

Abstract
Introduction
Duchenne muscular dystrophy (DMD) is a genetic muscle disorder that manifests during early childhood and is ultimately fatal. Recently approved treatments targeting the genetic cause of DMD are limited to specific subpopulations of patients, highlighting the need for therapies with wider applications. Pharmacologic inhibition of myostatin, an endogenous inhibitor of muscle growth produced almost exclusively in skeletal muscle, has been shown to increase muscle mass in several species, including humans. Taldefgrobep alfa is an anti-myostatin recombinant protein engineered to bind to and block myostatin signaling. Preclinical studies of taldefgrobep alfa demonstrated significant decreases in myostatin and increased lower limb volume in three animal species, including dystrophic mice.
Methods
This manuscript reports the cumulative data from three separate clinical trials of taldefgrobep alfa in DMD: a phase 1 study in healthy adult volunteers (NCT02145234), and two randomized, double-blind, placebo-controlled studies in ambulatory boys with DMD–a phase 1b/2 trial assessing safety (NCT02515669) and a phase 2/3 trial including the North Star Ambulatory Assessment (NSAA) as the primary endpoint (NCT03039686).
Results
In healthy adult volunteers, taldefgrobep alfa was generally well tolerated and resulted in a significant increase in thigh muscle volume. Treatment with taldefgrobep alfa was associated with robust dose-dependent suppression of free myostatin. In the phase 1b/2 trial, myostatin suppression was associated with a positive effect on lean body mass, though effects on muscle mass were modest. The phase 2/3 trial found that the effects of treatment did not meet the primary endpoint pre-specified futility analysis threshold (change from baseline of ≥ 1.5 points on the NSAA total score).
Conclusions
The futility analysis demonstrated that taldefgrobep alfa did not result in functional change for boys with DMD. The program was subsequently terminated in 2019. Overall, there were no safety concerns, and no patients were withdrawn from treatment as a result of treatment-related adverse events or serious adverse events.
Trial Registration
Supplementary Information
The online version contains supplementary material available at 10.1007/s40120-023-00570-w.
Keywords: Clinical trial, Duchenne muscular dystrophy, Taldefgrobep alfa, Neuromuscular disorder, Myostatin
Plain Language Summary
The goal of this program was to develop a treatment to improve muscle function in patients with Duchenne muscular dystrophy (DMD). Muscle weakness in patients with DMD is progressive, leading to the irreversible loss of walking ability and eventually death due to cardiorespiratory failure. One potential way of improving muscle function is to target a protein known as myostatin that acts in healthy muscle to regulate muscle size. Studies in animals have shown that blocking myostatin can increase muscle size. Taldefgrobep alfa is a drug designed to block myostatin and it was shown to induce muscle growth in animals. A study in healthy volunteers found that taldefgrobep alfa was able to increase muscle size in humans and was not associated with safety concerns. Following this, a study was conducted in boys with DMD who were either treated with taldefgrobep alfa or a placebo. This first study in patients found that treatment was able to reduce myostatin levels and had a small effect on muscle size, supporting a larger trial in more patients with DMD. The aim of the larger trial was to test if taldefgrobep alfa had a meaningful effect on muscle function in patients with DMD. Results from this key trial did not meet the targeted improvement in function and a decision was made to end the trial and halt the use of taldefgrobep alfa as a potential treatment for DMD. No patients stopped treatment with taldefgrobep alfa as a result of adverse safety effects and no safety concerns were identified.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40120-023-00570-w.
Key Summary Points
| Why carry out this study? |
| Duchenne muscular dystrophy (DMD) is a genetic muscle disorder that manifests during early childhood and is ultimately fatal |
| Approved therapies for DMD are only available for specific populations of patients and an unmet need remains for those patients who are not able to receive available treatments |
| The aim of the studies was to evaluate the safety and efficacy of the anti-myostatin taldefgrobep alfa as a potential treatment for patients with DMD |
| What was learned from the study? |
| The phase 2/3 trial of taldefgrobep alfa in boys with DMD did not meet the primary endpoint of a functional change assessed using the North Star Ambulatory Assessment and the trial was subsequently terminated |
| Although taldefgrobep alfa did not result in functional improvements in boys with DMD, there were no safety concerns identified from the treatment |
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked, recessive, degenerative neuromuscular disorder that affects 1 in 3500–5000 male births worldwide [1–3], and has a pooled global prevalence of 7.1 cases per 100,000 males and 2.8 cases per 100,000 in the general population [4]. DMD manifests in the early pre-school years [1]. Muscle weakness begins in proximal leg and pelvic muscles, resulting in loss of ambulation by the ages of 8–14 years. Later, muscle weakness progresses into the axial muscles and upper extremities, culminating in premature death in early adulthood due to cardiorespiratory failure [1].
DMD occurs as a result of an inherited or spontaneous loss-of-function mutation in the DMD gene, resulting in a lack of functional dystrophin protein [5, 6]. Dystrophin is a critical component of a protein complex that serves as a structural anchor connecting the muscle fiber cytoskeleton to the extracellular matrix through the cell membrane. This structure provides sarcolemmal strength and stability during the conformational changes involved in muscle contraction and relaxation. Absence of dystrophin destabilizes the complex, resulting in damage to the cell membrane during muscle contractions [7]. Damaged muscle is replaced with fibrous tissue and fatty deposits, leading to severe and progressive muscle wasting [8].
Current standard of care for DMD includes corticosteroid use, which allows modest functional improvements at the cost of multiple side effects [9, 10]. Approved therapeutic treatments targeting the genetic cause of DMD are currently limited to use in individuals who carry either nonsense mutations, present in 10–15% of boys with DMD [11, 12], specific mutations that are amenable to antisense oligonucleotide (ASO)-induced skipping of exons 51 [13, 14], 53 [15, 16], or 45 [17], present in only 13–14% [13, 14, 18], 8–10% [15, 18], and 8–9% [14, 17, 18] of boys with DMD, respectively, or to a restricted age range of patients [19]. Given the limitations of these treatments, researchers have sought alternative therapies with wider applications that could be relevant for the entire DMD population, irrespective of genotype, that could potentially be used as add-on therapies.
Myostatin is a member of the transforming growth factor-β superfamily [20]. It is an endogenous inhibitor of muscle growth with predominant expression in developing and adult skeletal muscle [20–22]. Myostatin is expressed as a precursor protein that undergoes processing to generate an N-terminal inhibitory peptide and a C-terminal peptide, which is biologically active as a dimer following proteolysis of inhibitory peptides [23, 24]. Mature myostatin acts upon muscle tissue by binding to the activin type IIB (ActRIIB) receptor [25]. Myostatin binding triggers activin-like kinase-4/5 (signaling receptor) recruitment and subsequent phosphorylation of Smad2 and Smad3, which dimerize and, in association with Smad4, inhibit genes responsible for myogenesis [25]. Overexpression of myostatin in skeletal muscle causes a significant loss of muscle mass in association with reduced expression of structural genes and myogenic transcription factors [26]. Conversely, limiting myostatin has been shown to increase muscle mass in animals as diverse as zebrafish to humans [21, 22, 27, 28]. Taken together, these results suggest that anti-myostatin therapy could be effective for the treatment of muscle-wasting diseases, such as DMD.
In the mdx mouse, a preclinical model of DMD, inhibition or loss of myostatin has been shown to increase muscle mass and muscle strength [29–32]. In addition, inhibition of myostatin has been shown to enhance muscle regeneration in mouse models of acute and chronic injury [33], and to reverse fibrosis in skeletal muscle tissue in mdx mice [34]. This evidence has led to the development of a variety of investigational anti-myostatin therapies designed to promote muscle growth and repair in muscular dystrophy.
Taldefgrobep alfa (RG6206, RO7239361, previously BMS-986089) is a fully human anti-myostatin adnectin recombinant protein that binds to the C-terminal of mature myostatin and the ActRIIB–myostatin complex. Taldefgrobep alfa binding prevents activin-like kinase-4/5 recruitment, resulting in the inhibition of downstream phosphorylated Smad2/3 signaling, which blocks myostatin activity.
In preclinical studies, taldefgrobep alfa inhibited phosphorylation of Smad2/3 in vitro in a concentration-dependent manner. It also produced statistically significant decreases in free myostatin and increases in lower limb volume across three animal species—severe combined immunodeficient mice, juvenile rats, and adolescent cynomolgus monkeys. These effects were dose- and time-dependent across all three species, with lower limb muscle volume increases of approximately 30% observed in rodents and increases of approximately 5% in cynomolgus monkeys. The data from these studies are reported in Sect. 3 of the Supplementary Material. These results supported the further evaluation of taldefgrobep alfa in clinical trials for DMD.
This manuscript reports the cumulative data from three separate clinical trials of taldefgrobep alfa in DMD: a phase 1 study in healthy adult volunteers, a phase 1b/2 clinical trial in ambulatory boys with DMD, and a phase 2/3 clinical trial in ambulatory boys with DMD. The clinical development program for taldefgrobep alfa was halted in 2019.
Methods
WN40225: A Placebo-Controlled, Double-Blind, Single- and Multiple-Ascending-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Taldefgrobep Alfa in Healthy Adults (NCT02145234)
Study Design
The phase 1 study of taldefgrobep alfa was a randomized, placebo-controlled, sequential, combined single ascending dose (SAD) and multiple ascending dose (MAD) study of subcutaneous (SC) administration of taldefgrobep alfa in healthy adults. The study was conducted at a single site at West Coast Clinical Trials (Langhorne, PA, USA).
Participants
The first participant was enrolled into the study on June 26, 2014 and the last participant completed the study on February 4, 2016. Eligible individuals were aged 18–55 years with no clinically significant deviation from normal medical history as determined by physical examination, electrocardiograms (ECGs), and laboratory evaluations. A full list of inclusion/exclusion criteria can be found in Sect. 4.1 of the Supplementary Material.
Study Endpoints
The primary objective of this phase 1 study was to assess the safety and tolerability of a single dose or multiple SC doses of taldefgrobep alfa in healthy volunteers. Secondary and exploratory objectives included assessment of the pharmacokinetics (PK), target engagement (free myostatin lowering), immunogenicity, and pharmacodynamics (PD) of taldefgrobep alfa. Exploratory PD endpoints included magnetic resonance imaging (MRI), measures of thigh muscle volume, and dual-energy x-ray absorptiometry (DXA) measures of lean body mass (LBM). A full list of study objectives can be found in Sect. 4.2 of the Supplementary Material.
Randomization and Dosing
Participants were randomized to receive a SAD (see Sect. 4.3 of the Supplementary Material for additional methodologic details of the SAD phase) or MADs (15, 45, 90, or 180 mg) of taldefgrobep alfa or placebo administered subcutaneously (Fig. 1).
Fig. 1.

Study design of the MAD phase of the healthy volunteers phase 1 study. Study design of the MAD phase comprising healthy adult subjects randomized 3:1 to receive taldefgrobep alfa or placebo administered subcutaneously. Dosing for the MAD phase occurred only after safety and tolerability were reviewed from at least three SAD panels. MAD multiple ascending dose, Q1W once-weekly dosing, Q2W once every 2 weeks dosing, SAD single ascending dose
Dosing in the MAD panels occurred only after the safety and tolerability data through day 49, and PK and free-myostatin concentrations through day 15 from at least three SAD panels were reviewed. Safety, PK, and PD data collected in SAD panels 1–3 were used to determine dosing and panel size in the MAD phase (see Sect. 4.3 of the Supplementary Material for SAD data). Ninety-seven healthy volunteers were randomized (3:1) to five double-blinded dose panels. In four panels, five doses of taldefgrobep alfa were administered by SC injection, once-weekly (Q1W) (days 1, 8, 15, 22, and 29; 15, 45, 90, or 180 mg). In a fifth panel, three 45 mg doses of taldefgrobep alfa were administered by SC injection every 2 weeks (Q2W) (days 1, 15, and 29). The sample size of the cohorts was determined to be sufficient to support assessment of adverse events (AEs) and a preliminary assessment in percentage change in LBM from baseline.
Randomization was conducted according to a computer-generated randomization scheme prepared by a randomization coordinator within the Drug Supply Management Department of BMS Research and Development. Enrolled individuals, including those not dosed, were assigned sequential subject numbers. Those enrolled individuals meeting inclusion and exclusion criteria were randomized.
