

Abstract
An approach to designing dihydropyridines that bind to adenosine receptors without binding to L-type calcium channels has been described. 1,4-Dihydropyridine derivatives substituted with β-styryl or phenylethynyl groups at the 4-position and aryl groups at the 6-position were synthesized and found to be selective for human A3 receptors. Combinations of methyl, ethyl, and benzyl esters were included at the 3- and 5-positions. Affinity was determined in radioligand binding assays at rat brain A1 and A2A receptors using [3H]-(R)-PIA [[3H]-(R)-N6-(phenylisopropyl)adenosine] and [3H]CGS 21680 [[3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine], respectively. Affinity was determined at cloned human and rat A3 receptors using [125I]AB-MECA [N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)-adenosine]. Structure-activity analysis indicated that substitution of the phenyl ring of the β-styryl group but not of the 6-phenyl substituent was tolerated in A3 receptor selective agents. Replacement of the 6-phenyl ring with a 3-thienyl or 3-furyl group reduced the affinity at A3 receptors by 4- and 9-fold, respectively. A 5-benzyl ester 4-trans-β-styryl derivative, 26, with a Ki value of 58.3 nM at A3 receptors, was >1700-fold selective vs either A1 receptors or A2A receptors. Shifting the benzyl ester to the 3-position lowered the affinity at A3 receptors 3-fold. A 5-benzyl, 3-ethyl ester 4-phenylethynyl derivative, 28, displayed a Ki value of 31.4 nM at A3 receptors and 1300-fold selectivity vs A1 receptors. The isomeric 3-benzyl, 5-ethyl diester was >600-fold selective for A3 receptors. Oxidation of 28 to the corresponding pyridine derivative reduced affinity at A3 receptors by 88-fold and slightly increased affinity at A1 receptors.
The physiological role of A3 adenosine receptors,1–4 the most recently cloned receptors of the adenosine,5,6 is under investigation, and potent and selective antagonists are needed as pharmacological probes and as potential therapeutic agents. Activation of the A3 receptor in the rat results in hypotension7 by promotion of release of inflammatory mediators from mast cells.8 IB-MECA, a selective A3 receptor agonist,4 was shown to inhibit the release of TNF-α in macrophages.9 A3 adenosine receptor antagonists are being sought as potential antiasthmatic,10 antiinflammatory,10 and cerebroprotective agents.11 Selective A3 receptor agonists at high concentrations were found to induce apoptosis in a HL-60 human leukemia cell line.12 There may also be an involvement of A3 receptors in the cytolytic activity of antitumor killer lymphocytes.13
1,4-Dihydropyridine blockers of L-type calcium channels14 are used extensively in the treatment of cardiovascular disorders as dilators of coronary arteries. We have found that, in addition to binding to calcium channels, this class of 1,4-dihydropyridines tends to bind to three subtypes of adenosine receptors, i.e. A1, A2A, and A3.15,16 For example, nifedipine (Figure 1, 1) is bound by these three adenosine receptors with micromolar affinity, although it is much more potent at L-type channels. A newer generation calcium channel blocker, nicardipine,17 2, within the family of adenosine receptors is actually selective for the A3 subtype. The (S)-enantiomer of niguldipine, 3, which is the more potent enantiomer at L-type channels, binds to human A3 adenosine receptors with a Ki value of 2.8 μM and is totally inactive at A1 and A2A receptors. Thus, with respect to adenosine receptors alone it is highly specific for the A3 subtype.
Figure 1.

Structures of 1,4-dihydropyridines as potent calcium channel antagonists (1-3) and an adenosine receptor antagonist (4). Ki values (μM) are shown for rat brain A1 receptors (rA1) and for cloned human A3 receptors (hA3).
We have recently described approaches to designing 1,4-dihydropyridines that bind to adenosine receptors without binding to L-type calcium channels.16 Furthermore, the moderate affinity for A3 adenosine receptors of readily available 1,4-dihydropyridines has provided leads for novel antagonists for that subtype. Inclusion of 4-trans-β-styryl and 6-phenyl substituents enhanced A3 receptor selectivity in an additive fashion and completely abolished recognition at L-type channels. Compound 4 (3,5-diethyl 2-methyl-6-phenyl-4-[2-phenyl-(E)-vinyl]-1,4-(±)-dihydropyridine-3,5-dicarboxylate, MRS 1097) was 55-fold selective for human A3 receptors vs rat A1 receptors and 44-fold selective vs rat A2A receptors. In a functional assay, compound 4 attenuated the A3 agonist-elicited inhibitory effect on adenylyl cyclase. Furthermore, whereas nicardipine, 2, displaced radioligand ([3H]-S-(4-nitrobenzyl)-6-thioinosine) from the Na+-independent adenosine transporter with an apparent affinity of 5.4 μM, compound 4 displaced less than 10% of total binding at a concentration of 100 μM. The selectivity of compound 4 for adenosine receptors was further demonstrated through its inactivity in binding assays at other receptors, ion channels, and second messenger components. The present study examines combinations and compatibility of dihydropyridine substituents found previously to enhance affinity at A3 receptors. New derivatives of exceptionally high potency and selectivity have been discovered.
Results
Synthesis.
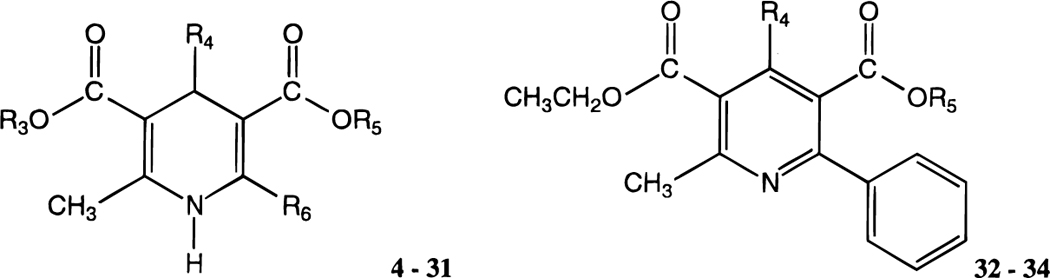
The structures of the 1,4-dihydropyridines (4-31) and related pyridine derivatives (32–34) tested for affinity in radioligand binding assays at adenosine receptors are shown in Table 1. New dihydropyridine derivatives were synthesized using standard methodology (Scheme 1),18–20 as reported in Table 2. The synthesis consisted of the Hantzsch condensation of a 3-amino-2-butenoate ester, 35a,b, an aldehyde, 36a-h, and a 3-ketopropionate ester derivative, 37a-i, that were dissolved in ethanol and refluxed under N2. In order to obtain substitution at the 4-position the aldehyde component, 36a-h, was varied. Substitution at the 6-position was achieved by varying the 3-ketopropionate ester component, 37e-g,i (Scheme 2), prepared according to method of Straley and Adams.21 In the synthesis of compounds 29-31, the precursor, ethyl 3-aminocinnamate was prepared from the imidate hydrochloride and Meldrum’s acid (Scheme 3).22 The yields of the 6-phenyl-1,4-dihydropyridines obtained using a ≤72 h reaction time varied between 6 and 63%. All of the dihydropyridines examined in this study were racemic mixtures at position C-4.
Table 1.
