

Abstract
GPIHBP1 plays an important role in the hydrolysis of triglyceride (TG) lipoproteins by lipoprotein lipases (LPLs). However, Gpihbp1 knockout mice did not develop hypertriglyceridemia (HTG) during the suckling period but developed severe HTG after weaning on a chow diet. It has been postulated that LPL expression in the liver of suckling mice may be involved. To determine whether hepatic LPL expression could correct severe HTG in Gpihbp1 deficiency, liver-targeted LPL expression was achieved via intravenous administration of the adeno-associated virus (AAV)-human LPL gene, and the effects of AAV-LPL on HTG and HTG-related acute pancreatitis (HTG-AP) were observed. Suckling Gpihbp1−/− mice with high hepatic LPL expression did not develop HTG, whereas Gpihbp1−/− rat pups without hepatic LPL expression developed severe HTG. AAV-mediated liver-targeted LPL expression dose-dependently decreased plasma TG levels in Gpihbp1−/− mice and rats, increased post-heparin plasma LPL mass and activity, decreased mortality in Gpihbp1−/− rat pups, and reduced the susceptibility and severity of both Gpihbp1−/− animals to HTG-AP. However, the muscle expression of AAV-LPL had no significant effect on HTG. Targeted expression of LPL in the liver showed no obvious adverse reactions. Thus, liver-targeted LPL expression may be a new therapeutic approach for HTG-AP caused by GPIHBP1 deficiency.
Keywords: lipoprotein lipase, GPIHBP1, hypertriglyceridemia, gene therapy, adeno-associated virus, acute pancreatitis
Graphical abstract
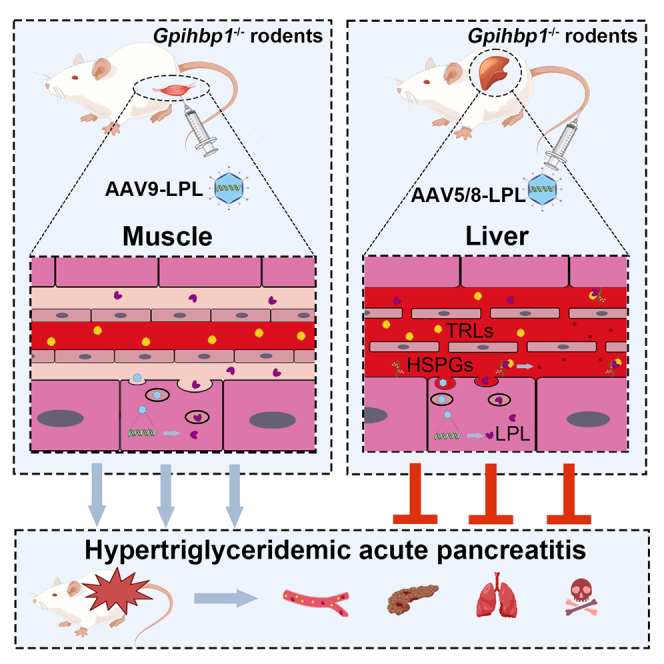
Li and colleagues develop an AAV vector encoding human LPL-S447X, which enable them to reduce high plasma TG levels and alleviate HTG-AP without discernible adverse effects in Gpihbp1−/− mice and rats. AAV-mediated liver-specific expression of human LPL serves as an effective treatment for HTG-AP caused by Gpihbp1 deficiency.
Introduction
Hypertriglyceridemia (HTG) is a multifactorial disease characterized by the abnormal synthesis or degradation of triglycerides (TGs), resulting in their excessive accumulation in the plasma.1,2 Lipoprotein lipase (LPL) is a key enzyme involved in TG hydrolysis. LPL hydrolyzes endogenous and exogenous TGs carried by plasma very-low-density lipoproteins (VLDL) and chylomicrons (CMs), respectively. Released free fatty acids (FFAs) are taken up by adipocytes to form TG storage or are utilized as energy in skeletal and cardiac muscle.1,3 The processes of LPL maturation, processing, transport, and hydrolysis of triglycerides are tightly controlled, with many accessory proteins helping it function. Lipase maturation factor 1 (LMF1), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), apolipoprotein A5 (ApoA5), and apolipoprotein C2 (ApoC2) are all involved in the activation and transport of LPL.4,5 Specifically, GPIHBP1, which is responsible for LPL transport to capillary endothelium,6 can bind LPL and chylomicrons simultaneously, providing a platform for LPL hydrolysis of TG and playing an important role in the lipolysis of chylomicrons.7,8,9
Familial chylomicronemia syndrome (FCS), which is mainly caused by LPL loss-of-function mutations, is an extremely rare group of monogenic diseases with a reported prevalence of approximately 1 per million.10 FCS is characterized by severe HTG and is frequently accompanied by recurrent acute pancreatitis (AP), which can be life threatening. Patients with GPIHBP1 mutations have been reported to develop severe HTG and recurrent AP, and their heterozygous family members experience mild fasting HTG.11
Although a deficiency in GPIHBP1 is well documented to result in severe HTG, Young et al.7 found that plasma TG levels in suckling Gpihbp1−/− mouse pups (<4 weeks of age) were only slightly higher than those in wild-type (WT) mice, but they developed severe HTG after a normal laboratory chow diet at 4 to 5 weeks of age. This is puzzling in contrast to the phenotype of all deaths due to severe HTG during the suckling period in Lpl knockout mice and ApoC2 knockout hamsters.12,13 Young et al.7 suggested that near-normal TG levels during the suckling period in Gpihbp1−/− mouse pups might be related to hepatic LPL expression during this stage, given previous findings that mouse liver Lpl genes were highly expressed during the suckling period, gradually decreased, and stopped completely after weaning. They also found that the levels of Lpl mRNA in the livers of suckling Gpihbp1−/− mice were more than 10 times higher than those in adult mice. Additionally, we generated Gpihbp1 gene-deficient rats, which, in contrast to Gpihbp1 knockout mice, developed severe HTG with high mortality during the suckling period (G.L., B.L., Y.X., et al. unpublished data). Therefore, it would be of interest to determine whether the livers of suckling Gpihbp1−/− rat pups exhibit LPL expression. If there is no or low hepatic LPL expression in suckling rat pups, it would suggest that the near-normal TG levels in Gpihbp1−/− mouse pups are likely to result from hepatic LPL expression. Moreover, if LPL expression could be induced in the liver in the presence of Gpihbp1 deficiency, it is plausible that severe HTG could be ameliorated.
Adeno-associated viruses (AAVs) delivered intravenously are known to be mainly expressed in the liver, and AAV vectors of serotypes 5 and 8 have a high specificity for liver targeting.14 Hence, the use of AAV vectors to mediate liver-targeted expression of LPL will verify whether normal TG levels in suckling Gpihbp1−/− mouse pups are determined by hepatic LPL. AAV is also an ideal vector for gene therapy for a variety of diseases.15 In 2012, the European Medicines Agency (EMA) approved the first gene therapies in Western countries, alipogene tiparvovec (Glybera), AAV1-LPL-S447X, as treatments for LPL gene-deficient patients who develop severe HTG and recurrent AP.16,17 Unfortunately, this drug was used in only one case after approval and has not been further utilized, probably because of issues such as high cost, intramuscular multipoint injection, and limited TG-lowering effects.18 However, no relevant clinical treatment has been reported for patients with GPIHBP1 deficiency. Therefore, it will be desirable to investigate whether AAV-mediated liver-targeted expression of LPL could serve as an effective intervention for mitigating severe HTG in both Gpihbp1−/− rats and mice. If it works as we expect, the liver-targeted expression of LPL may serve as a novel therapeutic strategy for addressing severe HTG in Gpihbp1-deficient patients.
