~ 40 nM) nucleosomes showed similar potencies [20]. These results suggest that these two tail positions are similarly accessible to the HDAC1 catalytic site.
It is conceivable that the nucleosome-bound CoREST complex adopts a unique structure with dynamics less catalytically favorable for H3K14 deacetylation as compared with other H3 acetylations. Interestingly, mutation of the preceding residue G13 into R (G13R) restores the CoREST deacetylase activity toward H3K14ac nucleosome substrates (V/[E] ~0.08 min−1 vs. H3K9ac V/[E] ~0.12 min−1) [19]. This activity enhancement by the polar residue substitution hints at the requirement of exquisitely precise electrostatic potential nearby the substrate entrance for rapid deacetylation. It is also interesting that H3K14ac slows down the demethylase activity of the CoREST complex, making the complex particularly sensitive to H3K14ac.
While the structure of HDAC1 ‘ready for deacetylation’ in the nucleosome-CoREST complex has not yet been solved, recent biophysical and structural characterization of nucleosome-CoREST complexes have been reported [28,38]. These structures reflect that LSD1 is ‘ready for demethylation’ both with and without HDAC1, hereon termed ternary and binary CoREST complexes, respectively. These structures highlight the variable modes by which the CoREST complex recognizes nucleosomes, and provide more clues about the CoREST complex’s unique sensitivity toward H3K14ac.
The nucleosome-bound ternary CoREST complex was captured through a covalent linkage between the flavin of LSD1 and H3 containing a propargylamine mimic of H3K4me [28]. This illustrates how the CoREST complex recognizes the nucleosome for demethylation while remaining poised for deacetylation. The outer portion of the LSD1 amine oxidase domain (LSD1AOD), known to serve as a docking platform for long non-coding RNA [29], makes direct contact with the dyad nucleosomal DNA to optimally orient LSD1AOD near the histone H3 tail for demethylation. In this structure HDAC1 sits at the far end of the tower domain of LSD1, adjacent to the SANT2 domain of CoREST1. Cross-linking mass spectrometry (XL-MS) studies show that the SANT2 domain makes close contact with Lys220 of HDAC1, at the second potassium ion binding site of HDAC1. In silico, it has been demonstrated that the second potassium ion-bound moiety is allosterically linked with the catalytic center of HDAC1 (about ~20 Å away) [30]. It is plausible that the ‘ready for deacetylation’ structure of the nucleosome-bound CoREST complex adopts unique structural dynamics through the HDAC1-SANT2 domain interaction that influence the orientation of the acetyl-Lys14 residue during binding, due to the absence of a polar moiety in the sidechain of the preceding residue (K9/18/27 are preceded by R, and K23 by T). Using Alphafold and Rosetta molecular modeling tools [31–34], we speculate that G13R substitution can cause subtle orientational changes of Phe205 and Tyr303 in HDAC1, critical residues for substrate entry and HDAC1 activity [35–37].
The key interaction between HDAC1 and the SANT2 domain of CoREST1 also hints at HDAC1’s potential role as a demethylase activity regulator. In the binary CoREST complex, the SANT2 domain interacts with the nucleosome core to position LSD1AOD in a catalytically productive conformation [38]. Substantial rate differences in demethylation of nucleosome substrates (binary CoREST complex, V/E > 0.025 min−1; ternary CoREST complex V/E ~0.001 min−1), could indicate that HDAC1 occludes the SANT2 domain-nucleosome core interaction through direct competition. It should be noted, however, that the nucleosome substrates rapidly demethylated by the binary CoREST complex contain a thialysine mimic of H3K4me, the lower pKa of which could contribute to more rapid demethylation as compared with the natural H3K4me used to study ternary CoREST complex. In addition, different constructs of LSD1 and CoREST were used to biochemically and biophysically characterize the CoREST complexes: N-terminally truncated LSD1 (aa171–852) and CoREST1 (aa286–440) in the binary CoREST complex [38], and N-terminally truncated CoREST1 (aa86–485) with full-length LSD1 and HDAC1 in the ternary complex [28].
Several synthetic compounds including 4SC-202, rodin, and corin have been reported to be selective for the CoREST complex relative to other class I HDAC complexes (Table 1). Of these, corin is the best characterized and is based on dual targeting of the LSD1 active site with a cyclopropylamine and the HDAC catalytic site with a benzamide Zn binding group. Corin shows enhanced residence time with the CoREST complex relative to monofunctional HDAC benzamide inhibitors and displays promise as an antineoplastic agent for melanoma, diffuse pontine glioma, and colorectal cancer. Corin also appears to enhance tumor immune surveillance [25,39–41].
MiDAC Complex
The mitotic deacetylase complex (MiDAC) modulates developmental gene expression, orchestrates chromosome alignment during mitosis, and is critical for embryogenesis, neural differentiation, and cancer cell survival [16,42]. Pull-down assays have shown that the MiDAC complex contains three core components: HDAC1, DNA/nucleosome binding DNTTIP1, and the scaffold protein/HDAC1-modulator MIDEAS (Figure 1). Recent structural studies have highlighted the interesting architecture of MiDAC, in which heterotrimers of MIDEAS, HDAC1, and DNTTIP1 form an X-shaped tetrameric complex through the dimerization domain of DNTTIP1 and the inositol phosphate-bridged SANT domain of MIDEAS. By anchoring its flexible DNA-binding domain of DNTTIP1 onto linker DNA, the MiDAC complex can position its four HDAC1 molecules >45 Å apart from each other, ready for deacylation of nucleosomal histone tails nearby.
