

Abstract
Background
Haloperidol is worldwide one of the most frequently used antipsychotic drugs with a very high market share. Previous narrative, unsystematic reviews found no differences in terms of efficacy between the various first‐generation (“conventional”, "typical") antipsychotic agents. This established the unproven psychopharmacological assumption of a comparable efficacy between the first‐generation antipsychotic compounds codified in textbooks and treatment guidelines. Because this assumption contrasts with the clinical impression, a high‐quality systematic review appeared highly necessary.
Objectives
To compare the efficacy, acceptability, and tolerability of haloperidol with other first‐generation antipsychotics in schizophrenia and schizophrenia‐like psychosis.
Search methods
In October 2011 and July 2012, we searched the Cochrane Schizophrenia Group’s Trials Register, which is based on regular searches of CINAHL, BIOSIS, AMED, EMBASE, PubMed, MEDLINE, PsycINFO, and registries of clinical trials. To identify further relevant publications, we screened the references of all included studies and contacted the manufacturers of haloperidol for further relevant trials and missing information on identified studies. Furthermore, we contacted the corresponding authors of all included trials for missing data.
Selection criteria
We included all randomised controlled trials (RCTs) that compared oral haloperidol with another oral first‐generation antipsychotic drug (with the exception of the low‐potency antipsychotics chlorpromazine, chlorprothixene, levopromazine, mesoridazine, perazine, prochlorpromazine, and thioridazine) in schizophrenia and schizophrenia‐like psychosis. Clinically important response to treatment was defined as the primary outcome. Secondary outcomes were global state, mental state, behaviour, overall acceptability (measured by the number of participants leaving the study early due to any reason), overall efficacy (attrition due to inefficacy of treatment), overall tolerability (attrition due to adverse events), and specific adverse effects.
Data collection and analysis
At least two review authors independently extracted data from the included trials. The methodological quality of the included studies was assessed using The Cochrane Collaboration`s 'Risk of bias' tool.
We analysed dichotomous outcomes with risk ratios (RR) and continuous outcomes with mean differences (MD), both with the associated 95% confidence intervals (CI). All analyses were based on a random‐effects model and we preferably used data on an intention‐to‐treat basis where possible.
Main results
The systematic review currently includes 63 randomised trials with 3675 participants. Bromperidol (n = 9), loxapine (n = 7), and trifluoperazine (n = 6) were the most frequently administered antipsychotics comparator to haloperidol. The included studies were published between 1962 and 1993, were characterised by small sample sizes (mean: 58 participants, range from 18 to 206) and the predefined outcomes were often incompletely reported. All results for the main outcomes were based on very low or low quality data. In many trials the mechanism of randomisation, allocation, and blinding was frequently not reported. In short‐term studies (up to 12 weeks), there was no clear evidence of a difference between haloperidol and the pooled group of the other first‐generation antipsychotic agents in terms of the primary outcome "clinically important response to treatment" (40 RCTs, n = 2132, RR 0.93 CI 0.87 to 1.00). In the medium‐term trials, haloperidol may be less effective than the other first‐generation antipsychotic group but this evidence is based on only one trial (1 RCT, n = 80, RR 0.51 CI 0.37 to 0.69).
Based on limited evidence, haloperidol alleviated more positive symptoms of schizophrenia than the other antipsychotic drugs. There were no statistically significant between‐group differences in global state, other mental state outcomes, behaviour, leaving the study early due to any reason, due to inefficacy, as well as due to adverse effects. The only statistically significant difference in specific side effects was that haloperidol produced less akathisia in the medium term.
Authors' conclusions
The findings of the meta‐analytic calculations support the statements of previous narrative, unsystematic reviews suggesting comparable efficacy of first‐generation antipsychotics. In efficacy‐related outcomes, there was no clear evidence of a difference between the prototypal drug haloperidol and other, mainly high‐potency first‐generation antipsychotics. Additionally, we demonstrated that haloperidol is characterised by a similar risk profile compared to the other first‐generation antipsychotic compounds. The only statistically significant difference in specific side effects was that haloperidol produced less akathisia in the medium term. The results were limited by the low methodological quality in many of the included original studies. Data for the main results were low or very low quality. Therefore, future clinical trials with high methodological quality are required.
Plain language summary
Haloperidol versus first‐generation antipsychotics for the treatment of schizophrenia
Haloperidol is one of the most frequently used antipsychotic drugs worldwide. It is a first‐generation antipsychotic drug. Haloperidol is highly effective in treating the ‘positive symptoms’ of schizophrenia, such as hearing voices, seeing things and having strange beliefs. However, haloperidol also has serious side effects such as involuntary shaking, blurred vision, having a dry mouth and causing strange postures. Psychiatrists and people with schizophrenia often face a trade‐off between protection against mental illness and coping with these severe side effects.
Previous small studies and unsystematic reviews have found no difference between the various first‐ generation antipsychotic drugs. This has led to the assumption that these drugs are similar in effectiveness (despite observations by psychiatrists and health professionals that these drugs do sometimes differ in their effectiveness and side effects). Because of high prescription‐rates, research on haloperidol is very important.
A search for randomised trials was run in 2012. This review includes 63 trials with 3675 participants. Haloperidol was compared with a large number of other first‐generation antipsychotic drugs (including bromperidol, loxapine and trifluoperazine) to assess its effectiveness, acceptability and tolerability. The findings of the review support the evidence of previous small, narrative studies and unsystematic reviews. There was no difference between haloperidol and other mainly high‐potency first‐generation antipsychotic drugs. In addition, haloperidol was characterised by a similar risk profile and side effects to other first‐generation antipsychotic drugs. People receiving haloperidol were less likely to experience akathisia in the medium term. Occurrence of other specific side effect such as tremor, dystonia, dyskinesia and rigor were all similar between treatment groups. Psychiatrists and people with schizophrenia should know that haloperidol and other first‐generation antipsychotic drugs are similar in their effectiveness and risk of side effects. These drugs should also be similar in their acceptability for people with schizophrenia.
However, results were limited due to the low quality of many of the included studies and low quality of evidence provided. Future studies of higher quality are required.
This plain language summary has been written by a consumer Ben Gray: Senior Peer Researcher www.mcpin.org.
Summary of findings
Summary of findings for the main comparison. Haloperidol versus first‐generation antipsychotics for schizophrenia (short term).
| Haloperidol versus first‐generation antipsychotics for schizophrenia | ||||||
|
Patient or population: people with schizophrenia Setting: inpatients and outpatients Intervention: haloperidol versus other first‐generation antipsychotics (short term) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| first‐generation antipsychotic drugs | haloperidol | |||||
| Clinically important response to treatment (short term) | Study population | RR 0.93 (0.87 to 1) | 2132 (40 studies) | ⊕⊕⊝⊝ low1,2 | ||
| 457 per 1000 | 425 per 1000 (398 to 457) | |||||
| Moderate | ||||||
| 532 per 1000 | 495 per 1000 (463 to 532) | |||||
| Leaving the study early due to any reason as a measure of overall acceptability (short term) | Study population | RR 1.04 (0.87 to 1.24) | 1299 (28 studies) | ⊕⊕⊝⊝ low1,2,3 | ||
| 201 per 1000 | 209 per 1000 (175 to 250) | |||||
| Moderate | ||||||
| 142 per 1000 | 148 per 1000 (124 to 176) | |||||
| Leaving the study early due to inefficacy of treatment as a measure of overall efficacy (short term) | Study population | RR 0.93 (0.4 to 2.16) | 507 (13 studies) | ⊕⊝⊝⊝ very low1,2,3,4 | ||
| 43 per 1000 | 40 per 1000 (17 to 94) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Leaving the study early due to adverse events as a measure of overall tolerability (short term) | Study population | RR 1 (0.42 to 2.35) | 640 (16 studies) | ⊕⊝⊝⊝ very low1,2,3,4 | ||
| 31 per 1000 | 31 per 1000 (13 to 73) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Adverse effects: number of participants with at least one adverse effect (short term) | Study population | RR 1.06 (0.94 to 1.2) | 693 (10 studies) | ⊕⊕⊝⊝ low1,2,3 | ||
| 615 per 1000 | 652 per 1000 (578 to 738) | |||||
| Moderate | ||||||
| 588 per 1000 | 623 per 1000 (553 to 706) | |||||
| Adverse effects: extrapyramidal side effects: number of participants with at least one extrapyramidal side effect (short term) | Study population | RR 1.12 (0.95 to 1.31) | 998 (17 studies) | ⊕⊝⊝⊝ very low1,2,3,5 | ||
| 365 per 1000 | 409 per 1000 (347 to 479) | |||||
| Moderate | ||||||
| 410 per 1000 | 459 per 1000 (389 to 537) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes6. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias ‐ rated 'very serious': many studies did not report the methods for sequence generation and/or allocation concealment and were not free from selective reporting. 2 Inconsistency ‐ rated 'no': there was no substantial level of heterogeneity (defined by an I2 greater than or equal to 50% accompanied by a statistically significant Chi2 test). The direction of the effect of almost all studies was the same. Therefore, the overall results are not challenged by inconsistency.
3Publication bias ‐ rated 'undetected': based on the largely symmetrical arrangement of the trials in the funnel plot the likelihood for the presence of a publication bias can be regarded as being low.
4 Imprecision ‐ rated 'very serious': the 95% confidence interval around the pooled effect size includes "no effect", "appreciable benefit", and "appreciable harm".
5 Imprecision ‐ rated 'serious': the 95% confidence interval around the pooled effect size includes both "no effect" and "appreciable harm". 6 The basis for the assumed risk was the risk in the pooled control group of the relevant studies.
Summary of findings 2. Haloperidol versus first‐generation antipsychotics for schizophrenia (medium term).
| Haloperidol versus first‐generation antipsychotics for schizophrenia | ||||||
|
Patient or population: people with schizophrenia Setting: inpatients and outpatients Intervention: haloperidol versus other first‐generation antipsychotics (medium term) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| first‐generation antipsychotic drugs | haloperidol | |||||
| Clinically important response to treatment (medium term) | Study population | RR 0.51 (0.37 to 0.69) | 80 (1 study) | ⊕⊕⊝⊝ low1,3,5 | ||
| 1000 per 1000 | 510 per 1000 (370 to 690) | |||||
| Moderate | ||||||
| 1000 per 1000 | 510 per 1000 (370 to 690) | |||||
| Leaving the study early due to any reason as a measure of overall acceptability (medium term) | Study population | RR 1.02 (0.75 to 1.38) | 137 (2 studies) | ⊕⊝⊝⊝ very low2,3,5,6 | ||
| 543 per 1000 | 554 per 1000 (407 to 749) | |||||
| Moderate | ||||||
| 548 per 1000 | 559 per 1000 (411 to 756) | |||||
| Leaving the study early due to adverse events as a measure of overall tolerability (medium term) | Not estimable | Not estimable | Not estimable | 80 (1 study) | ⊕⊕⊝⊝ low1,3,5 | |
| Adverse effects: number of participants with at least one adverse effect (medium term) | Study population | RR 1.07 (0.58 to 1.97) | 137 (2 studies) | ⊕⊝⊝⊝ very low2,4,5,6 | ||
| 657 per 1000 | 703 per 1000 (381 to 1000) | |||||
| Moderate | ||||||
| 708 per 1000 | 758 per 1000 (411 to 1000) | |||||
| Adverse effects: extrapyramidal side effects: number of participants with at least one extrapyramidal side effect (medium term) | Study population | RR 1.04 (0.62 to 1.75) | 80 (1 study) | ⊕⊝⊝⊝ very low1,3,5,6 | ||
| 405 per 1000 | 421 per 1000 (251 to 708) | |||||
| Moderate | ||||||
| 405 per 1000 | 421 per 1000 (251 to 709) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes7. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias ‐ rated 'very serious': many studies did not report the methods for sequence generation and/or allocation concealment and were not free from selective reporting. 2 Risk of bias ‐ rated 'very serious': many studies did not report the methods for sequence generation and/or allocation concealment and were not free from selective reporting. High risk of bias regarding other bias in two studies (Abuzzahab 1982, Engelhardt 1978) and in terms of blinding in one trial (Abuzzahab 1982).
3 Inconsistency ‐ rated 'no': there was no substantial level of heterogeneity (defined by an I2 greater than or equal to 50% accompanied by a statistically significant Chi2 test). The direction of the effect of almost all studies was the same. Therefore, the overall results are not challenged by inconsistency.
4 Inconsistency ‐ rated 'very serious': there was a substantial level of heterogeneity (defined by an I2 greater than or equal to 50% accompanied by a statistically significant Chi2 test).
5 Publication bias ‐ rated 'undetected': based on the largely symmetrical arrangement of the trials in the funnel plot the likelihood for the presence of a publication bias can be regarded as being low. 6 Imprecision ‐ rated 'very serious': the 95% confidence interval around the pooled effect size includes "no effect", "appreciable benefit", and "appreciable harm". 7 The basis for the assumed risk was the risk in the pooled control group of the relevant studies.
Background
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately one per cent of the population worldwide with little gender differences. Schizophrenia ranks among the seven most frequent causes listed by the World Health Organization (WHO) for loss of years of life due to disability. Its typical manifestations are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations), 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought, and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% not working (Marvaha 2004) and up to 10% dying by suicide (Tsuang 1978).
Description of the intervention
Antipsychotic drugs are the core treatment for schizophrenia. They can be classified according to their biochemical structure (e.g. butyrophenones, phenothiazines, thioxanthenes, etc.), their risk of producing movement disorders ('atypical' versus 'typical' antipsychotics) and the doses necessary for an antipsychotic effect (high‐potency versus low‐potency antipsychotics). They all have in common that they block to a greater or lesser extent, the transmission of dopamine in the brain. The classification into high‐potency and low‐potency compounds means that for low‐potency antipsychotic drugs higher doses are necessary to obtain the same dopamine receptor occupancy and efficacy than for high‐potency antipsychotic drugs (Seeman 1975).
Haloperidol, the intervention in the present study, is one of the most frequently used antipsychotic compounds and still has a very high market share (Lohse 2009). It is a first‐generation ('typical', 'conventional') antipsychotic drug and belongs chemically to the butyrophenone series of neuroleptic compounds. Due to its very high antidopaminergic properties, haloperidol can be classified as a high‐potency antipsychotic agent. Its mean elimination half‐life has been reported to range from 15 to 37 hours and its bioavailability is 60% to 70% (Kudo 1999), which indicates a high 'first‐pass‐effect'. Haloperidol is highly effective to treat schizophrenia, but on the other hand, it is associated with severe extrapyramidal adverse effects (EPS). The most predominant among these symptoms are dystonia, parkinsonian‐like syndrome, and tardive dyskinesia. Other side effects include anticholinergic effects (e.g. constipation, dry mouth, blurred vision, and urinary hesitancy), sexual dysfunction, elevations in serum prolactin, sedation and there could even be shown a relationship with sudden death.
Therefore, clinicians and people with schizophrenia often face a trade‐off between protection against psychotic episodes and adverse effects.
How the intervention might work
The theory is that schizophrenia is caused by hyperdopaminergic states in the limbic system (Berger 2003). All antipsychotic drugs block dopamine receptors. The assumption is that dopamine receptor blockade is mediating the antipsychotic effect. Therefore continuous treatment with antipsychotic compounds may be necessary to keep the dopaminergic tone low and to avoid psychotic relapses.
There are cortical dopamine‐D2‐pathways, which seem to play an important role regarding the therapeutic and adverse effects of conventional antipsychotics: the nigrostriatal dopamine pathway (responsible for the EPS side effects), the mesolimbic and mesocortical dopamine pathways (responsible for the improvement of the positive symptoms), and the tuberoinfundibular dopamine pathway (responsible for hyperprolactinaemia).
Because of its very high antagonism to dopamine‐D2‐receptors, haloperidol can be classified in the high‐potency antipsychotic agent group. On the other hand, compared to the other first‐generation antipsychotics, the affinity to dopamine‐D1‐receptors is relative low.
Why it is important to do this review
Haloperidol is one of the most frequently used antipsychotic drugs in Europe and the US (Kaye 2003; Paton 2003). Additionally, it has been used as a standard comparator in randomised trials for the introduction of many other antipsychotics including the newer second‐generation antipsychotics. Haloperidol is also on the list of essential drugs of the WHO (WHO 2009). Because of the high prescription‐rates, research on haloperidol is very important. First‐generation antipsychotic drugs are still the mainstay of treatment in countries that can not afford newer, expensive second‐generation antipsychotic drugs. But even in some industrialised countries such as Germany, conventional antipsychotic medications still have a very high market share (Lohse 2009). Recent reviews examining the more expensive second‐generation antipsychotics have also called their superiority into question (Duggan 2005; Essali 2009; Hunter 2003; Leucht 2009; Lieberman 2005; Srisurapanont 2004). Therefore, research on the older first‐generation agents is essential.
Previous narrative, unsystematic reviews found no differences in efficacy between conventional antipsychotics such as haloperidol (Davis 1978; Klein 1969). This caused the unproven psychiatric assumption, codified in textbooks and guidelines (Gaebel 2006; Lehman 2004), that ‐ with the exception of clozapine ‐ there is no difference in efficacy between the available compounds. Due to this lack of evidence, treatment guidelines make statements such as “all conventional antipsychotics if adequately dosed have comparable efficacy” (German National Schizophrenia Guideline (Gaebel 2006); or guideline of the World Federations of Societies of Biological Psychiatry (WFSBP) (Falkai 2005)).These guidelines contrast with the clinical impression that not all antipsychotic drugs are equally efficacious. This can be seen for example on the frequent selection of the high‐potency compound haloperidol for acutely ill schizophrenic patients.
To close the empirical gap concerning this topic, we compared the standard first‐generation antipsychotic drug haloperidol with a large number of other frequently used first‐generation antipsychotic compounds. We excluded the low‐potency first‐generation drugs, because these were already addressed in other Cochrane Reviews (Table 3). We also excluded the so‐called second‐generation ('atypical') antipsychotics as comparators, because these have been extensively compared with haloperidol in other systematic reviews (e.g. Essali 2009; Hunter 2003; Leucht 2009).
1. Other reviews in this series.
| Title | Reference |
| Haloperidol versus chlorpromazine | Leucht 2008 |
| Haloperidol vs low‐potency first‐generation antipsychotic drugs | Tardy 2011a |
| Perphenazine versus low‐potency first‐generation antipsychotic drugs | Tardy 2011b |
| Fluphenazine versus low‐potency first‐generation antipsychotic drugs | Tardy 2011c |
| Trifluoperazine versus low‐potency antipsychotic drugs | Tardy 2011d |
| Flupenthixol versus low‐potency first‐generation antipsychotic drugs | Tardy 2011e |
This review considers whether the statement of psychopharmacology is true that all antipsychotic drugs have the same efficacy. Thus, the findings have important impact on guidelines, clinical practise and our understanding of antipsychotic drugs. Additionally, it allows comparison of the different side effects of each compound on a large empirical basis.
Objectives
To compare the efficacy, acceptability, and tolerability of the high‐potency first‐generation antipsychotic agent haloperidol with other first‐generation antipsychotics (with the exception of the low‐potency antipsychotics chlorpromazine, chlorprothixene, levopromazine, mesoridazine, perazine, prochlorpromazine, and thioridazine) in the pharmacotherapy of schizophrenia and other similar psychotic disorders.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised trials. We excluded quasi‐randomised studies such as those using allocation by day of the week, date of birth, or alternate allocation. This decision was based on the evidence of a strong relationship between allocation concealment and direction of effect (Schulz 1995).
Where a trial was described as 'double‐blind', but it was implied that the study was randomised, we included the trial in a sensitivity analysis. If there was no substantive difference within the primary outcome (see Types of outcome measures) when these 'implied randomisation' studies were added, we included these studies in the final analysis. If there was a substantive difference, we only analysed clearly randomised trials and described the results of the sensitivity analysis in the text.
Types of participants
We included all randomised studies of participants with schizophrenia and other types of schizophrenia‐like psychoses (e.g. schizophreniform, schizoaffective, or delusional disorders), irrespective of the diagnostic system applied. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994). In accordance with the general strategy of the Cochrane Schizophrenia Group (see group module), we included studies that had used other diagnostic criteria than those of the International Statistical Classification of Diseases and Related Health Problems (ICD) and the Diagnostic and Statistical Manual of Mental Disorders (DSM). These diagnostic criteria are not meticulously used in clinical routine either, so broader inclusion criteria will enhance applicability of findings. Patients were included without a restriction concerning age, gender and whether they were suffering from other conditions.
If a study involved people with other diagnoses, we only included it if at least 75% of participants suffered from a schizophrenic syndrome or, if that was not the case, results regarding people exclusively with schizophrenia were reported.
We included studies with participants suffering from first episode schizophrenia as well as multiple episodes. Additionally, we included trials investigating treatment‐resistant participants. We considered these different conditions by applying subgroup analyses.
Types of interventions
1. Haloperidol
Any oral form (oral tablets, oral liquids) at any dose. We excluded depot formulations. Injections (i.m. ‐ intramuscular, or i.v. ‐ intravenous) were allowed for initial treatment, but we included data only if people were transferred to oral medication within the first week.
2. Control
Any other first‐generation antipsychotic agent that is currently available in at least one country worldwide with the exception of the low‐potency antipsychotics chlorpromazine, chlorprothixene, levopromazine, mesoridazine, perazine, prochlorpromazine, and thioridazine that were already compared with haloperidol in other Cochrane Reviews (Table 3) (Leucht 2008; Tardy 2011a). The medication could be administered in any oral form (oral tablets, oral liquids) at any dose. We excluded depot formulations. Injections (i.m. ‐ intramuscular, or i.v. ‐ intravenous) were allowed for initial treatment, but we included data only if people were transferred to oral medication within the first week.
Types of outcome measures
The outcomes were analysed for different lengths of follow‐up: up to three months (short term), six months (medium term) or more than six months (long term).
Primary outcomes
Global state: Clinically important response to treatment.
If presented, we used a cut‐off of at least 50% reduction of the baseline value of a rating scale such as the 'Positive and Negative Syndrome Scale' (PANSS; Kay 1987) or the 'Brief Psychiatric Rating Scale' (BPRS; Overall 1962), because studies showed that this definition is clinically meaningful (Leucht 2005a; Leucht 2005b; Leucht 2006). Otherwise we, used the definition of the original studies.
Secondary outcomes
1. Global state
1.1. Average score/change of the global state 1.2. Relapse ‐ as defined by each of the studies
2. Mental state
2.1. General ‐ average score/change of the general mental state 2.2. Specific ‐ depersonalisation 2.3. Specific ‐ depressive symptoms 2.4. Specific ‐ manic symptoms 2.5. Specific ‐ negative symptoms 2.6. Specific ‐ positive symptoms
3. Behaviour
3.1. General behaviour 3.2. Specific behaviour
4. Leaving the study early (‘drop‐out’)
4.1. Due to any reason ‐ as a measure of overall acceptability 4.2. Due to inefficacy of treatment ‐ as a measure of overall efficacy 4.3. Due to adverse events ‐ as a measure of overall tolerability
5. Adverse effects
5.1. General ‐ at least one adverse effect 5.2. Specific ‐ extrapyramidal/movement disorders 5.3. Specific ‐ death 5.4. Specific ‐ hypotension 5.5. Specific ‐ sedation 5.6. Specific ‐ weight gain
6. Satisfaction with care
6.1. Clinically important change in satisfaction 6.2. Average score/change in satisfaction
7. Quality of life
7.1. Clinically important change in quality of life 7.2. Average score/change of the quality of life score
8. Service utilisation
8.1. Days in hospital 8.2. Admitted
9. Functioning
9.1. Days requiring a sick certificate 9.2. Employed 9.3. Clinically important change in general functioning
10. Economic outcomes
10.1. Average change in total cost of medical and mental health care 10.2. Total indirect and direct costs
11. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008) and used GRADEPRO to import data from RevMan v5 to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study, the magnitude of effect of the examined interventions, and the sum of available data on all outcomes that we rated as important to patient‐care and decision making.
We anticipated including the following main outcomes in a 'Summary of findings' table.
Clinically important response to treatment
Acceptability of treatment ‐ leaving the study early due to any reason
Overall efficacy of treatment ‐ leaving the study early due to inefficacy
Tolerability of treatment ‐ leaving the study early due to adverse effects
Adverse effects ‐ number of participants with at least one adverse effect
Adverse effects ‐ number of participants with extrapyramidal/movement disorder
Quality of life ‐ improved to an important extent
Search methods for identification of studies
No language restriction was applied within the limitations of the search tools to avoid the problem of 'language bias' (Egger 1997b).
Electronic searches
1. Cochrane Schizophrenia Group’s Trials Register
In October 2011 and July 2012, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Registry of Trials using the following search strategy:
(*haloperi* or *R‐1625 or *haldol* or *alased* or *aloperidi* or *bioperido* or *buterid* or *ceree* or *dozic* or *duraperido* or *fortuna* or *serena* or *serenel* or *seviu* or *sigaperid* or *sylad* or *zafri*) in Interventions of STUDY
The Cochrane Schizophrenia Group’s Registry of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, EMBASE, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected the references of all identified studies for more trials.
2. Previous reviews
We searched the publication lists of the previous conventional reviews by Klein 1969 and Davis 1989, but if we identified other relevant reviews, these were also searched for publications fulfilling the inclusion criteria.
3. Personal contact
We tried to contact the first author of each included study for missing information.
4. Drug companies
We contacted the original manufacturers of haloperidol and asked them for further relevant studies and for missing information on identified studies.
Data collection and analysis
Selection of studies
Review author MD inspected all abstracts of studies identified as above and identified potentially relevant reports. In addition, to ensure reliability, a second review author (MS) inspected a random sample of these abstracts, comprising 25% of the total. Where disagreement occurred this was resolved by discussion, or where there was still doubt, the full article was acquired for further inspection. The full articles of relevant reports were acquired for reassessment and carefully inspected for a final decision on inclusion (see Criteria for considering studies for this review). Once the full articles were obtained, in turn MD and MS inspected all full reports and independently decided whether they met inclusion criteria. MD and MS were not blinded to the names of the authors, institutions, or journal of publication. Where difficulties or disputes arose, we asked review author SL for help and if it was impossible to decide, these studies were added to those awaiting assessment and the authors of the papers contacted for clarification.
Data extraction and management
1. Extraction
At least two review authors (MD, MS, CL) independently extracted data from all selected trials. When disagreement arose, we resolved it by discussion with a third review author (SL). Where this was not possible, we contacted the study authors to resolve the dilemma.
2. Management
2.1 Forms
Data were extracted on standard simple forms that were piloted based on a random sample of 10 studies.
2.2 Scale‐derived data
We included continuous data from rating scales only if: (a) the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and (b) the measuring instrument was not written or modified by one of the trialists for that particular trial. Ideally, the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). As this is not often reported clearly, we noted in Description of studies if this was not the case.
2.3. Endpoint versus change data
There are advantages to both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. Endpoint and change data were combined in the analysis as we used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: (a) data from studies of at least 200 participants were entered in the analysis irrespective of the following rules, because skewed data pose less of a problem in large studies; (b) endpoint data: when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggested a skew and the study was excluded. If this ratio was higher than 1 but below 2, there is suggestion of skew. We entered the study and tested whether its inclusion or exclusion substantially changed the results. If the ratio was larger than 2 the study was included, because skew is less likely (Altman 1996; Higgins 2011). (c) change data: when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We entered the study, because change data tend to be less skewed and because excluding studies would also lead to bias, because not all the available information was used.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week, or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
If possible, efforts were made to convert outcome measures to dichotomous data. This could be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
Assessment of risk of bias in included studies
Again at least two review authors (MD, MS, CL) worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data, and selective reporting.
If the raters disagreed, the final rating was made by consensus, with the involvement of another member of the review group (SL). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information. Non‐concurrence in quality assessment was reported, but if disputes arose as to which category a trial was to be allocated, again, resolution was made by discussion.
The level of risk of bias was noted in both the text of the review and in the Table 1.
Measures of treatment effect
1. Dichotomous data
For binary outcomes, we calculated a standard estimation of the random‐effects (Der‐Simonian 1986) risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. Where possible, efforts were made to convert outcome measures to dichotomous data.
2. Continuous data
For continuous outcomes, we estimated mean difference (MD) between groups. MDs were based on the random‐effects model as this takes into account any differences between studies, even if there is no statistically significant heterogeneity. We did not calculate standardised mean differences (SMD) measures. There was one exception to this rule, however. In the case of where scales were of such similarity to allow pooling, we calculated the SMD and, whenever possible, transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance is overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. Where clustering had been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect. If a cluster study had been appropriately analysed taking into account intra‐class correlation co‐efficient and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique, where the natural logarithm of the effect estimate (and standard errors) for all included trials for that outcome would be calculated and entered into RevMan along with the log of the effect estimate (and standard errors) from the cluster randomised trial(s). We would have used methods described in section 7.7.7.2 and 7.7.7.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to obtain standard errors.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, randomised cross‐over studies were eligible, but only data up to the point of the first cross‐over.
3. Studies with multiple treatment groups
If a trial investigated more than two treatment arms we only used data from these study arms that evaluated the antipsychotics drugs of interest for this systematic review. If two dose groups were analysed and data were binary, these were simply added and combined within the two‐by‐two table. If these data were continuous, we combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). The loss to follow‐up in randomised schizophrenia trials is often considerable calling the validity of the results into question. Nevertheless, it is unclear which degree of attrition leads to a high degree of bias. We did not exclude trials from outcomes on the basis of the percentage of participants completing them. We did, however, use the 'Risk of bias' tool described above to indicate potential bias when more than 25% of the participants left the studies prematurely, when the reasons for attrition differed between the intervention and the control group, and when no appropriate imputation strategies were applied.
2. Dichotomous data
Data were presented on a 'once‐randomised‐always‐analyse' basis, assuming an 'intention‐to‐treat' (ITT) analysis. If the authors applied such a strategy, we used their results. If the original authors presented only the results of the per‐protocol or completer population, we assumed that those participants lost to follow‐up would have had the same percentage of events as those who remained in the study.
3. Continuous data
3.1 General
Intention‐to‐treat data (ITT) were used when available. We anticipated that in some studies, in order to do an ITT analysis, the method of last observation carried forward (LOCF) was employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leon 2006; Leucht 2007). Therefore, where LOCF data have been used in the analysis, they have been indicated in the review.
3.2 Missing standard deviations
If standard deviations (SDs) were not reported, we primarily tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and confidence intervals available for group means, and either 'P' value or 't' value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): When only the SE was reported, SDs were calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, t or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we calculated the SDs according to a validated imputation method, which is based on the SDs of the other included studies (Furukawa 2006).
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies without any comparison to judge clinical heterogeneity.
We simply inspected all studies for clearly outlying situations or people which we had not predicted would arise. Where such situations or participant groups arose, these were fully discussed.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, these were fully discussed.
3. Statistical
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2statistic
Heterogeneity between studies was investigated by considering the I2 method alongside the Chi2 'P' value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2011). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. 'P' value from Chi2 test, or a confidence interval for I2).
We interpreted an I2 estimate greater than or equal to 50% accompanied by a statistically significant Chi2 statistic, as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, reasons for heterogeneity were explored.
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included randomised trials. If the protocol was available, outcomes in the protocol and in the published report were compared. If the protocol was not available, outcomes listed in the methods section of the trial report were compared with the reported results.
2. Funnel plots
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997a). These are again described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We employed a random‐effects model for analyses (Der‐Simonian 1986). We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. Therefore, the random‐effects model is usually more conservative in terms of statistical significance, although as a disadvantage, it puts added weight onto smaller studies, which can either inflate or deflate the effect size. Therefore, we examined in a sensitivity analysis whether using a fixed‐effect model markedly changed the results of the primary outcome.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analysis
We examined the subgroups of people with a first episode of schizophrenia and patients with treatment‐resistant schizophrenia (failure of response to at least one previous pharmacotherapy with antipsychotics in adequate dose and duration) to determine if their results for primary outcomes substantively differed from other participant groups'. Furthermore, we performed a stratification according to the different first‐generation antipsychotics administered as active comparator agent to haloperidol.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First, we investigated whether data had been entered correctly. Second, if the data were correct, we evaluated whether there were obvious reasons that caused the heterogeneity in the relevant studies. If we found appropriate reasons (e.g. different methods), we removed these trials from the pooled data analysis. If we did not find any obvious reason, the data of these studies were pooled but heterogeneity was thoroughly discussed.
Additional potential causes of high heterogeneity were explored by performing a random‐effects restricted maximum‐likelihood meta‐regression of the primary outcome. The following potential effect modifiers of the primary outcome were addressed: schizophrenia severity at baseline (BPRS or PANSS total score at baseline), haloperidol dose, comparator dose, ratio of haloperidol and comparator dose, study duration, pharmaceutical sponsor. We are aware that subgroup analyses are observational by nature and therefore consider the results to be exploratory and not explanatory.
Sensitivity analysis
All sensitivity analyses were applied only for the primary outcome.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcome we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then all data were employed from these studies.
2. Implication of non double‐blind trials
We excluded trials in a sensitivity analysis if they were not double‐blind.
3. Exclusion of cross‐over‐trials
We removed cross‐over‐trials in a sensitivity analysis to detect any substantive difference when these studies were not included in the analysis of the primary outcome.
4. Fixed‐effect versus random‐effects models
A sensitivity analysis was carried out to clarify, whether the use of a fixed‐effect model resulted in a substantial difference in the primary outcome compared to the random‐effects model.
Results
Description of studies
For substantive description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The electronic literature search identified a total of 2944 references, of which 158 citations were closely inspected. Sixty‐three studies (87 citations) were included and 64 studies (71 citations) were excluded. We did not identify any studies awaiting classification or ongoing studies (Figure 1).
1.
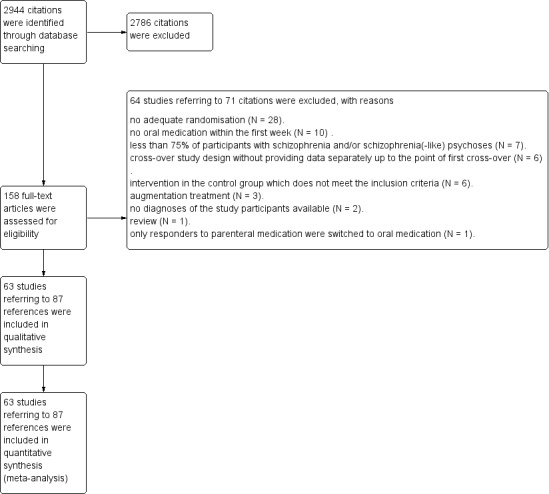
Study flow diagram.
Included studies
Sixty‐three trials published between 1962 and 1993 were included in this systematic review. Most of the studies were in English; six in Japanese (Itoh 1985; Kodama 1984; Kurihara 1983; Mori 1989; Nishimatu 1975; Okuda 1979), three in Italian (Cocchi 1971; Germana 1990; Mauri 1994), two in French (Darondel 1981; Giordana 1984), two in Spanish (Bueno 1979; Fuentenebro 1989), and one study in German (Mattke 1976).
1. Study design
All studies were randomised ("implied randomisation" in 12 trials) and most applied double‐blind methodology. Sixty studies were conducted using a parallel group design whereas three were designed as cross‐over studies (Gerlach 1985; Shalev 1993; Stewart 1969). Further details regarding methodological issues are displayed in the sections on allocation and blinding.
2. Trial duration
Fifty‐seven studies evaluated a short‐term trial duration (up to 12 weeks) whereas five studies (Abuzzahab 1982; Cosar 1999; Engelhardt 1978; Paprocki 1976; Teja 1975) investigated a medium‐term period (from more than 12 weeks to 26 weeks). One trial was carried out over a long‐term period (> 26 weeks) (Nishikawa 1984).
3. Participants
The 63 included trials comprised a total of 3675 participants. One study examined children (Faretra 1970) and two trials adolescents (Pool 1976; Versiani 1978). Forty‐six studies included only participants with schizophrenia or schizophrenia‐like psychosis. The remaining trials also enrolled persons with other diagnoses or ambiguities regarding the diagnoses remained. These studies were included because at least 75% of the whole trial sample had schizophrenia or a schizophrenia‐like psychosis. Parent 1983 included 40 participants, of which only 21 were diagnosed with schizophrenia or schizophrenia‐like psychosis, but the data used in this analysis refer exclusively to the 21 persons with schizophrenic disorders.
In 14 trials the diagnoses of the participants were based on international classification systems. One study applied the DSM‐II (Brannen 1981), six trials the DSM‐III (Dufresne 1993; Escobar 1985; Fuentenebro 1989; Nishikawa 1984; Shalev 1993; Tuason 1986), two the DSM‐III‐R (Kinon 1993; Mauri 1994), and one the DSM‐IV (Cosar 1999). In three investigations, participants were diagnosed according to ICD‐8 (Haas 1982; Mattke 1976; Nedopil 1981) and Baastrup 1993 used the ICD‐9 as diagnostic criterion.
