

Abstract
The role of different cell types and their interactions in Alzheimer’s disease (AD) is a complex and open question. Here, we pursued this question by assembling a high-resolution cellular map of the aging frontal cortex using single-nucleus RNA sequencing of 24 individuals with a range of clinicopa thologic characteristics. We used this map to infer the neocortical cellular architecture of 638 individuals profiled by bulk RNA sequencing, providing the sample size necessary for identifying statistically robust associations. We uncovered diverse cell populations associated with AD, including a somatostatin inhibitory neuronal subtype and oligodendroglial states. We further identified a network of multicellular communities, each composed of coordinated subpopulations of neuronal, glial and endothelial cells, and we found that two of these communities are altered in AD. Finally, we used mediation analyses to prioritize cellular changes that might contribute to cognitive decline. Thus, our deconstruction of the aging neocortex provides a roadmap for evaluating the cellular microenvironments underlying AD and dementia.
Over the past decade, our understanding of the molecular landscape of AD has advanced as new experimental and analytic methods have synergized to uncover the sequence of events that lead to AD dementia. While transcriptomic analyses have the power to capture the state of the target organ along disease progression, most previous large-scale efforts profiled RNA at the bulk tissue level (for example, refs. 1,2), averaging expression measures across a diversity of cells, which obscured finer distinctions and contributions of each cell subtype. Recent studies that profiled single nuclei from brain tissue of healthy and AD individuals have uncovered large interindividual diversity and specific glial subsets as well as RNA signatures associated with AD3–7. However, limited sample size and the moderate number of nuclei per subject in such studies yields an incomplete picture of the architecture of the aging neocortex and limits the statistical power of association analyses. In addition, while most studies focus on a cell-intrinsic view, cellular interactions and dependencies between cell types in the brain remain under-explored but could play crucial roles in disease pathology.
Several key questions remain open: (1) How distinct is the cellular architecture of AD brains? (2) Are transcriptional changes in certain cell types coordinated with or independent of one another? (3) How do changes in the cellular architecture relate to the causal chain of events leading to AD? Addressing these questions currently requires several new tools, including methods to relate detailed maps of single-nucleus profiles from selected individuals to large cohorts of deeply pheno-typed individuals with sufficient size to complete statistically robust association studies, as well as approaches that characterize both individual cells and multicellular communities.
Here, we have deployed a combined approach that integrates single-nucleus RNA sequencing (snRNA-seq) profiling of the dorso-lateral prefrontal cortex (DLPFC) tissue from a structured subgroup of 24 well-characterized individuals, together with bulk RNA profiles of a statistically well-powered set of 638 individuals1. All individuals are participants in longitudinal studies of cognitive aging with prospective autopsy and structured cognitive and neuropathologic assessments (the Religious Orders Study (ROS) or the Memory and Aging Project (MAP))8–11. The 24 participants were chosen to capture neocortical cellular diversity across a range of clinicopathologic states, while the 638 participants reflect the general distribution of characteristics seen in the older population, enabling robust statistical modeling (Fig. 1a). Our experimental strategy yielded an enhanced map of cell subtypes and cell states of the aging cortex3–5, which we extended with a computational approach, CelMod (Cellular Landscape Modeling by Deconvolution), to estimate proportions of cell populations across the 638 bulk RNA sequencing (RNA-seq) profiles. The 638 samples provided the statistical power to identify specific subpopulations of cells associated with the pathophysiology of AD, highlighting certain oligodendrocyte transcriptional programs and a relative decrease in somatostatin (SST) neurons. We also used our map to infer a network of coordinated multicellular communities across individuals, which may reflect microenvironments in the aging brain; two of these communities are anti-correlated and associated with both cognitive decline and tau pathology burden. Our model informs further investigations and therapeutic development by identifying those cellular factors that may most proximally and directly contribute to loss of cognitive function with advancing age and AD.
Fig. 1 |. A cellular–molecular map of the human aging DLPFC in 24 cognitively healthy and AD individuals.

a, Overview of the experimental scheme and analysis plan. Twenty-four individuals with clinicopathologic characteristics were profiled by snRNA-seq to generate a cellular map of the aging DLPFC brain region, and used as input to our CelMod deconvolution algorithm to estimate cellular compositions in an independent set of 638 individuals with bulk RNA-seq data. Network analysis uncovered cellular communities and cell subsets varying in a coordinated manner across individuals, and statistical modeling associated AD traits to cell subsets and to cellular communities. b, The 172,659 nuclear cDNA libraries were generated from the 24 postmortem samples of the DLPFC brain region of aging individuals. We report the number of cell profiles for each individual, ordered by the four major archetypes of the aging population: reference (nonimpaired individuals with minimal AD pathology), resilient (cognitively nonimpaired with a pathologic diagnosis of AD), AD group (both clinical and pathologic AD) and clinical AD only (AD dementia with minimal AD pathology). c, UMAP embedding of 172,659 single-nucleus RNA profiles from the DLPFC brain region of 24 individuals; colored by cell type. d, Diversity of cell type proportions across individuals. The proportions of cell types, color coded as in c, for each individual (rows). Exc., excitatory; Inh., inhibitory; OPC, oligodendrocyte precursor cells.
Results
A high-resolution cell atlas of the aging neocortex
To build a map of the aging DLPFC (BA9), we generated snRNA-seq profiles from 24 aged ROSMAP9,10 participants with deep cognitive and neuropathologic characterization (Supplementary Table 1). To sample a wide variety of cell states, we selected participants that represent four major archetypes of the aging population with diverse pathological and clinical manifestations (Fig. 1a and Supplementary Table 1; 6 individuals per group, 50% men/women): two groups of cognitively nonimpaired individuals with either minimal (reference group) or high (resilient) AD pathology, and two groups of individuals diagnosed with AD dementia with either high (AD group) or minimal AD pathology (clinical AD only). We retained 172,659 DLPFC nuclear transcriptomes for analysis, with an average of 7,194 nuclei per participant after quality control (Fig. 1b, Extended Data Fig. 1a and Methods).
Unsupervised clustering identified distinct groups of nuclei spanning all eight major expected cell types (Fig. 1c,d, Extended Data Fig. 1 and Supplementary Table 2), which we further analyzed separately, identifying finer distinctions in cellular diversity (Figs. 2 and 3, Extended Data Figs. 2 and 3 and Supplementary Tables 2 and 3).
Fig. 2 |. Diversity of neuronal subtypes across layers in the aging DLPFC.
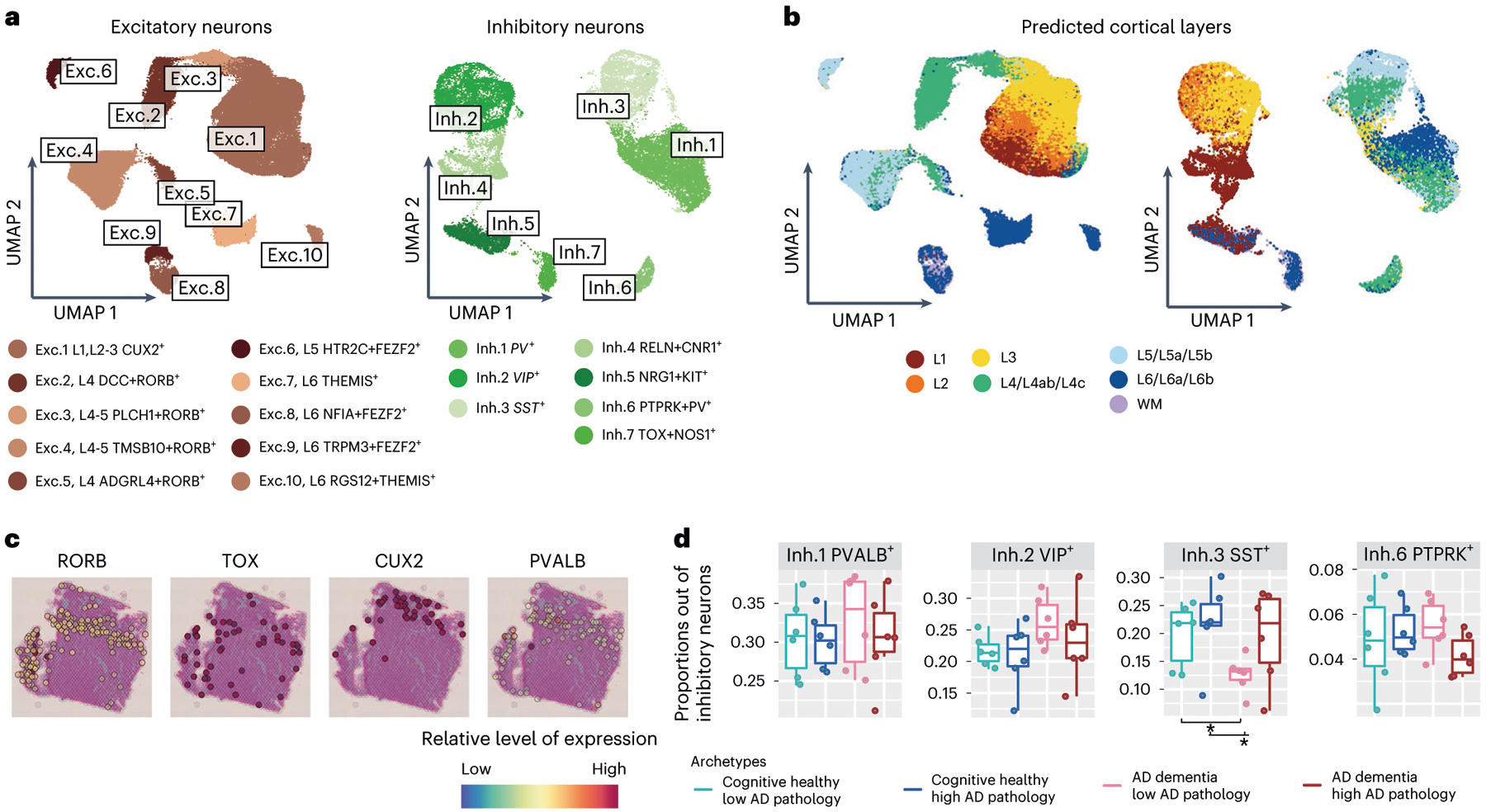
a, Neuronal diversity in the DLPFC. Left, UMAP embedding of excitatory (74,999 cells, 10 clusters) and, right, inhibitory neuronal subtypes (24,938 cells, 7 clusters), colored by clusters capturing distinct subtypes (n = 24 independent samples of men and women), annotated by known neuronal markers (Extended Data Fig. 2a) and mapped to previous annotations12. b, Neuronal diversity in the DLPFC includes neurons from the various cortical layers. UMAPs of neuronal subtypes colored by predicted cortical layers according to a classifier applied to annotated RNA profiles from the Allen Brain Atlas12 (Methods). c, Spatial transcriptomics using the Visium platform highlights layers in DLPFC slices using cortical neuronal markers (RORB, CUX2, TOX, PVALB). d, Proportion of SST GABAergic neuronal subtype varies in relation to cognitive decline. Box plots of the proportions of three GABAergic subtypes (out of total GABAergic neurons) are shown across the four major archetypes of the aging population: reference group, resilient, clinical and pathological AD, and clinical AD only (n = 24 independent samples, 6 per group). For box plots, the bottom and upper borders show the first and third quartiles. The central line indicates the median. The whiskers are extended to the extrema values (without accounting for outliers). Dots show individual samples.
Fig. 3 |. Diverse cell states of glial and endothelial cells in the aging DLPFC.

a,d,h, Diversity of non-neuronal cell states in the aging DLPFC. UMAP embedding of microglia with 2,837 nuclei (a), astrocytes with 29,486 nuclei (d) and endothelia with 2,296 nuclei (h), colored by clusters capturing distinct cell states. b,e, Dot plots of the z-scored mean expression level in expressing nuclei (color) and percentage of expressing nuclei (circle size) of selected marker genes (columns) across microglial (b) or astrocyte (e) subsets (rows). c,f,i, Enriched pathways (hypergeometric test, FDR-adjusted p-value < 0.05, blue color) in up-regulated gene signatures for each subset of microglial (c), astrocyte (f) or endothelial (i) nuclei (columns). g, Spatial transcriptomics using the Visium platform showing the position of Ast.3 cells by marker gene ID3 and reactive astrocytes marker GFAP. Oligodendrocyte marker MBP marks the white matter border. Bottom right, schematic annotation of the tissue. Additional tissues and genes are shown in Extended Data Fig. 5. j, Continuum of expression programs in oligodendrocyte cells inferred by topic modeling19–22. For each topic model panel: UMAP embedding of oligodendrocyte cells, colored by the weight of each topic per cell (right); the top scoring genes (colored by the score), computed as the Kullback–Leibler (KL) divergence between the expression level and the topic’s weight across cells (red color scale, left); and the cumulative distribution function of topic weights for cells split by the sample of origin to four major archetypes of the aging population (as in Fig. 2d).
Among neurons, we identified ten excitatory (glutamatergic) and seven inhibitory (GABAergic) subsets (74,999 and 24,938 nuclei, respectively; Fig. 2a and Extended Data Fig. 2a–c), capturing the diversity of the neocortex. Each subset expressed unique marker genes, such as the known inhibitory subtype markers SST (Inh.3 cluster) or parvalbumin (PVALB) (Inh.1) (Extended Data Fig. 2a). Our clusters aligned with previously annotated neuronal subtypes12 (Methods and Fig. 2a) found in different cortical layers (Fig. 2b), which we confirmed by spatial transcriptomics (Fig. 2c and Extended Data Fig. 2d). Notably, the relative proportions of the SST (Inh.3) neuronal cells were nominally lower in the AD group of individuals (Fig. 2d), although the sample size of our single-nucleus data was underpowered for statistical analysis.
Glial and endothelial cell states in the aging neocortex
The 2,837 microglial nuclei were partitioned into five major subsets (Fig. 3a–c and Extended Data Fig. 3), and they mapped clearly to previously defined subsets derived from single-cell RNA-seq of live human microglia from the DLPFC7 (Extended Data Fig. 4a). Subsets were annotated as: surveilling (Mic.1), stress response/anti-inflammatory (Mic.2), enhanced redox (Mic.3), interferon-response (Mic.4) and proliferative (Mic.5) microglia. Mic.2 aligned with two different reactive states seen in the living microglia7 (Extended Data Fig. 4a), probably due to the smaller number of captured nuclei (compared with cells). However, our snRNA-seq data identified all of the RNA signatures (subsets) present in live sorted microglia, suggesting that neither isolation protocol (sorting of live cells or preparation of nucleus suspensions from frozen tissue) misses microglial states. Finally, despite previously reported differences in microglial RNA profiles13,14, the nucleus-derived data captured expected microglial markers (for example, SPP1, APOE, TMEM163; Fig. 3b).