In vitro biochemical characterization of the MiDAC complex using designer nucleosome substrates revealed rapid enzymatic activity [11,19] that may promote precise chromatin remodeling during a narrow time window of cell cycle progression. The MiDAC complex displays the most rapid catalytic activity toward nucleosome substrates among the class I HDAC complexes deacetylating the histone H3 tail (H3K9ac/K14ac/K18ac/K23ac/K27ac) with V/[E] values 2- to 10-fold greater than other complexes at matched acetylation sites. The complex displays preferential deacetylation activity toward acetylations nearer the N-terminus of both H3 (V/[E] ~1.2 min−1 toward H3K9ac vs. V/[E] ~0.048 min−1 toward H3K23ac) and H2B tails (V/[E] ~ 2.4 min−1 toward H2BK11ac vs. V/[E] ~ 0.0022 min−1 toward H2BK20ac). MiDAC further distinguishes itself by its ability to remove lactyl- and β-hydroxybutryl- modifications of H2BK11 [11,19].
There are a few plausible explanations for MiDAC’s remarkable catalytic activity, relatively agnostic substrate specificity (H3 vs. and H2B; acetyl vs. lactyl and β-hydroxybutryl), and distinct site selectivity (N-terminal preference): 1) the flexible DNA/nucleosome-binding linker of DNTTIP1 provides the MiDAC complex conformational freedom to recognize histone tails that are positioned close to (H3) and far from (H2B) the dyad, (both tails are protruding from two DNA gyres); 2) thoroughly solvent-exposed, intact HDAC1 molecules maximize active site accessibility; 3) overall bulk of the tetrameric complex restrains HDACs from easily binding histone tail sequences nearer to the nucleosome core; 4) unique MIDEAS SANT-domain-inositol phosphate-HDAC1 interaction is further rigidified by its dimer that alters structure and dynamics of the catalytic site nearby.
SMRT/NCOR Complex
The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) complex and its paralog nuclear receptor corepressor (NCOR) complex are the only known class I HDAC complexes containing HDAC3. SMRT regulates gene expression involved in development, metabolism, inflammation, and circadian rhythms [43]. The SMRT/NCOR complex contains four core components: HDAC3, HDAC3-modulating and scaffolding protein SMRT/NCOR, transcription factor-binding protein GPS2, and oligomerization mediator TBL1X. Interestingly, TBL1X has been shown to associate with transcription activators and SCF ubiquitin ligase complex and E2 ubiquitin ligase Ubc5H, hinting at its role as a coregulator exchange factor to facilitate transcription activation by dissociating/degrading SMRT-HDAC3 molecules [44].
A high-resolution crystal structure of HDAC3 in complex with the deacetylase activating domain (DAD) of SMRT served as the first structural representation of a class I HDAC complex [91]. It provided atomic level detail on how the binding of inositol phosphate and the HDAC-scaffolding protein mediate activation of the class I HDAC complex through multiple electrostatic interactions. In silico studies have demonstrated that the formation of the ternary complex (SMRT DAD-HDAC3-inositol phosphate) greatly stabilizes the HDAC3 catalytic site, orienting Y298 residue inward to assist deacetylation [45].
Coupled with its ability to deacetylate nucleosome at a moderate rate, favoring H3K9ac and H3K27ac over K14, K18 and K23 (V/[E] ~ 0.01−1 min for the H3K14ac/K18ac/K23ac vs. V/[E] ~ 0.04 min−1 for H3K9ac/K27ac) [19], the SMRT complex’s ability to form a large complex (1.5 ~ 2 MDa in cell) [44] through the tight interaction between SMRT and HDAC3 are considered to be essential for SMRT complex-mediated gene repression in various cell types [46,47].
A multimeric structure reflecting the proposed stoichiometry of four TBL1 to two each of GPS2-SMRT-HDAC3 was simulated [44] to probe how the SMRT complex might recognize substrates (Figure 2). In the model, two HDAC3 molecules are tightly packed closely, bridged by two DAD. This compaction could limit accessibility of the nucleosome substrates to both HDAC3 active sites, which could explain the moderate in vitro deacetylation rate. The enhanced binding energetics of this compact interface could help maintain the integrity of such a large complex. Recent time-resolved FRET studies illustrate robust stability of the SMRT complex, and profound changes in conformation and dynamics of the HDAC3 active site when bound to inositol phosphates [48]. This observation is consistent with the model in which sandwiched DADs bridging two HDAC3s are further stabilized by the binding of inositol phosphate molecules.
NuRD
The nucleosome remodeling and deacetylase complex (NuRD) is involved in cell differentiation, lineage maintenance and the DNA damage response [49]. NuRD distinguishes itself among HDAC complexes through contribution to some gene activation events, though this may be primarily a function of its chromatin remodeling module.
The NuRD HDAC module is a dimer of MTA1/2/3 and HDAC1/2, with up to 4 associated RBBP4/7 histone binding proteins (Figure 1) [50,51]. Asymmetrically bound MBD2/3 spans the length of the HDAC dimer and binds the largely disordered GATAD2A/B through a coiled-coil interaction, which links the module to a nucleosome binding CHD3/4/5 chromatin remodeler and CDK2AP1 [52]. Absent CHD, the NuRD HDAC module with MBD alone, MBD and GATAD2, or with PWWP2A constitute discreet nuclear deacetylase complexes [53–55]. Through RBBP, the NuRD HDAC module interacts with a variety of primarily DNA-binding nuclear proteins that may recruit it to chromatin (Table 1) [56].
The in vitro deacylase activity of the NuRD HDAC module, relative to other HDAC complexes, is robust for histone proteins, but modest for nucleosomes. The complex deacetylates most sites on the H3 and H2B tails but shows little selectivity (~2-fold) between sites [19]. Deacylation of histones in chromatin requires disentangling tails from the DNA backbone, which could be facilitated by DNA binding domains. Cryo-EM and XL-MS place the DNA-binding domain of MBD near the HDAC active site, where it could help feed histone tails to HDAC (Figure 3). This raises the question of whether isoform-specific differences in the affinity of MBDs for methylated, hydroxymethylated and unmethylated DNA could regulate HDAC activity [96]. Deacylation of accessible histone tails may be further regulated by competition with the histone tail binding domains of RBBP and the BAH domain of MTA. Of these, the BAH domain appears positioned to crowd the HDAC active site, which could either cooperatively position tails for catalysis, or exclude them from the HDAC active site [50]. An argument against cooperation can be made by comparison to RERE, which has a BAH domain also predicted to crowd the HDAC active site and exhibits slow deacylation of histone proteins and slower deacylation of nucleosomes (Figure 3).
