

SUMMARY
The role that human papillomavirus (HPV) oncogenes play in suppressing responses to immunotherapy in cancer deserves further investigation. In particular, the effects of HPV E5 remain poorly understood relative to E6 and E7. Here, we demonstrate that HPV E5 is a negative regulator of anti-viral interferon (IFN) response pathways, antigen processing, and antigen presentation. Using head and neck cancer as a model, we identify that E5 decreases expression and function of the immunoproteasome and that the immunoproteasome, but not the constitutive proteasome, is associated with improved overall survival in patients. Moreover, immunopeptidome analysis reveals that HPV E5 restricts the repertoire of antigens presented on the cell surface, likely contributing to immune escape. Mechanistically, we discover a direct interaction between E5 and stimulator of interferon genes (STING), which suppresses downstream IFN signaling. Taken together, these findings identify a powerful molecular mechanism by which HPV E5 limits immune detection and mediates resistance to immunotherapy.
Graphical Abstract
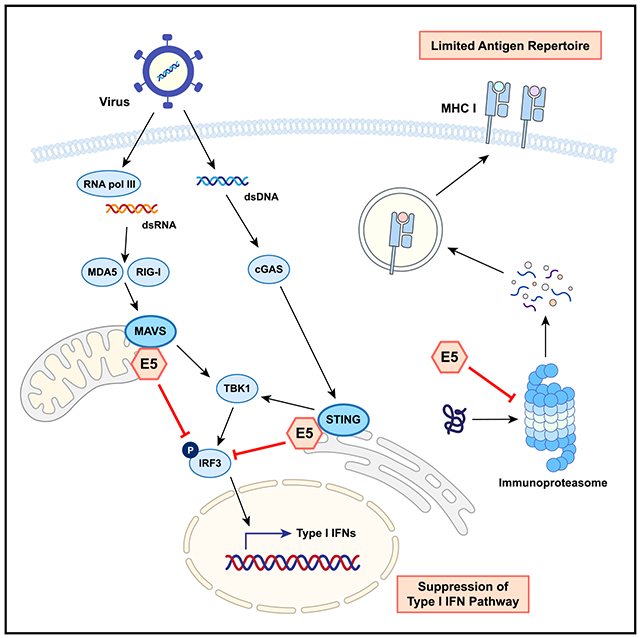
In brief
Miyauchi et al. report that the HPV E5 oncoprotein functions as a powerful negative regulator of anti-viral response pathways by blocking STING and MAVS and inhibiting the immunoproteasome. This study reveals a mechanism of action of E5 that enables virally infected cancer cells to escape host immune surveillance.
INTRODUCTION
Human papillomavirus (HPV) is estimated to be involved in approximately 5% of all cancers worldwide and is the causative agent for the vast majority of cervical cancers, oropharyngeal head and neck squamous cell carcinomas (HNSCCs), anal cancers, and other gynecologic and genitourinary malignancies.1 The incidence of HPV-associated HNSCC has increased dramatically over the past two decades, and oropharyngeal HNSCC is currently one of the fastest-rising cancers worldwide.2,3 Interestingly, HPV infection can occur decades prior to malignant transformation and cancer diagnosis.4 There are over 170 strains of HPV, with some strains carrying a greater risk for development of cancer. The HPV16 strain carries a particularly high cancer risk and is present in over 86% of HPV-positive cancers.2 The HPV16 genome is comprised of early genes (E2, E4, E5, E6, and E7) and late genes (L1 and L2). The functions of E6 and E7 have been extensively studied, and these genes are commonly expressed in many HNSCC models.5 However, the functions of E5 remain understudied, and HPV-associated cancer models do not widely feature E5. E5 is an 83-amino-acid-long small hydrophobic protein that localizes mainly in the endoplasmic reticulum (ER) and Golgi apparatus membranes. Although less is known about E5 compared with other oncoproteins, several functions of E5 have been reported, including effects on cell differentiation and cell cycle progression.6,7 Importantly, E5 can also modulate the host immune response via downregulation of major histocompatibility complex (MHC) class I,8,9 and inhibition of acidification of late endosomes by modulating H+ ATPase.10 Previously, we reported that HPV E5 mediates resistance to anti-PD-L1 blockade immune therapy in HNSCC11 and demonstrated the impact of E5 expression on treatment efficacy and patient outcomes. HNSCC patients with low expression of E5 and high expression of human leukocyte antigen (HLA) showed prolonged disease-free and overall survival.11 However, the broader role of E5 and the molecular mechanisms underlying the functions of E5 remain unclear.
