

Abstract
Six cytochrome P450 enzymes are involved in human steroidogenesis, converting cholesterol to sex steroids, mineralocorticoids, and glucocorticoids. While early work was accomplished with steroidogenic P450 orthologs from more accessible sources, knowledge of basic biochemistry through successful drug design have been greatly facilitated by recombinantly-expressed, highly purified human versions of these membrane proteins. Many membrane proteins are difficult to express and purify and are unstable. Membrane P450 expression in E. coli has been facilitated by modification and/or truncation of the membrane-interacting N-terminus, while metal-affinity resins and histidine-tagging greatly facilitates purification. However, substantial optimization is still frequently required to maintain protein stability. Over time, a generalized three-column purification scheme has been developed and tweaked to generate substantial quantities of fully-active, highly purified human cytochrome P450 enzymes that have made possible the application of many structural, biochemical, and biophysical techniques to elucidate the mysteries of these critical human enzymes.
Keywords: cytochrome P450, CYP11A1, CYP17A1, CYP21A2, CYP11B1, CYP11B2, steroidogenesis, recombinant expression, membrane proteins
1. Introduction
The ability to produce highly purified human (membrane) cytochrome P450 enzymes in recombinant systems has contributed significantly to our ability to characterize the functionality of these enzymes and has been an absolute requirement for many biophysical, biochemical, and structural studies. P450-dependent steroid hydroxylation was first demonstrated using bovine adrenal cortex mitochondrial fractions (Harding et al., 1964; Omura et al., 1966). Much of the early work occurred with bovine enzymes because of the accessibility of tissues from slaughterhouses. Work with human steroidogenic P450 enzymes suffered from both poor access to human tissues and low isolation yields. Therefore, recombinant expression is often essential, especially when an interrogating technique requires larger amounts of enzyme. A notable exception is CYP19A1 or aromatase, which was heroically isolated from human placenta in the purity and quantity required to support crystallographic work (Ghosh et al., 2009), but a subsequent structure from E. coli-generated aromatase was essentially identical (Lo et al., 2013) and the latter approach is more feasible and economical. While a number of human P450 enzymes have been expressed in insect cells (Patten & Koch, 1995; Zhan et al., 2016), yeast (Dhir et al., 2009; Kikuta et al., 1998), and E. coli, the latter is by far favored in terms of the quantity of high-quality protein required for biophysical and structural studies and the production cost.
Accessible general E. coli expression and purification protocols have been developed for generating many human P450 enzymes, including the six involved in human steroidogenesis. These include the three mitochondrial enzymes: CYP11A1 or side-chain cleavage enzyme; CYP11B1, the cortisol-generating 11β-hydroxylase; CYP11B2, the multi-step aldosterone generating enzyme; and the three microsomal enzymes: CYP17A1 17α-hydroxylase/17,20-lyase enzyme; CYP21A2 or 21-hydroxylase enzyme, and estrogen-generating CYP19A1 or aromatase enzyme. Thus, these P450 enzymes control diverse elements of normal human physiology and are often drug targets for hormone-fueled diseases from prostate and breast cancers to Cushing syndromes and hypertension. Steroidogenic P450 enzymes also display intriguing biochemical variety, ranging from hydroxylation to lyase chemistry, interacting with diverse NADPH-cytochrome P450 reductase or adrenodoxin redox partners, and performing single, double, or triple reaction cycles. Regardless of the need to study human steroidogenic enzymes to understand diverse biochemistry or for drug targeting, having access to the individual P450 enzyme is a critical step.
2. General strategies
2.1. Brief historical synopsis
While soluble P450 enzymes from other organisms were readily expressed in E. coli from early days of the recombinant protein revolution (e.g. (Unger et al., 1986), E. coli expression of full-length, unmodified human cDNAs to generate membrane P450 proteins such as those found in humans can be problematic in terms of yield of correctly folded, heme-incorporated protein (e.g. (Barnes et al., 1991)). This can be due to the presence of rare codons, problems with heme incorporation, protein folding and/or membrane insertion, much of which can be difficult to diagnose.
Early work by Barnes and colleagues discovered that the nucleotide sequence coding for the N-terminal sequence of the microsomal bovine CYP17A1 could be slightly modified to preferred codons and to minimize mRNA secondary structure, yielding significant amounts of holoprotein in E. coli (Barnes et al., 1991). This bovine CYP17A1 “Barnes” sequence coding for the amino acids MALLAVFL was subsequently applied to the N-terminus of a range of human P450 enzymes with good success (e.g. vitamin D3-metabolizing CYP27A1 (Axen et al., 1994), fatty-acid metabolizing CYP4F11 (Tang et al., 2010), and drug-metabolizing CYP3A4 or CYP2D6 (Pan et al., 2011)). For microsomal P450 enzymes this sequence is part of an N-terminal transmembrane helix that interacts with the endoplasmic reticulum signal recognition particle for co-translational insertion into the membrane (Sakaguchi et al., 1984). It is normally not cleaved in the mature P450 protein and its presence can significantly impair high yields of human P450 expression in E. coli.
The next most significant advance was to truncate this N-terminal region of bovine CYP17A1 (Sagara et al., 1993). Truncation still yielded a membrane-bound P450 protein with a normal spectrum and intact catalytic function but further increased the yield. It was later determined that the proline-rich linker immediately following the N-terminal transmembrane helix contributed to protein folding and should be preserved (Chen et al., 1998; Kusano, Kagawa, et al., 2001; Kusano, Sakaguchi, et al., 2001). Subsequent work on N-terminal truncation and modification of the new N-terminus to a hydrophilic sequence such as the rabbit CYP2C3 MAKKTSS (Cosme & Johnson, 2000; vonWachenfeldt et al., 1997), and appending C-terminal His tags were successful in generating membrane-bound drug metabolizing P450 proteins that could be crystallized, resulting in the first membrane P450 structures (Johnson et al., 2002; Wester et al., 2002). This general approach has now resulted in the ability to generate fully active forms of many microsomal human P450 enzymes in significant quantities and high purity, including human steroidogenic P450 enzymes CYP17A1 (DeVore & Scott, 2012) and CYP21A2 (Fehl et al., 2018). Mitochondrial P450 enzymes were initially expressed with a mitochondrial-targeting N-terminal sequence, which is proteolytically cleaved after membrane insertion. However, similar modifications of the mitochondrial P450 N-termini have similarly resulted in significant quantities of the remaining human steroidogenic P450 enzymes CYP11A1 (Strushkevich et al., 2011), CYP11B1 (Brixius-Anderko & Scott, 2019), and CYP11B2 (Brixius-Anderko & Scott, 2021).
The protocols outlined below reflect the experiences of the Scott laboratory expressing and purifying N-terminally truncated and modified, C-terminally His-tagged forms of five human steroidogenic P450 enzymes: CYP11A1, CYP11B1, CYP11B2, CYP17A1, and CYP21A2. These protocols built on concepts and approaches that worked well for generating human drug-metabolizing P450 enzymes and have been modified and adapted over time to incorporate technical advances, new information reported in the literature, and our own observations and experiences with each protein. It would be impossible to credit all the individuals or manuscripts on whose work the current protocols depend. The resulting purified proteins are fully catalytically active, producing the same products with the same regio- and stereospecificity as the native, full-length enzymes. However, these versions of the human steroidogenic P450 enzymes are obtained in higher quantity and are more stable and more soluble than the full-length enzymes. For some, the detergent used to initially solubilize the protein from the membrane can subsequently be withdrawn; others are more stable in the presence of a small amount of a specific detergent, likely interacting with the second membrane-binding region around the F′/G′ region. They interact well with their cognate redox partners and do not require the addition of lipid for catalysis. In fact, sometimes they metabolize substrates better than full-length, membrane-associated forms. The increased solubility facilitates and enables the application of a broad range of biochemical and biophysical techniques to interrogate functionality and is essential for the application of X-ray crystallography, which has generated all atomic-level human P450 structures to date.
2.2. Expression
Steroidogenic cytochrome P450 expression is not sensitive to the E. coli cell line employed. While increased production could be possible by trying multiple bacterial cell our experience has been that this is not usually worth the time or effort. All five of the human steroidogenic P450 enzymes discussed in the protocol below express well in simple DH5α and or JM109 cell lines, which are readily available commercially.
For membrane P450 enzymes, the cDNA or synthetic gene can be cloned into the pCWori+ vector. This vector contains isopropyl β-D-1-thiogalactopyranoside (IPTG) inducible tac promoters just upstream of the cloned P450 gene as well as an ampicillin-resistance gene. A common approach adopted in our laboratory is to coexpress human P450 genes with a chaperone to assist with folding. The pGro7 plasmid containing the GroEL/ES genes under the control of an arabinose-inducible promoter is used, in part because its chloramphenicol resistance is compatible with the ampicillin-resistance of the pCWori+ plasmid. Addition of the pGro7 plasmid and its induction can have no effect, moderate effect, or substantial effect on the yield of holoprotein obtained for other human P450 enzymes. The overexpressed chaperone proteins are purified away from the P450 during metal-affinity and ion exchange chromatography so there seems to be no detrimental effects to including it.
Growth profiles outlined are targeted toward growing to a moderate optical density at 600 nm (O.D.600), inducing chaperone and/or P450 with supplementation with δ-aminolevulinic acid as a heme precursor, then reducing the shaking and temperature to grow for 24-72 hours until harvesting. Shaking and amount of culture per flask can significantly affect expression, likely because oxygen availability does seem to be important, though baffled flasks vs. non-baffled flasks haven’t seemed to make a consistent difference in our experience. Harvested cells can typically be stored frozen, but once thawed and lysed it is usually best to work continuously through the purification process to obtain the highest quality protein at the end.