Figure 3. Substrate recognition mechanisms of HDAC complexes.
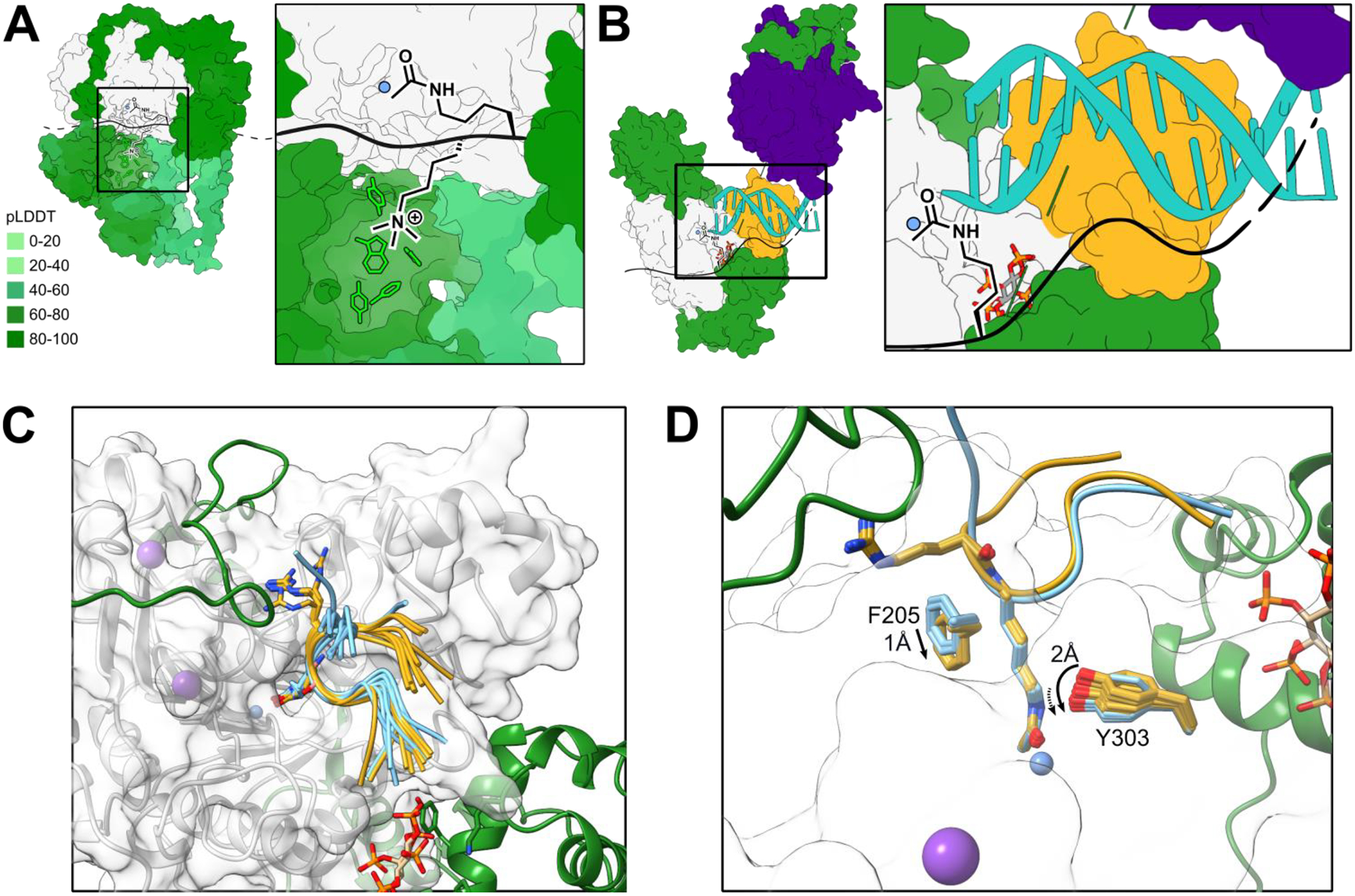
(A) RERE-HDAC1 (green-grey) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue). Inset: the methyllysine-binding aromatic cage (lime) of the BAH domain is depicted in complex with a theoretical methyllysine/acetyllysine substrate. Complex simulated with AlphaFold multimer (v 2.1.1); pLDDT quintiles depicted by green gradient with darker color corresponding to higher pLDDT. Top-scoring models were selected and potassium, zinc, and inositol hexaphosphate were added based on an existing model (PDBID: 5ICN), then subjected to Rosetta relax for further refinement. The lowest energy structure was selected for depiction. (B) Partial NuRD deacetylase module shown here with 1 each of MTA1-HDAC1-MBD2-RBBP4 (green-grey-gold-indigo; PDBID: 7AOA) complex with IP6 (red/orange/dark grey/white), potassium (purple) and zinc (blue), aligned MBD2-DNA complex (turquoise; PDBID: 7MWM). Inset: nucleic acid binding by the MBD domain could serve to position the HDAC active site near histone tails or other chromatin-bound proteins. (C-D) Histone H3 tail (gold and blue) docking to RCOR1elm2sant-HDAC1 (green-grey) complex illustrating conformational states dependent on the amino acid preceding the substrate lysine. Histone H3 peptide (S10-K18) was re-constructed from the existing peptide inhibitor-bound HDAC1 crystal structure (PDB 5ICN) using ChimeraX and Rosetta. Then, the reconstructed peptide was docked into the RCOR1elm2sant-HDAC1 complex structure generated by Alphafold multimer using Rosetta Flexpepdock [34]. Top 10 structures were superimposed for visual inspection (C). The lowest energy complex structure was further refined using Rosetta relax [31]. Sidechains of Phe205 and Tyr303 in the active site from all 50 relaxed structures were superimposed (D) to inspect conformational heterogeneity caused by G13R. (C) H3 tail sequences with either G13 preceding K14ac (blue) or R13 preceding K14ac (yellow) are shown. The conformational space surveyed by the arginine side chain contributes to a difference in the conformation of both HDAC and the C-terminal portion of the docked peptide. (D) Differences in active site residue positions and conformational heterogeneity dependent on the amino acid preceding the substrate lysine. Substitution of H3G13 for R results in a shift in the F205, a gatekeeper of the HDAC active site, and a change in dynamics of Y303, a hydrogen bond donor in catalysis. (A-D) All structures rendered with ChimeraX.