Antigen processing and presentation are critical for antigen-specific anti-viral and anti-tumor immune responses. The proteasome is an organelle and structural complex that cleaves and degrades proteins, including ubiquitinated proteins and unfolded or misfolded proteins.12 When activated, the constitutive proteasome can transform into the immunoproteasome, which efficiently generates immunogenic peptides and antigenic epitopes that can be loaded and anchored into MHC class I.13 Immunoproteasomes are commonly expressed in hematopoietic cells. In non-immune cells, stimulation with factors such as tumor necrosis factor alpha (TNF-α), interferon (IFN)-α/β, or IFN-γ can induce the immunoproteasome, resulting in subunits of the constitutive proteasome, β1 (proteasome subunit, beta [PSMB] 6), β2 (PSMB7), and β5 (PSMB5), being replaced with the immunoproteasome-specific subunits β1i (PSMB9/low molecular mass protein [LMP] 2), β2i (PSMB10/multicatalytic endopeptidase complex-like 1 [MECL1]), and β5i (PSMB8/LMP7), respectively.14,15 Each subunit has a different peptidase activity. β1i has a branched-chain amino acid-preferring activity, β2i has a trypsin-like activity, and β5i has a chymotrypsin-like activity. Mice deficient in all three immunoproteasome subunits showed greatly impaired and altered MHC class I epitope presentation, indicating that the immunoproteasome plays a critical role in antigen processing.16 Given that several viruses, including HPV, enter a latent phase in which they are dormant and go largely undetected by the immune system, the role that HPV oncogenes have in suppressing immune responses via the immunoproteasome deserves further investigation.
Here, we investigated the effects of HPV E5 on antigen processing, antigen presentation, responses to immunotherapies, and cancer patient outcomes. Our studies revealed that HPV E5 is a powerful negative regulator of anti-viral type I IFN response pathways and immunoproteasome expression and function, resulting in a decreased repertoire of presented peptides. Furthermore, we identified that E5 directly binds and blocks a mitochondrial antiviral-signaling protein (MAVS) and stimulator of IFN genes (STING), resulting in impaired anti-tumor immunity and immunotherapy activity. Thus, our studies reveal a mechanism of action of HPV E5, one that enables infected cancer cells to escape host immune surveillance and resist the activity of immunotherapies.
RESULTS
HPV E5 downregulates anti-virus and type I IFN responses
Despite accumulating evidence demonstrating the importance of E5 in the HPV viral life cycle, the detailed mechanisms of action of E5 remain poorly understood. To understand the global alteration of the transcriptome in E5-positive cells, we performed bulk RNA sequencing (RNA-seq) analysis on HPV E5-positive cell lines from two different origins: the human head and neck carcinoma cell line CAL-27 and the murine dendritic cell line DC2.4. HPV E5 was stably transfected in the cell lines, and mRNA expression levels were compared with the parental line with an empty vector control (Figure S1; Tables S1, S2, S3, and S4). In CAL-27 cells, a total of 184 genes were differentially expressed upon HPV E5 overexpression, with 114 genes downregulated and 70 genes upregulated (Figure 1A). This is consistent with HPV E5 functioning predominantly as a negative regulator. Results from DC2.4 cells also showed a greater number of genes downregulated versus upregulated by HPV E5 (Figure 1A). Despite these cell lines being from different species and different tissue origins, we identified 50 shared genes that were downregulated by HPV E5 in both cell lines (Figure 1B). Remarkably, further analysis revealed that 92% (46 of 50) of these shared downregulated genes were type I IFN-inducible genes (Figure 1B). Gene Ontology analysis of the downregulated genes revealed that almost every pathway involved regulation of viral processes or IFN responses, including viral life cycle, viral replication, viral defense responses, and regulation and cellular responses to type I IFN (Figure 1C). Results were similar in the analysis with Reactome (Figure S1B), and, as expected, HPV infection was one of the top hits in the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure S1C). To better visualize the impact of HPV E5 expression, we generated heat-maps demonstrating the broad downregulation of critical genes involved in type I IFN and viral responses compared with parental lines expressing empty vector (Figure 1D). Major type I IFN-inducible genes, including IFIT1, IFIT3, ISG15, MX1, and IRF7, were downregulated. Interestingly, certain pathways involved in keratinocyte and epidermal development were upregulated, likely secondary to distinct effects of E5 in early stages of the HPV viral life cycle in basal epithelial cells. Additionally, neutrophil and myeloid cell activation pathways were also upregulated, effects associated with compensatory impairment in adaptive immune responses. Because neutrophils and myeloid cells have immune-suppressive functions during tumor development,17–19 this upregulation is consistent with E5 suppressing antigen-specific host adaptive immune responses. These data demonstrate that HPV E5 downregulates anti-virus and type I IFN responses across species and cell origins.
