

Summary
The Montseny brook newt (Calotriton arnoldi), considered the most endangered amphibian in Europe, is a relict salamandrid species endemic to a small massif located in northeastern Spain. Although conservation efforts should always be guided by genomic studies, those are yet scarce among urodeles, hampered by the extreme sizes of their genomes. Here, we present the third available genome assembly for the order Caudata, and the first genomic study of the species and its sister taxon, the Pyrenean brook newt (Calotriton asper), combining whole-genome and ddRADseq data. Our results reveal significant demographic oscillations which accurately mirrored Europe’s climatic history. Although severe bottlenecks have led to depauperate genomic diversity and long runs of homozygosity along a gigantic genome, inbreeding might have been avoided by assortative mating strategies. Other life history traits, however, seem to have been less advantageous, and the lack of land dispersal has driven to exceptional levels of population fragmentation.
Subject areas: Genomics, Zoology, Ecology
Graphical abstract
Highlights
-
•
Third genome assembly of a urodele and first genomic study of the Montseny brook newt
-
•
Inferred demographic oscillations are accurately predicted by past climate changes
-
•
Bottlenecks have led to low genomic diversity and runs of homozygosity
-
•
Montseny brook newt populations are highly structured due to the lack of land dispersal
Genomics; Zoology; Ecology
Introduction
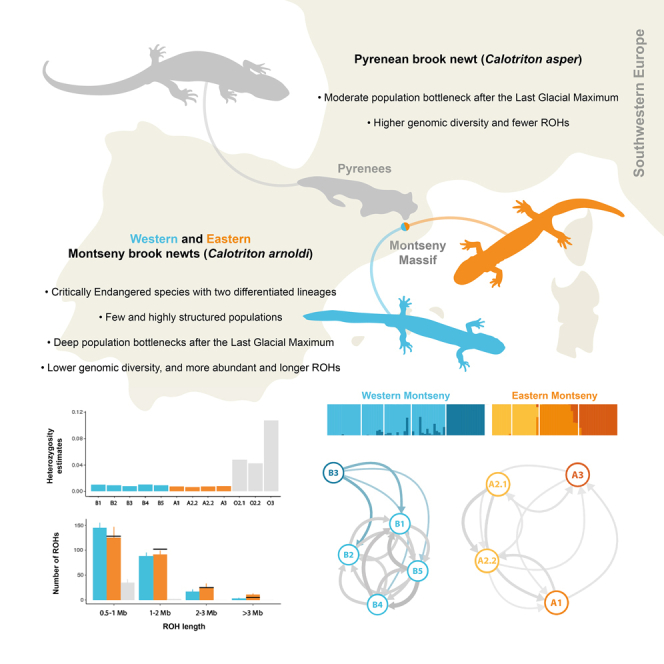
During the Quaternary, the shifting climate of Europe and its complex geography, resulted in an intricate dance of expanding and retracting species ranges.1 While warm periods fostered the recolonization of the continent by some thermophilic taxa expanding from diverse glacial refugia, alpine species ranges shattered into patches, and conversely during glaciations. Such fragmentation into the so-called alpine archipelago, resulted in copious examples of disjunct distributions and, ultimately, in vicariant sibling species.2 One example are the newts in the genus Calotriton, comprising the relatively widespread Pyrenean brook newt C. asper and the Montseny brook newt C. arnoldi. Whereas the former inhabits the Pyrenees and neighboring lesser mountain ranges, the latter is a Critically Endangered microendemism exclusive to the Montseny Massif (Catalonia, NE Spain; Figure 1).3
Figure 1.

Distribution of brook newts Calotriton spp. in Western Europe
First close-up (below) shows the Pyrenees and adjacent mountain ranges, including the Montseny Massif (dashed). Second close-up (top-right) shows the area from this massif to the neighboring Eastern Pyrenees, and the rivers Ter and Tordera (in white), which divide the ranges of the Pyrenean-Montseny brook newts, and Western-Eastern Montseny lineages, respectively. Montseny brook newt (Calotriton arnoldi) range in blue (Western lineage) and orange (Eastern lineage). Pyrenean brook newt (Calotriton asper) distribution in light gray, within dashed line. White circles identify the sampling localities used in this study. Sampling points at Montseny do not correspond to exact geographic localities for conservation reasons.
At this Eurosiberian exclave, the Montseny brook newt is thought to have been evolving in isolation for the last 1.76 million years (my).4 Currently, natural populations of this species are scattered across only eight streams, barely covering an occupancy area of 10 km2.5 Although this minute range might not seem prone to fragmentation, the deep Tordera River valley divides the Montseny into two submassifs from West to East, breaking apart two sets of populations. The connectivity between them is disrupted not only because of geographical barriers, but also due to inherent biological features of the species: whereas Calotriton asper undergoes a 2-year-long terrestrial stage following metamorphosis,6 dispersal in the Montseny brook newt is greatly hampered by the lack of functional lungs.3 This adaptation to avoid undesired buoyancy in strong-current brooks makes them fully dependent on groundwater during dry periods, and directly restricts dispersal to water-way routes. In turn, the 5 km width of the Tordera River valley represents a largely unsuitable >30 km water pathway, crossing unfavorable environments for this strictly aquatic amphibian. Consequently, genetic studies showed that the Western and Eastern C. arnoldi sets of populations constitute two differentiated lineages, isolated since the late Middle Pleistocene.4 The combination of long-term isolation and small population sizes has resulted in incipient morphological differentiation, including the fixation of distinct color patterns. However, contrary to expectations, genetic studies did not find reduced diversity compared to C. asper.4,7
Loss of genetic variability has been theoretically and empirically linked to decreases in the mean fitness and evolutionary potential of populations.8,9,10,11 In amphibians, genetic erosion has been found to be an important factor diminishing resistance to the parasitic fungus Batrachochytrium dendrobatidis in anurans.12 Batrachochytrium infections are one of the main global drivers of declines in amphibian biodiversity.13,14 While Batrachochytrium salamandrivorans wreaks havoc on urodele populations across Europe15 and already caused an outbreak in a Natural Park 15 km south of Montseny in 2018,16 its lethal effect in Calotriton arnoldi has been confirmed by lab trials.17 On the other hand, groundwater-depending amphibians are one of the most vulnerable components of global diversity due to rapid groundwater depletion at global and regional scales.18 Indeed, nearly one third of the total bottled water consumed in Spain is abstracted in the Montseny Massif and its surroundings.19 This groundwater overdraft has resulted in the desiccation of the brook headwaters, leading to the disappearance of the species in some of these areas.3,5 On top of that, despite being aquatic, the Montseny brook newt requires specific types of forests covering the streams. Unfortunately, some of these forests are being replaced by non-native timber plantations, while local species, such as beech trees, are shifting toward higher elevations due to the effects of global warming,20 limiting the potential habitat available for the Montseny brook newt.
Altogether, the menace of emergent diseases and habitat loss due to global warming and human activities impose significant pressure on these small, fragmented, and declining populations, whose size has been estimated at approximately 1,000–1,500 adult individuals in the wild.5 This situation led to the declaration of the Montseny brook newt as the only ‘Critically Endangered’ urodele species in Europe by the IUCN, and one of the most threatened amphibian species in Europe5 with the Karpathos frog (Pelophylax cerigensis) (but see Toli et al.21 for the current need of conservation reassessment in the later). As a result, two years after its description as a new species, a provisional conservation program by the Catalan government began with the purpose of guaranteeing its future,22 followed later by a LIFE project (LIFE15 NATO/SE/000757) in 2016. Thanks to the genetic studies corroborating the existence of two well differentiated lineages,3,4,7 representatives from both submassifs were chosen to be founders of an international ex situ breeding program. Since then, these lineages have been treated as different management units, allowing no admixture to preserve their distinctiveness, despite the lack of any genomic insight. Ideally, any conservation effort should be guided by genomic studies, which provide valuable information such as effective population size, inbreeding, demographic history, and population structure.23 However, those are yet scarce in urodeles, whose gigantic genomes24 render the sequencing of enough data for reference genomes challenging.
Here, we present the reference genome of the Montseny brook newt, the third for the order Caudata,25,26 as well as exhaustive population genomic analyses based on 22 low-coverage whole genomes (lcWGS) and ddRADseq data for 132 individuals from all Montseny populations and several populations of the closely related Pyrenean brook newt C. asper. Our results reveal a series of population bottlenecks during the Late Pleistocene and Holocene, coinciding with warm periods in Europe, as well as their ultimate signatures: long Runs of Homozygosity and depauperate diversity values in all extant populations. Overall, this study highlights both the urge to take conservation actions under the current global climate crisis and the deep-rooted vulnerability of Europe’s most threatened amphibian species.
Results
We generated the third de novo genome assembly for the order Caudata using two captive-bred siblings from one Eastern populations (A1) of the Montseny brook newt. This resulted in a genome assembly size of 22.89 Gb, N50 = 1.65 Mb and 82.4% of vertebrata ortholog completeness (Figures S1–S3; Tables S1 and S2) which was used to map both lcWGS and ddRADseq data from 21 to 132 wild animals, respectively. The sampling comprises all C. arnoldi natural populations and representatives of the five mitochondrial lineages of C. asper.
Brook newts’ evolutionary history was shaped by climatic oscillations
We found the effective population sizes of both brook newt species to be significantly dependent on climate oscillations along the Late Pleistocene and Holocene, showing negative correlations with waning ice sheets and, mainly, rising temperatures (Table S3), which explains up to 85% of C. arnoldi’s demographic trends. While large effective population sizes for both C. arnoldi and C. asper along the Late Pleistocene were estimated, population declines toward the present were also inferred (Figure 2). The Pyrenean brook newt shows a slow but constant decrease in effective population size, with the largest drop at the end of the Last Glacial Maximum (LGM, c. 26.5–19 kya). On the other hand, the Montseny brook newt suffered a more recent and severe bottleneck, coinciding with two warming periods: the Late Glacial Interstadial (LGI, c. 14.67–12.89 kya), and especially the sudden end of the Last Glacial Period (LGP, 110–11.65 kya), in which near-modern temperatures were reached. In between both events, the colder Young Dryas (YD, c. 12.89-11.65 kya) seems to have partially buffered their collapse. Nevertheless, during a brief warm period, the Roman Climatic Optimum (RCO, c. 2.25–1.55 kya), the effective population size reached its minimum with <500 effective individuals. Afterward, the populations recovered to some extent coinciding with the beginning of a locally colder and wetter period in modern Europe: the Little Ice Age (LIA, 14th −18th centuries).
Figure 2.