Affinities of Dihydropyridine and Pyridine Derivatives in Radioligand Binding Assays at A1, A2a, and A3 Receptorsa-f
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
|
Ki (μM) or % inhibitiond |
||||||||
| compd | R3 | R4 | R5 | R6 | rA1a | rA2Ab | hA3c | rA1/hA3 |
|
| ||||||||
| 5 e | CH3 | CH3 | CH2CH3 | CH3 | 32.6 ± 6.3 | 46.1 ± 6.8 | 32.3 ± 5.1 | 1.0 |
| 6 e | CH3 | CH3 | CH2Ph | CH3 | 6.45 ± 1.47 | 9.72 ± 0.63 | 2.78 ± 0.89 | 2.3 |
| 7 e | CH3 | CH2CH3 | CH2CH3 | CH3 | 7.52 ± 2.79 | 7.89 ± 2.87 | 13.6 ± 2.0 | 0.53 |
| 8 e | CH3 | PhCH=CH-(trans) | CH2CH3 | CH3 | 16.1 ± 0.5 | 49.3 ± 12.5 | 0.670 ± 0.195 | 24 |
| 9 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | CH3 | 4.65 ± 1.21 | 9.23 ± 3.60 | 0.887 ± 0.138 | 5.2 |
| 10 | CH2CH3 | PhCH=CH-(trans) | CH2Ph | CH3 | 13.7 ± 2.6 | 14 ± 4% (10−4) | 3.13 ± 0.51 | 4.4 |
| 11 e | CH2CH3 | CH3 | CH2CH3 | Ph | 25.9 ± 7.3 | 35.9 ± 15.3 | 7.24 ± 2.13 | 3.6 |
| 12 | CH2CH3 | CH2CH3 | CH2CH3 | Ph | 21.5 ± 2.7 | 14.5 ± 3.5 | 8.49 ± 1.74 | 2.5 |
| 13 | CH2CH3 | CH3 | CH2Ph | Ph | 26.0 ± 8.7 | 3.15 ± 0.96 | 1.75 ± 0.47 | 15 |
| 4 e | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | Ph | 5.93 ± 0.27 | 4.77 ± 0.29 | 0.108 ± 0.012 | 55 |
| 14 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 4-CH3Ph | 14.9 ± 4.9 | 37 ± 2% (10−4) | 9.13 ± 2.43 | 1.6 |
| 15 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 4-OCH3Ph | 9.49 ± 1.99 | d (10−4) | 1.43 ± 0.37 | 6.6 |
| 16 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 4-ClPh | 33.0 ± 7.5 | 12 ± 7% (10−4) | 0.785 ± 0.272 | 42 |
| 17 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 4-NO2Ph | 13.1 ± 1.6 | 40 ± 3% (10−4) | 4.14 ± 0.51 | 3.2 |
| 18 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 3-furyl | 5.49 ± 0.25 | 35 ± 6% (10−4) | 0.907 ± 0.307 | 6.1 |
| 19 | CH2CH3 | PhCH=CH-(trans) | CH2CH3 | 3-thienyl | 7.52 ± 1.38 | d (10−4) | 0.407 ± 0.066 | 18 |
| 20 | CH2CH3 | 2-OCH3PhCH=CH-(trans) | CH2CH3 | Ph | 25.3 ± 2.4 | 17 ± 12% (10−4) | 0.334 ± 0.059 | 76 |
| 21 | CH2CH3 | 2-NO2PhCH=CH-(trans) | CH2CH3 | Ph | 6.03 ± 1.39 | d (10−4) | 0.109±0.017 | 55 |
| 22 | CH2CH3 | 4-NO2PhCH=CH-(trans) | CH2CH3 | Ph | 23 ± 9% (10−4) | 33% (10−4) | 0.0585 ± 0.0164 | >1700 |
| 23 | CH2CH3 | 4-NH2PhCH=CH-(trans) | CH2CH3 | Ph | 31 ± 3% (10−4) | 26 ± 6% (10−4) | 0.198 ± 0.047 | >500 |
| 24 | CH2CH3 | (Ph)2C=CH- | CH2CH3 | Ph | 1.20 ± 0.14 | 12 ± 6% (10−4) | 1.42 ± 0.23 | 0.84 |
| 25 | CH2CH3 | PhC≡C- | CH2CH3 | Ph | 11.0 ± 0.1 | 26 ± 12% (10−4) | 0.0766 ± 0.0151 | 140 |
| 26 | CH2CH3 | PhCH=CH-(trans) | CH2Ph | Ph | 35 ± 3% (10−4) | 15 ± 3% (10−4) | 0.0583 ± 0.0124 | >1700 |
| 27 | CH2CH3 | 4-NO2PhCH=CH-(trans) | CH2Ph | Ph | 33 ± 1% (10−4) | d (10−4) | 0.0724 ± 0.0377 | >1300 |
| 28 | CH2CH3 | PhC≡C- | CH2Ph | Ph | 40.1 ± 7.5 | d (10−4) | 0.0314 ± 0.0028f | 1300 |
| 29 | CH2Ph | PhCH=CH-(trans) | CH2CH3 | Ph | d (10−4) | 16 ± 11% (10−4) | 0.142 ± 0.047 | >700 |
| 30 | CH2Ph | 4-NO2PhCH=CH-(trans) | CH2CH3 | Ph | d (10−4) | d (10−4) | 0.286 ± 0.038 | >400 |
| 31 | CH2Ph | PhC≡C- | CH2CH3 | Ph | 24 ± 4% (10−4) | d (10−4) | 0.169 ± 0.026 | >600 |
| 32 e | CH3 | CH2CH3 | 7.41 ± 1.29 | 28.4 ± 9.1 | 4.47 ± 0.46 | 1.7 | ||
| 33 | PhCH=CH-(trans) | CH2CH3 | 2.49 ± 0.47 | 2.40 ± 0.22 | 2.80 ± 1.78 | 0.85 | ||
| 34 | PhC≡C- | CH2Ph | 11.6 ± 4.8 | 43 ± 2% (10−4) | 2.75 ± 0.78 | 4.2 | ||
Displacement of specific [3H]-(R)-PIA binding in rat brain membranes, expressed as Ki ± SEM in μM (n = 3–5), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes, expressed as Ki ± SEM in μM (n = 3–6), or as a percentage of specific binding displaced at the indicated concentration (M).
Displacement of specific [125I]AB-MECA binding at human A3 receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in μM (n = 3–4).
Displacement of ≤ 10% of specific binding at the indicated concentration (M).
Values taken from van Rhee et al.16
Ki value at rat A3 receptors stably expressed in CHO cells1 found to be 3.53 ± 0.61 μM.
Scheme 1.

General Procedure for the Synthesis of 1,4-Dihydropyridine Derivatives 9-22 and 24-31
Table 2.
Characterization of Dihydropyridine and Pyridine Derivatives
| no. | Tm (°C) | formula | MS | analysis | yielda (%) | |
|---|---|---|---|---|---|---|
|
| ||||||
| 9 | 148–149 | C21H25NO4 | 355(CI) | C,H,N | 16.3 | |
| 10 | 115–117 | C26H27NO4 | 417 (CI) | C,H,N | 69.5 | |
| 12 | 106–108 | C20H25NO4 | 343 (CI) | C,H,N | 25.5 | |
| 13 | 112–114 | C24H25NO4 | 391 (EI) | C,H,N | 62.9 | |
| 14 | 129–131 | C27H29NO4 | 431 (CI) | C,H,N | 32.2 | |
| 15 | 137–139 | C21H33NO4 | 447 (CI) | C,H,N | 6.1 | |
| 16 | 149–151 | C26H26NClO4 | 451 (EI) | C,H,N | 31.2 | |
| 17 | oil | C26H26N2O6 | 462 (EI) | b | 15.3 | |
| 18 | 114–116 | C24H25NO5 | 407 (CI) | C,H,N | 25.5 | |
| 19 | oil | C24H25NSO4·0.50EtOH | 423 (EI) | C,H,N | 15.1 | |
| 20 | 130–132 | C27H29NO5 | 447 (EI) | C,H,N | 12.0 | |
| 21 | 175–176 | C26H26N2O6 | 462 (EI) | C,H,N | 8.0 | |
| 22 | 195–196 | C26H26N2O6 | 462 (EI) | C,H,N | 58.2 | |
| 23 | oil | C26H28N2O4 | 432 (EI) | c | 24.1 | |
| 24 | 164–165 | C32H31NO4 | 491 (EI) | C,H,N | 8.2 | |
| 25 | 132–134 | C26H25NO4 | 415 (EI) | C,H,N | 36.4 | |
| 26 | 161–163 | C31H29NO4 | 479 (EI) | C,H,N | 27.9 | |
| 27 | oil | C31H28N2O6 | 524 (EI) | d | 26.2 | |
| 28 | 156–158 | C31H27NO4 | 477 (CI) | C,H,N | 31.0 | |
| 29 | 149–151 | C31H29NO4 | 479 (EI) | C,H,N | 41.5 | |
| 30 | oil | C31H28N2O6 | 524 (EI) | e | 32.9 | |
| 31 | 124–125 | C31H27NO4 | 477 (CI) | C,H,N | 29.3 | |
| 33 | oil | C26H25NO4 | 415 (EI) | f | 29.8 | |
| 34 | oil | C31H25NO4 | 475 (EI) | g | 74.0 | |
Purification was achieved by thin layer chromatography, silica 60, 1000 μm layer thickness, using EtOAc-petroleum ether (pe) 35–60, 20:80 (v/v) as eluent.