The astrocytes (29,486 nuclei) included five major subsets (Fig. 3d–f and Extended Data Fig. 3), annotated as: homeostatic protoplasmic-like (Ast.1), nonhomeostatic (Ast.2; reactive markers GFAP, SERPINA3 (ref. 15)), interlaminar-like astrocytes (Ast.3; reactive markers GFAP, ID3 (ref. 15), meningeal/fibrous marker CD44 (ref. 16) and endfeet marker AQP4 (ref. 17)), stress (Ast.4; S100A6, MT1A) and interferon-responding (Ast.5; IFI44L, IFI6, detected in two individuals) (Fig. 3e,f). We validated the position of Ast.3 in proximity to the meninges by spatial transcriptomics (Fig. 3g and Extended Data Fig. 5). Notably, Ast.4 showed expression of genes previously proposed to have higher levels in AD in the human cortex (COL5A3, PDE4DIP (ref. 6), Extended Data Fig. 4b) and this population is enriched for AD genes (Q < 0.05, hypergeometric test; Fig. 3f). Ast.2 showed high expression of genes suggested to be reduced in AD (SERPINA3, Extended Data Fig. 4b). While both Ast.2 and Ast.3 expressed the reactive marker GFAP (Fig. 3e), they each contained genes involved in distinct pathways; for example, TGF-β signaling in Ast.2 and amyloid fibril formation in Ast.3 (Q < 0.05; Fig. 3f).
Endothelial cells (2,296 nuclei) included six major subsets, matching recently defined subsets18 (Fig. 3h and Extended Data Fig. 3e). They were annotated as: capillary (End.1, End.2 and End.3), venous (End.4), arterial (End.5) and smooth muscle (End.6) cells. Each subset of nuclei was associated with distinct pathways (Q < 0.05, hypergeometric test; Fig. 3i), specifically, End.2 with various stress response pathways and AD (Q < 0.05; Fig. 3i).
As oligodendrocytes (29,543 nuclei) exhibited gradients of expression without clearly discrete boundaries, we applied topic modeling19–22 to recover gene programs (called topics) based on covariation patterns of gene expression across cells (Methods). We found four major topics in oligodendrocyte nuclei, annotated by highly scoring genes (based on the Kullback–Leibler divergence21; Fig. 3j). For example, the cellular adhesion protein (SVEP1 (ref. 23)) was highly weighted in topic Oli.1, while a susceptibility gene for late-onset AD—clusterin (CLU)24—was highly weighted in Oli.4. Further, Oli.4 and Oli.2 topics included genes (for example, QDPR) previously reported to have higher expression in cortical oligodendrocytes in AD6, while Oli.1 and Oli.3 included genes (for example, MOG) previously reported to have lower expression in AD (Extended Data Fig. 4b). Finally, oligodendrocyte programs are distributed differently among the four archetype groups of individuals (Fig. 3j); however, the small sample size hinders robust statistical evaluation of this observation.
We could not detect robust subdivisions within oligodendrocyte precursor cells and pericytes due to their small numbers. Overall, the cell population structure that we have defined in our data is consistent with earlier clustering of frozen nuclei and live cells3,4,6,25 (Extended Data Fig. 4c).
Inferred cortical cellular compositions in 638 individuals
Our 24 archetypal participants profiled by snRNA-seq cannot by themselves capture the substantial heterogeneity in the clinicopathologic characteristics of aging individuals, nor is their number sufficient for robust statistical associations. To overcome these limitations, we used the snRNA-seq census of the cellular population structure to infer cell population proportions in bulk DLPFC RNA profiles from 638 ROSMAP participants1 (our 24 participants have the same bulk RNA profile; Supplementary Table 1). We developed and applied our CelMod method, which builds and validates a model of cell subtype and state proportions from participants with both snRNA-seq and bulk RNA-seq from the same tissue (Fig. 4a). CelMod then infers the population structure in any bulk RNA profile. CelMod relies on a consensus of gene-wise regression models, with cross-validation to estimate accuracy (Methods and Fig. 4a). CelMod is sensitive enough to detect the proportions of cell subsets (subtypes and states) within each broad cell class, and, beyond discrete cell populations, it can also determine the relative contributions of continuous expression programs (for example, topics). We applied CelMod at the broad cell class level and separately on subtypes within each broad class (Fig. 4a), inferring relative proportions of each of the 33 cell subsets and four oligodendrocyte topics in bulk RNA-seq from the 638 ROSMAP participants with bulk RNA-seq data1 (Supplementary Table 4).
Fig. 4 |. Proportions of cell subsets are associated with AD traits in a cohort of 638 individuals.

a, Scheme of the CelMod algorithm. Input: snRNA-seq-derived signatures of cell types and subsets and expression programs, as well as their proportions across individuals. A two-step algorithm estimates cell subset proportions in bulk RNA-seq samples, training on matching samples using a fivefold cross-validation approach (Methods). b, CelMod estimated cell subset proportions (y axis) match snRNA-seq measured proportions (x axis) (n = 24 independent samples; additional subsets and cell types in Extended Data Fig. 6a,b). R, Spearman correlation. c, Spearman correlations of the CelMod estimated proportions and the snRNA-seq measured proportions for each cell subset (n = 24 individuals). d, Validations of CelMod in an independent dataset. Correlations of CelMod estimated proportions and snRNA-seq from a published dataset are shown3. e, Immunohistochemistry in DLPFC sections of 48 individuals (24 healthy, 24 with cognitive decline), stained for markers for neurons (anti-NeuN, top) and reactive astrocytes (anti-GFAP, bottom). Left, representative immunofluorescence images. DAPI, nuclei. Scale bar, 100 μm. Right, Pearson correlation coefficients of CelMod and immunofluorescence-based estimations of proportions (out of the total number of cells) for all neurons and for GFAP+ astrocytes (Ast.2, Ast.3). f, Correlations of bulk cortica protein expression levels to CelMod estimates and to bulk RNA-seq in n = 196 individuals30. g–i, Association scores for the CelMod estimated proportions of all cell subsets (cell subtypes, states or topic models) to cognitive decline rate (g, x axis), tangle burden (h, x axis) and β-amyloid burden (i, x axis). Association score = −log(FDR) × sign(β), from multivariable linear regression analysis (Methods; n = 638 independent samples). Positively (purple) or negatively (turquoise) associated subsets are colored when statistically significant (FDR < 0.01). j, Correlation (color scale) of proportions of cell subsets from snRNA-seq to cognitive decline (n = 24 independent samples). Associations to additional AD traits in Extended Data Fig. 6g. k, Correlation (color scale) of protein levels (rows) to rate of cognitive decline, β-amyloid burden and tangle burden measured in n = 400 individuals. **FDR < 0.01. Left bar, direction of association of the CelMod estimated proportion with the traits (purple, positive; turquoise, negative).
CelMod accurately inferred proportions of most snRNA-seq-derived cell types, cell subsets and programs (topics) as validated by fivefold cross-validation to the matched, empirically measured proportions from snRNA-seq (mean r = 0.90, stdev = 0.05 across cell types and mean r = 0.86, stdev = 0.09 across cell subsets; Fig. 4b,c, Extended Data Fig. 6a,b and Methods). We also validated CelMod prediction accuracy by applying it to bulk data from pre-frontal cortex (BA10) of 48 donors, and comparing the estimates with previously published snRNA-seq-derived proportions from the same donors3; this showed that CelMod could accurately predict cell subtype and state proportions on independent datasets and expand the resolution of the published annotations (Fig. 4d, Extended Data Fig. 6c,d and Methods). CelMod showed higher accuracy in predicting snRNA-seq-derived cluster proportions compared with previous methods26–28 (Methods and Extended Data Fig. 6e). There was also agreement in the proportions inferred by CelMod and matched immunofluorescence data (from the contralateral fixed hemisphere29) in 48 ROSMAP participants (Methods), for neurons (NeuN+, r = 0.40) and reactive astrocytes (reactive marker GFAP+, r = 0.49) (Fig. 4e). We further validated the estimated CelMod proportions from matching proteomics measurements of key markers across 196 individuals (with bulk proteomics30 from the DLPFC), focusing on key cell subsets and topics (Methods, Fig. 4f and Extended Data Fig. 6d). For selected cell subsets, we confirmed the correlation of protein levels of marker genes across 196 individuals with both bulk RNA and protein data, showing correlations between the CelMod estimated proportions and the protein levels of the selected marker: the IGFBP7 protein for Ast.2, CD44 for Ast3, S100A6 for Ast.4, QDPR for Oli.4, SST for Inh.3 (R = 0.18–0.3). These correlations were in the same range as the correlations between bulk RNA and protein levels31 for these markers (R = 0.15–0.35; Fig. 4f). Of note, some cell subsets had lower prediction accuracy, including the rare Ast.5 enriched for interferon response and the redox-associated Mic.3 (Fig. 4b–f). Nonetheless, we include all inferred subsets in the next steps of the analysis, as poor inference due to noise would be unlikely to create false positive associations.
Cellular compositions are associated with AD-related traits
With this validated set of inferred cell subset proportions in 638 individuals, we tested for association of cell subset proportions with three central AD-related traits from the deep ante- and post-mortem characterization of ROSMAP participants8–10: quantitative measures of (1) β-amyloid and (2) tau proteinopathy, which are the two defining pathologic characteristics of AD; as well as (3) the slope of aging-related cognitive decline over up to 20 yr before death, which captures the progressive cognitive impairment that leads to dementia. β-amyloid generally accumulates earlier than tau proteinopathy, and tau burden is more closely associated with cognitive impairment and dementia32,33 (Extended Data Fig. 6d).
Linear regression analysis (with correction for age, RNA integrity (RIN) score and sex; Methods) revealed statistically significant associations (false discovery rate (FDR) < 0.01) between the proportions of neuronal and glial cell subsets (as measured within each cell class) and the AD-related traits (Fig. 4g–i and Supplementary Table 5). Most cell subsets/topics associated with tau pathology also showed significant associations with cognitive decline (FDR < 0.01; Fig. 4g): a positive association for Oli.4, Ast.4, Mic.3 and End.2; and a negative association for Oli.1, Inh.3 (SST+), End.4 and Ast.2 (GFAP+ SERPINA3+). A few subsets were only associated with cognitive decline: End.1 with slow decline and Inh.1/7 with rapid decline (FDR < 0.01). On the other hand, Oli.2 was positively associated with tau pathology (FDR < 0.01) and only marginally associated with cognitive decline (FDR < 0.036). Further, proportions of several cell subsets were significantly associated with β-amyloid pathology (FDR < 0.01), but not significantly associated with the rate of cognitive decline: a negative association for glutamatergic neuron subtypes Exc.4/5/6 (layer 4–5 pyramidal neurons), inhibitory neuron subtype Inh.6 (PTPRK+, inferred to be found predominantly in layer 4) and an endothelial subset (End.3); as well as positive associations with Ast.3 (GFAP+ CD44+), Exc.2 and Inh.2 (Fig. 4i). These findings are consistent with the stronger association between tau (versus β-amyloid) pathology and cognitive decline32,33, and the fact that β-amyloid pathology is only partially correlated with tau pathology (r = 0.48; Extended Data Fig. 6f). Thus, while we found a range of strong cellular associations (FDR < 0.01) for β-amyloid and tau pathology, only some of these were marginally correlated with both pathologic features (FDR < 0.05, for example, Oli.1, End.2, Ast.4 and Inh.3). The two pathologies therefore had a largely distinct set of associations with cell subtypes/states, consistent with our earlier report of distinct microglial transcriptional programs being associated with amyloid and tau pathology29.
The associations with cognitive decline and tau pathology showed that the inferred oligodendrocyte topic Oli.1 (SVEP1+) appears to be more prominent in nonimpaired individuals while topic Oli.4 (QDPR+) was enriched in individuals with cognitive decline. In parallel, the relative proportion of inhibitory neuronal subtype Inh.3 (SST+) was higher in healthy individuals, in contrast to inhibitory subtype Inh.1 (PVALB+), which was relatively higher in individuals with cognitive decline (Fig. 4g–i). This suggests a potential vulnerability of SST+ GABAergic neurons in AD. Additional subsets that are associated with cognitive decline include End.2 and Ast.4, which were enriched for genes related to stress response pathways such as oxidative stress (Fig. 3f,i).
Although our single-nucleus dataset itself has limited statistical power, we still found a clear trend of correlations between the proportions of certain cell subsets in the snRNA-seq data (n = 24, within each broad cell class) and cognitive decline, β-amyloid and tau burden (Fig. 4j and Extended Data Fig. 6g), which are consistent with our findings from the CelMod estimations in 638 samples.
Finally, we further validated the association between cell subtypes/subsets/topics and AD traits using independent bulk RNA-seq profiles and proteomic measurements. Applying the CelMod model to an independent cohort of 106 bulk RNA-seq profiles (from the Mount Sinai Brain Bank (MSBB34) (Methods), we replicated many results, with consistent associations to both cognitive decline and tau burden for most cellular subsets (except for Ast.2, End.4 and Mic.5 subsets; Extended Data Fig. 6h). Separately, we used proteomic measurements of markers for key prioritized cell subsets in 400 ROSMAP individuals30. Consistent with our RNA-based analyses, we found that the SST protein (Inh.3 marker), IGFBP7 (Ast.2) and S100A6 (Ast.4) are correlated with cognitive decline and tau burden. QDPR (Oli.4) is associated with cognitive decline, and CD44 (Ast.3 marker) is associated with amyloid burden (P < 0.01; Fig. 4k). The directions of these associations are consistent in the RNA- and proteome-based analyses. We found that, at the protein level, SST, IGFBP7 and S100A6 also associated with amyloid burden, and CD44 also positively correlated with cognitive decline and tangles (P < 0.01; Fig. 4k).
Multicellular communities of co-occurring cell subsets
Assessing the extent of interindividual variation in cell subset frequency revealed differences within each of the cell classes separately, which was robust across the 24 snRNA-seq measurements and the 638 CelMod inferred proportions (Fig. 5a,b and Supplementary Table 4). To further test how interindividual variation in cell subsets was coordinated across all cell types, we calculated the Spearman correlation coefficient for each pairwise combination of cell subset or topic proportions (where proportions are defined within a cell class; Fig. 5c), uncovering a covariation structure of cell subsets from multiple cell types captured by hierarchical clustering of these pairwise correlations (Fig. 5d,e and Methods). The covariation structures were significantly similar across the 24 empirically determined proportions (Fig. 5d) and the 638 inferred proportions (Fig. 5e, permutation test, P < 0.001, Fig. 5f and Methods), and they were confirmed in bulk RNA-seq profiles of 106 individuals from the independent MSBB cohort with cell subtype proportions inferred by CelMod (Methods and Extended Data Fig. 7a).
Fig. 5 |. Covariation structure of cell subset proportions across 638 individuals.