MIER
The Mesoderm induction early response proteins, MIER1/2/3, are HDAC-binding, ELM2-SANT motif proteins predicted to be largely unstructured [79]. All three are predominantly nuclear, although MIER2 displays cell type-specific cytoplasmic accumulation (~30%). Of the three proteins MIER1 is substantially more associated with both HDAC1 and 2 ex vivo [12].
While no structure of the MIER-HDAC complex has been reported, mutagenic approaches confirm that the ELM2 motif is anchored by a conserved tryptophan. The complex does not appear to dimerize by coimmunoprecipitation, likely because the ELM2 domain lacks a 12-residue helix (helix 2) that drives MTA dimerization [50]. Those SANT residues required for IP4 binding are retained in MIER, however, the complex does not appear to depend on IP4/6 for histone deacetylation in vitro [12]. We display a structural model in Figure 2.
The MIER complex displays particularly rapid histone deacetylation kinetics in vitro, though little activity toward nucleosomes [11]. In vitro evaluation of substrate selectivity suggests that H3 is preferred over H2B. Modeling the binary complex suggests a broad range of conformations within the disordered N- and C-termini of MIER [32,33]. The disordered termini are reliably predicted to have helical content (E48-E68, P82-S107, Y359-P376, P455-E473, P489-H501), and a region between the final helices may interact with the C-terminus of H2B [57]. Nonetheless, a clear mechanism of molecular recognition capable of explaining the dramatic deacetylation of H3K9ac protein (V/[E] ~ 28 min−1) remains elusive.
Summary and Outlook
Here we have summarized some of the structural and enzymatic features of a set of class I HDAC complexes that illustrate how important the particular subunits of each complex are in conferring their unique substrate efficiencies with chromatin and non-chromatin acetylated lysines. In the case of MiDAC, the high catalytic efficiencies with H3 and H2B N-terminal tails in nucleosomes stand out among the HDAC complexes. We propose that other complexes such as NuRD which shows very low rates with nucleosomal substrates likely depend on transcription factor recruitment to achieve targeted deacetylation. The data thus far suggests greater site-selectivity for tail deacetylation in chromatin versus free histone substrates. This emphasizes the more intricate molecular recognition that occurs when HDAC complexes encounter nucleosomes. Initial strides have been made toward selective inhibition of individual HDAC complexes with small molecules such as corin for the CoREST complex. We believe that such selective HDAC complex targeting offers promise for novel epigenetic therapies. Key challenges that remain in our understanding of HDAC complexes include elucidating the structural basis for site-specificity in chromatin, how these complexes are turned on and off in a cellular context, and what their functions are in physiological and disease processes. We believe that new chemical approaches will help tackle these challenges and pave the way to a richer portrait of HDAC complex roles in biology.
Acknowledgements
This work was supported by American Heart Association (Postdoctoral fellowship award 826614 to K.L.), American Cancer Society (PF-20-105-01-DMC to S.D.W), Leukemia and Lymphoma Society (SCOR to P.A.C.) and NIH (GM62437 to P.A.C.). Authors want to thank the Research Computing Group at Harvard Medical School for providing computing resource and helpful advice.
Footnotes
Declaration of competing interest
Philip Cole is a founder of Acylin Therapeutics and a consultant for Abbvie regarding p300 acetyltransferase inhibitors. He also is a co-inventor on a U.S. patent application for corin.
References
Papers of particular interest, published within the period of review, have been highlighted as: * of special interest
- 1.Basith S, Chang JH, Nithiyanandam S, Shin HT, Manavalan B, Lee G: Recent Trends on the Development of Machine Learning Approaches for the Prediction of Lysine Acetylation Sites. Current Medicinal Chemistry 2022, 29:235–250. [DOI] [PubMed] [Google Scholar]
- 2.Shvedunova M, Akhtar A: Modulation of cellular processes by histone and non-histone protein acetylation. Nature Reviews Molecular Cell Biology 2022. [DOI] [PubMed] [Google Scholar]
- 3.Wang ZA, Cole PA: The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chemical Biology 2020, 27:953–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taunton J, Hassig Christian A, Schreiber Stuart L: A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272:408–411. [DOI] [PubMed] [Google Scholar]
- 5.Porter NJ, Christianson DW: Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Current Opinion in Structural Biology 2019, 59:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R: Chemical phylogenetics of histone deacetylases. Nature Chemical Biology 2010, 6:238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hai Y, Shinsky SA, Porter NJ, Christianson DW: Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nature Communications 2017, 8:15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao J, Sun L, Aramsangtienchai P, Spiegelman Nicole A, Zhang X, Huang W, Seto E, Lin H: HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proceedings of the National Academy of Sciences 2019, 116:5487–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C, Qi S, Peng C, Cole Philip A, Roeder Robert G, et al. : The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Science Advances 2021, 7:eabe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Identified possible regulatory enzymes for β-hydroxybutyrylation in vitro and in cells. The study reported that HDAC1 and 2 show de-hydroxybutyrylation activity.