Demographic inference (above) coupled with climatic oscillations (below) along the Holocene and Late Pleistocene
Median effective population sizes (Ne x1,000) calculated with Stairwayplot2 for Calotriton arnoldi (orange line, below) and C. asper (gray line, above) are shown with corresponding 95% CI (areas shaded in orange or gray, respectively). Climatic oscillations are represented by the mean temperatures in the Northern Hemisphere (de Boer et al., 2013; red line) and West Mediterranean temperatures within 95% CI (red shaded area; Margaritelli et al., 2020). Eurasian ice sheet volume (blue area) is depicted as negative meters of the sea level in the corresponding time compared to the present. Time before present shown in logarithmic scale (x axis). The Pleistocene-Holocene transition and some other important climatic events are highlighted: the Little Ice Age, Roman Climatic Optimum (RCO), Last Glacial Period, Younger Dryas (YD), Late Glacial Interstadial (LGI) and Last Glacial Maximum (LGM). The two events associated with the main population bottlenecks in the brook newts are highlighted by dashed lines: 1) the beginning of the LGI for C. arnoldi and 2) the end of the LGM for C. asper.
Comparisons of simulations with different migration patterns between lineages support both recent migration between Montseny submassifs (marnoldi in Table S4) and, at higher rates, ancestral migration from the Pyrenees into the Montseny (masper; Figure S4; Tables S4 and S5). Effective population sizes were lower for the Eastern Montseny C. arnoldi than for its Western counterpart, and both were extremely reduced compared to C. asper’s current Ne (NA for current Eastern Montseny, NB for Western Montseny, and NO for the Pyrenean brook newt in Table S4). Large ancestral populations (NAB for ancestral C. arnoldi population before lineage divergence and NABO for ancestral Calotriton population before species divergence in Table S4) and bottlenecks are in accordance with the demographic inference results from Stairwayplot analyses and lead to the idea of a progressive rarefaction of the genus.
At a larger scale, we found divergence events within the genus Calotriton to match certain cold or warm periods (Figure 3). Montseny and Pyrenean newts shared their most recent common ancestor 1.69 mya (95% highest probability density [HPD] intervals: 1.05–2.35 mya), partially coinciding with the Donau glaciation (1.8–1 mya). Divergence between Calotriton asper lineages, around 160 kya (101–258 kya), overlaps with another cold period: the Riss glaciation (300–130 kya). In contrast, divergence between Eastern and Western Montseny lineages seems to have taken place later, around 111 kya (68–155 kya), roughly matching with a warmer period: the Riss-Würm Interglacial (130–115 kya).
Figure 3.

Genomic phylogeny of the genus Calotriton
Left, time-calibrated Bayesian phylogeny from ddRADseq data (cloudogram+consensus tree). The nodes are shown with associated posterior probabilities (in bold) and estimated mean times of divergence with their 95%CIs (horizontal gray bars). Three glacial periods (Donau, Riss and Würm/LGP) are highlighted in light blue, whereas the warmer Riss-Würm Interglacial is displayed as the red vertical line. Right, pictures of representative individuals of the three different brook newt lineages in the wild: from top to bottom, Eastern and Western Montseny brook newt Calotriton arnoldi and Pyrenean brook newt C. asper.
Genetic erosion and unexpected levels of fragmentation
Observed heterozygosity (HO) for the Montseny brook newt was estimated to be between 10.18 and 21.14 (from lcWGS; Table S6) and 4.45–17.23 (from ddRADseq; Table S7) times lower for the Montseny brook newt than for its sister species, the Pyrenean brook newt. Moreover, HO was slightly higher in the Western Montseny populations than in the Eastern ones according to both sequencing techniques, and at a finer scale, sparsely populated creeks (e.g., B3, A2.1) yielded lower values than populous well-connected demes (i.e., B1–2, B4–5). However, for all C. arnoldi populations (and two out of three C. asper demes), HO was always higher than expected (HE), suggesting absence of inbreeding (GIS) based on ddRADseq data (Figure 4A; Table S7). Low coverage WGS showed a higher number of ROHs in C. arnoldi compared to C. asper, including the presence of medium- and long-sized ROHs in the former, especially in the Eastern Montseny lineage (Figure 4B; Table S8).
Figure 4.

Genomic diversity, runs of homozygosity (ROHs) and fixation indices in genus Calotriton
(A) Population-level observed and expected heterozygosity estimates from 132 ddRADseq samples, including all Montseny and three Pyrenean populations. Observed heterozygosity is lower in Montseny brook newts than in Pyrenean brook newts, but is always higher than expected.
(B) Bar plot of the mean + standard deviation counts of ROHs found in the Western Montseny (blue, n = 10), Eastern Montseny (orange, n = 6), and Pyrenean (gray, n = 5; C. asper) brook newts from lcWGS sorted by four increasing length categories. Black bars show the respective counts obtained from one F1 captive-bred Eastern individual used in the reference genome assembly. Montseny brook newts show a higher number of short ROHs than Pyrenean brook newts and, moreover, exhibit medium- and long-sized ROHs in their genomes, as expected in bottlenecked and isolated taxa. Within Montseny, the Eastern lineage shows more long-sized ROHs than the Western lineage.
(C) Population-pairwise fixation indices (F’ST) among the 12 populations studied with ddRADseq. F’ST and significance values are shown in Table S9. Fixation of diversity between Western Montseny populations (in blue) and Eastern Montseny ones (orange) is more similar to fixation between C. arnoldi and C. asper (in gray) than between populations belonging to the same lineages.
Despite low levels of heterozygosity in the Montseny brook newt, the fixation of such diversity was unexpectedly high. The mean fixation index (F’ST; i.e., the percentage of shared diversity lost across a barrier in between demes) between Western and Eastern Montseny submassifs was higher (F’ST = 0.801; p < 0.001) than between Western and Eastern Pyrenees C. asper (F’ST = 0.554; p < 0.001), in spite of involving a much smaller geographical scale. Within Montseny lineages, population-pairwise fixation indices were relatively lower but still significant (Figure 4C; Table S9). Genomic PCAs showed agreeing results with both types of genomic data. First components of lcWGS and ddRADseq PCAs including C. asper explained a vast majority of the variance, sorting out the two sister species (Figures S5 and S6). Second and third components split lineages within C. asper Western (lineages O2–3) and Eastern Pyrenees (O1, O4–5) and C. arnoldi respectively. Within C. arnoldi, PCAs explained mostly the split between Western and Eastern submassifs, with A3 and B3 as the most divergent populations for each lineage.
Genus-level Admixture analysis with ddRADseq data recovered three clusters without admixture among them: Calotriton asper, East Montseny and West Montseny (reaching cross validation errors (CV) plateau at K3, Table S10; Figure 5A). Migration networks similarly supported the complete isolation between Montseny lineages, which represent two independent metapopulations without current migration between them (Figure 5B). Isolation-by-distance (IBD) alone did not fully explain genomic differences between Montseny submassifs (p < 0.001), despite considering water-restricted dispersal. However, structure within both Montseny lineages is concordant with the same IBD pattern (Figure S8). Although Admixture analysis within Montseny yielded the lowest CV errors at K = 2, separating again Eastern and Western Montseny lineages, K = 5 holds clear biological significance with a close CV mean value (Table S10), separating A1–A2 (which remain lumped at K = 4) and distinguishing the three Eastern inhabited brooks as different clusters (Figure 5A). Matching the high levels of isolation in the East, migration was found to be low within the metapopulation, even between A2.1–A2.2 (Figure 5B). These two subpopulations, despite belonging to the same creek and genomic cluster, show migration rates similar to other populations within the Eastern submassif, likely because a road, with the concomitant desiccation of a creek stretch, has artificially disconnected them. On the other hand, at the Western submassif, two putative clusters were identified: one population, B3, remains free from admixture with any other population, whereas the rest of the Western populations are merged into the same cluster (i.e., B1–2, B4–5), exhibiting high levels of connectivity (Figure 5B) and varying levels of admixture with B3 (Figure 5A). Migration from B3 to other Western populations appears to be significantly asymmetrical, being a source but not receiving migrant from other populations. This source-sink dynamic can be explained by the watershed architecture: B3 is physically isolated from the other populations by a large waterfall, rendering upstream dispersal impossible.
Figure 5.

Genome-wide population structure and migration networks inferred from ddRADseq data
(A) Admixture plots at K = 3 (with C. asper; 9,470 uSNPs) and K = 5 (without C. asper; 1,339 uSNPs) for 132 ddRADseq samples. K = 3 shows no admixture among the three taxa, whereas K = 5 depicts fine-scale fragmentation within C. arnoldi, especially at the Eastern Montseny.
(B) Left and right, networks of current migration in the two isolated Western and Eastern Montseny lineages; center, scheme of newt-inhabited creeks in the Tordera River basin. Calotriton arnoldi populations are encoded in circles connected by arrows that darken and increase in size as relative geneflow rates increase. Population B3 is isolated from other Western Montseny populations by a high waterfall. In turn, geneflow from B3 (colored arrows in the network) is significantly asymmetrical: i.e., newts can only disperse downstream of B3. Gene flow between populations A2.1 and A2.2 of the Eastern Montseny lineage, despite being sampled at different elevations within the same creek and belonging to the same genomic cluster, is relatively low. This may result from a barrier effect caused by a road that cuts across the stream between the two sampled populations.
Discussion
Evolutionary history
The tighter dependence of C. arnoldi population size on climatic oscillations than C. asper agrees with a narrower range of preferred body temperatures in the former,27,28 despite inhabiting more meridional and temperate places (Table S3; Figure 2). Such intimate relation between demographic trends and climatic oscillations seems to have shaped the evolutionary history of Calotriton spp. On the one hand, glacial periods (i.e., Donau and Riss) may have caused divergences in the Pyrenean–Montseny and the Western Pyrenees–Eastern Pyrenees species and lineages, respectively (Figure 3). Indeed, glaciations have been considered as drivers of transverse alpine speciation in temperate mountain ranges all around the globe, including the Pyrenees (see Wallis et al.29). Species survival during glacial periods at high-altitude refugia,30 is consistent with higher genomic diversity values in high altitude populations (Table S6). In addition, cold-adapted species are expected to thrive during glaciations and expand their ranges across ice-free suitable areas. Accordingly, our results infer larger population sizes for both species during the LGP (Figure 2). This is also supported by species distribution modeling (SDM) studies, which show, in turn, continuous potential distribution ranging from the Pyrenees to the Montseny Massif at the LGM.7 Moreover, a much wider range could vindicate a possible Calotriton fossil record quite beyond their current range (Cueva de las Hienas, Asturias31; see discussion in Lobo et al; 32). Putative expanding distributions and populations in the past would provide opportunities for ancestral inter- and intra-specific migration events that are in agreement with our results (Tables S4 and S5). However, evidence of periglacial events at the summit of the Montseny Massif at LGM33 led to the hypothesis that C. asper never reached the Tordera River basin after divergence from C. arnoldi due to the harsh alpine conditions, suggesting that both species never came into secondary contact again.7 Regardless, migrants between the two Montseny lineages were probably never common due to their limited dispersal capacity, which could explain why they remained differentiated after divergence from C. asper. In contrast, C. asper expanded and occupied a larger, mostly continuous range after divergence from C. arnoldi, resulting in reduced isolation among its main lineages despite their older divergence compared to the Montseny brook newt lineages.