Results
Lpl is expressed in the liver of suckling Gpihbp1 knockout mouse pups but not in similar rat pups
Previous studies have reported that plasma TG levels (120 ± 12 mg/dL) in Gpihbp1−/− mice are only slightly higher than WT mice during the suckling period (<4 weeks of age) and have a stable HTG phenotype when plasma TG levels exceed 1,000 mg/dL or even up to 5,000 mg/dL in adulthood.7 We first examined the expression of Lpl mRNA in the liver of suckling Gpihbp1−/− mice and found that, consistent with previous reports, endogenous Lpl was highly expressed in the liver 7 days after birth, followed by a gradual decline, with little Lpl expression in the liver at weaning (28 days) (Figure 1A).
Figure 1.

Correlation between Lpl mRNA expression and plasma lipid levels in suckling liver of Gpihbp1−/− rats and mice
(A and B) Lpl mRNA expression in the liver of Gpihbp1−/− mice (n = 6) and rats (n = 6) during suckling period. (C) Log2 of plasma TG and TC levels in WT (n = 4) and Gpihbp1−/− mice (n = 6) during suckling period. (D) Log2 of plasma TG and TC levels in WT (n = 4) and Gpihbp1−/− rats (n = 6) during suckling period. Data are presented as mean ± standard error of mean (SEM). Statistical comparisons were made using two-way ANOVA with Sidak correction. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. WT mice or WT rats.
Unlike Gpihbp1−/− mouse pups, Gpihbp1−/− rat pups exhibited severe HTG at birth, and only 60% of homozygous Gpihbp1−/− rats survived suckling without any intervention owing to spontaneous HTG-AP (G.L., B.L., Y.X., et al. unpublished data). We examined Lpl mRNA in the livers of Gpihbp1−/− rat pups and found Lpl mRNA remained low from day 7 through day 28 (Figure 1B), which might explain the difference in plasma TG and total cholesterol (TC) levels between Gpihbp1−/− mouse pups and rat pups (Figures 1C and 1D).
AAV mediates overexpression of EGFP and LPL in the liver
The structure of the recombinant AAV (serotype 5)-LPL viral vector is shown in Figure 2A. First, we evaluated the safety of AAV-LPL recombinant viral vectors in WT animals. Four weeks after tail vein injection of AAV-enhanced green fluorescent protein (EGFP) at 1 × 1013 vector genomes (vg)/kg and AAV-LPL at various doses in WT mice, no morphological abnormalities were observed in the heart, liver, lungs, kidneys, or pancreas stained by hematoxylin and eosin (H&E) (Figure S1A). Alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TBIL), creatinine (CERA), and blood urea nitrogen (BUN) levels were not significantly abnormal, and the values were within the normal range (Figures S1B–S1F).
Figure 2.

Construction and infection efficacy to liver of AAV-LPL recombinant viral vector
(A) Structure diagram of AAV-LPL recombinant viral vector. (B) Representative fluorescent images of livers from WT mice treated with different doses of AAV-LPL (n = 6 per group). Scale bar, 100 μm. (C) Representative fluorescent images of various organs from WT mice treated with 1 × 1013 vg/kg AAV-EGFP (n = 6). Scale bar, 100 μm. (D) Expression of human LPL mRNA relative to Gapdh in various tissues of WT mice treated with 1 × 1013 vg/kg AAV-LPL (n = 6). (E) Expression of human LPL mRNA relative to Gapdh in liver from WT mice treated with different doses of AAV-LPL (n = 6 per group). (F) Immunohistochemical analysis of LPL in the liver from WT mice treated with different doses of AAV-LPL (n = 6 per group). Scale bars, 200 and 50 μm. Data are presented as mean ± SEM.
To increase the specificity of AAV5-mediated transgene expression in the liver, a strong liver-specific promoter, named the LP promoter, was used in both LPL and EGFP gene expression cassettes (Figure 2A). As expected, the livers treated with AAV-EGFP expressed green fluorescent protein at a higher level than the AAV-LPL-treated livers (Figure 2B), while no obvious green fluorescence was detected in other organs from AAV-EGFP-treated mice (Figure 2C). These results were confirmed by qRT-PCR (Figure 2D). Although there was no endogenous mouse Lpl mRNA in the liver of adult WT mice, the expression level of human LPL mRNA in the livers increased with increasing doses of AAV-LPL. The hepatic LPL mRNA in the 1 × 1013 vg/kg AAV-LPL group increased approximately 8-fold compared with 1 × 1011 vg/kg AAV-LPL and was about 2-fold higher than that in the 1 × 1012 vg/kg AAV-LPL group (Figure 2E). The LPL protein in the liver also appeared to increase with the doses of the AAV-LPL vector, as detected by immunochemistry, which seemed to be located mainly in plasma membrane and cytosol (Figure 2F).
AAV-mediated liver expression of LPL increases post-heparin plasma LPL in Gpihbp1−/− mice and corrects severe HTG
S447X is the only LPL beneficial mutant found to date that promotes LPL secretion and increases the interaction of LPL with lipoproteins and cell surface receptors. Treatment with human LPL-S447X is significantly more effective in reducing TG levels and increasing LPL activity than normal human LPL gene therapy; hence, S447X was selected for gene therapy.19 In order to assess the LPL-S447X-specific plasma TG-lowering effect in Gpihbp1−/− mice, we conducted experiments involving the liver-specific expression of the LPL gene and its 447 mutant. Gpihbp1−/− mice were randomly divided into three groups: PBS, 1 × 1013 vg/kg AAV-LPL-S447X and 1 × 1013 vg/kg AAV-LPL-WT. Plasma TG and TC levels in the three groups were determined on days 7, 14, 21, and 28 after treatment. Within 28 days, AAV-mediated hepatic expression of LPL-WT and LPL-S447X at 1 × 1013 vg/kg resulted in a comparable reduction in plasma TG levels, whereas AAV-LPL-S447X further lowered plasma TG levels. Plasma TC levels also changed in parallel with TG levels but to a lesser extent, as shown in Figures 3A and 3B. Therefore, LPL-S447X was selected as the gene therapy regimen and named AAV-LPL for subsequent experiments.
Figure 3.

Comparison of lipid-lowering effects of AAV-LPL-S447X and AAV-LPL-WT in Gpihbp1−/− mice
(A and B) Plasma TG and TC levels at 7 days (7d), 14 days (14d), and 28 days (28d) after treatment with AAV-LPL-S447X and AAV-LPL-WT (n = 6 per group). Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared to PBS group, two-way ANOVA with Sidak correction.