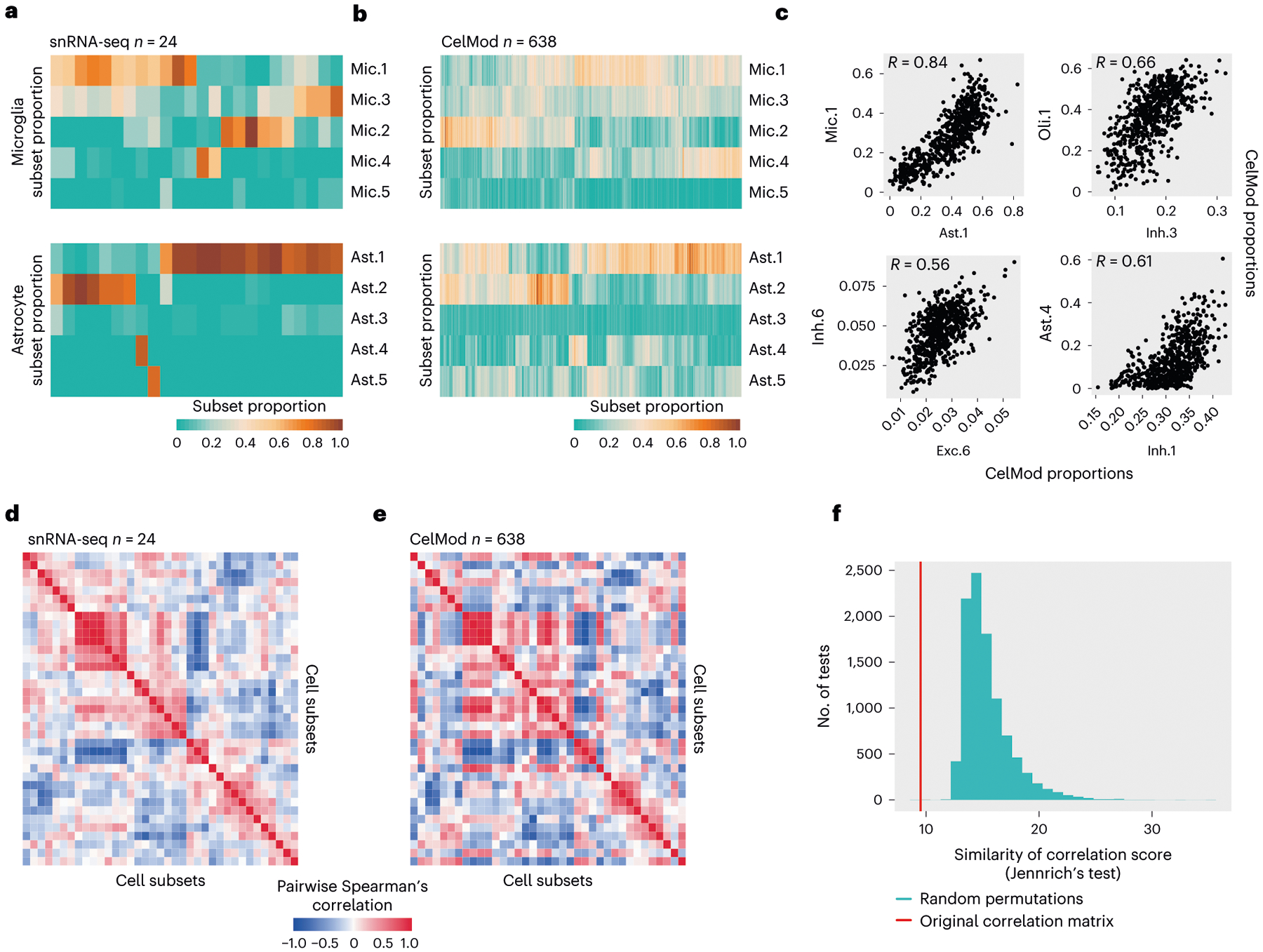
a,b, Proportions of cell subsets across individuals in astrocytes and microglia. The frequency (out of total cells in the class, color scale) of each cell subset (columns) in each individual (rows): from snRNA-seq (n = 24, in a) or CelMod estimations (n = 638, in b). The cell subsets in b are ordered as in a. c, Scatter plots of selected pairs of cell subsets from different cell classes, showing high correlations between proportions of subsets (n = 638). d,e, Coordinated changes in proportions of cell states and subtypes across individuals. A heatmap of the pairwise Spearman’s correlation coefficients of the proportions of all cell states and subtypes across 24 individuals (snRNA-seq measurements, d) and in 638 individuals (CelMod estimated proportions, e). We found a structure of mixed correlated and anti-correlated cellular subsets of mixed cell types. f, CelMod estimated pairwise correlations of cellular populations (n = 638 individuals) match the snRNA-seq measurements (n = 24 individuals). Similarity between the two correlation matrices (in d and e) is statistically significant (P = 0.001, by permutation test, one-sided; Methods). Histogram of the distribution of similarity scores (Jennrich’s test45) of correlation matrices in 10,000 random permutations of the cellular frequencies independently per cell type. Red, similarity score of the true matrices in d and e.
The uncovered covariation structures suggest the existence of distinct multicellular communities in the aging human brain. To model those, we built a network of cell subsets, with connections between each pair of subsets (nodes) that are significantly correlated or anti-correlated (signed edges) (R > 0.4 or R < −0.4, with P < 0.05; Methods and Fig. 6a,b). These networks highlighted an underlying structure of cellular communities (connected components of positively correlated cell subsets) comprising multiple neuronal, glial and endothelial cell subsets or topics, along with strongly opposing communities (negatively correlated cell subsets between communities) (Fig. 6b). A similar architecture was detected in the 24 snRNA-seq measurements-derived network (Extended Data Fig. 7b).
Fig. 6 |. Multicellular communities exist in the aging DLPFC brain region.

a, Scheme of the computational framework for estimating multicellular communities: proportions of cell subsets across individuals within each cell type are calculated and combined, and pairwise correlations between all cellular subsets are computed. A multicellular network is derived from the pairwise correlations, associated with AD traits by statistical analysis, and connected components are annotated as cellular communities (Methods). b, A network of cellular subsets reveals coordinated variation across individuals in multiple cell types. Network of coordinated and anti-coordinated cell subsets (nodes). Edges between pairs of subsets with statistically significant correlated proportions across individuals (R > 0.4, two-sided P value threshold = 0.05, solid red line) or anti-correlated (R < −0.4, dashed blue line) based on CelMod proportions (n = 638). snRNA-seq-based network is shown in Extended Data Fig. 7b. Nodes are colored by the cell type and numbered by the subset as in Figs. 2a and 3a,d,h. c, Correlation patterns of proteomic30 expression of signature genes across cell subsets match CelMod estimates. For selected cell subsets, pairwise correlations of snRNA-seq proportions (n = 24, left), CelMod proportions (n = 638, middle) and average protein expression levels of signature genes (n = 400, right). d–f, Cellular communities are linked to AD-associated traits. Cellular network (as in b) of coordinated and anti-coordinated cell subsets (nodes), colored by the associations (multivariable linear regression, FDR 0.01) with AD traits (purple, positive; green, negative association; gray, nonsignificant) for: cognitive decline (d), tangles burden (e) and β-amyloid burden (f). Bottom, each bar represents the connectivity score (no. of positive edges − no. of negative edges)/potential edges between groups of cells according to their association to each trait, showing that cell subsets associated with AD traits are highly connected in the network. Statistical significance for the connectivity score was calculated based on random permutations (one-sided, not adjusted) (Methods): *P = 0.05, **P = 0.01. a, amyloid load; c, cognitive decline; t, tangles load; neg, negatively associated; pos, positively associated; neutral, not associated.
We validated, at the protein level, the coordinated levels of cell subsets across individuals for subsets of different cell types using bulk proteomic data. Consistent with the two transcriptomic networks (CelMod estimated n = 638, and snRNA-seq measured n = 24), there were statistically significant correlations between the protein levels of signature genes of various cell subsets, validating the structure of two reciprocal communities: Ast.4, Oli.2, Oli.4 and Mic.3 (average R = 0.31); and Oli.3, Ast.2 and Inh.3 (average R = 0.41). There was also the expected anti-correlation between the two sets of cell subtypes (average R = −0.2). Notably, an additional correlation of Ast.2 and Ast.3 signatures with Mic.3 emerged only in the proteomic data (Fig. 6c), highlighting the need for multiomic data in future studies.
Multicellular communities associated with cognitive decline
We next showed that our network captured a coordinated set of changes across cell types (Fig. 6b), as cell subsets associated with AD traits formed connected communities in our networks (Methods and Fig. 6d–f). The cognitive decline-associated subsets segregated into two anti-correlated communities, each composed of different highly interconnected neuro–glial–endothelial cell subsets: (1) the cognitive decline community consisting of all cell subsets whose proportions are associated with more rapid cognitive decline and high tau burden (P < 0.01; Methods), including Oli.4 (QDPR+), Ast.4 (S100A6+), Mic.3, End.2, and Inh.1 and Inh.7 subtypes (PVALB+); (2) the cognition nonimpaired community consisting of all cell subsets whose proportions were associated with slower cognitive decline and low tau pathology (P < 0.01), including the Oli.1 signature, and the Inh.3 (SST+), Ast.2 (GFAP+ SERPINA3+) and End.1 subsets (Fig. 6d,e). For β-amyloid burden, the amyloid-low community included neuronal cell subsets that are negatively associated with β-amyloid burden: specifically, there was a loss of middle/deep layer glutamatergic neuronal subtypes Exc.3–6 (predominantly layers 4–5) and the Inh.6 subtype (PTPRTK+, layer 4, P < 0.001; Fig. 6f). This was consistent with previous work reporting increased neuronal vulnerability associated with AD pathology in RORB+ pyramidal neurons at middle/deep cortical layers25 (Figs. 2 and 4i and Extended Data Fig. 2a). As excitatory neuronal subsets largely formed two opposing connected components in the graph dependent on the cortical layer (layers 2–3 versus layers 4–6; Fig. 6b), we cannot exclude that our findings are driven by a dissection bias as we prepare samples for RNA extraction. However, this is unlikely as this association was captured independently by the single-nucleus and bulk RNA-seq (Fig. 5d,e and Extended Data Fig. 7a), and, notably, compositions of all other cell types were found to be independent of the cortical layer (Methods and Extended Data Fig. 7c). By a random permutation test, we also showed that subsets associated with each AD trait captured statistically significant (P < 0.01) connected components in the network, and reciprocal communities were significantly connected by negative edges (P < 0.01; Methods). We therefore identified several sets of coordinated cellular responses that are associated with distinct aspects of AD.
Shared pathways and signaling within the cognitive decline communities
The existence of cellular communities (Fig. 6) suggests shared functionalities and distinct signaling between cells within each community35,36. We searched for shared pathways and for ligand–receptor interactions within and between the two opposing communities—the cognition nonimpaired (Inh.3, Oli.1, Ast.2, End.1, End.4) and cognitive decline (Inh.1, Inh.7, Oli.4, Ast.3, Mic.3, End.2) communities.
Examining the differentially enriched pathways across all cell subsets within each community, we found overlapping pathways within the cognition nonimpaired (n = 14); separately, we found more shared pathways within the cognitive decline community (n = 275) (hypergeo-metric test, Q < 0.05, clustered pathways; Methods). Interestingly, the cognitive decline community cell subsets shared pathways related to known AD risk factors, including response to oxidative stress, hypoxic stress and DNA damage, as well as oxidative phosphorylation and neurodegenerative diseases-associated genes (including AD) (Q < 0.05; Fig. 7a). On the other hand, the cell subsets within the cognition nonim-paired community shared pathways related to axon development and cell–cell junctions, and synapses-related pathways (Q < 0.05; Extended Data Fig. 7d).
Fig. 7 |. The cellular environment of the AD and the cognitively nonimpaired brains.
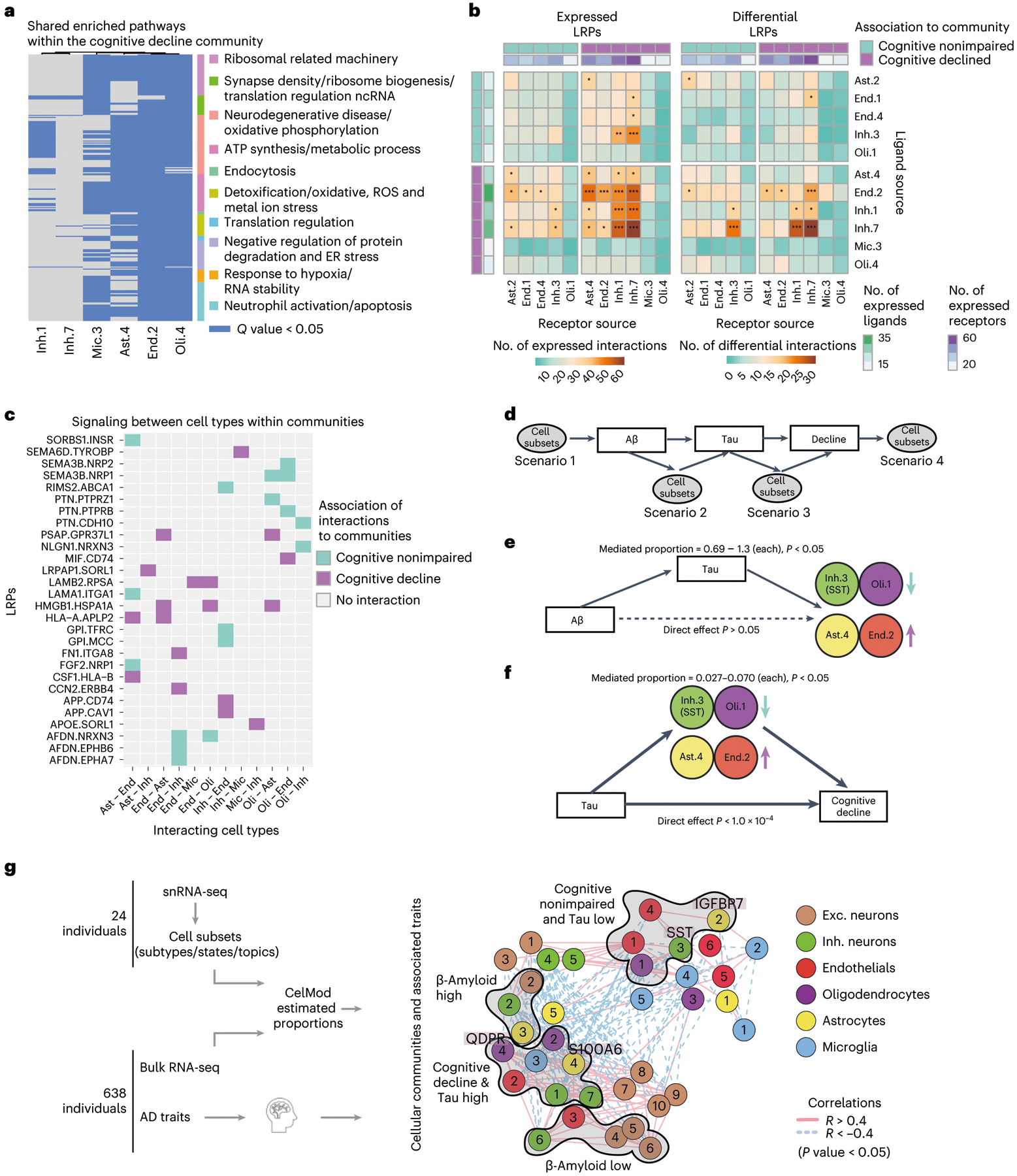
a, Shared pathways within the cognitive decline community. Enriched pathways (hypergeometric test, FDR Q < 0.05) in up-regulated genes within each cell subset, shared between at least three subsets within the community. Pathways clustered by shared genes (Methods). Nonimpaired community shared pathways are shown in Extended Data Fig. 7d. b, Increased LRPs between cell subsets of the cognitive decline community compared with the nonimpaired community. For each pair of cell subsets, showing the number of LRPs (colorbar, row, ligand; column, receptor) that are expressed (left) or differentially expressed in at least one of the subsets (right). Top and side bars, positive (purple) or negative (turquoise) association with cognitive decline, and the number of expressed LRPs (color scale). c, Examples of community-specific LRPs. For each LRP, marking the association to each pair of cell subsets with the cognitive decline community (purple) or the cognition nonimpaired community (turquoise) or no-association (gray). LRPs are associated to a community if the ligand and its receptor are positively differentially expressed in cell subsets within one community compared with the other. d, A scheme of underlying assumptions and four possible scenarios of the cell subset causal relationship with AD pathology and cognitive decline assessed by mediation analysis. e, Mediation analysis results showing that tau pathology burden is predicted to be upstream of changes in proportions of Inh.3, Oli.1, Ast.4 and End.2. Colored by cell type, the arrow indicates the direction of change in proportion in association with cognitive decline and tangles burden. Full results are shown in Extended Data Fig. 8c. f, Mediation analysis results showing effect of changes in proportions of Inh.3, Oli.1, Ast.4 and End.2 on cognitive decline independent of tau pathology burden. Colors and arrows as in e. Full results are shown in Extended Data Fig. 8d. g, A scheme of our proposed model of multicellular communities of the aging DLPFC brain region and their associations with AD traits. Cellular networks (as in a), nodes colored by the community assignments. The statistically significant enriched associations to AD traits (hypergeometric P value) are marked next to the graph.