On the other hand, warm periods seem to have historically compromised the survival of the brook newts. First, the divergence of Western and Eastern Montseny lineages is associated with the Riss-Würm Interglacial (Figure 3), in which temperatures were similar or higher than the present. In this suboptimal scenario, C. arnoldi’s range probably split up into at least two marginal refugia at the headwaters of the different watersheds, and distribution has remained disjoint to this day. Secondly, increasing temperatures triggered severe population bottlenecks in brook newts, especially in C. arnoldi (Figure 2). This is reflected in a higher number of ROHs in the Montseny species compared with its sister taxon C. asper (Figure 4B; Table S8), as expected in isolated and bottlenecked populations.34 Post-glacial population expansions after LGM and LGP have been inferred for many vertebrate groups,35,36,37,38,39,40,41,42 including Eurasian salamandrids from similar latitudes.43,44,45 However, well documented post-glacial bottlenecks are scarcer in the genomic literature and typically limited to either relict or extinct megafauna.37,46,47,48,49 Although paleodemographic modeling has statistically shown tight links between demography and climate oscillations for humans at the end of LGP from archaeological radiocarbon dates,39,50 to our knowledge this is the first time the dependence of a genomically inferred past demography on climate variables is proven, moreover, on a non-model species. Regarding future scenarios, a major decrease in the potential distribution of the Pyrenean brook newt and, concomitantly, of its genetic diversity and evolutionary history has been foreseen.51 In fact, the situation could be even more menacing for C. arnoldi, as climate change is expected to cause more grievous effects in species with low genetic diversity, small ranges, low effective population sizes, specific habitat requirements and limited dispersal capabilities,30 all characteristics exhibited by the Montseny brook newt.
Population genomics
Previous studies based on microsatellites concluded that Pyrenean and Montseny brook newts exhibited similar genetic diversity,4 in contrast with our genomic estimates, which show that diversity in all Montseny populations is much lower than in Pyrenean populations, supported both by lcWGS (Table S6) and ddRADseq data (Figure 4A; Table S7). Although traditional genetics do not always disagree with genomics, this illustrates how conservation can benefit from the advent of Next-Generation Sequencing and its superior resolution power.52,53,54,55 Within Montseny, results show higher genetic diversity and a lower number of long ROHs in the Western Montseny lineage relative to the Eastern (Figure 4B; Table S8), as expected in larger and admixed populations.34 Interestingly, the highest ddRADseq HO value at the Eastern Montseny lineage was found in population A3 (Table S7), which is a small population that could be virtually extinct at present. Despite exhaustive searches in different seasons, no newts have been found at this locality (previously disturbed by intense human activities) since the storm “Gloria” lashed Montseny in January 2020 and changed the geomorphology of the brook. Such dire event clearly evidences how the survival of small-ranged endemisms can be compromised by stochastic phenomena.56
Despite long ROHs and low genomic diversity in all C. arnoldi, we found no evidence of inbreeding. In fact, observed heterozygosity values higher than expected were already reported based on microsatellite data,4 leading to the hypothesis of putative assortative mating strategies to avoid the negative effects of breeding among close relatives. Assortative mating has been reported in a number of salamandrid species, either involving particular preferences by males57 or, more usually, by females.58,59,60 However, those strategies do not take place exclusively during courtship, as female salamandrids can store sperm from different males. Cryptic female choice has been described for at least three other salamandrid genera,61,62,63 with females preferring less-related males as the most compatible mates. This model of mating driven by genetic compatibility could shed light on why the most diverse studied population (O3 Western Pyrenees brook newt) would display the least evidence for assortative mating (i.e., the only positive GIS value; Table S7), and, more importantly, why half of the captive Montseny brook newt couples have never sired descendants in 15 years of ex situ breeding program (even a higher proportion regarding the Eastern Montseny lineage, whose heterozygosity is lower). Another singularity related to diversity affecting the breeding program could be the homomorphic embryonic arrest syndrome, originally described in Triturus,64 sister genus to Calotriton.3 This phenomenon, which implies a 50% of genetic load for all individuals, may occur in Calotriton as well, as fertilized eggs belonging to successful clutches have been reported to unexpectedly arrest their development in captivity. No feasible explanation for why this phenomenon has evolutionarily survived has been proposed, although it might also contribute to pumped-up heterozygosities.
Regarding population structure, genomic analyses have successfully revealed population structure at a finer level than previous genetic studies.4,7 Our genomic investigations accurately identified previously unnoticed differences between the A1–A2 clusters (Figure 5A) and revealed the asymmetrical admixture of B3 with the other Western demes (Figure 5B). Since the impact of genetic isolation needs time to become detectable, i.e., several generations for spatial genetic structure to arise,65,66 we can conclude that the isolation of Montseny Massif populations into five clusters is long enough to be attributed to natural reasons. In contrast, and regarding the artificial fragmentation of A2 creek, recent human impacts on the environment (i.e., most roads) are relatively young in terms of animal generations. Therefore, only weak genetic differences are expected, and only methods inferring contemporary migration are able to detect a barrier effect.67 The threatening impact of roads in amphibians is not new, and it has been linked to an increased mortality, reduced genetic diversity, habitat fragmentation and, ultimately, an increased risk of extinction by reducing demographic and genetic input from immigrants and decreasing the probability of recolonization after extinction,68,69,70,71 while road verges promote the expansion of invasive species67 and, thus, of potential pathogen vectors.
Differences across populations at the Montseny Massif are driven not only by natural- and human-related barriers, but also by simple distance effects. In contrast with previous studies,4 probably because of the higher resolution of genomic approaches, our results point to the existence of IBD within Montseny submassifs, but not in between: i.e., geographic distance is not enough to explain differences between the two lineages (Figure S7). Rivers are one of the main geographical barriers shaping amphibian distributions,70 in our case (Tordera River, Figure 1) due to unsuitable conditions in terms of temperature, pollution, and the presence of predatory fishes, but also because of the meagre dispersal abilities of C. arnoldi compared to C. asper (mean displacements of barely 7 m in a 2-year period72). Low dispersal rates and small population sizes lead to an accelerated differentiation,73 reflected in high fixation indices (Figure 4C; Table S9). F’ST from genetic data between Pyrenean brook newt populations had already shown values comparable to reptiles and amphibians inhabiting different islands within archipelagos.30 Furthermore, our genomic data shows even higher values of isolation between Montseny linages at a much smaller scale, comparable to values found in troglobiont amphibians across different cave systems,74 in desert-dwelling frogs from fragmented populations >300 km apart,75 or even in between Calotriton species (Figure 4C). Consequently, in view of their genomic and phenotypic differentiation, the Montseny brook newt lineages might need further taxonomic reassessment.
Limitations of the study
This study presents not only the third genome assembly in the order Caudata, but also the first population genomic study in the group based on whole-genome resequencing. Due to the large genome sizes in urodeles, the sequencing of these amounts of data is always challenging and, therefore, yields ultra-low coverages that compel to analyze genotype likelihoods instead of true genotypes. The lack of confidence in the called genotypes, added to mapping difficulties of short reads to a highly repetitive genome, could hamper the correct identification of ROHs. Furthermore, the fragmentary nature of the reference genome limits the amount of ROHs that can be detected, especially the longest ones. We prioritized the number of samples rather than their coverage for lcWGS, so that we could compare results not only between lineages but within groups and populations as well. In the same way, despite the lack of other similar whole-genome studies in urodeles to compare with, we ensured the soundness of the ultra-low coverage data by comparing it to ddRADseq results.
Regarding C. asper, the sampling includes fewer individuals, while its range is much wider. Although we have chosen representatives of the different mitochondrial lineages, other genomic clusters may remain undiscovered. Future studies on this species should improve the sampling by covering new areas and exploring the contact zone between the Western and Eastern Pyrenees to assess the extent of lineage isolation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| MagAttract HMW DNA kit | Qiagen | N/A |
| Deposited data | ||
| Calotriton arnoldi Reference genome assembly (aCalArn1) | This study | PRJEB57772 |
| ORG.one Nanopore data used for the assembly | This study | PRJEB57582 |
| Illumina data for lcWGS population genomic analyses and Nanopore and Illumina data generated outside ORG.one used for the assembly | This study | PRJEB57773 |
| ddRADseq data from 154 Calotriton arnoldi and C. asper individuals | This study | PRJNA1023478 |
| Software and algorithms | ||
| Cutadapt v3.2 | Martin76, 2011 | https://github.com/marcelm/cutadapt |
| Meryl v1.7 | Rhie et al.77, 2020 | https://github.com/marbl/meryl |
| Smudgeplot v.0.2.5 | Ranallo-Benavidez et al.78, 2020 | https://github.com/KamilSJaron/smudgeplot |
| KMC v.3.1.1 | Kokot et al.79, 2017 | https://github.com/refresh-bio/KMC |
| GenomeScope v2.0 | Ranallo-Benavidez et al.78, 2020 | https://github.com/schatzlab/genomescope |
| Filtlong v0.2.0 | N/A | https://github.com/rrwick/Filtlong |
| Flye v2.8.3 | Kolmogorov et al.80, 2019 | https://github.com/fenderglass/Flye |
| BWA v.0.7.15 | Li81, 2013 | https://github.com/lh3/bwa |
| HyPo v1.0.3 | Kundu et al.82, 2019 | https://github.com/kensung-lab/hypo |
| Minimap2 v2.17-r941 | Li83, 2018 | https://github.com/lh3/minimap2 |
| purge_dups v1.2.5 | Guan et al.84, 2020 | https://github.com/dfguan/purge_dups |
| BlobToolKit | Challis et al.85, 2020 | https://github.com/blobtoolkit/blobtoolkit |
| RepeatMasker v4.1.2 | Smit et al.86, 2013-2015 | https://www.repeatmasker.org/ |
| RepeatModeler v1.0.11 | Smit & Hubley87, 2008 | https://www.repeatmasker.org/RepeatModeler/ |
| FastQC v0.11.8 | Andrews88, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| fastp v0.23.1 | Chen et al.89, 2018 | https://github.com/OpenGene/fastp |
| PicardTools v2.8.2 | BroadInstitute,2021 | https://github.com/broadinstitute/picard |
| SAMtools v1.9 | Danecek et al.90, 2021 | https://github.com/samtools/samtools |
| ipyrad v0.9.78 | Eaton & Overcast91, 2020 | https://github.com/dereneaton/ipyrad |
| PLINK v2.0 | Purcell et al.92, 2007 | https://www.cog-genomics.org/plink/2.0/ |
| easySFS | N/A | https://github.com/isaacovercast/easySFS |
| Stairway Plot v2.1.1 | Liu & Fu93, 2020 | https://github.com/xiaoming-liu/stairway-plot-v2 |
| R v.3.6.3 | R Core Team94, 2021 | https://www.r-project.org/ |
| RRPP v. 1.3.1 | Collyer & Adams95, 2018 | https://github.com/mlcollyer/RRPP |
| nlme v3.1-140 | Pinheiro et al.96, 2016 | https://github.com/cran/nlme/blob/master/R/lme.R |
| fastsimcoal27 | Excoffier et al.97,98, 2021 | http://cmpg.unibe.ch/software/fastsimcoal27/ |
| RAxML-NG v1.0.2 | Kozlov et al.99, 2019 | https://github.com/amkozlov/raxml-ng |
| BEAST v2.6.4 | Bouckaert et al.100, 2019 | http://www.beast2.org/ |
| ANGSD v0.935 | Korneliussen et al.101, 2014 | http://www.popgen.dk/angsd/index.php/ANGSD |
| ngsF-HMM | Vieira et al.102, 2016 | https://github.com/fgvieira/ngsF-HMM |
| ggplot2 v3.4.0 | Wickham103, 2016 | https://ggplot2.tidyverse.org/ |
| GenoDive v3.06 | http://www.patrickmeirmans.com/software/Home.html | |
| Admixture v1.3 | Alexander et al.104, 2009 | https://dalexander.github.io/admixture/ |
| diveRsity | Keenan et al.105, 2013 | https://github.com/kkeenan02/diveRsity |
| prabclus | Hennig & Hausdorf106, 2019 | https://rdrr.io/cran/prabclus/ |
Resource availability
Lead contact
Further information can be requested via the lead contact, Adrián Talavera (adrian.talavera@csic.es).
