

Abstract

SOS1 and SOS2 are guanine nucleotide exchange factors that mediate RTK-stimulated RAS activation. Selective SOS1:KRAS PPI inhibitors are currently under clinical investigation, whereas there are no reports to date of SOS2:KRAS PPI inhibitors. SOS2 activity is implicated in MAPK rebound when divergent SOS1 mutant cell lines are treated with the SOS1 inhibitor BI-3406; therefore, SOS2:KRAS inhibitors are of therapeutic interest. In this report, we detail a fragment-based screening strategy to identify X-ray cocrystal structures of five diverse fragment hits bound to SOS2.
Introduction
The Son of Sevenless (SOS) family of rat sarcoma virus–guanine exchange factors (RAS–GEFs) couple activating signals from upstream receptor tyrosine kinases (RTKs) to downstream effector pathways to regulate cellular growth and survival.1−3 The two paralogs of SOS, SOS1, and SOS2, have a high degree of structural similarity and are approximately 75% identical in amino acid composition, with the greatest differences observed in the carboxyl-terminal region implicated in binding to the SH3 domains of the adaptor growth factor receptor-bound protein 2 (GRB2).4−8 As is the case for SOS1, SOS2 stimulates the release of guanosine diphosphate (GDP) from RAS, promoting the conversion of RAS from the inactive, GDP-bound state to the active, GTP-bound state.9 Although both SOS proteins are highly homologous and ubiquitously expressed, key differences in function have been observed, with SOS1 having a more dominant role over SOS2 in cellular proliferation, lymphopoiesis, inflammation, mitochondrial function, and overall organismal survival.10−14 Additionally, SOS1 was found to be essential for embryonic development, whereas the genetic knockout of SOS2 resulted in no defects in fertility and viability in adult mice.5,15 Notably, dual inactivation of SOS1 and SOS2 resulted in death within 2 weeks of treatment in a tamoxifen-inducible null mutant mouse model.10
As the prevalent SOS paralog implicated in RAS activation, SOS1 is subject to adaptive feedback mechanisms within the KRAS-mitogen-activated protein kinase (KRAS-MAPK) pathway.16,17 SOS1 gain-of-function mutations are present in 1–5% of cancers, including lung adenocarcinomas and endometrial tumors,17−19 and are also prevalent in RASopathies including Noonan’s syndrome (10% incidence) and hereditary gingival fibromatosis.2,20−24 Functional genomic screens have identified cancer cell lines addicted to KRAS signaling that are particularly sensitive to genetic perturbation or pharmacological inhibition of SOS1, which is anticipated to shift the tumor cell equilibrium of KRAS toward the inactive KRAS-GDP form, leading to MAPK pathway inhibition.2,25 Consistent with in vitro observations, the SOS1 inhibitor treatment of preclinical human xenograft models harboring KRAS, SOS1, PTPN11, or EGFR mutations results in MAPK pathway modulation and tumor growth inhibition.26 Of significance, SOS1 inhibition by MRTX0902,27 a potent and selective inhibitor of the SOS1:KRAS protein–protein interaction (PPI), leads to significant antitumor activity when administered in combination with the KRAS G12C inhibitor adagrasib in preclinical KRAS G12C mutant models and is currently being evaluated in a Phase 1/2 clinical trial.28
In contrast to SOS1, the SOS2 paralog is largely viewed as having a minimal role in KRAS-MAPK pathway activation, whereas SOS2–/– studies indicate that SOS2-dependent PI3K signaling plays an important role in mediating KRAS-phosphoinositide 3-kinase (KRAS-PI3K) pathway signaling for the survival of human KRAS mutant colorectal, pancreatic, and lung tumor cells.29,30 Moreover, in vitro data suggest that SOS2 can compensate for a decrease in SOS1 activity. In a recent study, Hofmann and colleagues observed more pronounced decreases in RAS-GTP and pERK levels and enhanced antiproliferative effect in KRAS G12C mutant cells harboring a SOS2 knockout when compared to the parental cell line following treatment with the SOS1 inhibitor BI-3406.26 Thus, inhibition of SOS2:KRAS PPI could be beneficial in downregulating aberrant KRAS-dependent signaling implicated in both cancer and RASopathies.
To date, no inhibitors of SOS2:KRAS PPI have been reported; therefore, we initiated a lead discovery program to identify chemical starting points that bound to SOS2 and also provided accompanying SOS2 X-ray cocrystal structural data that suggested the potential for the hit to be optimized into potent SOS2:KRAS PPI inhibitors.
Several reports have detailed the discovery of SOS1:KRAS PPI activators (1)31 and SOS1:KRAS PPI inhibitors (2–6),25−27,32,33 as shown in Figure 1. Compounds 1–6 bind to SOS1 in the binding site known as “site-A”34 adjacent to the Switch II region of KRAS. Compounds 2–6 share a closely related binding mode and make several productive interactions with the protein: π-stacking with His905, an H bond between the ligand’s aromatic N–H and Asn879, and the chiral α-methyl substituent fills a lipophilic cavity adjacent to Leu901, as seen in Figure 2A. The structures of compounds 2–6 can each be divided into two components, with one component represented by the bolded bonds and atoms and the other by the nonbolded bonds and atoms shown in Figure 1. The nonbolded bonds and atoms drive the binding affinity to SOS1, while the bolded components are responsible for the PPI disruption at the SOS1:KRAS interface. An overlay of the SOS1 X-ray cocrystal structure of MRTX0902 (6, PDB 7UKR) superimposed with the KRAS–SOS1 complex (PDB 6EPL) visualizes the disruption of the interaction between SOS1Tyr884 and KRASArg73 caused by the location of the morpholino group of MRTX0902 (6) protruding into the KRAS binding interface, while the remainder of MRTX0902 (6) is surrounded by the gray surface of the SOS1 binding pocket making productive binding interactions (Figure 2B).