We also searched for ligand–receptor pairs (LRPs)37,38 that putatively connect different cell subsets within and between the two communities (Methods). We first identified all expressed LRPs (P < 0.05, permutation test, CellPhoneDB39), connecting one cell subset expressing the ligand with another cell subset expressing the receptor (Fig. 7b), finding the highest number of expressed LRPs connecting two subsets within the cognitive decline community (1,005 expressed LRPs; Fig. 7b). Next, we searched for community-specific LRPs, defined as expressed LRPs in which at least the ligand or the receptor was differentially expressed (FDR < 0.01; Fig. 7b,c and Extended Data Fig. 8a,b). We identified 384 cognitive decline community-specific LRPs, such as the LAMB2 gene, encoding Laminin Subunit Beta 2, expressed by End.2; and RPSA, encoding the Laminin Receptor 1, expressed by both Mic.3 and Oli.4 (FDR < 0.01; Fig. 7c). We also identified 181 cognition nonimpaired community-specific LRPs, such as FGF2, encoding the Fibroblast Growth Factor 2, expressed by Ast.2; and NRP1, encoding the receptor Neuropilin40, expressed in End.1 (FDR < 0.01; Fig. 7c). We further identified pairs of cell subsets statistically significantly enriched in LRPs (permutation test, P < 0.01; Methods and Fig. 7b): we found that most such pairs of cell subsets were within the cognitive decline community (7 of 12 for community-specific LRPs and 14 of 26 for expressed LRPs; Fig. 7b).
AD-related traits are partially mediated by cell subsets
While our autopsy-based data cannot determine the causal chain of events leading to AD, modeling can propose a most likely scenario. To test whether cell proportion changes may mediate the association between AD pathologies and cognitive decline, we applied a causal modeling framework, aimed to align cell subset proportions along the widely assumed sequence of AD pathophysiology (Fig. 7d), which starts with β-amyloid accumulation, followed by tau with subsequent neurodegeneration and cognitive decline41–44 (consistent with our data; Fig. 4g–i).
We focused on the six tau-associated cell subsets that were also marginally associated with β-amyloid (P < 0.05, but FDR > 0.017): Inh.2, Oli.1, Oli.2, Ast.4, End.2, Inh.3 (SST). First, we tested three possible scenarios (Fig. 7d), where cell proportion changes are either (1) upstream drivers of β-amyloid levels; (2) mediators, downstream of β-amyloid but upstream of tau; or (3) downstream of tau. In each scenario, we tested the change in association between two variables once the third is added as a mediator. Ast.4, End.2, Inh.3 and Oli.1 were predicted to be downstream of tau, as tau burden mediated most (>69%) of the β-amyloid–subset associations, with no statistically significant direct amyloid effect (P > 0.05) (Fig. 7e and Extended Data Fig. 8c). However, we predict that Oli.2 might be affected by both amyloid and tau, and Inh.2 is upstream of tau (Extended Data Fig. 8c), suggesting that changes in these subsets may be occurring at an earlier stage of AD.
Next, as Ast.4, End.2, Inh.3 (SST+) and Oli.1 were positioned downstream of tau accumulation, we performed a second mediation analysis to predict their contribution to cognitive decline (Fig. 7d,f). Our analysis positioned all four subsets upstream of cognitive decline, as each of their proportions partially mediated the effect of tau on cognitive decline (2.7–7.0% of total effect) (Fig. 7f and Extended Data Fig. 8d). The proportions of the four subsets simultaneously explained 7.4% of the tau–cognition association, which is a small but meaningful fraction of the effect of tau proteinopathy on cognitive decline.
Discussion
In this study, we constructed a cellular map of the neocortex, capturing cellular diversity and expression programs at high resolution (compared with previous reports3,6; Figs. 1–3 and Extended Data Fig. 4), revealing insights about intra- and interindividual diversity in the aging brain (Fig. 5) and uncovering coordinated multicellular communities associated with cognitive impairment and AD pathologies (Figs. 6 and 7). With our analytical method (CelMod), we expanded the estimated cellular landscapes from a selected set of 24 individuals to a random set of 638 individuals in the same aging cohorts (ROSMAP). Our approach can be readily applied to other tissue-level datasets, as we have demonstrated its deployment in a published snRNA-seq dataset of 48 individuals3 and in 106 bulk RNA-seq datasets of participants from an independent cohort (MSBB) (Fig. 4 and Extended Data Fig. 6). The inferred cellular composition data were statistically powered for disease association analyses, which uncovered observations such as a decrease in SST (Inh.3) GABAergic neuron proportions, validated by proteomics of the same brain region in 400 individuals, suggesting that SST neurons may be more vulnerable than other neuronal subtypes in AD (Figs. 4 and 6). Next, we applied our computational framework to capture a multicellular view of cellular environments and organization, which led to the discovery of distinct cellular communities that we linked to AD-associated traits (Fig. 6).
In microglia, we found a good match between our snRNA-seq-based clusters and those found in single live microglial cells purified from fresh autopsy and surgically resected tissue7, addressing an important concern raised in an earlier study13. As a result, despite differences relating to the quantity, quality and nature of single-nucleus- and cell-derived RNA data, the same microglial subsets were captured in both datasets. Notably, the similarity in microglial diversity captured by live cells and frozen nuclei also substantially reduces the likelihood of artifacts from ex vivo manipulations.
Oligodendrocytes emerge as an interesting cell type for further evaluation for their strong association to AD (Fig. 4g–i). Oligodendrocytes also highlight the need to analytically accommodate the unique characteristics of each cell type (Fig. 3j). We represented their heterogeneity utilizing topic models19–22 to capture expression programs instead of discrete cell clusters, finding two programs strongly associated with tau pathology and cognitive decline, one positive (Oli.4) and one negative (Oli.1) (Figs. 4 and 6c).
A major innovation of our study is the definition of cellular communities defined by correlated changes in the frequency of different cell subsets across individuals (defined within each cell class) and their association to AD traits (Figs. 6 and 7g). Within the cognitive decline cellular community, we found LRPs linking different cell subsets (Fig. 7b,c) as well as multiple shared pathways that are enriched in different cell subsets within the community. Many of these pathways relate to known risk factors for AD, offering a further form of validation from earlier studies (Fig. 7a and Extended Data Fig. 7d). Overall, these results fit within our conceptual understanding that AD is a distributed pathophysiologic process involving multiple interacting cell types. Spatial transcriptomic methods will help to resolve whether these communities represent colocalized cells or distributed communities responding to a shared signal.
One strength of our analyses comes from the ROSMAP cohorts that have detailed, quantitative clinicopathologic measures which enable resolving changes related to pathologies from those involved in cognitive decline. This is nicely illustrated by cell subsets associated with β-amyloid but not with cognitive decline, and leads us to prioritize as therapeutic targets four cell populations that, in our mediation modeling, appear to contribute to the consequences of tau proteinopathy on cognitive decline.
Our study does have limitations: while we have statistically robust results due to CelMod estimates in bulk RNA-seq from 638 individuals, our small reference snRNA-seq dataset may not capture the full diversity of neocortical cells, especially in smaller cell populations such as microglia and pericytes. Moreover, we have a limited ability to reliably estimate the abundance of rare cell subsets such as Mic.3, End.4 and Ast.5. Further, while we have validated the estimated cellular architecture and association to traits using histology, RNA and proteomics data, the associations are limited by the detection limits of each method and by differences between RNA and protein expression levels. Validation of our results in larger-scale snRNA-seq studies is necessary to ensure the robustness of our cellular map and is likely to resolve additional rare cell subsets that may be important in AD. Finally, when profiling postmortem brain tissue we do not directly measure the temporal links between cellular communities, appearance of pathology and cognitive symptoms. Nonetheless, we explored this key issue using mediation analysis, a statistically rigorous method whose results suggest that the known link between tau pathology and cognitive decline is mediated in part through changes in the proportion of specific cell subsets (Fig. 7e,f), proposing testable hypotheses for future studies.
Overall, our work highlights the importance of a unified view of the cellular ecosystems of the brain in the study of AD and other disorders, demonstrating how a network approach can uncover new insights, such as cellular communities each involved in different aspects of AD.
Methods
Source of clinical, pathologic and omics data and ethics
Data were derived from subjects in one of two clinical-pathologic cohort studies of aging and dementia, the Religious Orders Study (ROS) or the Memory and Aging Project (MAP), collectively, ROSMAP8, both undergoing deep ante- and postmortem characterization. The ROS and MAP studies were approved by an Institutional Review Board of Rush University Medical Center and participants signed informed and repository consents to brain donation at the time of death and an Anatomic Gift Act.
The methods of assessing cognition and pathology have been extensively summarized in previous publication10,46–49. Briefly, cognitive decline was determined based on the uniform structured annual cognitive assessment of ROSMAP participants10,46–49. Scores from 17 cognitive performance tests were used to obtain a summary measure for global cognition. Cognitive decline is defined by person-specific random slopes of longitudinal modeling of global cognition with a mixed effects model, adjusting for age, sex and education50. The clinical diagnosis of AD at the time of death was determined by a neurologist specializing in dementia51, blinded to all neuropathologic data52, providing the most likely diagnosis as previously described in detail46,53,54. β-amyloid and tau pathology burdens were determined by quantification and estimation of the burden of parenchymal deposition of β-amyloid and the density of abnormally phosphorylated tau-positive neurofibrillary tangle (tau pathology) levels present at death10,46–49, in eight regions of the brain: the hippocampus, entorhinal cortex, anterior cingulate cortex, midfrontal cortex, superior frontal cortex, inferior temporal cortex, angular gyrus and calcarine cortex. A pathologic diagnosis of AD was determined by a board-certified neuropathologist blinded to age and all clinical data55, based on the scores of Braak stage for severity of neurofibrillary tangles and CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) estimates for burden of neuritic plaques, as previously described10,46–49.
In this study, we generated snRNA-seq profiles from 24 ROSMAP participants profiled (12 male and 12 female, average age: 87.9) from four groups (Supplementary Table 1): (1) a reference group of NCI (no cognitive impairment) with minimal AD pathology; (2) a high-resilience group of NCI with pathologic AD; (3) an AD group with both clinical AD dementia and pathologic AD; and (4) a low-resilience group of individuals diagnosed with clinical AD dementia with minimal AD pathology. We included only samples that had RIN > 5 with postmortem interval < 24 hours, and that had bulk RNA-seq in a previous study1 and whole-genome sequencing data8–10. In addition, we used bulk RNA-seq data of 638 ROSMAP participants1 (230 men and 408 women, average age: 88.69315(95% confidence interval, 88.17613 to 89.21017), including the 24 individuals). Finally, we used a published proteomic dataset of 400 ROSMAP participants30 (119 men and 281 women of advanced age). In addition, we used for validations published bulk RNA-seq data of 106 men and woman of advanced age from an independent cohort from the MSBB34.
Data randomization and sample size
In total, 638 bulk RNA samples and 400 proteomics samples were randomly selected in regard to AD traits, while the 24 snRNA-seq samples were chosen based on their AD traits. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications1,30,34. For bulk RNA1 and bulk proteomics30 datasets, the individuals were randomly assigned into batches and included both men and women. The snRNA-seq data were divided into four batches designed to be balanced for pathological and clinical diagnosis and sex. Samples size was based on availability and no statistical methods were used to determine the dataset sizes.
Single-nucleus isolation and RNA-seq
DLFPC tissue specimens were received frozen from the Rush Alzheimer’s Disease Center. Working on ice throughout56, we carefully dissected them to remove white matter and meninges (presence varied between specimens). Then, 50–100 mg of tissue was gently Dounced with Pestle A followed by 25 times with Pestle B (Sigma, cat no. D8938) in 2 ml of NP40 Lysis Buffer (0.1% NP40, 10 mM Tris, 146 mM NaCl, 1 mM CaCl2,21 mM MgCl2,40 U ml−1 RNAse inhibitor (Takara, 2313B)). Tissue was transferred to a 15-ml conical tube, adding 3 ml of PBS mix (PBS + 0.01% BSA (NEB, B9000S) and 40 U ml−1 RNAse inhibitor), then centrifuged immediately (swing bucket rotor at 500g for 5 min at 4 °C). The supernatant was removed, and the nuclei pellets were resuspended in 500 ml of PBS mix. Nuclei were filtered through 20-μm preseparation filters (Miltenyi, 130-101-812) and counted using the Nexcelom Cellometer Vision (Nexcelom, CHT4-SD100-002). Next, 20,000 nuclei in around 15–30-μl volume were run on the 10X Single Cell RNA-Seq Platform using the Chromium Single Cell 3’ Reagent Kits v2 (10x GENOMICS PN-120237). Libraries were made following the manufacturer’s protocol for v2 library construction. The amplified whole transcriptome (WTA) was diluted to <8 ng ml−1 for library construction, and amplified complementary DNA and libraries were assessed by Qubit HS DNA assay (Thermo Fisher Scientific, Q32851) and BioAnalyzer (Agilent, 5067-4626). Libraries from four channels were pooled and sequenced on one lane of an Illumina HiSeqX by the Broad Institute Genomics Platform, for a target coverage of around 1 million reads per channel.