2.3. Purification
The overall purification process consists of cell lysis, protein solubilization using detergent, and three chromatographic steps. We have experience with multiple types of mechanical lysis combined with lysozyme: sonication, French press, and microfluidizer. Depending on operation, all three can work reasonably well. Solubilization of protein requires empirical determination of the optimal detergent which maximizes yield of P450 isolated from the membrane yet maintains protein stability (typically as measured by a reduced-carbon monoxide difference spectrum). The detergent concentration is typically higher during P450 extraction from the membrane than during later purification steps. Solubilized P450 enzyme is then separated from membrane components by ultracentrifugation. Older protocols employed one ultracentrifugation spin before detergent addition to remove soluble contaminants, thorough resuspension of the pellet in a detergent-containing buffer, then a second ultracentrifugation step to pellet lipids from the solubilized P450 in the supernatant. While this strategy yields more highly purified protein prior to the chromatographic steps, it adds significant time and optimized chromatographic steps typically obviate the necessity for the first spin. However, if purity problems are encountered, it can be useful to reintroduce the additional first ultracentrifuge spin. Subsequent chromatographic steps include metal affinity, ion exchange, and size exclusion chromatography. Membrane P450 enzymes almost always require 20% glycerol but also tend to be more stable in detergent, in higher salt, with ligand, and sometimes in lower concentrations. Therefore, as compromises need to be made in one of these parameters as purification proceeds, it is often useful to compensate in another area. An example would be maintaining detergent when dropping the ionic strength to bind an ion exchange column. Obtaining absolute and reduced-carbon monoxide difference spectra and retaining samples for SDS-PAGE analysis throughout purification are invaluable for evaluating purity, stability, and liganded state to guide optimization. If highly purified protein is required for experiments like crystallography, then progressing through all three chromatographic steps is highly desirable. If one is making protein for enzymatic assays or ligand binding assays, then only the first two columns might be sufficient. This will increase the yield, though the amount of P420 present should be considered, especially for CYP17A1. However, using only the first metal-affinity chromatography step typically results in poor purity, P450 protein bound to the imidazole or histidine used for elution, and can yield significant P420 instead of P450 in a reduced-carbon monoxide difference spectrum. If protein is destined for crystallography with a particular ligand of interest, then ligand added to purification buffers and even to the expression media (depending on ligand cost/availability) can often increase P450 stability and increase yield. However, this is not a suitable approach if the protein is to be used for enzymatic or ligand binding assays, as one can never be completely sure that the ligand is completely removed even in the absence of a spectral shift.
2.4. Anticipated outcomes
Once purification is complete, the P450 protein should always be characterized by absolute spectra to provide information on the heme environment/liganded state and purity, by reduced-carbon monoxide difference spectra to determine the stability of the heme iron interaction with the proximal Cys, and by SDS-PAGE. Purity is evaluated in terms of the observed bands on an overloaded SDS-PAGE gel stained with Coomassie blue, but also by the ratio of absorbance of the heme Soret peak to protein absorbance at 280 nm. The final chromatographic step additionally provides information about the oligomeric state. While best practice is to use protein immediately for crystallization, protein can be frozen for assays and sometimes for successful crystallization. However, the protein should be frozen in thoughtful aliquot sizes because once thawed, the P450 protein should never be refrozen for later use.
3. Equipment and Reagents for the Protocols
3.1. Equipment
Dounce homogenizer (Fisher 50-194-5207)
CO Tank (Airgas) and pressure regulator Aldrich 21A00L464)
Refrigerated tabletop micro centrifuge (Eppendorf 5424 R)
Tabletop swinging bucket centrifuge (Eppendorf 5804 R)
−80 C freezer (ThermoFisher RDE60086FA)
SDS-PAGE gel box (BioRad Mini Protean 3 Cell)
Gel electrophoresis power source (BioRad PowerPac Basic)
Large centrifuge (Beckman Coulter Avanti J-E with JLA 10.5 fixed angle rotor and six 500 mL canisters)
500 mL centrifuge bottles (Beckman 361691)
Fisher sonic dismembrator (Fisher model 100; Horn: Qsonica Microson XL2000 microprobe, ¼” diameter) or 40 kpsi French Press with 35 mL piston (Glen Mills G-M) or Microfluidizer (Avestin C3)
Stir plate (e.g. Fisher 11-676-263 or SP88857200)
Ultracentrifuge (Beckman Optima L90K with Type 45 Ti fixed angle rotor)
Polycarbonate ultracentrifuge tubes, 70 mL (Beckman Coulter NC9708359)
Fast protein liquid chromatography system (Cytiva AKTA PureM with sample pump, S9)
Superloop of 150 mL capacity (Cytiva 18102385)
XK 16/20 empty chromatography column (Cytiva 28988937)
Scanning UV-visible spectrophotometer (Shimadzu UV-2700)
Large incubator shaker (Eppendorf Innova 44 R)
Small incubator shaker (Eppendorf Innova 4200)
OD600 reader (Implen OD600)
3.2. Reagents and Supplies
pCWori+ vector (NovoPro V001682))
2.8 L Fernbach flasks (PYREX 10-092)
500 mL Erlenmeyer flask (PYREX 10-040H)
1 mL polystyrene disposable cuvettes (Fisher 14-955-127)
1 mL quartz cuvettes (Starna 29B-Q-10)
BD syringes with Leur-lok tips, 1 mL and 5 mL (Fisher 14-823-52, 14-823-16D)
Sodium dithionite (Sigma 157953)
10-25 mL disposable serological pipettes (Fisher 1367811)
0.2 micron Steritop filter (Sigma Aldrich S2GPT01RE)
Ethanol (Fisher A-300-1)
Sodium hydroxide (Fisher S318-500)
4-20% SDS-PAGE gels (BioRad 4561095)
Simply Blue Safestain (Fisher 6065)
Liquid nitrogen (Cryogenic Gases)
pGro7 plasmid (Takara Bio USA, Inc. 3340)
Isopropyl b-D-1-thiogalactopyranoside (IPTG) (Fisher BP1755-100)
Lysogeny Broth (LB) (Fisher BP1426-500)
Bacto Tryptone (Fisher 211705)
Bacto Yeast Extract (Fisher 212750)
Chloramphenicol (GoldBio C-105)
Competent DH5a E. coli cells (Fisher FEREC0111)
Carbenicillin (GoldBio C-103)
L-arabinose (GoldBio A-300-1)
d-aminolevulinic acid (GoldBio A-680)
SIGMAFAST™ Protease Inhibitor Cocktail, EDTA free (Sigma S8830) or HALT protease inhibitor cocktail (Fisher PI78441)
Emulgen 913 (Desert Biologicals 2846)
Lysozyme (Fisher 89833)
DNase (Sigma Aldrich DN25-5G)
Filters: 0.8 μm filter (Fisher 450-0080)
Nickel-Nitrilotriacetic acid (Ni-NTA) Superflow resin (Qiagen 30410)
Guanidinium hydrochloride (Fisher AAA135430I)
HiLoad 16/600 Superdex 200 column (Cytiva 120 mL)
Amicon Ultra centrifugal filter units, MWCO 50 kDa (Fisher UFC905024)
Tween20 (Sigma Aldrich P1379)
SP Sepharose High-Performance cation exchange 5 mL prepacked column (Cytiva 17115101)
CM Carboxymethylcellulose Sepharose Fast Flow 5 mL prepacked column (Cytiva 17505601)
HiLoad 16/600 Superdex 200 prep grade column (Cytiva 17-1043-01)
EDTA (Fisher BP120-500)
Acetic Acid (Sigma A6283)
Potassium phosphate monobasic (Fisher P06620)
Potassium phosphate dibasic (Fisher P290-212)
Glycerol (Fisher G33)
Sodium Acetate (Fisher S210)
Sodium Cholate (Sigma C644)
Sodium Chloride (Fisher S271)
b-mercaptoethanol (Sigma M6250)
Histidine (Sigma H8000)
ATP (GoldBio A-081)
Glycine (Sigma G7126)
3.3. Solutions
Terrific Broth is composed of 12 g tryptone, 24 g yeast extract, 4 mL glycerol (or 5 ml 80% glycerol) in purified water up to 900 mL
20x potassium phosphate buffer (0.34 M KH2PO4, 1.44 M K2HPO4
Arabinose solution: 0.2-0.4 g/mL solution of L-arabinose is prepared in H2O and filter sterilized (solubility in water is ~0.8 g/mL, but warming the water will expedite solubilization).
1 M d-ALA prepared in water and filter sterilized.
1 M IPTG prepared in water and filter sterilized.
6M Guanidinium hydrochloride with 0.2 M acetic acid. (add 573.2 g guanidium hydrocholoride and 11.5 mL Acetic Acid to 200 mL of water, stir to dissolve, then dilute to a final volume 1 L)
100 mM nickel sulfate (26.3 g nickel sulfate in 1 L water)
20 mM KPi, 500 mM NaCl, 100 mM EDTA (0.37 g KH2PO4 3.01 g K2HPO4, 29.22 g NaCl, 37.22 g EDTA, pH 8.0 to a final volume of 1 L)
4. Protein engineering
4.1. Construct Design
Given both the engineering involved to generate the intended construct and the desire to use codon optimization for E. coli expression throughout, our typical practice is to determine the desired amino acid sequence, then back-translate this to preferred E. coli codons, using standard practices to minimize secondary structure, eliminate undesirable restriction sites, etc. Any number of commercial enterprises can be engaged to synthesize the desired coding region, which is cloned into the pCWori+ plasmid downstream of two IPTG-inducible tac promoters. Constructs coding for the N- and C-termini shown in Figure 1 have demonstrated excellent expression and purification in our hands. While the N-terminal modifications are generally based on prior experience, the length of the C-terminal purification tag is probably extendable or expendable?. For crystallography, one needs to minimize non-native sequences that might be flexible or unstructured. Four histidines suits that purpose while longer histidine tags can increase binding to metal-affinity chromatography resin if desired.
Figure 1.

Modifications of steroidogenic P450 enzyme N- and C-termini for high expression in E. coli and facile purification. Deletions are indicated by dashes. Hydrophilic residue substitutions are highlighted in cyan. Four to six histidines (yellow) are added at the C-terminus to facilitate purification. Prolines are shown in red.