Figure 1.

Summary of the structures of the SOS1:KRAS PPI activators and inhibitors. Bolded atoms and bonds indicate the substituents that prevent the binding of KRAS to SOS1.
Figure 2.
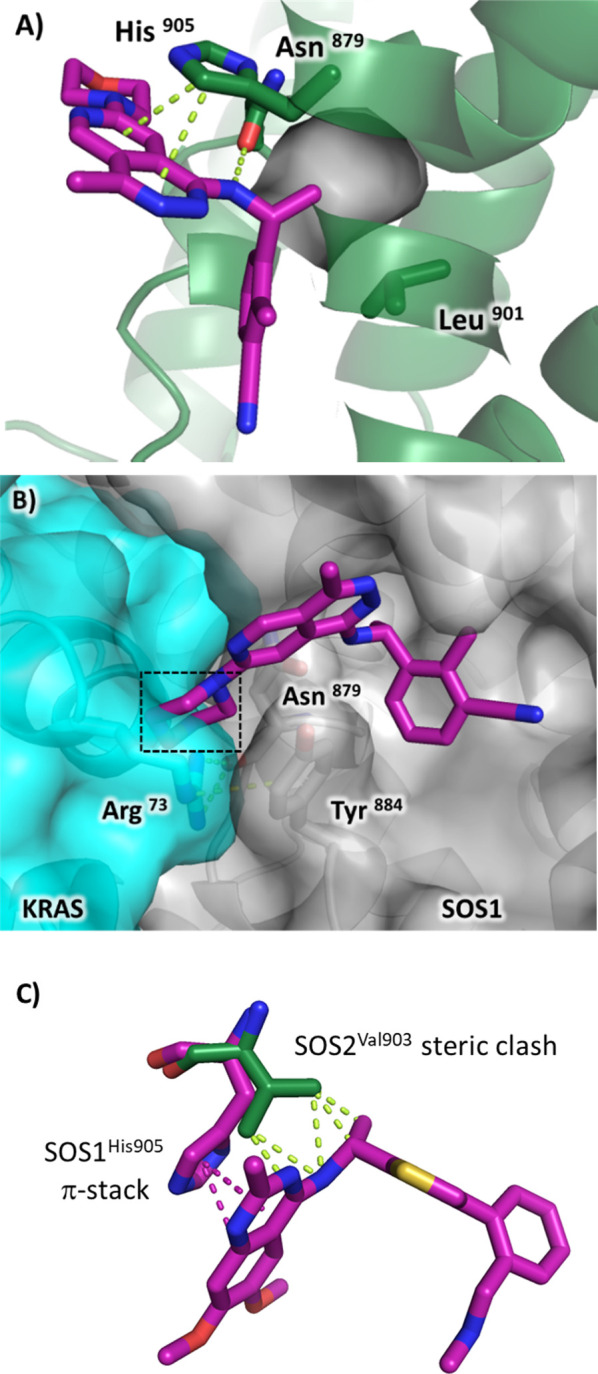
(A) SOS1 X-ray cocrystal structure of MRTX0902 (PDB 7UKR) bound in site-A. (B) SOS1 X-ray crystal structure of MRTX0902 (PDB 7UKR) superimposed with the KRAS–SOS1 complex (PDB 6EPL). (C) Overlay of SOS1/BAY-293 X-ray cocrystal structure (magenta, PDB 5OVI) with apo SOS2 X-ray crystal structure (green, PDB 6EIE).
BAY-293 (2), BI-3406 (3), and MRTX0902 (6) are reported to be highly selective for SOS1 over SOS2. The scientists at Bayer also reported an apo crystal structure of the catalytic domain of SOS2.25 The authors attributed the SOS1 selectivity over SOS2 of BAY-293 (2) to a nonconserved residue, SOS1His905, which is a valine in SOS2 (SOS2Val903), that resulted in removing the productive π-stacking interaction between SOS1His905 and the quinazoline scaffold of BAY-293 (2), shown in Figure 2C. We also reasoned that a SOS2Val903 residue would produce a steric clash between compounds 2 and 6 and prevent binding to SOS2. Considering these observations, we elected not to attempt the design of an SOS2 binder using the SOS1 scaffolds found in compounds 2–6. Instead, we focused on screening libraries against SOS2 to identify new compounds that bound to SOS2 and elected to initiate a SOS2 fragment-based lead discovery (FBLD) program to discover SOS2 fragment hits and characterize the hits using SOS2 X-ray cocrystallography.
FBLD has been extensively reviewed.35−37 In summary, FBLD is the screening of low-molecular-weight compounds (roughly 150–300 Da) in small libraries of approximately 300–15,000 compounds at high concentrations, typically in the range 10 μM–1 mM. After fragment hits are identified, potency is enhanced through fragment-growing strategies to create additional interactions with the protein supported by structure-based drug design (SBDD). AstraZeneca reported a fragment SOS1:HRas X-ray crystallographic screen of 1160 fragments in cocktails of four at 5 mM each. Three binding sites were reported: site-A, the SOS1 binding site where the SOS1 compounds 1–6 bind; site-B, a site on the SOS1:HRas interface; and site-C, a binding site located on HRas.34
Here, we report the fragment-based discovery of five hits with the accompanying SOS2 X-ray cocrystal structures. The fragment hits bind SOS2 at site-1 where the SOS2 site-1 is analogous to SOS1 site-A.