All study site investigators, except an unblinded pharmacist who prepared the doses of taldefgrobep alfa and placebo, and individuals participating in the study were blinded to the treatment allocation. The sponsor was also blinded to treatment; however, one member of the Bioanalytical Sciences section was unblinded to the treatment assignment in order to minimize unnecessary analysis of samples from participants in the control group. Additionally, a single pharmacokineticist and a single biostatistician were also unblinded to enable preparation of preliminary summaries of PK, PD, and safety data as needed before data were more generally unblinded. Data from completed treatment panels were masked until after documented completion of the corresponding AE and serious AE (SAE) case report forms, prior to the formal locking of the study database.
For all dose panels, dose escalation did not occur until the safety and tolerability for 75% of participants, through day 30 or day 15 respectively, from the first dose of the preceding dose panel and all safety and tolerability data from completed panels, were reviewed.
Participants in all MAD panels were instructed to maintain their usual level of physical activity throughout the study. Panels 1–5 included MRI measures of right thigh muscle volume on days 29 and 57, and DXA measure of LBM and body fat mass on days 15, 29, and 57.
Procedures
Participants were closely monitored throughout the study for AEs and SAEs, including all SAEs that occurred during the screening period and within 60 days of discontinuation of dosing. All individuals who received taldefgrobep alfa were included in safety analyses. Individuals who received placebo in any panel were pooled into a single placebo group for analysis. Anti-drug antibodies (ADAs) were assessed using a validated ligand binding assay. The immunogenicity analysis population included patients who received taldefgrobep alfa and had at least one post-treatment immunogenicity measurement.
PK and PD analyses were conducted on all participants who received taldefgrobep alfa and had adequate data available. Quantitative determination of total taldefgrobep alfa in human serum was performed using an automated microfluidic fluorescence immunoassay performed on the Gyrolab xP workstation [35]. Free myostatin levels in serum were analyzed using a commercially available enzyme-linked immunosorbent assay (ELISA) validated at QPS as a fit-for-purpose biomarker method. Total myostatin (myostatin drug complex) levels were assessed using a validated ligand binding assay.
DXA scans were performed at baseline and days 15, 29, and 57 to measure LBM and fat mass. MRI scans of right thigh volume were performed at baseline and days 29 and 57 (MAD panels) for the measurement of the thigh muscle total volume and volume of SC and intramuscular fat. VirtualScopics software (Rockville, MD, USA) was used for segmentation and quantification of thigh muscle volume semi-automatically. Right thigh images were acquired using a two-dimensional, multi-slice proton-density-weighted, fast-spin-echo pulse sequence to cover the entire thigh (knee to hip). Once boundaries and segments were automatically determined [36] and manually reviewed, tissue volumes were automatically calculated. Percentage change from baseline in LBM and right thigh muscle volume were calculated using Bayesian dose–response modeling with minimal-informative prior or prior based on preclinical data.
Statistical Analyses
Changes in right-thigh muscle volume and percentage change in LBM were compared with placebo using a mixed effects model that included treatment, visit, and treatment by visit interaction as fixed effects and measurements within subjects as repeated measures.
WN40226 (THUNDERJET): A Randomized, Placebo-Controlled, Double-Blind, Multiple-Dose Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of Taldefgrobep Alfa in Ambulatory Boys with DMD (NCT02515669)
Study Design
The phase 1b/2 study of taldefgrobep alfa was a multicenter, randomized, placebo-controlled, double-blind, MAD study that assessed safety, tolerability, and PK of weekly SC injections of taldefgrobep alfa in ambulatory boys with DMD.
Participants
Patients were recruited between December 23, 2015 and September 12, 2016 by principal investigators from across two sites in Canada and ten sites in the USA. Eligible patients were male, aged ≥ 5 to < 11 years at randomization with a confirmed diagnosis of DMD (i.e., onset of clinical signs or symptoms before 6 years of age with an elevated serum creatine kinase [CK] level). All were ambulatory without assistance, with a 4-stair climb (4SC) time of ≤ 8 s at screening. Patients must have been receiving corticosteroids (prednisone, prednisolone, or deflazacort) for at least 6 months prior to the start of study drug administration, with no significant change in dosage (> 0.2 mg/kg) or dosing regimen for at least 3 months prior to the start of study treatment, with the expectation that dosage and dosing regimen would not change significantly for the duration of the double-blind phase of the study. Key exclusion criteria included any injury, planned surgery, or behavioral or cognitive impairment that may have affected an individual’s ability to participate in functional testing. Patients receiving treatment with ataluren or any investigational drug (excluding deflazacort), or those who had received treatment within five half-lives of the aforementioned treatments prior to the start of study drug administration, were also excluded. A full list of inclusion/exclusion criteria can be found in Sect. 5.1 of the Supplementary Material.
Randomization and Masking
Patients were randomized 3:1 to receive either taldefgrobep alfa (three different dosing panels) or placebo administered weekly. The dose ranges used in this study were determined using free myostatin data collected at week 5 of the healthy volunteers study (WN40225). A body weight-tiered fixed dose strategy was used to select the three doses for the study panels. Doses targeted the moderate (> 50% suppression), high (> 85%), and near complete (> 95%) suppression of serum free myostatin levels for panels 1, 2, and 3, respectively (Fig. 2). Randomization was conducted following the screening visit using an automated interactive web response system. The patients, study site, and investigators were all blinded to treatment assignment. Members of the independent data monitoring committee were periodically unblinded to study data for safety/efficacy review and select sponsor personnel (a pharmacokineticist, biostatistician, and programmer) were unblinded after documented completion and review of case report forms to allow pre-specified interim analyses to enable adjustments in subsequent dosing panels and to select the dose for the subsequent SPITFIRE study. The BMS Bioanalytical Sciences section was also unblinded to treatment assignment to minimize unnecessary analysis of samples from participants within the control group. Except as noted above, other members of BMS Research and Development remained blinded to treatment assignment. Randomization assignment was released to the study sites by the final study report generated for the study.
Fig. 2.

Study design overview and dosing strategy of WN40226 (phase 1b/2 study). The study included four dosing panels: panels 1–3 and the expansion panel (a). Patients underwent sentinel dosing, with up to four patients randomized on a single day in panels 1 and 2. Once the third patient reached day 8, then the remaining patients in the panel could be randomized. Panels 2 and 3 (and the expansion) were randomized when a patient had completed 14 days on the lower dose in the preceding panel. The dosing strategy for panels 1–3 and the expansion panel is shown in b. *n numbers per protocol. †Expansion panel utilizes the same dose as dosing panel 3. ‡Panels 2 and 3 (and the expansion) were weight tiered. Once a patient reached > 45 kg, the dose was increased to 35 mg (panel 2) or 50 mg (panel 3 and expansion)
Initial safety data at week 4 supported the selection of the panel 3 dose for the treatment of an additional group of trial participants in an expansion panel.
Procedures
All patients in the placebo group, the treatment groups, and the expansion panel received blinded treatment for 24 weeks. The initial 24-week double-blind period was followed by a 48-week open-label (OL) phase where all patients received taldefgrobep alfa at the dose assigned to their panel (Fig. 2). After the OL phase, patients could continue treatment in the OL extension. On November 6, 2019, Roche discontinued the taldefgrobep alfa clinical trial program and the study was halted.
Study Endpoints
The primary endpoint of the study was the safety and tolerability of taldefgrobep alfa, including incidence of AEs, SAEs, AEs leading to discontinuation and death, as well as marked treatment-emergent abnormalities in clinical laboratory tests, vital sign measurements, ECGs, echocardiograms, and physical examinations across treatment conditions during 24 weeks of double-blind treatment. Secondary endpoints included maximum (Cmax) and minimum (Ctrough) serum concentrations of taldefgrobep alfa, serum concentration of free myostatin and drug–myostatin complex, percentage inhibition of free myostatin at trough, anti-taldefgrobep alfa antibodies, the change from baseline in LBM index (LBMI), and MRI measurements of changes from baseline in right thigh maximal cross-sectional area (CSAmax) during the 24-week, double-blind treatment period, compared with placebo. Exploratory efficacy endpoints included clinical assessments of motor function. A full list of study endpoints can be found in Sect. 5.2 of the Supplementary Material.
Analyses
PK/PD analyses included individual patient data from individuals who had dosing information available and at least one adequately documented and quantifiable serum taldefgrobep alfa or myostatin concentration. Quantification of total taldefgrobep alfa in serum was performed as described in WN40225. Serum samples were analyzed for free myostatin using a customized Quantikine® ELISA Human GDF-8/Myostatin chemiluminescent immunoassay kit (R&D Systems, Minneapolis, MN, USA) and for total myostatin (free and drug-bound) using a validated immunocapture-LC–MS/MS assay (Bristol-Myers Squibb, Princeton, NJ, USA).
The administration of taldefgrobep alfa to six patients (the smallest number of patients in a single treatment group) provided an 80% probability of observing at least one occurrence of any AE that would occur with 24% incidence in the population from which the sample is drawn. For imaging and efficacy endpoints, all patients who received at least one dose of the study drug or placebo were included in the analysis. Owing to the small number of patients included in each dose panel and the high variability between individuals, the results across all doses were pooled. Analyses were conducted on data available from all patients. To facilitate interpretation of the MRI data, analyses were conducted on the combined data from all muscles measured. Results are described descriptively.
WN40227 (SPITFIRE): A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy, Safety, and Tolerability of Taldefgrobep Alfa in Ambulatory Boys with DMD (NCT03039686)
Study Design
WN40227 was a multicenter, randomized, placebo-controlled, double-blind, phase 2/3 study that assessed efficacy, safety, and tolerability of two different weekly SC doses of taldefgrobep alfa in ambulatory boys with DMD.
Participants
Patients were recruited by principal investigators from hospitals across the USA (17 sites), Japan (five sites), and France (four sites); Australia and Italy (each with three sites); Canada, the Netherlands, Spain, and UK (each with two sites); Argentina, Belgium, Germany, and Sweden (each with one site). Patients were recruited between July 6, 2017 and November 1, 2019. Eligible patients were male, aged ≥ 6 to < 12 years at randomization with a weight ≥ 15 kg and a confirmed genetic diagnosis of DMD. Other inclusion and exclusion criteria were similar to the WN40226 (THUNDERJET) study. A full list of inclusion/exclusion criteria can be found in Sect. 6.1 of the Supplementary Material.
Randomization and Masking
Enrolled patients were stratified by age (6–7-year and 8–11-year age groups) and corticosteroid regimen (daily vs. intermittent, except for patients from Japan), and randomized to receive high-dose taldefgrobep alfa (35/50 mg), low-dose taldefgrobep alfa (7.5/15 mg), or placebo (1:1:1 across three treatment groups, Fig. 3) using an automated computer-generated interactive web response system. Low- and high-dose taldefgrobep alfa targeted moderate (≥ 70%) and high (90%) levels of suppression of serum free myostatin levels, respectively, and were administered according to patient weight.
Fig. 3.

WN40227 phase 2/3 (SPITFIRE) study design. The two dose panels were weight tiered. Once a patient reached > 40 kg, the dose of taldefgrobep alfa was increased to 15 mg (low dose) or 50 mg (high dose). DMD Duchenne muscular dystrophy, Q1W once-weekly dosing, R randomization
Employees of the sponsor involved in study management and data analysis, patients, and all clinical trial site staff in contact with patients were masked to treatment assignment until the announcement of program termination (November 6, 2019) (Fig. 3).
Procedures
Patients received either taldefgrobep alfa or placebo weekly for 48 weeks. After completion of the 48-week double-blind phase, patients could enter the OL phase in which all patients received taldefgrobep alfa. Patients who originally received taldefgrobep alfa continued to receive the dose to which they were originally randomized. Patients originally receiving placebo were randomized to receive high- or low-dose taldefgrobep alfa on day 1 of the OL phase. Dose assignment in the OL phase had the potential to be modified if any safety findings emerged. Patients entering the OL phase remained blinded to their original dose assignment. Patients who discontinued study treatment at any time point could enter the 24-week safety follow-up phase of the study.
Study Endpoints
The primary endpoint of the study was the change from baseline in the North Star Ambulatory Assessment (NSAA) total score (range 0–34, higher score indicates better function) [37] at week 48 in taldefgrobep alfa-treated patients compared with placebo-treated patients. Secondary endpoints included the incidence of AEs, SAEs, AEs leading to discontinuation, and laboratory abnormalities, and the efficacy of taldefgrobep alfa regarding motor function relative to placebo (e.g., 4SC velocity, stand from supine velocity, 10-m walk/run velocity, and 6-min walk distance [6MWD]). A full list of study endpoints can be found in Sect. 6.2 of the Supplementary Material.