17 pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.21; CH2Cl2-MeOH = 40:1 (v/v), Rf = 0.46; EI calcd 462.1806, found 462.1791.
23 (C26H28N2O4) insufficient quantity for CHN, pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.06; CHCl3-MeOH-CH3COOH = 94:6:1 (v/v/v), Rf = 0.47; EI calcd 432.2049, found 432.2042.
27 pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.23; CH2Cl2-MeOH = 40:1 (v/v), Rf = 0.68; EI calcd 524.1938, found 524.1947.
30 pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.21; CH2Cl2-MeOH = 40:1 (v/v), Rf = 0.71; EI calcd 524.1938, found 524.1928.
33 pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.62; CH2Cl2-MeOH = 40:1 (v/v), Rf = 0.69; EI calcd 415.1772, found 415.1783.
34 pure on analytical TLC (silica 60, 250 μm) EtOAc-pe = 20:80 (v/v), Rf = 0.52; CH2Cl2-MeOH = 40:1 (v/v), Rf = 0.75; EI calcd 475.1776, found 475.1783.
Scheme 2.

General Procedure for the Synthesis of β-Keto Esters
Scheme 3.

Procedure for the Synthesis of Ethyl 3-Aminocinnamate
An aromatic nitro group on the C-4 side chain of a dihydropyridine could be reduced selectively using zinc/acetic acid (Scheme 4). This was desired as a precursor for the potential radioiodination reaction for the preparation of an A3 receptor radioligand, as has been carried out for an agonist.27
Scheme 4.

Method for Reduction of the Nitro Group on 1,4-Dihydropyridines
Oxidation of 1,4-dihydropyridines (4 and 28) to the corresponding pyridine derivatives (33 and 34, respectively) was carried out using tetrachloro-1,4-benzoquinone (chloranil, 45) in tetrahydrofuran (Scheme 5).23
Scheme 5.

General Procedure for the Oxidation of 1,4-Dihydropyridine Derivatives Using Tetrachlorobenzoquinone, 45
Binding at Adenosine Receptors.
Ki values at A1 and A2A receptors were determined in radioligand binding assays in rat brain membranes vs [3H]-(R)-PIA[[3H]-(R)-N6-(phenylisopropyl)adenosine] or [3H]CGS 21680 [[3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine], respectively.24,25 Affinity at cloned human A3 receptors expressed in HEK-293 cells26 was determined using [125I]AB-MECA [N6-(4-amino-3-[125I]iodobenzyl)-5′-(N-methylcarbamoyl)adenosine].27,28
Structure-activity relationship (SAR) analysis of 1,4-dihydropyridine derivatives at adenosine receptors has indicated that increasing the size of the substituents from small alkyl either at the C-4 position or at the 5-ester position tends to increase binding affinity at A1, A2A, and A3 subtypes (Table 1).16 This was illustrated with the 5-benzyl ester, 6, and the 4-ethyl analogue, 7.16 The benzyl ester, 6, vs compound 5 provided a 12-fold enhancement of affinity at A3 receptors. The 4-β-styryl substituent in 8 provided an even greater enhancement of A3 receptor affinity (48-fold vs 5) and selectivity. The combination of the 5-benzyl ester and 4-β-styryl substituents in 10 was compatible in its affinity enhancement at A1 and A3, but not at A2A, subtypes.
The 6-phenyl substituent enhanced affinity particularly at A3 receptors, thus compound 11 was slightly A3 selective. Extending the 4-methyl group to ethyl in 12 did not result in an enhancement of affinity, as it did in the 6-methyl case (compound 7 vs 5). The combination of the 5-benzyl ester and 6-phenyl substituents in 13 was additive in affinity enhancement at A2A and A3, but not at A1, subtypes. The ratios of affinity of compound 13 vs the corresponding 5-ethyl ester, 11, were 11- and 4.1-fold at A2A and A3 receptors, respectively.
We have previously demonstrated that compound 4 is 55-fold selective for human A3 vs rat A1 receptors; thus the combination of the 4-β-styryl and 6-phenyl substituents is well tolerated at the A3 receptor.16 The compatibility of the 4-β-styryl group with other aromatic substituents at the 6-position was examined in the current study. In compounds 14-17, the 6-phenyl ring was substituted at the para position with methyl, methoxy, chloro, and nitro groups. All substitutions were less potent than 6-phenyl at A3 receptors, although the 4-chloro derivative, 16, retained 42-fold selectivity vs A1 receptors. The 4-methoxy derivative, 15, was 85-fold less potent than 4 at A3 receptors. Affinity at A2A receptors was particularly sensitive to substitution of the 6-phenyl ring. Substitution of the 6-phenyl ring with 3-furyl, 18, or 3-thienyl, 19, groups reduced the affinity at A3 receptors by 9- and 4-fold, respectively.
Substitutions of the phenyl ring of the β-styryl group, 20-23, suggested much more freedom in this region in binding at A1 and A3, but not A2A, subtypes. The 4-nitro analogue, 22 (MRS 1222), was slightly more potent at A3 receptors than 4. Phenyl substitution at the β-olefinic carbon in 24 resulted in relatively weak affinity at adenosine receptors (A1, A3 > A2A). Compound 24 was 5-fold more potent than 4 at A1 receptors and 13-fold less potent at A3 receptors. The affinity of the dehydro equivalent (4-phenylethynyl substituent) of 4, i.e. compound 25, was very similar to its affinity at A1 and A3 receptors.
Triple substitutions of the simple dihydropyridines were also probed in compounds 26-31. All contained the 6-phenyl substituent and either a 4-β-styryl or a 4-phenylethynyl group. Benzyl esters at the 3- (29–31) or 5-positions (26-28) were included. A 5-benzyl ester 4-trans-β-styryl derivative, 26, with a Ki value of 58.3 nM at A3 receptors, was >1700-fold selective vs either A1 receptors or A2A receptors. The enhanced selectivity was mainly the result of loss of affinity at A1 receptors. Shifting the benzyl ester from the 5- to the 3-position as in 29 lowered the affinity at A3 receptors 3-fold. The presence of a 4-nitro group on the β-styryl substituent, as in 27 and 30, had a negligible effect on affinity at any of the receptors studied. 3-Ethyl 5-benzyl 2-methyl-4-(phenylethynyl)-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 28 (MRS 1191), displayed a Ki value of 31.4 nM at A3 receptors and 1300-fold selectivity vs A1 receptors. Relative to the corresponding 5-ethyl ester derivative, 25, compound 28 was 2.4-fold more potent at A3 receptors and less potent at A1 receptors. A derivative isomeric to compound 28, the corresponding 3-benzyl 5-ethyl diester, 31, was >600-fold selective for A3 receptors. Thus, bulky groups at combinations of either 3-, 4-, and 6-positions or 4-, 5-, and 6-positions still resulted in high A3 receptor selectivity as a result of very low affinity at A1 and A2A receptors.