Results and Discussion
We initiated a SOS2 FBLD screening cascade to identify fragment hits with accompanying SOS2 X-ray cocrystal structures. A workflow to discover both dual SOS1/2 binders and selective SOS2 binders was created, and an SOS2 X-ray crystallography platform was developed. To enhance the probability of technical success, three biophysical screening technologies were applied simultaneously into the workflow: an SPR screen of 2187 fragments at 500 μM against SOS1 and SOS2, a target-immobilized NMR screen (TINS)38,39 of 1,068 fragments against SOS1 and SOS2, and third, an X-ray crystallographic screen of 300 fragments at 10 mM using a SOS2 crystal soaking method. SPR and NMR are among the most common screening technologies used for fragment screening, but there are fewer reports using X-ray crystallography screening due to technical challenges associated with the method. Technical challenges include the requirement to have high solubility, typically in the 10–200 mM range, the need for a robust soaking protocol, and time-consuming processes, such as crystal harvesting and data evaluation. Despite these challenges, an X-ray crystallographic screen was included for three reasons: (1) the feasibility of using the SOS family protein in an X-ray crystallographic fragment screen, as evidenced by the recent report from the team at AstraZeneca,34 (2) X-ray crystallographic screening is reported to produce high hit rates,40 and (3) the screening method was potentially the fastest path to identify ligand-bound SOS2 X-ray cocrystal structures and accelerate fragment growth and further elaboration strategies. A summary of the workflow and results is shown in Figure 3.
Figure 3.
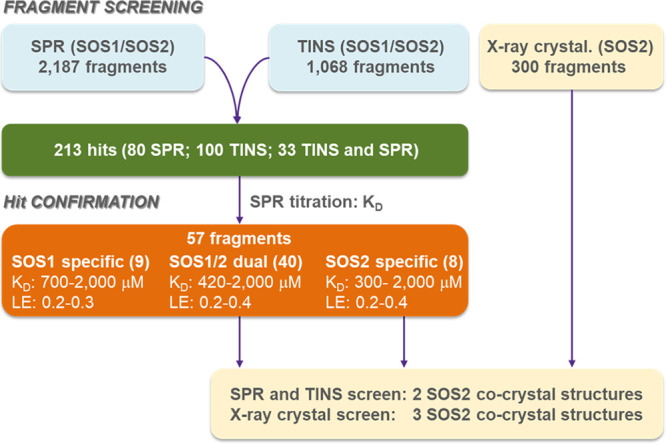
Flow scheme and summary results for the SPR, TINS, and X-ray crystallographic fragment screens.
SPR and TINS Screen
For the SPR screen, SOS1, or SOS2 was immobilized on a neutravidin-coated sensor chip, and the binding of 2187 fragments was investigated at 500 μM. A total of 113 hits were identified (5% hit rate), as defined by the normalized occupancy ≥40% and visual inspection of sensorgrams to select hits displaying fast-on/off kinetics.
The TINS screen was configured to select fragments that preferentially bound to both SOS1 and SOS2. In TINS,38 a Sepharose-based resin was loaded with SOS1 and SOS2 together and a reference protein loaded separately and placed in a dual cell sample holder. Cocktails of 3–5 fragments were injected into both samples simultaneously. Fragments that bound to SOS1 and SOS2 became temporarily immobilized, and the relaxation of the NMR signal was enhanced greater than 3 orders of magnitude, which resulted in a diminution of resonance peaks. Preferential binding to the target over the reference was determined through an examination of the ratio of peak amplitudes in each cell (T/R ratio). Fragments that bound to both SOS1 and SOS2 experienced a twofold higher concentration of protein and were therefore more completely relaxed, resulting in the lowest T/R ratio. A total of 1068 fragments were assayed, and by the analysis of the T/R screen profile (see Supporting Information), 133 fragments with the smallest T/R ratio were selected as hits (12% hit rate).
The combined hits from the SPR and TINS screens totaled 213, with 100 hits from TINS, 80 hits from SPR, and 33 compounds that were hits in both assays. The 213 hits were then submitted for KD determination by SPR; 57 hits were confirmed, as defined by saturable binding to SOS1 or SOS2, a reasonable occupancy in a 25–300% range and sensorgrams displaying fast on/off kinetics. We were encouraged to see that of the 57 confirmed hits, 40 were dual SOS1/SOS2 binders, 8 were SOS2-specific, and 9 were SOS1-specific.
Our experience at the early stage of the project indicated that SOS2 X-ray crystallography was challenging and not amenable to high-throughput methods; therefore, only five hits (compounds 7–11Table 1) were selected for SOS2 X-ray crystallography. The compounds were selected based on a combination of their structural diversity, solubility ≥500 μM, and SOS2 binding potency. Compound 7 was the weakest compound selected, with the SOS2 KD of approximately 2000 μM and nonsaturable binding against SOS1. Compound 8 demonstrated binding to SOS1 and SOS2, with KD values of 960 and 300 μM, respectively, and was the most potent SOS2 binder identified in the screen. Compounds 9 and 10 bound to SOS2 with a KD of 330 and 730 μM, respectively, but nonsaturable binding was detected in the SOS1 assay. SOS1 and SOS2 KDs were successfully determined for compound 11 and were found to be 490 and 430 μM, respectively. It was not clear what was causing the nonsaturable SOS1 binding for compounds 7, 9, and 10 in the SOS1 assay. Therefore, to rule out the compound quality control issues of samples originating from screening libraries, HPLC, MS, and 1H NMR data were collected on compounds 7–11. The compounds were found to be ≥95% pure by HPLC, with the molecular mass and 1H NMR consistent with the structure; thus, we speculated that the SOS1 nonsaturable binding may be caused by nonspecific binding to the immobilized protein. Despite the uncertainty with the SOS1 binding data, the SOS2 sensorgrams looked good; therefore, compounds 7–11 proceeded to SOS2 crystallography.