Preprocessing of snRNA-seq data
De-multiplexing, alignment to the hg38 transcriptome and unique molecular identifier-collapsing were performed using the Cell-ranger toolkit (v.2.1.1, chemistry V2, 10X Genomics, for Chemistry Single Cell 3’), and run using cloud computing on the Terra platform (https://Terra.bio). We used a genome reference with premessenger RNA annotations, accounting for exons and introns. Technical artifacts of ambient RNA were corrected by the CellBender package (remove-background function, with 300 epochs). We excluded nuclei with fewer than 400 detected genes and genes with less than 15 reads across all nuclei. Gene counts were log-normalized per nucleus (NormalizeData function from the Seurat57 package v.4, natural-log of the read counts divided by the total reads and multiplied by 10,000), and scaled and centered (ScaleData function) across the dataset, after selection of variable genes (FindVariableFeatures, selection. method = ‘vst’).
Dimensionality reduction, clustering and quality controls
Principal component analysis (with RunPCA function, npcs = 50) was run on the scaled expression matrix. The significant principal components (selected by the standard deviation of each principal component) were embedded using Uniform Manifold Approximation and Projection (UMAP)58. Clustering was performed by the Louvain community detection algorithm over a k-nearest neighbors graph of nuclei59 (FindNearestNeighbors and FindClusters functions). Clusters were manually matched to cell types based on the expression of known markergenes15,56.
Doublet cells removal.
For doublet detection and elimination, we ran DoubletFinder60 to score nuclei as doublets. We excluded high-confidence doublet cells. Next, we clustered our data at high resolution to identify and exclude small doublet clusters. A doublet cluster is defined as a cluster with over 70% of doublet nuclei, which we validated as also coexpressing markers of least two different cell populations. The automatic doublet analysis aided the identification of the doublet cluster and of individual doublet cells. Removal of nuclei with high content of cytoplasmic and low nuclear RNA: clusters with low nuclear RNA content and high cytoplasmic RNA content were removed. Cytoplasmic/nuclear RNA is defined as the top 400 differentially expressed genes between nuclear content and cellular content14.
Subclustering analysis and annotations
For cell types with sufficient numbers of cells, we performed subclustering analysis to reveal additional diversity: astrocytes, microglia, endothelial, inhibitory neurons and excitatory neurons (oligodendrocytes were analyzed by topic modeling). The pipeline described for all cells was applied separately to each cell type, with minor differences: the number of significant principal components used was adjusted per cell type (30 principal components for astrocytes and excitatory neurons, 25 for inhibitory neurons and 20 for microglial and endothelial cells). The resolution of the clustering ranged from 0.1 to 0.6. Additional filtration of low-quality nuclei was done at the cell type level, to overcome the differences in RNA content between brain cell types. Glial and endothelial subset annotations were done by known marker genes and profiles3,5–7,15. Inhibitory and excitatory neuronal subtype annotations and cortical layer were further predicted by a regression model (SingleR package) trained on a published cortical snRNA-seq dataset12 (using the ‘fine tuned’ label12 to determine the best cortical layer fit), and applied to our neuronal snRNA-seq data. Annotations were based on the highest scoring label, and validated by expression of known markers12,56.
To validate that subclusters were not driven by batch, RIN or sex, we examined the distribution of each covariate within the subcluster. Next, for astrocytes, microglia and endothelial cells separately, we regressed the effect of each covariate independently during the scaling of the expression matrix (Seurat ScaleData function), and followed the same downstream clustering analysis pipeline as described, matching the number of clusters in the standard analysis. The two sets of cluster assignments, the original and the regressed, were compared by the pairwise Jaccard index (cell overlap between clusters), showing one-to-one match between the two sets of clusters.
Topic modeling for oligodendrocyte cells
We modeled the cell state diversity in oligodendrocyte cells by topic modeling19–22, using Latent Dirichlet Allocation. Topic modeling was performed on the normalized data matrix, reduced to the oligodendrocyte variable genes. We used the CountClust package in R, which calculated the score of membership for four topics (using the GoM function, which was run on the scaled expression matrix with tolerance 0.01). The results were robust to the choice of topic numbers and tolerance levels, yet we favored a small number of topics given the number of cells and samples in our data. We used the Kullback-Leibler divergence of gene weights over topics to select genes highly associated with each topic (using the ExtractTopFeatures function with default parameters), and excluded genes negatively correlated to the related topics.
Density plots to visualize cellular populations and genes
To visualize cell attributes in dense two-dimensional graphs, we plotted the weighted average value of the attribute in the neighboring cells using the Gaussian kernel adjacencies in the two-dimensional embedding (Gauss kernel from the ‘KLRS’ R package, with sigma = 1.5). We filtered out cells with an adjacency measure >0.0005. We used this approach to plot the expression of marker genes and scores of the four topics of oligodendrocytes.
Differential expression and pathway analysis
We calculated the differentially expressed genes within each cluster within each cell type by the MAST (Model-based Analysis of Single Cell Transcriptomics) method, and corrected for multiple comparisons using Bonferroni correction (FindAllMarkers function, test. use = ‘MAST’). We defined differential genes as those with Q value threshold <0.05, expressed in at least 10% of nuclei in the given cluster, and with at least 0.25-fold average expression (compared with cells outside of the cluster). To calculate differentially expressed genes in oligodendrocytes, we used nuclei that scored more than 0.5 to one of the topics and addressed it as a regular hard assignment. The differential expression signatures were tested for enriched pathways and gene sets (compareCluster function in the clusterProfiler package in R), and corrected for multiple comparisons by FDR, using FDR Q < 0.05 for significance. Gene sets were taken from the KEGG and Gene Ontology (GO) resources61.
Shared pathways analysis.
Shared enriched pathways among a group of cell subsets were defined as statistically significant enriched pathways in the independent analysis of at least three subsets. To overcome the redundancy within the pathways databases, the shared enriched pathways were clustered by hierarchical clustering (pheatmap function) using the pairwise Pearson correlation distance computed over the gene space. Each pathway was represented by the differentially expressed genes linked to it. Pathway clusters were named manually.
Comparison of clusters with published datasets
We compared our cell clusters with four recently published human brain snRNA-seq datasets3,4,6,25, and with single-cell RNA-seq of live microglia cells from fresh autopsy and surgically resected human brain tissue7, by Canonical Correlation Analysis (CCA) (Seurat, with 3,000 genes and 20 canonical components). We predict the class membership of the CCA-transformed snRNA-seq nuclei profiles by a naive Bayes classifier trained on the other dataset in CCA space, and each nucleus cluster was assigned a published cluster by the maximum prediction value.
CelMod: estimating cells and topics proportions in bulk data
We developed a regression-based consensus model (CelMod) to extend our snRNA-seq-derived cell subset estimates to bulk data, leveraging matched bulk and snRNA-seq data from the same 24 donors.
We train the regression model as follows: (1) filter genes to include only those that have at least 100 counts across all nuclei of the cell type of interest, and a mean counts per million value > 10; (2) perform a linear regression on each gene separately for each cell cluster of interest, using its expression as the dependent variable and the proportion of that cluster in each snRNA-seq sample in the training set as the independent variable; (3) for each gene, use the regression model to calculate the predicted proportion of each cell type, normalizing their sum to 1; (4) rank genes by the 90th percentile of the absolute value of the error between predicted and training proportions, for each cell type; and (5) select the number of top-ranked genes (constant for each cell cluster) to use for deconvolving a new bulk RNA-seq sample; this number of genes, the only tunable parameter, is selected based on cross-validation, as described below.
CelMod identifies a large set of informative genes for each cell subset, ensuring that a small set of overlapping gene markers from different cell groups are not skewing the proportion estimates for broad cell classes as well as for subsets within each cell class. To determine the optimal number of genes to use for the prediction, we use fivefold cross-validation: 80% train and 20% validation set. The validation sets are mutually exclusive, such that after five runs, the proportions in every bulk sample have been ‘predicted’ once. This cross-validation is run using 3 to 100 ranked genes (from step 3 above), selecting the optimal gene number, which minimizes the mean of the 90th percentile errors for each cell group in all samples, and applied for deconvolution predictions in the larger bulk RNA-seq dataset.
We run the algorithm iteratively, starting at the ‘top level’, with the broad cell classes (glutamatergic neurons, GABAergic neurons, astrocytes, oligodendrocytes, oligodendrocyte precursor cells, microglia, endothelial cells and pericytes), and then again for the subtypes within each of the cell classes. For the broad cell classes, the proportions are based on the total nuclei per sample. For the subtypes/subsets, the proportions are normalized to the total nuclei from the broad class of interest. This allows us to directly model both the overall and subtype-level compositions of the bulk tissue, especially for cell types that comprise a small fraction of the overall population (such as endothelial and microglial cells). For the oligodendrocyte signatures, which are modeled as topics instead of discrete clusters, we sum the weights for each given signature over all nuclei from a given sample, and then normalize these sums so that they add up to 1 for a given sample. This reflects a ‘proportion of topic weights’ per sample, as opposed to a strict proportion that can be calculated for the discrete cell types. Finally, for microglia, we ensured the model training was robust, by only including donors with at least 25 total microglia.
The performance of this algorithm on the validation set (20% of the data, with fivefold cross-validation) is shown in Fig. 4c–f and Extended Data Fig. 6a–c, as well as the correlation structure between cell subsets in Fig. 5d,e and Fig. 6c.
We applied the inferred CelMod model to an independent externally published snRNA-seq dataset3 from 48 individuals of the prefrontal cortex (BA10 and not BA9 as our dataset), with matching bulk RNA-seq profiles. The data were downloaded and annotated using our higher-resolution cell clustering (compared with the published annotations), and RNA signatures and proportions were obtained for each of the clusters (as described above). We applied CelMod to the 48 bulk RNA-seq samples with matching snRNA-seq data, as described, to estimate the proportions of cell types and subsets in the bulk samples, and these estimates were used to validate the accuracy of CelMod.
We applied the inferred CelMod model to an independent external dataset of 106 bulk RNA-seq profiles from a different independent cohort of aged men and women from the MSBB34. The samples were preprocessed similarly to the ROSMAP dataset1, then we applied the CelMod model learned on our snRNA-seq data to estimate the major cell types, cell subsets and topics proportions in each bulk sample. The cell subset estimates from this independent MSBB cohort were used to validate the associations of cell subset proportions to AD-associated traits and the correlation structure of cellular subsets. The MSBB trait analysis matched the ROSMAP analysis, except that for the MSBB cohort the CDR (Clinical Dementia Rating) measurement was used instead of the cognitive decline rate, and the Braak score was used instead of the tau tangles load.
We compared the performance of CelMod with three deconvolution methods that use reference profiles of snRNA-seq clusters to estimate proportions in bulk data: DCQ, DeconRNAseq and dtangle, with standard parameters for each method. We ran each method to predict proportions of major cell classes and subclusters, and calculated Spearman correlations between the predicted proportions and the actual snRNA-seq-derived proportions.
Associating cell subsets and programs to AD-related traits
We analyzed three major pathological and cognition hallmarks of AD, collected for the ROSMAP cohorts: continuous measure of cognitive decline (the slope of the cognitive decline trajectory50) and continuous measures of tau tangle pathology density and β-amyloid burden (both averaged over multiple regions62), as detailed under Source of clinical, pathologic and omic data and ethics.
To test the statistical associations between AD phenotypes and proportions of cell types/subsets/topic models, we performed multivariable linear regressions, modeling cellular proportions as the outcome, and phenotypes as the independent variable, adjusted for age, sex and RIN score as covariates, and corrected for multiple comparisons by FDR. The analysis was done for each trait separately across all cell subsets (five astrocytes, six endothelial subsets, seven GABAergic neurons, ten glutamatergic neurons, five microglia subsets and four oligodendrocyte topics).
Multicellular communities
To find co-occurring cellular populations across individuals, we identified cellular communities, defined as a set of cell subsets and expression programs that have coordinated variation of proportions across individuals (for topics we use the weights). We identified cellular communities in the snRNA-seq dataset (n = 24 individuals) and the CelMod estimated proportions (n = 638 individuals), across six broad brain cell classes for which we had defined subsets or topic models (expression programs). To detect cellular communities, we followed these four steps: (1) Cellular proportions. Given a classification of single cells to subsets, we calculated per individual the proportion of each cell subset within each cell class (that is, out of the total number of cells in the cell class or sum of all topic weights). Next, we appended the cell subset proportions across all cell classes into a combined frequency matrix. For bulk data we used the estimated proportions by CelMod. (2) Correlation matrix. We calculated the pairwise Spearman correlation coefficient over the proportions of all cell subsets across individuals, clustered by hierarchical clustering (R pheatmap function with 1-Pearson correlation distance). (3) Cellular network. We built a graph where each cell subset was a node, and edges connected every two nodes if their absolute pairwise Spearman correlation value was at least 0.4 and P value < 0.05, and assigned a sign by the sign(R). The network layout was manually assigned over the igraph layout with positive edges (with layout = layout_with_fr), to maintain a common layout between the 24 and 638 derived networks. (4) Associating cellular communities to AD traits. To test the statistical significance of the association to AD traits (cognitive decline, tangles burden or amyloid burden), we calculated the connectivity score within three sets of nodes (nodes in the network): positively associated (FDR < 0.01, beta > 0), negatively associated (FDR < 0.01, beta < 0) or neutral (FDR > 0.01), to each trait. We defined the connectivity score for these sets of nodes in the network to be: the differences between the numbers of positive and negative edges divided by the total number of possible edges between them. To assess the statistical significance of high and low scores, we applied a permutation test on the subset labels within the network. The empirical P value is the proportion of permutations (of 10,000) that lead to a higher/lower score. Significant connected components associated with AD traits were termed communities.
Testing potential layer bias underlying the network analysis.