Statistical Analyses
For the primary endpoint, the desired sample size of 159 patients (53 patients per arm) provided 80% power for testing the null hypothesis of no difference between a dose of taldefgrobep alfa and placebo in the change from baseline in the NSAA total score at 48 weeks assuming a true treatment difference of 2.5 points, a standard deviation of 4.4 points, and a treatment discontinuation rate of 5%.
A pre-planned futility analysis was conducted after 30% of patients had completed the week 48 assessment. Analyses of efficacy endpoints were conducted on all available data from all patients. The futility analysis compared the placebo arm with the pooled active dose using mixed model repeated measures (MMRM) with treatment, visit, treatment by visit interaction, randomization stratification factors (age group [6–7 vs. 8–11 years], frequency of corticosteroid regimen [daily vs. intermittent, except for patients from Japan]), and baseline value as continuous covariates. The threshold for the futility analysis was set at 1.5 points for the placebo-corrected treatment difference (pooled taldefgrobep alfa doses minus placebo) in change from baseline in NSAA total score at week 48.
WN40226/WN40227 Safety Analyses
Safety data were reported in both the WN40226 and WN40227 studies using the following two approaches. A safety analysis population for the double-blind treatment period included all patients who received at least one dose of study drug or placebo. For both studies, this safety analysis population consisted of all randomized patients. Additional analyses were performed using a modified safety population of the whole study, which included all patients who received taldefgrobep alfa. This included (1) patients who were randomized to taldefgrobep alfa (low or high dose) at the start of the double-blind period, and (2) patients who received placebo at the start of the double-blind period and received at least one dose of taldefgrobep alfa in the OL period. Modified analyses included all safety data available from the double-blind phase in taldefgrobep alfa-treated patients (AEs reported during the double-blind phase in patients randomized to placebo were excluded), as well as data from all eligible patients from the OL periods.
WN40226/WN40227: Modeling of Muscle Mass and Functional Outcomes
A consistent linear relationship between muscle mass/contractile tissue fraction and DMD-specific functional outcomes has been reported in previous studies [38, 39]. Making the assumptions that the rate of taldefgrobep alfa-induced muscle growth is stable and that the muscle produced is as functional as natural muscle tissue, using a linear relationship, the observed amount of muscle growth can be used to provide an upper bound estimate for the potential of taldefgrobep alfa to improve functional outcomes in DMD.
Modeling was conducted using interim analysis data from the WN40226 study. Linear regression analyses were used to predict baseline functional outcomes (NSAA and 4SC) using baseline LBMI and CSA contractile estimates. Using the obtained beta coefficients, predicted functional changes were estimated using the change in LBMI and CSAmax contractile fractions observed between pooled taldefgrobep alfa and placebo data in the double-blind phase, as well as for the week 48 time point for WN40227.
Study Oversight of the Taldefgrobep Alfa Clinical Trials
The taldefgrobep alfa clinical trials were conducted in accordance with the principles of the 1964 Declaration of Helsinki and its later amendments, and in accordance with Good Clinical Practice guidelines. Approval from the institutional review board/independent ethics committee at each participating study site was obtained before each of the studies started and the studies were registered with ClinicalTrials.gov. Signed written informed consent was collected prior to study participation from participants, and, in the case of minors, from parents, guardians, or legally acceptable representatives.
Results
Taldefgrobep Alfa in Healthy Adult Volunteers (Phase 1 WN40225 Study)
A total of 736 healthy volunteers were enrolled in this phase 1 study. One hundred and forty participants were randomized into either the SAD or MAD phase of the study. Ninety-seven participants were randomized to receive a MAD of taldefgrobep alfa (15, 45, 90, or 180 mg) or placebo administered subcutaneously (Fig. 4). Baseline characteristics and demographics of these participants can be found in Table 1. Information and results from the 43 participants enrolled in the SAD phase of this study can be found in Sect. 4.4 of the Supplementary Material. (Fig. 4).
Fig. 4.

Trial profile of the phase I healthy volunteer study
Table 1.
Demographics and baseline characteristics of healthy volunteers (MAD phase)
| Placebo (n = 25) |
15 mg Q1W (n = 6) |
45 mg Q1W (n = 12) |
90 mg Q1W (n = 12) |
180 mg Q1W (n = 14) |
45 mg Q2W (n = 12) |
Any taldefgrobep alfa (n = 72)* |
All (N = 97)* |
||
|---|---|---|---|---|---|---|---|---|---|
| Age, years | Mean (SD) | 33.5 (8.13) | 38.0 (14.51) | 39.6 (7.25) | 37.6 (10.27) | 35.8 (8.85) | 35.5 (8.45) | 38.1 (9.50) | 36.9 (9.34) |
| Gender, n (%) | Male | 24 (96) | 5 (83) | 12 (100) | 12 (100) | 14 (100) | 12 (100) | 70 (97) | 94 (97) |
| Female | 1 (4) | 1 (17) | 0 | 0 | 0 | 0 | 2 (3) | 3 (3) | |
| Race, n (%) | White | 8 (32) | 4 (67) | 7 (58) | 9 (75) | 6 (43) | 6 (50) | 32 (44) | 40 (41) |
| Black/African American | 5 (20) | 1 (17) | 1 (8) | 1 (8) | 7 (50) | 2 (17) | 12 (17) | 17 (18) | |
| Japanese | 4 (16) | 0 | 0 | 0 | 0 | 1 (8) | 16 (22) | 20 (21) | |
| Asian other | 2 (8) | 0 | 0 | 0 | 0 | 2 (17) | 2 (3) | 4 (4) | |
| Native Hawaiian/ Other Pacific Islander | 0 | 0 |
1 (8) |
0 | 0 | 0 | 1 (1) | 1 (1) | |
| Other | 6 (24) | 1 (17) | 3 (25) | 2 (17) | 1 (7) | 1 (8) | 9 (13) | 15 (16) | |
| Weight, kg | Mean (SD) | 77.5 (13.61) | 84.80 (7.80) | 78.2 (19.06) | 84.2 (11.02) | 79.3 (9.88) | 77.8 (7.88) | 76.7 (13.35) | 76.9 (13.35) |
| BMI, kg/m2 | Mean (SD) | 25.2 (3.61) | 27.5 (2.63) | 25.7 (4.45) | 26.3 (2.60) | 25.4 (2.66) | 26.0 (2.62) | 25.0 (3.49) | 25.0 (3.50) |
*Totals include information from the Japanese 45 mg Q1W (n = 9) and 180 mg Q1W (n = 7) cohorts, values for the Japanese cohorts are not reported separately
BMI body mass index, MAD multiple ascending dose, Q1W once-weekly dosing, Q2W once every 2 weeks dosing, SD standard deviation
Safety in Healthy Adult Volunteers
Taldefgrobep alfa was generally well tolerated as a single dose and when given as multiple doses (see Table S3 in the Supplementary Material for SAD data). In the MAD phase, AEs were reported in 43 (60%) participants receiving taldefgrobep alfa and 11 (44%) participants receiving placebo (Table 2). There was no apparent relationship to dose in the incidence of AEs. The most frequently reported AEs in the study were injection-site erythema (n = 12 participants, 12%), followed by upper respiratory tract infection (n = 11 participants, 11%), and rash (n = 9 participants, 9%). All AEs were mild except for two unrelated moderate AEs of vomiting and acute bacterial sinusitis reported in the 15 mg Q1W and the 180 mg Q1W dose groups, respectively, and two related moderate AEs (one event of injection-site erythema and one event of injection-site hemorrhage) in the 90 mg Q1W dose group. There were no SAEs reported.
Table 2.
AEs reported in healthy volunteers (MAD phase)
| System organ class (%) Preferred term (%) |
Placebo (n = 25) |
15 mg Q1W (n = 6) |
45 mg Q1W (n = 12) |
90 mg Q1W (n = 12) |
180 mg Q1W (n = 14) |
45 mg Q2W (n = 12) |
Any taldefgrobep alfa (n = 72)* |
All (N = 97)* |
|---|---|---|---|---|---|---|---|---|
| Total subjects with AE | 11 (44.0) | 1 (16.7) | 9 (75.0) | 6 (50.0) | 10 (71.4) | 5 (41.7) | 43 (59.7) | 54 (55.7) |
| General disorders and administration-site conditions | 3 (12.0) | 0 | 5 (41.7) | 4 (33.3) | 5 (35.7) | 1 (8.3) | 19 (26.4) | 22 (22.7) |
| Injection-site erythema | 0 | 0 | 4 (33.3) | 3 (25.0) | 4 (28.6) | 0 | 12 (16.7) | 12 (12.4) |
| Injection-site bruising | 1 (4.0) | 0 | 0 | 2 (16.7) | 2 (14.3) | 0 | 4 (5.6) | 5 (5.2) |
| Injection-site reaction | 0 | 0 | 0 | 1 (8.3) | 2 (14.3) | 0 | 4 (5.6) | 4 (4.1) |
| Injection-site hemorrhage | 1 (4.0) | 0 | 0 | 2 (16.7) | 0 | 0 | 2 (2.8) | 3 (3.1) |
| Injection-site papule | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 2 (2.8) | 2 (2.1) |
| Injection-site rash | 0 | 0 | 2 (16.7) | 0 | 0 | 0 | 2 (2.8) | 2 (2.1) |
| Chest pain | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 1 (1.4) | 1 (1.0) |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Injection-site pruritus | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
| Skin and subcutaneous tissue disorders | 6 (24.0) | 0 | 5 (41.7) | 3 (25.0) | 3 (21.4) | 0 | 15 (20.8) | 21 (21.6) |
| Rash | 1 (4.0) | 0 | 3 (25.0) | 2 (16.7) | 0 | 0 | 8 (11.1) | 9 (9.3) |
| Ecchymosis | 0 | 0 | 0 | 1 (8.3) | 1 (7.1) | 0 | 3 (4.2) | 3 (3.1) |
| Acne | 2 (8.0) | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2.1) |
| Erythema | 0 | 0 | 0 | 0 | 2 (14.3) | 0 | 2 (2.8) | 2 (2.1) |
| Rash maculopapular | 1 (4.0) | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 2 (2.1) |
| Dermatitis | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Dry skin | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Papule | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
| Scab | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
| Skin lesion | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Infections and infestations | 1 (4.0) | 0 | 4 (33.3) | 1 (8.3) | 3 (21.4) | 2 (16.7) | 14 (19.4) | 15 (15.5) |
| Upper respiratory tract infection | 1 (4.0) | 0 | 3 (25.0) | 1 (8.3) | 2 (14.3) | 1 (8.3) | 10 (13.9) | 11 (11.3) |
| Gastroenteritis | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 1 (1.4) | 1 (1.0) |
| Otitis media | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Sinusitis bacterial | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Viral infection | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Gastrointestinal disorders | 1 (4.0) | 1 (16.7) | 1 (8.3) | 2 (16.7) | 2 (14.3) | 1 (8.3) | 9 (12.5) | 10 (10.3) |
| Abdominal discomfort | 1 (4.0) | 0 | 0 | 1 (8.3) | 0 | 0 | 2 (2.8) | 3 (3.1) |
| Abdominal pain | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (8.3) | 2 (2.8) | 2 (2.1) |
| Abdominal distension | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Abdominal tenderness | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Chapped lips | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
| Diarrhea | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Lip dry | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Vomiting | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Nervous system disorders | 2 (8.0) | 0 | 0 | 0 | 5 (35.7) | 1 (8.3) | 7 (9.7) | 9 (9.3) |
| Headache | 2 (8.0) | 0 | 0 | 0 | 2 (14.3) | 0 | 2 (2.8) | 4 (4.1) |
| Dizziness | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Paresthesia | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Presyncope | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 1 (1.4) | 1 (1.0) |
| Sensory disturbance | 0 | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (1.4) | 1 (1.0) |
| Somnolence | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Injury, poisoning, and procedural complications | 3 (12.0) | 0 | 2 (16.7) | 1 (8.3) | 0 | 0 | 5 (6.9) | 8 (8.2) |
| Contusion | 2 (8.0) | 0 | 1 (8.3) | 0 | 0 | 0 | 3 (4.2) | 5 (5.2) |
| Arthropod bite | 1 (4.0) | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (1.4) | 2 (2.1) |
| Eye injury | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Musculoskeletal and connective tissue disorders | 0 | 0 | 1 (8.3) | 3 (25.0) | 2 (14.3) | 1 (8.3) | 8 (11.1) | 8 (8.2) |
| Muscle spasms | 0 | 0 | 0 | 1 (8.3) | 2 (14.3) | 0 | 3 (4.2) | 3 (3.1) |
| Arthralgia | 0 | 0 | 1 (8.3) | 1 (8.3) | 0 | 0 | 2 (2.8) | 2 (2.1) |
| Back pain | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Myalgia | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Neck pain | 0 | 0 | 0 | 0 | 0 | 1 (8.3) | 1 (1.4) | 1 (1.0) |
| Pain in extremity | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Vascular disorders | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 2 (2.8) | 2 (2.1) |
| Hematoma | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Phlebitis | 0 | 0 | 1 (8.3) | 0 | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Eye disorders | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
| Blurred vision | 1 (4.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.0) |
|
Metabolism and nutrition disorders |
0 | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (1.4) | 1 (1.0) |
| Decreased appetite | 0 | 0 | 0 | 1 (8.3) | 0 | 0 | 1 (1.4) | 1 (1.0) |
AE adverse event, MAD multiple ascending dose, Q1W once-weekly dosing, Q2W once every 2 weeks dosing
*The totals include information from the Japanese 45 mg Q1W (n = 9) and 180 mg Q1W (n = 7) cohorts; values for the Japanese cohorts are not reported separately
In the MAD phase, no AEs related to clinical laboratory tests were reported. The most common marked laboratory abnormalities reported in > 10% of participants who received taldefgrobep alfa were high CK (n = 36 participants, 50%) and low leukocytes (n = 22 participants, 31%). These changes were transient, not associated with clinical symptoms, and were also observed in placebo-treated participants (high CK; n = 10, 40%; low leukocytes; n = 3, 12%).