Table 1. SPR KD's for SPR and TINS Fragment Hits 7–14 Submitted for SOS2 X-Ray Crystallography.

KD is based on n ≥ 2 with SEM within 0.2 units.
NS: nonsaturable binding.
Solubility measured by 1H NMR in DPBS buffer at pH 7.2.
Solubility measured by total light and dynamic light scattering in 1× PBS buffer at pH 7.4.
SOS2 X-Ray Crystallography
Fragment soaking and fragment cocrystallizations with compounds 7–11 were investigated across a range of conditions using the SOS2SB protein construct described by Hillig and colleagues.25 This resulted in the discovery of two SOS2 X-ray cocrystal structures with compounds 9 and 10 (Figures 4A,B). Despite multiple attempts, SOS2 X-ray cocrystal structures were not identified for compounds 7, 8, and 11.
Figure 4.

X-ray cocrystal structure of (A) 9 bound to SOS2 (PDB 8UC9) and (B) 10 bound to SOS2 (PDB 8UH0).
SOS2SB crystals grown in the space group P43, as described by Hillig et al.,25 were soaked with 10 mM of 9 for 15 min to obtain an X-ray cocrystal structure of 9 bound at site-1 (PDB 8UC9), as shown in Figure 4A. Compound 9 made a series of productive interactions with the protein, including a bifurcated H bond between the aromatic −NH2 and the side chain of Asp885 plus the backbone carbonyl of Tyr882, a π-stacking interaction between the aromatic bicyclic scaffold and Phe888, and an H bond between the protonated quinoline aromatic nitrogen and the Asp900 side chain.
An X-ray cocrystal structure of 10 bound at site-1 of SOS2 (PDB 8UH0, Figure 4B) was obtained by cocrystallizing SOS2SB with compound 10 at 9 mM. These conditions identified SOS2SB crystals in the P1 space group, a space group not previously reported. Compound 10 made productive interactions with SOS2, including a π-stacking interaction between the aromatic bicyclic scaffold and Phe888 and a productive H bond between the side chain of Asp900 and the phenolic substituent of 10.
SOS2 X-Ray Crystallography Fragment Screen
Crystallographic fragment screening via crystal soaking requires a large number of apo crystals that are stable under the soaking conditions and that consistently diffract well. Initial efforts to develop a robust soaking protocol with apo crystals in the P43 space group, as described by Hillig et al.,25 were hampered by the crystals either dissolving or cracking under a range of soaking conditions. Fortunately, when crystallographic conditions for compounds 7–11 were investigated, as described earlier, a new apo SOS2SB crystal form was discovered in the P1 space group. These apo SOS2SB crystals in the P1 space group were found to be more robust under soaking conditions than the previously reported P43 space group crystals. After further optimization, a SOS2SB apo structure was solved at 1.6 Å in the P1 space group (PDB 8UF2), and these crystals were selected for use in the SOS2 X-ray crystallographic screen. A total of 300 fragments were individually soaked at 10 mM for 18 h at 8 °C with a final DMSO concentration of 10%. Data collection returned 252 data sets with resolutions in the range of 1.7–2.3 Å. Analysis of the data sets, supported by PanDDA (Pan-Data set Density Analysis),41 revealed three hits bound in the site-1 pocket. The binding poses of the three hits, compounds 12–14, bound to the SOS2SB site-1 pocket, are shown in Figure 5A–C. A summary of the SOS1 and SOS2 KDs determined by SPR using the same procedure previously described is shown in Table 1.
Figure 5.

SOS2 X-ray cocrystal structures of (A) 12 (PDB 8T5G), (B) 13 (PDB 8T5R), and (C) 14 (PDB 8T5M) bound to site-1.
Compound 12 bound in a closely related binding mode to 9, making a bifurcated H bond between the aromatic −NH2 substituent and the side chain of Asp885 and the backbone carbonyl of Tyr882, a π-stacking interaction between the aromatic bicyclic portion of the 1,2,3,4-tetrahydroacridine scaffold and Phe888, and an H bond between the protonated aromatic nitrogen of the tricyclic scaffold and the Asp900 side chain, as shown in Figure 5A. Compound 12 also displayed similar binding KD's to 9, with nonsaturable binding detected for SOS1 and a KD of 550 μM for SOS2.
The SOS2 X-ray cocrystal structure with 13 revealed a bifurcated H bond between the side chain of Asp885 and the backbone carbonyl of Try882 with the basic primary amino group of 13, as shown in Figure 5B. This bifurcated interaction was also observed with compounds 9 and 12, but 13 made the interactions through its basic −NH2 group rather than via an aromatic −NH2 substituent, as observed with 9 and 12. The primary amino sulfonamide group of 13 also made a productive H bond interaction with the side chain of Asn877. In the SPR assay, nonsaturable SOS1 binding was detected for compound 13, and binding to SOS2 was weak with an approximate SOS2 KD of 2000 μM.