In the second set of experiments, four groups of Gpihbp1−/− mice aged 10–12 weeks received (1) PBS only, (2) AAV-EGFP, (3) AAV-LPL at 1 × 1011 vg/kg as a low dose, and (4) AAV-LPL at 1 × 1013 vg/kg as a high dose through tail vein injection. We evaluated the safety of AAV-LPL treatment and showed that AAV-mediated hepatic expression of LPL did not cause significant organ damage to Gpihbp1−/− mice (Figures S2A–S2F). Changes in plasma TG and TC levels are shown in Figures 4A–4C. Plasma TG and TC levels decreased significantly 7 days after AAV-LPL injection and remained at normal levels for a long time. After 2 months of treatment, plasma TG levels in the PBS group and AAV-EGFP control group were maintained at about 2,200 mg/dL, whereas the TG levels in the low- and high-dose AAV-LPL treatment groups decreased significantly to about 1,000 and 200 mg/dL, respectively. Plasma TC levels in the low- and high-dose AAV-LPL-treated groups decreased from 600 mg/dL to approximately 300 and 200 mg/dL, respectively, compared to the PBS and AAV-EGFP groups. After 2 months of treatment, there was no significant difference in post-heparin plasma LPL concentration and activity in the AAV-EGFP group compared to the PBS group, while the plasma LPL concentration increased to 100 ng/mL and the activity increased approximately 1.8-fold in the low-dose AAV-LPL treatment group. Meanwhile, LPL protein and activity increased to 500 ng/mL and 3-fold, respectively, in the high-dose AAV-LPL treatment group (Figures 4D and 4E). As shown in Figure 4F, the plasma samples from the PBS and AAV-EGFP groups remained milky, whereas the plasma from the low-dose AAV-LPL group was partially milky. In contrast, the plasma from the high-dose AAV-LPL group was nearly as transparent as that from WT mice.
Figure 4.

Effect of intravenous injection of AAV-LPL on plasma lipid levels in Gpihbp1−/− mice
(A and B) Dynamic changes of plasma TG and TC levels at different time points after different doses of AAV-LPL treatment. (C) Plasma TG and TC levels at 2 months after different doses of AAV-LPL treatment. (D and E) Post-heparin plasma LPL concentration and activity after 2 months of treatment with different doses of AAV-LPL. (F) Representative plasma sample appearance after 2 months of treatment with different doses of AAV-LPL. PBS, n = 6; 1 × 1013 vg/kg AAV-EGFP, n = 6; 1 × 1011 vg/kg AAV-LPL, n = 12; 1 × 1013 vg/kg AAV-LPL, n = 12. For (A–C), statistical comparisons were made using two-way ANOVA with Sidak correction. For (D) and (E), statistical comparisons were made using one-way ANOVA with LSD correction. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. PBS or 1 × 1013 vg/kg AAV-EGFP group.
AAV-mediated liver expression of LPL improves survival of suckling Gpihbp1−/− rat pups and corrects severe HTG in adult Gpihbp1−/− rats
As shown in Figures S3A–S3F, no significant organ damage was observed in AAV-LPL-treated Gpihbp1−/− rats. To reduce the high mortality during the suckling period in Gpihbp1−/− rat pups, we injected 1 × 1013 vg/kg AAV-LPL via the orbital vein at 7 days after birth into neonatal Gpihbp1−/− suckling pups. Similar Gpihbp1−/− pups were injected with PBS as controls. The experimental protocol is shown in Figure 5A. Compared to the control group, the survival rate of Gpihbp1−/− pups injected with AAV-LPL increased from 60% to approximately 80% (Figure 5B). The plasma TG levels decreased significantly to approximately 800 mg/dL 7 days after treatment, whereas plasma TG levels in pups of the PBS controls were above 3,000 mg/dL. With the growth and development of suckling rats and the change from breast milk to a chow diet, the plasma TG levels in AAV-LPL-treated pups gradually increased. By 8 weeks of age, the mean TG level reached 1,200 mg/dL but was still significantly lower than that of the PBS control group at approximately 2,500 mg/dL.
Figure 5.

Effect of intravenous injection of AAV-LPL on survival rate and plasma lipid levels in Gpihbp1−/− rats
(A) Experimental protocol of the administration of AAV-LPL in suckling and adult Gpihbp1−/− rats. Neonatal Gpihbp1−/− suckling pups were injected with 1 × 1013 vg/kg AAV5-LPL via orbital vein at 7 days of birth. Another serotype of AAV recombinant viral vector, AAV8-LPL, was administered in these animals at age 8 weeks in high (1 × 1013 vg/kg) and low dose (1 × 1011 vg/kg), respectively. Gpihbp1−/− rats receiving PBS served as controls. (B) Twenty-eight-day survival curves of suckling Gpihbp1−/− rats treated with 1 × 1013 vg/kg AAV5-LPL (n = 52) or PBS (n = 44). (C and D) Dynamic changes of plasma TG and TC levels at different time points after AAV5-LPL and AAV8-LPL gene therapy. (E and F) Post-heparin plasma LPL concentrations and activities after 2 weeks (10 weeks old) of the administration of AAV8-LPL at different doses. (G) Representative plasma sample appearance after 2 weeks (10 weeks old) of the administration of AAV8-LPL at different doses. PBS, n = 12; 1 × 1013 vg/kg AAV5-LPL, n = 32; 1 × 1011 vg/kg AAV8-LPL, n = 16; 1 × 1013 vg/kg AAV8-LPL, n = 16. Data are presented as mean ± SEM. Survival analysis was made using Kaplan-Meier method and log rank test. For (C) and (D), statistical comparisons were made using two-way ANOVA with Sidak correction. For (E) and (F), statistical comparisons were made using one-way ANOVA with LSD correction. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. PBS group.
In view of the relatively insufficient amount of virus in the adult liver of Gpihbp1−/− rats treated with AAV-LPL during the suckling period, we administered another serotype of AAV vector, AAV8, containing the same LPL-S447X to these animals at the age of 8 weeks in high (1 × 1013 vg/kg) and low dose (1 × 1011 vg/kg). The control group was administered only PBS (Figure 5A). As shown in Figures 5C and 5D, plasma TG levels decreased to approximately 100 and 500 mg/dL, respectively, in the high- and low-dose AAV8-LPL treatment groups. Plasma TC levels were also significantly reduced. The long-term (up to 16 weeks) lipid-lowering efficacy of AAV treatment was also observed (Figures S4A and S4B). Compared with the control group, the plasma LPL concentration increased to 300 ng/mL and the activity increased approximately 1.5-fold in the low-dose AAV8-LPL-treated rats. In the high-dose AAV8-LPL group, the LPL mass reached 600 ng/mL, whereas the activity increased approximately 3-fold in post-heparin plasma (Figures 5E and 5F). Consistent with the changes in plasma TG levels, the appearance of the plasma from the PBS control group remained milky but was entirely transparent in the group receiving the second treatment of high-dose AAV8-LPL after 2 weeks, whereas the plasma samples of the animals in the low-dose group were still cloudy or partially transparent as shown in Figure 5G.
AAV-mediated liver expression of LPL decreases the susceptibility and severity of Gpihbp1−/− mice and rats HTG-AP
To investigate the effect of AAV-mediated hepatic expression of LPL on the pathogenesis of HTG-AP, Gpihbp1−/− mice were treated with 1 × 1013 vg/kg AAV-LPL for 2 months. HTG-AP was induced by the intraperitoneal injection of caerulein (Cae). Very low doses of Cae (5 μg/kg) did not cause pancreatic injury in WT mice but resulted in significant increases in plasma amylase and lipase, and they caused mild edema plus certain inflammatory cell infiltration in the pancreas in control Gpihbp1−/− mice. AAV-LPL treatment significantly decreased plasma amylase and lipase levels and restored the normal pathological morphology of the pancreas (Figures 6A and 6B). In the experiment of HTG-AP induced by regular doses of Cae (50 μg/kg), the plasma amylase and lipase of Gpihbp1−/− mice in the AAV-LPL-treated group significantly decreased, and the pathological damage of pancreatic tissue was significantly reduced, as shown by decreased edema, decreased inflammatory cell infiltration, and acinar cell necrosis (Figures 6C and 6D). These results suggested that the susceptibility of Gpihbp1−/− mice to HTG-AP was greatly reduced after AAV-LPL treatment.
Figure 6.