5. Expression
5.1. Procedure
Day 0: Preparation:
To generate cells for steroidogenic P450 expression, chemically-competent DH5α E. coli are typically first transformed with the pGro7 chaperone-expressing plasmid and grown on Lysogeny Broth (LB) agar plates supplemented with 20 mg/mL chloramphenicol. The JM109 E. coli strain has also been substituted for DH5α for CYP11A1 (Strushkevich et al., 2011) and CYP17A1 (DeVore & Scott, 2012). Small scale cultures can be grown in liquid LB supplemented with the same antibiotic, made chemically competent, and used immediately or frozen at −80 °C.
Day 1: Transformation
In the afternoon, the DH5α/pGro7 competent cells are transformed with the desired pCWP450dH plasmid and spread on LB agar plates. This and all subsequent growth media are supplemented by both 20 mg/mL chloramphenicol and 100 mg/mL carbenicillin. Carbenicillin is typically substituted for ampicillin as it is stable in growth media for longer time periods, which is most relevant after induction. It is not recommended to transform both plasmids into E. coli simultaneously (Takara user manual). Plates are incubated at 37 °C overnight.
Day 2: Scaling up
For CYP17A1 and CYP21A2 expression, in the morning an individual colony is inoculated into 5 mL of LB media with carbenicillin and chloramphenicol and grown all day at 37°C with shaking (250 rpm). After 6-8 hours of growth, 100 or 200 μL of the starter culture is introduced to 200 mL of LB media containing antibiotics.
For expression of CYP11 enzymes, an individual colony from the LB plate can be transferred directly to 50 mL of LB containing antibiotics.
Alternatively, 100 or 200 μL of a previously-generated DH5α/pGro7/pCWP450dH glycerol stock may be used to inoculate 200 mL of LB containing antibiotics, though stocks >6 months old may result in decreased expression yield.
In all three cases, the culture is then grown at 37 °C and 250 rpm shaking overnight, about 14-18 hours.
Day 3: Scale up and Induction
The following morning, 8-20 mL of the overnight culture is introduced to 0.8-1 L of modified Terrific Broth (TB) media containing both antibiotics. Our practice is to grow a total of 4-6 flasks at a time and the protocols below are scaled to start from 4.8 L of culture for CYP11 enzymes or 3 L of culture for CYP17A1 and CYP21A2. The smaller media volume per flask increases aeration and is preferred, particularly for expression of the CYP11 enzymes. The TB media is modified from the original composition to double the buffering capacity and prevent acidification during longer incubation times. This isn’t required for CYP17A1 or CYP21A2, but it is routine for expression of all three CYP11 enzymes and has never been observed to have a deleterious effect on expression across even non-steroidogenic human P450 enzymes; one liter of modified Terrific Broth sterilized in a 2.8 L Fernbach flask, is buffered by the addition of 100 mL sterile 20x phosphate buffer.
These cultures are then incubated at 37 °C with shaking at 200-220 rpm, and the optical density at 600 nm is measured every hour to monitor growth. When the O.D.600 reaches ~0.3-0.4 for CYP17A1 and CYP21A2, expression of GroEL/ES is induced by addition of 10 mL of the L-arabinose solution to each flask, equivalent to 2-4 g/L. For CYP11 enzymes, this step occurs at the same time as P450 induction, when O.D.600 reaches 0.6.
When O.D.600 = 0.6-0.8 (or as high as 0.9 for CYP21A2), 1 mL of 1M δ-ALA and 1 mL of IM IPTG is added to each flask to induce P450 expression. Incubation temperature is then reduced to 25 °C for CYP21A2, to 27 °C for CYP11s, or to 28 °C for CYP17A1, though there is likely some flexibility. Shaking speed is also reduced to 120 rpm for CYP21A2, 140 rpm for CYP17A1, and 190 rpm for CYP11s. If co-expressing with a ligand, it should be added at this point, with the concentration depending on the ligand solubility, permeability, affinity, and cost.
Day 4: Continued growth
Day 5: Harvesting
Cultures are harvested after 48 hours (though CYP17A1 may express for up to 72 hours) via centrifugation at 10,500 x g for 10-15 minutes. The resulting pellet is weighed, then transferred to a plastic freezer bag or 50 mL conical tube and stored at −80 °C until purification. If purifying immediately after harvesting, lysis may be improved by flash-freezing the pellet in liquid nitrogen and allowed to thaw before proceeding.
5.2. Notes
It is often useful to express multiple L of culture at a time, subdividing the cell pellet as desired prior to freezing and storage.
6. Purification: Protein extraction from membranes
6.1. General comments
All buffers have a pH of 7.4 unless otherwise indicated. All steps are performed at 4 °C. It is convenient to make up concentrated potassium phosphate monobasic and dibasic stocks to use to create buffers at the desired pH. Glycerol added to 20% in almost all buffers is important for protein stability. It is convenient to make up an 80% glycerol stock and use this stock to generate purification buffers. Glycerol, Tween 20, and Emulgen are added (v/v). Sodium cholate is added (w/v).
6.2. Procedure
Cell Pellet Resuspension
Cell pellets corresponding to 4.8 L of the original cell culture for CYP11 enzymes or 3 L of culture for CYP17A1 and CYP21A2 are transferred to an appropriately sized beaker. One SIGMAFAST™ Protease Inhibitor Cocktail, EDTA-Free tablet is crushed, solubilized in a small amount of resuspension buffer (Table 1), and added to the thawing pellet. If SIGMAFAST tablets are not available, HALT protease inhibitor cocktail can be substituted. While some earlier reported purifications employed only phenylmethylsulfonyl fluoride (PMSF) as a serine protease inhibitor, PMSF is rapidly inactivated in aqueous solutions and has to be added regularly throughout purification. Resuspension buffer (Table 1) is added to bring the final volume up to 350 mL for CYP11 enzymes and 300 mL for CYP17A1 and CYP21A2. Using smaller volumes can result in solutions that do not lyse completely.
Table 1.
Resuspension buffer compositions.
| CYP11 Proteins | CYP17A1 | CYP21A2 |
|---|---|---|
|
| ||
| 50 mM KPi (pH 7.4) | 100 mM KPi (pH 7.4) | 500 mM KPi (pH 6.8) |
| 20% (v/v) glycerol | 20% (v/v) glycerol | 20% (v/v) glycerol |
| 500 mM sodium acetate | 300 mM sodium chloride | 300 mM sodium chloride |
| 1.5 % (w/v) sodium cholate | *1 mM β-mercaptoethanol | |
| 1.5 % (v/v) Tween 20 | ||
Inclusion of β-mercaptoethanol is optional, but should be added immediately prior to buffer use.
Cell Lysis and Detergent Extraction
For CYP17A1 and CYP21A2, 0.3 mg lysozyme is added per mL of the resuspended cells, which are then thoroughly homogenized using a Dounce homogenizer with both pestles A and then B. The sample can then be lysed through various methods. The first option is to sonicate with a Fisher sonic dismembrator. If sonication is used, the resuspended cells are divided into 50 mL aliquots and, while on ice, sonicated for six 30-second pulses at 10% amplitude with 30 seconds of rest between pulses. The second option is to put the sample through either a French press or microfluidizer at 10,000-15,000 psi for 2-3 passes while keeping cold. Two passes are typically sufficient for successful lysis, but if the sample viscosity remains high after the second pass, a third pass should be completed. After lysis, the detergent Emulgen 913 should be dissolved in ~20 mL of resuspension buffer so that the final concentration of detergent in the resuspended, lysed cells will be 2% and 1% v/v Emulgen 913 for CYP17A1 and CYP21A2, respectively. Samples are incubated with detergent for 1-2 hours with stirring.
For CYP11 enzymes, 105 mg of lysozyme and 40 mg DNAase are added to the resuspended cells and left to incubate with stirring for 45-60 min, followed by thorough homogenization using a Dounce homogenizer with pestles A and B. The solution is then divided into 50 mL aliquots and sonicated on ice 10 times for 30-second pulses at 10% amplitude with 30 seconds of rest between pulses. Because the resuspension buffer already contains detergent, the samples are then immediately centrifuged.
The lysed, detergent-extracted samples are then subjected to ultracentrifugation to pellet lipids and other components. This is accomplished at ~100,000 x g (maximum relative centrifugal force) for 1 hour at 4°C in 70 mL polycarbonate bottles. Discarding the lipid-containing pellet, the P450-containing supernatant is filtered through a 0.8 μm filter before proceeding to the chromatographic steps. A small sample (~50 µl) of the lysis mixture should be retained for an SDS-PAGE gel, but absorbance spectra are not usually collected due to high background.
6.3. Notes
Hearing protection is required if the sonicator is not set up inside a sound enclosure. French presses have pinch points and the manufacturer instructions should be closely followed. Ultracentrifugation used here to pellet the membranes requires careful balancing of tubes.
7. Purification: Immobilized Metal Affinity Chromatography
7.1. General
This and all subsequent chromatographic steps are most easily accomplished on an AKTA PureM chromatography system (Cytiva). This system not only allows precise control of flow rates and column pressures and facilitates complex run profiles, but the ability to measure both 280 nm for general protein detection and visible wavelengths for heme protein detection is particularly useful. A sample loading pump is immensely useful for loading large volumes that might not fit into a Superloop (Cytiva, 150 mL Superloop).
7.2. Procedure
Since all five protein constructs contain a C-terminal His-tag, binding to an immobilized metal affinity resin is the first chromatographic step. The stated resin capacity for 6xHis-tagged proteins is 5-10 mg/ml but for the scales described herein (3-4.8 L of culture) an XK 16/20 column is packed with ~30 mL of Nickel-Nitrilotriacetic acid (Ni-NTA) Superflow resin according to the manufacturer’s instructions. The manufacturer recommends equilibration with 5 CV, but at least two CV (60 mL) of Ni-NTA wash buffer 1 (Table 2) is usually sufficient in our experience. The filtered ultracentrifuge supernatant is then loaded onto the column at a flow rate of 1.5-2 mL/min. Typical volumes for loading are between 200-300 mL. As mentioned above, loading these larger volumes can be accomplished by using either a sample pump or by using a 150 mL superloop, though the latter usually requires multiple loadings. Concentrating the sample is not recommended. During loading a visible red band should accumulate at the top of the Ni-NTA Superflow resin. The loaded Ni-NTA column is then washed with Ni-NTA wash buffer 1, followed by wash buffer 2, and eluted with Ni-NTA elution buffer (Table 2) while collecting 4-5 mL fractions. All flow rates are 1.5-2 mL/min for the indicated volumes (Table 2). It is a good idea to collect the flow-through in a clean, separate bottle in case the protein does not bind well to the column and troubleshooting is required.