Materials availability
This study did not generate any new reagents.
Data and code availability
All data is publicly available as of the date of publication in the following NCBI BioProjects: aCalArn1 genome assembly (PRJEB57772), ORG.one Nanopore raw data used for the assembly (PRJEB57582), Illumina data for lcWGS population genomic analyses and Nanopore and Illumina data generated outside ORG.one used for the assembly (PRJEB57773), and demultiplexed ddRADseq data (PRJNA1023478).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (adrian.talavera@csic.es) upon request.
Experimental model and subject details
Material and methods
Sampling and ethics statement
The Montseny brook newt (Calotriton arnoldi) reference genome was sequenced from heart tissue samples of two F1 captive-bred siblings (OR192 & OR193) from the “Centre de Fauna de Torreferrussa-Generalitat de Catalunya” (Santa Perpètua de Mogoda, Spain), which were sacrificed due to their unsuitability to breed. These animals were direct descendants of founder individuals of the breeding programme (TOR16 & TOR18), from the Eastern Montseny lineage (type locality of the species). For population genomic analyses, a total of 154 tail samples were collected, including 134 C. arnoldi from all the known natural populations at the Montseny Massif (namely the Eastern Montseny lineage from populations A1, A2 and A3 and the Western Montseny lineage from populations B1, B2, B3, B4, B5), and 23 C. asper from six populations belonging to the five described mitochondrial lineages107 (namely lineages O1–5; Figure 1). Tissue samples (i.e., tail tips) were stored in 99% ethanol at -20°C. Due to the species’ susceptibility to chytridiomycosis, strict biosecurity measures were taken while sampling. New nitrile gloves were worn for each animal and all tools and equipment were disinfected with Virkon™ (Zotal).
Genomic data production
High molecular weight DNA extractions were performed using MagAttract HMW DNA kit (Qiagen) to sequence the reference genome. By using 10 different flowcells, we sequenced 562.08 Gbp of Oxford Nanopore Technologies (ONT) data (63.88 M reads, read N50 > 34 kbp and 98.2% of the reads with QV>7) from OR192 and OR193 individuals. Data was basecalled with Guppy versions v3.2.10, v4.3.4 or v5.0.11, using the most recent version available at the time data was generated. On the other hand, we built an Illumina library following the BEST protocol108 with minor changes and 588.3 Gbp of Illumina data (1.96x2 M 150-pb reads) was sequenced with HiSeq4000 from OR193 individual.
For lcWGS and ddRADseq libraries, DNA was extracted from tissue samples following standard high-salt protocols109 and quality-checked with 1% agarose gel electrophoresis. DNA concentrations were quantified with BroadRange Qubit 2.0 fluorometer (Invitrogen). Out of the 154 DNA samples, we selected one male and female of each known inhabited brook at the Montseny, plus one representative of each C. asper lineage, to build 21 libraries for lcWGS, following the BEST protocol and sequencing at low coverage on HiSeq4000 with 150 pb PE reads. On the other hand, we followed the ddRADseq protocol by Peterson et al.110 with minor changes and double-digested 154 samples of all C. arnoldi and three C. asper populations -belonging to two lineages- with the enzymes SbfI and MspI. Digested fragments were ligated with barcoded Illumina adapters and size-selected for 415-515 bp. Finally, ddRAD libraries were quality-checked with Bioanalyzer (Agilent) and sequenced with 75 bp SE reads using Illumina NextSeq 500.
Method details
De novo reference genome assembly
Illumina raw data was trimmed afterwards with Cutadapt v3.2.76 Then, we generated a k-mer database using Meryl v1.7. A 22-mer database (in complex mode) was built using the best_k.sh script from Merqury v.1.1,77 with the k-mer length decided based on the length of the axolotl genome assembly ASM291563v226 as an estimate of the genome size. Diploidy of C. arnoldi was assessed and confirmed with Smudgeplot v.0.2.578 based on another 22-mer database obtained with KMC v.3.1.1,79 adding the option –middle (Figure S1). GenomeScope v2.078 was employed to estimate the genome assembly haploid length (22.75–28.38 Gb), the proportion of repeats (15.46–18.04 Gb) and heterozygosity with the 22-mer Meryl database (98.75–100% of homozygosity; Figure S2).
We used Filtlong v0.2.0 (available at https://github.com/rrwick/Filtlong) to aim for the 340 Gb of the best ONT data available (aiming for 12-15X coverage), setting a minimum size of 4 kpb and looking for high-quality reads (-minlen 4000 -min_mean_q 80). The amount of ONT data obtained was ∼12x of the maximum length calculated by GenomeScope (=28.3 Gb). The genome was assembled with Flye v2.8.380 adding two internal rounds of polishing, with the ONT raw reads option and scaffolding enabled by default. The assembly process required almost 80 computing days to obtain a draft assembly in a node with 48 CPUs, reaching >1Tb of maximum memory consumption. Trimmed Illumina data was mapped to the draft assembly with BWA v.0.7.1581 and used to polish it with HyPo v1.0.3.82 We used Minimap2 v2.17-r94183 with the option “-x map-ont” to map the reads to the polished assembly and then run purge_dups v1.2.584 to remove haplotigs. We manually changed cutoffs based on the k-mer histogram from “5 40 120” to “5 21 150”. Finally, we used the Meryl k-mer database (k=22) to run Merqury in order to evaluate the k-mer completeness and base accuracy of the final assembly, yielding 35.2 QV of base accuracy and 97.54% of k-mer completeness. Functional completeness was assessed using BUSCO v5.2.2 (82.3% BUSCO(S) vertebrata odb10, n=3,354) and contiguity (scaffold N50=1.65 Mb; 47,257 scaffolds) metrics were summarized as a snail plot (Figure S3) using BlobToolKit85 as well as compared with other previously published assemblies in the order Caudata (Table S1) and Vertebrate Genome Project’s (VGP) standards (Table S2).
Finally, we used RepeatMasker v4.1.286 to mask the resulting genome assembly following Nowoshilow et al.26 First, we used the available Vertebrata, Tetrapoda and Amphibia repeat databases followed by the use of the repeat custom library used for the axolotl genome in Nowoshilow et al. (2018) provided by the authors. Afterwards, we ran RepeatModeler v1.0.1187 to create a Montseny brook newt specific repeat database and ran again RepeatMasker with the resulting database.
Processing of lcWGS data
Low-coverage WGS raw data, along with the high-coverage WGS raw data from the reference genome animal (OR193), was quality-checked using FastQC v0.11.888 and visualized with MultiQC.111 Data was trimmed using fastp v0.23.1,89 allowing deduplication, requiring a minimum read length of 30 bp, allowing for the detection of adapters in PE data for adapter removal and providing the adapter sequences. We used BWA81 with BWA-MEM algorithm to map the trimmed data to the de novo assembled reference genome. After mapping, we marked duplicates of the re-sequenced genomes with Picard v2.8.2.112 Finally, we filtered with SAMtools v1.9,90 removing unmapped reads, secondary alignments and previously marked PCR duplicates (-F 3332). We also removed any alignment with mapping quality <25.
Processing of ddRADseq data
A total of 3.34 x 108 reads were obtained from the ddRAD sequencing, with a mean of 2.17 x 106 reads per sample. ddRAD sequencing output was quality-checked using FastQC and MultiQC. Sample demultiplexing, alignment, SNP-calling and some basic filtering steps were carried out with ipyrad v0.9.78.91 We tested clustering thresholds of 0.85, 0.89, 0.92 and 0.95 for de novo assembly and reference-assisted assembly with the C. arnoldi reference genome scaffolds, retrieving a different number of loci (Table S11). For downstream analyses, only the reference-assisted method was chosen. PhredQ score offset was kept by default at 33 and minimum samples per locus at 4. The output from ipyrad was later filtered in different ways to produce a number of datasets. The final number of unlinked SNPs achieved for each dataset, the performed filtering steps and the analyses in which they were used are reported on Table S12. A main dataset (general, Table S12) was filtered by common markers, followed by an iterative filter which discards alternatively individuals and genotypes with higher missingness than a 2%-step-decreasing threshold, from 100% missing data to 75% (script from Burriel-Carranza et al.113, following recommendations by O’Leary et al.114). Later, we applied a hard filter of 15% genotype missingness, removed singletons (MAC = 1), monomorphic sites and heterozygote excess (command “--hwe 1e-50 midp keep-fewhet” of PLINK v2.092) and kept only biallelic SNPs. Finally, we used PLINK v1.9 to prune the dataset by linkage disequilibrium, allowing a maximum correlation between SNPs of 0.5 within 50-SNP sliding windows to achieve a dataset comprising 132 individuals and 9,470 unlinked SNPs. As the sampling was carried out along several years and given the tissue regeneration skills of urodeles, a last filtering step using PLINK v2.0 was applied to detect putative pseudo-replicates. For demographic inference analyses, we removed all 1st-degree or closer relatives by using PLINK v2.0 function –king-cutoff and applied the same above-mentioned filtering steps to create the dataset no_relatives, comprising 88 individuals and 8,841 unlinked SNPs.
Another dataset (no_outgroup) was created directly from the ipyrad output selecting just the C. arnoldi samples which passed the postprocessing filtering of the general dataset, and applying the same filters but allowing 30% genotype missingness; resulting in a dataset of 115 animals and 1,339 uSNPs. Also using ipyrad, we created a branch at step 3 to generate the dataset loci, allowing for 15% genotype missingness. Finally, the seven individuals with the least missingness, representing different pure genomic clusters from Admixture (see results) were used to create the dataset no_missingness, which was filtered allowing no missingness and, alike the dataset general, keeping just unlinked biallelic SNPs.
Quantification and statistical analyses
Demographic inference
The ddRADseq dataset no_relatives was downprojected by means of the easySFS script (https://github.com/isaacovercast/easySFS) into two sets of Site Frequency Spectrum (SFS) files. For the first one (2_spp_SFS), we split the individuals by species and chose the optimum number of retained haploid sequences without missing data based on the number of segregation points (C. arnoldi 124 sequences, 525 segregation points; C. asper 10 seq., 1,357 seg. points). The second set of SFS -3_lineages_SFS- files was created assuming there are 3 lineages: C. asper, Western C. arnoldi and Eastern C. arnoldi. 10, 70 and 60 sequences and 1,367, 245 and 208 segregation points were retrieved as the best option to downproject.