Effect of intravenous administration of AAV-LPL on the pathogenesis of HTG-AP in Gpihbp1−/− mice
(A) Gpihbp1−/− mice were injected intraperitoneally with a very low dose (5 μg/kg) of caerulein (Cae) after intravenous injection of 1 × 1013 vg/kg AAV-LPL, and the susceptibility to HTG-AP was observed by H&E staining of pancreatic tissue. WT + PBS, n = 4; Gpihbp1−/− + PBS, n = 4; Gpihbp1−/− + 1 × 1013 vg/kg AAV-LPL + PBS, n = 4; WT + Cae, n = 6; Gpihbp1−/− + Cae, n = 6; Gpihbp1−/− + 1 × 1013 vg/kg AAV-LPL + Cae, n = 6. (B) Plasma amylase and lipase levels in Gpihbp1−/− mice injected with very-low-dose (5 μg/kg) Cae-induced HTG-AP. (C) HTG-AP was induced by intraperitoneal injection of Cae (50 μg/kg) after intravenous injection of 1 × 1013 vg/kg AAV-LPL to Gpihbp1−/− mice. Pancreatic injury was observed by H&E staining. WT + PBS, n = 4; Gpihbp1−/− + PBS, n = 4; Gpihbp1−/− + 1 × 1013 vg/kg AAV-LPL + PBS, n = 4; WT + Cae, n = 6; Gpihbp1−/− + Cae, n = 6; Gpihbp1−/− + 1 × 1013 vg/kg AAV-LPL + Cae, n = 6. (D) Changes in plasma amylase and lipase levels after induction of HTG-AP by 50 μg/kg of Cae. Scale bar, 50 μm. Data are presented as mean ± SEM. For (B) and (D), statistical comparisons were made using two-way ANOVA with Sidak correction. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. WT + Cae or Gpihbp1−/− + Cae group.
We previously found that HTG-AP could be induced in adult Gpihbp1−/− rats by feeding them a high-fat diet (HFD) for 10–14 days, which could serve as another model of HTG-AP (G.L., B.L., Y.X., et al. unpublished data). Indeed, Gpihbp1−/− rats fed an HFD developed severe HTG with plasma TG levels reaching as high as 15,000 mg/dL in only 3 days and were maintained at the extreme HTG for 14 days. When the animals were sacrificed for pathological evaluation, extensive necrosis of the pancreatic tissue, massive inflammatory cell infiltration, and fibrous connective tissue hyperplasia were observed with only a few remaining pancreatic acinar cell structures. There were also manifestations of acute lung injury, including thickening of the alveolar septa and massive inflammatory cell infiltration, suggesting a severe systemic inflammatory response and multiple organ dysfunction. However, in Gpihbp1−/− rats receiving AAV-LPL in the suckling stage and the second intravenous injection of AAV8-LPL 2 months later (Figure 7A), there was a significant reduction in HFD-induced HTG along with reduced plasma TC (Figures 7B and 7C). AAV-LPL treatment also significantly attenuated the degree of pathological injury in pancreatic and lung tissues. As shown in Figures 7D and 7E, only tissue edema and a small amount of inflammatory cell infiltration were observed, and acinar cell necrosis was rare in the high-dose AAV-LPL-treated group, comparable to the mild injury state in Gpihbp1−/− rats under chow diet feeding. These results clearly indicate that AAV-LPL administration during suckling and secondary treatment with AAV8-LPL in adulthood markedly reduced the severity of spontaneous HTG-AP induced by HFD feeding in Gpihbp1−/− rats.
Figure 7.

Effect of intravenous administration of AAV-LPL on HFD-induced spontaneous HTG-AP in Gpihbp1−/− rats
(A) Experimental protocol of the administration of AAV-LPL on HFD-induced HTG-AP in Gpihbp1−/− rats. Seven-day-old Gpihbp1−/− suckling pups were injected with 1 × 1013 vg/kg AAV5-LPL, and subsequently AAV8-LPL was administered in these animals at age of 8 weeks in high (1 × 1013 vg/kg) and low dose (1 × 1011 vg/kg), respectively. Two weeks after AAV8 treatment, Gpihbp1−/− rats were fed with HFD to induce spontaneous HTG-AP. (B and C) The plasma TG and TC levels of Gpihbp1−/− rats in each group at different time points after HFD feeding. (D) Representative H&E images of pancreatic and lung tissues of Gpihbp1−/− rats in each group after 14 days of HFD feeding. WT + HFD, n = 8; Gpihbp1−/− + PBS + HFD, n = 18; Gpihbp1−/− + 1 × 1011 vg/kg AAV8-LPL+HFD, n = 16; Gpihbp1−/− + 1 × 1013 vg/kg AAV8-LPL + HFD, n = 16; Gpihbp1−/− + chow diet (CD), n = 16. Scale bars, 100 and 25 μm. (E) Pathological score of pancreatic injury in Gpihbp1−/− rats of each group after 14 days of HFD feeding. For (B) and (C), statistical comparisons were made using two-way ANOVA with Sidak correction. For (E), statistical comparisons were made using one-way ANOVA with LSD correction. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. Gpihbp1−/− + PBS + HFD or Gpihbp1−/− + 1 × 1011 vg/kg AAV8-LPL + HFD.
LPL expression in muscle has no effect on severe HTG with Gpihbp1 deficiency
The muscle is the site of physiological LPL expression. To compare the effects of AAV-mediated liver expression of human LPL-S447X with those of muscle expression following severe HTG treatment in Gpihbp1−/− mice and rats, an AAV9-LPL recombinant viral vector was designed and constructed. The liver-specific promoter in the AAV5-LPL vector was replaced with a muscle-specific strong promoter, the AAV9 vector with higher muscle tissue specificity was selected, the remaining construction process was unchanged. Gpihbp1−/− mice and rats were administered the same amount of PBS, 1 × 1013 vg/kg AAV9-EGFP and 1 × 1013 vg/kg AAV9-LPL, to evaluate the lipid-lowering effect of AAV9-LPL intramuscular administration. Muscles treated with AAV9-EGFP from Gpihbp1−/− mice and rats expressed high levels of green fluorescent protein compared to the AAV9-LPL-treated groups (Figures S5A and S5B). Two months after intramuscular injection of AAV9-LPL, the plasma TG and TC levels of these mice were not significantly different from those of the AAV9-EGFP control and the PBS groups. In addition, the milky appearance of the plasma remained unchanged (Figures 8A and 8G). Intramuscular AAV9-LPL administration did not increase LPL mass or activity in post-heparin plasma. There was no change in LPL activity, but there was a slight elevation in human LPL mRNA in muscle tissues in the group receiving an intramuscular injection of AAV9-LPL (Figures 8B and 8C). Similarly, no significant lipid-lowering effects were observed in Gpihbp1−/− rats receiving AAV9-LPL (Figures 8D–8G). These results suggest that intramuscular delivery of the LPL gene did not effectively increase circulating LPL mass and activity in Gpihbp1−/− mice and rats or alleviate severe HTG. Thus, hepatic expression of LPL has tissue-specific effects on the reduction of severe HTG in Gpihbp1 gene-deficient animals.
Figure 8.