Oxidation of a simple 6-phenyl-1,4-dihydropyridine, 11, to the corresponding pyridine derivative increased affinity at A1 but not A3 receptors. The presence of additional bulky substituents, e.g. at the 4- and 5-positions, caused the effects of oxidation to be less tolerated in binding, at A3 receptors in particular. Oxidation of 28 to the corresponding pyridine derivative, 34, reduced affinity at A3 receptors by 88-fold, while affinity at A1 receptors increased 4-fold.
Discussion
Among the most selective ligands in the present study were compounds 26-28, i.e. 5-benzyl esters having ≥ 1300-fold selectivity for A3 vs A1 adenosine receptors, and compounds 29-31, i.e. 3-benzyl esters having ≥ 400-fold selectivity. The para-substituted 4-styryl-6-phenyl derivatives 22 and 23 were also highly selective. Structure–activity analysis at A3 adenosine receptors indicated that sterically bulky groups in 1,4-dihydropyridines are tolerated at the 3-, 4-, 5-, and 6-positions. In the present study simultaneous substitution at two or three, but not all four, of these positions was carried out. In this study large substituents at the 4-(β-styryl or phenylethynyl) and 6-positions (phenyl) were combined with bulky esters at either the 3- or 5-position, and a beneficial effect on selectivity for human A3 receptors was observed. In combination with the 4- and 6-position substitutions mentioned above, a benzyl ester group preserved affinity (vs 4) at the A3 receptors to a greater extent when located at the 5-position (e.g. 26) than at the 3-position (e.g. 29). Nevertheless, it is surprising that combinations of bulky substituents at the 3-, 4-, and 6-positions result in receptor affinity profiles not radically different from those combinations at the 4-, 5-, and 6-positions. This suggests that the A3 receptor binding site is not highly sterically constrained. Also, similar enhancements of affinity at A3 receptors by either a 4-trans-β-styryl or a 4-phenylethynyl group point to the same conclusion. Both of these groups are highly rigid, yet occupy nonidentical spatial regions.
A series of 8-styrylxanthines30,31 were found to be subject to photoisomerization about the olefinic bond, which reduces their utility as pharmacological probes. One might expect a similar complication with the 4-styryldihydropyridine derivatives. Incorporation of a linear phenylethynyl group instead of the styryl group avoids this potential problem of isomerization.
Pyridine derivatives, which have a distinctly more planar geometry than that of the corresponding dihydropyridines, lost affinity for A3 adenosine receptors but, remarkably, gained affinity for A1 receptors. Numerous classes of adenosine receptor non-xanthine antagonists have been found for the A1 receptor, and all tend to have a planar geometry.29 It is possible that the A3 receptor does not share this preference for planar antagonists.
There appears to be more flexibility of substitution of the phenyl ring of 4-β-styryl derivatives (e.g. compounds 20-23) than of the 6-phenyl substituent in A3 receptor selective agents. Substitution of the 6-phenyl ring in the para position with a variety of electron-donating (methyl, methoxy) or -withdrawing (nitro, chloro) groups or its replacement with 3-thienyl or 3-furyl groups (e.g. compounds 14-19) all reduced the affinity at A3 receptors.
It will now be necessary to investigate the regio- (and stereo-) selectivity of the ligand receptor interaction more fully. Many of the dihydropyridines examined in our previous study16 are asymmetric only on account of nonequivalent ester substitutions at the 3- and 5-positions. One such pair of enantiomers, (R)- and (S)- niguldipine, was studied at adenosine receptors. The difference in affinity at human A3 receptors between (R)- and (S)-niguldipine, 3, was insignificant (Ki values of 1.9 and 2.8 μM, respectively). Thus, by analogy to the present set of mixed ethyl and benzyl esters, it is likely that there is no dramatic difference in A3 receptor affinity between (R)- and (S)-enantiomers of 3,4- or 4, 5-disubstituted dihydropyridines (i.e. in which methyl groups occur at both the 2- and 6-positions). The effects of 6-aryl substitution on stereoselectivity of binding at adenosine receptors have yet to be explored.
At the rat A3 receptor, the affinity of 1,4-dihydropyridines is considerably lower than at human A3 receptors.16 For example the Ki value of 28 at the rat A3 receptor was found to be 3.53 μM, while at the human A3 receptor the Ki value is only 31 nM. Although compound 28 is still somewhat selective for A3 receptors in the rat (11-fold), it is 110-fold less potent than at human A3 receptors. This species dependence of A3 receptor affinity has also been demonstrated for xanthines2,4 and other classes of adenosine antagonists, such as flavonoids,28 which tend to bind with lower Ki values at human vs rat A3 receptors.
It is expected that all of the 4-(arylalkyl)-6-phenyl-1,4-dihydropyridines prepared in the present study are selective for adenosine receptors vs L-type Ca2+ channels, since this principle was demonstrated in a previous study for compound 4.16 Moreover, compound 4 was also shown to have negligible affinity for the adenosine transporter.16 Furthermore, it was demonstrated that dihydropyridines such as compound 4 effectively attenuate the IB-MECA elicited inhibition of adenylyl cyclase in CHO cells expressing the cloned rat A3 adenosine receptor.16 Thus, the selective ligands introduced here should be useful as antagonists in probing the role of A3 receptors, especially for the human homologue of the receptor. These selective agents are now suitable for study in functional assays and in investigations of the physiological role of A3 receptors. With slight additional improvement of affinity, dihydropyridines may also provide affinity probes such as radioligands for human A3 receptors, although it has not been determined if the binding is competitive.
Experimental Section
Materials.
Ethyl acetoacetate (37a), acetaldehyde (36a), propionaldehyde (36b), ethyl 3-aminocrotonate (35a), and trans-cinnamaldehyde (36c) were obtained from Fluka (Ronkonoma, NY). Ethyl 3-aminocrotonate (35a), 4-nitro-trans-cinnamaldehyde (36d), 2-nitro-trans-cinnamaldehyde (36e), 2-methoxy-trans-cinnamaldehyde (36f), β-phenylcinnamaldehyde (36g), phenyl propargylaldehyde (36h), benzyl acetoacetate (37b), ethyl benzoylacetate (37c), and tetrachloro-1,4-benzoquinone (45) were from Aldrich (St. Louis, MO). Compounds 4-8 and 32 were prepared as described in van Rhee et al.16 (R)-PIA and 2-chloroadenosine were purchased from Research Biochemicals International (Natick, MA). All other materials were obtained from commercial sources.
Synthesis.
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and spectra were taken in DMSO-d6 or CHCl3-d. Chemical-ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer, and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Atlantic Microlab Inc. (Norcross, GA). All melting points were determined with a Unimelt capillary melting point apparatus (Arthur H. Thomas Co., PA) and were uncorrected.
General Procedure for the Preparation of 1,4-Dihydropyridine-3,5-dicarboxylate Esters (9–22, 24–31).
Equimolar amounts (0.5 mmol) of the appropriate 3-amino-2-propenoate ester (35a,b), aldehyde (36a-h), and 3-ketopropionate ester (37a-i) derivative were dissolved in 5 mL of absolute ethanol. The solution was sealed in a glass tube and heated to 100 °C (for volatile aldehydes) or was refluxed under N2 for at least 24 h, and at most 72h. The solvent was then evaporated, and products were purified either by crystallization, column chromatography (silica 60; 220–440 mesh; Fluka, Buchs, CH; 20% ethyl acetate-80% petroleum ether 35–60), or preparative TLC (silica 60; 1000 μm; Analtech, Newark, DE; 20% ethyl acetate-80% petroleum ether 35–60). All procedures were performed under nitrogen and low-light conditions to prevent oxidation of the products. The products were shown to be homogeneous by analytical TLC.
3,5-Diethyl 2,6-Dimethyl-4-[2-phenyl-(E)-vinyl]-1,4-(±)-dihydropyridine-3,5-dicarboxylate (9).