Figure 1. HPV E5 downregulated anti-virus and type I IFN response pathways.
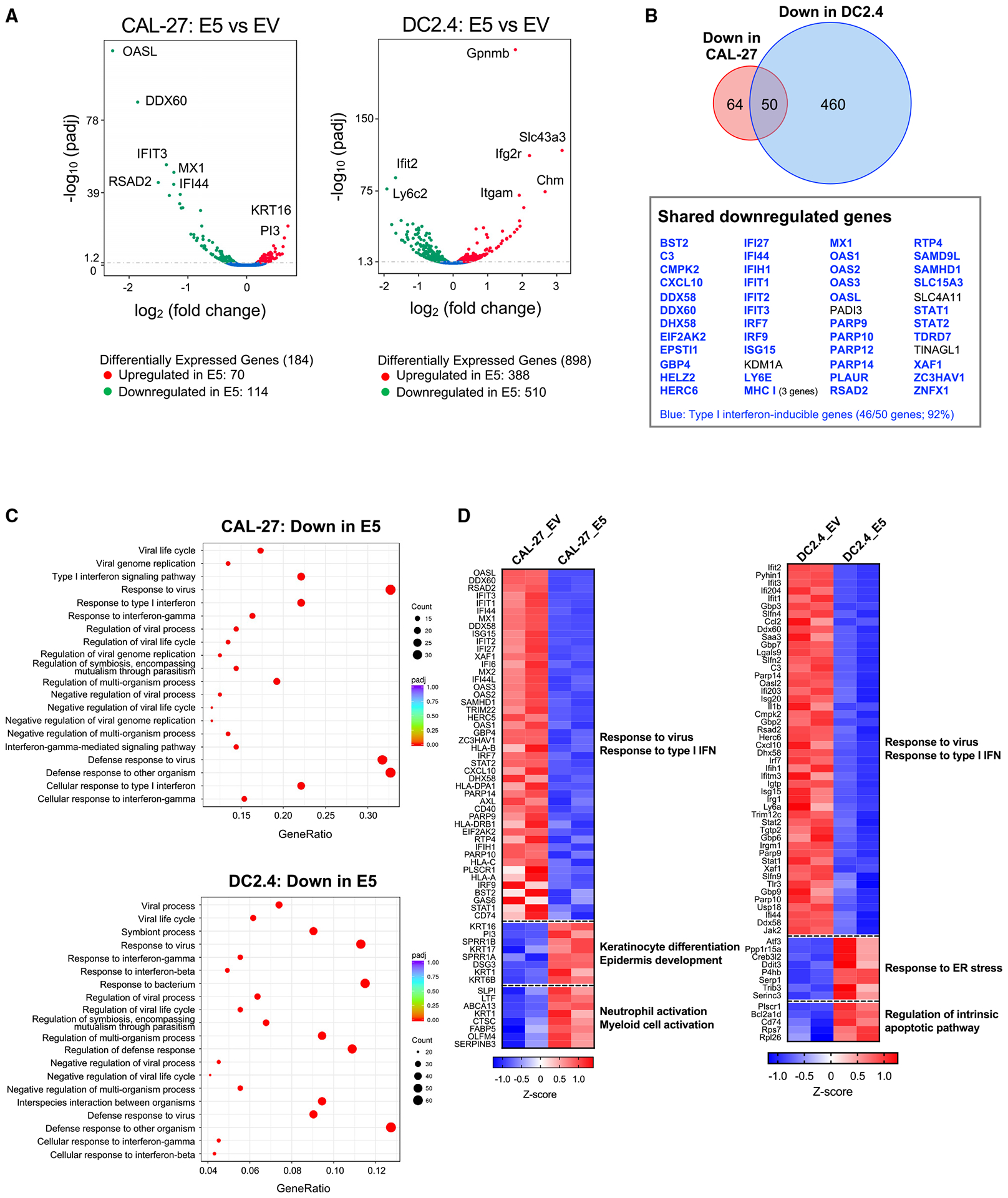
The transcriptomes of HPV E5-expressing CAL-27 and DC2.4 cells were analyzed by RNA-seq.
(A) Volcano plot of differentially expressed genes. Genes with p < 0.05 were defined as differentially expressed.
(B) Venn diagram of downregulated genes in CAL-27 and DC2.4 cells.
(C) Downregulated gene sets in CAL-27 and DC2.4 cells were analyzed by GO. The top 20 pathways are shown.