Effect of intramuscular injection of AAV9-LPL recombinant viral vector on plasma lipid levels of Gpihbp1−/− mice and rats
(A) Dynamic changes of plasma TG and TC levels at different time points after intramuscular injection of AAV9-LPL in Gpihbp1−/− mice (n = 6 per group). (B) Post-heparin plasma LPL concentration and activity after 2 months of AAV9-LPL treatment in Gpihbp1−/− mice (n = 6 per group). (C) Muscular human LPL mRNA expression level after 2 months of AAV9-LPL treatment in Gpihbp1−/− mice (n = 6 per group). (D) Dynamic changes of plasma TG and TC levels at different time points after intramuscular injection of AAV9-LPL in Gpihbp1−/− rats (n = 6 per group). (E) Post-heparin plasma LPL concentrations and activities after 2 months of AAV9-LPL treatment in Gpihbp1−/− rats (n = 6 per group). (F) Muscular human LPL mRNA expression level after 2 months of AAV9-LPL treatment in Gpihbp1−/− rats (n = 6 per group). (G) Representative plasma sample appearance after 2 months of AAV9-LPL treatment in Gpihbp1−/− mice and rats (n = 6 per group). (H) Immunofluorescence assay showed that liver LPL was colocalized with HSPG after 2 months of intravenous administration of AAV-LPL in Gpihbp1−/− mice and rats (n = 6 per group). Scale bar, 100 μm. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 vs. Gpihbp1−/− + PBS or Gpihbp1−/− + AAV9-EGFP group, one-way ANOVA and LSD test.
Colocalization of hepatic-targeted LPL expression with HSPG in the livers of Gpihbp1−/− mice and rats
Heparan sulfate proteoglycans (HSPGs) are similar to heparin in nature and contain polyanions that can bind to the positively charged arginine residues of LPL. HSPG is located on the surface of liver cells, binds to LPL with low affinity, and anchors LPL to hepatocytes. However, this effect is less pronounced than that of GPIHBP1, and HSPG may also have no effect on the transport of cell-surface LPL to the endothelial surface. Because of the large gap between hepatic endothelial cells and the presence of a Disse space between endothelial cells and hepatocytes, LPL anchored on the surface HSPG of hepatocytes can directly contact chylomicrons and VLDL in the plasma for TG hydrolysis. Therefore, we hypothesized that AAV-mediated LPL overexpression in the liver may effectively reduce TG levels in Gpihbp1 gene-deficient animals via similar pathways. We determined whether LPL and HSPG could co-localize by immunofluorescence staining. As shown in Figure 8H, the LPL signal was very low in the livers of untreated Gpihbp1−/− mice and rats, whereas the liver LPL fluorescence signal significantly increased after the intravenous delivery of AAV-LPL. Fluorescence signals of HSPG were observed in the livers of both Gpihbp1−/− mice and rats, and indeed colocalized with those of LPL on the liver cell membrane, which may be a critical site of action for LPL in the hydrolysis of plasma TG when GPIHBP1 is absent.
Discussion
Based on the finding of normal TG levels and high hepatic LPL expression in Gpihbp1 knockout mice during the suckling period and severe HTG, but no LPL expression in the liver of Gpihbp1 knockout rats during the suckling period, we found that AAV-mediated hepatic expression of LPL can effectively and safely correct severe HTG in Gpihbp1−/− mice and rats in adulthood. This finding implies that targeted LPL expression in the liver may serve as an effective treatment for patients with severe HTG with GPIHBP1 gene deficiency. The present study further confirmed that AAV-mediated LPL expressed in the liver could hydrolyze TG-rich lipoproteins in the plasma by binding to HSPG on the cell surface in the absence of the GPIHBP1 protein, whereas the muscular expression of LPL-S447X mediated by AAV9 had no effect on plasma TG. We compared LPL-WT and LPL-S447X beneficial mutants and found that both had essentially the same TG-lowering effects in mice deficient in Gpihbp1, indicating that this effect was not S447X specific. Given that LPL-S447X has been validated in phase 3 clinical trials as a therapeutic gene, we completed this study using LPL-S447X, laying the foundation for future translation into clinical trials.
Patients with severe HTG with GPIHBP1 deficiency often develop recurrent life-threatening AP; however, there is currently no effective treatment for severe HTG, especially without intervention to prevent the development of severe HTG-AP. Glybera, the commercial name of AAV1-LPL-S447X, as a gene therapy approved for marketing in 2012 by the EMA exhibited certain efficacy in severe HTG caused by LPL gene deficiency.20 Glybera was delivered to patients by multiple injections in the quadriceps femoris muscle. Although the viral load of Glybera at the injection site was high, the spread to the adjacent muscle tissue through the injection site was extremely limited. Hence, the efficacy was not ideal, and the plasma TG levels were still much higher in treated patients than in normal.18 Owing to the inconvenience of multiple intramuscular injections, treatment costs, and unsatisfactory lipid-lowering efficacy, Glybera is not widely accepted as a cost-effective therapeutic agent for the treatment of clinically severe HTG.
Our results suggest that only suckling Gpihbp1−/− mouse pups, but not rat pups, express LPL at high levels in the liver, which may be critical for maintaining the normal TG levels during suckling in Gpihbp1−/− mouse pups. Therefore, in this study, the liver was selected as the target organ for LPL-S447X gene expression. By selecting S447X as a therapeutic gene and optimizing the codon sequences to enhance efficacy, we added an liver-specific promoter to increase the specificity of liver infection. Furthermore, the AAV5 vector was chosen to mediate LPL expression in the liver via a single intravenous injection, which overcomes the disadvantages of multisite injections into the muscle, resulting in severe pain and poor infection. Thus, the combined effects of our gene delivery method greatly simplified the mode of administration and enabled the application of LPL gene delivery in clinical practice.
Our results showed that the expression level of LPL mRNA in liver was significantly increased after intravenous injection of AAV-LPL, and the expression of LPL mRNA was dose dependent. LPL protein levels and activity in post-heparin plasma were significantly increased, and plasma TG levels in Gpihbp1−/− mice and rats were markedly reduced. At the same time, we generated a spontaneous HTG-AP model using Gpihbp1−/− rats to better simulate the clinical characteristics of HTG-AP pathogenesis and pathophysiological processes. Our results showed that hepatic expression of AAV-LPL not only effectively reduced TG levels but also reduced the susceptibility and severity of spontaneous HTG-AP in Gpihbp1−/− rats.
Due to the severe HTG and high lethality in Gpihbp1−/− neonatal rat pups, we administered AAV-LPL intravenously to these new-born animals, which significantly reduced severe HTG and improved the survival rate during suckling. This was also consistent with the clinical characteristics of abnormally high fetal mortality in severe HTG infants,21,22 providing experimental evidence for future neonatal interventions. Replication-deficient AAV vectors administered during suckling are relatively inadequate in adult animals because of the increased liver size during development. Therefore, in this study, we carried out a second administration of LPL to adult Gpihbp1−/− rats that received AAV-LPL during suckling. Considering the immune response to AAV via the production of neutralizing antibodies against the primary AAV vector, thus affecting the therapeutic effect of second administration of AAV-LPL in adults, we switched to AAV8 vector and achieved further TG-lowering effect in the Gpihbp1−/− rats that received AAV-LPL at the neonatal stage, with a concomitant increase in LPL protein and activity in post-heparin plasma. This clearly demonstrates that, in AAV-mediated gene therapy regimens, it is possible to execute secondary dosing by replacing AAV vectors of different serotypes.