Low ADA titers (highest titer observed = 64) were observed in 45% of participants treated with taldefgrobep alfa in the SAD phase and in 28% of participants in the MAD phase. The presence of ADAs did not appear to impact taldefgrobep alfa exposure or free myostatin suppression. There was no correlation between ADA positivity and the occurrence of injection-site and other skin reactions, suggesting a lack of relationship between the presence of ADAs and risk of cutaneous AEs.
PK and Target Engagement in Healthy Adult Volunteers
For a healthy adult of 77 kg, the apparent clearance and the apparent central volume of taldefgrobep alfa were estimated as 0.515 L/day and 13.5 L/day, respectively. Because of the feedback control on endogenous myostatin production with a slow rate of moderator production (0.00585 L/day), the PK and PD steady state are reached after approximately 80 weeks of weekly administration of taldefgrobep alfa. The peak (Cmax) and overall exposure (area under curve [AUCtau]) of taldefgrobep alfa increased with dose with a slightly greater than proportional increase in Cmax and AUCtau (Fig. 5a).
Fig. 5.

PK /PD profile of taldefgrobep alfa in healthy volunteers (MAD phase). a The mean taldefgrobep alfa concentration profile versus time shows dose-dependent increase in taldefgrobep alfa exposure. b Change in free myostatin level versus time in the MAD phase. The maximum reduction (% of baseline) of free myostatin was ≥ 90% at all doses at day 22. c Change in mean myostatin drug complex concentration versus time in the MAD phase. Maximal total myostatin concentration ranged from 300 to 2000 ng/mL for the 15 mg Q1W and 180 mg Q1W dose groups, respectively. BL baseline, MAD multiple ascending dose, PD pharmacodynamics, PK pharmacokinetics, Q1W once weekly dosing, Q2W once every 2 weeks dosing
The maximum reduction (expressed as percentage of baseline) of free myostatin was ≥ 90% at all doses at day 22. Doses > 15 mg Q1W achieved this level of reduction following the first dose; the 15 mg Q1W dose group required three doses to achieve this level of target engagement (Fig. 5b). Maximal total myostatin concentration ranged from 300 to 2000 ng/mL for the 15 mg Q1W and 180 mg dose groups, respectively (Fig. 5c).
Fig. 6.
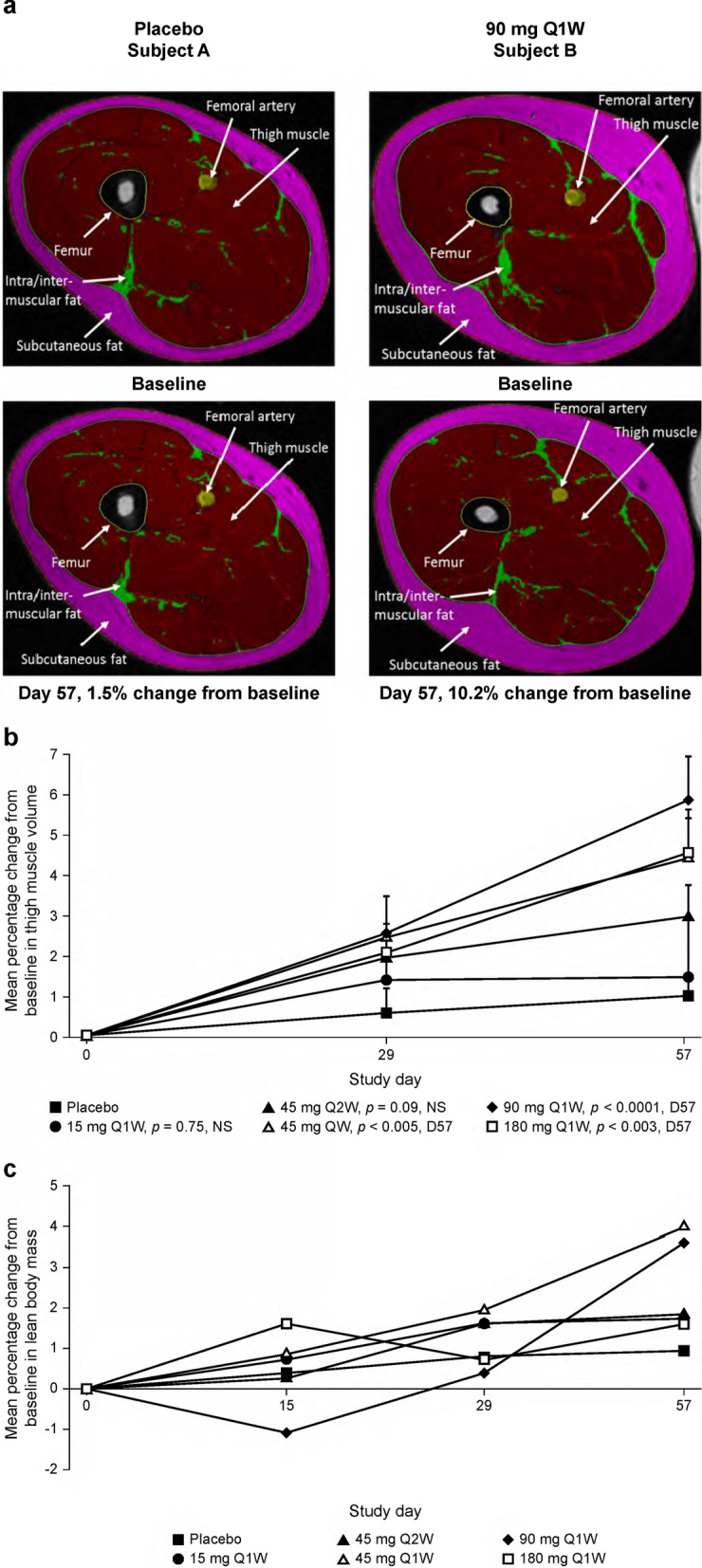
The effect of taldefgrobep alfa on right thigh volume and muscle mass in healthy volunteers (MAD phase). a Representative segmentations of MRI scans from two patients at baseline and day 57. b Change in thigh muscle volume over time. c Change in lean body mass over time. MAD multiple ascending dose, MRI magnetic resonance imaging, NS not significant, Q1W once-weekly dosing, Q2W once every 2 weeks dosing
Effects on LBM and Right Thigh Muscle Volume in Healthy Adult Volunteers
Right thigh muscle volume increased numerically with taldefgrobep alfa compared with placebo, as measured by percentage change from baseline at all doses and time points (Fig. 6a). Statistically significant increases in mean thigh muscle volume compared with placebo were observed in the 45 mg Q1W dose group (day 57, n = 11, mean = 3.41%, p = 0.0031), 90 mg Q1W group (day 29, n = 12, mean = 1.98%, p = 0.0469 and day 57, n = 9, mean = 4.75%, p < 0.0001), and 180 mg Q1W group (day 57, n = 10, mean = 3.52%, p = 0.0027; Fig. 6b). The mean percentage change in LBM from baseline increased numerically for most taldefgrobep alfa dose groups and time points. These increases were statistically significant relative to placebo for the 45 mg Q1W dose group (day 57, n = 12, mean = 2.69%, p = 0.0154) and the 90 mg Q1W dose group (day 57, n = 9, mean = 2.43%, p = 0.0347; Fig. 6c).
Taldefgrobep Alfa in DMD (Phase 1b/2 [THUNDERJET] and Phase 2/3 [SPITFIRE] Studies)
Safety and efficacy of taldefgrobep alfa were evaluated in 209 boys aged 5–11 years with DMD across two studies (WN40226 [phase 1b/2, THUNDERJET, n = 43] and WN40227 [phase 2/3, SPITFIRE; n = 166]). Trial profiles for each of the studies are shown in Figs. 7 and 8 for the THUNDERJET and SPITFIRE studies, respectively. Summary baseline characteristics are shown in Table 3.
Fig. 7.

Trial profile of the phase 1b/2 THUNDERJET study. *Patient discontinued because of visa issues. †Discontinued at the patient’s request
Fig. 8.

Trial profile of the phase 2/3 SPITFIRE study
Table 3.
Demographics and baseline characteristics in the WN40226 (phase 1b/2 THUNDERJET) and WN40227 (phase 2/3 SPITFIRE) studies
| WN40226 (phase 1b/2 study THUNDERJET) | WN40227 (phase 2/3 study [SPITFIRE]) | |||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | Panel 1 taldefgrobep alfa | Panel 2 taldefgrobep alfa | Panel 3 taldefgrobep alfa | Expansion panel taldefgrobep alfa | Placebo | Low-dose taldefgrobep alfa | High-dose taldefgrobep alfa | |
| n | 11 | 7 | 6 | 6 | 13 | 56 | 55 | 55 |
| Age, mean years (SD) |
8.8 (1.3) |
8.0 (2.2) |
8.0 (1.8) |
7.7 (2.3) |
8.2 (1.6) |
8.4 (1.7) |
8.5 (1.8) |
8.4 (1.5) |
| Weight, mean kg (SD) |
29.7 (7.6) |
26.1 (6.4) |
27.8 (6.6) |
27.5 (6.7) |
28.1 (9.0) |
27.7 (9.1) |
28.9 (7.9) |
30.3 (9.9) |
| NSAA total score, mean (SD) |
25.4 (6.04) |
22.1 (7.99) |
25.0 (6.99) |
24.0 (4.00) |
23.1 (7.40) |
23.1 (6.4) |
24.5 (5.5) |
22.7 (6.7) |
| 4SC time, mean s (SD) |
3.48 (2.29) |
4.29 (2.19) |
3.58 (1.51) |
3.39 (1.09) |
4.34 (1.88) |
3.81 (1.55) |
3.85 (1.61) |
3.92 (1.91) |
| Stand from supine time, mean s (SD) |
6.70 (6.10) |
8.11 (7.89) |
5.61 (2.57) |
5.18 (2.07) |
6.53 (3.50) |
6.28 (4.75) |
6.15* (4.07) |
7.24 (9.22) |
| 6MWD, mean m (SD) |
440.73 (74.57) |
364.89 (46.70) |
377.60 (92.01) |
389.75 (57.945) |
371.95 (106.20) |
388.33 (69.59) |
399.73* (68.35) |
370.73 (93.35) |
Baseline characteristics were recorded at screening in WN40226 and at the baseline assessment in WN40227. *Mean calculated from N = 54 patients due to missing data from one patient
4SC 4-stair climb, 6MWD 6-min walk distance, NSAA North Star Ambulatory Assessment, SD standard deviation
PK and Target Engagement of Taldefgrobep Alfa in Boys with DMD
Total taldefgrobep alfa and serum free myostatin concentrations were modeled simultaneously using a target-mediated drug disposition model, using the quasi-steady-state approximation, which assumes that the rate of taldefgrobep alfa binding to myostatin is balanced by the sum of association and dissociation rates. The disposition of taldefgrobep alfa is described by a one-compartment model with linear absorption and elimination. Taldefgrobep alfa binds to myostatin in the central compartment and is degraded with a first-order process. Free myostatin binds to taldefgrobep alfa in a second-order process. Additional details of the model can be found in Sect. 5.3 of the Supplementary Material. For a patient with DMD weighing 28 kg, the apparent clearance is 0.241 L/day, and the apparent central volume is 4.9 L. PK steady state was reached after approximately 12 weeks of Q1W administration. The PK of taldefgrobep alfa seems to be dose-proportional, considering the between-patient variability (Fig. 9a). PK parameters are summarized in Table 4. Overall, the exposure achieved in this patient population was higher than the exposure in healthy adults at similar doses.