4. Sample size
The number of participants included in each study ranged from 18 (Glazer 1990; Rubin 1971) to 206 (Itoh 1985).
5. Setting
Thirty‐nine trials included exclusively inpatients and six studies (Abuzzahab 1982; Engelhardt 1978; Glazer 1990; Luckey 1967; Nishikawa 1984; Tobin 1980) exclusively outpatient participants. Giordana 1984 as well as Kodama 1984 included both inpatients and outpatients. Four studies described the inclusion of mostly inpatients (Kariya 1983; Kurihara 1983; Mori 1989; Nishimatu 1975). In Escobar 1985, the participants were hospitalised at least during the injectable phase of the trial and in Spina 1992, they had to be inpatients for at least the first four weeks of treatment. Similarly, in Cocito 1970, the participants had to be inpatients at least for the beginning of the trial. There was no information regarding the setting for the remaining nine trials examined in this systematic review (Cosar 1999; Dufresne 1993; Fuentenebro 1989; Itoh 1985; Malfroid 1978; Mattke 1976; Pöldinger 1977; Stewart 1969; Ulmar 1990).
6. Interventions
According to the predefined inclusion criteria all included trials compared haloperidol with another first‐generation antipsychotic drug. One trial compared haloperidol with benperidol (Nedopil 1981), nine with bromperidol (Brannen 1981; Denijs 1980; Germana 1990; Itoh 1985; Kodama 1984; Malfroid 1978; Mauri 1994; Pöldinger 1977; Spina 1992), two with clopenthixol (Heikkilä 1981; Serafetinides 1972), two with droperidol (Cocchi 1971; Cocito 1970), one with flupenthixol (Parent 1983), three with fluphenazine (Faretra 1970; Hall 1968; Kinon 1993), seven with loxapine (Bueno 1979; Mattke 1976; Paprocki 1976; Pool 1976; Selman 1976; Tuason 1986; Versiani 1978), one with methylperidol (Nishimatu 1975), one with nemonapride (Mori 1989), one with mesoridazine (White 1981), four with molindone (Dufresne 1993; Fuentenebro 1989; Escobar 1985; Glazer 1990), three with perphenazine (Goldstein 1969; Kurihara 1983; Shalev 1993), three with pimozide (Gowardman 1973; Haas 1982; Silverstone 1984), three with pipotiazine (Bechelli 1983; Darondel 1981; Giordana 1984), one with propericuazine (Nishikawa 1984), five with sulpiride (Cassano 1975; Cosar 1999; Gerlach 1985; Okuda 1979; Rama Rao 1981), one with thiopropazate (Hollister 1962), five with thiothixene (Abuzzahab 1982; Engelhardt 1978; Howard 1974; Teja 1975; Tobin 1980), one with timiperone (Kariya 1983), six with trifluoperazine (Goldstein 1966; Luckey 1967; O´Brien 1974; Rubin 1971; Stewart 1969; Teja 1975), two with trifluperidol (Gallant 1967; Ulmar 1990), and two with zuclopenthixol (Baastrup 1993; Heikkilä 1992).
A fixed dose or dose scheme was applied in six studies (Cocchi 1971; Darondel 1981; Kinon 1993; Mauri 1994; Nedopil 1981; Nishikawa 1984). In most of the included trials flexible doses of antipsychotic agents could be administered. The mean final doses of haloperidol ranged from 6.6 mg/day (Pöldinger 1977) to 30.4 mg/day (Brannen 1981). In the long‐term study by Nishikawa 1984 evaluating maintenance pharmacotherapy, no participant received more than 6 mg/day haloperidol. The maximum allowed dose of haloperidol that could be administered was 100 mg/day in the research projects of Tuason 1986 and White 1981.
7. Outcomes
7.1 Clinically important response to treatment
The primary outcome "clinically important response to treatment" was reported only by a limited number of the included studies (n = 40). We prespecified a reduction of at least 50% in the PANSS or BPRS as a relevant cut‐off to define clinically important response to treatment. Only the study by Kodama 1984 provided data to calculate the 50% threshold of the BPRS. In all other cases, we had to employ the definition of the original studies, which was mainly based on the categorisation according to the Clinical Global Impression Scale (CGI, Guy 1976). We used a CGI rating of at least "much improved" to assume clinically important response to treatment.
7.2 Rating scales
Different rating scales were applied to assess clinical response and adverse effects. Details of scales that provided usable data are shown below. Reasons for exclusion of data from other rating instruments are provided under "outcomes" in the Characteristics of included studies table.
7.2.1 Global state
7.2.1.1 Clinical Global Impression ‐ CGI (Guy 1976) This rating instrument is commonly employed in studies on schizophrenia and enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually used with low scores indicating decreased severity and/or greater recovery. Escobar 1985, Mauri 1994, Spina 1992, and White 1981 reported data from this scale.
7.2.2 Mental state
7.2.2.1 Brief Psychiatric Rating Scale ‐ BPRS (Overall 1962) This brief rating scale is used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The scale has 18 items, and each item can be defined on a seven‐point scale varying from "not present" (1) to "extremely severe" (7). Scores range between 18 and 126. Twenty‐four studies (Abuzzahab 1982; Bechelli 1983; Brannen 1981; Darondel 1981; Dufresne 1993; Gerlach 1985; Haas 1982; Hall 1968; Heikkilä 1981; Heikkilä 1992; Hollister 1962; Itoh 1985; Kinon 1993; Luckey 1967; Mauri 1994; Parent 1983; Rama Rao 1981; Selman 1976; Serafetinides 1972; Spina 1992; Tobin 1980; Tuason 1986; Versiani 1978; White 1981) reported data from the BPRS scale.
7.2.2.2 Montgomery Rating Score ‐ MRS (Montgomery 1978) The MRS consists of 12 items and allows an evaluation in terms of schizophrenic first rank symptoms. Silverstone 1984 provided data from this scale.
7.2.2.3 Rating Scale for Quantification of Psychotic Symptom Severity ‐ RSQPSS (Goodrich 1953) The RSQPSS allows a methodical exploration of physical conditions (appetite, sleep, body weight, psychosomatic symptoms), sensorium (orientation, state of intellectual functions relative to schizophrenic deficiency), behaviour (personal care, activity, abnormal language, behaviour), emotional status (mood, tension, affectivity), and mental content (hallucination, delusion). A total sum score (final rating) ranging from 2.0 (security ward) to 4.0 (possible discharge from mental hospital) provides an overall evaluation of severity. The study by Cocito 1970 reported data from this scale.
7.2.2.4 Scale for the Assessment of Positive Symptoms ‐ SAPS (Andreasen 1983) This six‐point scale contains a global rating of the following positive symptoms: hallucinations, delusions, bizarre behaviour, and conceptual disorganisation. High scores indicate a high magnitude of symptoms. Mauri 1994 provided data from this scale.
7.2.2.5 Scale for the Assessment of Negative Symptoms ‐ SANS (Andreasen 1989) This scale was designed to assess negative symptoms in schizophrenic conditions. These negative symptoms include alogia, affective blunting, avolition‐apathy, anhedonia‐associality, and attention impairment. As in the SAPS, higher scores in this six‐point scale are associated with more severe symptoms. Mauri 1994 reported data from this scale.
7.2.2.6 Hamilton Depression Rating Scale ‐ HAM‐D (Hamilton 1960) The HAM‐D (Hamilton 1960) is a well‐established 17‐item scale for the measurement of depression and is sensitive to change. Abuzzahab 1982, Dufresne 1993, and Mauri 1994 trials provided data concerning the HAM‐D.
7.2.2.7 AMDP (AMDP 2007) The AMDP system consists of a glossary of psychopathological symptoms (AMDP manual) and rating scales. Altogether, these instruments provide standardised diagnostic findings. Giordana 1984 and Nedopil 1981 allocated data in terms of the AMDP system.
7.2.3 Behaviour
7.2.3.1 Nurses Observation Scale for Inpatient Evaluation ‐ NOSIE (Honigfeld 1965) The NOSIE is an 80‐item scale with items rated on a five‐point scale from zero (not present) to four (always present). Ratings are based on behaviour over the previous three days. The seven headings are social competence, social interest, personal neatness, co‐operation, irritability, manifest psychosis, and psychotic depression. The total score ranges from zero to 320 with high scores indicating a poor outcome. Brannen 1981, Heikkilä 1981, and Serafetinides 1972 reported data from this scale.
7.2.3.2 Wing`s Ward Behaviour Scale (Wing 1961) The Wing`s Ward Behaviour Scale comprises two subscales measuring social withdrawal (e.g. slowness, underactivity, or lack of conversation) and social embarrassing behaviour (e.g. threats of violence or odd mannerisms). The Wing`s Ward Behaviour Scale requires senior nurses as informants. Rama Rao 1981 provided data from this scale.
7.2.4 Adverse effects
Adverse effects (death, at least one adverse effect, at least one movement disorder, akathisia, akinesia, dyskinesia, dystonia, rigor, tardive dyskinesia, tremor, use of antiparkinson medication, hypotension, sedation, weight gain) were reported in a dichotomous manner in terms of the number of participants who experienced the relevant adverse effect.
7.2.5 Missing outcomes
No data were available for the outcomes "satisfaction with care", "quality of life", "service utilisation", "functioning", and economic consequences of treatment.
Excluded studies
We excluded 64 trials (see Characteristics of excluded studies table). Twenty‐eight studies were excluded because they were not appropriately randomised. In 10 trials the participants did not receive oral medication within the first week and in seven studies fewer than 75% of participants suffered from schizophrenia and/or schizophrenia‐like psychoses and data were not provided separately for people with schizophrenic disorders. Six trials were performed applying a cross‐over study design without providing data separately up to the point of first cross‐over and six studies administered an intervention in the control group that does not meet the inclusion criteria. Three research projects evaluated augmentation strategies and in two trials there was no information regarding the diagnoses of the participants available. One citation was a review and in one study after four days of parenteral administration of the investigated agents, only responders were switched to oral medication.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 2 and Figure 3. Full details of judgements for every single included study are presented in the 'Risk of bias' tables within the section Characteristics of included studies.
2.
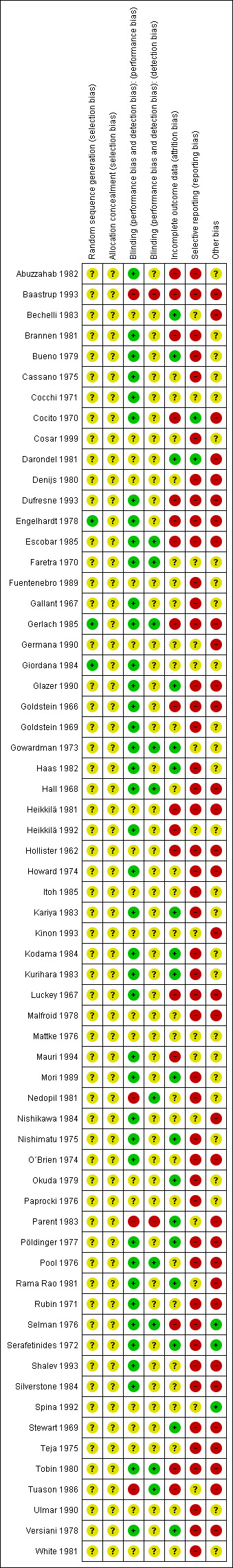
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.
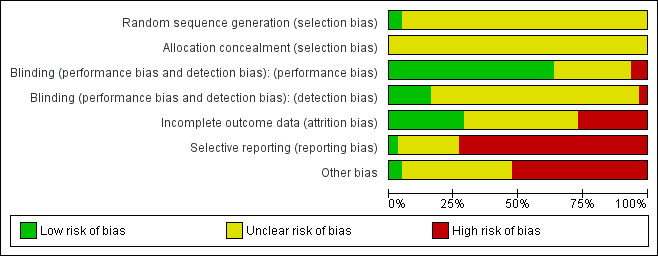
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Briefly, all studies were randomised, but only three of them described an adequate random sequence generation procedure. No study indicated adequate concealment of allocation. The risk for a performance bias was low in 40 studies and for detection bias in 10 trials. The risk of bias for incomplete outcome data reporting (attrition bias) was judged to be low in 18 trials, unclear in 28, and high in 17. Three of the 63 included studies appeared to be free of selective reporting and in 33 trials we found evidence for a high risk of other biases.
Allocation
Engelhardt 1978 used a computer‐generated randomisation scheme for sequence generation and Gerlach 1985 randomised referring to a random number table. In Giordana 1984 the participants were drawn by lots into two groups of treatment. These trials were given the quality score "low risk of bias". No further details on the randomisation method were provided in the remaining 60 studies. In 12 publications no randomisation was mentioned (Abuzzahab 1982; Bueno 1979; Fuentenebro 1989; Haas 1982; Heikkilä 1981; Itoh 1985; Mattke 1976; O´Brien 1974; Stewart 1969; Tobin 1980; Ulmar 1990; White 1981). Because the trials were described as "double‐blind", we implied that the studies were randomised. All these studies were rated with "unclear risk of bias". None of the included trials used a 'cluster randomisation'.
Regarding concealment of allocation, no study provided enough information to permit judgement of "low risk of bias" as well as "high risk of bias" in the quality score.
Blinding
Performance‐Bias: Most of the included studies were declared as "double‐blind". In 19 trials the authors provided no further information concerning the mechanism of blinding ("unclear risk of bias"), while 40 provided information to allow judgement with "low risk of bias" in the quality tool. Most of these studies used at least identically appearing capsules for blinding. Three trials (Baastrup 1993; Nedopil 1981; Parent 1983) were non‐blind and in Tuason 1986 the personnel administering the study medications was not blinded (classified as "high risk of bias").
Detection‐Bias: 51 publications did not provide enough information to allow classification of "high" or "low risk of bias". In 10 trials, we assessed a "low risk of bias" for a detection‐bias (Escobar 1985; Faretra 1970; Gerlach 1985; Gowardman 1973; Hall 1968; Nedopil 1981; Pool 1976; Selman 1976; Tobin 1980; Tuason 1986), while two studies appeared to have a "high risk" regarding this bias (Baastrup 1993; Parent 1983).
Incomplete outcome data
The overall‐attrition (participants who left the trials early for any indication) was low (< 10%) in 18 trials (rated as "low risk of bias") and moderate (10% to 25%) in 10 studies. Moderate attrition was classified as "unclear risk of bias" because in all relevant trials the authors of the original studies did not provide sufficient information to judge if the analysis‐methods were appropriate to deal with the missing data. In 17 trials the attrition could be considered as high (> 25%) (rated as "high risk of bias") and 18 studies did not address this outcome (judged as "unclear risk of bias"). In most of the research projects completers‐analyses were applied.
Selective reporting
The outcome‐data reporting was incomplete in 46 studies (rated as "high risk of bias"). In particular, standard deviations were often not indicated and had to be imputed from the other trials. In several instances the data had to be estimated from figures, which led to imprecision. Two trials appeared to be free of selective reporting (Cocito 1970; Darondel 1981) and were rated with "low risk of bias" in the quality score.
Other potential sources of bias
Three investigations appeared to be free of other potential sources of bias and received the rating "low risk of bias" (Selman 1976; Serafetinides 1972; Spina 1992). In 27 studies the "risk of bias" was considered as "unclear" due to a lack of available information in the publications. Thus, there was insufficient information to assess whether an important "risk of bias" exists. The remaining 33 studies were characterised by a "high risk of bias" in terms of other potential sources of bias. The reasons for rating "high risk of bias" are displayed in more detail in the Characteristics of included studies table for every included trial.
Effects of interventions
We analysed dichotomous outcomes with risk ratios (RR) and continuous outcomes with mean differences (MD), both with the associated 95% confidence intervals (CI)
1. Primary outcome: Clinically important response to treatment
1.1 Overall symptoms of schizophrenia (short term)
Four‐hundred and thirty out of 1045 (41.1%) participants in the pooled haloperidol group compared to 497 out of 1087 (45.7%) in the pooled group of the other first‐generation antipsychotics met the predefined criteria for the achievement of clinically important response to treatment. The pooled risk ratio (RR) revealed no statistically significant between‐group difference (40 RCTs, n = 2132, RR 0.93 CI 0.87 to 1.00).
1.2 Overall symptoms of schizophrenia (medium term)
Statistically significant less patients in the haloperidol group than in the control group achieved clinically important response to treatment (1 RCT, n = 80, RR 0.51 CI 0.37 to 0.69).
2. Global state
2.1 Mean Clinical Global Impression (CGI) score at endpoint (short term)
There was no statistically significant difference (4 RCTs, n = 151, MD ‐0.07 CI ‐0.39 to 0.25). The standard deviations had to be imputed for three trials (Escobar 1985; Mauri 1994; Spina 1992).
3. Mental state general
3.1 Mean Brief Psychiatric Rating Scale (BPRS) total score at endpoint (short term)
There was no statistically significant difference (23 RCTs, n = 998, MD 0.37 CI ‐1.66 to 2.39). The standard deviations had to be imputed for 12 trials (Brannen 1981; Dufresne 1993; Gerlach 1985; Haas 1982; Hall 1968; Heikkilä 1981; Hollister 1962; Luckey 1967; Selman 1976; Serafetinides 1972; Tobin 1980; Versiani 1978). An I² value of 91% accompanied by a statistically significant Chi2 statistic indicated a substantial level of heterogeneity.
3.2 Mean Brief Psychiatric Rating Scale (BPRS) total score at endpoint (medium term)
Because of missing standard deviations for the trial that contributed data to this outcome, no effect size could be calculated. No validated imputation method could be applied to obtain the missing standard deviations.
3.3 Mean Montgomery Rating Score (MRS) at endpoint (short term)
Because of missing standard deviations for the trial that contributed data to this outcome, no effect size could be calculated. No validated imputation method could be applied to obtain the missing standard deviations.
3.4 Mean Rating Scale for Quantification of Psychotic Symptom Severity (RSQPSS) at 60 days (short term)
There was no statistically significant difference (1 RCT, n = 12, MD 0.00 CI ‐0.04 to 0.04).
4. Mental state specific
4.1 Depersonalisation ‐ AMDP at endpoint (short term)
There was a statistically significant difference in favour of the other first‐generation antipsychotics (1 RCT, n = 30, MD 1.30 CI 0.62 to 1.98).
4.2 Depressive symptoms ‐ Hamilton Depression Rating Scale (HAM‐D) score at endpoint (short term)
There was no statistically significant difference (2 RCTs, n = 62, MD ‐0.46 CI ‐1.23 to 0.32). The standard deviations for Dufresne 1993 had to be imputed.
4.3 Depressive symptoms ‐ Hamilton Depression Rating Scale (HAM‐D) score at endpoint (medium term)
Because of missing standard deviations for all trials that contributed data to this outcome, no effect size could be calculated. No validated imputation method could be applied to obtain the missing standard deviations.
4.4 Negative Symptoms ‐ Scale for the Assessment of Negative Symptoms (SANS) score at endpoint (short term)
There was no statistically significant difference (1 RCT, n = 40, MD 0.30 CI ‐2.13 to 2.73).
4.5 Positive symptoms ‐ Conceptual disorganisation ‐ AMDP at endpoint (short term)
There was a statistically significant difference in favour of the other first‐generation antipsychotics (1 RCT, n = 30, MD 3.00 CI 2.07 to 3.93).
4.6 Positive symptoms ‐ Delusional symptoms ‐ AMDP at endpoint (short term)
There was a statistically significant difference in favour of the other first‐generation antipsychotics (1 RCT, n = 30, MD 3.30 CI 2.13 to 4.47).
4.7 Positive symptoms ‐ Positive symptoms (overall) measured by the Scale for the Assessment of Positive Symptoms (SAPS) score at endpoint (short term)
There was a statistically significant difference in favour of haloperidol (1 RCT, n = 40, MD ‐14.70 CI ‐17.42 to ‐11.98).
4.8 Positive symptoms‐ Hallucinatory symptoms ‐ AMDP at endpoint (short term)
The data of both studies were skewed and could therefore only be presented in an "other data" table.
4.9 Positive symptoms ‐ Paranoid symptoms ‐ AMDP at endpoint (short term)
The data of the study were skewed and could therefore only be presented in an "other data" table.
5. Behaviour
5.1 Mean Nurses Observation Scale for Inpatient Evaluation (NOSIE) score at endpoint (short term)
Because of missing standard deviations for all trials that contributed data to this outcome, no effect size could be calculated. No validated imputation method could be applied to obtain the missing standard deviations.
5.2 Mean Wing`s Ward Behaviour Scale score at endpoint (short term)
There was no statistically significant difference (1 RCT, n = 30, MD ‐1.27 CI ‐4.51 to 1.97).
6. Leaving the study early
6.1 Due to any reason as a measure of overall acceptability (short term)
There was no statistically significant difference (28 RCTs, n = 1299, RR 1.04 CI 0.87 to 1.24).
6.2 Due to any reason as a measure of overall acceptability (medium term)
There was no statistically significant difference (2 RCTs, n = 137, RR 1.02 CI 0.75 to 1.38).
6.3 Due to inefficacy of treatment as a measure of overall efficacy (short term)
There was no statistically significant difference (13 RCTs, n = 507, RR 0.93 CI 0.40 to 2.16).
6.4 Due to adverse effects as a measure of overall tolerability (short term)
There was no statistically significant difference (16 RCTs, n = 640, RR 1.00 CI 0.42 to 2.35).
6.5 Due to adverse effects as a measure of overall tolerability (medium term)
There was no statistically significant difference (1 RCT, n = 80, RR not estimable).
7. Adverse effects general
7.1 Number of participants with at least one adverse effect (short term)
There was no statistically significant difference (10 RCTs, n = 693, RR 1.06 CI 0.94 to 1.20).
7.2 Number of participants with at least one adverse effect (medium term) There was no statistically significant difference (2 RCTs, n = 137, RR 1.07 CI 0.58 to 1.97). An I² value of 86% accompanied by a statistically significant Chi2 statistic indicated a substantial level of heterogeneity.
8. Adverse effects specific
8.1 Extrapyramidal side effects: number of participants with akathisia (short term)
There was no statistically significant difference (22 RCTs, n = 1648, RR 1.05 CI 0.89 to 1.24).
8.2 Extrapyramidal side effects: number of participants with akathisia (medium term)
There was a statistically significant difference in favour of the haloperidol group (1 RCT, n = 57, RR 0.31 CI 0.16 to 0.60).
8.3 Extrapyramidal side effects: number of participants with akinesia (short term)
There was no statistically significant difference (2 RCTs, n = 235, RR 0.92 CI 0.31 to 2.68).
8.4 Extrapyramidal side effects: number of participants with at least one extrapyramidal side effect (short term)
There was no statistically significant difference (17 RCTs, n = 998, RR 1.12 CI 0.95 to 1.31).
8.5 Extrapyramidal side effects: number of participants with at least one extrapyramidal side effect (medium term)
There was no statistically significant difference (1 RCT, n = 80, RR 1.04 CI 0.62 to 1.75).
8.6 Extrapyramidal side effects: number of participants with dyskinesia (short term)
There was no statistically significant difference (11 RCTs, n = 807, RR 0.81 CI 0.48 to 1.35).
8.7 Extrapyramidal side effects: number of participants with dystonia (short term)
There was no statistically significant difference (15 RCTs, n = 1035, RR 1.34 CI 0.95 to 1.88).
8.8 Extrapyramidal side effects: number of participants with rigor (short term)
There was no statistically significant difference (13 RCTs, n = 940, RR 1.01 CI 0.81 to 1.26).
8.9 Extrapyramidal side effects: number of participants with rigor (medium term)
There was no statistically significant difference (1 RCT, n = 57, RR 0.97 CI 0.39 to 2.40).
8.10 Extrapyramidal side effects: number of participants with tardive dyskinesia (short term)
There was no statistically significant difference (2 RCTs, n = 207, RR 0.48 CI 0.06 to 3.57).
8.11 Extrapyramidal side effects: number of participants with tardive dyskinesia (medium term)
There was no statistically significant difference (1 RCT, n = 80, RR not estimable).
8.12 Extrapyramidal side effects: number of participants with tremor (short term)
There was no statistically significant difference (15 RCTs, n = 828, RR 1.00 CI 0.72 to 1.40).
8.13 Extrapyramidal side effects: number of participants with tremor (medium term)
There was no statistically significant difference (1 RCT, n = 57, RR 0.80 CI 0.28 to 2.34).
8.14 Extrapyramidal side effects: number of participants with use of antiparkinson medication (short term)
There was no statistically significant difference (21 RCTs, n = 949, RR 1.04 CI 0.89 to 1.20).
8.15 Number of participants with death (short term)
There was no statistically significant difference (1 RCT, n = 50, RR 0.33 CI 0.01 to 7.81).
8.16 Number of participants with hypotension (short term)
There was no statistically significant difference (14 RCTs, n = 580, RR 1.10 CI 0.31 to 3.91).
8.17 Number of participants with sedation (short term)
There was no statistically significant difference (5 RCTs, n = 306, RR 0.72 CI 0.45 to 1.18).
8.18 Number of participants with weight gain (short term)
There was no statistically significant difference (6 RCTs, n = 262, RR 0.67 CI 0.21 to 2.15). An I² value of 58% accompanied by a statistically significant Chi2 statistic indicated a substantial level of heterogeneity.
8.19 Number of participants with weight gain (medium term)
There was no statistically significant difference (1 RCT, n = 57, RR 0.48 CI 0.05 to 5.03).
9. Sensitivity analyses
All sensitivity analyses were performed only for the primary outcome (clinically important response to treatment).
9.1 Implication of randomisation
In 12 publications randomisation was not mentioned (Abuzzahab 1982; Bueno 1979; Fuentenebro 1989; Haas 1982; Heikkilä 1981; Itoh 1985; Mattke 1976; O´Brien 1974; Stewart 1969; Tobin 1980; Ulmar 1990; White 1981). Because the trials were described as "double‐blind" we implied that the studies were randomised. Removing these studies from the statistical analyses did not alter the meta‐analytic findings in terms of statistically significant differences between groups.
9.2 Exclusion of cross‐over‐trials
Removing the cross‐over trials (Gerlach 1985; Shalev 1993; Stewart 1969) did not change the meta‐analytic results in terms of statistically significant differences between groups.
9.3 Implication of non double‐blind trials
Exclusion of the non double‐blind trials (Baastrup 1993; Nedopil 1981; Parent 1983) and the studies with unclear blinding (Cosar 1999; Shalev 1993; Tuason 1986) did not convert the meta‐analytic findings regarding statistically significant between‐group differences.
9.4 Fixed‐effect versus random‐effects model
Applying a fixed‐effect model instead of a random‐effects approach did alter the meta‐analytic findings in terms of statistically significant differences between groups disfavouring haloperidol (40 RCTs, n = 2132, RR 0.90 CI 0.82 to 0.98).
10. Subgroup analyses
All subgroup analyses were performed only for the primary outcome (clinically important response to treatment).
10.1 Stratification according to the different first‐generation antipsychotics
Concerning the short‐term trials, a statistically significant difference between haloperidol and another antipsychotic drug administered as active comparator to haloperidol was only assessable for nemonapride which was significantly more effective than haloperidol in achieving clinically important response to treatment (1 RCT, n = 167, RR 0.55 CI 0.33 to 0.93). When analysing the studies with medium duration, thiothixene was significantly more effective than haloperidol (1 RCT, n = 80, RR 0.51 CI 0.37 to 0.69).
10.2 Trials investigating participants with treatment‐resistant schizophrenia
Four short‐term trials examined participants with treatment‐resistant schizophrenia (Kinon 1993; Hall 1968; Howard 1974; Teja 1975). There was no statistically significant difference between the pooled study groups of these trials in terms of the primary outcome (3 RCTs, n = 130, RR 1.46 CI 0.66 to 3.23). We could not detect any significant difference between the studies that investigated treatment‐resistant participants and the remaining trials (test for subgroup differences: P = 0.26).
10.3 Trials investigating participants with a first episode of schizophrenia
No study described the major inclusion criteria of participants with a first‐episode of schizophrenia. Therefore, this a priori planned subgroup analysis could not be conducted.
11. Assessment of heterogeneity
Statistical tests revealed substantial levels of heterogeneity (defined by an I2 greater than or equal to 50% accompanied by a statistically significant Chi2 test) for the outcomes "mental state general: mean BPRS total score at endpoint (short‐trial duration)", "adverse effects: number of participants with at least one adverse effect (medium‐trial duration)", and "adverse effects: number of participants with weight gain (short‐trial duration)". Possible reasons for the occurrence of substantial heterogeneity in the three outcomes are described in the context of the discussion.
12. Publication Bias
In the funnel plot the risk ratios of the studies are plotted against the standard errors (referring to the primary outcome) (Figure 4). Based on the largely symmetrical arrangement of the single trials around the pooled effect size as equivalence line, there is no evidence for the presence of publication bias.
4.
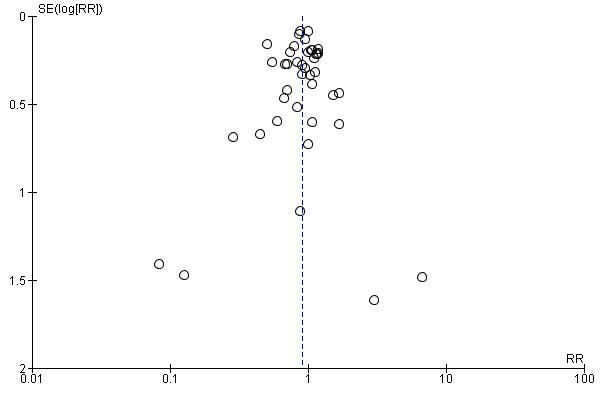
Funnel plot of comparison: 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, outcome: 1.1 Global state: 1. Clinically important response.
13. 'Summary of findings' table
The results of six outcomes ‐ "clinically important response to treatment", "leaving the study early due to any reason", "leaving the study early due to inefficacy", "leaving the study early due to adverse effects", "number of participants with at least one adverse effect", and "number of participants with at least one movement disorder" ‐ were considered more closely in a 'Summary of findings' table (see Table 1 for the studies with short duration and Table 2 for the trials with medium duration). The judgements derived from this instrument were taken into account for the discussion section of this systematic review. In none of the included studies did we find data for the predefined outcome "quality of life". This might be caused by the fact that the single original trials were conducted a number of years ago (published between 1962 and 1993). At that time, there was a stronger focus primarily on efficacy‐related outcomes, whereas in the last few years, attention has been directed more intensively to outcomes such as "quality of life".
Discussion
Summary of main results
1. General
The present systematic review includes 63 randomised controlled trials (involving 3675 participants) that compared haloperidol with another first‐generation antipsychotic drug (with the exception of the low‐potency antipsychotics chlorpromazine, chlorprothixene, levopromazine, mesoridazine, perazine, prochlorpromazine, and thioridazine that have been examined in another Cochrane Review (Tardy 2011a). Despite heterogeneity regarding the trial designs employed in the single original studies and the fact that the predefined outcomes were often incompletely reported, the results of the meta‐analytic calculations demonstrated consistently that there is no statistically significant difference between haloperidol and the other first‐generation antipsychotics in terms of overall efficacy, acceptability, and tolerability. As it is unfortunately typical for trials determining conventional antipsychotics in schizophrenia, the methods of randomisation, allocation concealment, and blinding were frequently not reported.
2. Clinically important response to treatment
Based on the data of 40 short‐term trials, we could not demonstrate any statistically significant difference in the achievement of clinically important response to treatment between haloperidol and the pooled group of the other first‐generation antipsychotic agents. In this context it must be considered that statistically significant difference in disfavour of haloperidol occurred in the sensitivity analysis applying a fixed‐effect model instead of a random‐effects approach. Investigating the studies with medium‐trial duration, the group of the other first‐generation antipsychotics was statistically significantly more effective than haloperidol in achieving treatment response. Because evidence for this outcome is based on only one trial (Engelhardt 1978), no empirical conclusions can be stated.
3. Global state
There was no statistically significant difference between haloperidol and the other first‐generation antipsychotic drugs assessing the mean CGI at endpoint. The analysis was limited by the fact that the standard deviations had to be imputed for three of four trials that contributed data to this outcome.
4. Mental state
Results regarding the participants' mental state were rarely reported and difficult to interpret, because different scales were applied. The widely used rating scale was the BPRS. Analysing the mean BPRS total score at endpoint, we did not detect any statistically significant between‐group differences. However, the results were highly heterogenous for the short‐term studies (I² = 97%) and for 12 of 23 trials standard deviations had to be imputed from the mean of the other studies. No statistically significant differences could be identified for the Montgomery Rating Score (MRS) and the Rating Scale for Quantification of Psychotic Symptom Severity (RSQPSS) score.
Regarding the specific mental state, a statistically significant superiority of haloperidol in reducing positive symptoms (measured by the SAPS) but not negative symptoms (SANS) could be verified. These findings were based on only one trial (Mauri 1994). There was no statistically significant difference between haloperidol and the other first‐generation antipsychotic drugs in terms of improving depressive symptoms (HAM‐D). Two trials evaluated schizophrenic symptoms by using the ADMP system (Giordana 1984; Nedopil 1981). The assessments revealed statistically significant difference in disfavour of haloperidol in terms of delusional symptoms, depersonalisation, and conceptual disorganisation.
5. Behaviour
Neither the assessment of the NOSIE score nor the Wing`s Ward Behaviour Scale showed any significant superiority of any study group. The analyses were limited by the fact that only four trials reported usable data on the behavioural state and three of them provided no standard deviations avoiding the calculation of the mean difference as effect size.
6. Leaving the study early
Altogether, 343 out of 1436 participants (24 %) terminated the studies prematurely due to any reason without any statistically significant difference between haloperidol and the pooled group of the other first‐generation antipsychotics. This result was consistent irrespectively of the trial duration (short‐ or medium‐trial duration). When specific reasons for leaving the study early, adverse effects or inefficacy of treatment, were indicated, there was again no significant difference between the study groups. Therefore, these findings suggest a comparable overall acceptability, overall efficacy, and overall tolerability of haloperidol and the various other first‐generation antipsychotics investigated in this systematic review.
7. Adverse effects
There was no statistically significant difference between haloperidol and the other first‐generation antipsychotic drugs, neither in terms of "at least one adverse effect", "at least one extrapyramidal/movement disorder", nor in terms of the specific adverse effects akathisia, akinesia, dyskinesia, dystonia, rigor, tardive dyskinesia, tremor, use of antiparkinson medication, death, hypotension, sedation, and weight gain. There was only one exception: significantly more participants in the other first‐generation antipsychotic group than in the haloperidol group experienced akathisia when analysing only the studies with medium‐trial duration. Because evidence for this outcome is based on only one trial, no empirical meta‐analytic conclusions can be stated. The incomplete reporting of adverse effects considerably limits the interpretation of these findings. Many comparisons for the single specific side effects were based on only one or two individual trials (for example, akathisia, akinesia, rigor, tardive dyskinesia, and death). Especially, there were not enough data available to appraise the risk profile of the pharmacotherapy in the long‐term treatment. In the same way, we found no data investigating adherence to the pharmacological treatment with the evaluated antipsychotic drugs. Future research projects are needed to assess this aspect of pharmacological treatment.
8. Missing outcomes
No included trial provided data that measured the a priori defined outcomes "satisfaction with care", "quality of life", "service utilisation", "functioning", and economic outcomes. Further clinical trials should consider these outcomes to allow an overall appraisal of a benefit‐risk‐ratio of a psychopharmacological intervention.
9. Subgroup analyses
Stratification according to the different antipsychotic drugs administered as comparator to haloperidol revealed that only a medication with nemonapride yielded a significantly higher effect size than haloperidol in terms of the primary outcome when analysing the short‐term trials. Regarding the studies with medium‐trial duration, thiothixene was statistically significantly superior to haloperidol. Because each of both findings is based on only one trial, no treatment recommendations can be deduced from the result of the meta‐analytic calculations.
10. Sensitivity analyses
The findings of the primary outcome did alter statistically significantly in disfavour of haloperidol if a fixed‐effect model instead of a random‐effects approach was applied but the direction of the effect was still the same. Thus, the interpretations of the results are not challenged. Exclusion of the studies with implied randomisation, non double‐blind mechanism, and cross‐over trial design did not convert the meta‐analytic findings in terms of statistically significant between‐group differences.
11. Heterogeneity
Substantial levels of heterogeneity were identified regarding the outcomes "mental state general: mean BPRS total score at endpoint (short term)" (I² = 97%), "adverse effects: number of participants with at least one adverse effect (medium term)" (I² = 86%), and "adverse effects: number of participants with weight gain (short term)" (I² = 58%). Concerning the continuous outcome "mental state general: mean BPRS total score at endpoint (short term)", the standard deviations had to be imputed for 12 of 23 trials. These imputations probably biased the pooled analysis and can be regarded as possible explanation for the high heterogeneity. As the number of trials (n = 2) and participants (n = 137) is still low for the outcome "adverse effects: number of participants with at least one adverse effect (medium term)", a part of this heterogeneity may be caused by this issue alone. In terms of the outcome "adverse effects: number of participants with weight gain (short term)", the difference between the various first‐generation antipsychotic drugs administered in the control groups of the included trials could be a possible reason for the substantial level of heterogeneity.