Prior to the discovery of the function of GPIHBP1 in interacting with LPL, both chylomicrons and LPL were considered to bind to HSPG on capillaries and cell surfaces.23 Relevant evidence includes the positively charged heparin-binding domain of LPL that binds to negatively charged HSPGs on the cell surface.24 Heparin injections release LPL from capillary endothelial cells. Heparin (strong anion) disrupts the electrostatic interaction between LPL and HSPG.25 In vitro experiments confirmed that normal endothelial cells bind more LPL than HSPG-null endothelial cells.26 Hepatocytes expressing HSPG similarly have a similar effect of anchoring LPL to the cell surface and vascular endothelium27 but are less pronounced than those expressing GPIHBP1. Although HSPG may not play a role in transporting cell-surface LPL to the vascular endothelial surface, there is a loose connection between the vascular endothelial cells of the liver and a Disse space between the vascular endothelial cells and hepatocytes. With such structures, plasma components can interact directly with hepatocytes. Therefore, we hypothesized that, when hepatocytes lack GPIHBP1, the expressed LPL in the liver may be anchored to HSPG on the surface of hepatocytes, which may directly contact chylomicrons and VLDL in the plasma and hydrolyze TG. Our results confirm this hypothesis; however, further mechanistic studies are required to elucidate the nature of such interactions.
The safety of AAV-mediated gene therapy is critical. Because FFAs resulting from rapid TG hydrolysis in muscle or liver of LPL transgenic mice have previously been found to cause damage to local tissues,28,29 whether liver-targeted expression of LPL causes liver injury is a key issue that requires urgent attention. Indeed, early studies on Glybera gene therapy selected the muscle as a target organ to avoid hepatotoxicity caused by the abnormal accumulation of excess FFAs during TG hydrolysis by LPL. The liver is an irreplaceable vital organ; once damaged, it can be life threatening. The quadriceps femoris muscle of the thigh can only impair the body’s ability to move even if it is injured. However, no significant muscle damage has been observed in experimental studies or clinical trials of AAV1-mediated LPL expression in muscle.16,18,30 Similarly, no severe liver damage has been observed in previous animal studies on gene therapy using adenoviral vectors to ectopically express LPL in the livers of mice and cats.31,32 These results differ markedly from those of previous studies in transgenic mice. One of the important reasons may be that, in transgenic mouse studies, LPL is expressed in the muscle or liver during early embryonic development, and the damage may be more severe than that caused by LPL expression in adulthood or the neonatal stage. In addition, transgenic mice generally have higher local levels of LPL expression in addition to pre-existing endogenous LPL expression, resulting in more pronounced damage. Indeed, skeletal muscle and myocardium normally have high LPL expression, but the FFAs produced are rapidly utilized or cleared without causing any damage.
AAV is non-pathogenic, has low immunogenicity, and is used widely as a gene delivery vehicle for long-term stable expression in vivo. Consistent with previous studies, we showed that AAV-LPL effectively infected the liver without causing obvious liver damage during long-term expression. In Gpihbp1−/− mice and rats, slight abnormal increases in liver enzymes and fatty liver changes may be due to long-term HTG, and no further aggravation of tissue damage was observed after AAV-LPL administration. This suggests that safety issues are not a major concern in AAV-LPL-mediated gene delivery of ectopically expressed LPL in the liver.
In conclusion, we report for the first time that AAV5 encoding human LPL-S447X can be safely administered to neonatal and adult Gpihbp1−/− mice and rats, thereby effectively reducing extremely high plasma TG levels and HTG-induced neonatal Gpihbp1−/− rat mortality. In addition, the long-term and effective reduction of TG levels can effectively prevent the occurrence of HTG-AP and reduce the susceptibility and severity of HTG-AP without obvious toxic and side effects. Our novel findings provide new evidence and perspectives for AAV5-mediated liver-specific expression of a beneficial mutant of human LPL, LPL-S447X, to alleviate severe HTG caused by GPIHBP1 gene deficiency. It is thus expected that this approach could also serve as an effective therapeutic approach for the treatment of HTG-AP caused by GPIHBP1 deficiency.
Materials and Methods
Construction of AAV5, AAV8 and AAV9 vectors
The AAV5-LPL-S447X vector was constructed based on the coding sequence of human LPL gene. Short introns were inserted into the optimized sequence to improve the transcriptional efficiency of the LPL gene. The coding sequence of LPL-S447X was divided into two parts (LPL-S447X CDS part1 and LPL-S447X CDS part2), and we further optimized the coding sequence of the LPL-S447X to enhance the protein translational efficiency after transcription. The liver-specific promoter was used in LPL gene expression cassette to increase AAV5-mediated transgene expression in the liver. To reduce the expression level of LPL-S447X gene in antigen-presenting cells and weaken the immune response, we introduced two miR-142-3p target sequences into the 3′ UTR. The constructed sequence was inserted into a double-chain AAV universal vector to obtain an AAV5-LPL-S447X recombinant vector. The coding sequence of the normal human LPL gene was used to construct the AAV5-LPL-WT recombinant vector, and the rest of the construction process was as described above. The coding sequence of human LPL gene was replaced with EGFP gene to construct the AAV5-EGFP vector, which was used to observe the infection efficiency. To compare the therapeutic effects of gene therapy with liver- and muscle-specific expression of LPL, we also designed and constructed an AAV9-LPL-S447X recombinant vector by replacing the liver-specific promoter with the muscle-specific strong promoter; the rest of the construction process was the same as above.
The helper plasmid pHelper, AAV coat plasmid pAAV-R2C9, and AAV vector plasmid were co-transfected into HEK293 cells. The cell precipitate and culture supernatant were harvested, and the packaged recombinant AAV was separated and purified by two cycles of CsCl density gradient ultracentrifugation. The AAV genome titer was determined by dot blot hybridization. The coating and purity of AAV were detected using SDS-PAGE. The genomic integrity and double-strand ratio of the packaged AAV were determined using Southern blotting.
Animals
WT Swiss Hauschka (ICR) mice and WT Sprague-Dawley rats were purchased from the Qinglongshan Animal Base (Nanjing, China). Gpihbp1 gene-deficient mice (C57BL/6) were purchased from the Mutant Mouse Resource and Research Centers (Bar Harbor, USA). Because ICR mice are more sensitive to pancreatic injury,33 we mated C57BL/6 Gpihbp1 gene-deficient mice with ICR WT mice for eight generations to obtain Gpihbp1 gene-deficient mice in ICR background (Gpihbp1−/− mice). To generate Gpihbp1 gene-deficient rats (Gpihbp1−/− rats), we designed a target site on exon 2 of Gpihbp1 using the TALEN technique and knocked out 102-bp fragments (G.L., B.L., Y.X., et al. unpublished data). Before the experiment, all animals were housed in the experimental animal center of Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University (General Hospital of Eastern Theater Command) in specific pathogen-free facilities, maintained under 12/12 h light-dark cycle at a controlled temperature (24°C ± 2°C), given water ad libitum, and fed standard laboratory chow. All animals were raised and operated on in accordance with the Principles of Laboratory Animal Care (NIH publication no. 85Y23, revised in 1996). All experimental protocols were approved by the Experimental Animal Ethics Committee of the Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University (no. 2018GKJDWLS-03-156).
Blood analysis
Orbital venous blood samples were collected for biochemical analyses. Plasma amylase, lipase, TG, and TC levels were determined using commercial kits according to the manufacturer’s protocols (Zhongsheng Beikong Biochemistry Company, Beijing, China).
Plasma LPL level and activity assay
Rats and mice were fasted overnight, and blood samples were collected 10 min after intravenous heparin injection. Plasma LPL levels were determined using a human LPL Elisa kit according to the manufacturer’s protocol (TSZ Biological Trade Company, San Francisco, USA). To measure LPL activity, total plasma lipase activity and hepatic lipase activity were first determined using an FFA release assay kit (Wako kit# NEFA-HR, Japan) with TG-rich plasma from Gpihbp1−/− mice as the lipolytic substrate.34 The subtraction of the hepatic lipase activity from the total lipase activity yielded a measure of plasma LPL activity.