Fig. 9.

PK/PD profile of taldefgrobep alfa in boys with DMD enrolled in WN40226 (phase 1b/2 study). a PK steady state was reached after approximately 12 weeks of Q1W administration. b Levels of free myostatin showed a rapid dose-dependent reduction following multiple doses of taldefgrobep alfa. At week 24 (denoted by the black arrow), boys in the placebo group switched to taldefgrobep alfa treatment at the dose of their assigned panel. c At steady state, suppression of serum free myostatin from baseline of approximately 77%, 92%, and 97% over the dosing interval was observed at doses of 4, 12.5/20, and 35/50 mg, respectively. DMD Duchenne muscular dystrophy, PD pharmacodynamics, PK pharmacokinetics, Q1W once-weekly dosing
Table 4.
PK parameter estimates in boys with DMD enrolled in WN40226 (phase 1b/2 study)
| Panel | Number | Dose |
Cmax (ng/mL) Geo. Mean (CV%) |
Tmax (h) Median (min–max) |
AUCtau (ng·day/mL) Geo. Mean (CV%) |
|---|---|---|---|---|---|
| 1 | 7 | 4 mg Q1W | 3217 (15.8%) | 28 (12–48) | 18,676 (23%) |
| 2 | 6 | 12.5 mg Q1W | 8490 (21.5%) | 24 (11–47) | 51,461 (18%) |
| 0 | 20 mg Q1W | – | – | – | |
| 3 | 18 | 35 mg Q1W | 24,242 (26.3%) | 30 (11–65) | 150,609 (24%) |
| 1 | 50 mg Q1W | 23,297 | 44 | 151,000 |
AUCtau area under the concentration–time curve from time zero to the time of next dosing, Cmax maximum observed serum concentration, CV coefficient of variation, DMD Duchenne muscular dystrophy, Geo. Mean geometric mean, PK pharmacokinetic, Q1W once-weekly dosing, Tmax time of maximum observed concentration
Free myostatin time-courses exhibit large variability, but after multiple doses of taldefgrobep alfa, a rapid, dose-dependent reduction in free myostatin levels was observed (Fig. 9b). At steady-state, taldefgrobep alfa maintains a reduction of serum free myostatin from baseline of approximately 77%, 92%, and 97% over the dosing interval of the 4, 12.5/20, and 35/50 mg weekly doses, respectively (Fig. 9c). PD steady state was reached after approximately 6 weeks (Fig. 9c).
Effects of Taldefgrobep Alfa on Muscle and Body Mass in Boys with DMD
In the WN40226 study, given the small number of patients included in each dose panel and the high variability between individuals, the results across all taldefgrobep alfa doses were pooled for analysis. Changes in right thigh muscle were evaluated by MRI; the changes from baseline in the CSAmax of both the contractile and non-contractile fraction were evaluated. Changes in LBMI were also evaluated.
In patients who received taldefgrobep alfa, MRI imaging showed that the CSAmax of the contractile fraction of the right thigh fluctuated over time from baseline (Fig. 10). At the end of the 24-week placebo-controlled phase, boys who received taldefgrobep alfa demonstrated a 5.45% increase from baseline in contractile CSA, compared with a 0.79% decrease from baseline in those receiving placebo. Boys who received taldefgrobep alfa through to week 168 demonstrated a 3.7% increase from baseline in contractile CSA, whilst those who originally received placebo decreased by 2.2% from baseline. For the CSAmax of the non-contractile fraction that increased in both groups at week 168, there was an increase of 9.7 cm2 from baseline in patients who received taldefgrobep alfa throughout the study and an increase of 10.5 cm2 in those who originally received placebo.
Fig. 10.

Percentage change from baseline in the CSA of contractile tissue of the right thigh in WN40226 (phase 1b/2 study). As a result of small sample sizes, and overlap between the dosing panels, taldefgrobep alfa-treated patients were pooled for analysis (blue line). Patients receiving placebo in the DB phase (red line) were also pooled across the dosing panels. At week 24, all placebo patients were switched to taldefgrobep alfa, receiving the dose assigned to their panel. Error bars represent 95% confidence interval of the mean. CSA cross-sectional area, DB double blind, OL open label
At the end of the 24-week placebo-controlled phase, boys who received taldefgrobep alfa demonstrated a 1.75% increase from baseline in LBMI, compared with a 1.38% decrease from baseline in boys receiving placebo (LBMI measured by DXA imaging). At week 168, patients who had received treatment with taldefgrobep alfa throughout the study showed an increase of 5.6% from baseline; an increase of 2.0% was seen in patients who originally received placebo (Fig. 11).
Fig. 11.

Percentage change from baseline in LBMI in WN40226 (phase 1b/2 study). The percentage change from baseline in LBMI as measured by DXA, across the total body (not including the head). As a result of the small sample sizes, and overlap between the dosing panels, taldefgrobep alfa-treated patients (blue line) were pooled for analysis. Patients receiving placebo in the DB phase (red line) were also pooled across the dosing panels. At week 24 (denoted by the arrow), all placebo patients were switched onto taldefgrobep alfa, receiving the dose assigned to their panel. Error bars represent 95% confidence interval of the mean. DB double blind, DXA dual-energy X-ray absorptiometry, LBMI lean body mass index, OL open label
Effects of Taldefgrobep Alfa on Functional Outcomes in Boys with DMD
In study WN40226, exploratory analysis of the effect of taldefgrobep alfa on functional endpoints was conducted. No direct impact of treatment on functional outcomes was observed at 24 or 72 weeks of treatment (see Sect. 5.4 of the Supplementary Material); however, as the number of study participants was low, the study was not powered to demonstrate effects on functional endpoints. The mean change from baseline in all pulmonary function tests fluctuated over time and did not demonstrate any specific trend.
To determine whether the observed LBM changes associated with taldefgrobep alfa could translate into a functional benefit in boys with DMD, the WN40227 study was conducted. The primary endpoint of this study was to evaluate the change from baseline in NSAA total score at week 48 in taldefgrobep alfa-treated participants relative to placebo-treated participants.
The futility analysis was conducted after 30% of patients had completed the week 48 assessment (clinical cutoff date October 1, 2019). The threshold for the futility analysis was set at 1.5 points for the MMRM placebo-corrected treatment difference in change from baseline to week 48 in NSAA total score; this condition was not met (Table 5). The difference in the least square mean (LSM) between the pooled taldefgrobep alfa results and placebo for the change from baseline in NSAA total score at week 48 was 0.12 (95% confidence interval – 1.65 to 1.89) (Table 5). Results from the intent-to-treat population for the secondary endpoints selected for inclusion in the supplementary interim analyses as specified in the interim analysis plan (timed function tests, 6MWD, DXA LBMI) did not demonstrate meaningful differences (Table 5). Efficacy results supported the decision to terminate taldefgrobep alfa development in DMD.
Table 5.
Futility analysis: MMRM analysis of change from baseline in primary and secondary endpoints in WN40227 (phase 2/3 SPITFIRE study)
| Variable | Placebo | Pooled active | Placebo | Pooled active | Treatment difference | ||
|---|---|---|---|---|---|---|---|
|
n (All) |
n (Week 48) |
n (All) |
n (Week 48) |
Week 48 LSM (SE) |
Week 48 LSM (SE) |
LSM (95% CI) | |
| NSAA total score* | 56 | 21 | 109 | 47 |
− 3.32 (0.78) |
− 3.21 (0.57) |
0.12 (− 1.65 to 1.89) |
| 4SC velocity (stairs/s) | 56 | 21 | 109 | 47 |
− 0.15 (0.06) |
− 0.16 (0.05) |
− 0.01 (− 0.15 to 0.14) |
| Stand from supine velocity (1/sec) | 56 | 19 | 109 | 45 |
− 0.05 (0.01) |
− 0.02 (0.01) |
0.02 (− 0.01 to 0.06) |
| 10-m walk/run velocity (m/s) | 56 | 21 | 108 | 45 |
− 0.22 (0.08) |
− 0.15 (0.06) |
0.07 (− 0.12 to 0.26) |
| 6MWD (m) | 55 | 21 | 108 | 42 |
− 46.5 (10.3) |
− 37.0 (7.7) |
9.5 (− 14.0 to 33.0) |
Data cutoff October 1, 2019
4SC 4-stair climb, 6MWD 6-min walk distance, CI confidence interval, LSM least squares mean, MMRM mixed model repeated measures, NSAA North Star Ambulatory Assessment, SE standard error
*Primary endpoint
A final analysis was conducted at week 48 using all available WN40227 data (Table S9 in the Supplementary Material). Results of this final analysis were consistent with the results of the futility analysis and supported the decision to terminate the study.
Safety of Taldefgrobep Alfa in Boys with DMD
AEs
Safety data were reported in the WN40226 and WN40227 studies (Tables 6 and 7). Among subjects exposed to taldefgrobep alfa in the modified safety population, 155 boys (86.1%) reported AEs: most were mild to moderate in intensity. The most common AEs were nasopharyngitis (reported in n = 44 boys, 24.4%), headache (n = 42, 23.3%), injection-site erythema (n = 39, 21.7%), pyrexia (n = 35, 19.4%), and vomiting (n = 32, 17.8%). Seventy-eight boys (43.3%) reported AEs considered as related to taldefgrobep alfa treatment. Treatment-related AEs were principally related to local effects at the injection site.
Table 6.
Summary of taldefgrobep alfa pooled safety in ambulatory boys with DMD
| WN40226 Total taldefgrobep alfa (N = 43) | WN40227 Total taldefgrobep alfa (N = 137)* | Total (N = 180) | |
|---|---|---|---|
| Number of patients with at least one, n (%) | |||
| Death | 0 | 1 (0.7)† | 1 (0.6) |
| AE | 42 (97.7) | 113 (82.5) | 155 (86.1) |
| AE leading to withdrawal | 0 | 1 (0.7)† | 1 (0.6) |
| AE leading to dose interruption | 2 (4.7) | 5 (3.6) | 7 (3.9) |
| SAE | 6 (14.0) | 6 (4.4) | 12 (6.7) |
| SAE leading to withdrawal | 0 | 1 (0.7)† | 1 (0.6) |
| SAE leading to dose interruption | 1 (2.32) | 2 (1.5) | 3 (1.7) |
| Related AE | 27 (62.8) | 51 (37.2) | 78 (43.3) |
| Related SAE | 0 | 1 (0.7)‡ | 1 (0.6) |
| Related AE leading to withdrawal | 0 | 0 | 0 |
| Related AE leading to dose interruption | 0 | 3 (2.2) | 3 (1.7) |
| Grade 3–5 AE | 5 (11.6) | 5 (3.6) | 10 (5.6) |
*A modified safety population that includes all patients who received at least one dose of taldefgrobep alfa (69 patients from the taldefgrobep alfa low-dose group and 68 patients from the high-dose group). AEs reported in patients randomized to placebo during the double-blind phase were excluded. †The patient experienced a severe cardiac arrest following cardiac ablation that was considered unrelated to the study treatment. ‡Hyperbilirubinemia
Investigator text for AEs encoded using the Medical Dictionary for Regulatory Activities version 23.0. Percentages are based on N in the column headings. Only treatment-emergent AEs are displayed. Multiple occurrences of the same AE in one individual are counted only once
WN40226 (phase 1b/2 THUNDERJET) LPLV: April 15, 2020. To LPLV, the duration of taldefgrobep alfa exposure ranges from 176 to 1426 days
WN40227 (phase 2/3 SPITFIRE) LPLV: April 28, 2020. Duration of taldefgrobep alfa exposure ranges from 121 to 457 days
AE adverse event, DMD Duchenne muscular dystrophy, LPLV last patient last visit, SAE serious AE
Table 7.