(D) Heatmap of genes assigned to the downregulated and upregulated pathways.
HPV E5 downregulates antigen-processing pathways
To further understand the impact of HPV E5 on immuneresponses, we analyzed processes downstream of type I IFN; namely, antigen processing and presentation pathways. The RNA-seq data revealed that major IFN-inducible transcriptional factors, including signal transducer and activator of transcription (STAT) 1, STAT2, and other IFN regulatory factors (IRFs), were downregulated in E5-expressing cell lines. One of the downstream pathways regulated by STAT1 is PSMB9 (LMP2), a subunit of the immunoproteasome. Upon stimulation by IFN, immunoproteasomes are generated from constitutive proteasomes by replacing three subunits. The immunoproteasome is highly efficient at catalyzing the production of peptides that bind MHC class I.13 We hypothesized that downregulation of IFN pathways in E5-expressing cell lines may impact the immunoproteasome. To investigate this, two major subunits of the immunoproteasome, PSMB9/LMP2 and PSMB8/LMP7, were analyzed. The mRNA expression of PSMB8/Psmb8 (coding LMP7) and PSMB9/Psmb9 (coding LMP2) was downregulated in E5-positive cell lines in CAL-27 and DC2.4 cells (Figure 2A). Similar results were observed in the bulk RNA-seq data. Expression of LMP2 and LMP7 was also significantly downregulated at the protein level (Figure 2B). Another key molecule for antigen processing is transporter associated with antigen processing (TAP). Because TAP1 expression is also regulated by STAT1, PSMB9 and TAP1 genes share a bidirectional promoter.20 We observed that TAP1/Tap1 and TAP2/Tap2 were also downregulated in E5-positive cell lines (Figure 2C). We then analyzed the impact of HPV E5 expression on the functional proteolytic activity of the immunoproteasome using fluorescently conjugated substrates. Three different substrates, Ac-Ala-Asn-Trp-7-Amino-4-methylcoumarin (Ac-ANW-AMC), Ac-Pro-Ala-Leu-AMC (Ac-PAL-AMC), and Ac-Lys-Gln-Leu-AMC (Ac-KQL-AMC), were used to quantify proteolytic activity, resulting in detectable fluorescence when cleaved by the proteasome. Chymotrypsin-like activity was analyzed with Ac-ANW-AMC, which is present in LMP7, and branched amino acid-preferring activity was analyzed with Ac-PAL-AMC, which is present in LMP2. Trypsin-like activity was analyzed with Ac-KQL-AMC, which can be cleaved by the immunoproteasome and the constitutive proteasome. Cell lysate was incubated with the substrates, and the fluorescence intensity was monitored. E5-positive cell lines showed significantly less cleavage activity than EV cell lines on all substrates (Figure 2D). The difference was smaller in the KQL substrate compared with the ANW and PAL substrates because KQL is not an immunoproteasome-specific substrate. Taken together, these data demonstrate that HPV E5 impairs the functional activity of the immunoproteasome, which is critical for generating high antigenic epitopes for adaptive immune responses.