- 10.Moreno-Yruela C, Zhang D, Wei W, Bæk M, Liu W, Gao J, Danková D, Nielsen Alexander L, Bolding Julie E, Yang L, et al. : Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Science Advances 2022, 8:eabi6696. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Characterized the de-lactylation activity of HDAC1–3 in vitro and in cells.
- 11.Wang ZA, Whedon SD, Wu M, Wang S, Brown EA, Anmangandla A, Regan L, Lee K, Du J, Hong JY, et al. : Histone H2B Deacylation Selectivity: Exploring Chromatin’s Dark Matter with an Engineered Sortase. Journal of the American Chemical Society 2022, 144:3360–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Systematically measured the deacylation activities of HDAC complexes and several sirtuins with semi-synthetic histone H2B proteins and nucleosomes. First deacylation study of H2B with N-terminal tail acylation with nucleosome substrates.
- 12.Derwish R, Paterno GD, Gillespie LL: Differential HDAC1 and 2 Recruitment by Members of the MIER Family. PLOS ONE 2017, 12:e0169338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudson GM, Watson PJ, Fairall L, Jamieson AG, Schwabe JWR: Insights into the Recruitment of Class IIa Histone Deacetylases (HDACs) to the SMRT/NCoR Transcriptional Repression Complex. Journal of Biological Chemistry 2015, 290:18237–18244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh T, Fairall L, Muskett FW, Milano CP, Watson PJ, Arnaudo N, Saleh A, Millard CJ, El-Mezgueldi M, Martino F, et al. : Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Research 2015, 43:2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Describes the components and unique structural features of the MiDAC complex, and its potential biological functions.
- 15.Kelly Richard DW, Cowley Shaun M: The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochemical Society Transactions 2013, 41:741–749. [DOI] [PubMed] [Google Scholar]
- 16.Turnbull RE, Fairall L, Saleh A, Kelsall E, Morris KL, Ragan TJ, Savva CG, Chandru A, Millard CJ, Makarova OV, et al. : The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nature Communications 2020, 11:3252. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Showed tetrameric structure of MiDAC at an atomic level detail and thoroughly demonstrated the MiDAC complex’s biological function / importance.
- 17.Vilhais-Neto GC, Fournier M, Plassat J-L, Sardiu ME, Saraf A, Garnier J-M, Maruhashi M, Florens L, Washburn MP, Pourquié O: The WHHERE coactivator complex is required for retinoic acid-dependent regulation of embryonic symmetry. Nature Communications 2017, 8:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schölz C, Weinert BT, Wagner SA, Beli P, Miyake Y, Qi J, Jensen LJ, Streicher W, McCarthy AR, Westwood NJ, et al. : Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nature Biotechnology 2015, 33:415–423. [DOI] [PubMed] [Google Scholar]
- 19.Wang ZA, Millard CJ, Lin C-L, Gurnett JE, Wu M, Lee K, Fairall L, Schwabe JWR, Cole PA: Diverse nucleosome Site-Selectivity among histone deacetylase complexes. eLife 2020, 9:e57663. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Systematically measured the deacetylation activities of HDAC complexes with semi-synthetic H3 proteins and nucleosomes with acetylation at five different acetylation sites in the H3 tail.
- 20.Wu M, Hayward D, Kalin JH, Song Y, Schwabe JWR, Cole PA: Lysine-14 acetylation of histone H3 in chromatin confers resistance to the deacetylase and demethylase activities of an epigenetic silencing complex. eLife 2018, 7:e37231. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Applied semi-synthetic histones and nucleosomes with site-specific PTMs for biochemical characterizations of CoREST complex. It pointed out the CoREST complex’s unique sensitivity toward H3K14ac for both deacetylase and demethylase activities.
- 21.Andrés María E, Burger C, Peral-Rubio María J, Battaglioli E, Anderson Mary E, Grimes J, Dallman J, Ballas N, Mandel G: CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proceedings of the National Academy of Sciences 1999, 96:9873–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, et al. : Regulation of Neuronal Traits by a Novel Transcriptional Complex. Neuron 2001, 31:353–365. [DOI] [PubMed] [Google Scholar]
- 23.Foster Charles T, Dovey Oliver M, Lezina L, Luo Jin L, Gant Timothy W, Barlev N, Bradley A, Cowley Shaun M: Lysine-Specific Demethylase 1 Regulates the Embryonic Transcriptome and CoREST Stability. Molecular and Cellular Biology 2010, 30:4851–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hino S, Kohrogi K, Nakao M: Histone demethylase LSD1 controls the phenotypic plasticity of cancer cells. Cancer Science 2016, 107:1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalin JH, Wu M, Gomez AV, Song Y, Das J, Hayward D, Adejola N, Wu M, Panova I, Chung HJ, et al. : Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nature Communications 2018, 9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin T, Ponn A, Hu X, Law BK, Lu J: Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29:4896–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, et al. : Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446:882–887. [DOI] [PubMed] [Google Scholar]
- 28.Song Y, Dagil L, Fairall L, Robertson N, Wu M, Ragan TJ, Savva CG, Saleh A, Morone N, Kunze MBA, et al. : Mechanism of Crosstalk between the LSD1 Demethylase and HDAC1 Deacetylase in the CoREST Complex. Cell Reports 2020, 30:2699–2711.e2698. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Biophysically characterized the ternary CoREST complex bound and unbound to nucleosome and systematically evaluated crosstalk between LSD1 and HDAC1. Crosslinking-MS and CryoEM data provided first model structure of the complex structure.