Excitatory neuronal subsets largely formed two opposing independent connected components in the graph, one consisting of upper cortical layer neuronal subsets (Exc.1, Exc.2, Exc.3, layers 1–4) and the other of lower layer neuronal subsets (layers 4–6). Given the intrinsic association between pyramidal neuron subsets and cortical layers, we cannot completely exclude the possibility that this partition is driven by a dissection bias of the cortical layer proportions. Yet, we can exclude a dissection bias for all other cell subsets by comparing the correlation structures of two distinct groups: high levels of Exc.1 (>0.5, n = 267 individuals) or low levels of Exc. 1 (≤0.5, n = 371 individuals). Next, we calculated the pairwise Spearman correlation coefficients of all cell subsets separately for the two groups, showing low differences in correlations between the groups, suggesting no dissection bias.
Comparison of the snRNA-seq network and the CelMod network.
To compare the snRNA-seq network (24 individuals) and the CelMod estimated cellular network (638 individuals), we calculated the statistical significance of the similarity between the two pairwise correlation matrices. We performed 10,000 random permutations of the CelMod estimated proportions of cell subsets in 638 individuals, within each cell cluster (total proportions within cell type = 1). We calculated an empirical P value based on the Jennrich’s score45 (R cortest.jennrich function) similarity between the snRNA-seq correlation matrix and each permutated matrix. The nonpermutation matrix consistently got the lowest score compared with the permuted matrix, that is, P < 0.001.
Analysis of proteomic data
We used published shotgun bulk proteomic data from 400 aging participants, both men and women, from the ROSMAP cohort30, with 196 individuals overlapping with the bulk RNA-seq dataset (log-normalized30 data). Protein markers of cell subsets of interest were used as validation for: (1) estimated proportions from bulk RNA-seq of cell subsets by CelMod (Fig. 4f and Extended Data Fig. 6d, using genes with matching RNA and protein profiles only); (2) associations between cell subset proportions and AD-associated traits (Fig. 4k); and (3) correlation structures between proportions of different cell subsets (Fig. 6c). In all analyses, we chose marker genes per snRNA-seq subset as differential genes with low expression outside of the subset, which were confidently measured in the proteomic dataset and had correlated bulk RNA and proteins levels. We excluded cell subsets lacking sufficient measured protein markers.
Immunohistochemistry and spatial transcriptomics
First, 6-μm sections of formalin-fixed paraffin-embedded tissue, obtained from Rush University Medical Center, from the frontal cortex were stained with NEF (Sigma, N2912). Heated-induced epitope retrieval was performed using citrate (pH 6) in a microwave (800 W, 30% power setting) for 25 min. The sections were blocked with blocking medium (3% BSA) for 30 min at room temperature, then incubated with primary antibody anti-NEF prepared in 1% BSA overnight at 4 °C. Sections were washed three times with PBS and incubated with fluochrome-conjugated secondary antibodies (Thermo Fisher) for 1 h at room temperature. Anti-fading reagent with DAPI (P36931, Life technology) was used for coverslipping. For each subject, 30 images of cortical gray matter at ×20 magnification (Zeiss Axio Observer.Z1 fluorescence microscope) were taken in a zigzag sequence along the cortical ribbon to ensure that all cortical layers were represented in the quantification in an unbiased manner. Dilutions: anti-NEF: 1/100; anti-GFAP: 1/300; anti-NeuN: 1/300. The acquired images were analyzed using CellProfiler and CellProfiler Analyst, developed by the Broad Institute. We estimated the proportion of a broad cell class or signature (neurons, microglia and GFAP+ astrocytes) from the images as the fraction of nuclei stained with the marker of interest (NeuN, IBA1 or GFAP, respectively, for neurons, microglia and GFAP+ astrocytes) out of all nuclei stained by DAPI.
For spatial transcriptomics with Visium, the cerebral cortex and the underlying white matter of fresh frozen brain tissue were obtained from the New York Brain Bank for six aging individuals and dissected on dry ice. The samples from each subject were prepared into 10 × 10-mm2 size tissue in cryo embedding matrix (OCT), and sectioned at 10-μm thickness in duplicate onto a slide containing capture probes. Sections were fixed with cold 100% methanol for 30 min and then stained with hematoxylin and eosin for 7 min at room temperature. Sections were scanned using a Leica microscope. After image capture, tissue sections were permeabilized to induce cDNA synthesis and a cDNA library was generated. The permeabilization time was optimized for the ST Visium platform and RNA quality, selecting sections with RIN > 7. Libraries were sequenced and aligned with the hematoxylin and eosin images using the Space Ranger software. The quantification and visualization of genes per spot was done using the Seurat package v.4.1.0.
Ligand-receptor analysis
We searched for ligand-receptor expression as an indication of signaling within and between cellular communities, focusing on the cognitive decline community (Inh.1, Inh.7, Oli.4, Ast.4, End.2 subsets) and the cognitive nonimpaired community (Inh.3, Oli.1, End.4, End.1, Ast.2 subsets). Ligand and receptor interactions were assembled from published resources37,38 and manually curated. We searched for statistically significant expressed LRPs between pairs of cell subsets using CellPhoneDB 2.0 (ref. 39). Briefly, CellPhoneDB computes the average expression of an LRP within a pair of subsets and computes an empirical P value for the specificity of this interaction by permuting cell subset identity (1,000 times). Community-specific differential LRPs were defined as statistically significant LRPs where the ligand or the receptor was found to be differentially expressed within these subsets compared with all other cells of the same type (differentially expressed genes as defined). Strict community-specific LRPs were defined as community-specific LRPs where both the ligand and the receptor were differentially expressed (Extended Data Fig. 8a).
Next, we found the significant interacting pairs of cell subsets based on their overall numbers of LRP interactions, community-specific LRPs or strict community-specific LRPs. We calculated an empirical P value by a permutation test (10,000 permutations) randomizing the ligand and receptor cell subset assignments (maintaining the total number of interactions), and calculated the number of LRPs per cell subset pair.
Causal modeling between cell subtype proportions and AD endophenotypes
To assess plausible causal relationships among CelMod inferred cell subtype proportions and AD endophenotypes, we performed a series of linear and mediation analyses. We built our models on the known sequence of AD progression which starts with β-amyloid accumulation, followed by tau aggregation and cognitive decline. We focused on cell subtypes associated with tau pathology that were also nominally associated with Aβ (P < 0.05).
We first tested the following linear model to assess the cell subtypes’ positions in the causal chain including β-amyloid and tau, testing different scenarios for the effect of each cell subset proportion: upstream of Aβ, between Aβ and tau, or downstream of tau:
If β1 is attenuated after including tau in the model, tau is either a confounder (independent effect on both β-amyloid and cell subtype proportion) or a mediator (relaying β-amyloid effect on cell subtypes)63. However, we assume that the direction of effect is from β-amyloid to tau based on the literature41,42, and thus tau will be inferred to be a mediator not a confounder, and cell subtype proportion will thus be inferred to be an outcome downstream of tau (the mediator). Since the first linear model indeed inferred the majority of subtype proportions to be downstream of tau, we therefore set the following mediation model in this case:
Here, β-amyloid is the independent causal variable, tau is the mediator and cell subtype proportion is the continuous outcome (dependent variable). We used R ‘mediation’ package64 to perform mediation analysis using this model. Mediated (indirect) effect, direct effect and proportion mediated were estimated using a non-parametric bootstrap mediation model in this casewith 10,000 simulations.
Similarly, for the cell subtype proportions that are likely downstream of tau, we assessed their impact on the downstream cognitive decline. Here, we assumed that cognitive decline is downstream of brain cell subtype proportion changes, as this is a more plausible direction of causation (rather than a model assuming that poor cognitive performance leads to brain cell subtype changes). Here, the mediation model we used is:
Here, tau is the independent causal variable, cell subset proportion is the mediator and cognitive decline (slope) is the continuous outcome (dependent variable). Mediation analysis was performed using the same package and setting as above.
Finally, we estimated the joint effect of cell subtype proportions that mediate tau effect on cognition, comparing β1 (effect size of tau) of the following two linear models:
Extended Data
Extended Data Fig. 1 |. A cellular-molecular map of the human aging DLPFC: Quality controls.
(a) High quality nuclei libraries generated across n = 24 postmortem independent samples of the DLPFC brain region of aging individuals. Nuclei (dots) colored by the doublet score (Methods). (b) Distribution of cell type frequencies across batches. The fraction of nuclei (y-axis) per cell type for each batch (x-axis, n = 3 batches). (c, d) Distribution of number of genes (c) and transcripts (d) across the 9 major cell types in the DLPFC. Violin plots showing the distribution per cell type (for n = 172,659 nuclei). (e, f) Distribution of cell type frequencies across sex (e, n = 24 independent samples, 12 per group) and archetypes of AD (f, n = 24, 6 per group). Boxplot showing the fraction of nuclei per cell type for males (blue, n = 12) and females (red, n = 12). For box plots, the bottom and upper borders show the first and third quartiles. The central line indicates the median. The whiskers are extended to the extrema values (without accounting for outliers). Dots show individual samples. Archetypes defined as in Fig. 1a, b.
Extended Data Fig. 2 |. Quality controls of neuronal subtypes and their cortical layer specificity.

(a) Distinct expression of known and de novo marker genes in excitatory neuronal subtypes (top) and inhibitory neuronal subtypes (bottom) as assigned by our clustering analysis. Mean expression level in expressing cells (color) and percent of expressing cells (circle size) of selected markers in each neuronal subtype (rows) of marker genes. (b-c) Distribution of neuronal subtype frequencies across sex or batch for n = 24 independent sample. Boxplots showing the fraction of nuclei per neuronal subtype for sex (b, n=12 per group) or batch (c, n=8 per group): For box plots, the bottom and upper borders show the first and third quartiles. The central line indicates the median. The whiskers are extended to the extrema values (without accounting for outliers). Dots show outliers samples. (d) Marker genes of neuronal subtypes exhibit a spatial organization at distinct layers within DLPFC slices. Spatial transcriptomics across 6 slices from 3 individuals for five marker genes (RORB, TOX, CUX2, PVALB, SLC17A7). Note variable orientation of slices. Complementary images to Fig. 2c.
Extended Data Fig. 3 |. Quality controls of glial subsets.
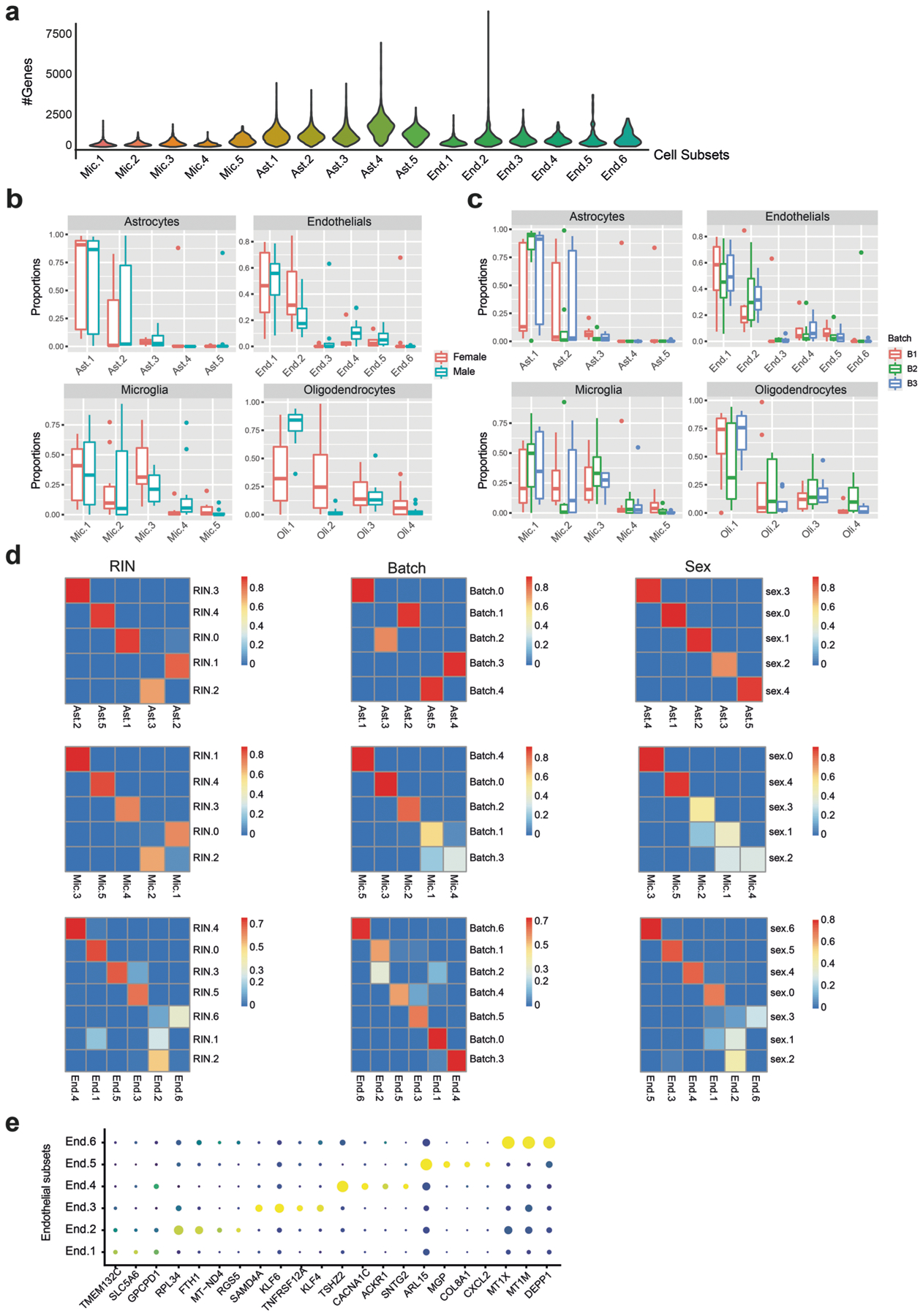
(a) Distribution of number of genes across microglia, astrocytes and endothelial clusters (denoted as subsets). (b, c) Distribution of non-neuronal subsets frequencies across sex and batch for n = 24 independent samples. Boxplot showing the fraction of nuclei per neuronal subtype for sex (b, n = 12 per group) or batch (c, n = 8 per group): For box plots, the bottom and upper borders show the first and third quartiles. The central line indicates the median. The whiskers are extended to the extrema values (without accounting for outliers). Dots show outlier samples. (d) RIN, sex and batch do not affect the sub-clustering of astrocytes, microglia and endothelial cells. Heatmaps of Jaccard score comparing overlaps of cells (color scales) between assignment of de-novo clusters after regression of the confounding variables from the expression matrix (rows) compared to the clusters in this study without such correction (columns) (Methods). (e) Endothelial subsets express unique markers. Dot plot of the mean expression level in expressing cells (color) and percent of expressing cells (circle size) of selected marker genes across endothelial subsets.
Extended Data Fig. 4 |. Comparison of cell clusters to previous studies.