1H NMR (CDCl3): δ 1.29 (t, 6H, J = 6.8 Hz, 3 and 5-CH2CH3), 2.33 (s, 6H, 2 and 6-CH3), 4.16 (m, 4H, 3 and 5-OCH2), 4.61 (d, 1H, J = 5.8 Hz, 4-H), 5.60 (br, 1H, NH), 6.17 (dd, 1H, J = 5.9, 16.6 Hz, C6H5C=CH), 6.25 (d, 1H, J = 16.6 Hz, C6H5CH=C), 7.16–7.33 (m, 5H, C6H5). MS (CI/NH3): m/z 356 (MH)+, 252 (M-C6H5CH=CH)+, base.
3-Ethyl 5-Benzyl 2,6-Dimethyl-4-[2-phenyl-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (10).
1H NMR (CDCl3): δ 1.29 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 2.34 (s, 3H, 2-CH3), 2.38 (s, 3H, 6-CH3), 4.22 (m, 2H, 3-OCH2), 4.70 (d, 1H, J = 5.8 Hz, 4-H), 5.20 (AB, 2H, J = 12.7 Hz, 5-OCH2), 6.16 (dd, 1H, J = 5.9, 15.8 Hz, C6H5C=CH), 6.21 (d, 1H, J = 15.8 Hz, C6H5CH=C), 7.20–7.44 (m, 10H, 2 × C6H5). MS (CI/NH3): m/z 418 (MH+), base.
3,5-Diethyl 2-Methyl-4-ethyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (12).
1H NMR (CDCl3): δ 0.88 (2t, 6H, J = 6.8 Hz, 3 and 5-CH2CH3), 1.32 (t, 3H, J = 7.0 Hz, 4-CH2CH3), 1.55 (m, 2H, 4-CH2CH3), 2.33 (s, 3H, 2-CH3), 3.90 (m, 2H, 3-OCH2), 4.06 (t, 1H, J = 6.5 Hz, 4-H), 4.22 (m, 2H, 5-OCH2), 5.63 (br, 1H, NH), 7.29–7.41 (m, 5H, 6-C6H5). MS (CI/NH3): m/z 361 (M + NH4+), base, 344 (MH+).
3-Ethyl 5-Benzyl 2,4-Dimethyl,6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (13).
1H NMR (CDCl3): δ 1.15 (d, 3H, J = 6.6 Hz, 4-CH3), 1.32 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 2.31 (s, 3H, 2-CH3), 4.02 (q, 1H, J = 6.7 Hz, 4-H), 4.21 (m, 2H, 3-OCH2), 4.95 (AB, 2H, J = 12.7 Hz, 5-OCH2), 5.70 (br, 1H, NH), 6.95–7.36 (m, 10H, 2 × C6H5). MS (EI): m/z 376 (M - CH3)+, 91 (C6H5CH2+), base.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(4-toluyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate (14).
1H NMR (DMSO-d6): δ 0.78 (t, 3H, J = 7.0 Hz, 3CH2CH3), 1.20 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.28 (s, 3H, toluyl-CH3), 2.34 (s, 3H, 2-CH3), 3.78 (q, 2H, J = 6.7 Hz, 3-OCH2), 4.10 (m, 2H, 5-OCH2), 4.49 (d, 1H, J = 7.3 Hz, 4-H), 6.20 (m, 2H, CH=CH), 7.18–7.36 (m, 9H, C6H4 and C6H5), 9.01 (br, 1H, NH). MS (CI/NH3): m/e 432 (MH+), 328 (M - C6H5CH=CH+), base.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(4-methoxyphenyl)-1,4-(R,S)-dihydropyridine-3,5-dicarboxylate (15).
1H NMR (CDCl3): δ 1.00 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 1.33 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.38 (s, 3H, 2-CH3), 3.85 (s, 3H, 4′-OCH3), 4.00 (q, 2H, J = 6.7 Hz, 3-OCH2), 4.22 (m, 2H, 5-OCH2), 4.74 (d, 1H, J = 6.1 Hz, 4-H), 5.76 (br, 1H, NH), 6.32 (dd, 1H, J = 6.6, 15.8 Hz, C6H5C=CH), 6.41 (d, 1H, J = 15.8 Hz, C6H5CH=C), 6.94 (d, 2H, J = 8.7 Hz, 2′- and 6′-H), 7.19–7.31 (m, 5H, C6H5), 7.37 (d, 2 H, J = 7.3 Hz, 3′- and 6′-H). MS (CI/NH3): m/z 448 (MH)+, 344 (M - C6H5CH=CH)+, base.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(4-chlorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate (16).
1H NMR (CDCl3): δ 0.99 (t, 3H, J = 7.2 Hz, 3-CH2CH3), 1.29 (t, 3H, J = 7.2 Hz, 5-CH2CH3), 2.37 (s, 3H, 2-CH3), 3.97 (q, 2H, J = 7.0 Hz, 3-OCH2), 4.22 (m, 2H, 5-OCH2), 4.75 (d, 1H, J = 6.3 Hz, 4-H), 5.71 (br, 1H, NH), 6.28 (dd, 1H, J = 6.6, 15.8 Hz, C6H5C=CH), 6.40 (d, 1H, J = 15.8 Hz, C6H5CH=C), 7.20–7.41 (m, 9H, C6H4 and 6-C6H5). MS (EI): m/z 451 (M)+, 422 (M - C2H5)+, 378 (M - CO2C2H5)+, base, 348 (M - C6H5CH=CH)+.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(4-nitrophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate (17).
1H NMR (CDCl3): δ 1.21 (t, 3H, J = 6.8 Hz, 3-CH2CH3), 1.28 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.38 (s, 3H, 2-CH3), 4.13 (q, 2H, J = 7.0 Hz, 3-OCH2), 4.25 (m, 2H, 5-OCH2), 4.77 (d, 1H, J = 5.9 Hz, 4-H), 5.67 (br, 1H, NH), 6.26 (dd, 1H, J = 6.6, 15.8 Hz, C6H5C=CH), 6.37 (d, 1H, J = 15.8 Hz, C6H5CH=C), 7.21–7.54 (m, 7H, 3′,5′-H and 6-C6H5), 8.28 (m, 2H, 2′ and 6′-H). MS (EI): m/z 462 (M)+, 433 (M - C2H5)+, 389 (M CO2C2H5)+, base, 359 (M - C6H5CH=CH)+.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(3-furyl)1,4-(±)-dihydropyridine-3,5-dicarboxylate (18).
1H NMR δ 1.13 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 1.31 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.37 (s, 3H, 2-CH3), 3.85 (s, 3H, 4′-OCH3), 4.08 (m, 2H, 3-OCH2), 4.21 (q, J = 7.1 Hz, 2H, 5-OCH2), 4.73 (d, 1H, J = 6.3 Hz, 4-H), 5.73 (br, 1H, NH), 6.24 (dd, 1H, J = 6.7, 15.8 Hz, C6H5C=CH), 6.35 (d, 1H, J = 15.8 Hz, C6H5CH=C), 6.52 (m, 1H, 3′-H), 7.19–7.37 (m, 5H, C6H5), 7.45 (m, 1 H, 2′-H), 7.65 (s, 1H, 5′-H). MS (CI/NH3): m/z 408 (MH)+, 304 (M - C6H5CH=CH)+, base.
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-(3-thienyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate (19).
1H NMR (CDCl3): δ 1.05 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 1.27 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.38 (s, 3H, 2-CH3), 4.05 (m, 2H, 3-OCH2), 4.22 (m, 2H, 5-OCH2), 4.73 (d, 1H, J = 6.2 Hz, 4-H), 5.88 (br, 1H, NH), 6.30 (dd, 1H, J = 6.5, 15.8 Hz, C6H5C=CH), 6.38 (d, 1H, J = 15.8 Hz, C6H5CH=C), 7.06 (m, 1H, 4′-H), 7.18–7.31 (m, 5H, C6H5), 7.35 (d, 1H, J = 5.2 Hz, 3′-H), 7.40 (d, 1H, J = 4.8 Hz, 5′-H). MS (EI): m/z 423 (M)+, 350 (M - CO2CH2CH3), base, 320 (M - C6H5CH=CH)+.