- 29.Hirschi A, Martin WJ, Luka Z, Loukachevitch LV, Reiter NJ: G-quadruplex RNA binding and recognition by the lysine-specific histone demethylase-1 enzyme. RNA 2016, 22:1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sixto-López Y, Bello M, Correa-Basurto J: Insights into structural features of HDAC1 and its selectivity inhibition elucidated by Molecular dynamic simulation and Molecular Docking. Journal of Biomolecular Structure and Dynamics 2019, 37:584–610. [DOI] [PubMed] [Google Scholar]
- 31.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D: Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Science 2014, 23:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, et al. : Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022:2021.2010.2004.463034. [Google Scholar]
- 33.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. : Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raveh B, London N, Schueler-Furman O: Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins: Structure, Function, and Bioinformatics 2010, 78:2029–2040. [DOI] [PubMed] [Google Scholar]
- 35.Seto E, Yoshida M: Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harbor Perspectives in Biology 2014, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wambua MK, Nalawansha DA, Negmeldin AT, Pflum MKH: Mutagenesis Studies of the 14 Å Internal Cavity of Histone Deacetylase 1: Insights toward the Acetate-Escape Hypothesis and Selective Inhibitor Design. Journal of Medicinal Chemistry 2014, 57:642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weerasinghe SVW, Estiu G, Wiest O, Pflum MKH: Residues in the 11 Å Channel of Histone Deacetylase 1 Promote Catalytic Activity: Implications for Designing Isoform-Selective Histone Deacetylase Inhibitors. Journal of Medicinal Chemistry 2008, 51:5542–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S-A, Zhu J, Yennawar N, Eek P, Tan S: Crystal Structure of the LSD1/CoREST Histone Demethylase Bound to Its Nucleosome Substrate. Molecular Cell 2020, 78:903–914.e904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, Alexandrescu S, Blanco MA, Giera S, Gillespie SM, Das J, et al. : Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36:528–544.e510. [DOI] [PubMed] [Google Scholar]
- 40.Wu M, Hanly A, Gibson F, Kuang K, Kalin J, Nocco S, Collard M, Cole M, Xiao A, Agus F, et al. : The CoREST Repressor Complex Mediates Phenotype Switching and Therapy Resistance in Melanoma. bioRxiv 2020:2020.2009.2030.320580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong Y, Wang L, Di Giorgio E, Akimova T, Beier UH, Han R, Trevisanut M, Kalin JH, Cole PA, Hancock WW: Inhibiting the coregulator CoREST impairs Foxp3+ Treg function and promotes antitumor immunity. The Journal of Clinical Investigation 2020, 130:1830–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mondal B, Jin H, Kallappagoudar S, Sedkov Y, Martinez T, Sentmanat MF, Poet GJ, Li C, Fan Y, Pruett-Miller SM, et al. : The histone deacetylase complex MiDAC regulates a neurodevelopmental gene expression program to control neurite outgrowth. eLife 2020, 9:e57519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mottis A, Mouchiroud L, Auwerx J: Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes & Development 2013, 27:819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oberoi J, Fairall L, Watson PJ, Yang J-C, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, et al. : Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nature Structural & Molecular Biology 2011, 18:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arrar M, Turnham R, Pierce L, de Oliveira CAF, McCammon JA: Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Science 2013, 22:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stengel KR, Barnett KR, Wang J, Liu Q, Hodges E, Hiebert SW, Bhaskara S: Deacetylase activity of histone deacetylase 3 is required for productive VDJ recombination and B-cell development. Proceedings of the National Academy of Sciences 2017, 114:8608–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Z, Feng D, Fang B, Mullican Shannon E, You S-H, Lim H-W, Everett Logan J, Nabel Christopher S, Li Y, Selvakumaran V, et al. : Deacetylase-Independent Function of HDAC3 in Transcription and Metabolism Requires Nuclear Receptor Corepressor. Molecular Cell 2013, 52:769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Payne NC, Mazitschek R: Resolving the deceptive isoform and complex selectivity of HDAC1/2 inhibitors. Cell Chemical Biology 2022. [DOI] [PubMed] [Google Scholar]; *Systematically evaluated the complex inhibitor selectivity and dynamics of HDAC isoforms as well as different HDAC complexes using time-resolved FRET.
- 49.Burgold T, Barber M, Kloet S, Cramard J, Gharbi S, Floyd R, Kinoshita M, Ralser M, Vermeulen M, Reynolds N, et al. : The Nucleosome Remodelling and Deacetylation complex suppresses transcriptional noise during lineage commitment. The EMBO Journal 2019, 38:e100788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Millard CJ, Fairall L, Ragan TJ, Savva CG, Schwabe JWR: The topology of chromatin-binding domains in the NuRD deacetylase complex. Nucleic Acids Research 2020, 48:12972–12982. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Reported the first structure of the core NuRD complex and provided model structures of the nucleosome-bound state.
- 51.Sharifi Tabar M, Mackay JP, Low JKK: The stoichiometry and interactome of the Nucleosome Remodeling and Deacetylase (NuRD) complex are conserved across multiple cell lines. The FEBS Journal 2019, 286:2043–2061. [DOI] [PubMed] [Google Scholar]
- 52.Spruijt CG, Gräwe C, Kleinendorst SC, Baltissen MPA, Vermeulen M: Cross-linking mass spectrometry reveals the structural topology of peripheral NuRD subunits relative to the core complex. The FEBS Journal 2021, 288:3231–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Link S, Spitzer RMM, Sana M, Torrado M, Völker-Albert MC, Keilhauer EC, Burgold T, Pünzeler S, Low JKK, Lindström I, et al. : PWWP2A binds distinct chromatin moieties and interacts with an MTA1-specific core NuRD complex. Nature Communications 2018, 9:4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Low JKK, Silva APG, Sharifi Tabar M, Torrado M, Webb SR, Parker BL, Sana M, Smits C, Schmidberger JW, Brillault L, et al. : The Nucleosome Remodeling and Deacetylase Complex Has an Asymmetric, Dynamic, and Modular Architecture. Cell Reports 2020, 33:108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Low JKK, Webb SR, Silva APG, Saathoff H, Ryan DP, Torrado M, Brofelth M, Parker BL, Shepherd NE, Mackay JP: CHD4 Is a Peripheral Component of the Nucleosome Remodeling and Deacetylase Complex. Journal of Biological Chemistry 2016, 291:15853–15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharifi Tabar M, Giardina C, Feng Y, Francis H, Moghaddas Sani H, Low JKK, Mackay JP, Bailey CG, Rasko JEJ: Unique protein interaction networks define the chromatin remodelling module of the NuRD complex. The FEBS Journal 2022, 289:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Analyzed the complex interaction between the MGCC module in the NuRD complex and revealed its unique biological functions.