Overall completeness and applicability of evidence
The 63 included studies were carried out in various settings (e.g. in‐ and outpatients), with different populations (e.g. participants in remission, participants with an acute schizophrenic episode, treatment‐resistant participants) and methods (e.g. outcome variables, diagnostic instruments). Because of this clinical and methodological diversity, we believe that the evidence is quite complete and applicable to routine care. Other limitations regarding this systematic review must be considered, for example, the trials were usually characterised by small sample sizes (medium sample size: 58 participants) and the outcomes were often incompletely reported thus not allowing their inclusion in the meta‐analytic calculations. In many cases missing standard deviations had to be imputed from the mean of the other studies. Because of variable trial designs and application of different outcome scales, pooling of the results was often impossible. Therefore, many comparisons were underpowered. One reason for the methodological diversity is probably that the studies were carried out during a long period of time (from 1962 to 1993).
Quality of the evidence
All included trials were randomised and most of them were described as being double‐blind but details were often not presented. Therefore, it appears unclear whether the studies were adequately randomised, whether treatment allocation was really concealed, and whether sufficient blinding could be assured over the whole trial period. Only in 18 out of 63 studies was the overall degree of attrition classified as being low (< 10%). Forty‐six of the 63 included trials were characterised by selective reporting, especially in terms of standard deviations. In 33 trials potential sources of bias such as extreme baseline imbalances etc. occurred. In summary, the overall quality of the studies according to these criteria can be regarded as low.
Potential biases in the review process
In general, it must be considered that meta‐analyses combine similar but not identical trials. The heterogeneous designs of the original studies (see Overall completeness and applicability of evidence) remain a critical issue concerning the interpretation of meta‐analytic findings. In this systematic review, we a priori decided to pool all first‐generation antipsychotics that were used as comparator drugs to haloperidol. This is justified for efficacy‐related outcomes because most antipsychotic compounds do not differ in efficacy and if differences exist between some antipsychotic drugs, these are not considerable (Davis 1978; Klein 1969; Leucht 2009). The decision to pool all studies irrespectively of the antipsychotic drug administered as comparator to haloperidol is more problematic for adverse effects because antipsychotics differ to a large extent in this regard. For example, the so‐called mid‐potency antipsychotic agents (described by a lower affinity to dopamine‐D2‐receptors than high‐potency antipsychotics) are typically characterised by more anticholinergic (e.g. dry mouth and obstipation), antiadrenergic (e.g. orthostatic dysregulation), and antihistaminic (e.g. sedation and weight gain) adverse effects compared to the so‐called high‐potency antipsychotics, which are usually associated with a higher number of movement disorders than low‐potency antipsychotics. Thus, any differences in a specific adverse effect between haloperidol and the pooled study group of other first‐generation antipsychotics cannot be generalised to all first‐generation antipsychotic drugs.
The study search was mainly based on the trial register of the Cochrane Schizophrenia Group, which primarily contains published literature. It is possible that unpublished studies we are not aware of exist. Therefore, a possibility of publication bias is present although the symmetrical funnel plot did not provide any evidence of the presence of publication bias.
We applied a random‐effects model for our meta‐analytic calculations to consider variability between the included studies (Huf 2011). The random‐effects approach does not assume that the populations from which the different trials are derived are comparable. This technique emphasizes the results from smaller trials and it is these studies that are likely to be most prone to bias. In this context, it must be taken into account that applying a fixed‐effect model instead of a random‐effects approach in a sensitivity analysis of the primary outcome generated statistically significant between‐group differences in disfavour of haloperidol without any alteration of the direction of the effect.
A major limitation of our meta‐analysis in terms of the methodology of the included original trials is that haloperidol was evaluated as active comparator drug in most of the studies. Therefore, the other first‐generation antipsychotics that were the control group in our systematic review were the drugs of interest in the original trials. Within this context it must be referred to the possibility of an industry sponsorship bias.
Agreements and disagreements with other studies or reviews
In the present systematic review, we determined the efficacy, acceptability, and tolerability of haloperidol compared with other first‐generation antipsychotics with the exception of the low‐potency antipsychotics chlorpromazine, chlorprothixene, levopromazine, mesoridazine, perazine, prochlorpromazine, and thioridazine. We did not examine these compounds because they have been evaluated in other Cochrane Reviews (Leucht 2008; Tardy 2011a). Similar to our findings, there were also no statistically significant differences between haloperidol and the investigated conventional antipsychotics.
Our main results are also in accordance with the findings of previous narrative, unsystematic reviews of Davis 1978 and Klein 1969 suggesting that there are no differences in terms of efficacy between the various first‐generation antipsychotic drugs. Applying a systematic approach and clearly better methodology, we could verify these findings in terms of the standard reference antipsychotic drug haloperidol that is characterised by comparable efficacy with that of the other first‐generation antipsychotics
Authors' conclusions
Implications for practice.
1. For clinicians
Based on the currently available randomised trial‐evidence, we found that haloperidol and the other evaluated first‐generation antipsychotic agents are characterised by a comparable efficacy for the treatment of schizophrenia and/or other psychotic disorders. Additionally, our results suggest a similar acceptability and tolerability of haloperidol and the other conventional antipsychotic drugs. However, the low methodological quality of many included studies probably confining the findings of our review should be taken into account.
2. For people with schizophrenia
People with schizophrenia should know that ‐ in terms of first‐generation antipsychotics ‐ not only haloperidol is effective for the pharmacological treatment of schizophrenia and/or other psychotic disorders. The application of other first‐generation antipsychotics than haloperidol can also improve schizophrenic symptoms to a comparable extent. They are generally associated with the occurrence of a comparable number of adverse effects to that of haloperidol. In our statistics, the only clear difference in terms of side effects was that haloperidol produced less akathisia in the medium‐term trials. However, it should be taken into account as a limitation of this review that many included individual studies were characterised by a low methodological quality. Therefore, our results should be interpreted very carefully. More research is needed to appraise differences in efficacy and tolerability between haloperidol and the other first‐generation antipsychotics more accurately. Therefore, future clinical trials with a higher methodological quality are required.
3. For managers and policy makers
The efficacy of other first‐generation antipsychotic drugs than haloperidol in schizophrenia and/or other psychotic disorders is supported by this systematic review.
Implications for research.
1. General
Any future trials should consider standards of measuring outcomes and of reporting data in order to enhance the comparability of the results of clinical trials. Therefore, strict adherence to the CONSORT statement (Schulz 2010) should be ensured in future research projects. Following the CONSORT statement very closely would help to increase the amount of data for further reviews on this topic considerably.
2. Specific
2.1 Reviews
Table 4, derived from the studies excluded for this review (please refer to Characteristics of excluded studies for details), contains suggestions for the elaboration of further reviews and for further comparisons probably relevant to reviews already existing.
2. Excluded studies and suggestions for relevant reviews.
| Excluded study | Comparison | Existing review |
| Al Haddad 1996, Chin 1998, Lamure 2003, Lublin 1991, Mosolov 2000, Taymeeyapradit 2002 | Haloperidol versus zuclopenthixol | Jayakody 2012 |
| Azima 1960, Crow 1986, Durost 1964, Simpson 1967 | Haloperidol versus placebo | Adams 2013 |
| Bechelli 1986 | Haloperidol versus pipothiazine palmitate | ‐ |
| Boyer 1987 | Amisulpride versus placebo // amisulpride dosage // amisulpride versus fluphenazine // amisulpride versus haloperidol | Silveira 2002; Sampford 2013 |
| Cole 1970, Gerlach 1978 | Haloperidol versus thioridazine | ‐ |
| Crow 1986 | Chlorpromazine versus placebo // flupenthixol versus placebo // pimozide versus placebo // trifluoperazine versus placebo | Adams 2014, Shen 2012, Mothi 2013, Koch 2014 |
| Costa 2007, de Jesus Mari 2004 | Antipsychotic drug versus olanzapine | Duggan 2005 |
| Fitzgerald 1969 | Haloperidol versus perphenazine | Hartung 2005 |
| Kelwala 1984, Levenson 1976, Stotsky 1977 | Haloperidol versus thiothixene | ‐ |
| Levenson 1976 | Fluphenazine versus haloperidol // fluphenazine versus thiothixene | ‐ |
| Lovett 1987 | Haloperidol versus trifluoperazine | ‐ |
| Paprocki 1977 | Haloperidol versus loxapine | Chakrabarti 2007 |
| Reznik 2000 | Fluvoxamine plus antipsychotic drug versus antipsychotic drug | ‐ |
| Saletu 1986 | Fluperlapine versus haloperidol | ‐ |
2.2 Trials
Although there were many randomised controlled trials carried out that compared haloperidol with other first‐generation antipsychotics, there is a need for more well‐designed randomised clinical studies. They are necessary because the methodological quality of the individual trials included in this review was low in many studies. Therefore, high‐quality trials are required to appraise the efficacy and tolerability of haloperidol in comparison to other first‐generation antipsychotics. For a suggested design of these studies please see Table 5.
3. Suggested design of study.
| Methods | Allocation: randomised, fully explicit description of methods of randomisation and allocation concealment. Blinding: blinded and independent raters. Duration: at least 52 weeks. |
| Participants | Diagnosis: schizophrenia (according to a diagnostic criteria). N = 300.* Age: adults. Sex: both. |
| Interventions | 1. Haloperidol. N = 150. 2. Other first‐generation antipsychotic. N = 150. |
| Outcomes | Global state: clinically important response to treatment, average score/change of the global state. General: time to all‐cause treatment failure marked by its discontinuation, relapse, general impression of clinician (CGI), carer/other, compliance with treatment, healthy days. Mental state: general measurement and specific domains (depressive symptoms, positive symptoms, negative symptoms) Leaving the study early (‘drop‐out’) due to any reason, due to inefficacy of treatment, and due to adverse events. Adverse events: any serious adverse event recorded. Service use: number of hospitalisation, days in hospital. Quality of life. Social functioning: return to everyday living for 80% of time.* Economic outcomes. Pharmacological interactions. |
| Notes | * Powered to be able to identify a difference of ˜ 20% between groups for primary outcome with adequate degree of certainty. |
CGI = Clinical Global Impression.
Acknowledgements
We thank the editorial team of the Cochrane Schizophrenia Group for its support. The Group produces and maintains standard text for use in the methods sections of their reviews. We have used this text as the basis and adapted it as required. We would like to thank Professor Nedopil for providing further information on his trials and Selim Asmer for peer reviewing this version.
Data and analyses
Comparison 1. HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Clinically important response | 41 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 short term | 40 | 2132 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.00] |
| 1.2 medium term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.37, 0.69] |
| 2 Global state: 2. Average score (CGI, endpoint, short term, high = poor) | 4 | 151 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.39, 0.25] |
| 3 Mental state: 1. General ‐ a. Average score (BPRS total, endpoint, high = poor) | 24 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 short term | 23 | 998 | Mean Difference (IV, Random, 95% CI) | 0.37 [‐1.66, 2.39] |
| 3.2 medium | 1 | 26 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Mental state: 1. General ‐ b. Average score ‐ short term (various scales, endpoint, high = poor) | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 MRS | 1 | 18 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 RSQPSS (at 60 days) | 1 | 12 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.04, 0.04] |
| 5 Mental state: 2. Specific ‐ a. Depersonalisation average score ‐ short term (AMDP, high = poor) | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 1.30 [0.62, 1.98] |
| 6 Mental state: 2. Specific ‐ b. Depressive symptoms average score (HAM‐D, high = poor) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 short term | 2 | 62 | Mean Difference (IV, Random, 95% CI) | ‐0.46 [‐1.23, 0.32] |
| 6.2 medium term | 1 | 44 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Mental state: 2. Specific ‐ c. Negative symptoms average score ‐ short term (SANS, endpoint, high = poor) | 1 | 40 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐2.13, 2.73] |
| 8 Mental state: 2. Specific ‐ d.i. Positive symptom average score ‐ short term | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 conceptual disorganisation (AMDP, endpoint, high = poor) | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 3.00 [2.07, 3.93] |
| 8.2 delusional symptoms (AMDP, endpoint, high = poor) | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 3.30 [2.13, 4.47] |
| 8.3 positive symptoms (SAPS, endpoint, high = poor) | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐14.7 [‐17.42, ‐11.98] |
| 9 Mental state: 2. Specific ‐ d.ii. Positive symptoms average score ‐ short term (AMDP, skewed or incomplete data) | Other data | No numeric data | ||
| 9.1 hallucinatory symptoms | Other data | No numeric data | ||
| 9.2 paranoid symptoms | Other data | No numeric data | ||
| 10 Behaviour: 1a. Average score ‐ short term (NOSIE, endpoint, high = poor) | 3 | 100 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Behaviour: 1b. Average score ‐ short term (Wing’s Ward Behaviour Scale, endpoint, high = poor) | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.27 [‐4.51, 1.97] |
| 12 Leaving the study early: 1. Due to any reason ‐ as a measure of overall acceptability | 30 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 short term | 28 | 1299 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.87, 1.24] |
| 12.2 medium term | 2 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.75, 1.38] |
| 13 Leaving the study early: 2. Due to inefficacy of treatment ‐ as a measure of overall efficacy (short term) | 13 | 507 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.40, 2.16] |
| 14 Leaving the study early: 3. Due to adverse events ‐ as a measure of overall tolerability | 17 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 short term | 16 | 640 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.42, 2.35] |
| 14.2 medium term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Adverse effects: 1. General ‐ at least one adverse effect | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 short term | 10 | 693 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.94, 1.20] |
| 15.2 medium term | 2 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.58, 1.97] |
| 16 Adverse effects: 2. Specific ‐ a. Movement disorders | 45 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 akathisia ‐ short term | 22 | 1648 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.89, 1.24] |
| 16.2 akathisia ‐ medium term | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.16, 0.60] |
| 16.3 akinesia ‐ short term | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.31, 2.68] |
| 16.4 at least one extrapyramidal/movement disorder ‐ short term | 17 | 998 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.95, 1.31] |
| 16.5 at least one extrapyramidal/movement disorder ‐ medium term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.62, 1.75] |
| 16.6 dyskinesia ‐ short term | 11 | 807 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.48, 1.35] |
| 16.7 dystonia ‐ short term | 15 | 1035 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.95, 1.88] |
| 16.8 rigor ‐ short term | 13 | 940 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.81, 1.26] |
| 16.9 rigor ‐ medium term | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.39, 2.40] |
| 16.10 tardive dyskinesia ‐ short term | 2 | 207 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.06, 3.57] |
| 16.11 tardive dyskinesia ‐ medium term | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 16.12 tremor ‐ short term | 15 | 828 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.72, 1.40] |
| 16.13 tremor ‐ medium term | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.28, 2.34] |
| 16.14 use of antiparkinson medication ‐ short term | 21 | 949 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.89, 1.20] |
| 17 Adverse effects: 2. Specific ‐ b. Various | 21 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 death ‐ short term | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.81] |
| 17.2 hypotension ‐ short term | 14 | 580 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.31, 3.91] |
| 17.3 sedation ‐ short term | 5 | 306 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.45, 1.18] |
| 17.4 weight gain ‐ short term | 6 | 262 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.21, 2.15] |
| 17.5 weight gain ‐ medium term | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.05, 5.03] |
1.1. Analysis.
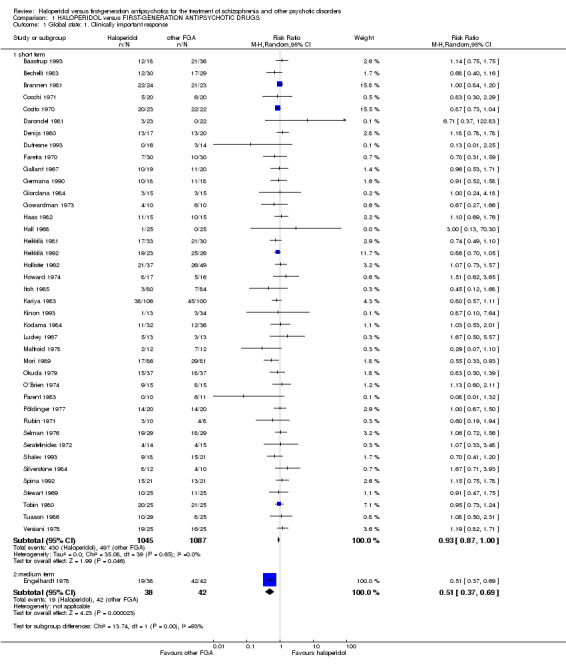
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 1 Global state: 1. Clinically important response.
1.2. Analysis.
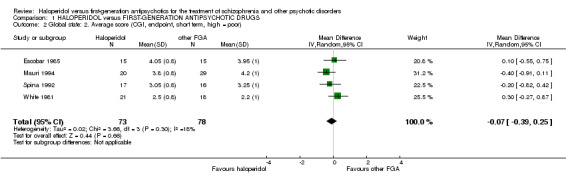
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 2 Global state: 2. Average score (CGI, endpoint, short term, high = poor).
1.3. Analysis.
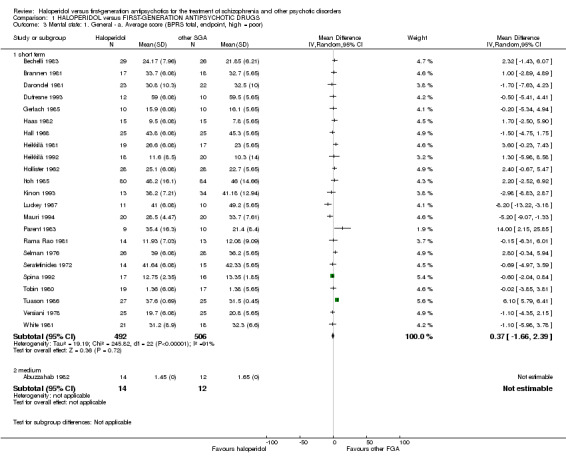
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 3 Mental state: 1. General ‐ a. Average score (BPRS total, endpoint, high = poor).
1.4. Analysis.
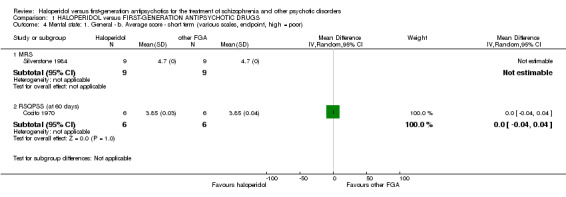
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 4 Mental state: 1. General ‐ b. Average score ‐ short term (various scales, endpoint, high = poor).
1.5. Analysis.
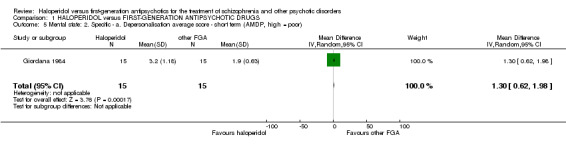
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 5 Mental state: 2. Specific ‐ a. Depersonalisation average score ‐ short term (AMDP, high = poor).
1.6. Analysis.
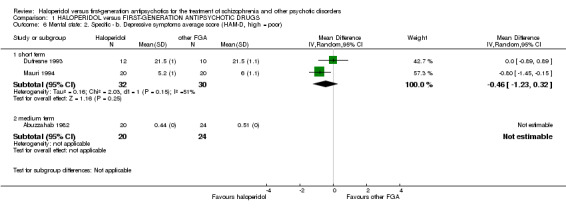
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 6 Mental state: 2. Specific ‐ b. Depressive symptoms average score (HAM‐D, high = poor).
1.7. Analysis.
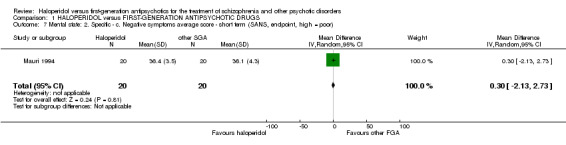
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 7 Mental state: 2. Specific ‐ c. Negative symptoms average score ‐ short term (SANS, endpoint, high = poor).
1.8. Analysis.
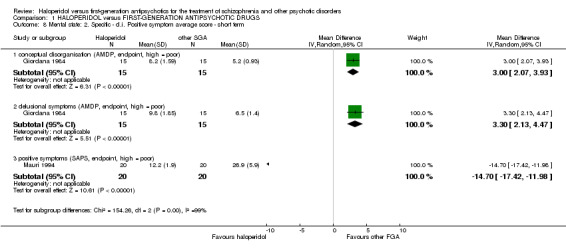
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 8 Mental state: 2. Specific ‐ d.i. Positive symptom average score ‐ short term.
1.9. Analysis.
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 9 Mental state: 2. Specific ‐ d.ii. Positive symptoms average score ‐ short term (AMDP, skewed or incomplete data).
| Mental state: 2. Specific ‐ d.ii. Positive symptoms average score ‐ short term (AMDP, skewed or incomplete data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | Number of participants (n) |
| hallucinatory symptoms | ||||
| Giordana 1984 | Haloperidol | 1.3 | not indicated | 15 |
| Giordana 1984 | other FGA | 1.3 | not indicated | 15 |
| Nedopil 1981 | Haloperidol | 1.05 | 2.15 | 14 |
| Nedopil 1981 | other FGA | 1.69 | 2.37 | 19 |
| paranoid symptoms | ||||
| Nedopil 1981 | Haloperidol | 3.25 | 3.5 | 14 |
| Nedopil 1981 | other FGA | 3.45 | 4.07 | 19 |
1.10. Analysis.

Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 10 Behaviour: 1a. Average score ‐ short term (NOSIE, endpoint, high = poor).
1.11. Analysis.
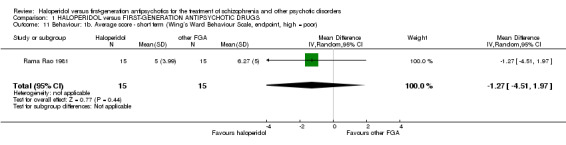
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 11 Behaviour: 1b. Average score ‐ short term (Wing’s Ward Behaviour Scale, endpoint, high = poor).
1.12. Analysis.
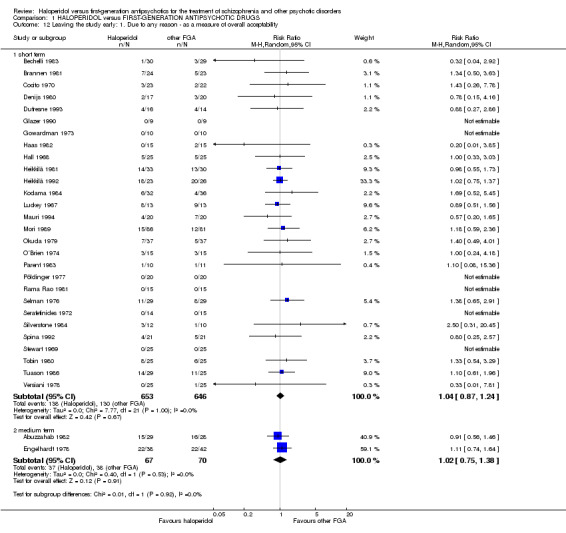
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 12 Leaving the study early: 1. Due to any reason ‐ as a measure of overall acceptability.
1.13. Analysis.
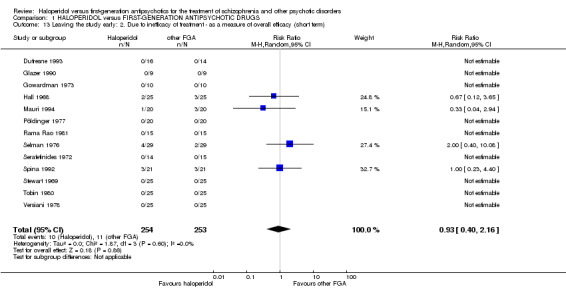
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 13 Leaving the study early: 2. Due to inefficacy of treatment ‐ as a measure of overall efficacy (short term).
1.14. Analysis.
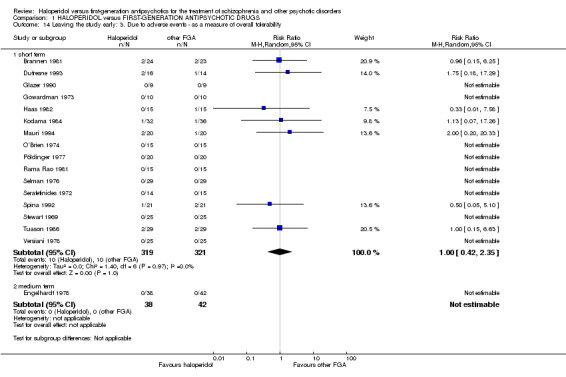
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 14 Leaving the study early: 3. Due to adverse events ‐ as a measure of overall tolerability.
1.15. Analysis.
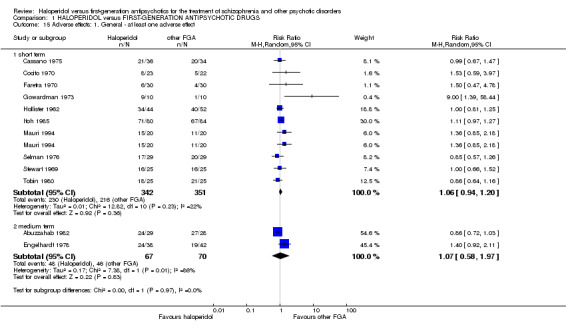
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 15 Adverse effects: 1. General ‐ at least one adverse effect.
1.16. Analysis.
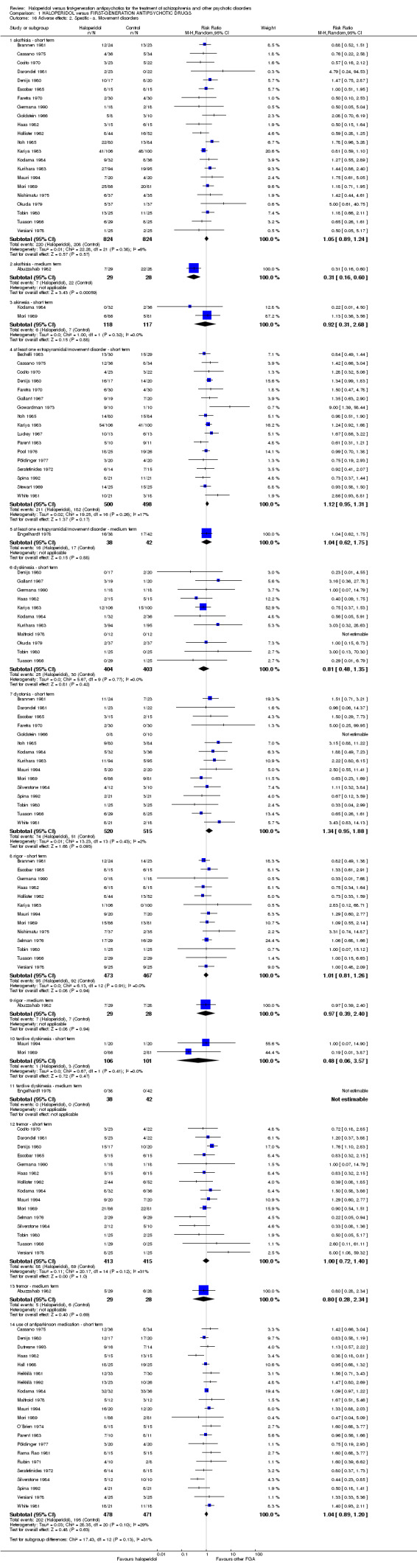
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 16 Adverse effects: 2. Specific ‐ a. Movement disorders.
1.17. Analysis.
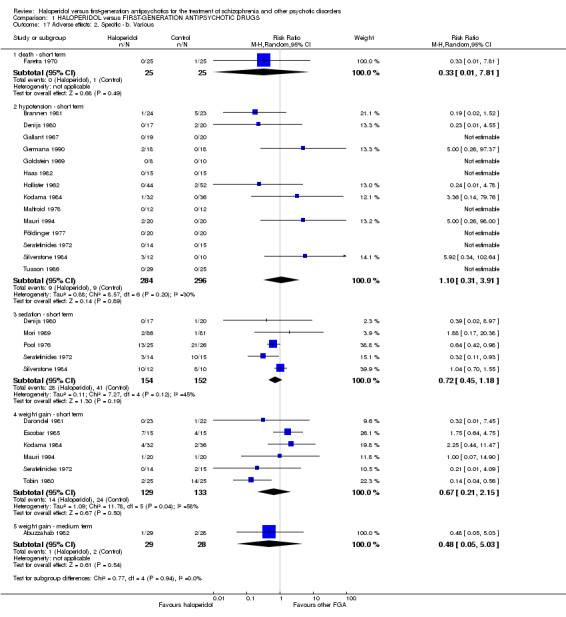
Comparison 1 HALOPERIDOL versus FIRST‐GENERATION ANTIPSYCHOTIC DRUGS, Outcome 17 Adverse effects: 2. Specific ‐ b. Various.
Comparison 2. Sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Sensitivity analysis ‐ Implication of randomisation, Outcome: overall symptoms of schizophrenia (short term) | 34 | 1745 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.01] |
| 2 Sensitivity analysis ‐ Exclusion of cross‐over‐trials, Outcome: overall symptoms of schizophrenia (short term) | 37 | 1989 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.87, 1.00] |
| 3 Sensitivity analysis ‐ Exclusion of non double‐blind trials, Outcome: overall symptoms of schizophrenia (short term) | 37 | 2018 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.00] |
| 4 Sensitivity analysis ‐ Fixed versus random‐effects models, Outcome: overall symptoms of schizophrenia (short term) | 40 | 2132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.82, 0.98] |
2.1. Analysis.
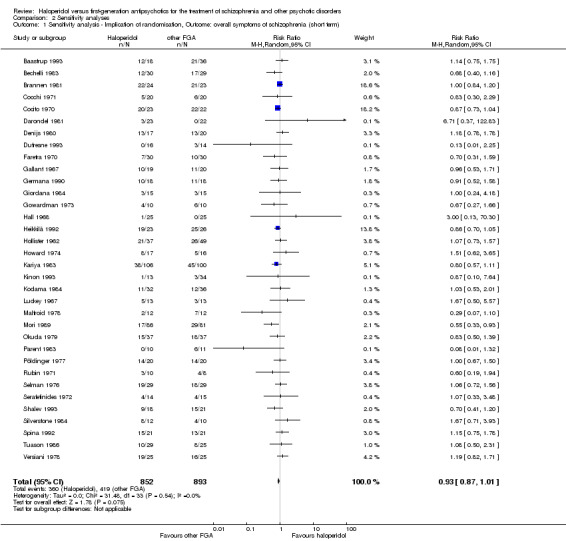
Comparison 2 Sensitivity analyses, Outcome 1 Sensitivity analysis ‐ Implication of randomisation, Outcome: overall symptoms of schizophrenia (short term).
2.2. Analysis.
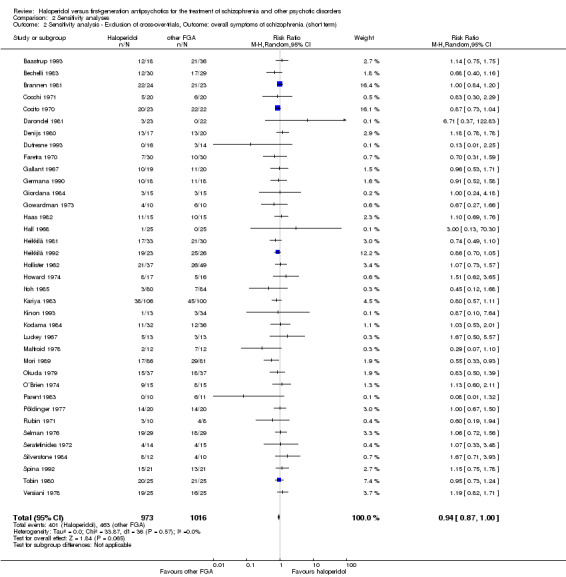
Comparison 2 Sensitivity analyses, Outcome 2 Sensitivity analysis ‐ Exclusion of cross‐over‐trials, Outcome: overall symptoms of schizophrenia (short term).
2.3. Analysis.
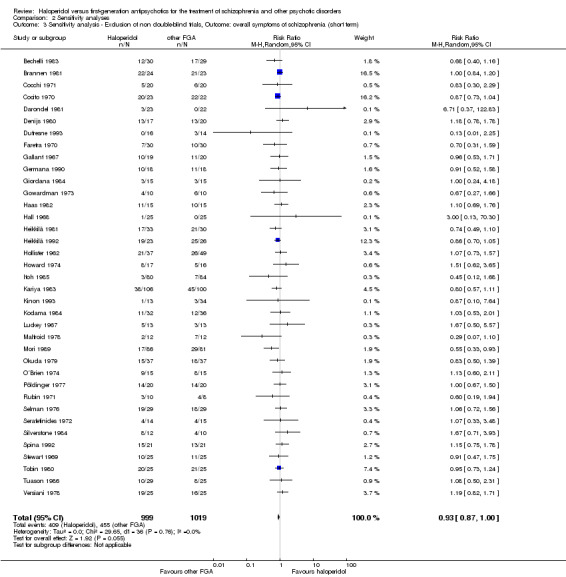
Comparison 2 Sensitivity analyses, Outcome 3 Sensitivity analysis ‐ Exclusion of non double‐blind trials, Outcome: overall symptoms of schizophrenia (short term).
2.4. Analysis.
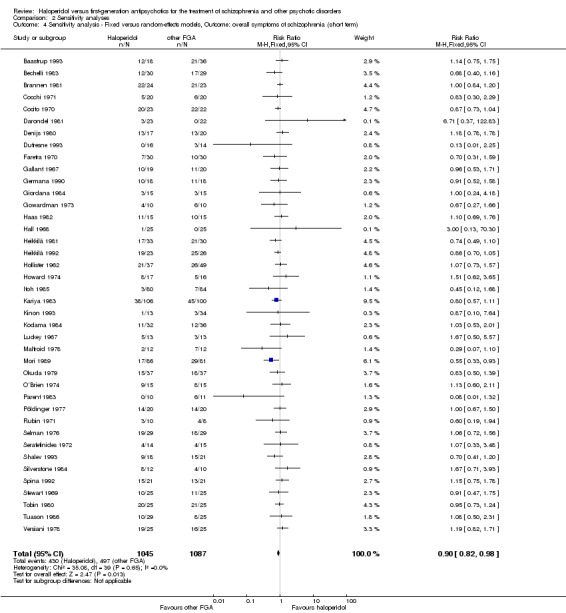
Comparison 2 Sensitivity analyses, Outcome 4 Sensitivity analysis ‐ Fixed versus random‐effects models, Outcome: overall symptoms of schizophrenia (short term).