Taldefgrobep alfa pooled safety in ambulatory boys with DMD: Most common AEs
| Preferred term Number of patients reporting an AE (%) |
WN40226 (N = 43) |
WN40227 (N = 137)* |
Total (N = 180) |
|---|---|---|---|
| Nasopharyngitis | 16 (37.2) | 28 (20.4) | 44 (24.4) |
| Headache | 16 (37.2) | 26 (18.9) | 42 (23.3) |
| Injection-site erythema | 12 (27.9) | 27 (19.7) | 39 (21.7) |
| Pyrexia | 13 (30.2) | 22 (16.1) | 35 (19.4) |
| Vomiting | 16 (37.2) | 16 (11.7) | 32 (17.8) |
| Cough | 13 (30.2) | 17 (12.4) | 30 (16.7) |
| Diarrhea | 13 (30.2) | 16 (11.7) | 29 (16.1) |
| Upper respiratory tract infection | 15 (34.9) | 14 (10.2) | 29 (16.1) |
| Pain in extremity | 9 (20.9) | 13 (9.5) | 22 (12.2) |
| Fall | 14 (32.6) | 7 (5.1) | 21 (11.7) |
| Injection-site bruising | 12 (27.9) | 8 (5.8) | 20 (11.1) |
| Arthralgia | 6 (14.0) | 14 (10.2) | 20 (11.1) |
| Contusion | 8 (18.6) | 11 (8.0) | 19 (10.6) |
| Rash | 6 (14.0) | 13 (9.5) | 19 (10.6) |
| Abdominal pain upper | 6 (14.0) | 12 (8.8) | 18 (10.0) |
| Back pain | 8 (18.6) | 10 (7.3) | 18 (10.0) |
| Nasal congestion | 9 (20.9) | 8 (5.8) | 17 (9.4) |
| Ligament sprain | 7 (16.3) | 8 (5.8) | 15 (8.3) |
| Ear infection | 8 (18.6) | 6 (4.4) | 14 (7.8) |
| Influenza | 5 (11.6) | 7 (5.1) | 12 (6.7) |
| Oropharyngeal pain | 5 (11.6) | 4 (2.9) | 9 (5.0) |
| Pharyngitis streptococcal | 5 (11.6) | 1 (0.7) | 6 (3.3) |
| Injection-site irritation | 5 (11.6) | 0 | 5 (2.8) |
AE adverse event, DMD Duchenne muscular dystrophy
*A modified safety population. Only treatment-emergent AEs are displayed. Events in patients randomized to placebo during the double-blind phase are excluded. Most frequently reported AEs are those reported in ≥ 10% of patients in either study. For frequency counts, multiple occurrences of the same AE in an individual are counted only once
Twelve boys (6.7%) reported SAEs. All except one (hyperbilirubinemia) were considered unrelated to taldefgrobep alfa. One death was reported in WN40227. The boy experienced a severe cardiac arrest following cardiac ablation that was unrelated to study treatment.
No related AEs were reported that led to discontinuation of treatment in either study. No boys had AEs that led to dose reduction.
Laboratory Parameters and Other Safety Tests
There were no clinically significant changes in vital signs, ECG parameters, or echocardiogram parameters in either study. There was no evidence that taldefgrobep alfa induced cardiac effects among patients with DMD. No clinically meaningful changes from baseline for chemistry parameters, iron levels, and urinalysis were observed over time, and there were no specific trends related to liver toxicity or iron overload.
Echocardiogram measures conducted in WN40226 at baseline were comparable at the end of the study (week 252), with the mean values comparable across the different treatment dose panels. Echocardiogram measures in WN402267 were also similar at baseline across both dose groups and up to the end of study termination.
The incidence of ADAs to taldefgrobep alfa was low. In WN40226, a positive ADA titer was reported in 1/43 (2%) patients. In WN40227, 12/166 (7%) patients had a positive ADA titer; one patient in the placebo group, six in the low-dose group, and five in the high-dose group. Sample results of “< 1” and “< 2” are classified as positive ADA titers.
Modeling of Muscle Mass and Functional Outcomes
A significant linear relationship was observed in WN40226 baseline data between muscle measures and functional outcomes (r values 0.35–0.51, all p < 0.05, Fig. 12). In the interim analysis, the approximately 4.9% increase in LBMI observed over 24 weeks for the pooled treatment versus placebo translated into a predicted change of 0.46 points in NSAA total score and 0.06 stairs/s in 4SC velocity. Similarly, a 2.5% change in CSAmax of the contractile fraction translated into a predicted change of 0.48 in NSAA total score and 0.06 stairs/s in 4SC velocity.
Fig. 12.

Modeling of muscle mass and functional outcomes. Scatter plots representing the relationship between a NSAA total score and CSA contractile tissue fraction; b 4SC velocity and CSA contractile tissue fraction; c NSAA total score and LBMI; d 4SC velocity and LBMI. Dotted lines represent the 95% CI. 4SC 4-stair climb, CI confidence interval, CSA cross-sectional area, LBMI lean body mass index, NSAA North Star Ambulatory Assessment
Assuming the muscle growth rate remained stable over the 48 weeks used for the futility analysis in WN40227, the above estimate would translate to an approximate 1-point improvement in NSAA total score versus placebo. These estimates indicate that the muscle growth observed in DMD with taldefgrobep alfa was not sufficient to provide a meaningful change in NSAA total score over 1 year. These data were used in conjunction with futility analysis data to support halting the taldefgrobep alfa program in DMD.
Discussion
Treatment with taldefgrobep alfa was generally well tolerated and was associated with robust, dose-dependent suppression of free myostatin in boys with DMD. The variability among patients and overlapping effect of taldefgrobep alfa doses in the WN40226 study emphasized a high level of myostatin suppression at all doses (≥ 77% at the lowest dose of 4 mg at week 24). Both low (12.5/20 mg/kg) and high (35/50 mg/kg) doses delivered > 90% myostatin suppression.
In the WN40226 phase 1b/2 study, myostatin suppression by taldefgrobep alfa was associated with a positive effect on LBM in boys with DMD, though effects on muscle were modest. However, further evaluation of the effect of treatment on motor function in this population in the WN40227 study showed that the effect of treatment did not achieve a clinically relevant treatment difference in the change in NSAA total score.
For the futility analysis of the WN40227 study, the threshold of a 1.5-point difference in NSAA total score was chosen on the basis of the expectation that observing a difference of at least this magnitude at the futility analysis would provide a sufficiently high conditional probability of meeting the minimum detectable difference (approximately 1.74 points) and the target treatment difference (2.5 points) at the final analysis. This target value was selected on the basis of NSAA distribution-based estimates (i.e., identifying measurement error in order to approximate meaningful change) [39, 40]. More recent analysis from six real-world data trials and eight clinical trials has demonstrated that the minimal detectable change thresholds for > 80% confidence in true change in NSAA total score range from 2.66 (individuals ≤ 7 years old) to 2.80 (individuals ≥ 7 years old) [41].
At the time of the WN40227 study futility analysis, an LSM difference of 0.12 was observed between the pooled taldefgrobep alfa results and placebo. Modeling of muscle mass and functional outcomes based on data from the WN40226 study estimated that muscle changes observed at the futility analysis of the WN40227 study would translate into a 1-point change in NSAA total score over 48 weeks in WN40227, further emphasizing that treatment with taldefgrobep alfa in WN40227 was unlikely to translate into a meaningful functional change in boys with DMD. Thus, on the basis of the results from the futility analysis of the WN40227 study, the decision was taken to discontinue the program.
In recent years, a number of other therapies that target myostatin have been developed, including anti-myostatin antibodies (MYO-029, LY2495655, domagrozumab, apitegromab [42–45]); an antibody that targets and inhibits binding to ActRIIB receptor (bimagrumab) [46]; a soluble form of the ActRIIB receptor used to sequester circulating myostatin (ACE-031 [47]); and follistatin overexpression [48–50]. In preclinical studies, these molecules caused cessation of downstream intracellular signaling, leading to removal of inhibition of myogenesis-promoting genes, and resulting in muscle cell growth and differentiation [32, 51–57]; however, the performance of myostatin inhibitors in published clinical trials has been mixed. With the exception of apitegromab in spinal muscular atrophy [58], and a few other small studies [46, 49, 50], collective clinical research has not demonstrated significant functional benefits of these treatments in a variety of muscle-wasting conditions [42, 43, 47, 59–61]. It is therefore important to consider why promising preclinical data have not translated into a benefit for patients.
Firstly, it is important to note that although a number of animal models of DMD have been developed (see [62] for review), the majority of evidence for anti-myostatin therapy in DMD is derived from studies of the mdx mouse model, with a nonsense mutation in exon 23 [63]. Disruption of the myostatin pathway in these mice improved dystrophic pathology [32, 53, 64, 65]; however, in the literature, circulating levels of myostatin have been reported to be 4- to 18-fold lower in monkeys, rats, and humans than in mice [66]. The significantly lower levels of circulating myostatin reported in humans (approximately tenfold reduction [67, 68]) may therefore provide one explanation for the disconnect between the results observed in the mdx mouse and the results reported in clinical trials of boys with DMD.
Results from taldefgrobep alfa and other published studies are consistent with these observations. Four weekly doses of taldefgrobep alfa increased lower limb muscle mass by 30% in a severe combined immunodeficient mouse model, but only a 5% increase was seen in monkeys (see Fig. S2 of the Supplementary Material). In healthy human volunteers, changes in right thigh muscle volume compared with placebo ranged from 3.41% to 4.75% at day 57. In published studies of domagrozumab, robust increases in muscle mass were observed in mdx mice (23–26%) and in monkeys (23.7–36.5%) [32]; however, effects were greatly reduced (4.49%) in healthy human volunteers [69]. Increases in muscle volume did not translate into a functional benefit for patients with DMD treated with domagrozumab [60] or taldefgrobep alfa.
Secondly, studies in a variety of neuromuscular diseases have suggested that decreased serum myostatin is associated with progression of muscle pathologies, with circulating myostatin levels particularly low in the DMD population studied [67]. Low levels of circulating myostatin in individuals with DMD may therefore limit the therapeutic value of myostatin inhibition. Serum myostatin levels in individuals with DMD and aged mdx mice have previously been reported to be around twofold lower than in healthy controls [67, 70], which may be related to the reduction in muscle mass associated with the DMD phenotype. However, although myostatin is downregulated in both DMD and the mdx mouse, proportionally, skeletal muscle myostatin in boys with DMD is suppressed to a greater extent relative to the mdx mouse model. Levels of skeletal muscle myostatin in the mdx model are approximately 25% of that of wild-type mice and, in contrast, boys with DMD typically have approximately 8% of the circulating myostatin found in healthy controls [68]. Together, these results may provide an explanation as to why taldefgrobep alfa and other anti-myostatin treatments have not elicited meaningful change in individuals with DMD.
Limitations
The mean age of patients in the WN40226 phase 1b/2 study and the WN40227 phase 2/3 study at baseline was approximately 8 years; at this age, previous studies indicate that significant muscle degeneration may have already occurred [1]. Myostatin suppression by taldefgrobep alfa was associated with a positive effect on LBM in the WN40226 study, though effects on muscle were modest. Modeling of muscle mass increases indicated that the small increases in muscle mass seen in these studies following treatment with taldefgrobep alfa were unlikely to lead to measurable functional benefits in patients with DMD; however, it is still not known what magnitude of an increase in muscle mass would be required to generate a detectable functional improvement. It is also possible that the treatment was given too late—earlier treatment may result in preservation/development of healthy muscle and may show a better functional result.
Another limitation of these studies potentially impacting efficacy in trials of taldefgrobep alfa was that all boys were treated with concomitant corticosteroids. Data from a recent study in the mdx mouse model suggest that steroid treatment disrupts potential muscle mass gains from myostatin inhibition [71]. Additionally, the established benefits of corticosteroids in improving muscle function [10] may make it difficult to clearly identify the true impact of anti-myostatin therapy on functional capacity. However, as corticosteroids form part of the accepted standard of care in DMD, it is common for inclusion criteria in clinical trials in DMD to allow for concomitant use of corticosteroids when given at a stable dose [13, 16, 17]. Inclusion criteria in trials of taldefgrobep alfa in DMD were consistent with those of other DMD trials.
Taldefgrobep alfa, as a recombinant adnectin protein (see [72] for review), was predicted to have a low immunogenicity profile due to the abundance of fibronectin within the body [73, 74]. Indeed, levels of ADAs in clinical studies were low, with only 13/211 (6%) patients developing a positive ADA titer in WN40226 and WN40227. However, it is important to note that in these two studies, the boys were treated with concomitant corticosteroids, and it is therefore possible that the corticosteroids blunted the immunogenic response. In the phase 1 study, where participants were not on corticosteroids, 28% of healthy adult volunteers in the MAD phase presented with a low ADA titer.
Conclusion
Overall, no patients in the taldefgrobep alfa clinical program were withdrawn from treatment as a result of treatment-related AEs and none of the SAEs except one (hyperbilirubinemia) were considered to be related to the study treatment. Although there were some concerns of non-specific inhibition of myostatin in one study with ACE-031 [47], there have been no safety signals reported in the literature that would prevent anti-myostatin treatment research in the future.