From 2_spp_SFS, both intraspecific and unidimensional folded SFS files were used as input for Stairway Plot v2.1.1.93 The number of total sites was estimated from the percentage of monomorphic and non-biallelic sites in each species. We assumed 4-year generation times and a mutational rate of 3.08x10-8 substitutions per site per generation.115 Singletons were considered only for C. asper analyses, as some populations retained just one individual after the downprojection. The medians of Ne estimates based on 200 runs per species were plotted for both species, along the respective 95% confidence intervals for the Montseny brook newt. Demographic inferences were coupled with reconstructions of the continental mean surface air temperature anomalies of the Northern Hemisphere (40º –80º N) and the total ice volume for the Eurasian ice sheet (in meters of sea level equivalent) for the last 200 ky (dataset from de Boer et al.,116 see STAR methods in de Boer et al.117). To account for local events, we included a Western Mediterranean Sea surface temperature reconstruction for the last 5 ky.118 Ultimately, some major and minor periods of climatic relevance were also highlighted. Afterward, we statistically tested for an association between Calotriton spp. median effective population sizes and either surface-air temperature or ice sheet volumes in Eurasia. We fitted four Generalized Least Squares (GLS) models over 10,000 permutations using a randomized residual procedure with ‘RRPP’ package v. 1.3.1.95 To account for temporal autocorrelation (i.e. the non-independence of the residuals), we previously built matrices of covariance for each variable to explain the decay of autocorrelation with time by means of a corAR1 model using ‘nlme’ package v3.1-14096 in R.94 The resulting matrices were incorporated in the structure of the respective error terms of each model, but, globally, little temporal autocorrelation was found (phi=0).
On the other hand, the multidimensional SFS from 3_lineages_SFS was used for demographic simulations and parameter inference under the composite-likelihood approach of fastsimcoal27.97 Four different models were designed, differing in the allowed migration matrices (Figure S4). The first model lacked migration at all, the second allowed for recent migration between C. arnoldi lineages, the third for ancient asymmetrical migration from C. asper to C. arnoldi; and the fourth model considered both migrations patterns. We chose not to test bidirectional migration between the two species since C. asper dispersal capacity is considerably higher than C. arnoldi’s. All models had the same topology, the resize option was permitted after whichever lineage split, and an extra instantaneous bottleneck was allowed along C. arnoldi history (TBOT; Table S4), so that the estimated divergence time between Montseny lineages (TDIV; Table S4) was less biased due to extreme changes in effective population changes. Eastern and Western C. arnoldi effective population sizes (NA and NB in Table S4, respectively) were bounded up to 12,000 haploid sequences (i.e., 6,000 diploid individuals) - the 95%-confidence maximum for the species at present according to Stairwayplot2 results. Generation time was set to 4 years and the C. asper-C. arnoldi split was fixed 440,000 generations ago (1.76 my, as the median of the mitochondrial estimate by Valbuena-Ureña et al.;4 95% HPD 1.24–2.44 my). For each scenario, 100 independent runs with different initial values were carried out and the best-fitting model was chosen based on Akaike’s Information Criterion (119), and the model normalized relative likelihood () based on Excoffier et al.98 For each run of each of the 4 models, we set 500,000 coalescent simulations, up to 50 ECM cycles in Brent optimization of parameters, with monomorphic sites masked.
Phylogenomic reconstructions
First, we constructed a Maximum Likelihood (ML) phylogeny based on the ddRADseq loci dataset of all individuals (Table S12) to assess the genomic monophyly of Calotriton spp. and their intra-specific lineages, namely Western Montseny, Eastern Montseny, Western Pyrenees and Eastern Pyrenees. To do so, we used RAxML-NG v1.0.299 with a GTRGAMMA model, starting from 50 random and 50 parsimony-based trees and with 1,000 bootstrap replicates (Figure S8). Then, we inferred a time-calibrated Bayesian Inference (BI) phylogeny based on SNPs under the coalescent model, which directly models incomplete lineage sorting, a common issue when dealing with divergence among young taxa.120 We ran SNAPP (implemented on BEAST v2.6.4100) on the ddRADseq dataset no_missingness of seven individuals with the least missingness, which were treated as belonging to one of the four monophyletic lineages (2 Western Montseny; 3 Eastern Montseny, 1 Eastern Pyrenees and 1 Western Pyrenees). The vcf file comprising 8,139 unlinked SNPs was converted to xml format following Stange et al.121 by using the script snapp_prep.rb (available at: https://github.com/mmatschiner/snapp_prep). Calotriton arnoldi and C. asper were constrained to be monophyletic and the divergence between them was set with a lognormal distribution in 1.76±0.2 mya following a previously published calibration.4 Analyses consisted of three independent runs with 6 million generations each. Tracer v1.7.2122 was used to check for stationarity (estimated sample sizes of all parameters ≥ 200) and convergence between runs. Converging log and tree files were combined using LogCombiner v1.10 with 10% burn-in. The cloudogram was visualized with DensiTree v. 2.6.4,123 and TreeAnnotator v2.6.3 was used to construct the ‘Maximum clade credibility’ tree and annotate nodes with the corresponding posterior probabilities. We plotted together the cloudogram, plus the consensus tree and the posterior probabilities of each node along with the mean divergence times and their 95% confidence intervals. Furthermore, important climatic events of each time frame were highlighted, specifically the glaciations of Donau (c. 1.8-1.0 mya), Riss (c. 300-130 kya) and Würm (c. 115-11.7 kya), and the interglacial period separating the latter two (130-115 kya).
Population genomic analyses and ROHs with lcWGS
We estimated the heterozygosity of the 22 Calotriton spp. individuals with WGS data (see Table S6 for details) using ANGSD v0.935101 on the downsampled (1.3x coverage) filtered bam files as follows: “angsd -i $file.bam -anc aCalArn_1.scaffolds.fa -dosaf 1 -gl 1”. Furthermore we applied additional filters (-remove_bads 1 -baq 1 -C 50 -minQ 20 -minMapQ 30) and calculated heterozygosity values for the largest scaffold and genome-wide, including and excluding repetitive regions, for each individual following the ANGSD manual.
ANGSD was used as well to call genotype likelihoods to perform Principal Component Analyses (PCAs) in order to explore population structure, and to estimate runs of homozygosity (ROHs, or identity-by-descent tracks) from all WGS samples, including the reference sample from the breeding center (an F1 Eastern-lineage individual). We calculated genotype likelihoods for five different datasets: all_wild&captivity (22 samples), all_wild (21 samples), only_arnoldi (16 samples), only_west (10 samples), only_east (6 samples); varying the number of samples and the minInd option (minInd = 18 for all_wild&captivity and all_wild; minInd = 15 for only_arnoldi, and minInd = (number of individuals – 1) for only_west and only_east). We inferred the minor and major alleles from likelihoods (-doMajorMinor 1), avoiding sites where the reference does not contain either the major or the minor alleles inferred (-skipTriallelic 1). Genotype likelihoods were reported as a beagle file (-doGlf2) and the allele frequency estimations were performed assuming the major allele inferred is known (-doMaf2). We removed sites with minor allele frequency lower than 0.04 (-minMaf 0.04) and sites with a p-value greater than 1e-6 (-SNP_pval 1e-6). Furthermore, different quality filters were applied to the imputed BAM files: we excluded non-primary alignments, and unmapped and duplicate reads (-remove_bads 1), discarded reads with multiple mappings (-uniqueOnly 1), kept only pairs of reads for which both were properly mapped (-only_proper_pairs 1), performed Base Alignment Quality calculation (-baq 1), avoided trimming (-trim 0), adjusted base quality for excessive mismatches (-C 50) and set a minimum mapping quality (-minMapQ 25). PCAs were performed using the output beagle files with PCAngsd v0.8. A total of 23.50M SNPs for all_wild, 2.08M SNPs for only_arnoldi, 1.55M for only_west and 0.75M for only_east datasets were used.
Finally, we used ngsF-HMM102 to estimate runs of homozygosity (ROHs, or identity-by-descent tracks) from the resulting beagle file for all_wild&captivity. ngsF-HMM uses an iterative approach that stops when a dual condition threshold is reached.102 We followed the authors’ recommendation of lowering this threshold to “--min_epsilon 1e-9”. Only the 50 largest scaffolds from our assembly (with lengths 11.6-7.1 Mb) were selected for this analysis. We compared the ROH counts between lineages (i.e. Western Montseny, Eastern Montseny and Pyrenees) at four different ranges in terms of ROH length (0.5-1, 1-2, 2-3 and >3 Mb) by one-way ANOVA tests and corresponding Bonferroni-adjusted post-hoc tests. Means and SD of ROH counts along those 50 largest scaffolds by lineage and by categories of ROH length were plotted in barplots with ggplot2 v3.4.0.103
Population genomic analyses with ddRADseq
GenoDive v3.06124 was used to estimate observed (HO) and expected heterozygosity (HS) and inbreeding coefficients (GIS) per population from the general dataset. The same software was also used for calculating the fixation indices, both lineage- and population-pairwise. Observed and expected heterozygosities per population from ddRAD data were plotted as a barplot and all the population-pairwise indices were plotted on a heatmap with ggplot2. To explore population structure, we performed two PCAs by means of PLINK v1.9, one at genus level (dataset general) and another at C. arnoldi species level (dataset no_outgroup), and then plotted results with ggplot2. Furthermore, to asess structure and admixture between populations, we run Admixture v1.3,104 again hierarchically, at genus- and species-level, for K values from to 1 to the number of sampling localities (i.e., twelve and nine at genus and species level, respectively) and calculated 20 cross-validation error (CV) values with 20 replicates. Optimal K values were chosen based on CV scores (see Table S10), the plateau profile of those values and the likelihood of the results in biological terms. Additionally, geneflow among C. arnoldi populations was estimated with the function divMigrate in the diveRsity package105 in R to obtain a relative migration network graph from the dataset no_outgroup. Geneflow, scaled to the largest magnitude estimated, was measured in Nm (i.e. effective number of migrants125,126) and the significance of asymmetric gene flow was tested with 1,000 bootstrap iterations and alpha = 0.05. Finally, the resulting matrix was plotted with the R package qgraph,127 and edges below 0.35 were disregarded (Figure 5B). Geneflow arrows which are significantly asymmetric are colored in these graphs, whereas the rest remain in gray scale depending on their intensity. Since the Montseny brook newt strictly disperses by water-way routes, migration networks were depicted with a scheme of brook connectivity within the Tordera river basin.