Comparison 3. Subgroup analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Subgroup analysis ‐ different antipsychotic drugs (short term) | 40 | 2132 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.00] |
| 1.1 Bromperidol | 8 | 458 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.88, 1.15] |
| 1.2 Clopenthixol | 2 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.52, 1.12] |
| 1.3 Droperidol | 2 | 85 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.73, 1.04] |
| 1.4 Flupenthixol | 1 | 21 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.01, 1.32] |
| 1.5 Fluphenazine | 3 | 157 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.37, 1.65] |
| 1.6 Loxapine | 3 | 162 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.87, 1.44] |
| 1.7 Molindone | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.25] |
| 1.8 Nemonapride | 1 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.33, 0.93] |
| 1.9 Perphenazine | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.7 [0.41, 1.20] |
| 1.10 Pimozide | 3 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.74, 1.61] |
| 1.11 Pipotiazine | 3 | 134 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.39, 2.02] |
| 1.12 Sulpiride | 1 | 74 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.50, 1.39] |
| 1.13 Tiopropazate | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.73, 1.57] |
| 1.14 Thiothixene | 2 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.70, 1.52] |
| 1.15 Timiperone | 1 | 206 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.57, 1.11] |
| 1.16 Trifluoperazine | 4 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.68, 1.50] |
| 1.17 Trifluperidol | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.53, 1.71] |
| 1.18 Zuclopenthixol | 2 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.70, 1.28] |
| 2 Subgroup analysis ‐ different antipsychotic drugs (medium term) | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.37, 0.69] |
| 2.1 Thiothixene | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.37, 0.69] |
| 3 Subgroup analysis ‐ treatment‐resistant participants (short term) | 40 | 2132 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.00] |
| 3.1 Trials with treatment‐resistant participants | 3 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.66, 3.23] |
| 3.2 Trials without treatment‐resistant participants | 37 | 2002 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.87, 1.00] |
3.1. Analysis.
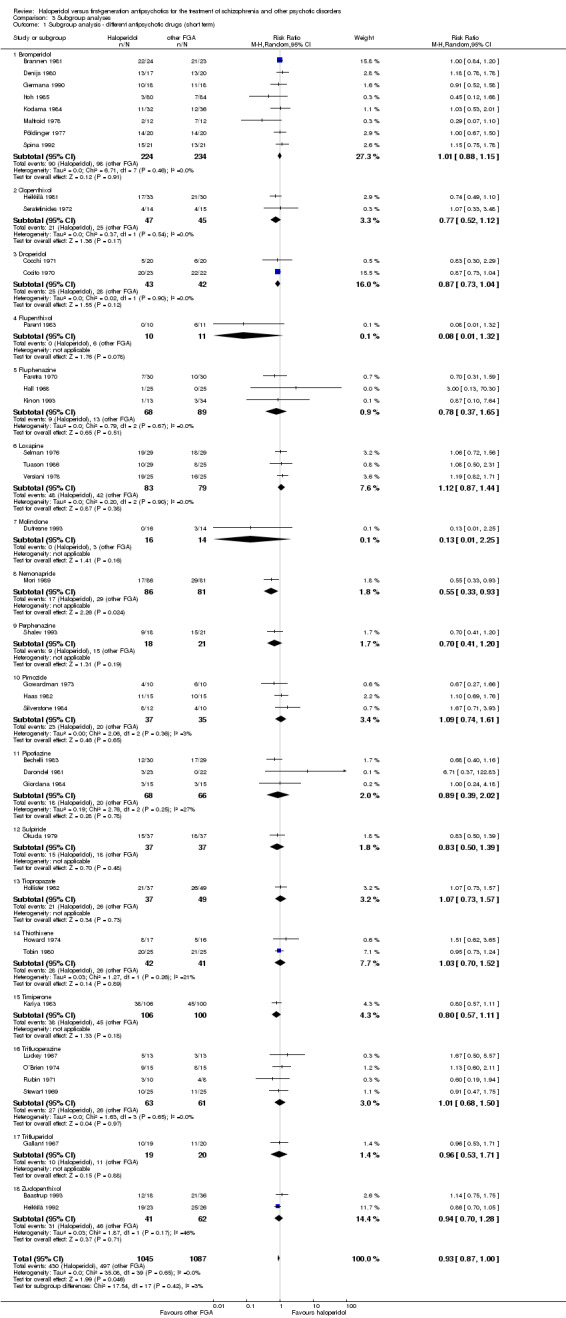
Comparison 3 Subgroup analyses, Outcome 1 Subgroup analysis ‐ different antipsychotic drugs (short term).
3.2. Analysis.
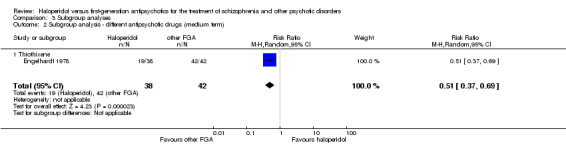
Comparison 3 Subgroup analyses, Outcome 2 Subgroup analysis ‐ different antipsychotic drugs (medium term).
3.3. Analysis.
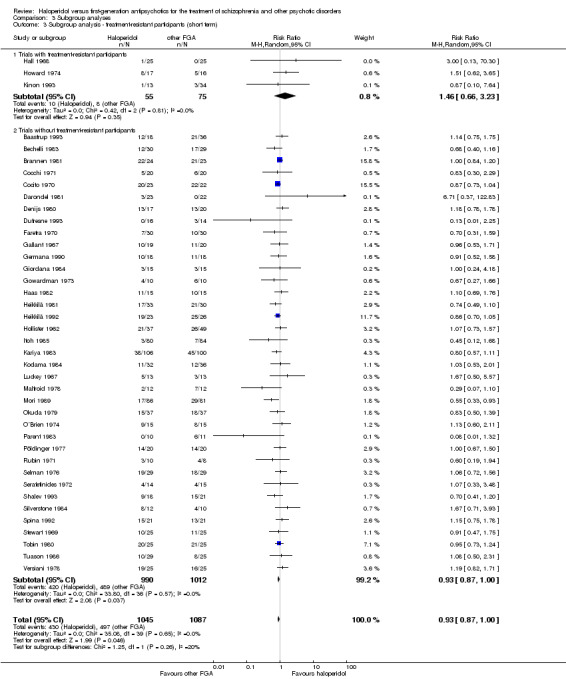
Comparison 3 Subgroup analyses, Outcome 3 Subgroup analysis ‐ treatment‐resistant participants (short term).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Abuzzahab 1982.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 24 weeks. Design: parallel. Location: not indicated. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia. N = 57. Gender (N = 46): 20M, 16F. Age: mean 35.5 years. History: duration stable: not indicated, duration of illness: mean 9 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 5 mg to 40 mg/day, mean dose: 17.5 mg/day. N = 22. 2. Thiothixene: fixed/flexible dose: not indicated, dose range: 10 mg to 80 mg/day, mean dose: 31.8 mg/day. N = 24. Rescue medication: “Antiparkinsonian drugs were permitted” |
|
| Outcomes | Examined: Leaving the study early due to any reason. Adverse effects: at least one adverse effect, akathisia, rigor, tremor, weight gain. Unable to use: Clinically important response to treatment: Investigator global evaluation (no raw data available). Global state general: Evaluation of Social Functioning (ESFR) (no total score available, no SDs available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available and no imputation method could be applied). Mental state specific: Hamilton Psychiatric Rating Scale for depression (HPRSD) (no SDs available and no imputation method could be applied). Mental state specific: ZUNG scale (self‐rating depression scale) (no raw data available). Behaviour: Nurses´ Observation Scale for Inpatient Evaluation (NOSIE) (mentioned in the abstract, but not in the methods and results section of the publication). |
|
| Notes | 57 participants were randomised, but participants were only included in the analyses if they “had at least 10 days of therapy.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus, it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “The study medications were administered as identical‐appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The overall‐attrition was high: 31 of 57 participants (54.4%) left the trial early. The trial authors indicated that 15 of 29 participants (51.7%) in the haloperidol‐group and 16 of 28 participants (57.1%) in the thiothixene‐group discontinued the drug treatment prematurely. Modified completers analyses were used (“all patients evaluated had at least 10 days of therapy”). |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no total score or SDs for the BPRS, HPRSD, ESFR, and investigator global evaluation; no raw data for the ZUNG scale; the NOSIE scale is mentioned in the abstract but not in the methods and results section of the publication). There was no information regarding the number of participants who received a medication with antiparkinson drugs in each group; only for the whole study sample of the 46 analysed participants. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Baastrup 1993.
| Methods | Randomisation: “randomly allocated”. Blinding: “open” study. Duration: 6 days. Design: parallel (three‐arm study also investigating zuclopenthixol acetate i.m.). Location: multicentre study (14 centres). Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia with exacerbation of psychotic state (ICD‐9: 295) (N = 35), schizophrenic psychosis (ICD‐9: 295) (N = 3), acute paranoid reaction (ICD‐9: 298.3) (N = 8), other and unspecified reactive psychosis (ICD‐9: 298.8) (N = 5), unspecified psychosis (ICD‐9: 298.9) (N = 4). N = 55. Gender (including manic participants, N = 65): 35M, 20F. Age: mean 37 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated, initial i.m. dose: 5‐10 mg. N = 19. 2. Zuclopenthixol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 15‐30 mg/day, initial i.m. dose: 10‐20 mg. N = 36. “Both in the haloperidol group and the zuclopenthixol group, the patients were switched to oral treatment as soon as possible.” Rescue medication: “An antiparkinson drug could be given in case of troublesome extrapyramidal symptoms, and in case of sleep disturbances a benzodiazepine hypnotic could be prescribed.” |
|
| Outcomes | Examined: Clinically important response to treatment: Brief Psychiatric Rating Scale (BPRS). Unable to use: Global state general: Clinical Global Impressions (CGI) severity of illness (no total score available, only subgroup analyses available). Mental state general: BPRS (no total score provided, only subgroup analyses available). Adverse effects: UKU Side Effect Rating Scale (only data regarding the whole study sample including manic participants available). Adverse effects: unspecific sedation as defined by Lingjaerde et al. (1987) (only data regarding the whole study sample including manic participants available). |
|
| Notes | “The patients were stratified into the 3 diagnostic categories: acute psychosis, mania and exacerbation of chronic psychosis……Within each category the patients were randomly allocated to treatment with either zuclopenthixol acetate, haloperidol, haloperidol or zuclopenthixol.” For the systematic review only data concerning the diagnostic categories “acute psychosis” and “exacerbation of chronic psychosis” as well as the pharmacological interventions with haloperidol and zuclopenthixol were included. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly allocated”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | High risk | “Open” study design. |
| Blinding (performance bias and detection bias) (detection bias) | High risk | “Open” study design. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | According to the number of participants on which the BPRS‐ and CGI‐ratings were based on, at least 6 of 19 participants (31.6%) left the trial early in the haloperidol group and at least 8 of 36 participants (22.2%) in the zuclopenthixol group. Therefore the overall attrition can be considered as being high (at least 14 of 55 participants; 25.5 %). |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no total score for the BPRS and CGI). |
| Other bias | High risk | “The distribution of the patients according to sex in the 3 treatment arms differed surprisingly much.” “The analysis of the baseline values for the primary outcome measures, the BPRS scores and the CGI severity of illness scores showed a certain inhomogenity.” |
Bechelli 1983.
| Methods | Randomisation: “distributed at random”. Blinding: double‐blind. Duration: 27 days (i.m. treatment with both agents during the first 3 days, afterwards 2 days wash‐out, than oral medication for the following 21 days). Design: parallel (three‐arm study also investigating placebo). Location: Psychiatric Hospital of Ribeirao Preto, Mental Health Division of the State Health Department (Sao Paulo, Brazil). Setting: inpatients. |
|
| Participants | Diagnosis: hebephrenic schizophrenia (N = 34), simple schizophrenia (N = 14), paranoid schizophrenia (N = 9), residual schizophrenia (N = 2). N = 59. Gender: 59M. Age: mean 28.9 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dosage scheme, dose range: not indicated, mean dose: 11.5 mg/day. N = 30. 2. Pipotiazine: fixed‐flexible dosage scheme, dose range: not indicated, mean dose: 21.4 mg/day. N = 29. “All patients received 50 mg/day chlorpromazine i.m. and 20 mg/day haloperidol i.m. during the first 3 days of the trial, followed by wash‐out for 2 days. During the subsequent 21 days they received the oral study medication.” “The dosage was adjusted to the clinical response to the patients.” Rescue medication: “Antiparkinson drugs were used only when extrapyramidal side effects appeared.” |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impressions (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Adverse effects: at least one movement disorder. |
|
| Notes | “All groups received 20 mg of haloperidol and 50 mg of chlorpromazine i.m. during the first 3 days, followed by wash‐out for 2 days and then oral pipotiazine, haloperidol or placebo for the following 21 days.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “The patients were assigned…in a random and probabilistic manner, after stratification had been performed on the basis of the following strata: I. simple schizophrenia, II. Hebephrenic schizophrenia, III. Paranoid schizophrenia, IV. Residual schizophrenia, V. Schizoaffective schizophrenia.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 of 30 participants (3.3%) left the trial early in the haloperidol‐group and 3 of 29 participants (10.3%) discontinued prematurely in the pipotiazine‐group. The overall‐attrition was rather low (4 of 59 participants; 6.8%). The analyses were based on completers‐only data, but due to the low attrition the risk of bias might be rather low. |
| Selective reporting (reporting bias) | Unclear risk | Many of the reported adverse effects data were not usable for this review. No information was available regarding the number of participants who received a medication with antiparkinson drugs. |
| Other bias | High risk | “All groups received 20 mg of haloperidol and 50 mg of chlorpromazine i.m. during the first 3 days, followed by wash‐out for 2 days and then oral pipotiazine, haloperidol or placebo for the following 21 days.” |
Brannen 1981.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 28 days (after a drug free wash‐out period of at least three days). Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: chronic undifferentiated schizophrenia (N = 21), paranoid schizophrenia (N = 18), acute schizophrenia (N = 3), schizoaffective (N = 3), hebephrenic schizophrenia (N = 2). Diagnoses according to Schneider`s first rank symptoms of schizophrenia and DSM‐II. N = 47. Gender: 23M, 24F. Age: mean 32 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 6 mg to 60 mg/day, mean final dose: 30.4 mg/day. N = 24. 2. Bromperidol: fixed/flexible dose: not indicated, dose range: 6 mg to 60 mg/day, mean final dose: 39.8 mg/day. N = 23. “fixed‐changing dosage schedule…..until a minimum reduction of 30% from baseline occurred in the total score of the BPRS, at which point no further increase in dosage was required”. Rescue medication: antiparkinsonian drugs for the control of extrapyramidal symptoms. Flurazepam or chloral hydrate were allowed for nighttime sedation. |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impressions (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to adverse effects. Adverse effects: akathisia, dystonia, rigor, hypotension. Unable to use: Behaviour: Nurses´ Observation Scale for Inpatient Evaluation (NOSIE) (no SDs available and no imputation method could be applied). |
|
| Notes | Study participants were 47 newly‐admitted schizophrenic patients. Each participant had “at least one of Schneider`s first rank symptoms of schizophrenia, and fulfilled criteria for a DSM‐II diagnosis of schizophrenia and had a minimum total score of 30 on the BPRS.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”; “All investigational medications were prepared in identical‐appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The attrition was high: 7 of 24 participants (29.2%) in the haloperidol group left the trial early and 5 of 23 participants (21.7%) in the bromperidol‐group. The overall‐attrition was 25.5% (12 of 47 participants). The trial authors did not mention which analysis method they used regarding the continuous data. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully reported (no SDs for the BPRS and NOSIE total score). Only the most prevalent adverse effects were reported. Data regarding the number of participants receiving rescue medication were missing. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Bueno 1979.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 6 weeks. Design: parallel. Location: multicentre. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 28), acute schizophrenic episode (N = 4), catatonic schizophrenia (N = 3), hebephrenic schizophrenia (N = 3). N = 38. Gender: 27M, 11F. Age: mean 28.8 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: 17 participants with no previous hospitalisations, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 5.4 mg/day. N = 19 (study completers). The number of participants randomised to this study group was not indicated. 2. Loxapine: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 58.3 mg/day. N = 19 (study completers). The number of participants randomised to this study group was not indicated. |
|
| Outcomes | Unable to use: Global state general: Clinical Global Impression (CGI) (no total score available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no total score available). Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no total score available). |
|
| Notes | Study participants with "established diagnosis of acute or chronic schizophrenia". 16 participants with first episode. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. The study medications were administered as identical‐appearing capsules. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With missing data of 2 of the 40 randomised participants (5%), the overall attrition was rather low. The analyses were based on completers‐only data, but due to the small drop‐out rate, the risk of bias might be considered as being low. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, CGI, and NOSIE). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Cassano 1975.
| Methods | Randomisation: “entirely randomised experimental design”. Blinding: double‐blind. Duration: 30 days. Design: parallel. Location: multicentre (Institute of Psychiatry, University of Pisa; Institute of Psychiatry, University of Pavia; Provincial Psychiatric Institutes, Ceremona; S. Lazzaro Psychiatric Institute, Reggio Emilia; Neuropsychiatric Hospital, Teramo; Provincial Neiropsychiatric Hospital, Varese). Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (paranoid and hebephrenic schizophrenia). N = 76. Gender: “both sexes”, no further details available. Age: mean 38 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 0.5 to 10.5 mg/day, mean dose: 5mg/day. N = 36 (number of participants included in the analyses, the number of participants randomised to this study group was not indicated). 2. Sulpiride: flexible dose, dose range: 100 mg to 2300 mg/day, mean dose: 1000mg/day. N = 34 (number of participants included in the analyses, the number of participants randomised to this study group was not indicated). Flexible dose: “The dosage scheme was flexible and individualised according to clinical response and tolerability.” Rescue medication: “an antiparkinson drug (orphenadrine) could be administered in the case of appearance of extrapyramidal signs.” “Each patient received an evening dose of a hypnotic (amobarbital 100mg).” |
|
| Outcomes | Examined: Adverse effects: at least one adverse effect, at least one movement disorder, akathisia, use of antiparkinson medication. Unable to use: Clinically important response to treatment: Clinical evaluation of the changes of global symptomatology (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Mental state general: Inpatient Multidimensional Psychiatric Scale (IMPS) (no raw data available). Leaving the study early due to inefficacy of treatment (number of participants randomised to the groups was not indicated). Leaving the study early due to adverse effects (number of participants randomised to the groups was not indicated). |
|
| Notes | “wash‐out period of treatment with placebo (2‐7 days).” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Entirely randomized experimental design”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “The compounds were contained in indistinguishable capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 19 of 76 participants (25%) left the trail early. “In 6 patients, the study was discontinued for reasons totally independent of the treatments; on this basis, they have not been included in the analysis of the results.” |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no raw data for the BPRS, IMPS, and the clinical evaluation of the changes of global symptomatology). The numbers of participants randomised to each treatment group (haloperidol or sulpiride) were not indicated. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Cocchi 1971.
| Methods | Randomisation: randomised. Blinding: double‐blind. Duration: 30 days. Design: parallel. Location: Clinica Psichiatrica dell´Università di Milano. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 17), hebephrenic schizophrenia (N = 12), pseudoneurotic schizophrenia (N = 4), schizophrenia simplex (N = 4), other types of schizophrenia (N = 3). N = 40. Gender: 24M, 16F. Age: mean 25.4 years. History: duration stable: not indicated, duration of illness: mean 2.58 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed dosage scheme (first day: 2 mg/day, increase of 2 mg/day every third day until reaching a plateau of 10 mg/day for 10 days followed by a dosage decrease of 2 mg/day every second day), dose range: not indicated, mean dose: not indicated. N = 20. 2. Droperidol: fixed dosage scheme (first day: 2 mg/day, increase of 2 mg/day every third day until reaching a plateau of 10 mg/day for 10 days followed by a dosage decrease of 2 mg/day every second day), dose range: not indicated, mean dose: not indicated. N = 20. Rescue medication: antiparkinson medication for all patients. |
|
| Outcomes | Examined: Clinically important response to treatment: Overall clinical judgement (“giudizio clinico espresso”). Unable to use: Mental state general: Scala dei Sintomi Bersaglio (scale not published). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised; no further detail. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. Identical appearing tablets. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects reporting was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Cocito 1970.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: “very flexible” (haloperidol‐group: mean duration: 51 days (range from 10 to 160 days); dehydrobenzperidol‐group: mean duration: 75 days (range from 12 to 160 days)) Design: parallel. Location: Ospedale Psichiatrico Provinciale di Genova, Istituto di Genova‐Quarto (Italy). Setting: inpatients (at least for the beginning of the trial). |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 22), hebephrenic schizophrenia (N = 10), catatonic schizophrenia (N = 2), atypical delusion syndrome (N = 1), other forms of schizophrenia (N = 10). N = 46. Gender: 46 M. Age: mean 34.5 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 6.0 mg/day. N = 23. 2. Dehydrobenzperidol (=Droperidol): fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 6.32 mg/day. N = 22. Rescue medication: “The basic neuroleptic treatment with either dehydrobenzperidol or haloperidol was supplemented with orphenadrine, levopromazine, and in about one half of the cases diazepam.” Antidepressants in two cases. |
|
| Outcomes | Examined: Clinically important response to treatment: Therapeutic results at 60 days. Mental state general: Rating Scale for Quantification of Psychotic Symptom Severity (RSQPSS). Adverse effects: at least one adverse effect, at least one movement disorder, akathisia, tremor. |
|
| Notes | Analysis was performed based on the data at 30 days (compromising altogether 40 participants) and 60 days (compromising altogether 12 participants). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”; The two drugs were administered “in identical bottles of the same potency and coded so as to be unrecognisable both to the experimentalists and to the patients.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Regarding the analysis at day 30, data of 40 of 46 randomised participants were available (13% missing data) and at day 60, data of only 12 of 46 randomised participants were provided (73.9% missing data). The trial authors used a completers‐only analysis. |
| Selective reporting (reporting bias) | Low risk | The outcomes have been reported in the pre‐specified way. |
| Other bias | High risk | “The basic neuroleptic treatment with either dehydrobenzperidol or haloperidol was supplemented with orphenadrine, levopromazine, and in about one half of the cases diazepam.” Antidepressants were administered in two cases. Significant between‐group difference regarding the average trial duration (51 versus 75 days). |
Cosar 1999.
| Methods | Randomisation: randomised. Blinding: not indicated. Duration: 90 days Design: parallel (four‐arm study also investigating clozapine and chlorpromazine). Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 80. Gender: not indicated. Age: not indicated. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 34.75 mg/day. N = 40. 2. Sulpiride: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 696 mg/day. N = 40. Rescue medication: not indicated. |
|
| Outcomes | Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). |
|
| Notes | Data of this trial were based only on a single conference abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | The study did not address this outcome. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | The study did not address this outcome. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no raw data for the BPRS). Adverse effects were not reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Darondel 1981.
| Methods | Randomisation: randomised. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: Hôpital de Lommelet, Saint‐André dans le Nord. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 20), hebephrenic schizophrenia (N = 9), schizophrenia simplex (N = 7), residual schizophrenia (N = 1), paranoia (N = 1), deliriant syndrome (N = 7). N = 55. Gender: 34M, 11F. Age: mean 40.8 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed dose, dose range: not indicated, mean dose: 15 mg/day. N = 23. 2. Pipotiazine: fixed dose, dose range: not indicated, mean dose: 15 mg/day. N = 22. Rescue medication: antiparkinson medication was used as well as trihexyphenidyle and levopromazine. |
|
| Outcomes | Examined: Clinically important response to treatment: Global improvement. Mental state general: Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Adverse effects: akathisia, dystonia, tremor, use of antiparkinson medication, weight gain. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With 2 of 45 participants leaving the study early (4.4%) the overall attrition was low. |
| Selective reporting (reporting bias) | Low risk | The outcomes have been reported in the pre‐specified way. |
| Other bias | High risk | Co‐administration with other antipsychotics was allowed. |
Denijs 1980.
| Methods | Randomisation: “according to a random scheme”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenic psychosis paranoid type (N = 15), schizophrenic psychosis catatonic type (N = 2), affective psychosis currently manic (N = 5), psychogenic paranoid psychosis (N = 4), excitative type (N = 6), psychogenic paranoid psychosis (N = 3), transient psychotic (N = 1), depressive type (N = 1). N = 37. Gender: 15M, 22F. Age: median 33.3 years. History: duration stable: not indicated, duration of illness: The duration of illness was fewer than 1 month in 15 participants, number of previous hospitalisations: 16 participants were admitted to hospital for the first time, age at onset: not indicated, severity of illness: 24 participants were classified as having "a severe degree of illness", baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 12 mg to 24 mg/day, mean dose: not indicated. N = 17. 2. Bromperidol: flexible dose, dose range: 9 mg to 20 mg/day, mean dose: not indicated. N = 20. Flexible dose: “The individual daily dose was determined according to clinical benefit and side‐effects.” Injections were allowed in “very disturbed, aggressive and non‐cooperating patients”. Rescue medication: orphenadrine (anticholinergic drug), if extrapyramidal side‐effects were present. Promethazine (H1‐receptor‐antagonist) as sedative medication. |
|
| Outcomes | Examined: Clinically important response to treatment: Global assessment about treatment effects. Leaving the study early due to any reason. Adverse effects: at least one movement disorder, akathisia, dyskinesia, tremor, use of antiparkinson medication, hypotension, sedation. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Behaviour: Nurses´ Observation Scale for Inpatient Evaluation (NOSIE) (no raw data available). Adverse effects: Simpson and Angus Scale (no raw data available). |
|
| Notes | All study participants were characterised by a "clear‐cut psychotic symptomatology". Acute form of schizophrenia in 15 participants, subacute form in 19 participants, and chronic form in 3 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study group allocation “according to a random scheme”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 of 19 participants (10.5%) left the trial early in the haloperidol group and 3 of 23 participants (13%) in the bromperidol group. With 5 of 42 participants (11.9%) the overall attrition was moderate. Regarding the primary outcome of the systematic review, data analyses based on the intention‐to‐treat (ITT) approach were provided. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, NOSIE, and Simpson and Angus Scale). |
| Other bias | High risk | “A lack of double‐blind medication” in one participant receiving bromperidol. “After breaking the double‐blind code it was noticed that the prescription and registration of promethazine have not been as careful as they should have been.” |
Dufresne 1993.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 6 weeks. Design: parallel (three‐arm study also investigating thioridazine). Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: chronic schizophrenia (DSM‐III). N = 30. Gender: 16M, 14F. Age (including participants of the thioridazine study group, only study completers, N = 35): mean 34 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: mean 7, age at onset: mean 21 years, severity of illness: BPRS total score of at least 30 with at least two moderately severe positive symptoms was the inclusion criteria, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum dose 40 mg/day, mean dose: not indicated. N = 16. 2. Molindone: flexible dose, dose range: maximum dose 200 mg/day, mean dose: not indicated. N = 14. The study protocol allowed “the psychiatrist to titrate dosage in a manner similar to good clinical practise.” Rescue medication: “amantadine was given for moderate to severe drug‐induced parkinsonism”; "chloral hydrate was allowed for insomnia or agitation." |
|
| Outcomes | Examined: Global State general: Clinical Global Impressions (CGI) (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Mental state specific: Hamilton Depression Rating Scale (HAM‐D) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: use of antiparkinson medication. Unable to use: Mental state specific: Concise Negative Symptoms Rating Scale (CNS‐RS) (no raw data available). Adverse effects: Abnormal Involuntary Movement Scale (AIMS) (no raw data available). Adverse effects: Reversible Extrapyramidal Symptom Rating Scale (REPS) (no raw data available). Adverse effects: Treatment Emergent Symptoms Scale (TESS) (no raw data available). |
|
| Notes | The schizophrenic study participants were "quite depressed; the mean depression rating on the HAM‐D was greater than 18 for all three treatment groups". Only participants who completed 6 weeks were included in the analyses. Randomisation “after a psychotropic‐free washout period of at least 1 week.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “identical capsules”. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 of 16 participants (25%) left the trial early in the haloperidol group and 4 of 14 participants (28.6%) in the molindone group. With 8 of 30 participants (26.7%) the overall attrition was high. Completers‐only analyses were used. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS and HAM‐D; no raw data for the CGI, CNS‐RS, AIMS, REPS, and TESS). The adverse effects were not fully addressed. |
| Other bias | High risk | “The majority of subjects assigned to the haloperidol and thioridazine groups were female, whereas those assigned to the molindone group were mostly male.” The high level of depression of the participants could be a risk of bias in terms of this systematic review. |
Engelhardt 1978.
| Methods | Randomisation: “computer‐generated randomisation scheme”. Blinding: double‐blind. Duration: 24 weeks. Design: parallel. Location: Psychopharmacology Research Unit of the State University of New York, Downstate Medical Center. Setting: outpatients. |
|
| Participants | Diagnosis (study completers, N = 36): chronic undifferentiated schizophrenia (N = 24), paranoid schizophrenia (N = 12). N = 80. Gender: 22M, 14F. Age: mean 35.3 years. History: duration stable: not indicated, duration of illness: mean 11.66 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 1.25 mg to 25 mg/day, mean dose: 5.7 mg/day. N = 38. 2. Thiothixene: flexible dose, dose range: 5 mg to 60 mg/day, mean dose: 16.0 mg/day. N = 42. “ratio of mean thiothixene‐to‐haloperidol dosage: 2,8:1” Rescue medication: antiparkinson medication was allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Improvement Rating. Leaving the study early due to any reason. Leaving the study early due to adverse effects. Adverse Effects: at least one adverse effect, at least one movement disorder, tardive dyskinesia. Unable to use: Mental state general: Katz Adjustment Scales (no total score available). Mental state general: Lipman‐Rickels Self‐Rating Symptom Scale (SRSS) (no raw data available). |
|
| Notes | 36 completers of the full 24 weeks. “4‐week placebo wash‐out period prior to active drug treatment.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Patients were assigned with a computer‐generated randomisation scheme stratified for sex and marital status.” |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Haloperidol and thiothixene were supplied in capsules of identical appearance.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Altogether 80 people with schizophrenia were assigned to treatment. “Of the 80 patients only 56 remained in treatment beyond the first 2 weeks.” Only 36 participants were able to remain in treatment for the full 24 weeks of the study period. So the overall‐attrition was high (44 of 80 participants; 55%). 22 of 38 participants (57.9%) left the trial early in the haloperidol‐group and 22 of 42 participants (52.4%) in the thiothixene‐group. The trial authors provided only completers‐analyses comprising altogether 36 participants. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data or total score regarding the Katz Adjustment Scales and SRSS). No information available regarding the number of participants who received a medication with antiparkinson drugs. |
| Other bias | High risk | Occurrence of baseline imbalances: the differences between the two treatment‐groups were statistically significant in terms of the mean age of the participants and the mean duration of the illness. Tobin 1980: "The test groups were not homogeneous at baseline. There was a significantly higher proportion of older, chronic patients with severe symptoms in the haloperidol group than in the thiothixene group." |
Escobar 1985.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: two‐centre study (Brentwood VA Medical Center, Los Angeles; Payne Whitney Clinic, New York Hospital‐Cornell Medical Center, New York). Setting: “Typically, patients remain in these acute wards for only a few days.” Hospitalisation at least during the injectable phase of the trial. No further details. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III). N = 35. Gender: 32M, 3F. Age: mean 37 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: mean 6, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum mean oral dose 32.4 mg/day, mean dose: not indicated. N = 15 (participants completing at least 1 week of the oral phase of the trial). 2. Molindone: flexible dose, dose range: maximum mean oral dose 160 mg/day, mean dose: not indicated. N = 15 (participants completing at least 1 week of the oral phase of the trial). “Dosages were clinically determined.” “Injections were administered only during the first 12‐72 hours.” |
|
| Outcomes | Examined: Global state general: Clinical Global Impressions (CGI) ‐ Symptom Severity (no SDs available). Adverse effects: akathisia, dystonia, rigor, tremor, weight gain. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no total score available). Adverse effects: Treatment Emergent Symptoms Scale (TESS) (no total score available). |
|
| Notes | “ongoing study”. The study drugs were “given for the first 2‐3 days of hospitalisation and then continued orally for up to 4 weeks.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “identical appearing tablets”. |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. “The rater was blind to the type of medication the subject received.” |
| Incomplete outcome data (attrition bias) All outcomes | High risk | “Only 30 subjects completed 1 week of treatment and 25 completed 2 weeks.” Thus, at least 10 of 35 participants (28.6%) left the trial early and therefore the overall attrition can be considered as being high. It is not explicitly mentioned how many patients were randomised to each study group (haloperidol or molindone group). “Analyses [were] based on the 30 subjects who completed at least the first week of the study.” |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no total score for the BPRS and TESS; no SDs for the CGI). |
| Other bias | High risk | “This study is limited by…..the relatively short evaluation period.” “Because of attrition, analyses for the oral portion of the study [were] limited to ratings at baseline, days 2‐3, and weeks 1 and 2.” |
Faretra 1970.
| Methods | Randomisation: randomly assigned. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: not indicated. Setting: inpatients (according to Engelhardt 1973). |
|
| Participants | Diagnosis: childhood schizophrenia (N = 52), psychosis with organic brain damage (N = 4), psychosis with mental deficiency (N = 3), primary behaviour disorder (N = 1). N = 60. Gender: 44M, 16F. Age: mean 9.9 years. History: duration stable: not indicated, duration of illness: mean 21.35 months, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 0.75 mg to 3.75 mg/day, mean dose: not indicated. N = 30. 2. Fluphenazine: fixed/flexible dose: not indicated, dose range: 0.75 mg to 3.75 mg/day, mean dose: not indicated. N = 30. Rescue medication: biperiden in case of extrapyramidal symptoms. |
|
| Outcomes | Examined: Clinically important response to treatment: “Overall change”. Adverse effects: at least one adverse effect, at least one movement disorder, akathisia, dystonia. |
|
| Notes | All study participants were children. "For 82 Percent of the patients, their psychiatric conditions were either static or deteriorating under the treatment they were receiving before the study." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Medications were prepared in identical appearing capsules”, "Capsules were scored in bottles identified only by a number", "Neither the patient nor the dispensing and rating physician knew which drug was given." |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “double‐blind”. "Neither the patient nor the dispensing and rating physician knew which drug was given." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes have been reported in the pre‐specified way with the exception of the number of participants in each study group that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Fuentenebro 1989.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 2 weeks. Design: parallel. Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III). N = 50. Gender: not indicated. Age: not indicated. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. Initial 5 mg injection, change to oral medication within the first 24 to 48 hours. N = not indicated. 2. Molindone: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. Initial 2 mg injection, change to oral medication within the first 24 to 48 hours. N = not indicated. |
|
| Outcomes | Unable to use: Global state general: Clinical Global Impression (CGI) (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no total score available for both study groups, no SDs available). |
|
| Notes | The primary aim of the study was to evaluate the patient`s response to antipsychotic agents as predictor of treatment response. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial authors did not explicitly mention a randomisation, but described a double‐blinding. Thus we implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | With missing data of 10 of the 50 randomised participants (20%), the overall attrition was moderate. The analyses were based on completers‐only data, but due to the moderate drop‐out rate, the risk of bias might be considered as being unclear. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no usable data and SDs for the BPRS; no raw data for the CGI). The numbers of participants randomised to each treatment group (haloperidol or molindone) were not indicated. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Gallant 1967.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 30 days. Design: parallel (three‐arm study with chlorpromazine as third treatment group). Location: Southeast Louisiana Hospital, Mandeville, Louisiana. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia. N = 39. Gender (including participants of the chlorpromazine study group, N = 58): 30M, 28F. Age (including participants of the chlorpromazine study group, N = 58): mean 33.4 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: maximum dose 16 mg/day, mean dose: not indicated. N = 19. 2. Trifluperidol: fixed/flexible dose: not indicated, dose range: maximum dose 4 mg/day, mean dose: not indicated. N = 20. Rescue medication: “use of anti‐parkinson medication (Artane) prophylactically for all subjects throughout the study”. Administration of benztropine (Cogentin, 2 mg i.m.) was allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Global Rating of Improvement. Adverse effects: at least one movement disorder, dyskinesia, hypotension. Unable to use: Mental state general: Beckomberga Rating Scale (no SDs available and no imputation method could be applied). Behaviour: MACC Behavioral Adjustment Scale (no raw data available). Behaviour: Tulane Test Battery (no raw data available). |
|
| Notes | In a third study arm of this trial chlorpromazine was investigated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “All drugs were supplied in identical capsules and were dispensed from individual medication bottles which were prepared and coded prior to the study.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully addressed (no raw data for the MACC Behavioral Adjustment Scale and the Tubane Test Battery; no SDs were provided for the Beckomberga Scale). Adverse effects reporting was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Gerlach 1985.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 12 weeks (first period of the cross‐over‐trial containing altogether two 12‐week periods). Design: cross‐over. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (based on the criteria of Feighner 1972). N = 28. Gender (N = 20): 17M, 3F. Age (N = 20): mean 34 years. History: duration stable: not indicated, duration of illness: mean 9 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 6 mg to 24 mg/day, mean final dose: 12 mg/day. N = 10. 2. Sulpiride: fixed/flexible dose: not indicated, dose range: 800 mg to 3200 mg/day, mean final dose: 2000 mg/day. N = 10. “During the first four to eight weeks, doses were gradually increased until an optimal therapeutic effect was attained. The optimal dose was maintained until the end of the 12‐week treatment period.” Rescue medication: allowed were “biperiden in case of extrapyramidal side‐effects, and diazepam or levomepromazine when sedation was required” |
|
| Outcomes | Examined: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Unable to use: Leaving the study early due to adverse effects (number of participants randomised to the groups was not indicated). Adverse effects: special checklist (only data regarding the whole trial duration available). |
|
| Notes | Only the 20 study completers were included in the analyses. “Following a wash‐out period of 1‐6 weeks (until clear treatment‐demanding symptoms had developed), patients were randomly assigned to either sulpiride or haloperidol, and treated for 12 weeks.” The mean duration of the neuroleptic treatment was 6 years. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Randomly assigned”. According to e‐mail correspondence with the first author: “Referring to a random number table.” |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “identically looking capsules”. |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. According to e‐mail correspondence with the first author the raters were blinded to treatments. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Regarding the whole trial duration, the overall attrition can be considered as being high (8 of 26 randomised participants). There were no information available, how many participants dropped out during the first phase of the trial. “Relatively small number of patients and the consequent Type II error.” The analyses were based on completers‐only data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS). The numbers of participants randomised to each treatment group (haloperidol or sulpiride) were not indicated. Concerning the adverse effects reporting there were only data for the whole trial duration (both 12‐week periods) provided but not separately for the first phase which was of interest for this systematic review. |
| Other bias | High risk | “The study was carried out as a double‐blind cross‐over trial.” ”Dampening effect of the relatively high doses [of sulpiride] employed in this study, and the sample of chronic, long‐term hospitalised patients which was studied.” |
Germana 1990.