Compound 14 made a series of productive interactions with SOS2 including an aryl–aryl edge-to-face interaction with Tyr882, a π-stacking interaction with Phe888, an ionic interaction with Asp885, and an H bond between the protonated basic secondary amine group and the backbone carbonyl of Tyr882 (Figure 5C). From the cohort of five SOS2 X-ray cocrystal structures described herein, 14 made the greatest number of interactions with the protein, and the geometry of the binding mode is notable in that 14 wraps around the Tyr882 side chain. However, nonsaturable binding was observed for 14 against SOS1 and SOS2 in the SPR assay. This was a confounding result, and therefore, to rule out questions of compound purity and integrity, and as described previously for compounds 7–11, HPLC, MS, and 1H NMR analyses were performed on compounds 12–14. All were found to be ≥95% pure by HPLC, with the molecular mass and 1H NMR consistent with the structure. The electron density for 14 bound to SOS2 was clear and unequivocal, thus demonstrating that 14 does indeed bind to SOS2. However, because a binding KD could not be determined in the SPR assay, this observation highlights a key challenge in FBLD, that is, differences in the end points of biophysical assays can generate nonoverlapping results. For example, SPR is a kinetic assay, while the crystallographic assay is a thermodynamic assay, and the top concentration used in the SPR assay was 1 mM, while the concentration used in the crystallographic assay was 10-fold higher at 10 mM. The observations with compound 14 highlight a potential challenge with crystallographic screening, in that a hit may be discovered and its binding mode determined unequivocally, but its binding KD may never be determined, and additional rounds of follow-up and optimization must be pursued at the risk of not being able to detect binding affinities to track the progress and SAR. A summary of data collection statistics and refinement for SOS2 X-ray cocrystal structure data of compounds 9, 10, 12–14 is presented in Tables SI-1 and SI-2.
Higher hit rates are described in the literature for X-ray crystallographic screening compared to other methods, and our data support these observations.42 Two SOS2 X-ray structures were obtained downstream of the SPR and TINS assays with an approximate hit rate of 0.1%, while three X-ray cocrystal structures were obtained from the X-ray crystallographic screen, producing a hit rate of 1%.
Conclusions
Here, we report for the first time liganded SOS2 structures that could form the basis of fragment growth and optimization strategies to discover SOS2:KRAS PPI inhibitors. SOS2–/– studies indicate that SOS2-dependent PI3K signaling plays an important role in mediating KRAS-phosphoinositide 3-kinase (KRAS-PI3K) pathway signaling for the survival of human KRAS mutant colorectal, pancreatic, and lung tumor cells, and in vitro data suggest that SOS2 can compensate for a decrease in SOS1 activity. Thus, inhibitors of SOS2:KRAS PPI could be beneficial in downregulating aberrant KRAS-dependent signaling implicated in both cancer and RASopathies. SOS1:KRAS PPI inhibitors are reported to be inactive against SOS2 due to the differences between several key residues in the SOS2 ligand binding site compared to SOS1. Therefore, we hypothesized that SOS1 compounds 1–6 would not make good starting points to initiate a SOS2 inhibitor program. Using a combination of SPR, TINS, and X-ray crystallographic fragment-based screens, five fragment hits with SOS2SB X-ray cocrystal structures were obtained (9, 10, and 12–14). Four of the hits (9, 10, 12, and 13) displayed SOS2 binding KDs in the range 0.3–2 mM, while SOS2 binding affinity could not be determined for compound 14. Subsequent optimization via X-ray cocrystal structure-supported fragment-growing strategies will be reported in due course.
Experimental Section
All test compounds were purchased from commercial suppliers and used as received, unless otherwise indicated. The purity of test compounds was determined to be ≥95% by high-performance liquid chromatography, and the confirmation of structure was determined by proton NMR and mass spectroscopy, as described below. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker Avance 400 MHz spectrometers. Chemical shifts are expressed in δ ppm and are calibrated to the residual solvent peak: proton. Coupling constants (J), when given, are reported in hertz. Multiplicities are reported using the following abbreviations: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, sxt = sextet, m = multiplet (range of multiplets is given), br = broad signal, and dt = doublet of triplets. The mass spectra and purity were obtained for all compounds by high-performance liquid chromatography–mass spectrometry (LC–MS) on an Agilent 1260 Infinity II instrument using electrospray ionization (ESI). HPLC conditions were as follows: Gemini NX-C18 LC column 4.6 mm × 150 mm, 3 μm, 5–95% ACN (0.0375% TFA) in water (0.01875% TFA), 10 min run, flow rate 1.5 mL/min, UV detection (λ = 220, 254 nm) or Agilent C18 LC column 4.6 mm × 150 mm, 3 μm, 70:30 ACN (0.0375% TFA):water (0.01875% TFA), 20 min run, flow rate 1.5 mL/min, UV detection (λ = 214 nm).
Acknowledgments
The X-ray crystallography work presented herein is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract No. DE-AC02-06CH11357 and the CMCF-ID beamline at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.43
Glossary
Abreviations
- FBLD
fragment-based lead discovery
- GEF
guanine nucleotide exchange factor
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- KRAS
Kirsten rat sarcoma virus
- PanDDA
pan-data set density analysis
- PPI
protein–protein interaction
- RAS
rat sarcoma virus
- RTK
receptor tyrosine kinase
- SOS
Son of Sevenless
- SPR
surface plasmon resonance
- TINS
target-immobilized NMR screen
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c02140.