Histological study
Paraffin-embedded tissue sections were stained with H&E. Two independent pathologists who were blinded to the experimental treatment examined the severity of pancreatic injury and evaluated the degree of edema, inflammation, and necrosis as we described previously.35
Immunohistochemistry analysis
For the immunohistochemical staining of LPL, paraffin sections of the liver tissue were boiled in EDTA antigen repair buffer solution. After natural cooling, the slices were incubated with a 3% hydrogen peroxide solution at room temperature for 15 min to block endogenous peroxidase activity. The slices were incubated with anti-LPL antibody (1:50 dilution, Santa Cruz, CA, USA) at 4°C overnight. The slices were then incubated with a biotinylated secondary antibody (1:200 dilution) for 1 h, after which the prepared diaminobenzidine substrate and hematoxylin were applied.
Immunofluorescence analysis
The fresh liver tissue samples were fixed in 4% neutral paraformaldehyde for 2 h, dehydrated with 30% sucrose overnight, embedded with optimal cutting temperature (OCT) compound, and then the slices were incubated with anti-human LPL polyclonal antibody (sc-73646, 1:50 dilution, Santa Cruz, CA, USA) and anti-mouse HSPG antibody (sc-73646, 1:50 dilution, Santa Cruz, CA, USA) at 4°C overnight. After washing three times with PBS, the slices were incubated in the dark with the red-violet fluorescent dye Fluorophore 620 (Akoya Biosciences FP1495001KT, 1:100 dilution) and green-yellow fluorescent dye Fluorophore 570 (Akoya Biosciences FP1488001KT, 1:100 dilution) for 2 h. After washing with PBS, slices were stained with 1 μg/mL DAPI (1:1,000) for 10 min.
qRT-PCR analysis
Briefly, total RNA was extracted using the total RNA extraction kit (TIANGEN Biotechnology, Beijing, PR China). RNA was reverse transcribed according to the manufacturer’s instructions. Gapdh was used as an endogenous control. The primers were as follows: H-LPL forward, 5′-ACACTTGCCACCTCATTCC-3′, H-LPL reverse, 5′-ACCCAACTCTCATACATTCCTG-3′; M-Lpl forward, 5′-TTGCCCTAAGGACCCCTGAA-3′, M-Lpl reverse, 5′-TTGAAGTGGCAGTTAGACACAG-3′; R-Lpl forward, 5′-CTTAGGGTACAGTCTTGGAGC-3′, R-Lpl reverse, 5′-CATCAGGAGAAAGGCGACTAG-3′; M-Gapdh forward, 5′-CTTTGTCAAGCTCATTTCCTGG-3′, M-Gapdh reverse, 5′-TCTTGCTCAGTGTCCTTGC-3′; R-Gapdh forward, 5′-TCCAGTATGACTCTACCCACG-3′, R-Gapdh reverse, 5′-CACGACATACTCAGCACCAG-3′.
Statistical analysis
GraphPad Prism 8.0 (GraphPad, San Diego, CA) was used for the statistical analysis. The numerical variables were expressed as mean ± standard error (SEM). The comparison between multiple groups was evaluated by one-way ANOVA, and the difference between the two groups was further compared by least significant difference (LSD) test. The comparison between multiple groups at different time points was evaluated using two-way ANOVA with Sidak correction. The Kaplan-Meier method was used for survival analysis, and the log rank test was used to compare survival rates between the two groups. p < 0.05 (two tail) was considered statistically significant (∗<0.05; ∗∗< 0.01; ∗∗∗< 0.001 vs. control group).
Data and code Availability
Data, further information, and requests for resources and reagents supporting the findings of this study are available from the corresponding authors, Weiqin Li (liweiqindr@nju.edu.cn), Baiqiang Li (li_baiqiang@aliyun.com), and George Liu (georgeliu@bjmu.edu.cn).
All AAV vectors employed in this study were meticulously designed, developed, produced, and provided by Beijing GeneCradle Therapeutics with exclusive intellectual property rights and are available by contacting the corresponding author and co-authors of Beijing GeneCradle Therapeutics (info@bj-genecradle.com).
Acknowledgments
This work was supported by funding from the National Natural Science Foundation of China (81801970, 81870441, and 82070669), Jiangsu Province Social Development Project, General Program (BE2022824), and Natural Science Foundation of Jiangsu Province of China, General Program (BK20211135). A graphical abstract was created using Figdraw (www.figdraw.com).
Author contributions
C.Y., Y. Xu, G. Lu, and Y.H. performed the experiments, analyzed the data, and wrote the manuscript. W.M. analyzed the data. Y. Xia, S.M., X.D., X.W. prepared the AAV recombinant viral vectors. W.L., B.L., G. Liu, and G. Lu designed and supervised the experiments and revised the manuscript. L.K., Z.T., and X.X. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.11.018.
Contributor Information
George Liu, Email: georgeliu@bjmu.edu.cn.
Baiqiang Li, Email: li_baiqiang@aliyun.com.
Weiqin Li, Email: liweiqindr@nju.edu.cn.
Supplemental information
References
- 1.Simha V. Management of hypertriglyceridemia. BMJ. 2020;371:m3109. doi: 10.1136/bmj.m3109. [DOI] [PubMed] [Google Scholar]
- 2.Pirillo A., Casula M., Olmastroni E., Norata G.D., Catapano A.L. Global epidemiology of dyslipidaemias. Nat. Rev. Cardiol. 2021;18:689–700. doi: 10.1038/s41569-021-00541-4. [DOI] [PubMed] [Google Scholar]
- 3.Cai S.J., Wong D.M., Chen S.H., Chan L. Structure of the human hepatic triglyceride lipase gene. Biochemistry. 1989;28:8966–8971. doi: 10.1021/bi00449a002. [DOI] [PubMed] [Google Scholar]
- 4.Wu S.A., Kersten S., Qi L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021;32:48–61. doi: 10.1016/j.tem.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young S.G., Fong L.G., Beigneux A.P., Allan C.M., He C., Jiang H., Nakajima K., Meiyappan M., Birrane G., Ploug M. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019;30:51–65. doi: 10.1016/j.cmet.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies B.S.J., Beigneux A.P., Barnes R.H., 2nd, Tu Y., Gin P., Weinstein M.M., Nobumori C., Nyrén R., Goldberg I., Olivecrona G., et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. doi: 10.1016/j.cmet.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beigneux A.P., Davies B.S.J., Gin P., Weinstein M.M., Farber E., Qiao X., Peale F., Bunting S., Walzem R.L., Wong J.S., et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young S.G., Davies B.S.J., Fong L.G., Gin P., Weinstein M.M., Bensadoun A., Beigneux A.P. GPIHBP1: an endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr. Opin. Lipidol. 2007;18:389–396. doi: 10.1097/MOL.0b013e3281527914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gin P., Yin L., Davies B.S.J., Weinstein M.M., Ryan R.O., Bensadoun A., Fong L.G., Young S.G., Beigneux A.P. The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J. Biol. Chem. 2008;283:29554–29562. doi: 10.1074/jbc.M802579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg A., Garg V., Hegele R.A., Lewis G.F. Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice. Lancet Diabetes Endocrinol. 2019;7:880–886. doi: 10.1016/S2213-8587(19)30156-1. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Hegele R.A. Homozygous missense mutation (G56R) in glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650) Lipids Health Dis. 2007;6:23. doi: 10.1186/1476-511X-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X., Qi R., Xian X., Yang F., Blackstein M., Deng X., Fan J., Ross C., Karasinska J., Hayden M.R., Liu G. Spontaneous atherosclerosis in aged lipoprotein lipase-deficient mice with severe hypertriglyceridemia on a normal chow diet. Circ. Res. 2008;102:250–256. doi: 10.1161/CIRCRESAHA.107.156554. [DOI] [PubMed] [Google Scholar]