| Methods | Randomisation: “randomised”. Blinding: double‐blind. Duration: mean duration: 19 days (range from 5 days to 33 days); haloperidol‐group: mean duration: 18.8 days; bromperidol‐group: mean duration: 19.4 days. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 27), deliriant syndrome (N = 2), various types of psychosis (N = 7). N = 36. Gender: 24M, 12F. Age: mean 36 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dose, dose range: 5 mg to 6 mg/day, mean dose 5.3 mg/day. N = 18. 2. Bromperidol: fixed‐flexible dose, dose range: 5 mg to 7 mg/day. mean dose: 5.5 mg/day. N = 18. Variable dosages from day 5 on. Rescue medication: anticholinergics were allowed |
|
| Outcomes | Examined: Clinically important response to treatment: Global Clinical Judgement. Adverse effects. akathisia, dyskinesia, rigor, tremor, hypotension. Unable to use: Mental state general: Symptom Rating Scale (no SDs available and no imputation method could be applied, scale not published). |
|
| Notes | Exclusively hospitalised patients. No fix endpoint of the trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomised”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | The outcome data were not fully addressed (no SDs for the Symptom Rating Scale). |
| Other bias | High risk | The trial was not characterised through a definite endpoint the data‐analyses were based on. Seven participants in the bromperidol‐group received already medication with anticholinergics when entering the trial. |
Giordana 1984.
| Methods | Randomisation: randomised (participants were drawn by lots into two groups of treatment, with always 4 participants balanced). Blinding: double‐blind. Duration: 3 weeks. Design: parallel. Location: not indicated. Setting: inpatients and outpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 16), hebephrenic schizophrenia (N = 5), simple schizophrenia (N = 5), dysthymic schizophrenia (N = 4). N = 30. Gender: male and female, no further details available. Age: mean 36.8 years. History: duration stable: not indicated, duration of illness: mean 11 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: mean BPRS at baseline 65, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: "initial dose 15 mg/day", mean dose: not indicated. N = 15. 2. Pipotiazine: flexible dose, dose range: "initial dose 15 mg/day", mean dose: not indicated. N = 15. |
|
| Outcomes | Examined: Clinically important response to treatment: Global evaluation of the efficacy of both drugs. Mental state specific: Psychopathology according to the AMDP‐system (depersonalisation, conceptual disorganisation, delusional and hallucinatory syndrome). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised (participants were drawn by lots into two groups of treatment, with always 4 participants balanced). |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. indistinguishable medication. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | The outcome data were not fully addressed (no SDs were available regarding the hallucinatory syndrome measured by the AMDP system). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Glazer 1990.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: at least 2 weeks. Design: parallel. Location: not indicated. Setting: outpatients. |
|
| Participants | Diagnosis: chronic or subchronic schizophrenia or schizoaffective disorder (research diagnostic criteria) and occurence of tardive dyskinesia. N = 18. Gender: 8M, 10F. Age: mean 47 years. History: duration stable: not indicated, duration of illness: mean 2.58 years, number of previous hospitalisations: mean 4.6, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 19.3 mg to 34.3 mg/day, mean dose: not indicated. N = 9. 2. Molindone: flexible dose, dose range: 75 mg to 145 mg/day, mean dose: not indicated. N = 9. “Medication dosing was determined by the occurrence of side effects or psychiatric symptoms.” |
|
| Outcomes | Examined: Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Unable to use: Global state general: Clinical Global Impression (CGI) (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Adverse effects: Abnormal Involuntary Movement Scale (AIMS) (no information available to calculate the values for both study group). Adverse effects: Treatment Emergent Symptoms Scale (TESS) (no raw data available). Adverse effects: Webster Parkinsonism Rating Scale (no raw data available). |
|
| Notes | Inclusion criteria: participants had to 1. “met research diagnostic criteria for chronic or subchronic schizophrenia or schizoaffective disorder”; 2. “meet diagnostic criteria for TD [tardive dyskinesia]” and 3. “have at least 12 months exposure to neuroleptic medication” other than molindone or haloperidol. 31 subjects agreed to participate in this study, but only 18 participants fulfilled subsequently the criterion for withdrawal‐exacerbated TD during the drug‐free period and were randomised to the study medications. “Neuroleptic medications were tapered over a 7‐10 day period and then withdrawn, with single‐blind substitution of placebo for 7‐14 days.” The participants who met the criterion for withdrawal‐exacerbated TD (18 0f 31 participants) during the drug‐free period “were then admitted to a masking phase in which they were randomly assigned to receive either molindone or haloperidol.” “If the patient experienced no side effects or psychiatric symptoms, the dose was raised during the first week from 100% to the second week when about 200% dose equivalency of the patient’s prestudy neuroleptic medication was given . |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Medication was supplied in identical‐appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Of the 18 randomised participants, who fulfilled the criterion for withdrawal‐exacerbated TD nobody left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the CGI, BPRS, TESS, and the Webster Parkinsonism Rating Scale). Adverse effects reporting was incomplete. |
| Other bias | High risk | Baseline imbalance between the two study groups in terms of post hospitalisation duration. The trial was not characterised through a definite endpoint. |
Goldstein 1966.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: not indicated. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: “symptoms of acute psychosis”. N = 21. Gender: not indicated. Age: 21‐55 years, no further details available. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: "no psychiatric hospitalisation during the previous six months", age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 10 mg to 22 mg/day, mean dose: not indicated. N = 8 (study completer). 2. Trifluoperazine: flexible dose, dose range: 7 mg to 20 mg/day, mean dose: not indicated. N = 10 (study completer). Rescue medication: antiparkinson or sleeping medication was allowed if indicated. |
|
| Outcomes | Examined: Adverse effects: akathisia, dystonia, hypotension. Unable to use: Mental state general: Inpatient Multidimensional Psychiatric Scale (IMPS) (no total score available). Behaviour: Ward Behaviour Rating Scale (WBRS) (no total score available). |
|
| Notes | All study participants were “newly admitted to the hospital". In the publication there was no information regarding the duration of the trial available. 18 of the 21 included patients “completed an assessable course of treatment”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned to one of the two treatment groups at random.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Trifluoperazine and haloperidol were packed in identical Number 2 gelatin capsules.” “The physicians were told that one capsule was equivalent to any of the following: 100mg chlorpromazine, 100mg thioridazine, 4mg perphenazine, 2mg trifluoperazine, 2mg haloperidol, or 1mg fluphenazine.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | With 3 drop‐outs (of 21 participants) the overall attrition was moderate (14.3%). It is not explicitly mentioned how many patients were randomised to each study group (haloperidol or trifluoperazine group). It can be assumed that the results were based on a completers‐only analysis. |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no total scores for the IMPS and WBRS). |
| Other bias | High risk | No information concerning the duration of the trial available. |
Goldstein 1969.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 5 weeks (first phase of the trial). Design: parallel. Location: Psychiatric Inpatient Service at the Jackson Memorial Hospitalin Miami, Florida, USA. Setting: inpatients. |
|
| Participants | Diagnosis: psychosis. N = 250. Gender: not indicated. Age: 21‐65 years, no further details available. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: "no psychiatric hospitalisation during the previous six months", age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 16 mg/day “at the peak of treatment”. N = not indicated. 2. Perphenazine: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 64 mg/day “at the peak of treatment”. N = not indicated. “a relatively fixed dosage schedule”. Rescue medication: “Side effects were treated with anti‐parkinson medication or reduction of dosage.” |
|
| Outcomes | Unable to use: Clinically important response to treatment: Global assessment of the degree of improvement (no raw data available). Global state general: Global assessment of the severity of illness (no raw data available). Mental state general: revised form of the Inpatient Multidimensional Psychiatric Scale (IMPS) (no raw data available). Mental state specific: Clyde Mood Scale (no raw data available). Behaviour: Relatives Rating Scale (no raw data available). Behaviour: Ward Behaviour Rating Scale (WBRS) (no raw data available). |
|
| Notes | “250 psychotic inpatients”; “newly admitted” to the hospital. Inclusion criterion: "Presence of two or more of the following symptoms or behaviours: thinking or speech disturbances, catatonic motor behaviour, paranoid ideation, hallucination, delusional thinking other than paranoid, blunted or inappropriate emotion, disturbance of social behaviour and interpersonal relations”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “identical appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The overall attrition was moderate (41 of 250 participants (16.4%) in both study arms). |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no raw data for the global assessment of the severity of illness, global assessment of the degree of improvement, IMPS, WBRS, Relatives Rating Scale, and Clyde Mood Scale). Adverse effects reporting was incomplete. No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Gowardman 1973.
| Methods | Randomisation: “randomly allotted”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 15 according to the main text, N = 14 according to table 1), hebephrenic schizophrenia (N = 3 according to the main text, N = 4 according to table 1), catatonic schizophrenia (N = 1), schizoaffective psychosis (N = 1). N = 20. Gender: not indicated. Age: mean 48.6 years. History: duration stable: not indicated, duration of illness: mean 20.6 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: maximum dose 14 mg/day, mean dose: 10.15 mg/day. N = 10. 2. Pimozide: fixed/flexible dose: not indicated, dose range: maximum dose 6 mg/day, mean dose: 5.1 mg/day. N = 10. “Having achieved a rapid control of psychotic symptoms, the drugs were suitably increased in individual cases to see if further benefit accrued, or until extrapyramidal side effects were noted.” Rescue medication: “patients exhibiting extrapyramidal side effects were treated with benztropine mesylate (Cogentin) 2 mg.” |
|
| Outcomes | Examined: Clinically important response to treatment: Global evaluation. Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one adverse effect, at least one movement disorder. |
|
| Notes | Study participants were 20 “chronic institutionalised and withdrawn schizophrenics”. “All patients had severe disorder of thinking, persecutory delusions, auditory hallucinations at some time, disturbed affect and social behaviour.” “short drug‐free interval” before the first administration of the study medications. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly allotted by the hospital pharmacist”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “The drugs were supplied in identical capsules.” “The capsules were indistinguishable in outward appearance.” “The investigators did not know who was receiving which drug.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. “The investigators did not know who was receiving which drug.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “All patients completed the trial period of three months.” |
| Selective reporting (reporting bias) | Unclear risk | The adverse effects were not fully addressed. No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Haas 1982.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 30 days. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis (ICD‐8): schizophrenia: paranoid‐hallucinatory type (ICD 295.3) (N = 19), schizophrenia: chronic undifferentiated type (ICD 295.0) (N = 10), schizophrenia: schizoaffective type (ICD 295.7) (N = 1). N = 30. Gender: 13M, 17F. Age: mean 39 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: not indicated, mean final dose: 23.75 mg/day. N = 15. 2. Pimozide: flexible dose, dose range: not indicated, mean final dose: 20.36 mg/day. N 15. Allowed dose range: up to 60 mg/day for both drugs. “Initial dose for both drugs was 10–40 mg/day. This was increased up to the fifth day to 60 mg and then continued according to clinical needs.” Rescue medication: “Chloral hydrate (1,5/day) or, if necessary, promethazine (100mg/day) were given as sleep medication. Biperiden (2mg tablets was given if extrapyramidal signs were observed)." |
|
| Outcomes | Examined: Clinically important response to treatment: Overall clinical assessment at day 30. Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to adverse effects. Adverse effects: akathisia, dyskinesia, rigor, tremor, use of antiparkinson medication, hypotension. Unable to use: Global state general: Clinical Global Impressions (CGI, modified severity scale) (modified version of the scale, no raw data available). Mental specific: ADMP (no results were provided). |
|
| Notes | Study participants were 30 “acutely hospitalised schizophrenic patients” (28 completers). No participant “received depot neuroleptics at least three weeks prior to hospitalisation”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial authors did not explicitly mention a randomisation, but described a double‐blinding. Thus we implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Presentation of the drugs was identical in liquid form” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”; No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no drop‐outs in the haloperidol‐group, but 2 of 15 (13.3%) participants left the trial early in the pimozide‐group. The overall‐attrition was 6.7% (2 of 30 participants). Altogether the attrition was rather low and the risk of bias might be considered as being low. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS; no results for the ADMP‐rating; no raw data for the CGI). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Hall 1968.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 50. Gender: 50M. Age: mean 45 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: not indicated, mean final dose: 6.95 mg/day. N = 25. 2. Fluphenazine: flexible dose, dose range: not indicated, mean final dose 17.86 mg/day. N = 25. Rescue medication: benztropine mesylate (Cogentin). |
|
| Outcomes | Examined: Clinically important response to treatment: Global judgement of the amount of change. Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Adverse effects: death, use of antiparkinson medication (17‐item side effect check list). Unable to use: Mental state general: Psychotic Inpatient Profile (PIP) (no raw data available). |
|
| Notes | Study participants were 50 male “chronic treatment‐resistant schizophrenics”. “The length of hospitalisation….ranged from three months to forty years, with a median of 50 months. Twenty of these patients had been hospitalised continuously for ten years or longer.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Medications for each patient were supplied by the pharmacy in an individual bottle with only the patient’s name and study number appearing on the bottle label. Both compounds dispensed in capsules identical in appearance. Therefore, the investigator who cared for the patients and all other raters did not know which of the two treatments any patient was receiving.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. “Medications for each patient were supplied by the pharmacy in an individual bottle with only the patient’s name and study number appearing on the bottle label….Therefore, the investigator who cared for the patients and all other raters did not know which of the two treatments any patient was receiving.” |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | With 5 of 25 participants (20%) leaving the study early in each treatment group the attrition was moderate. Regarding the dichotomous data, the trial authors provided an ITT‐analysis. The analyses‐methods used for the continuous data were not explicitly mentioned in the publication. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS; no raw data for the PIP). Adverse effects reporting was incomplete. |
| Other bias | High risk | “The patients were discontinued from any psychoactive drugs they had been receiving and placed directly on project medications with no intervening washout period.” “The patient sample (chronic) tended to make the finding of differences unlikely.” |
Heikkilä 1981.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: “at least 8 and in most cases 12 weeks”. Design: parallel. Location: three Finnish psychiatric hospitals (multicentre). Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 58), "various other diagnosis, like paranoic state, depressive or personality disorders” (N = 5). N = 63. Gender: 41M, 22F. Age: mean 42.7 years. History: duration stable: not indicated, duration of illness: "40 patients had been ill more than 10 years and a further 11 more than 5 years.", number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: “Previous neuroleptic treatment consisted of 14 different neuroleptics. About two‐third of the patients received more than one neuroleptic.” |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 2 mg to 24 mg/day, mean final dose: 10 mg/day. N = 33. 2. Cis(Z)‐clopenthixol: fixed/flexible dose: not indicated, dose range: 10 mg to 75 mg/day, mean final dose: 40 mg/day. N = 30. Rescue medication: antiparkinson drugs and hypnotics/sedatives were allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impression (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Adverse effects: use of antiparkinson medication. Unable to use: Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE‐30) (no SDs available and no imputation method could be applied). |
|
| Notes | Study participants were 63 “chronic schizophrenic in‐patients or other psychotic in‐patients”. “Test treatment was maintained for 8 weeks in 54 (26 [cis(Z)‐clopenthixol] +28 [haloperidol]) patients and for 12 weeks in 36 (17 [cis(Z)‐clopenthixol] +19 [haloperidol]) patients.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind, double‐dummy”. “One set of tablets was active while the other set was placebo.” No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind, double‐dummy”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 9 of 63 participants (14.3%) left the trial early until 8 weeks and 27 participants (42.6%) until week 12. Therefore the overall‐attrition was considered as high. A completers‐only analysis was used concerning the CGI. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully addressed (no SDs for the BPRS and NOSIE‐30). Adverse effects reporting was incomplete. |
| Other bias | High risk | “A planned wash‐out period preceding the start of test treatment was given up because of the severity of illness of the patients.” “Test treatment according to plan was given for only 8 weeks in 14 patients.” “Clopenthixol and haloperidol were administered rather frequently in the pre‐trial period, since 11 patients received clopenthixol and 16 haloperidol.” |
Heikkilä 1992.
| Methods | Randomisation: “randomised”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: four Finnish mental hospitals (multicentre). Setting: inpatients. |
|
| Participants | Diagnosis (38 completers of at least 4 weeks): chronic schizophrenia (N = 34), paranoid states (N = 2), reactive paranoid psychosis (N = 2). N = 49. Gender: 19M, 19F. Age: mean 36 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 2 mg to 30 mg/day, mean dose: 10.3 mg/day. N = 23. 2. Zuclopenthixol: flexible dose, dose range: 10 mg to 75 mg/day, mean dose: 33.5 mg/day. N = 26. “The doses were chosen on the basis of the condition of the patients.” “According to the protocol, the doses should be individually adjusted according to the patient´s response to treatment.” Rescue medication: “Biperiden could be prescribed in case of extrapyramidal side‐effects and nitrazepam or chloral hydrate could be given as a hypnotic.” |
|
| Outcomes | Examined: Clinically important response to treatment: “a slightly modified version of the Clinical Global Impressions Scale (CGI)”. Mental state general: 16‐item Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Adverse effects: use of antiparkinson medication. Unable to use: Global state general: CGI (modified version). Adverse effects: UKU Side‐Effekt Rating Scale (no information regarding the occurrence of adverse effects over the whole trial duration). |
|
| Notes | Study participants were 49 participants “with acute psychotic states” (38 completers of at least 4 weeks drug treatment). Criteria for study inclusion were “diagnosis of acute schizophrenia or an exacerbation of chronic schizophrenia, paranoid states or reactive paranoid psychosis” and a 16‐item BPRS total score >25. “All patients treated for at least 4 weeks were included in the statistical analyses of the results.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomised”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “tablets of identical appearance” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The overall‐attrition was high: 38 of 49 participants (77.6%) left the trial early. The trial authors indicated that 18 of 23 participants (78.3%) in the haloperidol‐group and 20 of 26 participants (76.9%) in the zuclopenthixol‐group discontinued the drug treatment prematurely. |
| Selective reporting (reporting bias) | Unclear risk | The reported adverse effects data were not usable for this systematic review. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Hollister 1962.
| Methods | Randomisation: “randomised blocks of 24”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: five Veterans Administration hospitals (multicentre). Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 59), undifferentiated schizophrenia (N = 23), catatonic schizophrenia (N = 7), hebephrenic schizophrenia (N = 3), other classes of schizophrenia (N = 4). N = 112. Gender: 112M. Age: mean 36 years. History: duration stable: not indicated, duration of illness: mean 7 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dosage scheme, dose range: not indicated, mean final dose: 8 mg/day. N = 52 (study completers of at least 6 weeks). 2. Thiopropazate: fixed‐flexible dosage scheme, dose range: not indicated, mean final dose: 80 mg/day. N = 44 (study completers of at least 6 weeks). “A fixed dose of drug was used during the first two weeks of treatment and flexible doses thereafter.” Rescue medication: “Adjunctive treatment with anticholinergics was allowed for those patients developing extrapyramidal syndromes.” |
|
| Outcomes | Examined: Clinically important response to treatment: Global Rating Form. Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Adverse Effects (at 6 weeks): at least one adverse effect, akathisia, rigor, tremor, hypotension. Unable to use: Behaviour: Nurse´s Evaluation Form (scale not published, no SDs available). |
|
| Notes | Study participants were “112 newly admitted schizophrenic men”. “Treatment was started with active medication from seven to fourteen days after admission; during this initial period, patients were treated with two placebo tablets daily so their switch to active medication would be inapparent.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomised blocks of 24”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 112 participants “were admitted to the study, for whom data were complete for six weeks of treatment in 96 and for twelve weeks of treatment in 56.” “During the entire twelve‐week period of the study, 56 patients or half the original sample dropped from the study.” The analyses were based on completers‐only data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS and the Nurse’s Evaluation Form). Adverse effects were provided only for the first six weeks of treatment. No information concerning the number of participants that received a medication with anticholinergic drugs. |
| Other bias | High risk | “Incomplete data collection” regarding the global judgment by clinicians. 96 participated in the study for at least 6 weeks. According to table 2 in the publication 52 participants were in the haloperidol‐group and 44 participants in the thiopropazate‐group. But according to table 5 after 6 weeks of treatment the proportion was conversely (44 participants in the haloperidol‐group and 52 participants in the thiopropazate‐group). |
Howard 1974.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: maximum trial duration 12 weeks (“Patients were considered treatment failures and dropped from the double‐blind study, when, in the opinion of the investigator, continued treatment was not likely to elicit further improvement.”) Design: parallel. Location: Eastern State Hospital, Williamsburg, Virginia. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (N = 26), manic depression (N = 4), psychotic reaction secondary to trauma (N = 1), psychosis with mental deficiency (N = 1), involutional psychosis (N = 1). N = 33. Gender: 33F. Age: mean 46 years. History: duration stable: not indicated, duration of illness: mean 20 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum dose 200 mg/day, mean dose: not indicated. N = 17. 2. Thiothixene: flexible dose, dose range: maximum dose 200 mg/day, mean dose: not indicated. N = 16. “Initial dosage and subsequent titration of the medication were individually determined according to the judgement of the investigator….Maximum daily dosage did not exceed 200 mg of either active compound.” “On the average, patients in active drug groups had been receiving 670 mg daily (range 200 mg – 2450 mg) (ATE, approximate thorazine equivalent) of neuroleptic medication prior to the study, and were receiving 3694 mg (range 600 mg – 8000 mg) (ATE) daily at the time of discharge.” “High dose neuroleptic therapy for refractory chronic patients.” Rescue medication: “therapeutic, but not prophylactic, administration of antiparkinson medication for the control of extrapyramidal reactions” |
|
| Outcomes | Examined: Clinically important response to treatment: “all‐or‐none” response to the study medications. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Mental state general: Mental Status Checklist (no raw data available). Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no raw data available). Adverse Effects (no data concerning the haloperidol group available). |
|
| Notes | Study participants were 33 (together with the placebo‐group 46) women characterised as “treatment resistant, and hopeless chronic psychotics”. No fix endpoint of the trial. Release from hospital was “achieved after an average of 6 weeks of treatment with haloperidol and 5, 4 weeks of treatment with thiothixene.” “Prior to initiation of the study medications, all patients received placebo during a two‐week washout…..Only those patients whose clinical status remained stable or who regressed during the washout period were advanced to the double‐blind portions of the study. Patients in whom dramatic improvement occurred as a result of discontinuation previous medication were not included.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “The study medications were prepared in identical appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, NOSIE, and the Mental Status Checklist). No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | High risk | The trial was not characterised through a definite endpoint the data‐analyses were based on. |
Itoh 1985.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenia. N = 164. Gender: not indicated. Age: not indicated. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: "fixed‐flexible dosage schedule", dose range: maximum dose 18 mg/day, mean dose: not indicated. N = 80. 2. Bromperidol: “fixed‐flexible dosage schedule", dose range: maximum dose 18 mg/day, mean dose: not indicated. N = 84. |
|
| Outcomes | Examined: Clinically important response to treatment: General Improvement Rating. Mental state general: Brief Psychiatric Rating Scale (BPRS). Adverse effects: at least one adverse effect, at least one movement disorder, akathisia, dystonia. Unable to use: Global state general: Global Usefulness Rating (scale not published). Mental state general: Keio University Psychiatric Rating Scale for Schizophrenia (no SDs available and no imputation method could be applied). Adverse effects: Overall Safety Rating (scale not published). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the Keio University Psychiatric Rating Scale). Adverse effects reporting was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Kariya 1983.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: “multi‐clinic” (29 institutes). Setting: “mainly inpatients”. |
|
| Participants | Diagnosis: schizophrenia. N = 212. Gender (N = 206): 109M, 97F. Age: not indicated. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: "fixed‐flexible method", dose range: maximum dose 18 mg/day, mean dose: not indicated. N = 106 (study completers). 2. Timiperone: "fixed‐flexible method", dose range: maximum dose 12 mg/day, mean dose: not indicated. N = 100 (study completers). Rescue medication: anti‐Parkinsonian and hypnotics were allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Global improvement rating. Adverse effects: at least one movement disorder, akathisia, dyskinesia, rigor. Unable to use: Mental state general: Keio University Psychiatric Symptoms Rating Scale (no raw data available). |
|
| Notes | Of the 212 participants “206 cases were finally subjected to the statistical analysis”. "The patients were relatively fresh cases with the clinical state of deficiency of initiative, blunted affect, hallucinations and delusions”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned at random to either the timiperone or the haloperidol group”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Both drugs were confirmed as being indistinguishable from each other, having an identical appearance, and identical colour shades and weights, by two controllers.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With missing data of 6 of the 212 randomised participants (2.8%), the overall attrition was rather low. The analyses were based on completers‐only data, but due to the small drop‐out rate, the risk of bias might be considered as being low. |
| Selective reporting (reporting bias) | High risk | The numbers of participants randomised to each treatment group (haloperidol or timiperone) were not indicated. These data were only available for the completers of the trial. The outcome data were not fully addressed (no raw data for the Keio University Psychiatric Symptoms Rating Scale). No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Kinon 1993.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: Hillside Hospital, Long Island Jewish Medical Center, Glen Oaks, New York. Setting: inpatients (newly admitted). |
|
| Participants | Diagnosis: schizophrenia, schizoaffective disorder, or schizophrenian disorder (DSM‐III‐R). N = 58. Gender: not indicated. Age: mean: 29.4 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed dose, dose range: not indicated, mean dose: 20 mg/day. N = 13. 2. Fluphenazine: fixed dose, dose range: not indicated, mean dose: 20 mg/day. N = 18. 3. Fluphenazine: fixed dose, dose range: not indicated, mean dose: 80 mg/day. N = 16. Rescue medication: concurrent benztropine for all participants. |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impressions (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS). Unable to use: Modified Scale for the Assessment of Negative Symptoms (SANS) (modified version of the scale). Modified Simpson‐Angus Extrapyramidal Scale (SAEPS) (modified version of the scale). |
|
| Notes | “DSM‐III‐R diagnosis of schizophrenia, schizoaffective disorder, or schizophrenian disorder”; “symptom severity of at least moderate on at least one of the four BPRS psychotic symptom items”. A total of 58 nonresponders to a 4‐week open‐treatment with fluphenazine 20 mg/day entered the double‐blind phase of this study” (45 completer). “Inpatients were treated openly with fluphenazine (FPZ) 20 mg/day and with prophylactic benztropine for 4 weeks. Those subjects who failed to meet a priori criteria for substantial therapeutic response [“rating of mild or better on each of the four BPRS psychotic items and a rating of much improved or better on the CGI”] at the end of week 4 were randomised to receive double‐blind treatment for an additional 4 weeks.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 13 of 58 participants (22.4%) left the investigated double‐blind phase of the trail early. The numbers of participants randomised to each treatment group (haloperidol or fluphenazine) were not indicated. The trial authors provided completers‐only analyses, but in terms of the outcome “response status” the responders who left the trial prematurely were included in the results but not the drop‐outs who were classified as non‐responder. |
| Selective reporting (reporting bias) | Unclear risk | Adverse effects reporting was incomplete. |
| Other bias | High risk | Only non‐responders to an open 4‐week fluphenazine 20 mg/day trial were included in the randomised phase of the study. This can be considered as potential source of bias. Only 58 of the 78 non‐responders to the open fluphenazine trial entered the double‐blind phase of the study. |
Kodama 1984.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: “multi‐clinic” (11 institutes). Setting: inpatients and outpatients. |
|
| Participants | Diagnosis: schizophrenia. N = 68. Gender (N = 66): 26M, 40F. Age: "most of the participants were between 30 and 49 years", no further details available. History: duration stable: not indicated, duration of illness: 5‐15 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: "fixed‐flexible method", dose range: 2 mg to 30 mg/day, mean dose: not indicated. N = 32. 2. Bromperidol: "fixed‐flexible method", dose range: 3 mg to 30 mg/day, mean dose: not indicated. N = 34. Rescue medication: "anti‐parkinsonian and hypnotics were allowed." |
|
| Outcomes | Examined: Clinically important response to treatment: Global improvement rating. Leaving the study early due to any reason. Leaving the study early due to adverse effects. Adverse effects: akathisia, akinesia, dyskinesia, dystonia, tremor, use of antiparkinson medication, hypotension, weight gain. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). |
|
| Notes | "66 cases were finally subjected to the statistical analysis. 2 cases left the study after 2 days and 3 days from the beginning, which were unable to evaluate the effect.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned at random to either the bromperidol or the haloperidol group”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Both drugs were confirmed as being indistinguishable from each other, having an identical appearance, and identical colour shades and weights.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of participants randomised to each treatment group were 36 for Bromperidol and 32 for Haloperidol. 2 cases left the study after 2 days and 3 days from the beginning. Therefore, the data of 2 cases from the 68 randomised participants were unable to evaluate the effect and were not subjected to statistic analysis. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS) |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Kurihara 1983.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel (three‐arm study also investigating clocapramin). Location: “multi‐clinic” (42 institutes). Setting: “mainly inpatients”. |
|
| Participants | Diagnosis: schizophrenia. N = 189. Gender: 105M, 84F. Age: "most of the participants were between 30 and 49 years", no further details available. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: “fixed‐flexible method”, dose range: 3 mg to 12 mg/day, mean dose: not indicated. N = 94. 2. Perphenazine: “fixed‐flexible method”, dose range: 9 mg to 36 mg/day, mean dose: not indicated. N = 95. Rescue medication: anti‐Parkinsonian and hypnotics were allowed. |
|
| Outcomes | Examined: Adverse effects: akathisia, dyskinesia, dystonia. Unable to use: Global state general: Global improvement rating (no raw data available). Mental state general: Keio University Psychiatric Symptoms Rating Scale (no raw data available). |
|
| Notes | “The patients were relatively fresh cases with the clinical state of deficiency of initiative, blunted affect, hallucinations and delusions”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned at random to the clocapramine, perphenazine and the haloperidol group”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Both drugs were confirmed as being indistinguishable from each other, having an identical appearance, and identical colour shades and weights.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 32 patients who left the study early, but all data of the 286 randomised participants were used. |
| Selective reporting (reporting bias) | High risk | The number of participants randomised to each treatment group (haloperidol, clocapramine, or perphenazine) was not indicated. These data were only available for the completers of the trial. The outcome data were not fully addressed (no raw data for the Global improvement rating and the Keio University Psychiatric Symptoms Rating Scale). No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Luckey 1967.
| Methods | Randomisation: “random assignment”. Blinding: double‐blind. Duration: two‐week washout period (with placebo); followed by 12 weeks on active drug. Design: parallel. Location: Minneapolis Veterans Administration Outpatient Clinic. Setting: outpatients. |
|
| Participants | Diagnosis (study completers, N = 9): paranoid schizophrenia (N = 4), schizoaffective schizophrenia (N = 3), undifferentiated schizophrenia (N = 2). N = 26. Gender: “most were male”, no further details available. Age: not indicated. History: duration stable: not indicated, duration of illness: "at least one year immediately prior the study", number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum dose 15 mg/day, mean dose: not indicated. N = 13. 2. Trifluoperazine: flexible dose, dose range: maximum dose 30 mg/day, mean dose: not indicated. N = 13. dosage scheme: “a ratio of 2mg trifluoperazine to 1mg haloperidol” “we expected to increase the initial dosage by one capsule [2,5mg haloperidol or 5mg trifluoperazine] at each evaluation time until the dosage was four capsules [10mg haloperidol or 20mg trifluoperazine].” Rescue medication: benztropine mesylate (Cogentin) (1 to 2mg/day). |
|
| Outcomes | Examined: Clinically important response to treatment: Global clinical judgement. Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Adverse effects: checklist of side effects: at least one movement disorder. Unable to use: Behaviour: Minnesota Multiphasic Personality Inventory (MMPI) (no raw data available). |
|
| Notes | Study participants were 26 chronic schizophrenic outpatients. “Of the 26 patients who began the study, 21 completed one month or more of active medication.” “we made use of data on any patient who completed one month or more of active medication”. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Random assignment”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Haloperidol, trifluoperazine and placebo were prepared in identical capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The overall‐attrition was very high (17 of 26 participants; 65.4%). 8 of 13 participants left the trial early in the haloperidol‐group (61.5%) and 9 of 13 participants in the trifluoperazine‐group (69.2%). A completers‐only analysis was used regarding the main outcome (global clinical judgment) and for the other outcomes the analysis was based on the results of one month active drug treatment. |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no SDs for the BPRS; no raw data for the MMPI). Only the most prevalent adverse effects were reported. |
| Other bias | High risk | “Due to the large number of dropouts and the overall limitations imposed by the small population, none of the results were statistically significant.” “The dosage ratio of 2mg trifluoperazine to 1mg haloperidol was probably too low and may have put haloperidol at a disadvantage.” |
Malfroid 1978.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 12), catatonic schizophrenia (N = 9), chronic psychosis (N = 3). N = 24. Gender: 24M. Age: mean 48.6 years. History: duration stable: not indicated, duration of illness: mean 25.3 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 6‐8 mg/day. N = 12. 2. Bromperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 4‐8 mg/day: N = 12. Rescue medication: anticholinergics were allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Global efficacy scale. Adverse effects: dyskinesia, use of antiparkinson medication, hypotension. Unable to use: Mental state general: Psychiatric rating scale (designed by the “Wirtschafts‐Mathematik Zürich”) (no raw data available). |
|
| Notes | Study participants were 24 “chronic psychotic patients”. “Before the trial period, all patients were on a maintenance neuroleptic treatment”. No participant with first‐episode schizophrenia was enrolled. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully addressed (no raw data for the psychiatric rating scale). Adverse effects reporting was incomplete. BPRS is mentioned in the abstract but no results were presented. |
| Other bias | High risk | “Before the trial period, all patients were on a maintenance neuroleptic treatment.” |
Mattke 1976.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: Max Planck Institut of Psychiatry, Munich, Germany. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenic psychosis (ICD‐8: 295.1, 295.2, 295.3, and 295.8). N = 40. Gender: 17M, 23F. Age: mean 29 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dose, dose range: not indicated, mean dose: not indicated. N = 18. 2. Loxapine: fixed‐flexible dose, dose range: not indicated, mean dose: not indicated. N = 22. Rescue medication: chloralhydrate in the case of agitation or sleep disturbances. |
|
| Outcomes | Unable to use: Measurement of the pupillary diameter. |
|
| Notes | 4‐day drug‐free phase before first administration of the study medication. The aim of this research project was to study pupillomotorics under the influence of neuroleptic medication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. |
| Other bias | Unclear risk | The aim of this research project was to study pupillomotorics under the influence of neuroleptic medication. Insufficient information to assess whether an important risk of bias exists. |
Mauri 1994.
| Methods | Randomisation: “randomised”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 18), indifferent schizophrenia (N = 10), disorganised schizophrenia (N = 6), schizophreniform disorder (N = 4), residual schizophrenia (N = 2), schizoaffective disorder (N = 2). N = 40. Gender: 23M, 17F. Age: mean 33.7 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: "15 subjects in each group received a pharmacotherapy with antipsychotics before study entry." |
|
| Interventions | 1. Haloperidol: fixed dose, dose range: not indicated, mean dose: 10 mg/day. N = 20. 2. Bromperidol: fixed dose, dose range: not indicated, mean dose: 10 mg/day. N = 20. “The initial dose of both drugs, 10 mg/day, was maintained for the eight weeks of study.” Rescue medication: antiparkinson medication if necessary. |
|
| Outcomes | Examined: Global state general: Clinical Global Impressions (CGI) (no SDs available). Mental state general: Brief Psychiatric Rating Scale (BPRS). Mental state specific: Scale for the Assessment of Positive Symptoms (SAPS). Mental state specific: Scale for the Assessment of Negative Symptoms (SANS). Mental state specific: Hamilton Depression Rating Scale (HAM‐D). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one adverse effect, akathisia, dystonia, rigor, tardive dyskinesia, tremor, use of antiparkinson medication, hypotension, weight gain. |
|
| Notes | 19 participants with "an exacerbation of a chronic schizophrenic disorder", Three subjects with first episode. One week wash‐out phase with placebo before administration of the study drugs. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomised”. Ratio bromperidol to haloperidol 1:1. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. Identical appearing capsules. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 of 20 participants (20%) left the trial early in the haloperidol‐group and 7 of 20 participants (35%) discontinued prematurely in the bromperidol‐group. The overall‐attrition was rather high (11 of 40 participants; 27.5%). The trial authors used a split‐plot ANOVA for the data‐analyses. |
| Selective reporting (reporting bias) | Unclear risk | The outcome data were not fully reported (no SDs for the CGI). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Mori 1989.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: not indicated. Setting: “mainly inpatients” (inpatients and outpatients). |
|
| Participants | Diagnosis: schizophrenia. N = 167. Gender: 101M, 66F. Age: not indicated. History: duration stable: not indicated, duration of illness: "most of the participants have schizophrenia for more than 10 years", number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: maximum dose 20 mg/day, mean dose: not indicated. N = 86. 2. Nemonapride: fixed/flexible dose: not indicated, dose range: maximum dose 30 mg/day, mean dose: not indicated. N = 81. Rescue medication: anti‐Parkinsonian, hypnotics, antipsychotics, antidepressant, and anti‐anxiety drugs were allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Global Improvement rating. Leaving the study early due to any reason. Adverse effects: akathisia, akinesia, dystonia, rigor, tardive dystonia, tremor, use of antiparkinson medication, sedation. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Mental state specific: Scale for the Assessment of Negative Symptoms (SANS) (no raw data available). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned at random to either the YM‐09151 or the Haloperidol group”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Both drugs were confirmed as being indistinguishable from each other, having an identical appearance, and identical colour shades and weights, by two controllers.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of participants randomised to each treatment group were 81 for YM‐09151 and 86 for Haloperidol. 27 participants left the study early, but the uncompleted data were also subjected to statistic analysis. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS and SANS). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Nedopil 1981.