The Supporting Information is available free of charge on the ACS Publications Web site. NMR spectra and HPLC trace of compounds 7–14; SOS2 protein expression and purification; SPR experimental procedures; TINS experimental procedures; cocrystal structure determination procedures; X-ray crystallographic fragment screen procedure; and X-ray data collection and refinement statistics for SOS2SB crystal structures (PDF)
SMILES data for compounds 7–14 (CSV)
Accession Codes
Atomic coordinates for the SOS2SB X-ray structures of 9 (PDB 8UC9), 10 (PDB 8UH0), apo (PDB 8UF2), 12 (PDB 8T5G), 13 (PDB 8T5R), and 14 (PDB 8T5M).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Baltanas F. C.; Zarich N.; Rojas-Cabaneros J. M.; Santos E. SOS GEFs in health and disease. Biochim. Biophys. Acta Rev. Cancer 2020, 1874 (2), 188445 10.1016/j.bbcan.2020.188445. [DOI] [PubMed] [Google Scholar]
- Jeng H. H.; Taylor L. J.; Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun. 2012, 3, 1168. 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo M. M.; Rana S.; Spencer-Dene B.; Nye E.; Stamp G.; Downward J. Disruption of the interaction of RAS with PI3-Kinase induces regression of EGFR-mutant-driven lung cancer. Cell Rep. 2018, 25 (13), 3545–3553. 10.1016/j.celrep.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chardin P.; Camonis J. H.; Gale N. W.; van Aelst L.; Schlessinger J.; Wigler M. H.; Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 1993, 260 (5112), 1338–1343. 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- Esteban L. M.; Fernandez-Medarde A.; Lopez E.; Yienger K.; Guerrero C.; Ward J. M.; Tessarollo L.; Santos E. Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol. Cell. Biol. 2000, 20 (17), 6410–6413. 10.1128/MCB.20.17.6410-6413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero C.; Rojas J. M.; Chedid M.; Esteban L. M.; Zimonjic D. B.; Popescu N. C.; Font de Mora J.; Santos E. Expression of alternative forms of Ras exchange factors GRF and SOS1 in different human tissues and cell lines. Oncogene 1996, 12 (5), 1097–1107. [PubMed] [Google Scholar]
- Pierre S.; Bats A. S.; Coumoul X. Understanding SOS (Son of Sevenless). Biochem. Pharmacol. 2011, 82 (9), 1049–1056. 10.1016/j.bcp.2011.07.072. [DOI] [PubMed] [Google Scholar]
- Vigil D.; Cherfils J.; Rossman K. L.; Der C. J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy?. Nat. Rev. Cancer 2010, 10 (12), 842–857. 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimnual A.; Bar-Sagi D. The two hats of SOS. Sci. STKE 2002, 145, pe36 10.1126/stke.2002.145.pe36. [DOI] [PubMed] [Google Scholar]
- Baltanas F. C.; Perez-Andres M.; Ginel-Picardo A.; Diaz D.; Jimeno D.; Liceras-Boillos P.; Kortum R. L.; Samelson L. E.; Orfao A.; Santos E. Functional redundancy of Sos1 and Sos2 for lymphopoiesis and organismal homeostasis and survival. Mol. Cell. Biol. 2013, 33 (22), 4562–4578. 10.1128/MCB.01026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Navas R.; Liceras-Boillos P.; Gomez C.; Baltanas F. C.; Calzada N.; Nuevo-Tapioles C.; Cuezva J. M.; Santos E. Critical requirement of SOS1 RAS-GEF function for mitochondrial dynamics, metabolism, and redox homeostasis. Oncogene 2021, 40 (27), 4538–4551. 10.1038/s41388-021-01886-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liceras-Boillos P.; Garcia-Navas R.; Ginel-Picardo A.; Anta B.; Perez-Andres M.; Lillo C.; Gomez C.; Jimeno D.; Fernandez-Medarde A.; Baltanas F. C.; Santos E. Sos1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene 2016, 35 (50), 6389–6402. 10.1038/onc.2016.169. [DOI] [PubMed] [Google Scholar]
- Liceras-Boillos P.; Jimeno D.; Garcia-Navas R.; Lorenzo-Martin L. F.; Menacho-Marquez M.; Segrelles C.; Gomez C.; Calzada N.; Fuentes-Mateos R.; Paramio J. M.; Bustelo X. R.; Baltanas F. C.; Santos E. Differential role of the RasGEFs Sos1 and Sos2 in mouse skin homeostasis and carcinogenesis. Mol. Cell. Biol. 2018, 38 (16), e00049 10.1128/MCB.00049-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suire S.; Baltanas F. C.; Segonds-Pichon A.; Davidson K.; Santos E.; Hawkins P. T.; Stephens L. R. Frontline Science: TNF-α and GM-CSF1 priming augments the role of SOS1/2 in driving activation of Ras, PI3K-γ, and neutrophil proinflammatory responses. J. Leukoc. Biol. 2019, 106 (4), 815–822. 10.1002/JLB.2HI0918-359RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X.; Esteban L.; Vass W. C.; Upadhyaya C.; Papageorge A. G.; Yienger K.; Ward J. M.; Lowy D. R.; Santos E. The Sos1 and Sos2 Ras-specific exchange factors: differences in placental expression and signaling properties. EMBO J. 2000, 19 (4), 642–654. 10.1093/emboj/19.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalan-Garcia S.; Yang S. S.; Degenhardt K. R.; Bar-Sagi D. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol. 1996, 16 (10), 5674–5682. 10.1128/MCB.16.10.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu D. K.; Nissley D. V.; McCormick F. RAS proteins and their regulators in human disease. Cell 2017, 170 (1), 17–33. 