Recent studies in mdx mice have demonstrated that inhibiting myostatin alongside the restoration of dystrophin using an exon-skipping ASO resulted in greater dystrophin levels than with ASO treatment alone [75, 76]. Although recent clinical trials of anti-myostatin monotherapy in DMD, including those reported here, have failed to reproduce the promising results observed in preclinical studies [68, 77], it is possible that there is still a role for myostatin inhibitors in the treatment of DMD, in combination therapy with dystrophin replacement therapies, or in milder forms of muscular dystrophy in which higher levels of residual myostatin have been found [78].
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Taldefgrobep Alfa Study Group: Irvith Carvajal, Anjaneya Chimalakonda, Jochem Gokemeijer, Michael Gulianello, Nicole Hellbach, Alexander Kozhich, Daniel Kukral, Harold Malone, Jere E. Meredith, Jr, Mathew Pletcher, Ginger Rakestraw, Lumelle Schneeweis, Joanna Swain, Frank Zambito, Ming Chang, Lora Hamuro, Feng Luo, Jon E. Peterson, Peter Hocknell, Zhen Lou, Malavi Madireddi, Clifford M. Bechtold, Michael K. Ahlijanian, Ming Chang, Lora Hamuro, Leslie K. Jacobsen, Feng Luo, Jon E. Peterson, Heidemarie Kletzl, Alberto L Dubrovsky, Lilia Mesa, Fernando Chloca, Agustin Jauregu, Kristi Jones, Craig Campbell, Jean Mah, Alice Ho, Angela Chiu, Vanessa D’Souza, Raymy Sadowski, Julie Dao, Michaela Grice, Tiffany Price, Erick Sell, Anna McCormick, Teresa Gidaro, Andrea Seferian, Yann Péréon, Armelle Magot, Carole Vuillerot, Ulrike Schara-Schmidt, Valerie Sansone, Emilio Albamonte, Alessandra Di Bari, Jasmine Refran, Francesca Salmin, Giuseppe Vita, Gian Luca Vita, Chiara Consulo, Hirofumi Komaki, Akihiko Ishiyama, Tsuyoshi Matsumura, Toshio Saito, Kana Ichihara, Naoki Hayashi, Kouji Terada, Kenji Takehara, Nobuko Hayashi, Yasuhiro Takeshima, Andrés Nascimiento, Daniel Natera, Laura Carrera, Jesica Exposito, Carlos Ortez, Julita Medina, Obdulia Moya, Sandra Roca, Alicia Rodriguez, Maria Valle, Imelda JM de Groot, Erik H Niks, Marjolein J. van Heur-Neuman, Menno van der Holst, Mariacristina Scoto, Chiara Brusa, Abidha Afazal, Eveline Miller, Linda Cripe, Richard S Finkel, Peter Heydemann, Katherine Matthews, Chandra Miller, Katie Laubsher, Shelley Mockeler, Han Phan, Kumaraswamy Sivakumar, Kristy Osgood, Jeffrey Statland, Cuixia Tian, Doris Leung, Genila Bibat, Nikia Stinson, Laurent Servais, Eugenio Mercuri, Tina Duong, Paul Strijbos, Klaas Veenstra. The authors would like to thank all the patients who participated in the taldefgrobep alfa clinical trial program, their families, and the clinical trial sites and staff around the world.
Medical Writing and Editorial Assistance.
The authors thank Jack Curran, PhD, and Lauren Walmsley, PhD, of Nucleus Global, UK, for providing medical writing support and Megan Speakman, of Nucleus Global, UK for providing medical editing support. Medical writing support and editing support were conducted in accordance with Good Publication Practice (GPP 2022) guidelines (https://www.ismpp.org/gpp-2022). Funding for medical writing and editorial assistance was provided by F. Hoffmann-La Roche Ltd. (Basel, Switzerland).
Authorship.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Data were collected by Francesco Muntoni, Barry J. Byrne, Hugh J. McMillan, Monique M. Ryan, Brenda L. Wong, and Kathryn R. Wagner. All authors were involved in data interpretation, discussing the results, drafting, and critically reviewing the manuscript for intellectual content, and provided final approval of the submitted version.
Funding
Preclinical studies were funded by Bristol Myers Squibb. The clinical studies were funded by F. Hoffmann-La Roche Ltd. The funder of the clinical studies (F. Hoffmann-La Roche) provided study drug, study management, medical monitoring, drug safety management and analysis, data management, and statistical analysis. The Rapid Service Fee and the Open Access fee for publication were sponsored by F. Hoffmann-La Roche Ltd.
Data Availability
Biohaven Pharmaceuticals is currently developing taldefgrobep alfa. Biohaven will provide access to de-identified patient-level data that underlie the results in this article in response to scientifically valid research proposals. Researchers may request access and send research proposals by emailing Cliff Bechtold (cliff.bechtold@biohavenpharma.com). Data from these studies will be made available after the publication of this article. Biohaven will consider requests from qualified researchers for access to the data. Biohaven will review the request using an internal committee composed of Biohaven staff, including a clinician, a statistician, and a data-sharing professional. Biohaven will make reasonable efforts to fulfill all data requests for legitimate research purposes, but there might be instances in which retrieval or delivery of data is not feasible, such as those involving, for example, patient privacy, requirements for permissions, contractual obligations, and conflicts of interest. All those receiving access to data will be required to enter into a data use agreement provided by Biohaven which will contain the terms under which the data will be provided.
Conflict of Interest
Francesco Muntoni was a site principal investigator in the trial and received support for the trial conduct. He has also received consultancy payments from F. Hoffmann-La Roche Ltd for participation in scientific advisory boards. Barry J. Byrne was a site principal investigator in the trial and received support for the trial conduct. Hugh J. McMillan was a site principal investigator in the trial and has received support for the trial conduct. He has also received consultancy payments from F. Hoffmann-La Roche Ltd for participation in scientific advisory boards. Monique M. Ryan was a site principal investigator in the trial and has received support for the trial conduct. Brenda L. Wong was a site principal investigator in the trial and received support for the trial conduct. Jurgen Dukart is a former employee of and received consultancy fees from F. Hoffmann–La Roche Ltd. Amita Bansal is a former employee of F. Hoffmann-La Roche Ltd. New affiliation: Novartis International AG, Basel, Switzerland. Roxana Dreghici is a former employee and shareholder of F. Hoffmann-La Roche Ltd. New affiliation: Solid Biosciences Inc., Charlestown, MA, USA. Valerie Cosson is an employee and shareholder of Roche Products Ltd. Maitea Guridi is an employee and shareholder of F. Hoffmann-La Roche Ltd. Michael Rabbia is an employee and shareholder of Genentech, Inc, a member of the Roche Group. Hannah Staunton is an employee and shareholder of Roche Products Ltd. Giridhar S Tirucherai is a former employee and shareholder of Bristol Myers Squibb. New affiliation: Scholar Rock Inc., Cambridge, MA, USA. Karl Yen is a former employee and shareholder of F. Hoffmann-La Roche Ltd. New affiliation: Sanofi SA, Basel, Switzerland. Xiling Yuan is an employee and shareholder of Bristol Myers Squibb. Kathryn R. Wagner was a site principal investigator in the trial and received support for the trial conduct. She is a former employee of F. Hoffmann-La Roche Ltd. New affiliation: Novartis International AG, Basel, Switzerland.
Ethical Approval
The taldefgrobep alfa clinical trials were conducted in accordance with the principles of the 1964 Declaration of Helsinki and its later amendments, and in accordance with Good Clinical Practice guidelines. Approval from the institutional review board/independent ethics committee at each participating study site was obtained before each of the studies started and the studies were registered with ClinicalTrials.gov. Signed written informed consent was collected prior to study participation from participants, and, in the case of minors, from parents, guardians, or legally acceptable representatives.
Footnotes
Prior Presentation: Aspects of this work have previously been presented at the 21st International Annual Congress of the World Muscle Society, October 4–8, 2016; the 70th Annual Meeting of the American Academy of Neurology, April 21–27, 2018; the 2018 Parent Project Muscular Dystrophy—2018 Annual Conference, June 28–30, 2018; the 15th International Congress on Neuromuscular Diseases, July 6–10, 2018; and the 23rd International Annual Congress of the World Muscle Society, October 2–6, 2018.
The Taldefgrobep Alfa Study Group authors are listed in the Acknowledgements.
Contributor Information
Kathryn R. Wagner, Email: kathrynraewagner@gmail.com
the Taldefgrobep Alfa Study Group:
Irvith Carvajal, Anjaneya Chimalakonda, Jochem Gokemeijer, Michael Gulianello, Nicole Hellbach, Alexander Kozhich, Daniel Kukral, Harold Malone, Jere E. Meredith, Jr, Mathew Pletcher, Ginger Rakestraw, Lumelle Schneeweis, Joanna Swain, Frank Zambito, Ming Chang, Lora Hamuro, Feng Luo, Jon E. Peterson, Peter Hocknell, Zhen Lou, Malavi Madireddi, Mathew Pletcher, Clifford M. Bechtold, Michael K. Ahlijanian, Ming Chang, Lora Hamuro, Leslie K. Jacobsen, Alexander Kozhich, Feng Luo, Jon E. Peterson, Frank Zambito, Heidemarie Kletzl, Alberto L. Dubrovsky, Lilia Mesa, Fernando Chloca, Agustin Jauregu, Kristi Jones, Monique Ryan, Craig Campbell, Jean Mah, Alice Ho, Angela Chiu, Vanessa D’Souza, Raymy Sadowski, Julie Dao, Michaela Grice, Tiffany Price, Hugh McMillan, Erick Sell, Anna McCormick, Teresa Gidaro, Andrea Seferian, Yann Péréon, Armelle Magot, Carole Vuillerot, Ulrike Schara-Schmidt, Valerie Sansone, Emilio Albamonte, Alessandra Di Bari, Jasmine Refran, Francesca Salmin, Giuseppe Vita, Gian Luca Vita, Chiara Consulo, Hirofumi Komaki, Akihiko Ishiyama, Tsuyoshi Matsumura, Toshio Saito, Kana Ichihara, Naoki Hayashi, Kouji Terada, Kenji Takehara, Nobuko Hayashi, Yasuhiro Takeshima, Andres Nascimiento, Daniel Natera, Laura Carrera, Jesica Exposito, Carlos Ortez, Julita Medina, Obdulia Moya, Sandra Roca, Alicia Rodriguez, Maria Valle, Imelda J. M. de Groot, Erik H. Niks, Marjolein J. van Heur-Neuman, Menno van der Holst, Mariacristina Scoto, Chiara Brusa, Abidha Afazal, Eveline Miller, Barry J. Byrne, Linda Cripe, Richard S. Finkel, Peter Heydemann, Katherine Matthews, Chandra Miller, Katie Laubsher, Shelley Mockeler, Han Phan, Kumaraswamy Sivakumar, Kristy Osgood, Jeffrey Statland, Cuixia Tian, Kathryn R. Wagner, Doris Leung, Genila Bibat, Nikia Stinson, Laurent Servais, Eugenio Mercuri, Tina Duong, Mariacristina Scoto, Craig Campbell, Paul Strijbos, and Klaas Veenstra
References
- 1.Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12:79. doi: 10.1186/s13023-017-0631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han S, Xu H, Zheng J, et al. Population-wide duchenne muscular dystrophy carrier detection by CK and molecular testing. BioMed Res Int. 2020;2020:1–12. doi: 10.1155/2020/8396429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53:145–151. doi: 10.1136/jmedgenet-2015-103387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifiro G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. 2020;15:141. doi: 10.1186/s13023-020-01430-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015;20:18168–18184. doi: 10.3390/molecules201018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annexstad EJ, Lund-Petersen I, Rasmussen M. Duchenne muscular dystrophy. Tidsskr Nor Laegeforen. 2014;134:1361–1364. doi: 10.4045/tidsskr.13.0836. [DOI] [PubMed] [Google Scholar]
- 7.Allikian MJ, McNally EM. Processing and assembly of the dystrophin glycoprotein complex. Traffic. 2007;8:177–183. doi: 10.1111/j.1600-0854.2006.00519.x. [DOI] [PubMed] [Google Scholar]
- 8.Bettica P, Petrini S, D'Oria V, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26:643–649. doi: 10.1016/j.nmd.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Gloss D, Moxley R, III, Ashwal S, Oskoui M. Practice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy. Neurology. 2016;86:465–472. doi: 10.1212/WNL.0000000000002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur A. Corticosteroids for the treatment of Duchenne muscular dystrophy (Review). The Cochrane database of systematic reviews 2016:CD003725. [DOI] [PMC free article] [PubMed]
- 11.Pichavant C, Aartsma-Rus A, Clemens PR, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830–840. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laing NG, Davis MR, Bayley K, Fletcher S, Wilton SD. Molecular diagnosis of Duchenne muscular dystrophy: past, present and future in relation to implementing therapies. Clin Biochem Rev. 2011;32:129–134. [PMC free article] [PubMed] [Google Scholar]
- 13.Lim KRQ, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Dev Ther. 2017;11:533–545. doi: 10.2147/DDDT.S97635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aartsma-Rus A, Straub V, Hemmings R, et al. Development of exon skipping therapies for Duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017;27:251–259. doi: 10.1089/nat.2017.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anwar S, Yokota T. Golodirsen for Duchenne muscular dystrophy. Drugs Today (Barc) 2020;56:491–504. doi: 10.1358/dot.2020.56.8.3159186. [DOI] [PubMed] [Google Scholar]
- 16.Clemens PR, Rao VK, Connolly AM, et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. 2020;77:982–991. doi: 10.1001/jamaneurol.2020.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner KR, Kuntz NL, Koenig E, et al. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: a randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve. 2021;64:285–292. doi: 10.1002/mus.27347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.US Food and Drug Administration. ELEVIDYS™ Highlights of prescribing information. 2023. https://www.fda.gov/media/169679/download. Accessed July 2023.