3,5-Diethyl 2-Methyl-4-[2-(2-methoxyphenyl)-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (20).
1H NMR (CDCl3): δ 0.93 (t, 3H, J = 7.3 Hz, 3-CH2CH3), 1.33 (t, 3H, J = 6.8 Hz, 5-CH2CH3), 2.35 (s, 3H, 2-CH3), 3.82 (s, 3H, 2′-OCH3), 3.92 (m, 2H, 3-OCH2), 4.20 (m, 2H, 5-OCH2), 4.75 (d, 1H, J = 5.9 Hz, 4-H), 5.76 (br, 1H, NH), 6.26 (dd, 1H, J = 5.9, 15.6 Hz, C6H5C=CH), 6.84 (d, 1H, J = 16.6 Hz, C6H5-CH=C), 7.15–7.48 (m, 9H, 4-C6H4 and 6-C6H5). MS (EI): m/z 447 (M)+, 402 (M - OC2H5)+, 374 (M - CO2C2H5)+.
3,5-Diethyl 2-Methyl-4-[2-(2-nitrophenyl)-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (21).
1H NMR (CDCl3): δ 0.91 (t, 3H, J = 7.3 Hz, 3-CH2CH3), 1.33 (t, 3H, J = 7.3 Hz, 5-CH2CH3), 2.37 (s, 3H, 2-CH3), 3.96 (q, 2H, J = 7.2 Hz, 3-OCH2), 4.27 (q, 2H, J = 7.2 Hz, 5-OCH2), 4.78 (d, 1H, J = 5.9 Hz, 4-H), 5.93 (br, 1H, NH), 6.34 (dd, 1H, J = 5.9, 15.6 Hz, C6H5C=CH), 6.90 (d, 1H, J = 16.6 Hz, C6H5-CH=C), 7.30–7.48 (m, 8H, 4-C6H3 and 6-C6H5), 7.90 (d, 1H, J = 9.8 Hz, 3′-H). MS (EI): m/z 462 (M)+, 417 (M - OC2H5)+, 389 (M - CO2C2H5)+.
3,5-Diethyl 2-Methyl-4-[2-(4-nitrophenyl)-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (22).
1H NMR (CDCl3): δ 0.89 (t, 3H, J = 7.3 Hz, 3-CH2CH3), 1.32 (t, 3H, J = 7.3 Hz, 5-CH2CH3), 2.39 (s, 3H, 2-CH3), 3.94 (q, 2H, J = 6.8 Hz, 3-OCH2), 4.24 (q, 2H, J = 6.8 Hz, 5-OCH2), 4.80 (d, 1H, J = 4.9 Hz, 4-H), 5.82 (br, 1H, NH), 6.47 (dd, 1H, J = 3.9, 16.1 Hz, C6H5C=CH), 7.32–7.51 (m, 8H, C6H5CH=C and 4-C6H2 and 6-C6H5), 8.15 (d, 2H, J = 8.8 Hz, 3′- and 5′-H). MS (EI): m/z 462 (M)+, 417 (M - OC2H5)+, 389 (M - CO2C2H5)+.
3,5-Diethyl 2-Methyl-4-[2-(4-aminophenyl)-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (23).
Compound 23 was prepared by the catalytic reduction of compound 22 with zinc and acetic acid as described previously.30 Compound 22 (23 mg, 0.05 mmol) was dissolved in 1.5 mL of glacial acetic acid. Zn powder (0.15 mmol, 10 mg) was added to the solution, and the reaction mixture was stirred with a magnetic stirring bar at room temperature. Six hours after the start of the reaction, another batch of zinc powder was added. At 9 h reaction time, TLC (silica 60; petroleum ether 35–60-EtOAc = 80:20) analysis of the reaction mixture indicated that all starting material had been converted. The reaction mixture was diluted with 30 mL of water and neutralized with saturated NaHCO3 solution. The aqueous solution was extracted three times with 15 mL of chloroform, and the organic phase was washed once with 20 mL of water. The organic phase was separated and dried over anhydrous MgSO4. The product was purified by preparative TLC (1000 μm of silica 60; petroleum ether 35–60-EtOAc, 90:10) to yield 5.2 mg (24%) of a slightly yellow oil, which was shown to be pure by HPLC (OD-5–60, C-18 column, Separation Methods Technologies, Inc, Newark, DE; 0.1 M triethylammonium acetate buffer-CH3CN, 40:60 gradient to 10:90 in 20 min; 1 mL/min flow; retention time = 4.45 min), and analytical TLC (silica 60; petroleum ether 35–60-EtOAc, 80:20, R f = 0.06; CHCl3-MEOH-HOAc, 94:6:1, R f = 0.47). 1H NMR (CDCl3): δ 0.91 (t, 3H, J = 6.8 Hz, 3-CH2CH3), 1.31 (t, 3H, J = 6.8 Hz, 5-CH2CH3), 2.34 (s, 3H, 2-CH3), 3.67 (br, 2H, 4′-NH2), 3.92 (m, 2H, 3-OCH2), 4.17 (m, 2H, 5-OCH2), 4.70 (d, 1H, J = 6.8 Hz, 4-H), 5.78 (br, 1H, NH), 6.08 (dd, 1H, J = 6.8, 15.6 Hz, C6H5C=CH), 6.30 (d, 1H, J = 15.6 Hz, C6H5CH=C), 6.60 (d, 2H, J = 8.8 Hz, 3′- and 5′-H), 7.18 (d, 2H, J = 8.8 Hz, 2′- and 6′-H), 7.32–7.42 (m, 5H, 6-C6H5). MS (EI): m/z 432 (M)+, 387 (M - OC2H5)+, 359 (M - CO2C2H5)+.
3,5-Diethyl 2-Methyl-4-(2,2-diphenylvinyl)-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (24).
1H NMR (CDCl3): δ 0.84 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 1.10 (t, 3H, J ) 7.2 Hz, 5-CH2CH3), 2.32 (s, 3H, 2-CH3), 3.70–4.18 (m, 4H, 3-OCH2 and 5-OCH2), 4.95 (d, 1H, J = 9.9 Hz, 4-H), 5.82 (br, 1H, NH), 6.02 (d, 1H, J = 10.3 Hz, C6H5C=CH), 7.17–7.46 (m, 15H, 4-C6H5 and 4-C6H5 and 6-C6H5). MS (EI): m/z 491 (M)+, 446 (M - OC2H5)+, 418 (M - CO2C2H5)+.
3,5-Diethyl 2-Methyl-4-(phenylethynyl)-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (25).
1H NMR (CDCl3): δ 0.95 (t, 3H, J = 7.0 Hz, 3-CH2CH3), 1.32 (t, 3H, J ) 6.8 Hz, 3-CH2CH3), 2.37 (s, 3H, 2-CH3), 3.98 (m, 2H, 3-OCH2), 4.27 (m, 2H, 3-OCH2), 5.12 (s, 1H, 4-H), 5.92 (br, 1H, NH), 7.23–7.43 (+m, 10H, 2 × C6H5). MS (EI+ ): m/z 415 (M)+, 386 (M - C2H5) , 342 (M - CO2C2H5) , base.
3-Ethyl 5-Benzyl 2-methyl-4-[2-phenyl-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (26).