| Methods | Randomisation: randomised (according to e‐mail correspondence with the first author). Blinding: “Raters of this study were blind to the actual dose of benperidol given.” No further details. Duration: 20 days. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (ICD‐8: 295.3) (N = 21), hebephrenec schizophrenia (ICD‐8: 295.1) (N = 6), catatonic schizophrenia (ICD‐8: 295.2) (N = 3), schizoaffective schizophrenia (ICD‐8: 295.7) (N = 2), coenesthetic schizophrenia (ICD‐8: 295.8) (N = 1). N = 33. Gender: 16M, 17F. Age: mean 32 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed dose, dose range: not indicated, mean dose: 14 mg/day. N = 14. 2. Benperidol: fixed dose, dose range: not indicated, mean dose: 3 mg/day. N = 10. 3. Benperidol: fixed dose, dose range: not indicated, mean dose: 12 mg/day. N = 9. Rescue medication: not indicated. |
|
| Outcomes | Examined: Mental state specific: Psychopathology according to the AMDP‐system (paranoid and hallucinatory syndrome). |
|
| Notes | Study participants were "33 newly admitted schizophrenic patients, who displayed among their symptoms both delusions and hallucinations.“ The participants were randomised to either 14 mg/day haloperidol, 3 mg/day benperidol or 12 mg/day benperidol. The main aim of this trial was to evaluate “the initial improvement after the onset of neuroleptic treatment……for its predictive value” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | High risk | No blinding of participants or personnel mentioned in the publication. |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Raters of this study were blind to the actual dose of benperidol given.” No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs were available for all data of the AMDP‐ratings). Adverse effects were not reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Nishikawa 1984.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: maximum 1 year. “The assigned drug was administered until relapse signs or adverse effects appeared.” Design: parallel (also investigating placebo). Location: Seiwakai Nishikawa Hospital, Hamada, Japan. Setting: outpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III). N = 74. Gender: 44M, 30F. Age: mean ˜ 39 years. History: duration stable: "all subjects in remission"not indicated, duration of illness: mean ˜ 8.7 years, number of previous hospitalisations: mean ˜ 3.8, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed dose, dose range: 1 mg to 6 mg/day, mean dose: 3.27 mg/day. N = 37. 2. Propericiazine: fixed dose, dose range: 10 mg to 60 mg/day, mean dose: 32.7 mg/day. N = 37. Rescue medication: “Each drug was combined with nitrazepam 10 mg and biperidine 6 mg to prevent insomnia and drug‐induced parkinsonism, respectively.” |
|
| Outcomes | Unable to use: Relapse (no predefined outcome of interest) |
|
| Notes | 74 “schizophrenic outpatients who satisfied the diagnosis criteria of DSM‐III for the recovery stage of remission or residual phase.” “The number of symptom‐free days for each patient was recorded and the trial was terminated after 1 year.” No participant with a first episode. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Drug appearance, with respect to powder color, taste and volume, was made identical by adding a gastric aid, SMP.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Because the main outcome of this trial was the evaluation of the relapse‐rates in both medication groups (haloperidol and propericiazine) it is not possible to judge the risk of bias in terms of the addressing of incomplete outcome data. |
| Selective reporting (reporting bias) | Unclear risk | The main outcome of the trial was not of interest for this systematic review. The adverse effects were not fully addressed. |
| Other bias | High risk | “All patients….received ordinary, brief psychotherapy every 2 weeks by a psychiatrist”. Baseline imbalance concerning the sex distribution between the haloperidol‐groups and the propericiazine‐groups. |
Nishimatu 1975.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: “multi‐clinic” (10 institutes). Setting: “mainly inpatients”. |
|
| Participants | Diagnosis: schizophrenia. N = 82. Gender: 51M, 21F. Age: mean 29.8 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dose, dose range: minimum dose 6 mg/day, mean dose: not indicated. N = 37. 2. Methylperidol: fixed‐flexible dose, dose range: minimum dose 30 mg/day, mean dose: not indicated. N = 35. Rescue medication: anti‐Parkinsonian and hypnotics were allowed. |
|
| Outcomes | Examined: Adverse effects: akathisia, rigor. Unable to use: Global state general: Global improvement rating (no raw data available). Mental state general: Keio University Psychiatric Symptoms Rating Scale (no raw data available). |
|
| Notes | “The patients were relatively fresh cases with the clinical state of deficiency of initiative, blunted affect, hallucinations and delusions”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were assigned at random to either the methylperidol or the haloperidol group”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Both drugs were confirmed as being indistinguishable from each other, having an identical appearance, and identical colour shades and weights.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | With missing data of 10 of the 82 randomised participants. The analyses were based on completers‐only data, the risk of bias might be considered as being low. |
| Selective reporting (reporting bias) | High risk | The number of participants randomised to each treatment group (haloperidol or methylperidol) was not indicated. These data were only available for the completers of the trial. The outcome data were not fully addressed (no raw data for the Global improvement rating and the Keio University Psychiatric Symptoms Rating Scale). No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Okuda 1979.
| Methods | Randomisation: 2 participants built one pair and 2 medications were assigned at random to each pair. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: “multi‐clinic” (3 institutes). Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (34 participants with acute schizophrenia and 40 with chronic schizophrenia.). N = 74. Gender: 36M, 28F. Age: 14‐54 years, no further details available. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dose, dose range: 3 mg to 10mg/day, mean dose: not indicated. N = 37. 2. Sulpiride: fixed‐flexible method, dose range: 300 mg to 1200mg/day, mean dose: not indicated. N = 37. Rescue medication: “Anti‐Parkinsonian, hypnotics and intestinal medicine were allowed.” |
|
| Outcomes | Examined: Clinically important response to treatment: General improvement rating. Leaving the study early due to any reason. Adverse effects: akathisia, dyskinesia. Unable to use: Mental state general: Keio University Psychiatric Symptoms Rating Scale (no raw data available). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Two participants built one pair and two medications were assigned at random to each pair. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of participants randomised to each treatment group was 37 for Sulpiride and 37 for Haloperidol. 12 participants left the study early, but the uncompleted data were also subjected to statistical analysis. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the Keio University Psychiatric Symptoms Rating Scale). |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
O´Brien 1974.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 3 weeks. Design: parallel. Location: Hospital of the University of Pennsylvania, Philadelphia Naval Hospital (two‐centre). Setting: inpatients. |
|
| Participants | Diagnosis (N = 24): paranoid schizophrenia (N = 20), paranoid personality (N = 2), paranoid state (N = 1), explosive personality (N = 1). N = 30. Gender: 22M, 2F. Age: mean 29.5 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum dose 20 mg/day, mean dose: not indicated. N = 15. 2. Trifluoperazine: flexible dose, dose range: maximum dose 48 mg/day, mean dose: not indicated. N = 15. Rescue medication: “benztropine was given only when necessary to control extra pyramidal reactions.” “If parenteral medication was required, intramuscular sodium amytal was given.” |
|
| Outcomes | Examined: Clinically important response to treatment: Global clinical outcome rating. Leaving the study early due to any reason. Adverse effects: use of antiparkinson medication. Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available; only the average improvement was provided, no SDs available). Mental state specific: Hamilton Depression Rating Scale (HAM‐D) (no raw data available). Mental state specific: Global Hostility Scale (GH) (no raw data available; only the average improvement was provided, no SDs available). Mental state specific: Global Paranoia Scale (GP) (no raw data available; only the average improvement was provided, no SDs available). |
|
| Notes | Study participants were characterised as “hostile, suspicious, uncooperative patients” and a “population of hostile suspicious patients”. “Starting sample was 30 patients but 6 …. were dropped from study”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind fashion”. “identically appearing pink capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind fashion”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The attrition was moderate (3 of 15 participants (20%) in both study arms). Completers‐only analyses were used in the study. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, GH, GP, and HAM‐D). Adverse effects were not fully addressed. |
| Other bias | High risk | “Milieu therapy, group therapy, and individual sessions with a psychiatrist” were allowed during the trial. |
Paprocki 1976.
| Methods | Randomisation: “administered at random”. Blinding: double‐blind. Duration: 90 days. Design: parallel. Location: State hospital “Instituto Raul Soares”, Belo Horizonte, Brasil. Setting: inpatients. |
|
| Participants | Diagnosis: acute schizophrenia. N = 50. Gender: 50F. Age: not indicated. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. N = 25. 2. Loxapine: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. N = 25. |
|
| Outcomes | Unable to use: Global state general: Clinical Global Impression (CGI) (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no raw data available). Adverse effects: Treatment Emergent Symptoms Scale (TESS) (no raw data available). |
|
| Notes | Study participants were 50 “acute newly hospitalized” female schizophrenic participants. "overall study of data obtained from three different trials." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Administered at random”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully addressed (no raw data for the BPRS, CGI, NOSIE, and TESS). Adverse effects reporting was incomplete. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Parent 1983.
| Methods | Randomisation: “treated at random”. Blinding: “open” study (not double‐blind). Duration: 28 days. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenic psychosis: simple type (N = 11), paranoid type (N = 4), schizo‐affective type (N = 2), acute schizophrenic episode (N = 1), latent schizophrenia (N = 1), residual schizophrenia (N = 1), specific reading retardation (N = 1). N = 21. Gender (N = 40): 21M, 19F. Age: mean 43.4 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: "a score of 23 or more on the Psychiatrists´ Clinical Global Impression Rating”, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 8 mg to 50 mg/day, mean dose: 21.1 mg/day. N = 10. 2. Flupenthixol: fixed/flexible dose: not indicated, dose range: 32 mg to 192 mg/day, mean dose: 111.3 mg/day. N = 11. Rescue medication: “If there were any extrapyramidal side‐effects, procyclidine might be given, and, if necessary, a benzodiazepine might be administered at night.” “During the first period of treatment dosage was titrated to the optimum. Initial dosage was determined by the severity of disease and by the age of the patient.” |
|
| Outcomes | Examined: Clinically important response to treatment: “modification of the Psychiatrists´Clinical Global Impression”. Mental state general: Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Adverse effects: at least one movement disorder, use of antiparkinson medication. Unable to use: Behaviour: Nurses` Clinical Impression (no raw data available). |
|
| Notes | The whole sample size of the trial included 40 participants (21 with schizophrenic psychosis, 13 with manic‐depressive psychosis and 6 with paranoid states). The results of the psychiatric assessments were provided separately for the 21 schizophrenic participants. Study participants were 21 participants with acute schizophrenic psychosis (“acutely psychotic patients"). 7 participants with first episode. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Allocation through “randomisation”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | High risk | “The study was open”. |
| Blinding (performance bias and detection bias) (detection bias) | High risk | “The study was open”. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The overall‐attrition was rather low (2 of 21 participants, 9.5%). One participant in each treatment group left the trial early. Although it was not explicitly mentioned which type of analysis was used for the BPRS, it was not considered as bias because of the low drop‐out rate in each treatment group. |
| Selective reporting (reporting bias) | Unclear risk | Raw data of the Nurses` Clinical Impression scale were not reported; but this was not relevant for the outcomes of interest in this systematic review. Adverse effects reporting was incomplete. |
| Other bias | High risk | A high dosage of flupenthixol was compared to conventional dosages of haloperidol. This can be considered as potential bias. Administration of benzodiazepines in 9 participants of each treatment group. “1 patient in each group was treated with an additional neuroleptic.” “There was some imbalance between the treatment groups with regard to previous acute episodes.” |
Pool 1976.
| Methods | Randomisation: “random assignment”. Allocation: “prearranged randomised procedure”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel (three‐arm study also investigating placebo). Location: Southeast Louisiana Hospital Adolescent Unit, Mandeville, Lousiana. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia. N = 51. Gender: 28M, 23F. Age: mean 15.7 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dosage scheme, dose range: not indicated, mean dose 9.8 mg/day. N = 25. 2. Loxapine: fixed‐flexible dosage scheme, dose range: not indicated, mean dose 87.5 mg/day. N = 26. Fixed‐flexible dosage scheme: After day 15 “the dosage regimen was then made flexible and could be regulated according to individual patient response” Rescue medication: ”antiparkinson agents when necessary for control of extrapyramidal side reactions and sodium amobarbital for night‐time sedation in extremely agitated patients or for severe insomnia.” |
|
| Outcomes | Examined: Adverse effects: at least one movement disorder, sedation. Unable to use: Global state general: Clinical Global Impression (CGI) (no raw data available). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no raw data available). |
|
| Notes | Study participants were 51 newly‐admitted adolescent participants with a diagnosis of schizophrenia, acute or chronic with exacerbation. They had an “undisputed diagnosis of schizophrenia associated with a gross disorder of thought associations and/or hallucinations at the time of admission.” “Selected subjects were maintained without psychotropic drugs for at least five days prior to entering this study.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Random assignment”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | “Prearranged randomised procedure”. No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “Medication was prepared in capsules of identical appearance and was supplied in individual bottles to each of the study subjects. These bottles were labeled with each subject’s study number, thus assuring that evaluating personnel would be unable to determine the drug group to which a given patient belonged, or even that any given patients were receiving the same drug.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. “Medication was prepared in capsules of identical appearance and was supplied in individual bottles to each of the study subjects. These bottles were labeled with each subject’s study number, thus assuring that evaluating personnel would be unable to determine the drug group to which a given patient belonged, or even that any given patients were receiving the same drug.” |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No drop‐out rates were provided; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, CGI, and NOSIE). Only the most prevalent adverse effects were reported. No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | High risk | “Patients who failed to complete four weeks of daily medication because of voluntary withdrawal or for administrative reasons were not included in the analyses of efficacy ratings and were replaced by new patients.” Extreme imbalance regarding sex distribution between the haloperidol‐group and loxapine‐group. |
Pöldinger 1977.
| Methods | Randomisation: “assigned at random”. Blinding: double‐blind. Duration: 28 days. Design: parallel. Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: schizophrenic syndromes (N = 33), excitable personality (N = 4), paranoid syndrome (N = 1), reactive excitement (N = 1), hypochondrial neurosis (N = 1). N = 40. Gender: not indicated. Age: mean 49.7 years. History: duration stable: not indicated, duration of illness: mean 14.35 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 5 mg to 9 mg/day, mean final dose: 6.6 mg/day. N = 20. 2. Bromperidol: flexible dose, dose range: 5 mg to 12 mg/day, mean final dose: 6.6 mg/day. N = 20. Flexible dose: “a uniform initial dose of 5 mg/day was chosen; this dose level could be increased or reduced in the further course of the treatment according to the patients´ individual needs.” Rescue medication: administration of biperiden HCI (Akineton) was allowed. “Administration of other psychotropic drugs was permitted in very urgent cases only.” |
|
| Outcomes | Examined: Clinically important response to treatment: Global evaluation of the effectiveness of both drugs. Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one movement disorder, use of antiparkinson medication, hypotension. Unable to use: Mental state general: “standard case report forms” (“among others – a 29‐item scale for the evaluation of the patients´ psychic conditions”) (scales not published). |
|
| Notes | “All but 3 patients had been on other neuroleptics before the study; the effect of the previous treatment was scored moderate in 19 cases, insufficient in 17 cases, and poor in 1 case.” 5 participants with first episode. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Assigned at random”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “identical‐looking tablets”. Tablets “were supplied in coded packages for patients´use”. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “The treatment scheduled for 28 days was completed by all patients.” |
| Selective reporting (reporting bias) | High risk | No explicit description of the outcomes in the methods section of the publication. The outcome data were not fully addressed (no SDs available). |
| Other bias | High risk | “All but 3 patients had been on other neuroleptics before the study; the effect of the previous treatment was scored moderate in 19 cases, insufficient in 17 cases, and poor in 1 case.” Comment: The review authors assumed that many of the participants included in this trial were non‐responders to previous medications. |
Rama Rao 1981.
| Methods | Randomisation: “randomly allocated”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 30. Gender: 30F. Age: mean 60 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: “All patients had been previously stabilised on haloperidol for at least 6 months". |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated (“the patients continued to receive the same dose as before the trial”). N = 15. 2. Sulpiride: fixed dose, dose range: not indicated, mean dose: 1200 mg/day. N = 15. Rescue medication: “15‐30 mg/day procyclidine was given where necessary” |
|
| Outcomes | Examined: Mental state general: Brief Psychiatric Rating Scale (BPRS). Behaviour: Wing’s Ward Behaviour Scale. Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: use of antiparkinson medication. |
|
| Notes | Study participants were “30 female patients who had been hospitalised for an average of more than 20 years. All had been diagnosed as suffering from schizophrenia”. Before the randomisation to either haloperidol or sulpiride all participants were stabilised on “the dosage of haloperidol which produced optimum therapeutic response.” The participants randomised to haloperidol “continued to receive the same dose as before the trial.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly allocated”. “Patients were stratified so that age and baseline morbidity were constant in each group.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “A double‐dummy technique was used.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. “A double‐dummy technique was used.” No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “Three patients were not sufficiently accessible to be rated on the BPRS.” According to the tables in the publication, no participants left the trial early. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes have been reported in the pre‐specified way with the exception of the adverse effects that were not fully addressed. The study report fails to include results for the primary outcome of the review. |
| Other bias | High risk | “The patients who received active sulpiride were in fact at a disadvantage compared to the haloperidol group who continued to receive the drug in therapeutically optimum dosage.” The trial authors “feel that a flexible dosage regime could have been even more favourable for sulpiride treatment.” |
Rubin 1971.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: ranges from 5 to 79 days (“the patient was discharged from the hospital or we had concluded the drug was not effective”). Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 13), undifferentiated schizophrenia (N = 4), manic depressive disorder (N = 1). N = 18. Gender: 18M. Age: mean 37.9 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: 2 mg to 20 mg/day, mean dose: not indicated. N = 10. 2. Trifluoperazine: flexible dose, dose range: 6 mg to 60 mg/day, mean dose: not indicated. N = 8. Rescue medication: benztropine. |
|
| Outcomes | Examined: Clinically important response to treatment: Mental Status Schedule ‐> change in this scale was used to assign the overall effectiveness. Adverse effects: use of antiparkinson medication. |
|
| Notes | Study participants were “18 newly admitted male psychiatric patients, most of whom were schizophrenics”. They were described as “acute psychotic patients”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”; “Drug was administered in identically‐appearing capsules” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the Mental Status Schedule). Adverse effects reporting was incomplete. |
| Other bias | High risk | “One patient receiving trifluoperazine began group therapy about the same time he started on drug. Two others receiving trifluoperazine began group therapy while on drug but not until the drug effect had been established.” Duration of the trial ranged from 5 to 79 days. “Because of our small sample size, the difference between the two drugs was significant only at the 0,10 level.” |
Selman 1976.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel (three‐arm study also investigating placebo). Location: Terrell StateHospital, Terrell, Texas, USA. Setting: inpatients. |
|
| Participants | Diagnosis: acute schizophrenia. N = 58. Gender: 47M, 11F. Age: mean 32.3 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: not indicated, mean final dose: 8.8 mg/day. N = 29. 2. Loxapine: flexible dose, dose range: not indicated, mean final dose: 110 mg/day. N = 29. Flexible dose: “The dosage schedule was adjusted weekly depending on the patients` clinical response” Rescue medication: “antiparkinsonism agents for extrapyramidal side effects and chloral hydrate or paraldehyde for sleep” were allowed. |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impression (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Adverse effects: at least one adverse effect, rigor, tremor. Unable to use: Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no raw data available). |
|
| Notes | Study participants with “acute symptoms of schizophrenia or an acute exacerbation of chronic schizophrenia". “For purpose of data analysis, any patients who received study medication for less than four weeks were considered to have had an inadequate trial” and were excluded on this basis (affected 4 participants). “All of the patients were completely off medication for 2 weeks before receiving the study medication.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “A double‐blind process was used in which neither patient nor investigator knew what medication was received until the study was completed.” “All medication was administered in identically appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “A double‐blind process was used in which neither patient nor investigator knew what medication was received until the study was completed.” |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The overall‐attrition was high: 19 of 58 participants (32.8%) left the trial early. The trial authors indicated that 11 of 29 participants (37.9%) in the haloperidol‐group and 8 of 29 participants (27.6%) in the loxapine‐group discontinued the drug treatment prematurely. Modified completers analyses were used (any patients who received study medication for less than 4 weeks were excluded). |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS; no raw data for the NOSIE). No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Serafetinides 1972.
| Methods | Randomisation: “randomly assigned”. Blinding: double‐blind. Duration: 12 weeks. Design: parallel (four‐arm study; additionally investigating chlorpromazine and placebo). Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 29. Gender: 12M, 17F. Age: mean 42.2 years. History: duration stable: not indicated, duration of illness: mean 14 years, number of previous hospitalisations: mean 12, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 3 mg to 15 mg/day, mean dose: 12.3 mg/day. N = 14. 2. Clopenthixol: fixed/flexible dose: not indicated, dose range: 50 mg to 250 mg/day, mean dose: 205 mg/day. N = 15. Rescue medication: “concomitant medication for Parkinsonism or bedtime sedation, when necessary, was allowed.” |
|
| Outcomes | Examined: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Clinically important response to treatment: Clinical Global Impression (CGI) Improvement. Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one movement disorder, use of antiparkinson medication, hypotension, sedation, weight gain. Unable to use: Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no SDs available and no imputation method could be applied). Behaviour: Global clinical impression by the research nurse (no raw data available). Behaviour: Oklahoma Behavior Rating Scale (OBRS) (no total score available). |
|
| Notes | Study participants were 29 “chronic schizophrenic subjects” (ill 2 years or longer). “12‐week dry‐out period” to dissipate the effects of previous treatment. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “All medications were prepared in identically appearing capsules.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. In the two investigated (haloperidol and clopenthixol) study arms no participant left the trial early. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the NOSIE and OBRS; no SDs for the BPRS). |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Shalev 1993.
| Methods | Randomisation: “randomly assigned”. Blinding: “The treating psychiatrists were blind to the sequence” of drug administration. No further details. Duration: minimum 4 weeks (first phase of the trial up to the point of first cross‐over). Design: cross‐over study also investigating levomepromazine. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐III). N = 39. Gender (including participants of the levomepromazine study group, N = 60): 35M, 25F. Age (N = 60): mean 33 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: mean 4.2, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 29.3 mg/day. N = 18 (study completers). 2. Perphenazine: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 35.8 mg/day. N = 21 (study completers). “Therapists were instructed…to reach average daily doses (defined as….32 mg/day of perphenazine and 20 mg/day of haloperidol) within one week and remain within 50% of that dose for another 3 weeks.” “Neuroleptics were administered orally. In cases of severe agitation, intramuscular administration was allowed for no more than 2 days…..Rapid increase in doses during the first days (rapid neurolpetization) was strictly avoided.” Rescue medication: “An anti‐parkinsonian drug (trihexyphenidyl, up to 10 mg per day) was used according to the patient’s condition.” |
|
| Outcomes | Examined: Clinically important response to treatment: Assessment of therapeutic success (“a decrease in the patients BPRS score of at least 30% [Psychometric criterion] and improvement of the patient’s clinical state to the point that allows the patient’s return to the community [clinical criterion]"). Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data were provided for each study group separately). |
|
| Notes | “The minimal duration of the illness required for inclusion in this study was….6 months.” “Newly hospitalised acutely exacerbated schizophrenics” were included in this study. Three antipsychotics (haloperidol, perphenazine and levomepromazine) “were administered one after the other, for 4 weeks each, in randomly determined order.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “The treating psychiatrists were blind to the sequence in which the 3 drugs were to be given to the patient.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were only data regarding the whole study sample (treated with haloperidol, perphenazine and levomepromazine) available: 15 of 75 participants (20%) left the trial early before the termination of the investigated first cross‐over‐phase. Thus the overall attrition can be considered as being moderate. Completers‐only analyses were used. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (in terms of the BPRS no raw data were provided for each study group separately). The adverse effects were not reported. Data regarding the number of participants receiving rescue medication with antiparkinson drugs were missing. |
| Other bias | High risk | Cross‐over study design. “The primary goal of this study was the evaluation of drug responsiveness in the natural clinical environment and not a comparison between drugs.” Trial duration was depended on the degree of response. |
Silverstone 1984.
| Methods | Randomisation: “randomly allocated”. Blinding: double‐blind. Duration: 28 days. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: acute schizophrenia. N = 22. Gender: 11M, 11F. Age: mean 38.5 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean final dose: 22.2 mg/day. N = 12. 2. Pimozide: fixed/flexible dose: not indicated, dose range: not indicated, mean final dose 21.6 mg/day. N = 10. Rescue medication: i.m. chlorpromazine “when the clinical situation demanded”. “Extrapyramidal side effects were treated with procyclidine. Temazepam or nitrazepam was prescribed if night sedation was required.” |
|
| Outcomes | Examined: Leaving the study early due to any reason. Adverse effects: dystonia, tremor, use of antiparkinson medication, hypotension, sedation. Unable to use: Mental state general: Montgomery Rating Scale (MRS) (no SDs available and no imputation method could be applied). Leaving the study early due to adverse effects (data only for the haloperidol group available). |
|
| Notes | Study participants were “22 patients with acute schizophrenic illness” (18 completers). 8 participants with first episode. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly allocated”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “pimozide or haloperidol in matching capsules”. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 3 drop‐outs in the haloperidol‐group (25%), and 1 participant left the trial early in the pimozide‐group (10%). The overall‐attrition was 18.2% (4 of 22 participants). Altogether the attrition was moderate. The trial authors provided the results of the completers‐only analysis. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the MRS). No usable data were provided in terms of premature discontinuation of the trial due to adverse effects. |
| Other bias | High risk | Additional antipsychotic pharmacological treatment with i.m. chlorpromazine was allowed (used in 3 participants). |
Spina 1992.
| Methods | Randomisation: “randomly allocated”. Blinding: double‐blind. Duration: 8 weeks. Design: parallel. Location: not indicated. Setting: “Patients had to be in‐patients for the first 4 weeks of treatment, and could then become out‐patients.” |
|
| Participants | Diagnosis: residual schizophrenia (N = 19), paranoid schizophrenia (N = 11), disorganised schizophrenia (N = 7), undifferentiated schizophrenia (N = 4 ), catatonic schizophrenia (N = 1). All diagnosis according to DSM‐III‐R. N = 42. Gender: 31M, 11F. Age: mean 43 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 4 mg to 9 mg/day, mean final dose: 6.7 mg/day. N = 21. 2. Bromperidol: fixed/flexible dose: not indicated, dose range: 5 mg to 15 mg/day, mean final dose: 7.1 mg/day. N = 21. “The medication started with 5 mg/day for both bromperidol and haloperidol. Dosages were then increased stepwise until maximum control of symptoms was achieved.” Rescue medication: “Anticholinergic drugs (biperiden or orphenadrine) were administered only to treat any extrapyramidal symptom that appeared.” |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impression (CGI) Improvement. Global state general: Clinical Global Impression (CGI) Severity of Illness (no SDs available). Mental state general: Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one movement disorder, dystonia, use of antiparkinson medication. Unable to use: Adverse effects: Simpson and Angus Scale (no raw data available). |
|
| Notes | “1‐week placebo washout period” before randomisation to either haloperidol or bromperidol. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Patients were randomly and blindly assigned.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 4 of 21 participants (19.0%) left the trial early in the haloperidol‐group and 5 of 21 participants (23.8%) in the bromperidol‐group. The overall‐attrition was moderate (9 of 42 participants; 21.4%). A completers‐only analysis was used. |
| Selective reporting (reporting bias) | Unclear risk | The outcomes have been reported in the pre‐specified way with the exception of the SDs in terms of the CGI Severity of Illness score and the results of the Simpson and Angus Scale. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Stewart 1969.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: not indicated. Design: cross‐over. Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: chronic schizophrenia. N = 50. Gender: 34M, 16F. Age: mean 48.6 years. History: duration stable: not indicated, duration of illness: mean 21.15 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: mean symptom severity of 1.9 (1 = mild, 2 = moderate, and 3 = severe), baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 10.4 mg/day. N = 25. 2. Trifluoperazine: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: 14.3 mg/day. N = 25. Rescue medication: antiparkinson drugs were administered to control extrapyramidal side effects. |
|
| Outcomes | Examined: Global Response assessment (senior staff psychiatrist). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: at least one adverse effect, at least one movement disorder. Unable to use: Mental state general: modified Brief Psychiatric Rating Scale (BPRS) (mentioned in the abstract, but not in the methods and results section of the publication). Mental state general: Inpatient Multidimensional Psychiatric Scale (IMPS) (mentioned in the abstract, but not in the methods and results section of the publication). Mental state general: Rockland‐Pollin scales (mentioned in the abstract, but not in the methods and results section of the publication). Behaviour: MACC rating scale (no total score available). |
|
| Notes | Study participants were “patients with chronic schizophrenia of long duration". “A drug‐free and placebo period preceded the course of treatment with the first drug.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial authors did not explicitly mention a randomisation, but described a double‐blinding. Thus we implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “All 50 patients completed all phases of the evaluation.” “Absence of any attrition in the study population.” |
| Selective reporting (reporting bias) | High risk | The duration of the trial was not indicated. No information regarding the number of participants that received a medication with antiparkinson drugs. Some rating scales (BPRS, IMPS, and Rockland‐Pollin scales) were mentioned in the abstract of the publication but no results of these scales were provided. |
| Other bias | High risk | Cross‐over study design. Because of epileptic convulsions the drug administration was temporally discontinued in one participant receiving haloperidol. |
Teja 1975.
| Methods | Randomisation: randomised. Blinding: double‐blind. Duration: 16 weeks (Phase II of the whole 36‐week trial (from the start of week 5 to the end of week 20)). Design: parallel (five‐arm study investigating haloperidol, trifluoperazine, thiothixene, chlorpromazine, and placebo). Location: Western State Hospital at Staunton, Verginia, USA. Setting: inpatients. |
|
| Participants | Diagnosis: chronic schizophrenia (hospitalised for a minimum of 2 years). N = 42. Gender (including all participants of the trial, N = 66): 30M, 36F. Age (N = 66): mean 38 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: “hospitalised from 2 to 20 years with a mean stay of 10,.1 years”, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed‐flexible dose, dose range: maximum dose 45 mg/day, mean dose: ˜ 25 mg/day ("standard dose"). N = 13. 2. Trifluoperazine: fixed‐flexible dose, dose range: maximum dose 90 mg/day, mean dose: ˜ 50 mg/day ("standard dose"). N = 15. 3. Thiothixene: fixed‐flexible dose, dose range: maximum dose 90 mg/day. mean dose: ˜ 50 mg/day ("standard dose"). N = 14. Rescue medication: “The only other drugs used during the trial were a hypnotic, if needed; benzotropine mesylate (cogentin) for alleviation of limiting extrapyramidal side effects, and parenteral or oral chlorpromazine in dosages of 25 to 50 mg, if the patient became an acute management problem during any phase of the trial.” |
|
| Outcomes | Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Mental state general: Mental status and global clinical change‐assessment (no raw data available). Behaviour: Burdock´s Ward Behavior Rating Scale (no raw data available). |
|
| Notes | Three different phases of the trial: “Following a 4 week placebo period [phase 1], high dose tranquilizers were given for 16 weeks [phase 2] and amitriptyline was added for the following 16 weeks [phase 3].” “It was the intent of the present investigators to employ fairly high dosages of the various tranquilizers for the treatment of this resistant chronic schizophrenic population.” “The four active tranquilizers investigated were all significantly more effective than placebo......no significant differences in efficacy were observed among the 4 tranquilizers however.” Study participants were “treated with a variety of medications, including the various major tranquilizers given singly or in combination. Sixteen had received ECT in addition.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “randomised”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the BPRS, Mental status and global clinical change‐assessment, and Burdock´s Ward Behavior Rating Scale). Adverse effects reporting was incomplete. No information regarding the number of participants that received a medication with antiparkinson drugs. |
| Other bias | High risk | The data were only “analyzed by comparing the placebo group with the [combined] active tranquilizer groups.” Study sample: “resistant chronic schizophrenic population”. Additionally to the randomised study medications, the administration of “parenteral or oral chlorpromazine in dosages of 25 to 50 mg, if the patient became an acute management problem during any phase of the trial”, was allowed. |
Tobin 1980.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 12 weeks. Design: parallel. Location: not indicated. Setting: outpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 33), schizoaffective schizophrenia (N = 7), undifferentiated schizophrenia (N = 5), hebephrenic schizophrenia (N = 3), catatonic schizophrenia (N = 2). N = 50. Gender: 18M, 32F. Age: mean 32.5 years. History: duration stable: not indicated, duration of illness: range 6 months to 42 years, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 1 mg to 15 mg/day, mean dose: ˜ 4 mg/day. N = 25. 2. Thiothixene: fixed/flexible dose: not indicated, dose range: 2 mg to 30 mg/day, mean dose: ˜ 8 mg/day. N = 25. “doses required to control symptoms based on clinical judgement. Various regimens were prescribed.” Rescue medication: “Antiparkinsonian agents were given as required to control extrapyramidal reactions.” |
|
| Outcomes | Examined: Clinically important response to treatment: Global rating. Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Adverse effects: at least one adverse effect, akathisia, dyskinesia, dystonia, rigor, tremor, weight gain. Unable to use: Mental state general: Adaptation of the Katz Adjustment Scales (not published adaptive version of this rating scale). Functioning: Evaluation of Social Functioning Rater (ESFR) (“was found unsatisfactory as an adequate measure of social functioning”). |
|
| Notes | Exclusively outpatients included. “They were admitted to the study only if they had three or more of the following signs of schizophrenia: flat affect, thought disorder, delusions, auditory hallucinations, or catatonia.” Baseline antipsychotic dose: 16 of the 50 participants “had been treated previously with psychotropic medications. Only 3 of the 16 had received antipsychotic compounds (not haloperidol or thiothixene).” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “The test medications were supplied in identical capsules.” “Only the dispenser knew the actual drug assignment. And the primary investigator remained blind until all the analyses were completed.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Double‐blind”. “Only the dispenser knew the actual drug assignment. And the primary investigator remained blind until all the analyses were completed.” |
| Incomplete outcome data (attrition bias) All outcomes | High risk | “Thirty‐six of the 50 patients completed the study.” Thus the overall attrition can be considered as being high: 28% (14 of 50 participants). 6 of 25 participants (24%) in the haloperidol group left the trial early and 8 of 25 participants (32%) in the bromperidol‐group. Modified completers‐only analyses were used. |
| Selective reporting (reporting bias) | High risk | Outcome data were not fully reported (no SDs for the BPRS; no usable results concerning the Katz Adjustment Scales). Data regarding the number of participants receiving rescue medication were missing. |
| Other bias | High risk | “44 patients had a history of psychiatric illness not necessarily diagnosed as schizophrenia.” “In one patient in each group, treatment was discontinued because of akathisia and was resumed when the akathisia abated.” |
Tuason 1986.