Table 2.
Buffer compositions and volumes for metal-affinity chromatography.
| Buffer | CYP11 Proteins | CYP17A1 | CYP21A2 |
|---|---|---|---|
|
| |||
| Ni-NTA | 50 mM KPi (pH 7.4) | 100 mM KPi (pH 7.4) | 100 mM KPi (pH 6.8) |
| Wash | 20% (v/v) glycerol | 20% (v/v) glycerol | 20% (v/v) glycerol |
| Buffer 1 | 500 mM sodium acetate | 300 mM NaCl | 200 mM sodium chloride |
| 1% (w/v) sodium cholate | 0.2% (v/v) Emulgen 913 | 0.2% (v/v) Emulgen 913 | |
| 1% (v/v) Tween 20 | *1 mM p-mercaptoethanol | ||
| 5 CV | 4 CV | 4 CV | |
|
| |||
| Ni-NTA | 50 mM KPi (pH 7.4) | 50 mM KPi (pH 7.4) | 100 mM KPi (pH 6.8) |
| Wash | 20% (v/v) glycerol | 20% (v/v) glycerol | 20% (v/v) glycerol |
| Buffer 2 | 1% (w/v) sodium cholate | 300 mM sodium chloride | 200 mM sodium chloride |
| 1% (v/v) Tween 20 | 0.2% (v/v) Emulgen 913 | 0.2% (v/v) Emulgen 913 | |
| 4 mM histidine | 100 mM glycine | 12 mM histidine | |
| 0.1 mM ATP | *1 mM p-mercaptoethanol | ||
| 5 CV | 8 CV | 5 CV | |
|
| |||
| Ni-NTA | 20 mM KPi (pH 7.4) | 10 mM KPi (pH 7.2) | 10 mM KPi (pH 6.8) |
| Elution | 20% (v/v) glycerol | 20% (v/v) glycerol | 20% (v/v) glycerol |
| Buffer | 1% (w/v) sodium cholate | 300 mM sodium chloride | 100 mM sodium chloride |
| 1% (v/v) Tween 20 | 0.2% (v/v) Emulgen 913 | 0.2 % (v/v) Emulgen 913 | |
| 80 mM histidine | 100 mM glycine | 80 mM histidine | |
| 80 mM histidine | 2 mM EDTA | ||
| *1 mM p-mercaptoethanol | |||
| 5 CV | 6 CV | 8 CV | |
Inclusion of β-mercaptoethanol is optional, but should be added immediately prior to buffer use.
While running the Ni-NTA column, recording of both 280 and 418 nm wavelengths will facilitate monitoring the success of the chromatographic step and assist in troubleshooting. Representative chromatograms for CYP17A1, CYP21A2, and CYP11B1 (Figure 2, panels A, C, and E) initially demonstrate high absorbance at 280 nm and 418 nm during the loading phase as many other proteins and free heme pass unimpeded through the column. During wash out of unbound proteins with Ni-NTA wash buffer 1, the absorbance at both wavelengths should return to baseline, while washing with Ni-NTA wash buffer 2 typically results primarily in the removal of non-specifically bound proteins as observed in an increase in absorbance at 280 nm. Notably, CYP17A1 binds to the Ni-NTA resin less avidly than the others, so glycine is used in wash buffer 2 to remove non-specifically-bound proteins instead of histidine, which is a stronger competitor for the Ni-NTA resin. Finally, the histidine-containing elution buffer results in an elution peak characterized by increasing absorbance at both 280 nm and 418-424 nm. Histidine is used in the elution buffer rather than the more typical imidazole because it is less likely to bind to the active site heme iron than imidazole. Imidazole can be substituted, but is a much stronger eluting reagent, so significantly lower concentrations should be used. Elution fractions should be pooled by selecting those with the highest ratio of A417/A280 as evaluated using a spectrophotometer rather than the FPLC UV detector.
Figure 2. Representative chromatograms and spectra for steroidogenic P450 enzymes at the metal-affinity (Ni-NTA) chromatographic step.
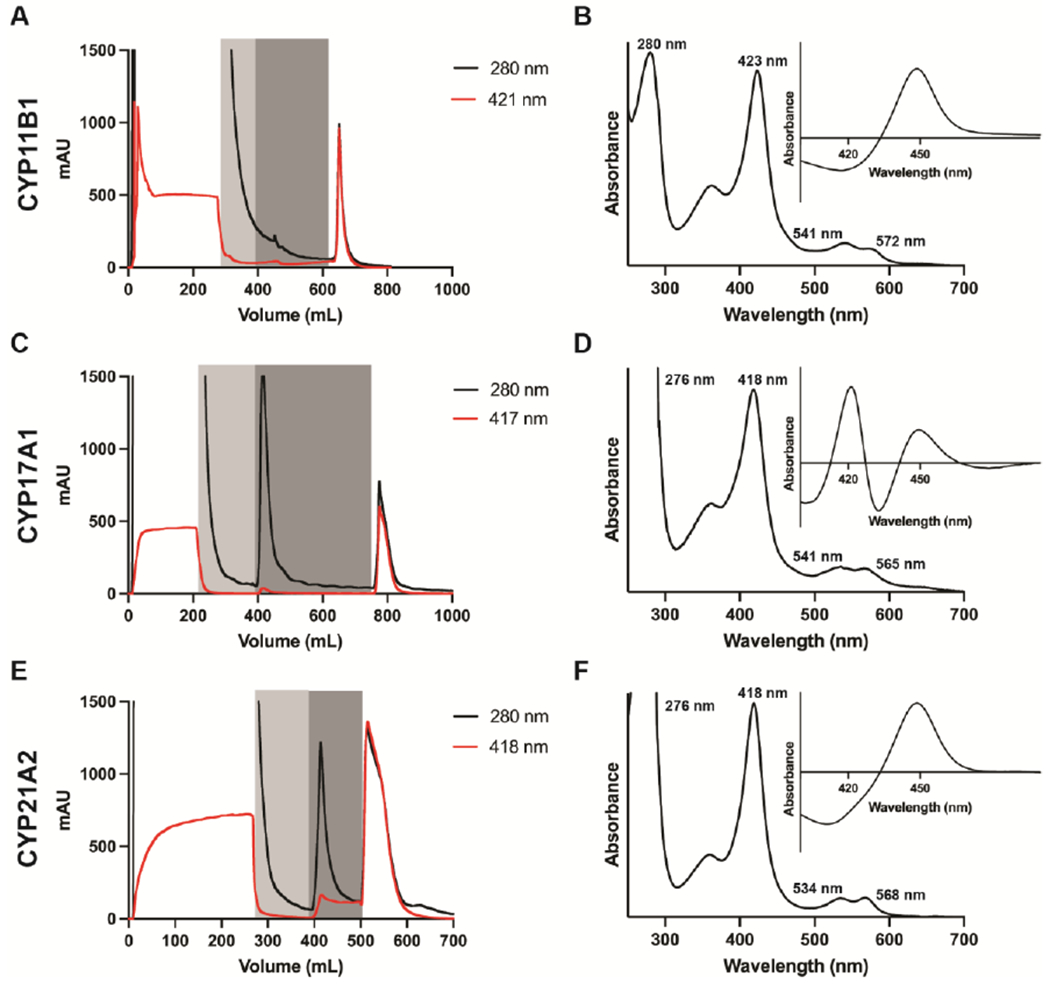
Chromatograms (panels A, C, and E) track the absorbance of the Soret peak (red), the absorbance of at 280 nm (black) during column loading, washing (grey shading), and gradient elution. The absolute spectra (panels B, D, and F) show the Soret peak wavelength and relative height compared to the 280 nm peak, while the reduced carbon monoxide difference spectra (inset) show the ratio of P450 to P420 at this stage of the purification.
7.3. Analysis
After this and each subsequent chromatographic step, ~50 µl of the pooled elution peak should be retained for an SDS-PAGE gel and characterized by collecting both the absorbance spectrum between 700 and 250 nm and a reduced carbon-monoxide difference spectrum (vide infra). Samples from the loading flow-through and wash steps can also be evaluated to provide clues for troubleshooting if needed. At this step, the eluted P450 absorbance spectrum can demonstrate a Soret peak shifted to or towards 424 nm (Figure 2B), consistent with histidine binding as a weak type II iron-coordinating ligand, which is readily removed in subsequent purification steps. An initial evaluation of the purity of the protein is made by calculating the absorbance of the Soret peak reporting on the heme content relative to the ~280 nm peak reporting primarily on tryptophans in proteins. Typical values at this stage are shown in Figure 2B, D, and F. However, preparations containing Emulgen or other conjugated compounds that have intrinsic absorbance in this region skew the ratio, causing the protein to look less pure than it actually is. Ideally, the reduced carbon monoxide difference spectra should have a peak at 450 nm—consistent with appropriately-incorporated heme—and do not have a peak at 420 nm—consistent with incorrect heme interactions (P420). This is typically true after the Ni-NTA column for all three CYP11 enzymes, but CYP21A2 can have some P420 and CYP17A1 can be mostly P420 at this stage (Figure 2B, D, F).
7.4. Notes
The Ni-NTA Superflow resin loses some of the coordinated nickel during this process, as evidenced by a fading of the blue color. The column can be washed and recharged with the resin in place for multiple uses for significant cost savings. Columns are regenerated by washing with 2-3 CV of water, followed by 2-3 CV of 6 M guanidinium hydrochloride with 0.2 M acetic acid to denature proteins. Then 2-3 CV of water are followed by 2-3 CV of 20 mM potassium phosphate, 500 mM sodium chloride, and 100 mM EDTA to strip the resin of any remaining nickel, resulting in white resin. The column is again washed with 2-3 CV of water, followed by 2-3 CV of 100 mM nickel sulfate in order to regenerate the column, restoring the blue color. The column is finally washed with 2-3 CV of water, after which it can be equilibrated with buffer for immediate use or stored in 20% ethanol at 4 °C.