1H NMR (CDCl3): δ 1.28 (t, 3H, J = 6.8 Hz, 3-CH2CH3), 2.38 (s, 3H, 2-CH3), 4.22 (m, 2H, 3-OCH2), 4.78 (d, 1H, J = 6.2 Hz, 4-H), 4.99 (AB, J = 12.7 Hz, 5-OCH2), 5.76 (br, 1H, NH), 6.30 (dd, 1H, J = 6.5, 16.2 Hz, C6H5C=CH), 6.38 (d, 1H, J = 16.2 Hz, C6H5CH=C), 6.98 (m, 2H, 2′- and 6′-H), 7.21–7.38 (m, 13H, 2 × C6H5, 3′-+, 4′-, and 5′-H). MS (EI): m/z+ 479 (M)+, 388 (M - C6H5CH2) , 376 (M - C6H5CH=CH) , 344 (M - CO2CH2-CH3), 91 (C6H5CH2)+, base.
3-Ethyl 5-Benzyl 2-Methyl-4-[2-(4-nitrophenyl)-E)-vinyl]-6-phenyl-1,4-(R,S)-dihydropyridine-3,5-dicarboxylate (27).
1H NMR (CDCl3): δ 1.31 (t, 3H, J = 6.9 Hz, 3-CH2CH3), 2.38 (s, 3H, 2-CH3), 4.24 (m, 2H, 3-OCH2), 4.84 (d, 1H, J = 5.8 Hz, 4-H), 5.02 (AB, J = 12.7 Hz, 5-OCH2), 5.82 (br, 1H, NH), 6.43 (dd, 1H, J = 6.5, 16.0 Hz, NO2C6H4C=CH), 6.45 (d, 1H, J = 16.0 Hz, NO2C6H4CH=C), 6.96 (dd, 2H, J = 2.0, 7.7 Hz, 3′- and 5′-H), 7.19–7.39 (m, 5H, C6H5), 8.13 (d, 2H, J = 8.8 Hz, 2′- and 6′-H). MS (EI): m/z 524 (M)+, 389 (M - CO2C6H5CH2)+, 376 (M - NO2C6H5CH=CH)+, 91 (C6H5CH2)+, base.
3-Ethyl 5-Benzyl 2-methyl-4-phenylethynyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (28).
1H NMR (CDCl3): δ 1.35 (t, 3H, J = 6.8 Hz, 3-CH2CH3), 2.37 (s, 3H, 2-CH3), 4.27 (m, 2H, 3-OCH2), 5.10 (AB, J = 12.7 Hz, 5-OCH2), 5.19 (s, 1H, 4-H), 5.87 (br, 1H, NH), 7.08+ −7.39 (m, 15H, 3 × C6H5). MS (CI/NH3): m/z 478 (MH) , 376 (M - C6H5CdC)+, 242 (M - CO2C6H5CH2)+, base.
3-Benzyl 5-Ethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (29).
1H NMR (CDCl3): δ 0.91 (t, 3H, J = 7.0 Hz, 5-CH2CH3), 2.38 (s, 3H, 2-CH3), 3.92 (q, 2H, J = 6.7 Hz, 5-OCH2), 4.81 (d, 1H, J = 5.9 Hz, 4-H), 5.18 (AB, J = 12.7 Hz, 2H, 3-OCH2), 5.78 (br, 1H, NH), 6.31 (dd, 1H, J = 5.9, 16.0 Hz, C6H5C=CH), 6.36 (d, 1H, J = 16.0 Hz, C6H5CH=C), 7.20–7.42 (m, 10H, 2 × C6H5). MS (EI): m/z 406 (M - CO2CH2)+, 376 (M - C6H5CH=CH)+, 91 (C6H5CH2)+, base.
3-Benzyl 5-Ethyl 2-Methyl-4-[2-(4-nitrophenyl)-(E)-vinyl]-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (30).
1H NMR (CDCl3): δ 0.88 (t, 3H, J = 6.8 Hz, 5-CH2CH3), 2.39 (s, 3H, 2-CH3), 4.09 (q, 2H, J = 6.8 Hz, 3-OCH2), 4.84 (d, 1H, J = 5.8 Hz, 4-H), 5.30 (AB, J = 12.2 Hz, 3-OCH2), 5.87 (br, 1H, NH), 6.40 (d, 1H, J = 16.0 Hz, NO2C6H4CH=C), 6.50 (dd, 1H, J = 5.9, 16.6 Hz, NO2C6H4C=CH), 6.96 (dd, 2H, J = 2.0, 7.7 Hz, 3′- and 5′-H), 7.19–7.39 (m, 5H, C6H5), 8.14 (d, 2H, J = 7.8 Hz, 2′- and 6′-H). MS (EI): m/z 524 (M)+, 389 (M - CO2C6H5CH2)+, 376 (M - NO2C6H5CH=CH)+, 91-(C6H5CH2)+, base.
3-Benzyl 5-Ethyl 2-Methyl-4-(phenylethynyl)-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (31).
1H NMR (CDCl3): δ 0.96 (t, 3H, J = 6.8 Hz, 5-CH2CH3), 2.38 (s, 3H, 2-CH3), 4.00 (q, 2H, J = 6.8 Hz, 5-OCH2), 5.21 (s, 1H, 4-H), 5.30 (AB, J = 13.6 Hz, 3-OCH2), 5.87 (br, 1H, NH), 7.30–7.49 (m, 15H, 3+× C6H5). MS (CI/NH3): m/z 478 (MH)+, 376 (M - C6H5CdC) , base.
Procedure for Oxidation of 1,4-Dihydropyridine-3,5-dicarboxylate Esters (33–34).
Equimolar amounts (0.25 mmol) of the 1,4-dihydropyridine-3,5-dicarboxylate ester (4, 28) and tetrachloro-1,4-benzoquinone (45) in tetrahydrofuran (2 mL) were mixed and refluxed for up to 4 h. The solvent was then evaporated, and products were purified by preparative TLC (silica 60; 1000 μm; Analtech, Newark, DE; 20% ethyl acetate-80% petroleum ether 35–60).
3,5-Diethyl 2-Methyl-4-[2-phenyl-(E)-vinyl]-6-phenylpyridine-3,5-dicarboxylate (33).
1H NMR (CDCl3): δ 0.96 (t, 3H, J = 6.9 Hz, 3-CH2CH3), 1.26 (t, 3H, J = 6.8 Hz, 5-CH2CH3), 2.67 (s, 3H, 2-CH3), 4.07 (q, 2H, J = 6.8 Hz, 3-OCH2), 4.35 (q, 2H, J = 6.8 Hz, 5-OCH2), 6.88 (d, 1H, J = 16.6 Hz, C6H5C=CH), 6.88 (d, 1H, J = 16.6 Hz, C6H5CH=C), 7.31–7.61 (2m, 10H, 2 × C6H5). MS (EI): m/e+ 415 (M)+, base, 370 (M - OC2H5)+, 358 (386 - CH2=CH2)+.
3-Ethyl 5-Benzyl 2-Methyl-4-(phenylethynyl)-6-phenylpyridine-3,5-dicarboxylate (34).
1H NMR (CDCl3): δ 1.42 (t, 3H, J = 6.8 Hz, 3-CH2CH3), 2.68 (s, 3H, 2-CH3), 4.48 (q, 2H, J = 6.8 Hz, 3-OCH2), 5.17 (s, 2H, 5- OCH2), 7.10–7.60 (4m, 15H, 3 × C6H5). MS (EI):+ m/e 475 (M)+, 438 (M - OC2H5)+, 384 (M - C6H5CH2)+, 356 (M - CH2=CH2)+, base.
Pharmacology. Radioligand Binding Studies.
Binding of [3H]-(R)-N6-(phenylisopropyl)adenosine ([3H]-(R)-PIA) to A1 receptors from rat cerebral cortex membranes and of [3H]-2[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine ([3H]CGS 21680) to A2A receptors from rat striatal membranes was performed as described previously.24,25 Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in a preincubation of 30 min at 30 °C, and during the incubation with the radioligands.