| Methods | Randomisation: “randomly assigned”. Blinding: "modified double‐blind"; according to the description in the publication the study was single‐blind. Duration: 10 days. Design: parallel. Location: not indicated. Setting: inpatients (emergency room). |
|
| Participants | Diagnosis: schizophrenia (DSM‐III). N = 54. Gender: 33M, 19F. Age: mean 33.9 years. History: duration stable: not indicated, duration of illness: < 1 year: 7 participants, 1‐10 years: 26 participants, > 10 years: 17 participants, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: maximum dose 100 mg/day, highest mean dose: 47 mg/day. N = 29. 2. Loxapine: flexible dose, dose range: maximum dose 250 mg/day, highest mean dose: 143 mg/day. N = 25. Dosages “were flexible within prescribed limits”; “Dosages were.......titrated according to clinical response.” “The loxapine‐to‐haloperidol dose ratio ranged from a minimum of 2.7:1 to a maximum of 4.4:1. Throughout the study, the dosages for both drugs were within recommended therapeutic ranges.” Rescue medication: “Chloral hydrate was prescribed as needed for sleep. Trihexyphenidyl HCI or benztropine mesylate was given for extrapyramidal side effects if needed.” |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impression (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS). Leaving the study early due to any reason. Leaving the study early due to adverse effects. Adverse effects: akathisia, dyskinesia, dystonia, rigor, tremor, hypotension. |
|
| Notes | Study participants were 54 “hostile and aggressive acutely schizophrenic patients”. “Each patient`s behaviour was characterised, at least in part, as hostile, aggressive, uncooperative, or “unmanageable.” Pretreatment scores on the hostility and uncooperativeness items of the BPRS had to total at least 8.” “The typical study patient was a white man with a diagnosis of chronic paranoid schizophrenia with acute exacerbation.” “54 acutely psychotic schizophrenics were given loxapine or haloperidol parenterally for 24 to 72 hours, then orally for a total study period of up to 10 days.” The trial stopped prematurely in case of earlier discharge. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomly assigned”. No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | High risk | “Personnel administering the study medications were not blinded.” |
| Blinding (performance bias and detection bias) (detection bias) | Low risk | “Those responsible for evaluating medication effects, however, remained blinded throughout.” |
| Incomplete outcome data (attrition bias) All outcomes | High risk | “56% of the loxapine and 41 % of the haloperidol patients completed the 10‐day study.” “Eleven of 25 (44%) loxapine patients and 14 of 27 (52%) haloperidol patients did not complete the study.” Therefore the overall attrition can be considered as being high. The trial authors provided both, intention‐to‐treat analyses and completers‐only analyses. |
| Selective reporting (reporting bias) | Unclear risk | The pre‐specified outcomes that are of interest in the review have been reported in the pre‐specified way with the exception of the data regarding the number of participants receiving rescue medication in each treatment group. |
| Other bias | High risk | “Two patients were excluded from all efficacy analyses because their final diagnosis was not schizophrenia.” “On day 3 19% of the haloperidol patients were withdrawn from the study while all loxapine patients remained.” “The haloperidol patients tended to be older by about 6 years (p=.056) and had a longer (p=.037) duration of schizophrenic illness.” The trial stopped prematurely in case of earlier discharge. |
Ulmar 1990.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 28 days. Design: parallel. Location: not indicated. Setting: not indicated. |
|
| Participants | Diagnosis: acute schizophrenia. N = 70. Gender: "male and female patients", no further details available. Age: 18‐65 years, no further details available. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. N = not indicated. 2. Trifluperidol:fixed/flexible dose: not indicated, dose range: not indicated, mean dose: not indicated. N = not indicated. |
|
| Outcomes | Unable to use: Mental state general: Brief Psychiatric Rating Scale (BPRS) (no raw data available). Mental state specific: AMDP‐rating (no raw data available). Adverse effects: Dosage Record Treatment Emergent Symptom Scale (DOTES) (no raw data available). |
|
| Notes | Data of this trial were based only on a single conference abstract. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | Outcome data reporting was incomplete (no raw data for the BPRS, AMDP, and DOTES). The numbers of participants randomised to each treatment group (haloperidol or trifluperidol) were not indicated. Adverse effects were not reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
Versiani 1978.
| Methods | Randomisation: “randomised”. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: hebephrenic schizophrenia (N = 17), catatonic schizophrenia (N = 17), paranoid schizophrenia (N = 9), residual schizophrenia (N = 3), simple schizophrenia (N = 2), schizoaffective schizophrenia (N = 2). N = 50. Gender: 48M, 2F. Age: mean 16.1 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: flexible dose, dose range: not indicated, mean dose: 7.6 mg/day. N = 25. 2. Loxapine: flexible dose, dose range: not indicated, mean dose: 70.4 mg/day. N = 25. Flexible dose: “The dosage was flexibly adjusted according to individual response.” “The two drugs were compared according to the ratio of loxapine 10: haloperidol 1.” Rescue medication: “In case of extrapyramidal signs and symptoms trihexyphenidyl (Artane) 5mg/day was employed.” |
|
| Outcomes | Examined: Clinically important response to treatment: Clinical Global Impression (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS) (no SDs available). Leaving the study early due to any reason. Leaving the study early due to inefficacy of treatment. Leaving the study early due to adverse effects. Adverse effects: akathisia, rigor, tremor, use of antiparkinson medication. Unable to use: Behaviour: Nurses´ Observation scale for Inpatient Evaluation (NOSIE) (no raw data for the total score available). |
|
| Notes | 34 participants with first‐episode schizophrenia. “The treatment was initiated after a washout period of two weeks with 2 to 4 capsules per day.” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | “Randomised according to the consecutive admission criterion.” No further details. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Low risk | “Double‐blind”. “capsules of identical appearance.” |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | “There was a drop‐out in the loxapine group” and no participant left the trial early in the haloperidol‐group. Altogether the attrition was rather low and the risk of bias might be considered as being low. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no SDs for the BPRS; no raw data for the NOSIE total score). |
| Other bias | High risk | At baseline “a good response to neuroleptic treatment was predicted in 20 cases in the loxapine group and in 11 cases in the haloperidol group.” “The presence of more cases of bad prognosis in the haloperidol‐group than in the loxapine‐group, as assessed by the raters at baseline, might diminish the value of conclusions drawn from the comparable efficacy of the two drugs, in all rated parameters.” “The sample was not evenly distributed along the range allowed by protocol.” |
White 1981.
| Methods | Randomisation: implied randomisation. Blinding: double‐blind. Duration: 4 weeks. Design: parallel. Location: not indicated. Setting: inpatients. |
|
| Participants | Diagnosis: paranoid schizophrenia (N = 17), non‐paranoid schizophrenia (N = 22). N = 39. Gender: 18M, 21F. Age: mean 28.3 years. History: duration stable: not indicated, duration of illness: not indicated, number of previous hospitalisations: not indicated, age at onset: not indicated, severity of illness: not indicated, baseline antipsychotic dose: not indicated. |
|
| Interventions | 1. Haloperidol: fixed/flexible dose: not indicated, dose range: 2 mg to 100 mg/day, mean dose: 28 mg/day. N = 21. 2. Mesoridazine: fixed/flexible dose: not indicated, dose range: 100 mg to 800 mg/day, mean dose: 421 mg/day. N = 18. “The protocol assumed a dosage equivalency of 2 mg haloperidol to 25 mg mesoridazine.” Rescue medication: concomitant antiparkinsonism medications were allowed. |
|
| Outcomes | Examined: Global state general: Clinical Global Impression (CGI). Mental state general: Brief Psychiatric Rating Scale (BPRS). Adverse effects: at least one movement disorder, dystonia, use of antiparkinson medication. Unable to use: Mental state specific: Dysphoric Response Index (DRI) calculated from the BPRS (no raw data available). |
|
| Notes | Study participants were “39 recently hospitalized inpatients, diagnosed schizophrenic according to the criteria of Feighner 1972, with modification to include illness less than 6 months in duration.” 19 participants had a process schizophrenia and twenty participants had a reactive schizophrenia. “Subjects received medication intramuscularly for the first 24h and orally thereafter.” The main aim of the study was “to test the hypothesis that a BPRS‐derived index of dysphoria may predict overall clinical outcome on such a neuroleptic trial” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No randomisation mentioned in the publication, but the trial was described as “double‐blind.” Thus it was implied that the study was randomised. |
| Allocation concealment (selection bias) | Unclear risk | No further details. |
| Blinding (performance bias and detection bias) (performance bias) | Unclear risk | “Double‐blind”. No further details. |
| Blinding (performance bias and detection bias) (detection bias) | Unclear risk | “Double‐blind”. No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The study did not address this outcome; insufficient information to permit judgment concerning incomplete outcome data. |
| Selective reporting (reporting bias) | High risk | The outcome data were not fully addressed (no raw data for the DRI). Only the “commonest side effects” were reported. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists. |
General abbreviations:
DSM‐II, ‐III, ‐III‐R, ‐IV = various versions of the Diagnostic and Statistical Manual of Mental Disorders; F = female; ICD = International Classification of Diseases; i.m. = intramuscular; M = male; n = number of participants; mg = milligram; SD = standard deviation; SE = standard error.
Rating scales:
AIMS = Abnormal Involuntary Movement Scale; BPRS = Brief Psychiatric Rating Scale; CGI = Clinical Global Impression; CNS‐RS = Concise Negative Symptoms Rating Scale; DOTES = Dosage Record Treatment Emergent Symptom Scale; DRI = Dysphoric Response Index; ESFR = Evaluation of Social Functioning Rater; GH = Global Hostility Scale; GP = Global Paranoia Scale; HAM‐D = Hamilton Depression Rating Scale; HPRSD = Hamilton Psychiatric Rating Scale for depression; IMPS = Inpatient Multidimensional Psychiatric Scale; MMPI = Minnesota Multiphasic Personality Inventory; MRS = Montgomery Rating Scale; NGI = Nurses´ Global Impressions; NOSIE = Nurses Observation Scale for Inpatient Evaluation; OBRS = Oklahoma Behavior Rating Scale; PIP = Psychotic Inpatient Profile; REPS = Reversible Extrapyramidal Symptom Rating Scale; RSQPSS = Rating Scale for Quantification of Psychotic Symptom Severity; SANS = Scale for Assessment of Negative Symptoms; SAPS = Scale for Assessment of Positive Symptoms; SRSS = Lipman‐Rickels Self‐Rating Symptom Scale; TESS = Treatment Emergent Symptom Scale; WBRS = Ward Behaviour Rating Scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Al Haddad 1996 | Randomisation: "randomly allocated". Participants: "patients with mania, acute psychosis or exacerbation of a chronic psychosis". Intervention: haloperidol (i.m. followed by oral medication) versus zuclopenthixol i.m. |
| Alpert 1995 | Randomisation: no randomisation mentioned in the publication. |
| Azima 1960 | Randomisation: implied randomisation. Participants: with "mental syndromes". Intervention: haloperidol versus placebo. |
| Barch 2005 | Randomisation: no randomisation between haloperidol and fluphenazine mentioned in the publication. |
| Baro 1972 | Randomisation: no randomisation mentioned in the publication. |
| Bechelli 1986 | Randomisation: implied randomisation. Participants: "recently admitted schizophrenic patients". Intervention: haloperidol i.m. followed by oral administration versus pipothiazine palmitate i.m. followed by placebo administration (glass of milk). |
| Boyer 1987 | Randomisation: "randomly assigned". Participants: "patients meeting the DSM III criteria of schizophrenia (subtypes: disorganized, catatonic or residual)". Intervention: haloperidol versus amisulpride. |
| Brook 1998 | Randomisation: "random allocation". Participants: Of the total sample (n = 44) 27 participants had a psychosis secondary to substance abuse and 23 participants had a positive urine cannabis. |
| Chin 1998 | Randomisation: "randomisation". Participants: people with "schizophrenian disorder or an exacerbation of chronic schizophrenia". Intervention: haloperidol versus zuclopenthixol acetate (no oral medication). |
| Classen 1988 | Randomisation: no randomisation mentioned in the publication. |
| Cole 1970 | Randomisation: implied randomisation. Participants: "acute and chronic subjects with geriatric diagnosis". Intervention: haloperidol versus thioridazine. |
| Costa 2007 | Randomisation: no randomisation between the first‐generation antipsychotics ("conventional group"). |
| Crow 1986 | Randomisation: "patients were randomised". Participants: people "who were suffering from a first psychotic episode". Intervention: haloperidol versus placebo (assessment of relapse). |
| Davies 2007 | Randomisation: no randomisation between the first‐generation antipsychotics. |
| de Jesus Mari 2004 | Randomisation: no randomisation between the first‐generation antipsychotics mentioned in the publication. |
| Digo 1967 | Randomisation: no randomisation mentioned in the publication. |
| Dubin 1985 | Randomisation: "randomized assignment table". Participants: only 48,4% with the diagnosis of schizophrenia. |
| Durost 1964 | Randomisation: implied randomisation. Participants: less than 50% of the participants were diagnosed as psychotic. Intervention: haloperidol versus placebo. |
| Ehmann 1987 | Randomisation: "randomly assigned". Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Eitan 1992 | Randomisation: no randomisation mentioned in the publication. |
| Engelhardt 1973 | Randomisation: sequence generation was at high risk of bias. “Children were randomly assigned to one of the two drug groups, unless their treatment history indicated recent use of either of the drugs (in which case the child was assigned to the drug not previously received).” |
| Fitzgerald 1969 | Randomisation: "randomly assigned". Participants: people with "acute psychiatric episodes". Intervention: haloperidol versus perphenazine (parenteral administration). |
| Galderisi 1994 | Randomisation: no randomisation mentioned in the publication. |
| Gerlach 1978 | Randomisation: no randomisation mentioned in the publication. Participants: "geronto‐psychiatric patients". |
| Gillis 1977 | Randomisation: no randomisation between haloperidol and the other first‐generation antipsychotics. |
| Harris 1992 | Randomisation: "randomized". Design: no information regarding the diagnosis of the participants available. |
| Holden 1970 | Randomisation: implied randomisation. Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Huang 2005 | Randomisation: no randomisation between haloperidol and the other first‐generation antipsychotics. |
| Hyugano 1986 | Randomisation: no randomisation mentioned in the publication. |
| Itil 1975 | Randomisation: "randomly assigned". Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Jones 2006 | Randomisation: no randomisation between the different first‐generation antipsychotics. |
| Karsten 1981 | Randomisation: "randomized allocation". Participants: "mentally retarded patients". |
| Kazamatsuri 1972 | Randomisation: "random divided". Design: no information regarding the diagnosis of the participants available. |
| Kelwala 1984 | Randomisation: "random assignment". Participants: more manic (n = 23) than schizophrenic (n = 21) participants. Intervention: parenteral administration of the antipsychotic compounds. |
| Kurt 2007 | Randomisation: no randomisation between haloperidol and the other first‐generation antipsychotics. |
| Lamure 2003 | Randomisation: "randomised". Participants: people "with DSM‐III‐R diagnosed schizophrenia and with an acute exacerbation". Intervention: haloperidol versus zuclopenthixol; 48,6% in the haloperidol group and 71,9% in the zuclopenthixol group were treated with depot form. |
| Lehmann 1967 | Randomisation: "randomly" allocated. Participants: "chronic schizophrenic patients". Intervention: investigation of add‐on medications. |
| Levenson 1976 | Randomisation: "randomly assigned". Participants: "acute schizophrenics". Intervention: parenteral administration of the antipsychotic compounds. |
| Liu 1996 | Randomisation: no randomisation mentioned in the publication. |
| Lovett 1987 | Randomisation: "randomly assigned". Participants: "senile psychosis.....in elderly patients". |
| Lublin 1991 | Randomisation: "randomised". Participants: "psychotic psychiatric in‐patients". Design: cross‐over trial. Intervention: haloperidol + basic medication with haloperidol versus zuclopenthixol + basic medication with haloperidol. |
| Malt 1995 | Randomisation: "randomised". Participants: "ICD‐10 diagnosis of F79‐79 (mental retardation)". |
| Mosolov 2000 | Randomisation: randomised. Participants: people with schizophrenia and schizoaffective disorder. Intervention: haloperidol versus zuclopenthixol (both agents were administered i.m. for 7 days before a switch to oral medication was undertaken). |
| Nahunek 1982 | Randomisation: "both neuroleptics were applied alternately". |
| Nordic 1986 | Randomisation: "cross‐over design in randomized order". Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Onodera 1984 | Randomisation: no randomisation mentioned in the publication. |
| Paprocki 1977 | Randomisation: “randomised”. Blinding: double‐blind. Duration: 4 weeks of oral medication (4 days parenteral administration of the investigated agents before only responders were switched to the 4‐week period of oral medication). |
| Pedros Rosello 2004 | Randomisation: no randomisation mentioned in the publication. |
| Reimold 2007 | Randomisation: no randomisation between haloperidol and flupentixol mentioned in the publication |
| Reznik 2000 | Randomisation: "randomly assigned". Participants: people with schizophrenia. Intervention: SSRI + antipsychotic versus antipsychotic. |
| Saletu 1986 | Randomisation: "randomly assigned". Participants: "acute or exacerbated schizophrenic patients". Intervention: haloperidol versus fluperlapine. |
| Samuels 1961 | Randomisation: no randomisation mentioned in the publication. |
| Serban 1984 | Randomisation: "assigned blindly.....according to a computer‐generated randomized code". Participants: 30,8% of the participants with borderline personality disorder. |
| Simpson 1967 | Randomisation: "randomly assigned". Participants: "chronic schizophrenic male subjects". Intervention: haloperidol versus placebo. |
| Singh 1976 | Randomisation: "cross‐over design". Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Smith 1984 | Randomisation: no randomisation mentioned in the publication. |
| Stotsky 1977 | Randomisation: "randomly assigned". Participants: "acutely excited, agitated psychotic patients". Intervention: parenteral administration of the antipsychotic compounds. |
| Su 2002 | Randomisation: no randomisation mentioned in the publication. Design: cross‐over study without separate results for the first study phase, before the point of the first cross‐over. |
| Taymeeyapradit 2002 | Randomisation: "randomly assigned". Participants: "The patients had diagnosis of acute psychosis (schizophrenia with acute exacerbation, mania and other forms of psychosis)" Intervention: parenteral haloperidol versus parenteral zuclopenthixol acetate. |
| Terminska 1989 | Randomisation: not randomised. |
| van Lommel 1974 | Randomisation: no randomisation mentioned in the publication. |
| van Putten 1984 | Randomisation: no randomisation mentioned in the publication. |
| van Putten 1986 | Intervention: review of dose‐comparison studies. |
| Zuoning 1999 | Randomisation: no randomisation mentioned in the publication. |
DSM III = Diagnostic and Statistical Manual of Mental Disorders; ICD = International Classification of Diseases; i..m = intramuscular; SSRI = Selective serotonin reuptake inhibitor
Differences between protocol and review
We changed the title from “Haloperidol versus first‐generation antipsychotics for schizophrenia” to “Haloperidol versus first‐generation antipsychotics for the treatment of schizophrenia and/or other psychotic disorders”.
In terms of the subgroup analyses, we added the following sentence within the methods section: "Furthermore, we performed a stratification according to the different first‐generation antipsychotics administered as active comparator agent to haloperidol."
To ensure harmonisation of the nomenclature of the secondary outcomes in terms of premature discontinuation (drop‐outs) we used the term "leaving the study early" within the whole text.
Contributions of authors
Markus Dold: protocol development, study selection, data extraction, statistical analyses, writing the report including the first draft of the manuscript. Myrto Samara: study selection, data extraction. Chunbo Li: study selection, data extraction. Magdolna Tardy: protocol development. Stefan Leucht: protocol development, study selection, data extraction, writing the report.
Sources of support
Internal sources
No sources of support supplied
External sources
-
Bundesministerium für Bildung und Forschung, Germany.
FKZ: 01KG1026
Declarations of interest
Markus Dold: has received a travel grant from Janssen‐Cilag. Myrto Samara: none to declare. Chunbo Li: none to declare. Magdolna Tardy: none to declare. Stefan Leucht has received honoraria for lectures from Lilly, Lundbeck, Pfizer, Janssen, BMS, Janssen, Johnson and Johnson, Lundbeck, Roche, SanofiAventis, ICON, Abbvie; for consulting from Roche, Janssen, Lundbeck and Lilly; and for the preparation of Educational Material and publications from the Lundbeck Institute and Roche. EliLilly has provided medication for a clinical trial led by SL as principal investigator.
New
References
References to studies included in this review
Abuzzahab 1982 {published data only}
- Abuzzahab FS, Zimmerman RL. Psychopharmacological correlates of post psychotic depression: a double‐blind investigation of haloperidol vs thiothixene in outpatient schizophrenia. Journal of Clinical Psychiatry 1982;43:105‐10. [PubMed] [Google Scholar]
Baastrup 1993 {published data only}
- Baastrup PC, Alhfors UG, Bjerkenstedt L, Dencker SJ, Fensbo C, Gravem A, et al. A controlled Nordic multicentre study of zuclopenthixol acetate in oil solution, haloperidol and zuclopenthixol in the treatment of acute psychosis. Acta Psychiatrica Scandinavica 1993;87:48‐58. [DOI] [PubMed] [Google Scholar]
- Fensbo C. Zuclopenthixol acetate, haloperidol, and zuclopenthixol in the treatment of acutely psychotic patients ‐ a controlled multicenter study [Zuclopenthixol acetat, haloperidol og zuclopenthixol i behandling af akut psykotiske patienter. En kontrolleret multicenterundersogelse]. Nordisk Psykiatrisk Tidsskrift 1990;44:295‐7. [Google Scholar]
Bechelli 1983 {published data only}
- Bechelli LP, Ruffino‐Netto A, Hetem G. A double‐blind controlled trial of pipotiazine, haloperidol and placebo in recently‐hospitalized acute schizophrenic patients. Brazilian Journal of Medical and Biological Research 1983;16:305‐11. [PubMed] [Google Scholar]
Brannen 1981 {published data only}
- Brannen JO, McEvoy JP, Wilson WH, Petrie WM, Ban TA, Berney SA, et al. A double‐blind comparison of bromperidol and haloperidol in newly admitted schizophrenic patients. Pharmacopsychiatria 1981;14:139‐40. [DOI] [PubMed] [Google Scholar]
Bueno 1979 {published data only}
- Bueno JR. A double‐blind comparative clinical trial with loxapine succinate and haloperidol in the treatment of Schizophrenia [Ensaio clinico comparativo e duplo‐cego com succina de loxapina e haloperidol no tratamento da esquizofrenia]. Folha Medica 1979;78:47‐52. [Google Scholar]
Cassano 1975 {published data only}
- Cassano GB, Castrogiovanni P, Conti L. The computer diagnosis in a multicenter study of psychoactive agents. Psychopharmacology Bulletin 1976;12:22‐4. [PubMed] [Google Scholar]
- Cassano GB, Castrogiovanni P, Conti L, Bonollo L. Sulpiride ‐ an antipsychotic agent: comparative trial vs. haloperidol. Psychopharmacology Bulletin 1977;13:41‐3. [PubMed] [Google Scholar]
- Cassano GB, Castrogiovanni P, Conti L, Bonollo L. Sulpiride versus haloperidol in schizophrenia: a double‐blind comparative trial. Current Therapy Research, Clinical and Experimental 1975;17:189‐201. [PubMed] [Google Scholar]
- Castrogiovanni P, Cassano GB, Conti L, Maggini C, Bonollo L, Sarteschi P. An automated diagnostic process (PDA) in clinical psychopharmacology. An exemplification of its use in a sulpiride versus haloperidol comparative trial. International Pharmacopsychiatry 1976;11:74‐83. [DOI] [PubMed] [Google Scholar]
Cocchi 1971 {published data only}
- Cocchi A, Fonda P, Perosino N. Droperidol: a double‐blind clinical study [Droperidol: studio clinico in doppio cieco]. Rivista Sperimentale di Freniatria e Medicina Legale Delle Alienazioni Mentali 1971;95:1109‐25. [PubMed] [Google Scholar]
Cocito 1970 {published data only}
- Cocito E, Ambrosini G, Arata A, Bevilacqua P, Tortora E. Clinical evaluation in 112 psychiatric patients of a butyrophenone neuroleptic, dehydrobenzperidol (R 4749). A controlled study in 45 patients of dehydrobenzperidol versus haloperidol. Arzneimittelforschung 1970;20:1119‐25. [PubMed] [Google Scholar]
Cosar 1999 {published data only}
- Cosar B, Candansayar S, Taner E, Isik E. Comparison of efficacy of clozapine, sulpiride, chlorpromazine and haloperidol in chronic schizophrenic patients therapy. European Neuropsychopharmacology 1999;9:287. [Google Scholar]
Darondel 1981 {published data only}
- Darondel A, Balthazard JC, Brun JP. A double‐blind comparison between pipotiazine and haloperidol [Bilan d'une etude controlee a double insu pipotiazine versus haloperidol]. Psychologie Medicale 1981;13:171‐80. [Google Scholar]
Denijs 1980 {published data only}
- Denijs EL. Clinical evaluation of bromperidol versus haloperidol in psychotic patients. International Pharmacopsychiatry 1980;15:309‐17. [DOI] [PubMed] [Google Scholar]
Dufresne 1993 {published data only}
- Dufresne RL, Valentino D, Kass DJ. Thioridazine improves affective symptoms in schizophrenic patients. Psychopharmacology Bulletin 1993;29:249‐55. [PubMed] [Google Scholar]
Engelhardt 1978 {published data only}
- Engelhardt DM, Rudorfer L, Rosen B. Haloperidol and thiothixene in the long‐term treatment of chronic schizophrenic outpatients in an urban community: social and vocational adjustment. Journal of Clinical Psychiatry 1978;39:834‐40. [PubMed] [Google Scholar]
Escobar 1985 {published data only}
- Escobar JI, Mann JJ, Keller J, Wilkins J, Mason B, Mills MJ. Comparison of injectable molindone and haloperidol followed by oral dosage forms in acutely ill schizophrenics. Journal of Clinical Psychiatry 1985;46:15‐9. [PubMed] [Google Scholar]
Faretra 1970 {published data only}
- Faretra G, Dooher L, Dowling J. Comparison of haloperidol and fluphenazine in disturbed children. American Journal of Psychiatry 1970;126:1670‐3. [DOI] [PubMed] [Google Scholar]
Fuentenebro 1989 {published data only}
- Fuentenebro F, Escobar JI, Keller J, Lopez de Ochoa EF, Vazquez C. Prediction of the therapeutic response to neuroleptics in schizophrenia [Prediccion de la respuesta al tratamiento neuroleptico en la esquizofrenia]. Psiquis Revista de Psiquiatria, Psicologia y Psicosomatica 1989;10:22‐8. [Google Scholar]
Gallant 1967 {published data only}
- Gallant DM, Bishop M, Guerrero‐Figueroa R. Effects of two butyrophenone compounds on acute schizophrenic patients: speculation on the neurophysiologic sites of action. International Journal of Neuropsychiatry 1967;3(Suppl. 1):53‐7. [PubMed] [Google Scholar]
- Pratt JP, Bishop MP, Gallant DM. Trifluperidol and haloperidol in the treatment of acute schizophrenia. American Journal of Psychiatry 1964;121:592‐4. [DOI] [PubMed] [Google Scholar]
Gerlach 1985 {published and unpublished data}
- Gerlach J, Behnke K, Heltberg J, Munk‐Andersen E, Nielsen H. Dogmatil and haloperidol for the treatment of schizophrenia. Double blind cross‐over study of therapeutic effectiveness,side effects and plasma concentrations. [article in french]. Semaine des Hopitaux 1985;61:1309‐16. [Google Scholar]
- Gerlach J, Behnke K, Heltberg J, Munk‐Anderson E, Nielsen H. Sulpiride and haloperidol in schizophrenia: a double‐blind cross‐over study of therapeutic effect, side effects and plasma concentrations. British Journal of Psychiatry 1985;147:283‐8. [DOI] [PubMed] [Google Scholar]
- Munk‐Andersen E, Behnke K, Heltberg J, Nielsen H, Gerlach J. Sulpiride versus haloperidol in the treatment of schizophrenia: A brief preliminary report [article in danish]. Nordisk Psykiatrisk Tidsskrift 1984;38:223‐8. [Google Scholar]
- Munk‐Andersen E, Behnke K, Heltberg J, Nielsen H, Gerlach J. Sulpiride versus haloperidol, a clinical trial in schizophrenia. A preliminary report. Acta Psychiatrica Scandinavica. Supplementum 1984;311:31‐41. [DOI] [PubMed] [Google Scholar]
Germana 1990 {published data only}
- Germana B, Rapisarda V. Clinical therapeutic experience with bromperidol ‐ a double‐blind study [Esperienze clinico terapeutiche con bromperidolo ‐ prova in doppio cieco e in chiaro]. Rivista di Psichiatria 1990;4:233‐5. [Google Scholar]
Giordana 1984 {published data only}
- Giordana JY, Frenay S. Comparative study of pipotiazine (in its non‐esterified form) and of haloperidol in acute or subacute psychotic states [Etude comparative de la pipothiazine (dans sa forme non esterifiee) et de l'haloperidol au cours d'etats psychotiques aigus et subaigus]. Psychologie Medicale 1984;16:1803‐15. [Google Scholar]
Glazer 1990 {published data only}
- Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophrenia Research 1990;3:315‐20. [DOI] [PubMed] [Google Scholar]
- Glazer WM, Hafez HM, Benarroche CL. Molindone and haloperidol in tardive dyskinesia. Journal of Clinical Psychiatry 1985;46:4‐7. [PubMed] [Google Scholar]
Goldstein 1966 {published data only}
- Goldstein BJ, Clyde DJ. Haloperidol in controlling the symptoms of acute psychoses. Part II. A double‐blind evaluation of haloperidol and trifluoperazine. Current Therapeutic Research, Clinical and Experimental 1966;8:236‐40. [PubMed] [Google Scholar]
Goldstein 1969 {published data only}
- Goldstein BJ, Brauzer B, Clyde DJ, Caldwell JM. The differential prediction of response to two anti‐psychotic drugs. Psychosomatics 1969;10:193‐7. [DOI] [PubMed] [Google Scholar]
Gowardman 1973 {published data only}
- Gowardman M, Barrer B, Brown RA. Pimozide (R6238) in chronic schizophrenia: double‐blind trial. New Zealand Medical Journal 1973;78:487‐91. [PubMed] [Google Scholar]
Haas 1982 {published data only}
- Haas S, Beckmann H. Pimozide and haloperidol in a double‐blind study in acutely schizophrenic patients [Pimozid und Haloperidol im Doppelblind‐Versuch bei akut schizophrenen Patienten]. Arzneimittelforschung 1982;32:888. [Google Scholar]
- Haas S, Beckmann H. Pimozide versus Haloperidol in acute schizophrenia. A double blind controlled study. Pharmacopsychiatria 1982;15:70‐4. [DOI] [PubMed] [Google Scholar]
Hall 1968 {published data only}
- Hall WB, Vestre ND, Schiele BC, Zimmermann R. A controlled comparison of haloperidol and fluphenazine in chronic treatment‐resistant schizophrenics. Diseases of the Nervous System 1968;29:405‐8. [PubMed] [Google Scholar]
Heikkilä 1981 {published data only}
- Heikkilä L, Laitinen J, Vartiainen H. Cis(Z)‐clopenthixol and haloperidol in chronic schizophrenic patients ‐ a double‐blind clinical multicentre investigation. Acta Psychiatrica Scandinavica 1981;294(Suppl. 1):30‐8. [PubMed] [Google Scholar]
Heikkilä 1992 {published data only}
- Heikkilä L, Eliander H, Vartiainen H, Turunen M, Pedersen V. Zuclopenthixol and haloperidol in patients with acute psychotic states. A double‐blind, multi‐centre study. Current Medical Research Opinion 1992;12:594‐603. [DOI] [PubMed] [Google Scholar]
Hollister 1962 {published data only}
- Hollister LE, Overall JE, Caffey E Jr, Bennett JL, Meyer F, Kimbell I Jr, et al. Controlled comparison of Haloperidol with thiopropazate in newly admitted schizophrenics. Journal of Nervous and Mental Disorders 1962;135:544‐9. [DOI] [PubMed] [Google Scholar]
Howard 1974 {published data only}
- Howard JS. Haloperidol for chronically hospitalized psychotics: a double‐blind comparison with thiothixene and placebo; a follow‐up open evaluation. Diseases of the Nervous System 1974;35:458‐63. [PubMed] [Google Scholar]
Itoh 1985 {published data only}
- Ito H, Anno H, Fujii Y, Hasegawa Y, Ikegami H, Ishigooka J, et al. Clinical evaluation of bromperidol, a new investigational butyrophenone derivative, in schizophrenia, a double‐blind comparison of bromperidol and haloperidol. Rinsho Hyoka (Clinical Evaluation) 1985;13:105‐36. [Google Scholar]
- Itoh H. A comparison of the clinical effects of bromperidol, a new butyrophenone derivative, and haloperidol on schizophrenia using a double‐ blind technique. Psychopharmacology Bulletin 1985;21:120‐2. [PubMed] [Google Scholar]
Kariya 1983 {published data only}
- Kariya T, Shimazono Y, Itoh H, Mori A, Murasaki M, Sugano K, et al. A comparison of the clinical effects of timiperone, a new butyrophenone derivative, and haloperidol on schizophrenia using a double‐blind technique. Journal of International Medical Research 1983;11:66‐77. [DOI] [PubMed] [Google Scholar]
- Kariya T, Shimazono Y, Yamashita I. Comparison of clinical effects of timiperone and haloperidol on schizophrenia using double‐blind technique. Seishin Igaku (Clinical Psychiatry) 1981;10:1281‐301. [Google Scholar]
Kinon 1993 {published data only}
- Kinon BJ, Kane JM, Chakos M, Munne R. Possible predictors of neuroleptic‐resistant schizophrenic relapse ‐ influence of negative symptoms and acute extrapyramidal side effects. Psychopharmacology Bulletin 1993;29:365‐9. [PubMed] [Google Scholar]
- Kinon BJ, Kane JM, Johns C, Perovich R, Ismi M, Koreen A, et al. Treatment of neuroleptic‐resistant schizophrenic relapse. Psychopharmacology Bulletin 1993;29:309‐14. [PubMed] [Google Scholar]
Kodama 1984 {published data only}
- Kodama H, Sarai K, Nakahara T. The clinical efficacy of bromperidol for schizophrenia: comparison with haloperidol by the double‐blind method. Igaku to Yakugaku 1984;12:269‐86. [Google Scholar]
Kurihara 1983 {published data only}
- Kurihara M, Ito H, Kato N. Clinical evaluation of clocapramine (clofekton) in schizophrenia: a double blind comparison of clocapramine, haloperidol and perphenazine. Rinsho Seishin Igaku (Japanese Journal of Clinical Psychiatry) 1983;12:519‐38. [Google Scholar]
Luckey 1967 {published data only}
- Luckey WT, Schiele BC. A comparison of haloperidol and trifluoperazine. (A double‐blind, controlled study on chronic schizophrenic outpatients). Diseases of the Nervous System 1967;28:181‐6. [PubMed] [Google Scholar]
Malfroid 1978 {published data only}
- Malfroid M, Hens L, Roosen P, Dom R. Double‐blind‐ evaluation of bromperidol versus haloperidol treatment in chronic psychotic patients. Acta Psychiatrica Belgica 1978;78:147‐54. [PubMed] [Google Scholar]
Mattke 1976 {published data only}
- Mattke DJ, Mombour W, Glotzner FL. The assessment of neuroleptogenic extrapyramidal syndroms in psychopharmacological research [Erfassung neuroleptisch bedingter extrapyramidaler Syndrome in einem psychopharmakologischen Forschungsprojekt]. Pharmakopsychiatrie und Neuropsychopharmakologie 1975;1:36‐44. [DOI] [PubMed] [Google Scholar]
- Mattke DJ, Wisuschil W. A method for studying pupilomotorics under the influence of neuroleptic medication [Eine Methode zur Untersuchung der Pupillomotorik unter dem Einfluss neuroleptischer Medikation]. Arzneimittelforschung 1976;26:946‐50. [PubMed] [Google Scholar]
Mauri 1994 {published data only}
- Mauri M, Cassano GB, Canova L, Cocconcelli C, Faravelli C, Kemali D, et al. Schizophrenic disorder in active phase: double‐blind study between bromperidol and haloperidol [Disturbo schizofrenico in fase attiva: studio in doppia cecita tra Bromperidolo e Aloperidolo]. Giornale di Neuropsicofarmacologia 1994;16:187‐93. [Google Scholar]
Mori 1989 {published data only}
- Mori A, Kazamatsuri H, Kaneno S, Kamijima K, Kariya T, Murasaki M, et al. A double‐blind comparison of a new benzamide compound YM 09151 with haloperidol in the treatment of schizophrenia. Rinsho Hyoka 1989;17:349‐77. [Google Scholar]
Nedopil 1981 {published and unpublished data}
- Nedopil N, Ruther E. Initial improvement as predictor of outcome of neuroleptic treatment. Pharmacopsychiatria 1981;14:205‐7. [DOI] [PubMed] [Google Scholar]
Nishikawa 1984 {published data only}
- Nishikawa T, Tsuda A, Tanaka M, Hoaki Y, Koga I, Uchida Y. Prophylactic effect of neuroleptics in symptom‐free schizophrenics ‐ a comparative dose‐response study of haloperidol and propericiazine. Psychopharmacology 1984;82:153‐6. [DOI] [PubMed] [Google Scholar]
Nishimatu 1975 {published data only}
- Nishimatu O, Horigudchi J, Akita K, Nomura S, Sarai K, Kodama H. Comparison of the clinical efficacy of methylperidol (luvatren) and haloperidol for schizophrenia by the double‐blind method. Seishin Igaku (Clinical Psychiatry) 1975;4:973‐85. [Google Scholar]
O´Brien 1974 {published data only}
- O'Brien CP, DiGiacomo JN, Webb W. Management of hostile, suspicious patients ‐ trifluoperazine versus haloperidol. Diseases of the Nervous System 1974;35:75‐8. [PubMed] [Google Scholar]
Okuda 1979 {published data only}
- Okuda O, Akaue Y, Okishio Y. The efficacy of sulpiride in the treatment of schizophrenia by the double‐blind method. Japanese Pharmacology and Therapeutics [Yakuri to Chiryo] 1979;7:439‐58. [Google Scholar]
Paprocki 1976 {published data only}
- Paprocki J, Barcala Peixoto MP, Andrade NM. A controlled double‐blind comparison between loxapine and haloperidol in acute newly hospitalized schizophrenic patients. Psychopharmacology Bulletin 1976;12:32‐4. [PubMed] [Google Scholar]
Parent 1983 {published data only}
- Parent M, Toussaint C. Flupenthixol versus haloperidol in acute psychosis. Pharmatherapeutica 1983;3:354‐64. [PubMed] [Google Scholar]
- Parent M, Toussaint C. Flupenthixol versus haloperidol in acute psychotic episodes [article in french]. Acta Psychiatrica Belgica 1982;82:617‐31. [PubMed] [Google Scholar]
Pöldinger 1977 {published data only}
- Bures E, Poldinger W. Introductory words to the exchange of results on the clinical effects of bromperidol. Results of open and double‐blind research [Einleitende Worte zum Austausch der Erfahrungen über die klinische Wirkung von Bromperidol. Mitteilung über eigene Ergebnisse im offenen und Doppelblindversuch]. International Pharmacopsychiatry 1978;13:45‐52. [PubMed] [Google Scholar]
- Pöldinger W. Clinical experiences in an open and a double‐blind trial. Acta Psychiatrica Belgica 1978;78:96‐101. [PubMed] [Google Scholar]
- Pöldinger W, Bures E, Haage H. Double‐blind study with two butyrophenone derivatives: bromperidol vs. haloperidol. International Pharmacopsychiatry 1977;12:184‐92. [DOI] [PubMed] [Google Scholar]
Pool 1976 {published data only}
- Pool D, Bloom W, Mielke DH, Roniger JJ Jr, Gallant DM. A controlled evaluation of loxitane in seventy‐five adolescent schizophrenic patients. Current Therapeutic Research, Clinical and Experimental 1976;19:99‐104. [PubMed] [Google Scholar]
Rama Rao 1981 {published data only}
- Rama Rao V, Bailey J, Bishop M, Coppen A. A clinical and pharmacodynamic evaluation of sulpiride. Psychopharmacology 1981;73:77‐80. [DOI] [PubMed] [Google Scholar]
Rubin 1971 {published data only}
- Rubin R. A double‐blind comparison of the onset of activity of haloperidol and trifluoperazine. Alabama Journal of Medical Sciences 1971;8:414‐8. [PubMed] [Google Scholar]
Selman 1976 {published data only}
- Selman FB, McClure RF, Helwig H. Loxapine succinate: a double‐blind comparison with haloperidol and placebo in acute schizophrenics. Current Therapeutic Research, Clinical and Experimental 1976;19:645‐52. [PubMed] [Google Scholar]
- Selman FB, McClure RF, Helwig H. Loxapine succinate: a double‐blind comparison with haloperidol and placebo in acute schizophrenics [article in french]. Psychologie Médicale 1979;11:1533‐40. [PubMed] [Google Scholar]
Serafetinides 1972 {published data only}
- Serafetinides EA. Consistency and similarity of drug EEG responses in chronic schizophrenic patients. International Pharmacopsychiatry 1973;8:214‐6. [DOI] [PubMed] [Google Scholar]
- Serafetinides EA. Voltage laterality in the EEG of psychiatric patients. Diseases of the Nervous System 1973;34:190‐1. [PubMed] [Google Scholar]
- Serafetinides EA, Clark ML. Psychological effects of single dose antipsychotic medication. Biological Psychiatry 1973;7:263‐7. [PubMed] [Google Scholar]
- Serafetinides EA, Collins S, Clark ML. Haloperidol, clopenthixol, and chlorpromazine in chronic schizophrenia. Chemically unrelated antipsychotics as therapeutic alternatives. Journal of Nervous and Mental Disease 1972;154:31‐42. [DOI] [PubMed] [Google Scholar]
- Serafetinides EA, Willis D. A method of quantifying EEG for psychopharmacological research. International Pharmacopsychiatry 1973;8:245‐7. [DOI] [PubMed] [Google Scholar]
- Serafetinides EA, Willis D, Clark ML. Haloperidol, clopenthixol, and chlorpromazine in chronic schizophrenia. II. The electroencephalographic effects of chemically unrelated antipsychotics. Journal of Nervous and Mental Disease 1972;155:366‐9. [DOI] [PubMed] [Google Scholar]
Shalev 1993 {published data only}
- Shalev A, Hermesh H, Rothberg J, Munitz H. Poor neuroleptic response in acutely exacerbated schizophrenic patients. Acta Psychiatrica Scandinavica 1993;87:86‐91. [DOI] [PubMed] [Google Scholar]
Silverstone 1984 {published data only}
- Silverstone T, Cookson J, Ball R, Chin CN, Jacobs D, Lader S, et al. The relationship of dopamine receptor blockade to clinical response in schizophrenic patients treated with pimozide or haloperidol. Journal of Psychiatric Research 1984;18:255‐68. [DOI] [PubMed] [Google Scholar]
Spina 1992 {published data only}
- Spina E, Domenico P, Bonanzinga M, D'Agostino MA, Gitto C, Rosa AE, et al. A double‐blind comparative study of bromperidol versus haloperidol in schizophrenic patients. New Trends in Experimental and Clinical Psychiatry 1992;7:67‐71. [Google Scholar]
Stewart 1969 {published data only}
- Stewart A, Lafave HG, Segovia G. Haloperidol ‐ new addition to the drug treatment of schizophrenia. Behavioral Neuropsychiatry 1969;1:23‐8. [PubMed] [Google Scholar]
Teja 1975 {published data only}
- Teja JS, Grey WH, Clum JM, Warren C. Tranquilizers or anti‐depressants for chronic schizophrenics: a long term study. Australian and New Zealand Journal of Psychiatry 1975;9:241‐7. [DOI] [PubMed] [Google Scholar]
Tobin 1980 {published data only}
- Tobin JM, Robinson GM. Double blind comparison of haloperidol and thiothixene with after care treatment evaluation in psychiatric outpatients with schizophrenia. Psychiatric Journal of the University of Ottowa 1980;5:168‐74. [Google Scholar]
Tuason 1986 {published data only}
- Tuason VB. A comparison of parenteral loxapine and haloperidol in hostile and aggressive acutely schizophrenic patients. Journal of Clinical Psychiatry 1986;47:126‐9. [PubMed] [Google Scholar]
Ulmar 1990 {published data only}
- Ulmar G, Edler R, Hagen M. Trifluperidol vs haloperidol – a double‐blind trial. Proceedings of the 17th Collegium Internationale Neuro‐Psychopharmacologicum Congress; 1990 Sep 10‐14; Kyoto, Japan. 1990:296.