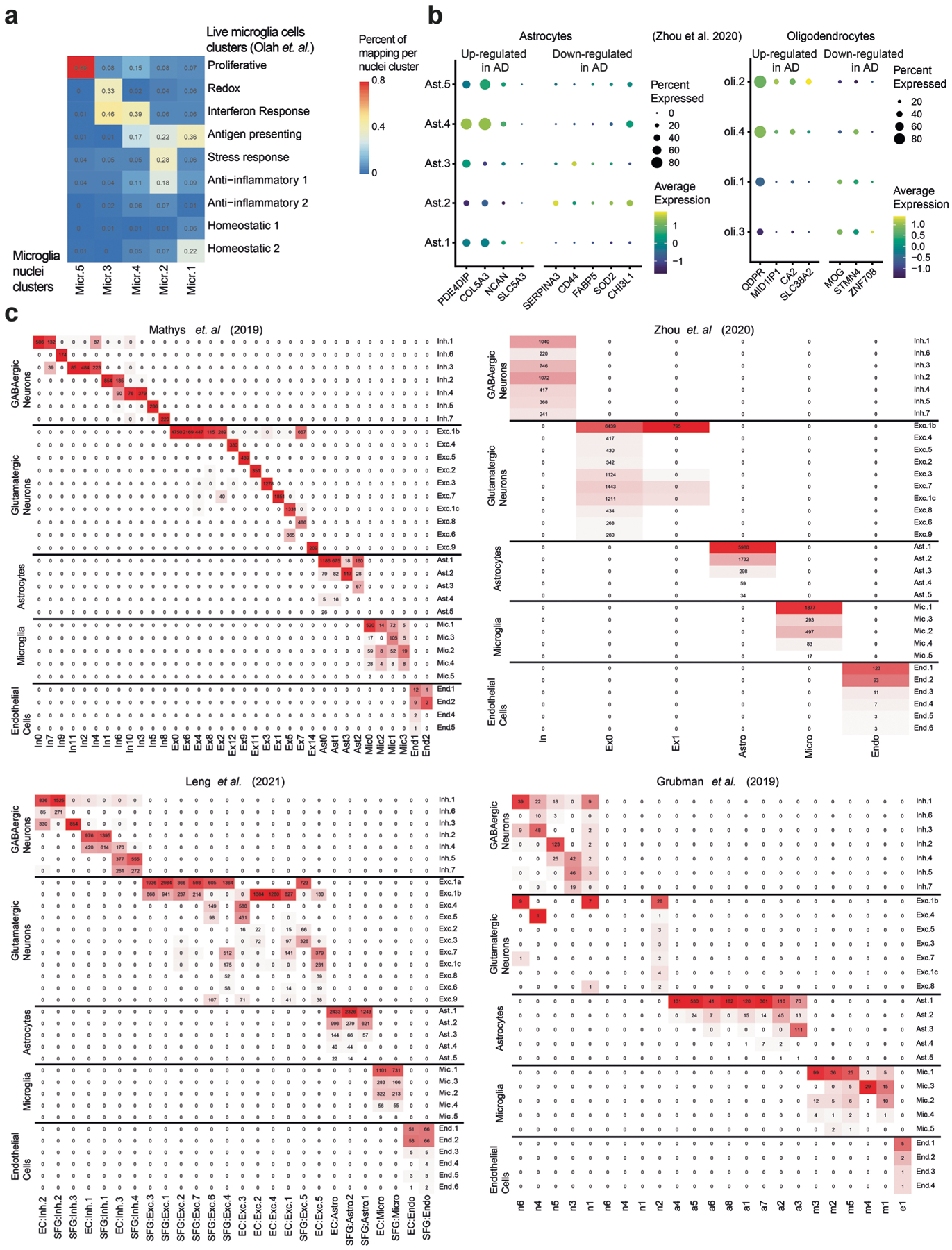
(a) Clusters of microglia nuclei from snRNA-seq match published live microglia cell clusters from scRNA-seq. The proportions (color scale, scaled per column) of nuclei per cluster (columns) mapped to each scRNA-seq cell cluster according to the best prediction (rows, Methods). (b) Mean expression level in expressing cells (color) and percent of expressing cells (circle size) across cell subsets (rows) of previously described up-regulated and down-regulated genes in AD brains compared to healthy individuals, as defined by Zhou et al.6 for astrocytes (left) and oligodendrocytes (right). (c) Nuclear-derived model is consistent with earlier, lower-resolution models across different cell types. Heatmaps (color scale) of assignment of nuclei from 24 individuals (rows) to published subsets (Methods) from 4 previous snRNA-seq derived cortical models3,5,6,25.
Extended Data Fig. 5 |. Spatial transcriptomics of glial markers.

Spatial transcriptomics of glial cell type and cell states markers, exhibit a spatial pattern across cortical layers matching DLPFC and white-matter anatomy. MBP (oligodendrocyte marker), GFAP (reactive astrocyte marker), ID3, CD44 (Ast.3 marker). Complementary to Fig.3g.
Extended Data Fig. 6 |. CelMod evaluation and comparison to other methods and datasets.
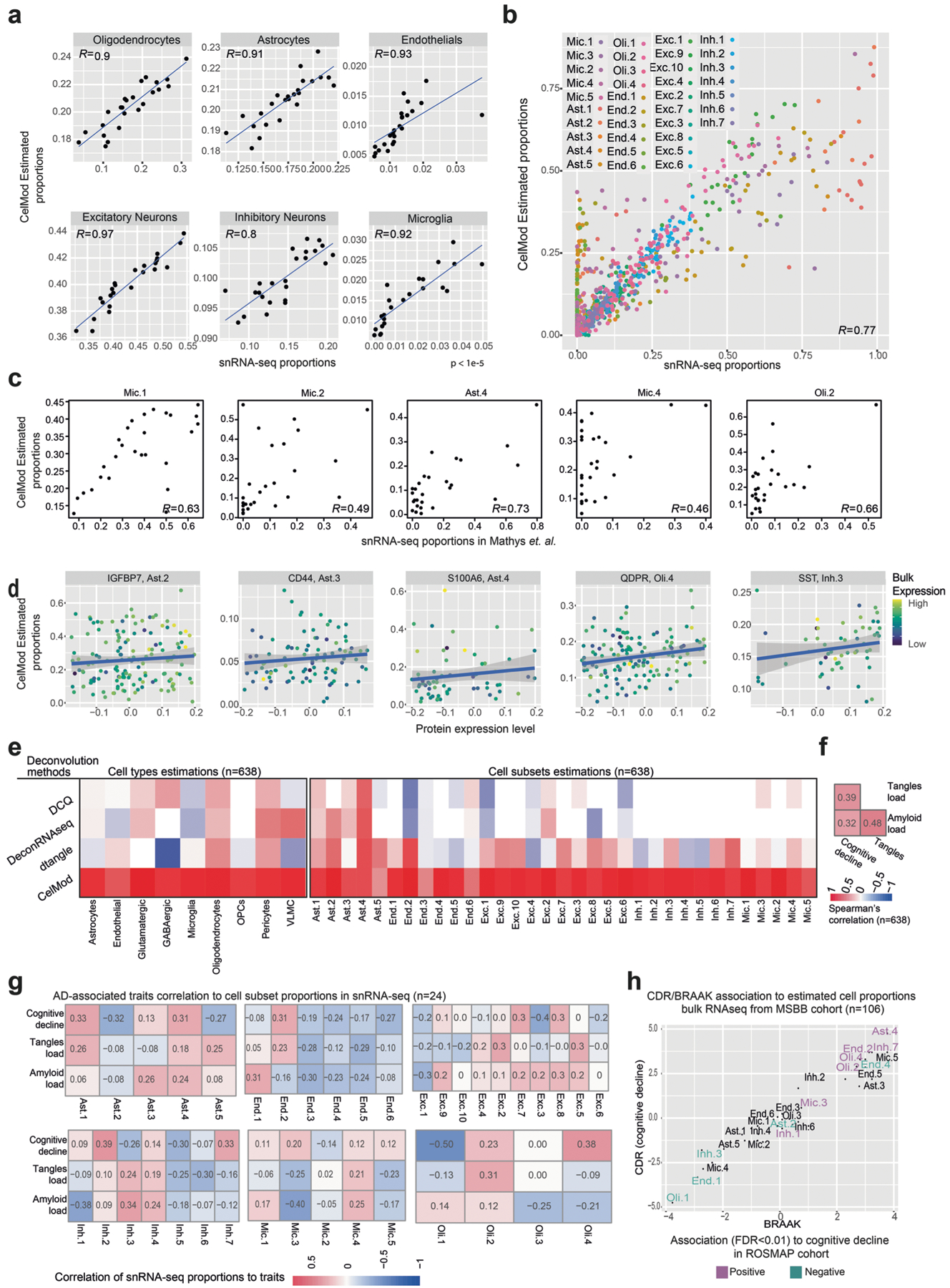
(a, b) CelMod estimated proportions match sn-RNAseq measured proportions. Scatter plots of CelMod proportions (Y-axis) compared to snRNA-seq proportions (X-axis), for 6 major cell classes (a) and all cell subsets and topic models (b, colored by subset). Line: linear regression. Each point is a sample (n = 24 independent samples). (c) Validation of CelMod in an independent dataset. CelMod estimated proportions of cell subsets (Y-axis) compared to snRNA-seq proportions (X-axis) measured across n = 48 independent samples of the prefrontal cortex Brodmann area 10 (Mathys et al.3). Each point is a single individual. Cell subset annotations are based on our cell atlas (Methods). R=Correlation. (d) Protein expression of selected markers reflect cell subsets abundance. Scaled protein expression levels (X-axis) compared to CelMod estimated proportions of the related cell subset, colored by the bulk RNA-seq (proteomics30 in n = 196 independent individuals with matching bulk RNA profiles). Each point is a single individual. Line: linear regression fit with confidence interval (grey). (e) CelMod outperforms previous methods. Spearman correlation scores (color scale) of the snRNA-seq measured proportions of each cell type (left) and cell subset (right), compared to the estimated proportions by CelMod and three previous models. (f) Correlations between AD traits within our data. Pairwise correlations (color scale) of the three traits across n = 638 independent individuals. (g) Measured proportions of cellular subsets from snRNA-seq in n = 24 individuals correlate with AD pathology and cognitive decline. Correlation (color scale) of the proportions of each cell subset (columns) to AD-traits (rows). The proportions are calculated over the total number of nuclei per individual (n = 24) within each cell. (h) Cellular proportions associations to AD-ranking in the independent MSBB32 cohort matches the ROSMAP1 cohort. Association scores −log(FDR) ×sign (β), by multivariable linear regression) of proportions of cell subsets to two measures of AD ranking: BRAAK stage (tangles load) and the CDR (level of cognitive decline), in n = 106 independent samples32 (Methods). Cell subsets colored by the statistical significance of associations to the cognitive decline rate in the ROSMAP cohort1 (n = 638).
Extended Data Fig. 7 |. Evaluation of multi-cellular communities.
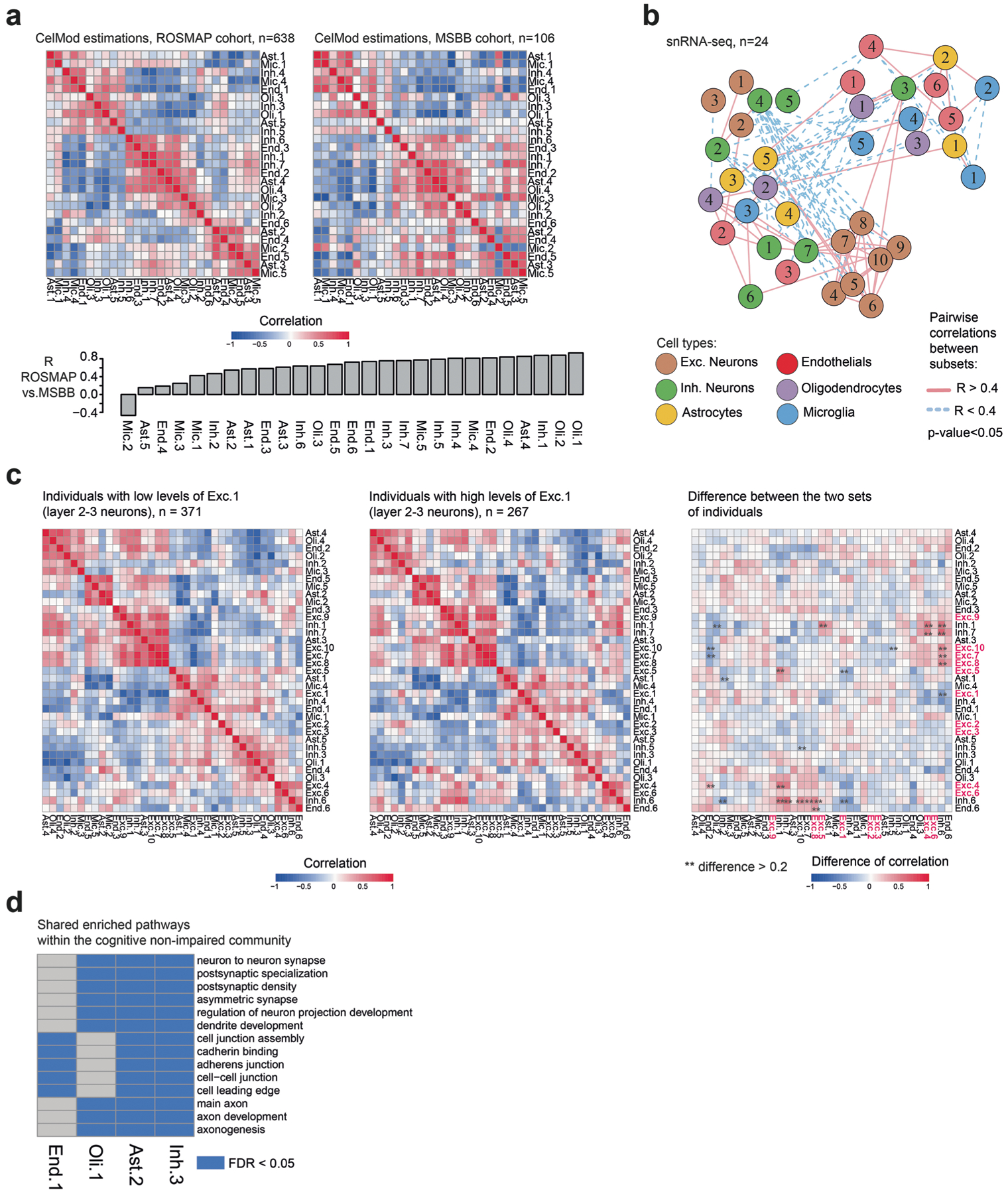
(a) Similar structure of coordinated changes between proportions of cell subsets and neuronal subtypes across individuals found in two independent cohorts. Pairwise Spearman correlation coefficients of the proportions of cell states and subtypes across individuals estimated by CelMod in bulk RNA-seq data of ROSMAP1 (left) and MSBB32 (right) cohorts. Bottom: Correlation between the pairwise association pattern of the ROSMAP and the MSBB cohorts per cell subset. (b) A network of cellular subsets reveals coordinated variation across individuals in multiple cell types. Network of coordinated and anti-coordinated cell subsets (nodes). Edges between pairs of subsets with statistically significant correlated proportions across individuals (r>0.4, p-value threshold=0.05, solid red line) or anti-correlated (r < −0.4, dashed blue line) based on snRNA-seq proportions (n = 24 independent samples, Spearman correlation, two sided, not adjusted for multiple comparison). Celmod based network in n = 638 individuals in Fig. 6b. Nodes are colored by the cell type and numbered by the subset as in Fig. 2a and Fig. 3a, d, h). (c) Coordinated changes in proportions of cell states and subtypes across individuals is independent of the cortical layer, except for excitatory neurons. Pairwise Spearman correlation coefficient of the CelMod proportions of all cell subsets and neuronal subtypes across individuals with low levels of Exc.1 (n = 371 individuals, left) or high levels of Exc.1 (n = 267 individuals, middle). Right: The differences in pairwise correlations between the Exc.1-high and Exc.1-low groups of individuals. Showing the partition mainly affects excitatory neurons (red). (d) Shared pathways within the cognitive non-impaired community. Enriched pathways (hypergeometric test, FDR q-value < 0.05, blue) in up-regulated genes for each cell subset within the cognitively non-impaired community (End.1, Oli.1, Ast.2, and Inh.3). Displaying shared enriched pathways between at least three subsets.