Binding of [125I]-N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine ([125I]AB-MECA) to membranes prepared from HEK-293 cells stably expressing the human A3 receptor (Receptor Biology, Inc., Baltimore, MD) or to membranes prepared from CHO cells stably expressing the rat A3 receptor was performed as described.27,28 The assay medium consisted of a buffer containing 50 mM Tris, 10 mM Mg2+, and 1 mM EDTA, at pH 8.0. The glass incubation tubes contained 100 μL of the membrane suspension (0.3 mg of protein/mL, stored at −80 °C in the same buffer), 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of a solution of the proposed antagonist. Nonspecific binding was determined in the presence of 200 μM NECA.
All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%.
Incubations were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each.
At least five different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in the InPlot program (Graph-PAD, San Diego, CA), were converted to apparent Ki values using the Cheng-Prusoff equation32 and Kd values of 1.0 and 14 nM for [3H]-(R)-PIA and [3H]CGS 21680, respectively, and 0.59 nM for binding of [125I]AB-MECA at human A3 receptors, respectively.
Acknowledgment.
We thank Gilead Sciences (Foster City, CA) and the Cystic Fibrosis Foundation (Silver Spring, MD) for financial support. We thank Prof. Gary L. Stiles and Dr. Mark Olah of Duke University School of Medicine (Durham, NC) for providing samples of [125I]-AB-MECA.
Abbreviations:
- [125I]AB-MECA, [125I]
N 6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- CGS 21680
2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine
- CHO cells
Chinese hamster ovary cells
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- HEK cells
human embryonic kidney cells
- IB-MECA
N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- K i
equilibrium inhibition constant
- NECA
5′-(N-ethylcarbamoyl)adenosine
- (R)-PIA
(R)-N 6-(phenylisopropyl)adenosine
- SAR
structure-activity relationship
- Tris
tris(hydroxymethyl)aminomethane
References
- (1).Zhou QY; Li CY; Olah ME; Johnson RA; Stiles GL; Civelli O. Molecular cloning and characterization of an adenosine receptor - The A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Linden J. Cloned adenosine A3 receptors - pharmacological properties, species -differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306. [DOI] [PubMed] [Google Scholar]
- (3).Jacobson KA; Nikodijevic O; Shi D; Gallo-Rodriguez C; Olah ME; Stiles GL; Daly JW. A role for central A3-adenosine receptors: Mediation of behavioral depressant effects. FEBS Lett. 1993, 336, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jacobson KA; Kim HO; Siddiqi SM; Olah ME; Stiles G; von Lubitz DKJE. A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future 1995, 20, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stiles GL. Adenosine receptor subtypes: New insights from cloning and functional studies. Wiley: New York, in press. [Google Scholar]
- (6).Jacobson KA; van Galen PJM; Williams M. Adenosine receptors - pharmacology, structure activity relationships, and therapeutic potential. J. Med. Chem. 1992, 35, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hannon JP; Pfannkuche HJ; Fozard JR. A role for mastcells in adenosine A3 receptor-mediated hypotension in the rat. Br. J. Pharmacol. 1995, 115, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ramkumar V; Stiles GL; Beaven MA; Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993, 268, 16887–16890. [PubMed] [Google Scholar]
- (9).Sajjadi FG; Takabayashi K; Foster AC; Domingo RC; Firestein GS. Inhibition of TNF-R expression by adenosine. J. Immunol. 1996, 156, 3435–3442. [PubMed] [Google Scholar]
- (10).Beaven MA; Ramkumar V; Ali H. Adenosine-A3 receptors in mast-cells. Trends Pharmacol. Sci. 1994, 15, 13–14. [DOI] [PubMed] [Google Scholar]
- (11).von Lubitz DKJE; Lin RCS; Popik P; Carter MF; Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol. 1994, 263, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kohno Y; Sei Y; Koshiba M; Kim HO; Jacobson KA. Induction of apoptosis in HL-60 human promyelocytic leukemia cells by selective adenosine A3 receptor agonists. Biochem. Biophys. Res. Commun. 1996, 219, 904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).MacKenzie WM; Hoskin DW; Blay J. Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A3 receptor. Cancer Res. 1994, 54, 3521–3526. [PubMed] [Google Scholar]
- (14).Triggle DJ. Drugs acting on ion channels and membranes. In Comprehensive Medicinal Chemistry; Emmett JC, Ed.; Pergamon Press: London, 1985; Vol. 3, pp 1047–1099. [Google Scholar]
- (15).Hu PS; Lindgren E; Jacobson KA; Fredholm BB. Interaction of dihydropyridine calcium channel agonists and antagonists with adenosine receptors. Pharmacol. Toxicol. 1987, 61, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).van Rhee AM; Jiang J.-l.; Melman N; Olah ME; Stsiles GL; Jacobson KA. Interaction of 1,4-dihydropyridine and pyridine derivatives with adenosine receptors: selectivity for A3 receptors. J. Med. Chem. 1996, 39, 2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sorkin EM; Clissold SP. Nicardipine: A review of its pharmacological and pharmacokinetic properties and therapeutic efficacy, in the treatment of angina pectoris, hypertension, and related cardiovascular disorders. Drugs 1987, 33, 296–345. [DOI] [PubMed] [Google Scholar]
- (18).Stout DM; Myers AI. Recent advances in the chemistry of dihydropyridines. Chem. Rev. 1982, 82, 223–243. [Google Scholar]
- (19).Singer A; McElvain SM. 2,6-Dimethylpyridine. Org. Synth. 1934, 14, 30–33. [Google Scholar]
- (20).Rathke MW; Deitch J. The reaction of lithium ester enolates with acid chlorides. A convenient procedure for the preparation of β-keto esters. Tetrahedron Lett. 1971, 31, 2953–2956. [Google Scholar]
- (21).Straley JM; Adams AC. Ethyl benzoylacetate. Organic Syntheses; Wiley: New York, 1963; Collect. Vol. IV, pp 415–417. [Google Scholar]
- (22).Celerier J-P; Deloisy E; Kapron P; Lhommet G; Maitte P. Imidate Chemistry. Synthesis 1981, 130–133. [Google Scholar]
- (23).Braude EA; Hannah J; Linstead R. Hydrogen transfer. Part XVI. Dihydrides of nitrogenous heterocycles as hydrogen donors. J. Chem. Soc. 1960, 3249–3257. [Google Scholar]
- (24).Schwabe U; Trost T. Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980, 313, 179–187. [DOI] [PubMed] [Google Scholar]
- (25).Jarvis MF; Schutz R; Hutchison AJ; Do E; Sills MA; Williams M. [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther. 1989, 251, 888–893. [PubMed] [Google Scholar]
- (26).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Olah ME; Gallo-Rodriguez C; Jacobson KA; Stiles GL. [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- (28).Karton Y; Jiang J -l.; Ji, X. d.; Melman, N.; Olah, M. E.; Stiles, G. L.; Jacobson, K. A. Synthesis and biological activities of flavonoid derivatives as A3 adenosine receptor antagonists, J. Med. Chem. 1996, 39, 2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Siddiqi SM; Ji XD; Melman N; Olah ME; Jain R; Evans P; Glashofer M; Padgett WL; Cohen LA; Daly JW; Stiles GL; Jacobson KA. A survey of non-xanthine derivatives as adenosine receptor ligands. Nucleosides Nucleotides 1996, 15, 693–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jacobson KA; Gallo-Rodriguez C; Melman N; Fischer B; Maillard M; van Bergen A; van Galen PJM; Karton Y. Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J. Med. Chem. 1993, 36, 13331342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nonaka Y; Shimada J; Nonaka H; Koike N; Aoki N; Kobayashi H; Kase H; Yamaguchi K; Suzuki F. Photoisomerization of a potent and selective adenosine A2 antagonist, (E)-1,3-Dipropyl-8-(3,4-dimethoxystyryl)-7-methylxanthine. J. Med. Chem. 1993, 36, 3731–3733. [DOI] [PubMed] [Google Scholar]
- (32).Cheng YC; Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem . Pharmacol. 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]