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D.; Choi P. S.; Gelbard M.; Meyerson M. Identification and characterization of oncogenic SOS1 mutations in lung adenocarcinoma. Mol. Cancer Res. 2019, 17 (4), 1002–1012. 10.1158/1541-7786.MCR-18-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F.; Mina M.; Armenia J.; Chatila W. K.; Luna A.; La K. C.; Dimitriadoy S.; Liu D. L.; Kantheti H. S.; Saghafinia S.; Chakravarty D.; Daian F.; Gao Q.; Bailey M. H.; Liang W. W.; Foltz S. M.; Shmulevich I.; Ding L.; Heins Z.; Ochoa A.; Gross B.; Gao J.; Zhang H.; Kundra R.; Kandoth C.; Bahceci I.; Dervishi L.; Dogrusoz U.; Zhou W.; Shen H.; Laird P. W.; Way G. P.; Greene C. S.; Liang H.; Xiao Y.; Wang C.; Iavarone A.; Berger A. H.; Bivona T. G.; Lazar A. J.; Hammer G. D.; Giordano T.; Kwong L. N.; McArthur G.; Huang C.; Tward A. D.; Frederick M. J.; McCormick F.; Meyerson M.; Van Allen E. M.; Cherniack A. D.; Ciriello G.; Sander C.; Schultz N.; Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173 (2), 321–337. 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeddu V.; Yin J. C.; Gunnarsson C.; Virtanen C.; Drunat S.; Lepri F.; De Luca A.; Rossi C.; Ciolfi A.; Pugh T. J.; Bruselles A.; Priest J. R.; Pennacchio L. A.; Lu Z.; Danesh A.; Quevedo R.; Hamid A.; Martinelli S.; Pantaleoni F.; Gnazzo M.; Daniele P.; Lissewski C.; Bocchinfuso G.; Stella L.; Odent S.; Philip N.; Faivre L.; Vlckova M.; Seemanova E.; Digilio C.; Zenker M.; Zampino G.; Verloes A.; Dallapiccola B.; Roberts A. E.; Cave H.; Gelb B. D.; Neel B. G.; Tartaglia M. Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan Syndrome. Hum. Mutat. 2015, 36 (11), 1080–1087. 10.1002/humu.22834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T. C.; Zhang Y.; Gorry M. C.; Hart P. S.; Cooper M.; Marazita M. L.; Marks J. M.; Cortelli J. R.; Pallos D. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am. J. Hum. Genet. 2002, 70 (4), 943–954. 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. E.; Araki T.; Swanson K. D.; Montgomery K. T.; Schiripo T. A.; Joshi V. A.; Li L.; Yassin Y.; Tamburino A. M.; Neel B. G.; Kucherlapati R. S. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 2007, 39 (1), 70–74. 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- Swanson K. D.; Winter J. M.; Reis M.; Bentires-Alj M.; Greulich H.; Grewal R.; Hruban R. H.; Yeo C. J.; Yassin Y.; Iartchouk O.; Montgomery K.; Whitman S. P.; Caligiuri M. A.; Loh M. L.; Gilliland D. G.; Look A. T.; Kucherlapati R.; Kern S. E.; Meyerson M.; Neel B. G. SOS1 mutations are rare in human malignancies: implications for Noonan Syndrome patients. Genes Chromosomes Cancer 2008, 47 (3), 253–259. 10.1002/gcc.20527. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Pennacchio L. A.; Zhao C.; Yadav K. K.; Fodale V.; Sarkozy A.; Pandit B.; Oishi K.; Martinelli S.; Schackwitz W.; Ustaszewska A.; Martin J.; Bristow J.; Carta C.; Lepri F.; Neri C.; Vasta I.; Gibson K.; Curry C. J.; Siguero J. P.; Digilio M. C.; Zampino G.; Dallapiccola B.; Bar-Sagi D.; Gelb B. D. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007, 39 (1), 75–79. 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- Hillig R. C.; Sautier B.; Schroeder J.; Moosmayer D.; Hilpmann A.; Stegmann C. M.; Werbeck N. D.; Briem H.; Boemer U.; Weiske J.; Badock V.; Mastouri J.; Petersen K.; Siemeister G.; Kahmann J. D.; Wegener D.; Bohnke N.; Eis K.; Graham K.; Wortmann L.; von Nussbaum F.; Bader B. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (7), 2551–2560. 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann M. H.; Gmachl M.; Ramharter J.; Savarese F.; Gerlach D.; Marszalek J. R.; Sanderson M. P.; Kessler D.; Trapani F.; Arnhof H.; Rumpel K.; Botesteanu D. A.; Ettmayer P.; Gerstberger T.; Kofink C.; Wunberg T.; Zoephel A.; Fu S. C.; Teh J. L.; Bottcher J.; Pototschnig N.; Schachinger F.; Schipany K.; Lieb S.; Vellano C. P.; O’Connell J. C.; Mendes R. L.; Moll J.; Petronczki M.; Heffernan T. P.; Pearson M.; McConnell D. B.; Kraut N. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discovery 2021, 11 (1), 142–157. 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketcham J. M.; Haling J.; Khare S.; Bowcut V.; Briere D. M.; Burns A. C.; Gunn R. J.; Ivetac A.; Kuehler J.; Kulyk S.; Laguer J.; Lawson J. D.; Moya K.; Nguyen N.; Rahbaek L.; Saechao B.; Smith C. R.; Sudhakar N.; Thomas N. C.; Vegar L.; Vanderpool D.; Wang X.; Yan L.; Olson P.; Christensen J. G.; Marx M. A. Design and discovery of MRTX0902, a potent, selective, brain-penetrant, and orally bioavailable inhibitor of the SOS1:KRAS protein-protein interaction. J. Med. Chem. 2022, 65 (14), 9678–9690. 10.1021/acs.jmedchem.2c00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A phase 1/2 study of MRTX0902 in solid tumors with mutations in the KRAS MAPK pathway. https://clinicaltrials.gov/ct2/show/NCT05578092.