- 20.Reference NGH. MSTN gene. 2019. https://ghr.nlm.nih.gov/gene/MSTN. Accessed July 2023.
- 21.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 22.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S-J, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfman NM, McPherron AC, Pappano WN, et al. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hitachi K, Tsuchida K. Role of microRNAs in skeletal muscle hypertrophy. Front Physiol. 2013;4:408. doi: 10.3389/fphys.2013.00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durieux AC, Amirouche A, Banzet S, et al. Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology. 2007;148:3140–3147. doi: 10.1210/en.2006-1500. [DOI] [PubMed] [Google Scholar]
- 27.Acosta J, Carpio Y, Borroto I, González O, Estrada MP. Myostatin gene silenced by RNAi show a zebrafish giant phenotype. J Biotechnol. 2005;119:324–331. doi: 10.1016/j.jbiotec.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 29.Bogdanovich S, Krag TO, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 30.Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 31.Muramatsu H, Kuramochi T, Katada H, et al. Novel myostatin-specific antibody enhances muscle strength in muscle disease models. Sci Rep. 2021;11:2160. doi: 10.1038/s41598-021-81669-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.St Andre M, Johnson M, Bansal PN, et al. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet Muscle. 2017;7:25. doi: 10.1186/s13395-017-0141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102:2519–2524. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bo Li Z, Zhang J, Wagner KR. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci. 2012;125:3957–3965. doi: 10.1242/jcs.090365. [DOI] [PubMed] [Google Scholar]
- 35.Liu R, Hoffpauir B, Chilewski SD, et al. Accelerating regulated bioanalysis for biotherapeutics: case examples using a microfluidic ligand binding assay platform. AAPS J. 2017;19:82–91. doi: 10.1208/s12248-016-0006-z. [DOI] [PubMed] [Google Scholar]
- 36.Positano V, Christiansen T, Santarelli MF, Ringgaard S, Landini L, Gastaldelli A. Accurate segmentation of subcutaneous and intermuscular adipose tissue from MR images of the thigh. J Magn Reson Imaging. 2009;29:677–684. doi: 10.1002/jmri.21699. [DOI] [PubMed] [Google Scholar]
- 37.Scott E, Eagle M, Mayhew A, et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. 2012;17:101–109. doi: 10.1002/pri.520. [DOI] [PubMed] [Google Scholar]
- 38.Skalsky AJ, Han JJ, Abresch RT, Shin CS, McDonald CM. Assessment of regional body composition with dual-energy X-ray absorptiometry in Duchenne muscular dystrophy: correlation of regional lean mass and quantitative strength. Muscle Nerve. 2009;39:647–651. doi: 10.1002/mus.21212. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt S, Hafner P, Klein A, et al. Timed function tests, motor function measure, and quantitative thigh muscle MRI in ambulant children with Duchenne muscular dystrophy: a cross-sectional analysis. Neuromuscul Disord. 2018;28:16–23. doi: 10.1016/j.nmd.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Wong B, Signorovitch J, Staunton H, et al. P.196 Estimating clinically meaningful change thresholds in the NORTH STAR ambolatory assessment (NSAA) and four-stair climb (4SC) in Duchenne muscular dystrophy (DMD). Neuromuscul Disord 2019;29:S106.
- 41.Muntoni F, Signorovitch J, Sajeev G, et al. Minimal detectable changes in functional measures in duchenne muscular dystrophy: a study of multiple centers, networks and trial arms. Presented at the Muscular Dystrophy Association Virtual Clinical & Scientific Conference, March 15–18, 2021
- 42.Woodhouse L, Gandhi R, Warden SJ, et al. A phase 2 randomized study investigating the efficacy and safety of myostatin antibody LY2495655 versus placebo in patients undergoing elective total hip arthroplasty. J Frailty Aging. 2016;5:62–70. doi: 10.14283/jfa.2016.81. [DOI] [PubMed] [Google Scholar]
- 43.Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 44.Today MDN. Domagrozumab (PF-06252616). 2019. 10 July 2019. https://musculardystrophynews.com/domagrozumab/. Accessed July 2023.
- 45.Place A, Barrett D, Nomikos G, et al. A phase 2 study to evaluate the efficacy and safety of SRK-015 in patients with later-onset spinal muscular atrophy (TOPAZ): a study update (1963). Neurology. 2021;95:supplement.
- 46.Amato AA, Sivakumar K, Goyal N, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014;83:2239–2246. doi: 10.1212/WNL.0000000000001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell C, McMillan HJ, Mah JK, et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2017;55:458–464. doi: 10.1002/mus.25268. [DOI] [PubMed] [Google Scholar]
- 48.Al-Zaidy SA, Sahenk Z, Rodino-Klapac LR, Kaspar B, Mendell JR. Follistatin gene therapy improves ambulation in Becker muscular dystrophy. J Neuromuscul Dis. 2015;2:185–192. doi: 10.3233/JND-150083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendell JR, Sahenk Z, Al-Zaidy S, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther. 2017;25:870–879. doi: 10.1016/j.ymthe.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mendell JR, Sahenk Z, Malik V, et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol The. 2015;23:192–201. doi: 10.1038/mt.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lach-Trifilieff E, Minetti GC, Sheppard K, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol. 2014;34:606–618. doi: 10.1128/MCB.01307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long KK, O'Shea KM, Khairallah RJ, et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. 2019;28:1076–1089. doi: 10.1093/hmg/ddy382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iskenderian A, Liu N, Deng Q, et al. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet Muscle. 2018;8:34. doi: 10.1186/s13395-018-0180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dantas SM, Weckstein JD, Bates JM, et al. Molecular systematics of the new world screech-owls (Megascops: Aves, Strigidae): biogeographic and taxonomic implications. Mol Phylogenet Evol. 2016;94:626–634. doi: 10.1016/j.ympev.2015.09.025. [DOI] [PubMed] [Google Scholar]
- 55.Singh P, Rong H, Gordi T, Bosley J, Bhattacharya I. Translational pharmacokinetic/pharmacodynamic analysis of MYO-029 antibody for muscular dystrophy. Clin Transl Sci. 2016;9:302–310. doi: 10.1111/cts.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith RC, Cramer MS, Mitchell PJ, et al. Myostatin neutralization results in preservation of muscle mass and strength in preclinical models of tumor-induced muscle wasting. Mol Cancer Ther. 2015;14:1661–1670. doi: 10.1158/1535-7163.MCT-14-0681. [DOI] [PubMed] [Google Scholar]
- 57.Cadena SM, Tomkinson KN, Monnell TE, et al. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 1985;2010(109):635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Place A. Apitegromab, a novel high-affinity anti-promyostatin monoclonal antibdy for treating spinal muscular strophy: results of a phase 2 interim analysis. Presented at MDA 2021 2021.
- 59.Wagner KR, Abdel-Hamid HZ, Mah JK, et al. Corrigendum to "Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy" [Neuromuscular Disorders, Vol. 30(6), 2020, pp. 492–502] Neuromuscul Disord. 2021;31:167–168. doi: 10.1016/j.nmd.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 60.Wagner KR, Abdel-Hamid HZ, Mah JK, et al. Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul Disord. 2020;30:492–502. doi: 10.1016/j.nmd.2020.05.002. [DOI] [PubMed] [Google Scholar]
- 61.Hanna MG, Badrising UA, Benveniste O, et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019;18:834–844. doi: 10.1016/S1474-4422(19)30200-5. [DOI] [PubMed] [Google Scholar]
- 62.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dumonceaux J, Marie S, Beley C, et al. Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol Ther. 2010;18:881–887. doi: 10.1038/mt.2009.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bechir N, Pecchi E, Vilmen C, et al. ActRIIB blockade increases force-generating capacity and preserves energy supply in exercising mdx mouse muscle in vivo. FASEB J. 2016;30:3551–3562. doi: 10.1096/fj.201600271RR. [DOI] [PubMed] [Google Scholar]
- 66.Latres E, Mastaitis J, Fury W, et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. 2017;8:15153. doi: 10.1038/ncomms15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burch PM, Pogoryelova O, Palandra J, et al. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J Neurol. 2017;264:541–553. doi: 10.1007/s00415-016-8379-6. [DOI] [PubMed] [Google Scholar]
- 68.Wagner KR. The elusive promise of myostatin inhibition for muscular dystrophy. Curr Opin Neurol. 2020;33:621–628. doi: 10.1097/WCO.0000000000000853. [DOI] [PubMed] [Google Scholar]
- 69.Bhattacharya I, Pawlak S, Marraffino S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of domagrozumab (PF-06252616), an antimyostatin monoclonal antibody, in healthy subjects. Clin Pharmacol Drug Dev. 2018;7:484–497. doi: 10.1002/cpdd.386. [DOI] [PubMed] [Google Scholar]
- 70.Mariot V, Joubert R, Hourde C, et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun. 2017;8:1859. doi: 10.1038/s41467-017-01486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hammers DW, Hart CC, Patsalos A, et al. Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight. 2020;5:e133276. doi: 10.1172/jci.insight.133276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S. Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J Mol Biol. 2012;415:393–405. doi: 10.1016/j.jmb.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.AlDeghaither D, Smaglo BG, Weiner LM. Beyond peptides and mAbs–current status and future perspectives for biotherapeutics with novel constructs. J Clin Pharmacol. 2015;55(Suppl 3):S4–20. doi: 10.1002/jcph.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu-Nguyen NB, Jarmin SA, Saleh AF, Popplewell L, Gait MJ, Dickson G. Combination antisense treatment for destructive exon skipping of myostatin and open reading frame rescue of dystrophin in neonatal mdx mice. Mol Ther. 2015;23:1341–1348. doi: 10.1038/mt.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu-Nguyen N, Ferry A, Schnell FJ, et al. Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet. 2019;28:3091–3100. doi: 10.1093/hmg/ddz125. [DOI] [PubMed] [Google Scholar]
- 77.Rybalka E, Timpani CA, Debruin DA, Bagaric RM, Campelj DG, Hayes A. The failed clinical story of myostatin inhibitors against Duchenne muscular dystrophy: exploring the biology behind the battle. Cells. 2020;9:2657. [DOI] [PMC free article] [PubMed]
- 78.Mariot V, Joubert R, Hourdé C, et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun. 2017;8:1859. doi: 10.1038/s41467-017-01486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Biohaven Pharmaceuticals is currently developing taldefgrobep alfa. Biohaven will provide access to de-identified patient-level data that underlie the results in this article in response to scientifically valid research proposals. Researchers may request access and send research proposals by emailing Cliff Bechtold (cliff.bechtold@biohavenpharma.com). Data from these studies will be made available after the publication of this article. Biohaven will consider requests from qualified researchers for access to the data. Biohaven will review the request using an internal committee composed of Biohaven staff, including a clinician, a statistician, and a data-sharing professional. Biohaven will make reasonable efforts to fulfill all data requests for legitimate research purposes, but there might be instances in which retrieval or delivery of data is not feasible, such as those involving, for example, patient privacy, requirements for permissions, contractual obligations, and conflicts of interest. All those receiving access to data will be required to enter into a data use agreement provided by Biohaven which will contain the terms under which the data will be provided.