Extended Data Fig. 8 |. Signaling within and between multi-cellular communities and causal modeling.
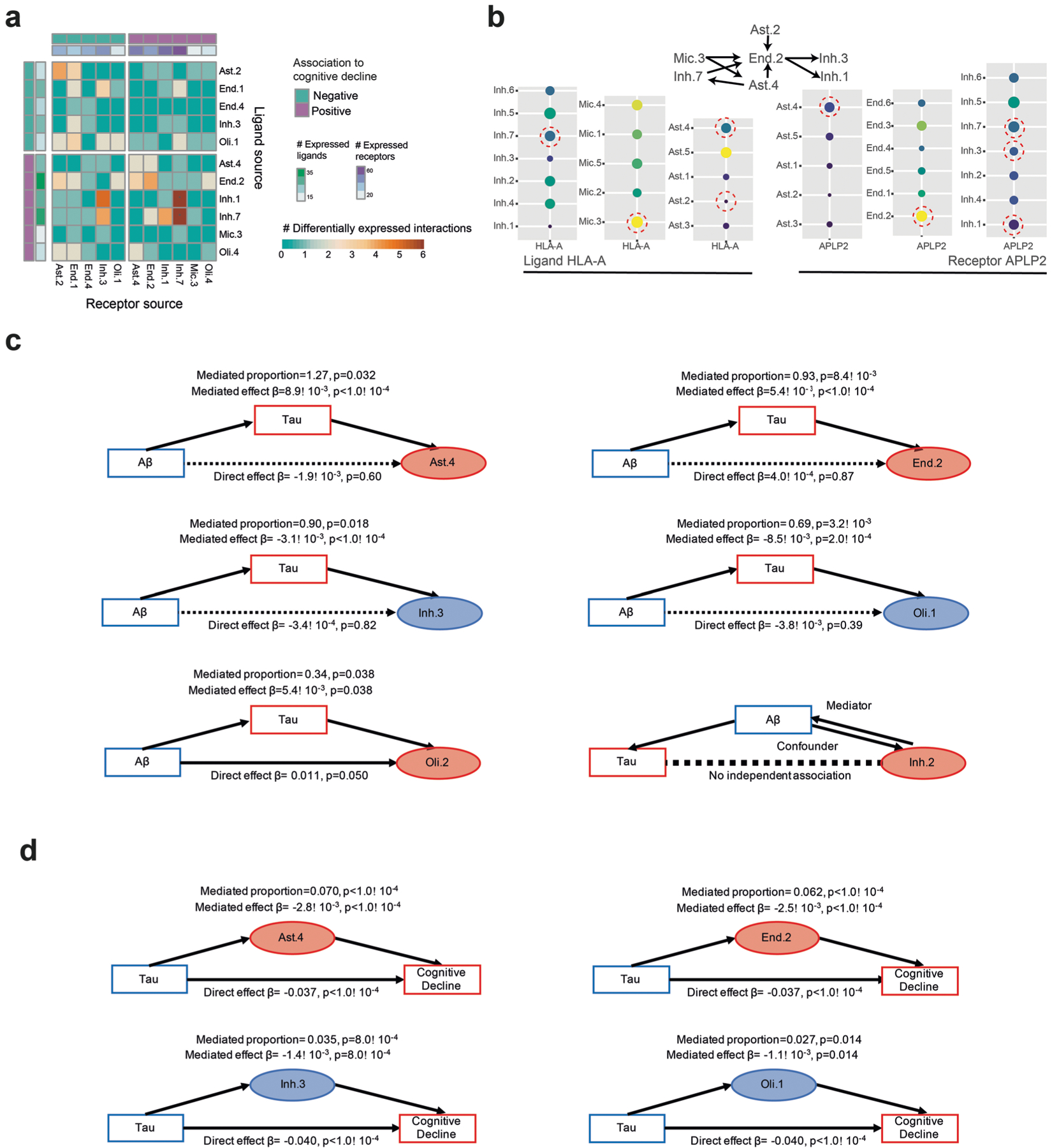
(a) Cell subsets positively associated with cognitive decline have an increased expression of ligand-receptor pairs compared to the negatively associated subsets. For each pair of cell subsets showing the number (color scale) of ligand-receptor pairs (LRP, row: ligand, column: receptor) where both the ligand and the receptor are differentially expressed in the relevant subsets. Top and side bar marking: subsets positively (purple) or negatively (turquoise) associated with cognitive decline, and the total number of ligands and receptors expressed (color scale). (b) Expression of the HLA-A - APLP2 ligand-receptor pair across subtypes of different cell types. Dot plot of the mean expression level in expressing cells (color) and percent of expressing cells (circle size) of HLA-A and of APLP2 across subsets of selected cell types. (c) Mediation analysis results showing tangle pathology burden (tau) is predicted to be upstream of changes in proportion of Inh.3, Oli.1, Oli.2, Ast.4, and End.2, but not Inh.2. (d) Mediation analysis results showing partial effect of changes in proportion of Inh.3, Olig.1, Ast.4, and End.2 on cognitive decline independent of tau pathology burden.
Supplementary Material
Acknowledgements
We thank the individuals who have generously donated their brain to research through the RUSH University Alzheimer’s Disease Center. This work was supported by the National Institute of Aging (NIA), grant nos. AMP-AD U01AG046152, U01AG061356 and RF1AG036042 (P.L.D. and D.A.B); grant nos. R01AG066831 and R21AG075754 and SenNet (grant no. U54AG076040) (V.M.); grant nos. P30AG10161, P30AG72975, R01AG15819 and R01AG17917 (ROSMAP, P.L.D.); the Chan Zuckerberg Initiative (grant no. CS-02018-191971) (P.L.D. and V.M.); Israel Science Foundation (ISF) grant no. 1709/19 (N.H.); the European Research Council grant no. 853409 (N.H.); grant no. MOST/IL 3-15687 (N.H.); and the Klarman Cell Observatory (A.R.). N.H. is a Goren Khazzam senior lecturer in neuroscience and is support by the Myers Foundation. F.Z. and A.R. are investigators of the Howard Hughes Medical Institute.
Footnotes
Code availability
The code for the CelMod deconvolution algorithm is available at: https://github.com/MenonLab/Celmod. Other code used within this study is available at: https://github.com/naomihabiblab/HumanDLPFC24.
Competing interests
A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until 31 August 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. From 1 August 2020, A.R. is an employee of Genentech, a member of the Roche Group. The remaining authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-023-01356-x.
Extended data is available for this paper at https://doi.org/10.1038/s41593-023-01356-x.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-023-01356-x.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
snRNA-seq data were deposited in the AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn16780177) and processed data can be browsed online at:https://vmenon.shinyapps.io/rosmap_snrnaseq24/. The bulk RNA-seq dataset can be accessed through the Synapse database (https://www.synapse.org/#!Synapse:syn3388564). The proteomic dataset can be accessed through the Synapse database (https://www.synapse.org/#!Synapse:syn17015098). All datasets are available for general research use according to the following requirements for data access and data attribution: https://adknowledgeportal.synapse.org/#/DataAccess/Instructions. Other ROSMAP resources can be requested at the RADC Resource Sharing Hub at https://www.radc.rush.edu. Source data are provided with this paper.
References
- 1.Mostafavi S et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci 21, 811–819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wan Y-W et al. Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 32, 107908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del-Aguila JL et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res. Ther 11, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grubman A et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olah M et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun 10.1038/s41467-020-19737-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett DA et al. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers Dis 64, S161–S189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett DA et al. Overview and findings from the Rush Memory and Aging Project. Curr. Alzheimer Res 9, 646–663 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett DA, Schneider JA, Arvanitakis Z & Wilson RS Overview and findings from the Religious Orders Study. Curr. Alzheimer Res 9, 628–645 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Jager PL et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 180142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thrupp N et al. Single-nucleus RNA-seq is not suitable for detection of microglial activation genes in humans. Cell Rep. 32, 108189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakken TE et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 13, e0209648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girgrah N et al. Localization of the CD44 glycoprotein to fibrous astrocytes in normal white matter and to reactive astrocytes in active lesions in multiple sclerosis. J. Neuropathol. Exp. Neurol 50, 779–792 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Leitão RA et al. Aquaporin-4 as a new target against methamphetamine-induced brain alterations: focus on the neurogliovascular unit and motivational behavior. Mol. Neurobiol 55, 2056–2069 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Yang AC et al. A human brain vascular atlas reveals diverse mediators of Alzheimera’s risk. Nature 603, 885–892 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pritchard JK, Stephens M & Donnelly P Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blei DM, Ng A & Jordan MI Latent Dirichlet allocation. J. Mach. Learn. Res 3, 993–1022 (2003). [Google Scholar]
- 21.Dey KK, Hsiao CJ & Stephens M Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet. 13, e1006599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bielecki P et al. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592, 128–132 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker KA et al. Large-scale plasma proteomic analysis identifies proteins and pathways associated with dementia risk. Nat. Aging 1, 473–489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster EM, Dangla-Valls A, Lovestone S, Ribe EM & Buckley NJ Clusterin in Alzheimer’s disease: mechanisms, genetics, and lessons from other pathologies. Front. Neurosci 10.3389/fnins.2019.00164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leng K et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci 24, 276–287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altboum Z et al. Digital cell quantification identifies global immune cell dynamics during influenza infection. Mol. Syst. Biol 10, 720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong T & Szustakowski JD DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA-Seq data. Bioinformatics 29, 1083–1085 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Hunt GJ, Freytag S, Bahlo M & Gagnon-Bartsch JA dtangle: accurate and robust cell type deconvolution. Bioinformatics 35, 2093–2099 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Patrick E et al. Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. PLoS Comput. Biol 16, e1008120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson ECB et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26, 769–780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edfors F et al. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol 12, 883 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farfel JM, Yu L, De Jager PL, Schneider JA & Bennett DA Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 37, 19–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett DA et al. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J. Neurol. Neurosurg. Psychiatry 76, 1194–1199 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neff RA et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv 7, eabb5398 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jha MK, Jo M, Kim J-H & Suk K Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25, 227–240 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Vainchtein ID & Molofsky AV Astrocytes and microglia: in sickness and in health. Trends Neurosci. 43, 144–154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiao W et al. Intercellular network structure and regulatory motifs in the human hematopoietic system. Mol. Syst. Biol 10, 741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramilowski JA et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun 10.1038/ncomms8866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 40.West DC et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J. Biol. Chem 280, 13457–13464 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Jack CR Jr et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanseeuw BJ et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sperling RA et al. The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Ann. Neurol 85, 181–193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jennrich RI An asymptotic Χ2 test for the equality of two correlation matrices. J. Am. Stat. Assoc 65, 904 (1970). [Google Scholar]
- 46.Bennett DA et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 10.1212/01.wnl.0000219668.47116.e6 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Wilson RS et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol. Aging 17, 179–193 (2002). [PubMed] [Google Scholar]
- 48.Wilson RS, Barnes LL & Bennett DA Assessment of lifetime participation in cognitively stimulating activities. J. Clin. Exp. Neuropsychol 25, 634–642 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Wilson RS et al. Conscientiousness, dementia related pathology, and trajectories of cognitive aging. Psychol. Aging 30, 74–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Jager PL et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol. Aging 33, 1017.e1–1017e. 15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKhann G et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- 52.Schneider JA, Arvanitakis Z, Bang W & Bennett DA Mixed brain pathologies account for most dementia cases in communitydwelling older persons. Neurology 69, 2197–2204 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Bennett DA et al. Natural history of mild cognitive impairment in older persons. Neurology 59, 198–205 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Bennett DA et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 27, 169–176 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Hyman BT & Trojanowski JQ Editorial on consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer Disease. J. Neuropathol. Exp. Neurol 56, 1095–1097 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mclnnes L, Healy J, Saul N & Großberger L UMAP: uniform manifold approximation and projection. J. Open Source Softw 3, 861 (2018). [Google Scholar]
- 59.Lancichinetti A & Fortunato S Benchmarks for testing community detection algorithms on directed and weighted graphs with overlapping communities. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 80, 016118 (2009). [DOI] [PubMed] [Google Scholar]
- 60.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol. Aging 18, S1–S2 (1997). [PubMed] [Google Scholar]
- 63.MacKinnon DP, Krull JL & Lockwood CM Equivalence of the mediation, confounding and suppression effect. Prev. Sci 1, 173–181 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tingley D, Yamamoto T, Hirose K, Keele L & Imai K mediation: R package for causal mediation analysis. J. Stat. Softw 59, 1–38 (2014).26917999 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
snRNA-seq data were deposited in the AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn16780177) and processed data can be browsed online at:https://vmenon.shinyapps.io/rosmap_snrnaseq24/. The bulk RNA-seq dataset can be accessed through the Synapse database (https://www.synapse.org/#!Synapse:syn3388564). The proteomic dataset can be accessed through the Synapse database (https://www.synapse.org/#!Synapse:syn17015098). All datasets are available for general research use according to the following requirements for data access and data attribution: https://adknowledgeportal.synapse.org/#/DataAccess/Instructions. Other ROSMAP resources can be requested at the RADC Resource Sharing Hub at https://www.radc.rush.edu. Source data are provided with this paper.