To explore the differences in population structure between Western and Eastern Montseny lineages, we tested for isolation-by-distance (IBD) in C. arnoldi populations following Hausdorf & Hennig128 These IBD tests check if genetic distances between individuals or, in our case, populations, belonging to two different lineages (Eastern and Western Montseny as candidate species sensu Hausdorf & Hennig128 or primary species hypotheses sensu Bamberger et al.129) are not larger than expected based on their geographical distances and within-group IBD patterns (null hypothesis sensu Hausdorf & Hennig128 or secondary species hypothesis sensu Bamberger et al.129). Genetic distances were measured as F’ST/(1-F’ST)130 based on previous results. We measured shortest water-way linear distances between populations in meters and then log-transformed values. We used R package prabclus106 to perform a Jackknife-based test for equality of two independent within-lineage regressions (i.e. within the Western and the Eastern Montseny lineages) to confirm that IBD evolves alike within both metapopulations, and thus that they can be merged into a single joint within-lineages regression (intercept p=0.802; slope p=0.745). Second, another Jackknife-based test was run to check for the equality of the latter and another regression over all comparisons, both within- and between Montseny lineages. The non-equality of both regressions would imply that the observed genetic differences cannot be explained by IBD alone.
Acknowledgments
We must thank to an anonymous reviewer for their constructive comments provided on a previous version of the manuscript, as well as Loukia Spilani, Maria Estarellas, Juliana Tabares and Laia Pérez-Sorribes for their help and support, Albert Català for his valuable insights into palaeoclimatic registers, and Elly Margaret Tanaka and Francisco Javier Falcon Chavez for providing the axolotl custom repeat library. We also want to thank the sequencing, assembly and annotation teams of CNAG (Center Nacional d'Anàlisi Genòmica) for their help during the sequencing and sequencing data management. AT is supported by “la Caixa” doctoral fellowship programme (LCF/BQ/DR20/11790007). MP-F is supported by “la Caixa” doctoral fellowship programme (LCF/BQ/DR20/11790032). BB-C was funded by FPU grant from Ministerio de Ciencia, Innovación y Universidades, Spain (FPU18/04742). GM-R was funded by an FPI grant from the Ministerio de Ciencia, Innovación y Universidades, Spain (PRE2019-088729). HT-C is supported by a Juan de la Cierva – Formación postdoctoral fellowship (FJC2021-046832-I) funded by MCIN/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. The work was funded by the grants PGC2018-098290-B-I00 (MCIU/AEI/FEDER, UE), Spain, and PID2021-128901NB-I00 (MCIN/AEI/10.13039/501100011033 and by ERDF, A way of making Europe), Spain to SC. We would like to thank Dan Forham and Oxford Nanopore Technologies for providing technical and fungible support for this project. Sample collection was carried out with the authorization of the governments of Catalonia, Aragon (Spain) and Andorra. License for the sacrifice of two captive-bred individuals for the genome assembly were issued by the Departament d’Acció Climàtica, Alimentació i Agenda Rural of the Catalan Government with code MBCFS-05-22. Fieldwork was authorized in Catalonia by the same institution with the permission numbers SF/298 and SF/469 for C. arnoldi and DF/90 and SF/429 for C. asper, in Aragon by Instituto Aragonés de Gestión Ambiental, Area II-Biodiversidad of the Aragonese Government with the permission numbers 24/2010/901 and 24/2012/661 and in Andorra by Departament de Medi Ambient i Sostenibilitat of the Andorran Government with the permission number EXP2200747.
Author contributions
A.T., T.M.-B., and S.C. conceived and designed the study. F.A., E.V.-U., S.C., A.T., B.B.-C., G.R., F.C., A.S.-M., and D.G. performed sample collection. M.P.-F. performed genome assembly and analyzed data. A.T. carried out laboratory work, analyzed the data, designed figures and wrote the manuscript with input from all other authors, who approved the paper in its final form.
Declaration of interests
The authors declare no competing interests.
Published: December 12, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108665.
Supplemental information
References
- 1.Schmitt T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 2007;4:11. doi: 10.1186/1742-9994-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varga Z.S., Schmitt T. Types of oreal and oreotundral disjunctions in the western Palearctic. Biol. J. Linn. 2008;93:415–430. [Google Scholar]
- 3.Carranza S., Amat F. Taxonomy, biogeography and evolution of Euproctus (Amphibia: Salamandridae), with the resurrection of the genus Calotriton and the description of a new endemic species from the Iberian Peninsula. Zool. J. Linn. Soc. 2005;145:555–582. [Google Scholar]
- 4.Valbuena-Ureña E., Soler-Membrives A., Steinfartz S., Orozco-TerWengel P., Carranza S. No signs of inbreeding despite long-term isolation and habitat fragmentation in the critically endangered Montseny brook newt (Calotriton arnoldi) Heredity. 2017;118:424–435. doi: 10.1038/hdy.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.IUCN SSC Amphibian Specialist Group . The IUCN Red List of Threatened Species 2022: e.T136131A89696462. 2022. Calotriton arnoldi. [Google Scholar]
- 6.Montori A. Universidad de Barcelona; 1988. Estudio sobre la Biología y Ecología del Tritón Pirenaico Euproctus asper Dugès, 1852 en la Cerdaña. PhD Thesis. [Google Scholar]
- 7.Valbuena-Ureña E., Amat F., Carranza S. Integrative Phylogeography of Calotriton Newts (Amphibia, Salamandridae), with Special Remarks on the Conservation of the Endangered Montseny Brook Newt (Calotriton arnoldi) PLoS One. 2013;8:e62542. doi: 10.1371/journal.pone.0062542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reed D.H., Frankham R. Correlation between Fitness and Genetic Diversity. Conserv. Biol. 2003;17:230–237. [Google Scholar]
- 9.Altizer S., Harvell D., Friedle E. Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol. Evol. 2003;18:589–596. [Google Scholar]
- 10.Spielman D., Brook B.W., Frankham R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA. 2004;101:15261–15264. doi: 10.1073/pnas.0403809101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radwan J., Biedrzycka A., Babik W. Does reduced MHC diversity decrease viability of vertebrate populations? Biol. Conserv. 2010;143:537–544. doi: 10.1016/j.biocon.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luquet E., Garner T.W.J., Léna J.P., Bruel C., Joly P., Lengagne T., Grolet O., Plénet S. Genetic erosion in wild populations makes resistance to a pathogen more costly. Evolution. 2012;66:1942–1952. doi: 10.1111/j.1558-5646.2011.01570.x. [DOI] [PubMed] [Google Scholar]
- 13.Fisher M.C., Garner T.W.J., Walker S.F. Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu. Rev. Microbiol. 2009;63:291–310. doi: 10.1146/annurev.micro.091208.073435. [DOI] [PubMed] [Google Scholar]
- 14.Stuart S.N., Chanson J.S., Cox N.A., Young B.E., Rodrigues A.S.L., Fischman D.L., Waller R.W. Status and Trends of Amphibian Declines and Extinctions Worldwide. Science. 2004;306:1783–1786. doi: 10.1126/science.1103538. [DOI] [PubMed] [Google Scholar]
- 15.Stegen G., Pasmans F., Schmidt B.R., Rouffaer L.O., Van Praet S., Schaub M., Canessa S., Laudelout A., Kinet T., Adriaensen C., et al. Drivers of salamander extirpation mediated by Batrachochytrium salamandrivorans. Nature. 2017;544:353–356. doi: 10.1038/nature22059. [DOI] [PubMed] [Google Scholar]
- 16.Kelly M., Pasmans F., Muñoz J.F., Shea T.P., Carranza S., Cuomo C.A., Martel A. Diversity, multifaceted evolution, and facultative saprotrophism in the European Batrachochytrium salamandrivorans epidemic. Nat. Commun. 2021;12:6688. doi: 10.1038/s41467-021-27005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martel A., Vila-Escale M., Fernández-Giberteau D., Martinez-Silvestre A., Canessa S., Van Praet S., Pannon P., Chiers K., Ferran A., Kelly M., et al. Integral chain management of wildlife diseases. Conserv. Lett. 2020;13 [Google Scholar]
- 18.Devitt T.J., Wright A.M., Cannatella D.C., Hillis D.M. Species delimitation in endangered groundwater salamanders: Implications for aquifer management and biodiversity conservation. Proc. Natl. Acad. Sci. USA. 2019;116:2624–2633. doi: 10.1073/pnas.1815014116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roigé X., Estrada F. In: Social and Ecological History of the Pyrenees. State, Market, and Landscape. Vaccaro I., Beltran O., editors. Left Coast; 2010. The Montseny Natural Park. Management and Evolution of the Landscape. [Google Scholar]
- 20.Peñuelas J., Boada M. A global change-induced biome shift in the Montseny mountains (NE Spain) Global Change Biol. 2003;9:131–140. [Google Scholar]
- 21.Toli E.A., Bounas A., Christopoulos A., Pafilis P., Sotiropoulos K. Phylogenetic analysis of the critically endangered Karpathos water frog (Anura, Amphibia): Conservation insights from complete mitochondrial genome sequencing. Amphibia-Reptilia. 2023;1 [Google Scholar]
- 22.Valbuena-Ureña E., Soler-Membrives A., Steinfartz S., Alonso M., Carbonell F., Larios-Martín R., Obon E., Carranza S. Getting off to a good start? Genetic evaluation of the ex situ conservation project of the Critically Endangered Montseny brook newt (Calotriton arnoldi) PeerJ. 2017;5:e3447. doi: 10.7717/peerj.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hohenlohe P.A., Funk W.C., Rajora O.P. Population genomics for wildlife conservation and management. Mol. Ecol. 2021;30:62–82. doi: 10.1111/mec.15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Canapa A., Biscotti M.A., Barucca M., Carducci F., Carotti E., Olmo E. Shedding light upon the complex net of genome size, genome composition and environment in chordates. Eur. Zool. J. 2020;87:192–202. [Google Scholar]