Versiani 1978 {published data only}
- Versiani M, Silva JA, Frota LH, Mundim FD. Double‐blind comparison between loxapine and haloperidol in the treatment of adolescent schizophrenic patients. Current Therapeutic Research 1978;24:559‐66. [Google Scholar]
White 1981 {published data only}
- Eaton EM, Busk J, Maloney MP, Sloane RB, Whipple K, White K. Hemispheric dysfunction in schizophrenia: assessment by visual perception tasks. Psychiatry Research 1979;1:325‐32. [DOI] [PubMed] [Google Scholar]
- White K, Busk J, Eaton E, Gomez G, Razani J, Sloane RB. Dysphoric response to neuroleptics as a predictor of treatment outcome with schizophrenics. A comparative study of haloperidol versus mesoridazine. International Pharmacopsychiatry 1981;16:34‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Al Haddad 1996 {published data only}
- Al Haddad MK, Kamel C, Sequeria RP, Mawgood MA. Zuclopenthixol versus haloperidol in the initial treatment of schizophrenic psychoses, affective psychoses and paranoid states: a controlled clinical trial. Arab Journal of Psychiatry 1996;7:44‐54. [Google Scholar]
Alpert 1995 {published data only}
- Alpert M, Pouget ER, Sison C, Yahia M, Allan E. Clinical and acoustic measures of the negative syndrome. Psychopharmacology Bulletin 1995;31:321‐6. [PubMed] [Google Scholar]
Azima 1960 {published data only}
- Azima H, Durost H, Arthurs D. The effect of R‐1625 (haloperidol) in mental syndromes: a multiblind study. American Journal of Psychiatry 1960;117:546‐7. [DOI] [PubMed] [Google Scholar]
Barch 2005 {published data only}
- Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophrenia Research 2005;77:43‐58. [DOI] [PubMed] [Google Scholar]
Baro 1972 {published data only}
- Baro F, Lommel R, Dom R, Mesmaecker L. Pimozide treatment of chronic schizophrenics as compared with haloperidol and penfluridol maintenance treatment. A multidisciplinary approach. Acta Psychiatrica Belgica 1972;72:199‐214. [PubMed] [Google Scholar]
Bechelli 1986 {published data only}
- Bechelli LP, Navas‐Filho F. Short term double‐blind trial of pipothiazine palmitate and haloperidol in the acute phase of schizophrenia. L'Encephale 1986;12:121‐5. [PubMed] [Google Scholar]
Boyer 1987 {published data only}
- Boyer P, Puech AJ. Determinants for clinical activity of neuroleptic drugs: chemical substances, doses, assessment tools [Modalities d'action clinique des neuroleptiques: substances, doses, instruments de mesure utilises]. Psychiatrie and Psychobiologie 1987;2:296‐305. [Google Scholar]
Brook 1998 {published data only}
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomised controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. Proceedings of the 10th European College of Neuropsychopharmacology Congress; 1997 Sep 13‐17; Vienna, Austria. 1997.
- Brook S, Berk M, Selemani S, Kolloori J, Nzo I. A randomized controlled double blind study of zuclopenthixol acetate compared to haloperidol in acute psychosis. Human Psychopharmacology 1998;13:17‐20. [Google Scholar]
Chin 1998 {published data only}
- Chin CN, Hamid AR, Philip G, Ramlee T, Mahmud M, Zulkifli G, et al. A double‐blind comparison of zuclopenthixol acetate with haloperidol in the management of acutely disturbed schizophrenics. Medical Journal of Malaysia 1998;53:365‐71. [PubMed] [Google Scholar]
Classen 1988 {published data only}
- Classen W, Laux G. Sensorimotor and cognitive performance of schizophrenic inpatients treated with haloperidol, flupenthixol, or clozapine. Pharmacopsychiatry 1988;21:295‐7. [DOI] [PubMed] [Google Scholar]
Cole 1970 {published data only}
- Cole JO, Tsuang MM. Haloperidol in geriatrics. Psychopharmacology Bulletin 1970;6:86‐8. [Google Scholar]
Costa 2007 {published data only}
- Costa AMN, Lima MS, Faria M, Filho SR, Oliveira IR, Jesus Mari J. A naturalistic, 9‐month follow‐up, comparing olanzapine and conventional antipsychotics on sexual function and hormonal profile for males with schizophrenia. Journal of Psychopharmacology 2007;21:165‐70. [DOI] [PubMed] [Google Scholar]
Crow 1986 {published data only}
- Crow TJ, MacMillan JF, Johnson AL, Johnstone EC. A randomised controlled trial of prophylactic neuroleptic treatment. British Journal of Psychiatry 1986;148:120‐7. [DOI] [PubMed] [Google Scholar]
- MacMillan JF, Crow TJ, Johnson AL, Johnstone EC. British Journal of Psychiatry. Short‐term outcome in trial entrants and trial eligible patients 1986;148:128‐33. [DOI] [PubMed] [Google Scholar]
Davies 2007 {published data only}
- Davies LM, Lewis S, Jones PB, Barnes TRE, Gaughran F, Hayhurst K, et al. Cost‐effectiveness of first‐ v second‐generation antipsychotic drugs: results from a randomised controlled trial in schizophrenia responding poorly to previous therapy. British Journal of Psychiatry 2007;191:14‐22. [DOI] [PubMed] [Google Scholar]
de Jesus Mari 2004 {published data only}
- Jesus Mari J, Lima MS, Costa AN, Alexandrino N, Rodrigues‐Filho S, Oliveira IR, et al. The prevalence of tardive dyskinesia after a nine month naturalistic randomised trial comparing olanzapine with conventional treatment for schizophrenia and related disorders. European Archives of Psychiatry and Clinical Neuroscience 2004;254:356‐61. [DOI] [PubMed] [Google Scholar]
Digo 1967 {published data only}
- Digo R, Karavokyros D, Mlynarski JC. Use of a new butyrophenone in psychiatric service. Outline of a differential study [Utilisation d'une nouvelle butyrophenone dans un service psychiatrique. Esquisse d'une etude differentielle]. L'Encephale 1967;56:75‐91. [PubMed] [Google Scholar]
Dubin 1985 {published data only}
- Dubin WR, Waxman HM, Weiss KJ, Ramchandani D, Tavani‐Petrone C. Rapid tranquilization: the efficacy of oral concentrate. Journal of Clinical Psychiatry 1985;46:475‐8. [PubMed] [Google Scholar]
Durost 1964 {published data only}
- Durost H, Lee H, Arthurs D. An early evaluation of haloperidol. In: Lehmann HE, Ban TA editor(s). The Butyrophenones in Psychiatry. Quebec Psychopharmacological Research Association, 1964:70‐3. [Google Scholar]
Ehmann 1987 {published data only}
- Ehmann TS, Delva NJ, Beninger RJ. Flupenthixol in chronic schizophrenic inpatients: a controlled comparison with haloperidol. Journal of Clinical Psychopharmacology 1987;7:173‐5. [PubMed] [Google Scholar]
Eitan 1992 {published data only}
- Eitan N, Levin Y, Ben Artzi E, Levy A, Neumann M. Effects of antipsychotic drugs on memory functions of schizophrenic patients. Acta Psychiatrica Scandinavica 1992;85:74‐6. [DOI] [PubMed] [Google Scholar]
Engelhardt 1973 {published data only}
- Engelhardt DM, Polizos P, Waizer J, Hoffman SP. A double‐blind comparison of fluphenazine and haloperidol in outpatient schizophrenic children. Journal of Autism and Childhood Schizophrenia 1973;3:128‐37. [DOI] [PubMed] [Google Scholar]
Fitzgerald 1969 {published data only}
- Fitzgerald CH. A double‐blind comparison of haloperidol with perphenazine. Current Therapeutic Research 1969;11:515‐9. [PubMed] [Google Scholar]
Galderisi 1994 {published data only}
- Galderisi S, Maj M, Mucci A, Bucci P, Kemali D. QEEG alpha1 changes after a single dose of high‐potency neuroleptics as a predictor of short‐term response to treatment in schizophrenic patients. Biological Psychiatry 1994;35:367‐74. [DOI] [PubMed] [Google Scholar]
Gerlach 1978 {published data only}
- Gerlach J, Simmelsgaard H. Tardive dyskinesia during and following treatment with haloperidol, haloperidol + biperiden, thioridazine, and clozapine. Psychopharmacology 1978;59:105‐12. [DOI] [PubMed] [Google Scholar]
Gillis 1977 {published data only}
- Gillis JS. The effects of selected antipsychotic drugs of human judgment. Current Therapeutic Research, Clinical and Experimental 1977;21:224‐32. [PubMed] [Google Scholar]
Harris 1992 {published data only}
- Harris MJ, Panton D, Caligiuri MP, Krull AJ, Tran Johnson TK, Jeste DV. High incidence of tardive dyskinesia in older outpatients on low doses of neuroleptics. Psychopharmacology Bulletin 1992;28:87‐92. [PubMed] [Google Scholar]
Holden 1970 {published data only}
- Holden JM, Itil TM, Keskiner A. Assessment and significance of changes in laboratory values with haloperidol and fluphenazine hydrochloride therapy. Biological Psychiatry 1970;2:173‐82. [PubMed] [Google Scholar]
Huang 2005 {published data only}
- Huang TL, Chen JF. Serum lipid profiles and schizophrenia: effects of conventional or atypical antipsychotic drugs in Taiwan. Schizophrenia Research 2005;80:55‐9. [DOI] [PubMed] [Google Scholar]
Hyugano 1986 {published data only}
- Hyugano H. The clinical psychopharmacological efficacy of bromperidol for schizophrenia comparison with haloperidol by the single‐blind cross‐over method. Shinryo to Shinyaku 1986;21:1296‐306. [Google Scholar]
Itil 1975 {published data only}
- Itil TM, Marasa J, Saletu B, Davis S, Mucciardi AN. Computerized EEG: predictor of outcome in schizophrenia. Journal of Nervous and Mental Disease 1975;160:188‐203. [PubMed] [Google Scholar]
Jones 2006 {published data only}
- Jones PB, Barnes TRE, Davies L, Dunn G, Lloyd H, Hayhurst KP, et al. Randomized controlled trial of the effect on quality of life of second‐ vs first‐generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Archives of General Psychiatry 2006;63:1079‐87. [DOI] [PubMed] [Google Scholar]
Karsten 1981 {published data only}
- Karsten D, Kivimaki T, Linna SL, Pollari L, Turunen S. Neuroleptic treatment of oligophrenic patients. A double‐blind clinical multicentre trial of cis(z)‐clopenthixol and haloperidol. Acta Psychiatrica Scandinavica Supplementum 1981;294:39‐45. [PubMed] [Google Scholar]
Kazamatsuri 1972 {published data only}
- Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. II. Short‐term efficacy of dopamine‐blocking agents haloperidol and thiopropazate. Archives of General Psychiatry 1972;27:100‐3. [DOI] [PubMed] [Google Scholar]
Kelwala 1984 {published data only}
- Kelwala S, Ban TA, Berney SA, Wilson WH. Rapid tranquilization ‐ a comparative study of thiothixene and haloperidol. Progress in Neuropsychopharmacology and Biological Psychiatry 1984;8:77‐83. [DOI] [PubMed] [Google Scholar]
Kurt 2007 {published data only}
- Kurt E, Akman B, Alata G, Dadelen S, Oral T. Comparison of cardiac influences of antipsychotic drugs in patients with schizophrenia [Sizofreni tanili hastalarda antipsikotik laclarin kardiyak etkilerinin karilatirilmasi]. Klinik Psikofarmakoloji Bülteni 2007;17:155‐61. [Google Scholar]
Lamure 2003 {published data only}
- Lamure M, Toumi M, Chabannes JP, Dansette GY, Benyaya J, Hansen K. Zuclopenthixol versus haloperidol: an observational randomised pharmacoeconomic evaluation of patients with chronic schizophrenia exhibiting acute psychosis. International Journal of Psychiatry in Clinical Practice 2003;7:177‐85. [Google Scholar]
Lehmann 1967 {published data only}
- Lehmann HE, Ban TA, Lee H. The effectiveness of combined phenothiazine and butyrophenone treatment in chronic schizophrenic patients. Current Therapeutic Research, Clinical and Experimental 1967;9:36‐7. [PubMed] [Google Scholar]
Levenson 1976 {published data only}
- Levenson AJ, Burnett GB, Nottingham JD, Sermas CE, Thornby JI. Speed and rate of remission in acute schizophrenia: a comparison of intramuscularly administered fluphenazine HC1 with thiothixene and haloperidol. Current Therapeutic Research, Clinical and Experimental 1976;20:695‐700. [PubMed] [Google Scholar]
Liu 1996 {published data only}
- Liu K, Lung FW. Difference in prolactin response of schizophrenic patients to equivalent doses of haloperidol, remoxipride and sulpiride. Kao Hsiung I Hsueh Ko Hsueh Tsa Chih Kaohsiung (Journal of Medical Sciences) 1996;12:685‐90. [PubMed] [Google Scholar]
Lovett 1987 {published data only}
- Lovett WC, Stokes DK, Taylor LB, Young ML, Free SM, Phelan DG. Management of behavioral symptoms in disturbed elderly patients: comparison of trifluoperazine and haloperidol. Journal of Clinical Psychiatry 1987;48:234‐36. [PubMed] [Google Scholar]
Lublin 1991 {published data only}
- Lublin H, Gerlach J, Hagert U, Meidahl B, Molbjerg C, Pedersen V, et al. Zuclopenthixol, a combined dopamine D1/D2 antagonist, versus haloperidol, a dopamine D2 antagonist, in tardive dyskinesia. European Neuropsychopharmacology 1991;1:541‐8. [DOI] [PubMed] [Google Scholar]
Malt 1995 {published data only}
- Malt UF, Nystad R, Bache T, Noren O, Sjaastad M, Solberg KO, et al. Effectiveness of zuclopenthixol compared with haloperidol in the treatment of behavioural disturbances in learning disabled patients. British Journal of Psychiatry 1995;166:374‐7. [DOI] [PubMed] [Google Scholar]
Mosolov 2000 {published data only}
- Mosolov SN, Molodetskikh AV, Eryomin AV, Graikov GM, Nourislamov SV. Effect and tolerability of clopixol and haloperidol in patients with acute symptoms of schizophrenia: comparative randomised investigation. Sotsial'naia I Klinicheskaia Psikhiatria 2000;10:47‐52. [Google Scholar]
Nahunek 1982 {published data only}
- Nahunek K, Svestka J, Ceskova E, Rysanek R. Blind comparison of oxyprothepin and haloperidol in six‐week treatment periods in schizophrenia. Activitas Nervosa Superior 1982;24:219‐20. [Google Scholar]
Nordic 1986 {published data only}
- Gerlach J. Tardive dyskinesia: pathophysiological mechanisms and clinical trials. L'Encephale 1988;14:227‐32. [PubMed] [Google Scholar]
- Nordic Dyskinesia Study Group. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video‐controlled multicenter study with chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden. Psychopharmacology 1986;90:423‐9. [PubMed] [Google Scholar]
- Povlsen UJ, Noring U, Meidahl B, Korsgaard S, Waehrens J, Gerlach J. The effects of neuroleptics on tardive dyskinesias. A video‐controlled, randomized study of chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden [En videokontrolleret, randomiseret undersogelse med klorprotixen, perfenazin, haloperidol og haloperidol + biperiden]. Ugeskrift for Laeger 1987;25:1682‐5. [PubMed] [Google Scholar]
Onodera 1984 {published data only}
- Onodera I, Ito K, Ito K. The double‐blind comparative study of bromperidol and haloperidol in the treatment of schizophrenic inpatients. Kiso to Rinsho 1984;18:3349‐71. [Google Scholar]
Paprocki 1977 {published data only}
- Paprocki J, Versiani M. A double‐blind comparison between loxapine and haloperidol by parenteral route in acute schizophrenia. Current Therapeutic Research, Clinical and Experimental 1977;21:80‐100. [PubMed] [Google Scholar]
Pedros Rosello 2004 {published data only}
- Pedros Rosello A, Tenias JM. Neuroleptics election and clinical outcomes in acute psychosis: a comparative study of risperidone versus other neuroleptics [Eleccion de neuroleptico y respuesta clinica en psicosis aguda: un estudio comparativo de risperidona frente a otros neurolepticos]. Anales de Psiquiatria 2004;20:167‐71. [Google Scholar]
Reimold 2007 {published data only}
- Reimold M, Solbach C, Noda S, Schaefer JE, Bartels M, Beneke M, et al. Occupancy of dopamine D(1), D (2) and serotonin (2A) receptors in schizophrenic patients treated with flupentixol in comparison with risperidone and haloperidol. Psychopharmacology 2007;190:241‐9. [DOI] [PubMed] [Google Scholar]
Reznik 2000 {published data only}
- Reznik I, Sirota P, Psych M. Obsessive and compulsive symptoms in schizophrenia: a randomized controlled trial with fluvoxamine and neuroleptics. Journal of Clinical Psychopharmacology 2000;20:410‐6. [DOI] [PubMed] [Google Scholar]
Saletu 1986 {published data only}
- Saletu B, Kufferle B, Grunberger J, Anderer P. Quantitative EEG, SPEM, and psychometric studies in schizophrenics before and during differential neuroleptic therapy. Pharmacopsychiatry 19;1986:434‐7. [DOI] [PubMed] [Google Scholar]
Samuels 1961 {published data only}
- Samuels AS. A controlled study of haloperidol: the effects of small dosages. American Journal of Psychiatry 1961;11:253‐4. [DOI] [PubMed] [Google Scholar]
Serban 1984 {published data only}
- Serban G, Siegel S. Response of borderline and schizotypal patients to small doses of thiothixene and haloperidol. American Journal of Psychiatry 1984;141:1455‐8. [DOI] [PubMed] [Google Scholar]
Simpson 1967 {published data only}
- Simpson GM, Angus JW, Edwards JG. A controlled study of haloperidol in chronic schizophrenia. Current Therapeutic Research, Clinical and Experimental 1967;9:407‐12. [PubMed] [Google Scholar]
- Simpson GM, Cooper TB, Braun GA. Further studies on the effect of butyrophenones on cholesterol synthesis in humans. Current Therapeutic Research, Clinical and Experimental 1967;9:413‐8. [PubMed] [Google Scholar]
Singh 1976 {published data only}
- Singh MM. Dysphoric response to neuroleptic treatment in schizophrenia and its prognostic significance. Diseases of the Nervous System 1976;37:191‐6. [PubMed] [Google Scholar]
- Singh MM. Dysphoric response to neuroleptic treatment in schizophrenia and its prognostic significance. Journal of the Bronx State Hospital 1974;2:173‐83. [PubMed] [Google Scholar]
Smith 1984 {published data only}
- Smith RC, Baumgartner R, Ravichandran GK, Mauldin M, Burd A, Vroulis G, et al. Lateral ventricular enlargement and clinical response in schizophrenia. Psychiatry Research 1985;14:241‐53. [DOI] [PubMed] [Google Scholar]
Stotsky 1977 {published data only}
- Stotsky BA. Relative efficacy of parenteral haloperidol and thiothixene for the emergency treatment of acutely excited and agitated patients. Diseases of the Nervous System 1977;38:967‐73. [PubMed] [Google Scholar]
Su 2002 {published data only}
- Su KP, Chen KP, Shen WW, Tsai SY. QTc prolongation associated with sulpiride and haloperidol use: a pilot crossover‐design study. European Neuropsychopharmacology. 2000; Vol. 10(Suppl. 3):311‐2.
- Su KP, Shen WW, Chuang CL, Chen KP, Chen CC. A pilot cross‐over design study on QTc interval prolongation associated with sulpiride and haloperidol. Schizophrenia Research 2002;59:93‐4. [DOI] [PubMed] [Google Scholar]
Taymeeyapradit 2002 {published data only}
- Taymeeyapradit U, Kuasirikul S. Comparative study of the effectiveness of zuclopenthixol acetate and haloperidol in acutely disturbed psychotic patients. Journal of the Medical Association of Thailand 2002;85:1301‐8. [PubMed] [Google Scholar]
Terminska 1989 {published data only}
- Terminska K, Mrowiec W. Comparison of influence of perazine, fluphenazine, trifluoroperazine, chloropromazine and haloperidol on primary and deficit schizophrenic symptoms in patients first hospitalized because of paranoid schizophrenia [Badanie porownawcze wplywu perazyny, flufenazyny, trifluoroperazyny, chloropromazny i haloperydolu na objawy pierwotne i deficytowe pierwszego zachorowania na schizofrenie paranoidalna]. Psychiatria Polska 1989;1:24‐30. [PubMed] [Google Scholar]
van Lommel 1974 {published data only}
- Lommel R, Baro F, Dom R. The influence of haloperidol and penfluridol on the learning capacity of the schizophrenic. Arzneimittelforschung 1974;24:1072‐4. [PubMed] [Google Scholar]
van Putten 1984 {published data only}
- Putten T, May PR, Marder SR. Akathisia with haloperidol and thiothixene. Archives of General Psychiatry 1984;41:1036‐9. [DOI] [PubMed] [Google Scholar]
van Putten 1986 {published data only}
- Putten T, Marder SR. Low‐dose treatment strategies. Journal of Clinical Psychiatry 1986;47:12‐6. [PubMed] [Google Scholar]
Zuoning 1999 {published data only}
- Zuoning J, Huang SZ, Qin YF, Yamamoto M. Comparative clinical study of nemonapride, a benzamide compound with dopamine D‐2‐like receptor blocking activity, and haloperidol in schizophrenic patients. Research Communications in Biological Psychology and Psychiatry 1999;24:35‐46. [Google Scholar]
Additional references
Adams 2013
- Adams CE, Bergman H, Irving CB, Lawrie S. Haloperidol versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD003082.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2014
- Adams CE, Awad GA, Rathbone J, Thornley B, Soares‐Weiser K. Chlorpromazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD000284.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
AMDP 2007
- AMDP. Das AMDP‐System. Manual zur Dokumentation psychiatrischer Befunde. 8. Göttingen: Hogrefe, 2007. [Google Scholar]
Andreasen 1983
- Andreasen NC. The Scale for the Assessment of Positive Symptoms (SAPS). Iowa City, IA: The University of Iowa, 1983. [Google Scholar]
Andreasen 1989
- Andreasen NC. Scale for the assessment of negative symptoms. British Journal of Psychiatry 1989;155:53‐8. [PubMed] [Google Scholar]
Berger 2003
- Berger M. Psychische Erkrankungen. Klinik und Therapie. 2nd edition. München: Urban & Fischer, 2003. [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54:405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Chakrabarti 2007
- Chakrabarti A, Bagnall A, Chue P, Fenton M, Palaniswamy V, Wong W, et al. Loxapine for schizophrenia. Cochrane Database of Systematic Reviews 2007, Issue 10. [DOI: 10.1002/14651858.CD001943.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 1978
- Davis JM, Garver DL. Neuroleptics: clinical use in psychiatry. In: L. Iversen, S Iversen, S. Snyder editor(s). Handbook of Psychopharmacology. New York: Plenum Press, 1978. [Google Scholar]
Davis 1989
- Davis JM, Barter JT, Kane JM. Antipsychotic drugs. Comprehensive Textbook of Psychiatry. Baltimore: Williams and Wilkins, 1989. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Der‐Simonian 1986
- Der‐Simonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Duggan 2005
- Duggan L, Fenton M, Rathbone J, Dardennes R, El‐Dosoky A, Indran S. Olanzapine for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD001359.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997a
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997b
- Egger M, Zellweger‐Zähner T, Schneider M, Junker C, Lengeler C, Antes G. Language bias in randomised controlled trials published in English and German. Lancet 1997;350:326‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31:140‐9. [DOI] [PubMed] [Google Scholar]
Essali 2009
- Essali A, Al‐Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD000059.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Falkai 2005
- Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Möller HJ, et al. World Federation of Societies of Biological Psychiatry (WFSBP) ‐ Guidelines for biological treatment of schizophrenia, part 1: Acute treatment of schizophrenia. World Journal of Biological Psychiatry 2005;6:132‐91. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA Jr, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26:57‐63. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59:7‐10. [DOI] [PubMed] [Google Scholar]
Gaebel 2006
- Gaebel W, Falkai P, Weinmann S. Behandlungsleitlinie Schizophrenie. Darmstadt: Steinkopff, 2006. [Google Scholar]
Goodrich 1953
- Goodrich W. Rating Scale for Quantification of Psychotic Symptom Severity (RSQPSS). American Journal of Psychiatry 1953;110:334. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU Assessment Manual for Psychopharmacology (DOTES: Dosage Record and Treatment Emergent Symptom Scale). Rockville: National Institute of Mental Health, 1976. [Google Scholar]
Hamilton 1960
- Hamilton, M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hartung 2005
- Hartung B, Wada M, Laux G, Leucht S. Perphenazine for schizophrenia. Cochrane Database of Systematic Reviews 2005, (1). [DOI: 10.1002/14651858.CD003443.pub2] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Honigfeld 1965
- Honigfeld G, Gillis RD, Klett CJ. Nurses' observation scale for inpatient evaluation: a new scale for measuring improvement in chronic schizophrenia. Journal of Clinical Psychology 1965;21:65‐71. [DOI] [PubMed] [Google Scholar]
Huf 2011
- Huf W, Kalcher K, Pail G, Friedrich ME, Filzmoser P, Kasper S. Meta‐analysis: fact or fiction? How to interpret meta‐analyses. World Journal of Biological Psychiatry 2011;12:188‐200. [DOI] [PubMed] [Google Scholar]
Hunter 2003
- Hunter R, Kennedy E, Song F, Gadon L, Irving CB. Risperidone versus typical antipsychotic medication for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 2. [DOI: 10.1002/14651858.CD000440] [DOI] [PubMed] [Google Scholar]
Jayakody 2012
- Jayakody K, Gibson RC, Kumar A, Gunadasa S. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD000525.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Kay 1987
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia Bulletin 1987;13:261‐76. [DOI] [PubMed] [Google Scholar]
Kaye 2003
- Kaye JA, Bradbury BD, Jick H. Changes in antipsychotic drug prescribing by general practitioners in the United Kingdom from 1991 to 2000: a population‐based observational study. British Journal of Clinical Pharmacology 2003;56:569‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Klein 1969
- Klein DF, Davis JM. Klein DF, Davis JM. Diagnosis and drug treatment of psychiatric disorders. Baltimore: Williams and Wilkins, 1969. Diagnosis and Drug Treatment of Psychiatric Disorders. Baltimore: Williams and Wilkins, 1969. [Google Scholar]
Koch 2014
- Koch K, Mansi K, Haynes E, Adams CE, Sampson S, Furtado VA. Trifluoperazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010226] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kudo 1999
- Kudo S, Ishizaki T. Pharmacokinetics of haloperidol: an update. Clinical Pharmacokinetics 1999;37:435‐56. [DOI] [PubMed] [Google Scholar]
Lehman 2004
- Lehman AF, Lieberman JA, Dixon LB, McGlashan TH, Miller AL, Perkins DO, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. American Journal of Psychiatry 2004;161:1‐56. [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59:1001‐5. [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. What does the PANSS mean?. Schizophrenia Research 2005;79:231‐8. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2006
- Leucht S, Kane JM, Etschel E, Kissling W, Hamann J, Engel RR. Linking the PANSS, BPRS, and CGI: clinical implications. Neuropsychopharmacology 2006;31:2318‐25. [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2008
- Leucht C, Kitzmantel M, Kane J, Leucht S, Chua WLLC. Haloperidol versus chlorpromazine for schizophrenia. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD004278.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2009
- Leucht S, Corves C, Arbter D, Engel R, Li C, Davis JM. A meta‐analysis comparing second‐generation and first‐generation antipsychotics for schizophrenia. Lancet 2009;373:31‐41. [DOI] [PubMed] [Google Scholar]
Lieberman 2005
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New England Journal of Medicine 2005;353:1209‐23. [DOI] [PubMed] [Google Scholar]
Lohse 2009
- Lohse MJ, Müller‐Oerlinghausen B. Psychopharmaka. In: U. Schwabe, D. Pfaffrath editor(s). Arzneimittelverordnungsreport. Heidelberg: Springer, 2009:767‐810. [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marvaha S, Johnson S. Schizophrenia and employment ? a review. Social Psychiatry and Psychiatric Epidemiology 2004;39:337‐49. [DOI] [PubMed] [Google Scholar]
Montgomery 1978
- Montgomery SA, Taylor P, Montgomery D. Development of a schizophrenia scale sensitive to change. Neuropsychopharmacology 1978;17:1061‐2. [DOI] [PubMed] [Google Scholar]
Mothi 2013
- Mothi M, Sampson S. Pimozide for schizophrenia or related psychoses. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD001949.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Paton 2003
- Paton C, Lelliott P, Harrington M, Okocha C, Sensky T, Duffett R. Patterns of antipsychotic and anticholinergic prescribing for hospital inpatients. Journal of Psychopharmacology 2003;17:223‐9. [DOI] [PubMed] [Google Scholar]
Sampford 2013
- Sampford J, Sampson S. Fluphenazine (oral) versus atypical antipsychotics for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD010832] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. The Journal of the American Medical Association 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Trials 2010;11:32. [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, et al. Grading Quality of Evidence and Strength of Recommendations for Diagnostic Tests and Strategies. BMJ 2008;336:1106‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Seeman 1975
- Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975;188:1217‐9. [DOI] [PubMed] [Google Scholar]
Shen 2012
- Shen X, Xia J, Adams CE. Flupenthixol versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD009777.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Silveira 2002
- Silveira da Mota Neto JI, Soares BGO, Silva de Lima M. Amisulpride for schizophrenia. Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI: 10.1002/14651858.CD001357] [DOI] [PMC free article] [PubMed] [Google Scholar]
Srisurapanont 2004
- Srisurapanont M, Maneeton B, Maneeton N, Lankappa S, Gandhi R. Quetiapine for schizophrenia. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD000967.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2011a
- Tardy M, Huhn M, Kissling W, Engel RR, Leucht S. Haloperidol versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD009268] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2011b
- Tardy M, Huhn M, Engel RR, Leucht S. Perphenazine versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD009369] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2011c
- Tardy M, Huhn M, Engel RR, Leucht S. Fluphenazine versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 8. [DOI: 10.1002/14651858.CD009230] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2011d
- Tardy M, Dold M, Engel RR, Leucht S. Trifluoperazine versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD009396] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tardy 2011e
- Tardy M, Dold M, Engel RR, Leucht S. Flupenthixol versus low‐potency first‐generation antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD009227] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐5. [DOI] [PubMed] [Google Scholar]
WHO 2009
- World Health Organization (WHO). Essential drugs. WHO Drug Information 2009;13:249‐92. [Google Scholar]
Wing 1961
- Wing JK. A simple and reliable subclassification of chronic schizophrenia. Journal of Mental Science 1961;107:862‐76. [DOI] [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33:254‐7. [Google Scholar]