8. Purification: Ion exchange chromatography
8.1. General
Many of the N-terminally truncated human P450 enzymes are solubilized from E. coli membranes using detergent, but can be removed at this step, facilitating usage in downstream applications. While most truncated human P450 enzymes are most stable in high salt and detergent, the detergent can often be removed if higher salt is maintained. This is true for CYP17A1 and CYP21A2. However, the CYP11 proteins, are purified using two detergents in the Ni-NTA step and only one of them (Tween 20) is depleted in the ion exchange step. Thus, ion exchange chromatography is not only a purification step, but often useful to thoroughly deplete detergent. When co-expressing with GroEL/ES, these chaperones are also effectively removed at this stage.
8.2. Procedure
All five of the steroidogenic P450 enzymes discussed herein have a positive isoelectic point (CYP17A1 pI=8.78, CYP21A2 pI=8.11, CYP11A1 pI=8.62, CYP11B1 pI=9.28, CYP11B2 pI=9.30) and are therefore compatible with a cation exchange column. CYP17A1 and CYP21A2 bind well to the weak cation exchanger Carboxymethylcellulose Sepharose Fast Flow resin but CYP11 enzymes interact better with the strong cation exchanger SP Sepharose High-Performance resin (Cytiva). In both cases, the Cytiva HiTrap 5 mL prepacked columns work quite effectively, with multiple columns connected in series as needed to provide sufficient binding capacity. On the scales described herein, this usually means three columns for CYP21A2 and two columns for CYP17A1 and CYP11 proteins. Before usage, the column is regenerated with 5 CV of 1 M sodium chloride according to manufacturer instructions and is then equilibrated with 5 CV of ion exchange loading buffer (Table 3).
Table 3:
Buffer compositions and volumes for metal-affinity chromatography.
| Buffer | CYP11 Proteins | CYP17A1 | CYP21A2 |
|---|---|---|---|
|
| |||
| Ion | 20 mM KPi (pH 7.4) | 5 mM KPi (pH 7.2) | 5 mM KPi (pH 6.8) |
| Exchange | 20% glycerol | 20% glycerol | 20% glycerol |
| Loading | 1% sodium cholate | 100 mM glycine | 1 mM EDTA |
| Buffer | 0.1 % Tween 20 | ||
| 15 CV | 10 CV | 20 CV | |
|
| |||
| Ion | 50 mM KPi (pH 7.4) | 50 mM KPi (pH 7.4) | 50 mM KPi (pH 6.8) |
| Exchange | 20% glycerol | 20% glycerol | 20% glycerol |
| Elution | 1% sodium cholate | 100 mM glycine | 350 NaCl |
| Buffer | 0.1% Tween 20 | 500 mM NaCl | 2 mM EDTA |
| 500 mM NaCl | |||
To bind to the ion exchange columns, the conductivity of the protein sample must be low. Since the CYP11 Ni-NTA elution buffer does not contain salt, the pooled Ni-NTA elution fractions from these proteins can be loaded directly from the NiNTA to the ion exchange column. As a result, the volume loaded onto the HiTrap SP Sepharose HP column is relatively small (~50 mL) and can be managed with Superloop of 50 or 150 mL capacity. However, the Ni-NTA elution buffers for both CYP17A1 and CYP21A2 contain significant amounts of salt and require dilution or salt removal prior to loading. For CYP17A1, the ion exchange loading buffer (Table 3) is supplemented with enough Emulgen 913 that the final detergent concentration for the total volume including the elution fractions will be 2% (v/v). This is then used to dilute the pooled Ni-NTA fractions 3-5-fold by volume. For CYP21A2, the same approach is used to dilute the pooled Ni-NTA fractions CYP21A2 5-fold by volume. As a result the loading volume for CYP17A1 and CYP21A2 is much higher (~200-500 mL), so use of the sample loading pump is convenient. However, sample could also be loaded using a large Superloop (with multiple loadings if necessary) or sample volume reduced by ultrafiltration prior to loading.
The prepared protein is then loaded onto the ion exchange column using a flow rate of 1.5-2 mL/min, taking care to stay under the pre-column pressure limit of 0.5 MPa and delta pressure limit of 0.3 MPa. As the column is loaded, a dark red band of P450 protein should initially form at the top of the column, growing larger as the resin is saturated. If this band approaches the end of the last column, simply pause the run and add another pre-equilibrated 5-mL HiTrap column after the last one before resuming the run. As with the Ni-NTA column, absorbance at 280 nm and the Soret peak should be monitored during the run to visualize the progress of the chromatography, assist in any troubleshooting, and guide elution fractions to pool. Again, it is a good idea to collect the flow-through in a clean, separate bottle in case the protein does not bind well to the column and troubleshooting is required.
After loading, the column is washed with 10-20 CV of ion exchange loading buffer (Table 3). This step not only purifies away other proteins (including GroEL/ES chaperones), but also depletes detergent for CYP17A1 and CYP21A2. The temptation to reduce the column volumes in this step should be avoided, even if it appears by monitoring the absorbance at 280 nm that little other protein is being removed.
Elution from the ion exchange column should be accomplished by a short linear gradient from loading to elution buffer (Table 3). For CYP17A1 and CYP21A2, a 10 CV gradient from loading buffer to elution buffer is followed by a 10 CV hold at 100% elution buffer. For the CYP11 proteins, a 30 CV gradient from loading to elution buffer is used. Fractionation volumes of 2-3 mL typically provide enough resolution for this stage. After elution, P450-containing fractions are identified by the increased Soret absorbance and by their visible red color and pooled, avoiding any fractions from contaminating peaks.
8.3. Analysis
The chromatograms (Figure 3A, C, E) should show relatively high background at 280 nm during sample loading, as well as some lower absorbance corresponding to the Soret peak, but the loading flow-through rarely represents significant P450 loss and is typically colorless. In contrast, the extended wash step should be characterized by nearly baseline absorbance at both wavelengths. As the elution gradient proceeds, a peak with increased absorbance at both 280 nm and 418 nm should be observed. As with the Ni-NTA column, a small sample (~50 µL) of the pooled P450 protein (and loading flow-through and wash steps, if desired) should be retained for SDS-PAGE analysis and characterization by UV-vis spectroscopy. At this step, the absorbance spectrum for the pooled elution fractions should indicate a Soret peak at 418-420 nm (Figure 3B, D, F), consistent with the removal of histidine used to elute from the nickel column and isolation of the water-Fe form. For CYP17A1, it is normal to see a small to moderate peak at 420 nm in the reduced carbon monoxide difference spectrum (Figure 3D) and does not signify a problem with the sample.
Figure 3. Representative chromatograms and spectra for steroidogenic P450 enzymes at the ion exchange chromatographic step.
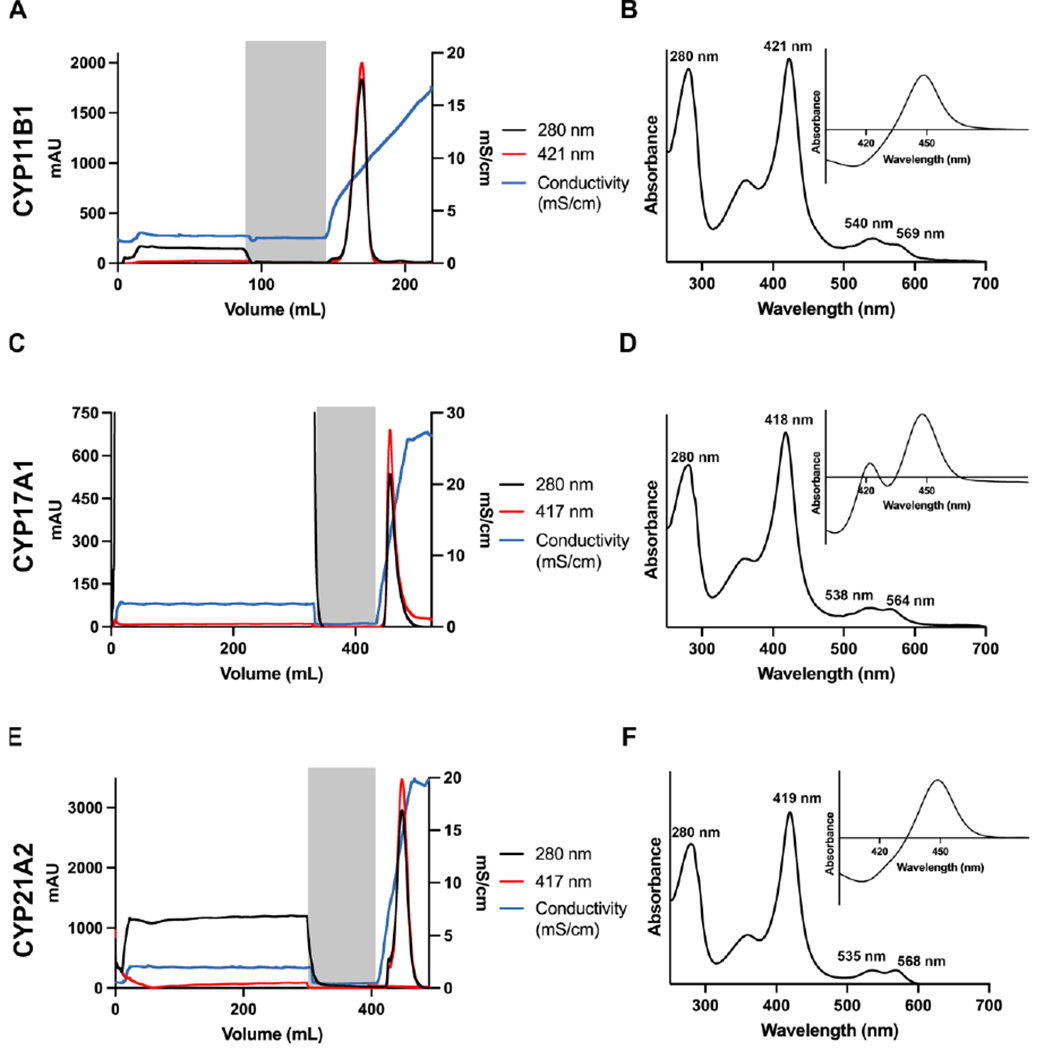
Chromatograms (panels A, C, E) track the absorbance of the Soret peak (red), the absorbance of at 280 nm (black) and the conductivity (green) during column loading, washing (grey shading), and gradient elution. The absolute spectra (panels B, D, and F) show the Soret peak wavelength and relative height compared to the 280 nm peak, while the reduced carbon monoxide difference spectra (inset) show the ratio of P450 to P420 at this stage of the purification.