- 25.Elewa A., Wang H., Talavera-López C., Joven A., Brito G., Kumar A., Hameed L.S., Penrad-Mobayed M., Yao Z., Zamani N., et al. Reading and editing the Pleurodeles waltl genome reveals novel features of tetrapod regeneration. Nat. Commun. 2017;8:2286. doi: 10.1038/s41467-017-01964-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowoshilow S., Schloissnig S., Fei J.F., Dahl A., Pang A.W.C., Pippel M., Winkler S., Hastie A.R., Young G., Roscito J.G., et al. The axolotl genome and the evolution of key tissue formation regulators. Nature. 2018;554:50–55. doi: 10.1038/nature25458. [DOI] [PubMed] [Google Scholar]
- 27.Contreras Cisneros J. Master’s thesis, Universitat de Barcelona; 2018. Temperatura crítica máxima, tolerancia al frío y termopreferendum del tritón del Montseny (Calotriton arnoldi)www.ub.edu/fem/docs/treballs/TFM_JeniferContreras.pdf [Google Scholar]
- 28.Trochet A., Dupoué A., Souchet J., Bertrand R., Deluen M., Murarasu S., Calvez O., Martinez-Silvestre A., Verdaguer-Foz I., Darnet E., et al. Variation of preferred body temperatures along an altitudinal gradient: A multi-species study. J. Therm. Biol. 2018;77:38–44. doi: 10.1016/j.jtherbio.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Wallis G.P., Waters J.M., Upton P., Craw D. Transverse alpine speciation driven by glaciation. Trends Ecol. Evol. 2016;31:916–926. doi: 10.1016/j.tree.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Milá B., Carranza S., Guillaume O., Clobert J. Marked genetic structuring and extreme dispersal limitation in the Pyrenean brook newt Calotriton asper (Amphibia: Salamandridae) revealed by genome-wide AFLP but not mtDNA. Mol. Ecol. 2010;19:108–120. doi: 10.1111/j.1365-294X.2009.04441.x. [DOI] [PubMed] [Google Scholar]
- 31.Sanchiz B., Martín C. Libro de Resúmenes. 8° Congreso Luso-Español y 12° Congreso Español de Herpetología. 2004. Salamándridos pleistocenos de la Cueva de las Hienas (Asturias) pp. 47–134. [Google Scholar]
- 32.Lobo J.M., Martínez-Solano I., Sanchiz B. A review of the palaeoclimatic inference potential of Iberian Quaternary fossil batrachians. Paleobiodivers. Paleoenviron. 2016;96:125–148. [Google Scholar]
- 33.Llobet S. IX. Revista de Geografía; 1975. Materiales y depósitos periglaciales en el macizo del Montseny. Antecedentes y resultados; pp. 35–58. [Google Scholar]
- 34.Ceballos F.C., Joshi P.K., Clark D.W., Ramsay M., Wilson J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018;19:220–234. doi: 10.1038/nrg.2017.109. [DOI] [PubMed] [Google Scholar]
- 35.Rato C., Sillero N., Ceacero F., García-Muñoz E., Carretero M.A. A survival story: evolutionary history of the Iberian Algyroides (Squamata: Lacertidae), an endemic lizard relict. Biodivers. Conserv. 2021;30:2707–2729. [Google Scholar]
- 36.Herman J.S., Searle J.B. Post-glacial partitioning of mitochondrial genetic variation in the field vole. Proc. Biol. Sci. 2011;278:3601–3607. doi: 10.1098/rspb.2011.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dussex N., Alberti F., Heino M.T., Olsen R.A., van der Valk T., Ryman N., Laikre L., Ahlgren H., Askeyev I.V., Askeyev O.V., et al. Moose genomes reveal past glacial demography and the origin of modern lineages. BMC Genom. 2020;21:854. doi: 10.1186/s12864-020-07208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nuñez J.J., Wood N.K., Rabanal F.E., Fontanella F.M., Sites J.W. Amphibian phylogeography in the Antipodes: Refugia and postglacial colonization explain mitochondrial haplotype distribution in the Patagonian frog Eupsophus calcaratus (Cycloramphidae) Mol. Phylogenet. Evol. 2011;58:343–352. doi: 10.1016/j.ympev.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 39.Fernández-López de Pablo J., Gutiérrez-Roig M., Gómez-Puche M., McLaughlin R., Silva F., Lozano S. Palaeodemographic modelling supports a population bottleneck during the Pleistocene-Holocene transition in Iberia. Nat. Commun. 2019;10:1872. doi: 10.1038/s41467-019-09833-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moura A.E., Janse van Rensburg C., Pilot M., Tehrani A., Best P.B., Thornton M., Plön S., De Bruyn P.J.N., Worley K.C., Gibbs R.A., et al. Killer whale nuclear genome and mtDNA reveal widespread population bottleneck during the Last Glacial Maximum. Mol. Biol. Evol. 2014;31:1121–1131. doi: 10.1093/molbev/msu058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Younger J.L., Clucas G.V., Kooyman G., Wienecke B., Rogers A.D., Trathan P.N., Hart T., Miller K.J. Too much of a good thing: Sea ice extent may have forced emperor penguins into refugia during the last glacial maximum. Global Change Biol. 2015;21:2215–2226. doi: 10.1111/gcb.12882. [DOI] [PubMed] [Google Scholar]
- 42.Malyarchuk B., Derenko M., Denisova G. Phylogeny and genetic history of the Siberian salamander (Salamandrella keyserlingii, Dybowski, 1870) inferred from complete mitochondrial genomes. Mol. Phylogenet. Evol. 2013;67:348–357. doi: 10.1016/j.ympev.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Mattoccia M., Marta S., Romano A., Sbordoni V. Phylogeography of an Italian endemic salamander (genus Salamandrina): glacial refugia, postglacial expansions, and secondary contact. Biol. J. Linn. 2011;104:903–992. [Google Scholar]
- 44.Malekoutian M., Sharifi M., Vaissi S. Mitochondrial DNA sequence analysis reveals multiple Pleistocene glacial refugia for the Yellow-spotted mountain newt, Neurergus derjugini (Caudata: Salamandridae) in the mid-Zagros range in Iran and Iraq. Ecol. Evol. 2020;10:2661–2676. doi: 10.1002/ece3.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shu X.X., Hou Y.M., Cheng M.Y., Shu G.C., Lin X.Q., Wang B., Li C., Song Z.B., Jiang J.P., Xie F. Rapid genetic divergence and mitonuclear discordance in the Taliang knobby newt (Liangshantriton taliangensis, Salamandridae, Caudata) and their driving forces. Zool. Res. 2022;43:129–146. doi: 10.24272/j.issn.2095-8137.2021.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palkopoulou E., Mallick S., Skoglund P., Enk J., Rohland N., Li H., Omrak A., Vartanyan S., Poinar H., Götherström A., et al. Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr. Biol. 2015;25:1395–1400. doi: 10.1016/j.cub.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Manuel M., Barnett R., Sandoval-Velasco M., Yamaguchi N., Garrett Vieira F., Zepeda Mendoza M.L., Liu S., Martin M.D., Sinding M.H.S., Mak S.S.T., et al. The evolutionary history of extinct and living lions. Proc. Natl. Acad. Sci. USA. 2020;117:10927–10934. doi: 10.1073/pnas.1919423117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lord E., Dussex N., Kierczak M., Díez-Del-Molino D., Ryder O.A., Stanton D.W.G., Gilbert M.T.P., Sánchez-Barreiro F., Zhang G., Sinding M.H.S., et al. Pre-extinction demographic stability and genomic signatures of adaptation in the woolly rhinoceros. Curr. Biol. 2020;30:3871–3879.e7. doi: 10.1016/j.cub.2020.07.046. [DOI] [PubMed] [Google Scholar]
- 49.Hansen C.C.R., Hvilsom C., Schmidt N.M., Aastrup P., Van Coeverden de Groot P.J., Siegismund H.R., Heller R. The muskox lost a substantial part of its genetic diversity on its long road to Greenland. Curr. Biol. 2018;28:4022–4028.e5. doi: 10.1016/j.cub.2018.10.054. [DOI] [PubMed] [Google Scholar]
- 50.McLaughlin T.R., Gómez-Puche M., Cascalheira J., Bicho N., Fernández-López de Pablo J. Late Glacial and Early Holocene human demographic responses to climatic and environmental change in Atlantic Iberia: LG-EH Demography Atlantic Iberia. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021;376:20190724. doi: 10.1098/rstb.2019.0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Pous P., Montori A., Amat F., Sanuy D. Range contraction and loss of genetic variation of the Pyrenean endemic newt Calotriton asper due to climate change. Reg. Environ. Change. 2016;16:995–1009. [Google Scholar]
- 52.Kardos M., Taylor H.R., Ellegren H., Luikart G., Allendorf F.W. Genomics advances the study of inbreeding depression in the wild. Evol. Appl. 2016;9:1205–1218. doi: 10.1111/eva.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leroy G., Carroll E.L., Bruford M.W., DeWoody J.A., Strand A., Waits L., Wang J. Next-generation metrics for monitoring genetic erosion within populations of conservation concern. Evol. Appl. 2018;11:1066–1083. doi: 10.1111/eva.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flanagan S.P., Jones A.G. The future of parentage analysis: From microsatellites to SNPs and beyond. Mol. Ecol. 2019;28:544–567. doi: 10.1111/mec.14988. [DOI] [PubMed] [Google Scholar]
- 55.Lemopoulos A., Prokkola J.M., Uusi-Heikkilä S., Vasemägi A., Huusko A., Hyvärinen P., Koljonen M.L., Koskiniemi J., Vainikka A. Comparing RADseq and microsatellites for estimating genetic diversity and relatedness — Implications for brown trout conservation. Ecol. Evol. 2019;9:2106–2120. doi: 10.1002/ece3.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allendorf F.W., Luikart G. Blackwell; 2007. Conservation and the Genetics of Populations. [Google Scholar]
- 57.Takahashi, M.K., Takahashi, Y.Y., and Parris, M.J. (2010). On the Role of Sexual Selection in Ecological Divergence: A Test of Body-Size Assortative Mating in the Eastern Newt Notophthalmus viridescens.