- 13.Gao M., Yang C., Wang X., Guo M., Yang L., Gao S., Zhang X., Ruan G., Li X., Tian W., et al. ApoC2 deficiency elicits severe hypertriglyceridemia and spontaneous atherosclerosis: A rodent model rescued from neonatal death. Metabolism. 2020;109 doi: 10.1016/j.metabol.2020.154296. [DOI] [PubMed] [Google Scholar]
- 14.Pañeda A., Vanrell L., Mauleon I., Crettaz J.S., Berraondo P., Timmermans E.J., Beattie S.G., Twisk J., van Deventer S., Prieto J., et al. Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum. Gene Ther. 2009;20:908–917. doi: 10.1089/hum.2009.031. [DOI] [PubMed] [Google Scholar]
- 15.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rip J., Nierman M.C., Sierts J.A., Petersen W., Van den Oever K., Van Raalte D., Ross C.J.D., Hayden M.R., Bakker A.C., Dijkhuizen P., et al. Gene therapy for lipoprotein lipase deficiency: working toward clinical application. Hum. Gene Ther. 2005;16:1276–1286. doi: 10.1089/hum.2005.16.1276. [DOI] [PubMed] [Google Scholar]
- 17.Stroes E.S., Nierman M.C., Meulenberg J.J., Franssen R., Twisk J., Henny C.P., Maas M.M., Zwinderman A.H., Ross C., Aronica E., et al. Intramuscular administration of AAV1-lipoprotein lipase S447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler. Thromb. Vasc. Biol. 2008;28:2303–2304. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 18.Gaudet D., Méthot J., Déry S., Brisson D., Essiembre C., Tremblay G., Tremblay K., de Wal J., Twisk J., van den Bulk N., et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rip J., Nierman M.C., Ross C.J., Jukema J.W., Hayden M.R., Kastelein J.J.P., Stroes E.S.G., Kuivenhoven J.A. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler. Thromb. Vasc. Biol. 2006;26:1236–1245. doi: 10.1161/01.ATV.0000219283.10832.43. [DOI] [PubMed] [Google Scholar]
- 20.Bryant L.M., Christopher D.M., Giles A.R., Hinderer C., Rodriguez J.L., Smith J.B., Traxler E.A., Tycko J., Wojno A.P., Wilson J.M. Lessons learned from the clinical development and market authorization of Glybera. Hum. Gene Ther. Clin. Dev. 2013;24:55–64. doi: 10.1089/humc.2013.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinclair R., Schindler T., Lui K., Bolisetty S. Hypertriglyceridaemia in extremely preterm infants receiving parenteral lipid emulsions. BMC Pediatr. 2018;18:348. doi: 10.1186/s12887-018-1325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai E., Czuzoj-Shulman N., Malhamé I., Abenhaim H.A. Maternal and neonatal outcomes in women with disorders of lipid metabolism. J. Perinat. Med. 2021;49:1129–1134. doi: 10.1515/jpm-2021-0028. [DOI] [PubMed] [Google Scholar]
- 23.Parthasarathy N., Goldberg I.J., Sivaram P., Mulloy B., Flory D.M., Wagner W.D. Oligosaccharide sequences of endothelial cell surface heparan sulfate proteoglycan with affinity for lipoprotein lipase. J. Biol. Chem. 1994;269:22391–22396. [PubMed] [Google Scholar]
- 24.Korn E.D. Clearing factor, a heparin-activated lipoprotein lipase. I. Isolation and characterization of the enzyme from normal rat heart. J. Biol. Chem. 1955;215:1–14. [PubMed] [Google Scholar]
- 25.Goldberg I.J. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J. Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- 26.Sendak R.A., Bensadoun A. Identification of a heparin-binding domain in the distal carboxyl-terminal region of lipoprotein lipase by site-directed mutagenesis. J. Lipid Res. 1998;39:1310–1315. [PubMed] [Google Scholar]
- 27.Adeyo O., Goulbourne C.N., Bensadoun A., Beigneux A.P., Fong L.G., Young S.G. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 and the intravascular processing of triglyceride-rich lipoproteins. J. Intern. Med. 2012;272:528–540. doi: 10.1111/joim.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merkel M., Weinstock P.H., Chajek-Shaul T., Radner H., Yin B., Breslow J.L., Goldberg I.J. Lipoprotein lipase expression exclusively in liver. A mouse model for metabolism in the neonatal period and during cachexia. J. Clin. Invest. 1998;102:893–901. doi: 10.1172/JCI2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levak-Frank S., Radner H., Walsh A., Stollberger R., Knipping G., Hoefler G., Sattler W., Weinstock P.H., Breslow J.L., Zechner R. Muscle-specific overexpression of lipoprotein lipase causes a severe myopathy characterized by proliferation of mitochondria and peroxisomes in transgenic mice. J. Clin. Invest. 1995;96:976–986. doi: 10.1172/JCI118145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira V., Twisk J., Kwikkers K., Aronica E., Brisson D., Methot J., Petry H., Gaudet D. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPL(S447X)) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum. Gene Ther. 2014;25:180–188. doi: 10.1089/hum.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Excoffon K.J., Liu G., Miao L., Wilson J.E., McManus B.M., Semenkovich C.F., Coleman T., Benoit P., Duverger N., Branellec D., et al. Correction of hypertriglyceridemia and impaired fat tolerance in lipoprotein lipase-deficient mice by adenovirus-mediated expression of human lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 1997;17:2532–2539. doi: 10.1161/01.atv.17.11.2532. [DOI] [PubMed] [Google Scholar]
- 32.Liu G., Ashbourne Excoffon K.J., Wilson J.E., McManus B.M., Rogers Q.R., Miao L., Kastelein J.J., Lewis M.E., Hayden M.R. Phenotypic correction of feline lipoprotein lipase deficiency by adenoviral gene transfer. Hum. Gene Ther. 2000;11:21–32. doi: 10.1089/10430340050016120. [DOI] [PubMed] [Google Scholar]
- 33.Yang X., Yao L., Fu X., Mukherjee R., Xia Q., Jakubowska M.A., Ferdek P.E., Huang W. Experimental Acute Pancreatitis Models: History, Current Status, and Role in Translational Research. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.614591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Q., Pu N., Li X.Y., Shi X.L., Chen W.W., Zhang G.F., Hu Y.P., Zhou J., Chen F.X., Li B.Q., et al. Digenic Inheritance and Gene-Environment Interaction in a Patient With Hypertriglyceridemia and Acute Pancreatitis. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.640859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt J., Rattner D.W., Lewandrowski K., Compton C.C., Mandavilli U., Knoefel W.T., Warshaw A.L. A better model of acute pancreatitis for evaluating therapy. Ann. Surg. 1992;215:44–56. doi: 10.1097/00000658-199201000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data, further information, and requests for resources and reagents supporting the findings of this study are available from the corresponding authors, Weiqin Li (liweiqindr@nju.edu.cn), Baiqiang Li (li_baiqiang@aliyun.com), and George Liu (georgeliu@bjmu.edu.cn).
All AAV vectors employed in this study were meticulously designed, developed, produced, and provided by Beijing GeneCradle Therapeutics with exclusive intellectual property rights and are available by contacting the corresponding author and co-authors of Beijing GeneCradle Therapeutics (info@bj-genecradle.com).