8.4. Notes
If the first one or two P450-containing fractions eluted from the ion exchange column are cloudy, this suggests that the salt content was not high enough to keep these initial, more concentrated fractions from aggregating. Adding a few drops of higher-salt buffer usually reverses the cloudiness, but it is unknown if this aggregation is completely reversible on the molecular level. A better approach is to adjust the linear gradient to be more gradual next time, which usually prevents the cloudiness in the first place.
After use, the ion exchange column should be washed with 5 CV water, then 5 CV 20% ethanol, and stored at 4 °C for future usage. These columns can be re-equilibrated and reused until issues arise, such as protein precipitation or high pressure issues that don not resolve with cleaning. Precipitated protein can often be removed from the column by washing with 0.5 M NaOH using reverse flow. If the issues don’t resolve, the column should be replaced.
9. Purification: Size Exclusion chromatography
9.1. General
If the P450 protein is to be used for enzymatic studies, the purity is usually sufficient at this point and the size exclusion step may be omitted, as it only reduces yield without increasing protein functionality for most P450 enzymes, though less so for CYP17A1. An appropriate test is to evaluate biological activity before and after size exclusion chromatography to discern if the final column is warranted. However, if crystallization or other biophysical characterization is intended, then size exclusion provides both a further increase in purity that can be critical, as well as an evaluation of the oligomeric state.
9.2. Procedure
Size exclusion chromatography is accomplished using a HiLoad 16/600 Superdex 200 column. The column is equilibrated with 1.2-1.5 CV (manufacturer recommends 2 CV) of size exclusion buffer (Table 4) using a flow rate of 0.7-1 mL/min, taking care to stay under the pre- and delta-column pressure limits of 0.5 MPa and 0.3 MPa, respectively. It is important to start pre-equilibrating the column as soon as possible so that it will be ready by the time the sample is ready to be loaded onto the column. The goal here is to minimize the time that the P450 protein is in a highly concentrated state.
Table 4:
Buffer compositions and volumes for size exclusion chromatography.
| Buffer | CYP11 Proteins | CYP17A1 | CYP21A2 |
|---|---|---|---|
|
| |||
| Size | 50 mM KPi (pH 7.4) | 50 mM KPi (pH 7.4) | 50 mM KPi (pH 6.8) |
| Exclusion | 20% (v/v) glycerol | 20% (v/v) glycerol | 20% (v/v) glycerol |
| Buffer | 500 mM NaCl | 500 mM NaCl | 350 mM NaCl |
| 100 mM glycine | 1 mM EDTA | ||
No buffer exchanges are necessary to prepare CYP17A1 and CYP21A2 for size exclusion chromatography, as the same buffer is used for ion exchange elution and size exclusion. However the CYP11 proteins are exchanged to the size exclusion buffer (Table 4) by concentrating the pooled ion exchange fractions to ~2 mL by ultrafiltration (e.g. Amicon Ultra centrifugal filter units, MWCO 50 kDa) and then diluting 1:1 with size exclusion buffer. This concentration and dilution process is repeated five times.
In size exclusion buffer, all three CYP11 proteins and CYP17A1 are concentrated to 1.5-3 mL and CYP21A2 to 5-8 mL. Per manufacturer recommendations, the sample loaded on the HiLoad 16/600 Superdex 200 column should not exceed 600 µM in concentration and 6 mL in volume. While this is not an issue for the CYP11 proteins and CYP17A1, the high yield of CYP21A2 often necessitates two size exclusion column runs since loading higher concentrations or larger volumes is not recommended. While there isn’t typically any visible precipitation after concentration, the sample is nonetheless centrifuged at ~4,000 x g for 10 minutes to remove any precipitate.
Concentrated samples are best loaded by transfer into a small syringe and injection onto the FPLC port to a sample loop. Sample loss can be reduced by using a capillary loop at least 1 mL larger than the sample volume. The isocratic column is run with 1.2 CV of size exclusion buffer with a 0.25 ml/min flow rate. Useful fraction sizes are ~2 mL. Fractions containing the P450 are identified by the absorbance of the Soret peak and pooled, avoiding any shouldering peaks (containing other oligomeric states) and contaminating peaks with higher 280 nm absorbance.
9.3. Analysis
The chromatograms from the size exclusion columns should not demonstrate significant peaks with only absorbance at 280 nm. Typically a single peak elutes with absorbance at both 280 nm and a wavelength corresponding to the Soret peak (Figure 4A, C, E). Comparison of the elution volume with calibration standards is used to estimate the molecular weight. Typical oligomeric states and Kav values are provided (Figure 4A, C, E). As with the previous chromatography steps, a ~50 µL sample should be collected for SDS-PAGE and spectral analysis. These final absolute spectra should have Soret peaks of 418-421 nm, corresponding to heme iron coordinated to water (Figure 4B, D, F). The initial reduced carbon monoxide difference spectra rarely show P420 for CYP11B enzymes, but some can be observed for unliganded CYP17A1 and CYP21A2 (Figure 4B, D, F insets). As the reduced state is less stable it is normal to see a small shoulder at 420 nm that develops in the reduced CO difference spectrum at longer times for all five P450 enzymes.
Figure 4. Representative chromatograms and spectra for steroidogenic P450 enzymes at the size exclusion chromatographic step.
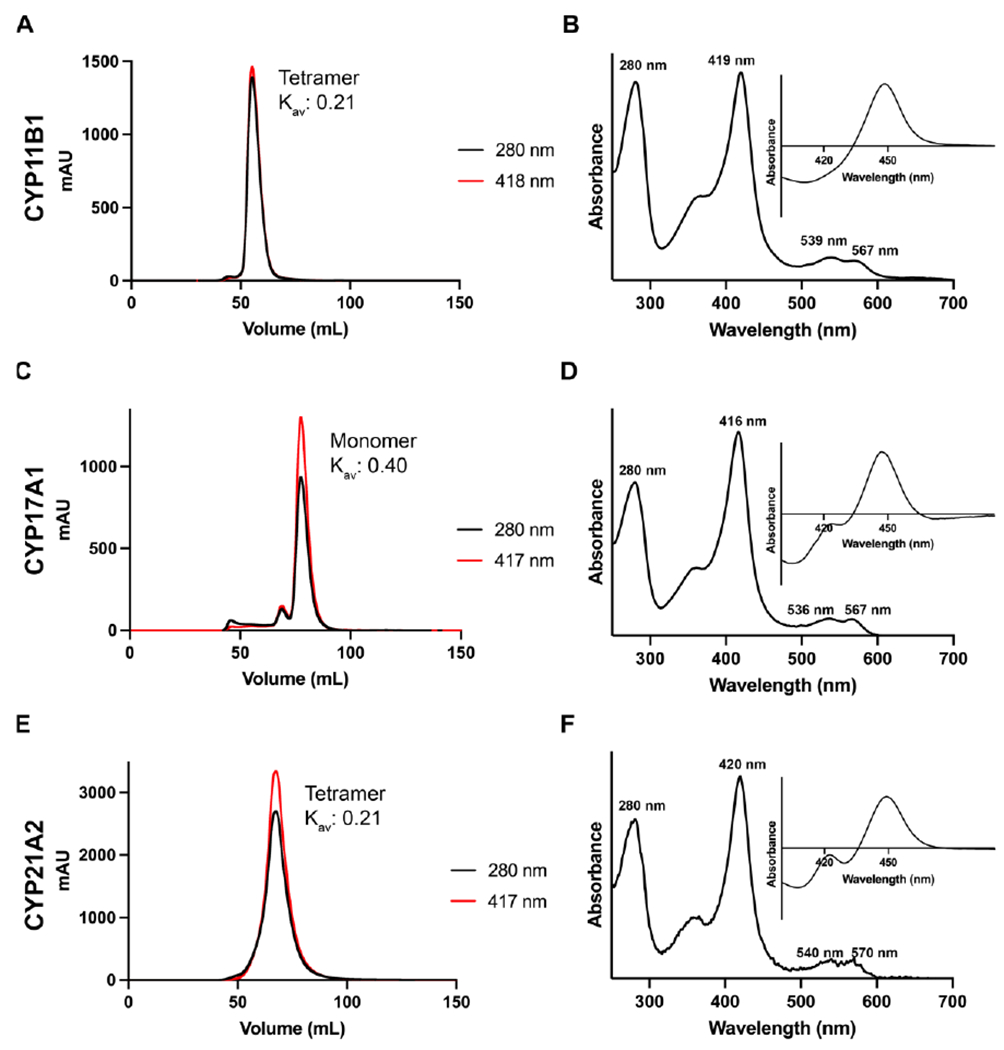
Chromatograms (panels A, C, E) track the absorbance of the Soret peak (red), the absorbance of at 280 nm (black) during an isocratic run. While the specific elution volumes vary between columns, the Kav should be relatively consistent between different columns calibrated with different standards. The absolute spectra (panels B, D, and F) show the Soret peak wavelength and relative height compared to the 280 nm peak, while the reduced carbon monoxide difference spectra (B, D, and F insets) show the relative amounts of P450 to P420 at this stage of the purification.
9.4. Notes
The HiLoad size exclusion column should be washed with 1 CV of water, then 1 CV of 20% ethanol at 1 ml/min and stored at 4 °C. Size exclusion columns can be cleaned according to the manufacturer instructions, typically after every 10 uses or any protein precipitation. If pressure issues persist after this, the column can be repacked using fresh resin and recalibrated using known protein standards.