- 57.Fasci D, van Ingen H, Scheltema RA, Heck AJR: Histone Interaction Landscapes Visualized by Crosslinking Mass Spectrometry in Intact Cell Nuclei. Molecular & Cellular Proteomics 2018, 17:2018–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fuller NO, Pirone A, Lynch BA, Hewitt MC, Quinton MS, McKee TD, Ivarsson M: CoREST Complex-Selective Histone Deacetylase Inhibitors Show Prosynaptic Effects and an Improved Safety Profile To Enable Treatment of Synaptopathies. ACS Chemical Neuroscience 2019, 10:1729–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarroux J, Foretek D, Bertrand C, Gabriel M, Szachnowski U, Saci Z, Guo S, Londoño-Vallejo A, Pinskaya M, Morillon A: HOTAIR lncRNA promotes epithelial–mesenchymal transition by redistributing LSD1 at regulatory chromatin regions. EMBO reports 2021, 22:e50193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan B, Liu H, Pan X, Dong X, Qu L-F, Sun J, Pan L-L: LSD1 downregulates p21 expression in vascular smooth muscle cells and promotes neointima formation. Biochemical Pharmacology 2022, 198:114947. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Huang T, Zhai H, Peng W, Zhou Y, Li Q, Yang H: Inhibition of lysine-specific demethylase 1A suppresses neointimal hyperplasia by targeting bone morphogenetic protein 2 and mediating vascular smooth muscle cell phenotype. Cell Proliferation 2020, 53:e12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luka Z, Pakhomova S, Loukachevitch LV, Calcutt MW, Newcomer ME, Wagner C: Crystal structure of the histone lysine specific demethylase LSD1 complexed with tetrahydrofolate. Protein Science 2014, 23:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chagraoui J, Girard S, Spinella J-F, Simon L, Bonneil E, Mayotte N, MacRae T, Coulombe-Huntington J, Bertomeu T, Moison C, et al. : UM171 Preserves Epigenetic Marks that Are Reduced in Ex Vivo Culture of Human HSCs via Potentiation of the CLR3-KBTBD4 Complex. Cell Stem Cell 2021, 28:48–62.e46. [DOI] [PubMed] [Google Scholar]
- 64.Subramaniam A, Žemaitis K, Talkhoncheh MS, Yudovich D, Bäckström A, Debnath S, Chen J, Jain MV, Galeev R, Gaetani M, et al. : Lysine-specific demethylase 1A restricts ex vivo propagation of human HSCs and is a target of UM171. Blood 2020, 136:2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE: Locus-specific editing of histone modifications at endogenous enhancers. Nature Biotechnology 2013, 31:1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inui K, Zhao Z, Yuan J, Jayaprakash S, Le LTM, Drakulic S, Sander B, Golas MM: Stepwise assembly of functional C-terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Science 2017, 26:997–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wobser M, Weber A, Glunz A, Tauch S, Seitz K, Butelmann T, Hesbacher S, Goebeler M, Bartz R, Kohlhof H, et al. : Elucidating the mechanism of action of domatinostat (4SC-202) in cutaneous T cell lymphoma cells. Journal of Hematology & Oncology 2019, 12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barnes CE, English DM, Broderick M, Collins MO, Cowley SM: Proximity-dependent biotin identification (BioID) reveals a dynamic LSD1–CoREST interactome during embryonic stem cell differentiation. Molecular Omics 2022, 18:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BVL, Schwabe JWR: Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nature Communications 2016, 7:11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vinogradova EV, Zhang X, Remillard D, Lazar DC, Suciu RM, Wang Y, Bianco G, Yamashita Y, Crowley VM, Schafroth MA, et al. : An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182:1009–1026.e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Millard CJ, Varma N, Saleh A, Morris K, Watson PJ, Bottrill AR, Fairall L, Smith CJ, Schwabe JWR: The structure of the core NuRD repression complex provides insights into its interaction with chromatin. eLife 2016, 5:e13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walavalkar NM, Gordon N, Williams DC Jr.: Unique Features of the Anti-parallel, Heterodimeric Coiled-coil Interaction between Methyl-cytosine Binding Domain 2 (MBD2) Homologues and GATA Zinc Finger Domain Containing 2A (GATAD2A/p66α;). Journal of Biological Chemistry 2013, 288:3419–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allen Hillary F, Daze Kevin D, Shimbo T, Lai A, Musselman Catherine A, Sims Jennifer K, Wade Paul A, Hof F, Kutateladze Tatiana G: Inhibition of histone binding by supramolecular hosts. Biochemical Journal 2014, 459:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Daze KD, Ma MCF, Pineux F, Hof F: Synthesis of New Trisulfonated Calix[4]arenes Functionalized at the Upper Rim, and Their Complexation with the Trimethyllysine Epigenetic Mark. Organic Letters 2012, 14:1512–1515. [DOI] [PubMed] [Google Scholar]
- 75.Xiong Y, Donovan KA, Eleuteri NA, Kirmani N, Yue H, Razov A, Krupnick NM, Nowak RP, Fischer ES: Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chemical Biology 2021, 28:1514–1527.e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Describes the design and synthesis of HDAC degraders and characterized their selective binding and influence on various HDAC complexes.