- Sheffels E.; Sealover N. E.; Theard P. L.; Kortum R. L. Anchorage-independent growth conditions reveal a differential SOS2 dependence for transformation and survival in RAS-mutant cancer cells. Small GTPases 2021, 12 (1), 67–78. 10.1080/21541248.2019.1611168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffels E.; Sealover N. E.; Wang C.; Kim D. H.; Vazirani I. A.; Lee E.; Terrell E. M.; Morrison D. K.; Luo J.; Kortum R. L. Oncogenic RAS isoforms show a hierarchical requirement for the guanine nucleotide exchange factor SOS2 to mediate cell transformation. Sci. Signal. 2018, 11 (546), eaar8371 10.1126/scisignal.aar8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. C.; Sun Q.; Daniels R. N.; Camper D.; Kennedy J. P.; Phan J.; Olejniczak E. T.; Lee T.; Waterson A. G.; Rossanese O. W.; Fesik S. W. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (9), 3401–3406. 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckl A.; Quintana E.; Lee G. J.; Shifrin N.; Zhong M.; Garrenton L. S.; Montgomery D. C.; Stahlhut C.; Zhao F.; Whalen D. M.; Thompson S. K.; Tambo-ong A.; Gliedt M.; Knox J. E.; Cregg J. J.; Aay N.; Choi J.; Nguyen B.; Tripathi A.; Zhao R.; Saldajeno-Concar M.; Marquez A.; Hsieh D.; McDowell L. L.; Koltun E. S.; Bermingham A.; Wildes D.; Singh M.; Wang Z.; Hansen R.; Smith J. A.; Gill A. L.. Discovery of a potent, selective, and orally bioavailable SOS1 inhibitor, RMC-023, an in vivo tool compound that blocks RAS activation via disruption of the RAS-SOS1 interaction, Proceedings of the American Association for Cancer Research Annual Meeting 2022; Cancer Research, 2021; p 1273.
- He H.; Zhang Y.; Xu J.; Li Y.; Fang H.; Liu Y.; Zhang S. Discovery of orally bioavailable SOS1 inhibitors for suppressing KRAS-driven carcinoma. J. Med. Chem. 2022, 65 (19), 13158–13171. 10.1021/acs.jmedchem.2c00986. [DOI] [PubMed] [Google Scholar]
- Winter J. J.; Anderson M.; Blades K.; Brassington C.; Breeze A. L.; Chresta C.; Embrey K.; Fairley G.; Faulder P.; Finlay M. R.; Kettle J. G.; Nowak T.; Overman R.; Patel S. J.; Perkins P.; Spadola L.; Tart J.; Tucker J. A.; Wrigley G. Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. J. Med. Chem. 2015, 58 (5), 2265–2274. 10.1021/jm501660t. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. A ‘rule of three’ for fragment-based lead discovery?. Drug Discovery Today 2003, 8 (19), 876–877. 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Fesik S. W.; Hubbard R. E.; Jahnke W.; Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discovery 2016, 15 (9), 605–619. 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M.; Burrows J. N.; Griffen E. J. Fragment screening: an introduction. Mol. Biosyst. 2006, 2 (9), 430–446. 10.1039/b610069b. [DOI] [PubMed] [Google Scholar]
- Vanwetswinkel S.; Heetebrij R. J.; van Duynhoven J.; Hollander J. G.; Filippov D. V.; Hajduk P. J.; Siegal G. TINS, target immobilized NMR screening: an efficient and sensitive method for ligand discovery. Chem. Biol. 2005, 12 (2), 207–216. 10.1016/j.chembiol.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Marquardsen T.; Hofmann M.; Hollander J. G.; Loch C. M.; Kiihne S. R.; Engelke F.; Siegal G. Development of a dual cell, flow-injection sample holder, and NMR probe for comparative ligand-binding studies. J. Magn. Reson. 2006, 182 (1), 55–65. 10.1016/j.jmr.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Schiebel J.; Radeva N.; Krimmer S. G.; Wang X.; Stieler M.; Ehrmann F. R.; Fu K.; Metz A.; Huschmann F. U.; Weiss M. S.; Mueller U.; Heine A.; Klebe G. Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: a case study. ACS Chem. Biol. 2016, 11 (6), 1693–1701. 10.1021/acschembio.5b01034. [DOI] [PubMed] [Google Scholar]
- Pearce N. M.; Krojer T.; Bradley A. R.; Collins P.; Nowak R. P.; Talon R.; Marsden B. D.; Kelm S.; Shi J.; Deane C. M.; von Delft F. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123 10.1038/ncomms15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn M. J.; Murray C. W.; Cleasby A.; Frederickson M.; Tickle I. J.; Jhoti H. Fragment-based lead discovery using X-ray crystallography. J. Med. Chem. 2005, 48 (2), 403–413. 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]
- Grochulski P.; Fodje M. N.; Gorin J.; Labiuk S. L.; Berg R. Beamline 08ID-1, the prime beamline of the Canadian Macromolecular Crystallography Facility. J. Synchrotron Radiat. 2011, 18 (Pt 4), 681–684. 10.1107/S0909049511019431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.