10. Expected Results
10.1. SDS-PAGE
Success of the protein purification is evaluated using SDS-PAGE. The above protocol typically yields a single major band for all five proteins even when overloaded (Figure 5). Notably, the GroEL band at ~60 kDa is significantly reduced after the ion exchange column.
Figure 5. Representative SDS-PAGE gels for purification of steroidogenic human cytochrome P450 enzymes.
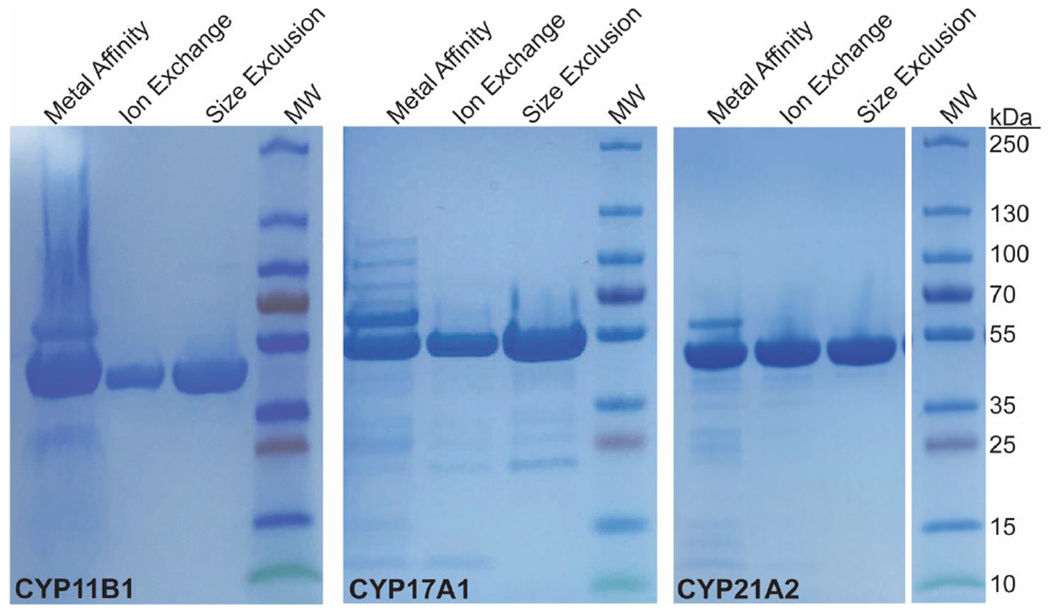
Elution fractions from each column are loaded at fairly high concentrations to increase awareness of secondary bands. Each P450 runs just below the 55 kDa molecular weight marker. Notably, much of the GroEL (60 kDa) is removed during ion exchange. According to such gels, the protein purity doesn’t appear to change significantly between the ion exchange and size exclusion steps and the protein can be used for enzymatic assays after ion exchange, but for crystallography the subsequent size exclusion step is highly recommended.
10.2. Spectral characterization
Typical purification statistics are shown below for CYP11A1, CYP11B1, CYP11B2, CYP17A1, and CYP21A2 as measured after the Ni-NTA, ion exchange, and size exclusion columns (Table 5). At each step, the λmax is the wavelength corresponding to the Soret peak in the UV-visible spectrum. The absorbance of the Soret peak is used to calculate nanomoles of P450, and the yield is calculated as the nanomoles of P450 at each step per liter of culture. The ratio of absorbance of the Soret peak vs. the absorbance of the peak near 280 nm can be a general estimate of purity, and should increase over the course of the purification. The percentage of P420 relative to P450 is an estimate from the values at the respective wavelengths in the reduced-carbon monoxide difference spectrum at the earliest time point, such that the lower the number the better. The oligomeric state is determined by using a size exclusion calibration curve to estimate the molecular weight of the protein from its elution volume.
Table 5:
Purification statistics
| CYP11A1 | CYP11B1 | CYP11B2 | CYP17A1 | CYP21A2 | ||
|---|---|---|---|---|---|---|
| Ni-NTA Column | Soret λmax (nm) | 418 | 423 | 424.5 | 418 | 418 |
| ASoret/A280 | 0.78 | 0.91 | 0.76 | N.A.* | N.A.* | |
| Yield (nmoles/L)† | 177 | 385 | 200 | 162-277 | 1100-1700 | |
| %P420 | 0% | 0% | 0% | ~75% | 0% | |
| Ion Exchange Column | Soret λmax (nm) | 418 | 421 | 421.5 | 418 | 419 |
| ASoret/A280 | 1.05 | 1.05 | 0.92 | 1.2 | 1.2 | |
| Yield (nmoles/L)† | 380 | 110 | 140 | 100-200 | 800-1200 | |
| %P420 | 0% | 0% | ~2% | ~30% | 0% | |
| Size Exclusion Column | Soret λmax (nm) | 417 | 419 | 421.5 | 416-417 | 419-420 |
| ASoret/A280 | 1.09 | 1.15 | 1.07 | 1.3-1.5 | 1.2-1.3 | |
| Yield (nmoles/L)† | 240 | 93 | 82 | 80-130 | 700-950 | |
| %P420 | 0% | 0% | ~5% | ~5% | ~5-10% | |
| Oligomeric state | Tetramer | Tetramer | Tetramer | Monomer | Trimer-tetramer |
N.A, not applicable. Rz values are not useful at this stage because the presence of Emulgen 913 in the buffer obscures the 280 nm peak from protein.
per liter of expression culture
11. Storage and Usage
11.1. Storage
Purified enzyme can be concentrated by filtration or diluted to the desired concentration and should be divided into single-use aliquots of sensible volumes depending on the intended use. These aliquots are flash frozen in liquid nitrogen or a dry ice/ethanol slurry before transfer to a −80 °C freezer for long term storage. Best practice is that aliquots should only be thawed once for immediate use and never refrozen.
11.2. Usage for Enzymology Applications
Protein purified through the ion exchange step is usually of sufficient purity for enzymology applications. The thawed protein is usually diluted into a reaction buffer with lower salt and no glycerol for enzymology studies. They do not require reconstitution with lipid to be fully active, which both simplifies usage and yields a more homogeneous reaction mixture. As for the full-length enzymes, reconstitution with NADPH-cytochrome P450 is required, in which a P450:reductase ratio of 1:4 is typically optimal. For the CYP11B enzymes, the highest activity is usually obtained at a P450:adrenodoxin:adrenodoxin reductase ratio of 1:40:1. These truncated, His-tagged, human steroidogenic cytochrome P450 proteins are enzymatically active, with the same substrate selectivity, producing products in the same ratios and with the same regio- and stereospecificity as the full length enzymes.
11.3. Usage for Structural Biology Applications
As a structural technique, NMR is not as sensitive to low concentrations of contaminants and thus P450s might be sufficiently pure after ion exchange for NMR applications. Crystallography is much more sensitive to small amounts of contaminating proteins, so size exclusion is highly recommended. If detergent is required for crystallization, the protein should be slightly over-concentrated before detergent is added so that the protein is diluted to the desired final concentration. If a particular liganded state is to be examined, expression and/or purification with ligand in the growth media and purification buffers can both increase the yield and further increase homogeneity.
12. Spectral analysis
12.1. Generation of the P450 UV-visible spectrum
Collecting a UV-visible spectrum of each sample from 250-700 nm will inform on sample purity, active site occupation, and yield. A scanning UV-visible spectrophotometer (Shimadzu) is set to scan from 700 to 250 nm, with the sampling interval set to measure absorbance every 0.5-1 nm. In a 1 mL quartz cuvette, 950-980 μL of elution buffer is added to measure the initial baseline. To the same cuvette, 20-50 μL (such that the final volume = 1 mL) of protein sample is added and inverted gently to mix. P450 concentrations are calculated from absorbance magnitude of the heme Soret peak using Beer’s law and extinction coefficient ε = 100 mM−1cm−1 (Peterson, 1971). The position of the Soret peak informs on the coordination of the heme iron. In general, a Soret peak at ~417 nm indicates that the iron is hexacoordinated with water as the sixth ligand. A Soret peak at ~393 nm indicates that the heme iron is pentacoordinated, often associated with substrate binding in the active site and displacing the water molecule. The Soret peak shifts to ~424 nm (sometimes higher) when a molecule binds within the active site and forms a coordinate-covalent bond with the iron (often through a nitrogen’s lone pair of electrons), replacing water as the sixth ligand. For biophysical studies dependent on the total amount of protein such as ligand binding experiments, X-ray crystallography and NMR spectroscopy, this type of spectrum is best for protein quantitation.
12.2. Generation of a reduced-carbon monoxide difference spectrum
To evaluate integrity of the heme-thiolate bond, a reduced carbon-monoxide-bound difference spectrum is collected. Using the same cuvette as before, a few grains of solid sodium dithionite (stored in small aliquots in an air-tight, desiccated container to prevent oxidation) are added, then gently mixed by inverting. The spectrophotometer is set to scan from 400-500 nm, with sampling intervals of 0.5-1 nm. A baseline of the reduced protein sample is collected so that only changes in absorbance are measured. Next, a gentle stream of individual bubbles of carbon monoxide gas (see safety footnote) is bubbled directly into the cuvette, then the cuvette is sealed and returned to the spectrophotometer. The resulting spectrum (e.g. Figures 2-4, right inset) will show only the difference in absorbance of the Soret peak, ideally with a peak at 450 nm and a trough at 420 nm (P450). Some proteins demonstrate some absorbance at 420 nm, called P420. If multiple spectra are collected over time, one often sees an increase in P420 as the reduced form is less stable than the oxidized form. Notably, such spectra collected after early purification steps often contain a strong absorbance at 420 nm which often resolves as purification proceeds. Our experience with many human P450 proteins suggests that this occurs not by purifying away protein that is the P420, but by conversion between different states in different buffers/conditions. Thus P420 can be converted to P450, at least under some conditions. It is frequently observed that saturation of the enzyme with ligand can improve the ratio of P450:P420. A particularly striking example is CYP8B1, whose reduced CO difference assay is almost all P420 in the absence of substrate and all P450 in the presence of substrate (Liu et al., 2022). The reduced carbon monoxide difference spectrum and the extinction difference between 450 and 490 nm of 91 cm−1 mM−1 (Omura & Sato, 1963) is used to quantitate enzyme concentration for metabolism assays.