- 58.Acord M.A., Anthony C.D., Hickerson C.A.M. Assortative mating in a polymorphic salamander. Copeia. 2013;2013:676–683. [Google Scholar]
- 59.Dreiss A.N., Guillaume O., Clobert J. Diverging cave- and river-dwelling newts exert the same mate preference in their native light conditions. Ethology. 2009;115:1036–1045. [Google Scholar]
- 60.Gabor C.R., Halliday T.R. Sequential mate choice by multiply mating smooth newts: females become more choosy. Behav. Ecol. 1997;8:162–166. [Google Scholar]
- 61.Garner T.W.J., Schmidt B.R. Relatedness, body size and paternity in the alpine newt, Triturus alpestris. Proc. Biol. Sci. 2003;270:619–624. doi: 10.1098/rspb.2002.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jehle R., Sztatecsny M., Wolf J.B.W., Whitlock A., Hödl W., Burke T. Genetic dissimilarity predicts paternity in the smooth newt (Lissotriton vulgaris) Biol. Lett. 2007;3:526–528. doi: 10.1098/rsbl.2007.0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rovelli V., Randi E., Davoli F., Macale D., Bologna M.A., Vignoli L. She gets many and she chooses the best: polygynandry in Salamandrina perspicillata (Amphibia: Salamandridae) Biol. J. Linn. Soc. 2015;116:671–683. [Google Scholar]
- 64.Macgregor H.C. In: Amphibian Cytogenetics and Evolution. Green D.M., Sessions S.K., editors. Academic Press, Inc; 1991. Chromosome Heteromorphism in Newts (Triturus) and Its Significance in Relation to Evolution and Development; pp. 175–196. (San Diego). [Google Scholar]
- 65.Barton N.H., Wilson I. Genealogies and geography. Phil Trans R Soc Lond B. 1995;349:49–59. doi: 10.1098/rstb.1995.0090. [DOI] [PubMed] [Google Scholar]
- 66.Murphy M.A., Evans J.S., Cushman S.A., Storfer A. Representing genetic variation as continuous surfaces: An approach for identifying spatial dependency in landscape genetic studies. Ecography. 2008;31:685–697. [Google Scholar]
- 67.Holderegger R., Di Giulio M. The genetic effects of roads: A review of empirical evidence. Basic Appl. Ecol. 2010;11:522–531. [Google Scholar]
- 68.Lesbarrères D., Primmer C.R., Lodé T., Merilä J. The effects of 20 years of highway presence on the genetic structure of Rana dalmatina populations. Ecoscience. 2006;13:531–538. [Google Scholar]
- 69.Buskirk J. Permeability of the landscape matrix between amphibian breeding sites. Ecol. Evol. 2012;2:3160–3167. doi: 10.1002/ece3.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emel S.L., Storfer A. A decade of amphibian population genetic studies: synthesis and recommendations. Conserv. Genet. 2012;13:1685–1689. [Google Scholar]
- 71.Cushman S.A. Effects of habitat loss and fragmentation on amphibians: A review and prospectus. Biol. Conserv. 2006;128:231–240. [Google Scholar]
- 72.Guinart D., Solórzano S., Amat F., Grau J., Fernández-Guiberteau D., Montori A. Habitat Management of the Endemic and Critical Endangered Montseny Brook Newt (Calotriton arnoldi) Land. 2022;11:449. [Google Scholar]
- 73.Frankham R., Ballou J.D., Briscoe D.A. Conservation Genetics. Cambridge University Press; 2002. [Google Scholar]
- 74.Vörös J., Ursenbacher S., Jelić D. Population genetic analyses using 10 new polymorphic microsatellite loci confirms genetic subdivision within the olm, Proteus anguinus. J. Hered. 2019;110:211–218. doi: 10.1093/jhered/esy067. [DOI] [PubMed] [Google Scholar]
- 75.Forester B.R., Murphy M., Mellison C., Petersen J., Pilliod D.S., Van Horne R., Harvey J., Funk W.C. Genomics-informed delineation of conservation units in a desert amphibian. Mol. Ecol. 2022;31:5249–5269. doi: 10.1111/mec.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:10–12. [Google Scholar]
- 77.Rhie A., Walenz B.P., Koren S., Phillippy A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:245. doi: 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ranallo-Benavidez T.R., Jaron K.S., Schatz M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kokot M., Dlugosz M., Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33:2759–2761. doi: 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- 80.Kolmogorov M., Yuan J., Lin Y., Pevzner P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 81.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 Preprint at. 1303.3997v2. [Google Scholar]
- 82.Kundu R., Casey J., Sung W.-K. HyPo: Super Fast & Accurate Polisher for Long Read Genome Assemblies. bioRxiv. 2019 Preprint at. [Google Scholar]
- 83.Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guan D., McCarthy S.A., Wood J., Howe K., Wang Y., Durbin R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Challis R., Richards E., Rajan J., Cochrane G., Blaxter M. BlobToolKit - interactive quality assessment of genome assemblies. G3 (Bethesda) 2020;10:1361–1374. doi: 10.1534/g3.119.400908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smit A.F.A., Hubley R., Green P. 2013. RepeatMasker Open-4.0.http://www.repeatmasker.org [Google Scholar]
- 87.Smit A.F.A., Hubley R. 2008. RepeatModeler Open-1.0.http://www.repeatmasker.org [Google Scholar]
- 88.Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data.
- 89.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. giab008–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eaton D.A.R., Overcast I. Ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics. 2020;36:2592–2594. doi: 10.1093/bioinformatics/btz966. [DOI] [PubMed] [Google Scholar]
- 92.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., De Bakker P.I.W., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X., Fu Y.X. Stairway Plot 2: demographic history inference with folded SNP frequency spectra. Genome Biol. 2020;21:280. doi: 10.1186/s13059-020-02196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.R Core Team . R Foundation for Statistical Computing; 2021. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
- 95.Collyer M.L., Adams D.C. RRPP: An r package for fitting linear models to high-dimensional data using residual randomization. Methods Ecol. Evol. 2018;9:1772–1779. [Google Scholar]
- 96.Pinheiro J., Bates C., Debroy S., Heisterkamp S., van Willigen B. Package “nlme.”. 2016. https://cran.r-project.org/web/packages/nlme/nlme.pdf
- 97.Excoffier L., Marchi N., Marques D.A., Matthey-Doret R., Gouy A., Sousa V.C. Fastsimcoal2: Demographic inference under complex evolutionary scenarios. Bioinformatics. 2021;37:4882–4885. doi: 10.1093/bioinformatics/btab468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Excoffier L., Dupanloup I., Huerta-Sánchez E., Sousa V.C., Foll M. Robust Demographic Inference from Genomic and SNP Data. PLoS Genet. 2013;9:e1003905. doi: 10.1371/journal.pgen.1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kozlov A.M., Darriba D., Flouri T., Morel B., Stamatakis A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bouckaert R., Vaughan T.G., Barido-Sottani J., Duchêne S., Fourment M., Gavryushkina A., Heled J., Jones G., Kühnert D., De Maio N., et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019;15:e1006650. doi: 10.1371/journal.pcbi.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Korneliussen, T.S., Albrechtsen, A., and Nielsen, R. (2014). ANGSD: Analysis of Next Generation Sequencing Data. [DOI] [PMC free article] [PubMed]
- 102.Vieira F.G., Albrechtsen A., Nielsen R. Estimating IBD tracts from low coverage NGS data. Bioinformatics. 2016;32:2096–2102. doi: 10.1093/bioinformatics/btw212. [DOI] [PubMed] [Google Scholar]
- 103.Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis.https://ggplot2.tidyverse.org [Google Scholar]
- 104.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keenan K., Mcginnity P., Cross T.F., Crozier W.W., Prodöhl P.A. DiveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013;4:782–788. [Google Scholar]
- 106.Hennig C., Hausdorf B. Package “prabclus”, version 2.3-1. Functions for Clustering of Presence-Absence, Abundance and Multilocus Genetic Data. 2019. http://cran.r-project.org/web/packages/prabclus/
- 107.Lucati F., Miró A., Ventura M. Conservation of the endemic Pyrenean newt (Calotriton asper) in the age of invasive species: Interlake dispersal and colonisation dynamics. Amphib. Reptil. 2020;41:281–282. [Google Scholar]
- 108.Carøe C., Gopalakrishnan S., Vinner L., Mak S.S.T., Sinding M.H.S., Samaniego J.A., Wales N., Sicheritz-Pontén T., Gilbert M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2018;9:410–419. [Google Scholar]
- 109.Sambrook J., Fritsch E.F., Maniatis T. 2nd ed. Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 110.Peterson B.K., Weber J.N., Kay E.H., Fisher H.S., Hoekstra H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Broad Institute . Broad Inst. GitHub Repos; 2021. PicardTools. [Google Scholar]
- 113.Burriel-Carranza B., Estarellas M., Riaño G., Talavera A., Tejero-Cicuéndez H., Els J., Carranza S. Species boundaries to the limit: Integrating species delimitation methods is critical to avoid taxonomic inflation in the case of the Hajar banded ground gecko (Trachydactylus hajarensis) Mol. Phylogenet. Evol. 2023;186 doi: 10.1016/j.ympev.2023.107834. [DOI] [PubMed] [Google Scholar]
- 114.O’Leary S.J., Puritz J.B., Willis S.C., Hollenbeck C.M., Portnoy D.S. These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 2018;27:3193–3206. doi: 10.1111/mec.14792. [DOI] [PubMed] [Google Scholar]
- 115.Wu Y., Wang Y., Jiang K., Hanken J. Significance of pre-Quaternary climate change for montane species diversity: Insights from Asian salamanders (Salamandridae: Pachytriton) Mol. Phylogenet. Evol. 2013;66:380–390. doi: 10.1016/j.ympev.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 116.de Boer B., Lourens L.J., van de Wal R.S.W. NOAA National Centers for Environmental Information; 2015. NOAA/WDS Paleoclimatology - Global 5 Million Year Sea Level, Temperature, and d18Osw Reconstructions. [Google Scholar]
- 117.de Boer B., van de Wal R.S.W., Lourens L.J., Bintanja R., Reerink T.J. A continuous simulation of global ice volume over the past 1 million years with 3-D ice-sheet models. Clim. Dynam. 2013;41:1365–1384. [Google Scholar]
- 118.Margaritelli G., Cacho I., Català A., Barra M., Bellucci L.G., Lubritto C., Rettori R., Lirer F. Persistent warm Mediterranean surface waters during the Roman period. Sci. Rep. 2020;10:10431. doi: 10.1038/s41598-020-67281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akaike H. A New Look at the Statistical Model Identification. IEEE Trans. Automat. Control. 1974;19:716–723. [Google Scholar]
- 120.Edwards S.V., Xi Z., Janke A., Faircloth B.C., McCormack J.E., Glenn T.C., Zhong B., Wu S., Lemmon E.M., Lemmon A.R., et al. Implementing and testing the multispecies coalescent model: A valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 2016;94:447–462. doi: 10.1016/j.ympev.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 121.Stange M., Sánchez-Villagra M.R., Salzburger W., Matschiner M. Bayesian Divergence-Time Estimation with Genome-Wide Single-Nucleotide Polymorphism Data of Sea Catfishes (Ariidae) Supports Miocene Closure of the Panamanian Isthmus. Syst. Biol. 2018;67:681–699. doi: 10.1093/sysbio/syy006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Suchard M.A., Lemey P., Baele G., Ayres D.L., Drummond A.J., Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bouckaert R.R. DensiTree: Making sense of sets of phylogenetic trees. Bioinformatics. 2010;26:1372–1373. doi: 10.1093/bioinformatics/btq110. [DOI] [PubMed] [Google Scholar]
- 124.Meirmans P.G. genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 2020;20:1126–1131. doi: 10.1111/1755-0998.13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sundqvist L., Keenan K., Zackrisson M., Prodöhl P., Kleinhans D. Directional genetic differentiation and relative migration. Ecol. Evol. 2016;6:3461–3475. doi: 10.1002/ece3.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alcala N., Goudet J., Vuilleumier S. On the transition of genetic differentiation from isolation to panmixia: What we can learn from Gst and D. Theor. Popul. Biol. 2014;93:75–84. doi: 10.1016/j.tpb.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 127.Epskamp S., Cramer A.O.J., Waldorp L.J., Schmittmann V.D., Borsboom D. qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Software. 2012;48:1–18. [Google Scholar]
- 128.Hausdorf B., Hennig C. Species delimitation and geography. Mol. Ecol. Resour. 2020;20:950–960. doi: 10.1111/1755-0998.13184. [DOI] [PubMed] [Google Scholar]
- 129.Bamberger S., Xu J., Hausdorf B. Evaluating species delimitation methods in radiations: the land snail Albinaria cretensis complex on Crete. Syst. Biol. 2022;71:439–460. doi: 10.1093/sysbio/syab050. [DOI] [PubMed] [Google Scholar]
- 130.Weir B.S., Cockerham C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is publicly available as of the date of publication in the following NCBI BioProjects: aCalArn1 genome assembly (PRJEB57772), ORG.one Nanopore raw data used for the assembly (PRJEB57582), Illumina data for lcWGS population genomic analyses and Nanopore and Illumina data generated outside ORG.one used for the assembly (PRJEB57773), and demultiplexed ddRADseq data (PRJNA1023478).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (adrian.talavera@csic.es) upon request.