- 76.Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E, Shi Y-J, Barretina J, Liu J, Howley PM, et al. : CDYL Bridges REST and Histone Methyltransferases for Gene Repression and Suppression of Cellular Transformation. Molecular Cell 2008, 32:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang L, Charroux B, Kerridge S, Tsai C-C: Atrophin recruits HDAC1/2 and G9a to modify histone H3K9 and to determine cell fates. EMBO reports 2008, 9:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng M, Hu Y, Song W, Duan S, Xu Q, Ding Y, Geng J, Zhou J: MIER3 suppresses colorectal cancer progression by down-regulating Sp1, inhibiting epithelial-mesenchymal transition. Scientific Reports 2017, 7:11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding Z, Gillespie LL, Mercer FC, Paterno GD: The SANT Domain of Human MI-ER1 Interacts with Sp1 to Interfere with GC Box Recognition and Repress Transcription from Its Own Promoter. Journal of Biological Chemistry 2004, 279:28009–28016. [DOI] [PubMed] [Google Scholar]
- 80.Hein Marco Y, Hubner Nina C, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak Igor A, Weisswange I, Mansfeld J, Buchholz F, et al. : A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163:712–723. [DOI] [PubMed] [Google Scholar]
- 81.Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM: The Conserved microRNA MiR-8 Tunes Atrophin Levels to Prevent Neurodegeneration in Drosophila. Cell 2007, 131:136–145. [DOI] [PubMed] [Google Scholar]
- 82.Hou R, Sibinga NES: Atrophin Proteins Interact with the Fat1 Cadherin and Regulate Migration and Orientation in Vascular Smooth Muscle Cells. Journal of Biological Chemistry 2009, 284:6955–6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim BJ, Zaveri HP, Jordan VK, Hernandez-Garcia A, Jacob DJ, Zamora DL, Yu W, Schwartz RJ, Scott DA: RERE deficiency leads to decreased expression of GATA4 and the development of ventricular septal defects. Disease Models & Mechanisms 2018, 11:dmm031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yanagisawa H, Bundo M, Miyashita T, Okamura-Oho Y, Tadokoro K, Tokunaga K, Yamada M: Protein binding of a DRPLA family through arginine-glutamic acid dipeptide repeats is enhanced by extended polyglutamine. Human Molecular Genetics 2000, 9:1433–1442. [DOI] [PubMed] [Google Scholar]
- 85.Banks CAS, Zhang Y, Miah S, Hao Y, Adams MK, Wen Z, Thornton JL, Florens L, Washburn MP: Integrative Modeling of a Sin3/HDAC Complex Sub-structure. Cell Reports 2020, 31:107516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Farias Eduardo F, Petrie K, Leibovitch B, Murtagh J, Chornet Manuel B, Schenk T, Zelent A, Waxman S: Interference with Sin3 function induces epigenetic reprogramming and differentiation in breast cancer cells. Proceedings of the National Academy of Sciences 2010, 107:11811–11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Clark MD, Zhang Y, Radhakrishnan I: Solution NMR Studies of an Alternative Mode of Sin3 Engagement by the Sds3 Subunit in the Histone Deacetylase-Associated Sin3L/Rpd3L Corepressor Complex. Journal of Molecular Biology 2015, 427:3817–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Becher I, Dittmann A, Savitski MM, Hopf C, Drewes G, Bantscheff M: Chemoproteomics Reveals Time-Dependent Binding of Histone Deacetylase Inhibitors to Endogenous Repressor Complexes. ACS Chemical Biology 2014, 9:1736–1746. [DOI] [PubMed] [Google Scholar]
- 89.Laitaoja M, Tossavainen H, Pihlajamaa T, Valjakka J, Viiri K, Lohi O, Permi P, Jänis J: Redox-dependent disulfide bond formation in SAP30L corepressor protein: Implications for structure and function. Protein Science 2016, 25:572–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie T, He Y, Korkeamaki H, Zhang Y, Imhoff R, Lohi O, Radhakrishnan I: Structure of the 30-kDa Sin3-associated Protein (SAP30) in Complex with the Mammalian Sin3A Corepressor and Its Role in Nucleic Acid Binding. Journal of Biological Chemistry 2011, 286:27814–27824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watson PJ, Fairall L, Santos GM, Schwabe JWR: Structure of HDAC3 bound to corepressor and inositol tetraphosphate. Nature 2012, 481:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong S-H, Yang Z, Privalsky Martin L: Arsenic Trioxide Is a Potent Inhibitor of the Interaction of SMRT Corepressor with Its Transcription Factor Partners, Including the PML-Retinoic Acid Receptor α Oncoprotein Found in Human Acute Promyelocytic Leukemia. Molecular and Cellular Biology 2001, 21:7172–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alrfaei BM, Vemuganti R, Kuo JS: microRNA-100 Targets SMRT/NCOR2, Reduces Proliferation, and Improves Survival in Glioblastoma Animal Models. PLOS ONE 2013, 8:e80865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gurard-Levin ZA, Kim J, Mrksich M: Combining Mass Spectrometry and Peptide Arrays to Profile the Specificities of Histone Deacetylases. ChemBioChem 2009, 10:2159–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riester D, Hildmann C, Grünewald S, Beckers T, Schwienhorst A: Factors affecting the substrate specificity of histone deacetylases. Biochemical and Biophysical Research Communications 2007, 357:439–445. [DOI] [PubMed] [Google Scholar]
- 96.Liu K, Xu C, Lei M, Yang A, Loppnau P, Hughes TR, Min J: Structural basis for the ability of MBD domains to bind methyl-CG and TG sites in DNA. Journal of Biological Chemistry 2018, 293:7344–7354 [DOI] [PMC free article] [PubMed] [Google Scholar]