13. Safety considerations and standards
Carbon monoxide used to generate the reduced carbon monoxide difference spectra is a flammable gas that is an inhalation hazard. Best practice is to use in a hood with appropriate precautions. Use of a sonicator for cell lysis should employ either a closed sound-dampening chamber or ear protection.
Acknowledgments
The technical knowledge represented herein was developed over many years, with input from many sources, but was primarily funded by the National Institutes of Health research grants to the Scott lab (R37 GM076343, R01 GM135346, R01 GM102505, R01 GM130997), training grants supporting two authors (T32 GM132046 and T32 GM007767), and a fellowship supporting a third author (F31 HD111338).
Abbreviations include:
- BME
β-mercaptoethanol
- CV
column volume
- EDTA
ethylenediaminetetraacetic acid
- KPi
potassium phosphate.
References
- Axen E, Postlind H, Sjoberg H, & Wikvall K (1994). Liver mitochondrial cytochrome P450 CYP27 and recombinant-expressed human CYP27 catalyze 1 alpha-hydroxylation of 25-hydroxyvitamin D3. Proc Natl Acad Sci U S A, 91(21), 10014–10018. 10.1073/pnas.91.21.10014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes HJ, Arlotto MP, & Waterman MR (1991). Expression and enzymatic activity of recombinant cytochrome P450 17 alpha-hydroxylase in Escherichia coli. Proc Natl Acad Sci U S A, 88(13), 5597–5601. 10.1073/pnas.88.13.5597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brixius-Anderko S, & Scott EE (2019). Structure of human cortisol-producing cytochrome P450 11B1 bound to the breast cancer drug fadrozole provides insights for drug design. J Biol Chem, 294(2), 453–460. 10.1074/jbc.RA118.006214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brixius-Anderko S, & Scott EE (2021). Structural and functional insights into aldosterone synthase interaction with its redox partner protein adrenodoxin. J Biol Chem, 100794. 10.1016/j.jbc.2021.100794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Doray B, & Kemper B (1998). A conserved proline-rich sequence between the N-terminal signal-anchor and catalytic domains is required for assembly of functional cytochrome P450 2C2. Arch Biochem Biophys, 350(2), 233–238. 10.1006/abbi.1997.0524 [DOI] [PubMed] [Google Scholar]
- Cosme J, & Johnson EF (2000). Engineering microsomal cytochrome P450 2C5 to be a soluble, monomeric enzyme. Mutations that alter aggregation, phospholipid dependence of catalysis, and membrane binding. J Biol Chem, 275(4), 2545–2553. 10.1074/jbc.275.4.2545 [DOI] [PubMed] [Google Scholar]
- DeVore NM, & Scott EE (2012). Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001 [Research Support, N.I.H., Extramural]. Nature, 482(7383), 116–119. 10.1038/nature10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir V, Reisch N, Bleicken CM, Lebl J, Kamrath C, Schwarz HP, Grötzinger J, Sippell WG, Riepe FG, Arlt W, & Krone N (2009). Steroid 17alpha-hydroxylase deficiency: functional characterization of four mutations (A174E, V178D, R440C, L465P) in the CYP17A1 gene. J Clin Endocrinol Metab, 94(8), 3058–3064. 10.1210/jc.2009-0172 [DOI] [PubMed] [Google Scholar]
- Fehl C, Vogt CD, Yadav R, Li K, Scott EE, & Aube J (2018). Structure-based design of inhibitors with improved selectivity for steroidogenic cytochrome P450 17A1 over cytochrome P450 21A2. J Med Chem, 61(11), 4946–4960. 10.1021/acs.jmedchem.8b00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Griswold J, Erman M, & Pangborn W (2009). Structural basis for androgen specificity and oestrogen synthesis in human aromatase [Research Support, N.I.H., Extramural]. Nature, 457(7226), 219–223. 10.1038/nature07614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding BW, Wong SH, & Nelson DH (1964). Carbon Monoxide-Combining Substances in Rat Adrenal. Biochim Biophys Acta, 92, 415–417. 10.1016/0926-6569(64)90206-8 [DOI] [PubMed] [Google Scholar]
- Johnson EF, Wester MR, & Stout CD (2002). The structure of microsomal cytochrome P450 2C5: a steroid and drug metabolizing enzyme. Endocr Res, 28(4), 435–441. 10.1081/erc-120016820 [DOI] [PubMed] [Google Scholar]
- Kikuta Y, Kusunose E, Sumimoto H, Mizukami Y, Takeshige K, Sakaki T, Yabusaki Y, & Kusunose M (1998). Purification and characterization of recombinant human neutrophil leukotriene B4 omega-hydroxylase (cytochrome P450 4F3). Arch Biochem Biophys, 355(2), 201–205. 10.1006/abbi.1998.0724 [DOI] [PubMed] [Google Scholar]
- Kusano K, Kagawa N, Sakaguchi M, Omura T, & Waterman MR (2001). Importance of a proline-rich sequence in the amino-terminal region for correct folding of mitochondrial and soluble microbial p450s. J Biochem, 129(2), 271–277. 10.1093/oxfordjournals.jbchem.a002854 [DOI] [PubMed] [Google Scholar]
- Kusano K, Sakaguchi M, Kagawa N, Waterman MR, & Omura T (2001). Microsomal p450s use specific proline-rich sequences for efficient folding, but not for maintenance of the folded structure. J Biochem, 129(2), 259–269. 10.1093/oxfordjournals.jbchem.a002853 [DOI] [PubMed] [Google Scholar]
- Liu J, Carlson HA, & Scott EE (2022). The structure and characterization of human cytochrome P450 8B1 supports future drug design for nonalcoholic fatty liver disease and diabetes. J Biol Chem, 298(9), 102344. 10.1016/j.jbc.2022.102344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo J, Di Nardo G, Griswold J, Egbuta C, Jiang W, Gilardi G, & Ghosh D (2013). Structural basis for the functional roles of critical residues in human cytochrome p450 aromatase. Biochemistry, 52(34), 5821–5829. 10.1021/bi400669h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sanders E, Estabrook RW, Cooper DY, & Rosenthal O (1966). Isolation from adrenal cortex of a nonheme iron protein and a flavoprotein functional as a reduced triphosphopyridine nucleotide-cytochrome P-450 reductase. Arch. Biochem. Biophys, 117(3), 660–673. 10.1016/0003-9861(66)90108-1 [DOI] [Google Scholar]
- Omura T, & Sato R (1963). Fractional solubilization of haemoproteins and partial purification of carbon monoxide-binding cytochrome from liver microsomes. Biochim Biophys Acta, 71, 224–226. 10.1016/0006-3002(63)91015-1 [DOI] [PubMed] [Google Scholar]
- Pan Y, Abd-Rashid BA, Ismail Z, Ismail R, Mak JW, & Ong CE (2011). Heterologous expression of human cytochromes P450 2D6 and CYP3A4 in Escherichia coli and their functional characterization. Protein J, 30(8), 581–591. 10.1007/s10930-011-9365-6 [DOI] [PubMed] [Google Scholar]
- Patten CJ, & Koch P (1995). Baculovirus expression of human P450 2E1 and cytochrome b5: spectral and catalytic properties and effect of b5 on the stoichiometry of P450 2E1-catalyzed reactions. Arch Biochem Biophys, 317(2), 504–513. 10.1006/abbi.1995.1194 [DOI] [PubMed] [Google Scholar]
- Peterson JA (1971). Camphor Binding by Pseudomonas putida Cytochrome P-450. Arch Biochem. Biophys, 144, 678–693. [DOI] [PubMed] [Google Scholar]
- Sagara Y, Barnes HJ, & Waterman MR (1993). Expression in Escherichia coli of functional cytochrome P450c17 lacking its hydrophobic amino-terminal signal anchor. Arch Biochem Biophys, 304(1), 272–278. 10.1006/abbi.1993.1349 [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Mihara K, & Sato R (1984). Signal recognition particle is required for co-translational insertion of cytochrome P-450 into microsomal membranes. Proc Natl Acad Sci U S A, 81(11), 3361–3364. 10.1073/pnas.81.11.3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, & Park HW (2011). Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc Natl Acad Sci U S A, 108(25), 10139–10143. 10.1073/pnas.1019441108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Salamanca-Pinzon SG, Wu ZL, Xiao Y, & Guengerich FP (2010). Human cytochrome P450 4F11: heterologous expression in bacteria, purification, and characterization of catalytic function. Arch Biochem Biophys, 494(1), 86–93. 10.1016/j.abb.2009.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger BP, Gunsalus IC, & Sligar SG (1986). Nucleotide sequence of the Pseudomonas putida cytochrome P-450cam gene and its expression in Escherichia coli. J Biol Chem, 261(3), 1158–1163. https://www.ncbi.nlm.nih.gov/pubmed/3003058 [PubMed] [Google Scholar]
- vonWachenfeldt C, Richardson TH, Cosme J, & Johnson EF (1997). Microsomal P450 2C3 is expressed as a soluble dimer in Escherichia coli following modifications of its N-terminus. Arch Biochem Biophys, 339(1), 107–114. https://doi.org/DOI 10.1006/abbi.1996.9859 [DOI] [PubMed] [Google Scholar]
- Wester MR, Stout CD, & Johnson EF (2002). Purification and crystallization of N-terminally truncated forms of microsomal cytochrome P450 2C5. Methods Enzymol, 357, 73–79. http://www.ncbi.nlm.nih.gov/pubmed/12424899 [DOI] [PubMed] [Google Scholar]
- Zhan YY, Liang BQ, Wang H, Wang ZH, Weng QH, Dai DP, Cai JP, & Hu GX (2016). Effect of CYP2D6 variants on venlafaxine metabolism in vitro. Xenobiotica, 46(5), 424–429. 10.3109/00498254.2015.1089364 [DOI] [PubMed] [Google Scholar]