Figure 2. HPV E5 downregulated key antigen-processing molecules, immunoproteasome, and TAP.
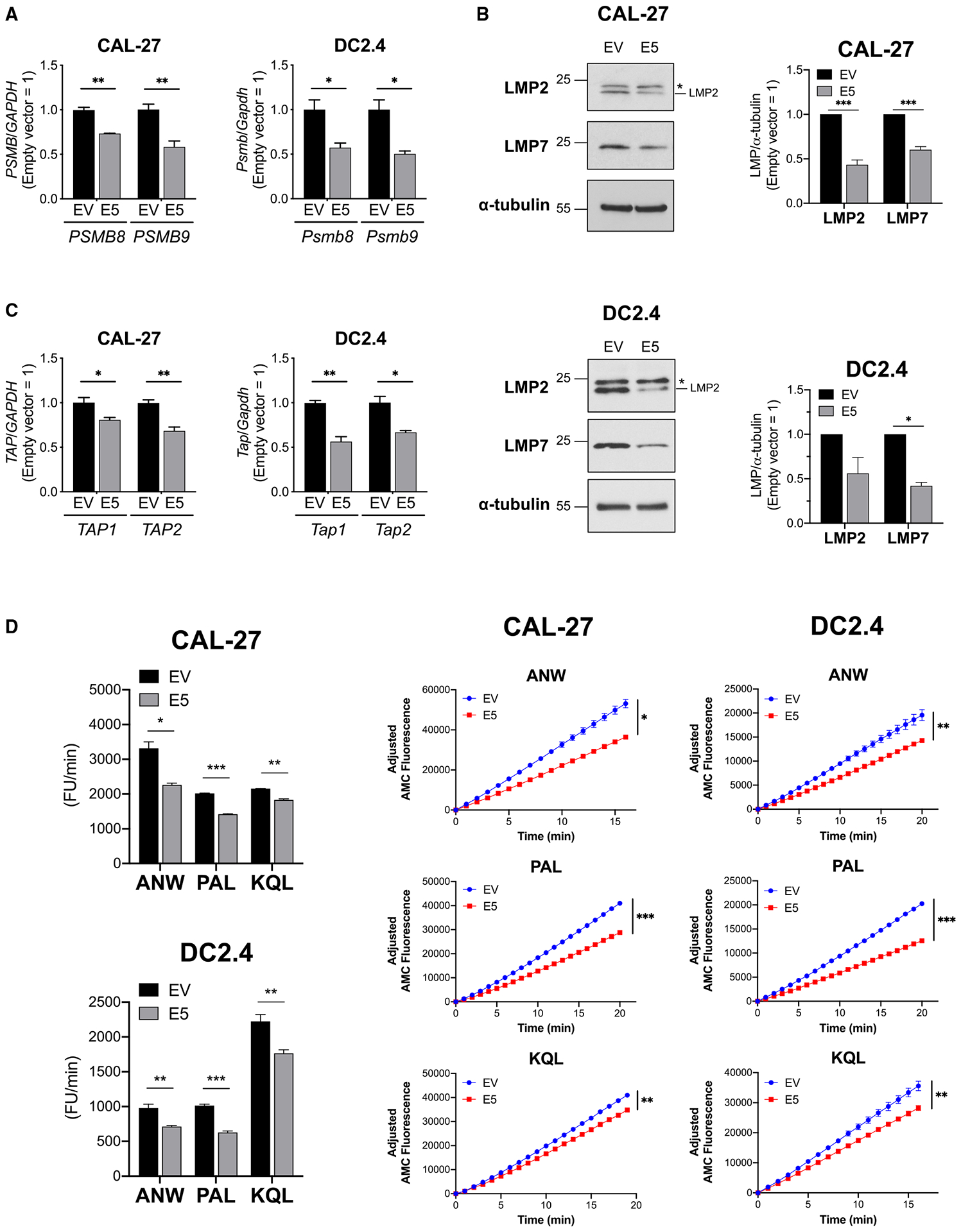
(A–C) Transcripts and proteins in HPV E5-expressing CAL-27 and DC2.4 cells were analyzed.
(A) mRNA expression of immunoproteasome subunits analyzed by qPCR.
(B) Western blot of immunoproteasome subunits. An asterisk indicates a non-specific signal.
(C) mRNA expression of TAP analyzed by qPCR.
(D) Immunoproteasome activities in HPV E5-expressing CAL-27 and DC2.4 cells were analyzed using fluorescence-conjugated substrates. Left: average of fluorescent units (FUs) per minute throughout the time monitored. Right: fluorescence kinetics of three substrates. Data are represented as mean ± SEM (n = 3).
Patients with high expression of HPV E5 and low expression of PSMB have worse clinical outcomes
Next, to analyze the impact of HPV E5 in patients, we analyzed a cohort of 36 HPV-positive HNSCC patients and 25 healthy volunteers from Johns Hopkins University (JHU).21 Clinical demographics and specimen details have been described in detail in our earlier work.11 The patients were divided into 2 groups, E2/E4/E5 high or E6/E7 high, based on their HPV gene expression profile,22 and we compared the expression of immunoproteasome subunits and patient outcomes between groups. We observed that patients with E2/E4/E5 high expression had lower expression of PSMB8 and PSMB9 (Figure 3A). Additionally, we observed a trend toward an inverse correlation between HPV E5 and PSMB8 (Figure 3B). These data suggest a stronger correlation between HPV E5 and PSMB8 compared with other immunoproteasome subunits in human HNSCC. We then determined whether expression of immunoproteasome subunits impacts HNSCC patient outcomes. Patients were grouped according to median expression level of PSMB8 or PSMB9. Notably, patients with high expression of PSMB9 had better disease-free survival and overall survival compared with patients with low expression of PSMB9 (Figure 3C). A similar trend was observed with PSMB8. Remarkably however, expression of the constitutive proteasome subunits PSMB5 and PSMB6 had no effect on survival outcomes, indicating that differential expression of the immunoproteasome, but not the constitutive proteasome, impacts patient outcomes (Figure 3C).
Figure 3. Patients with high HPV E5 expression had lower immunoproteasome expression, which is associated with worse clinical outcomes.
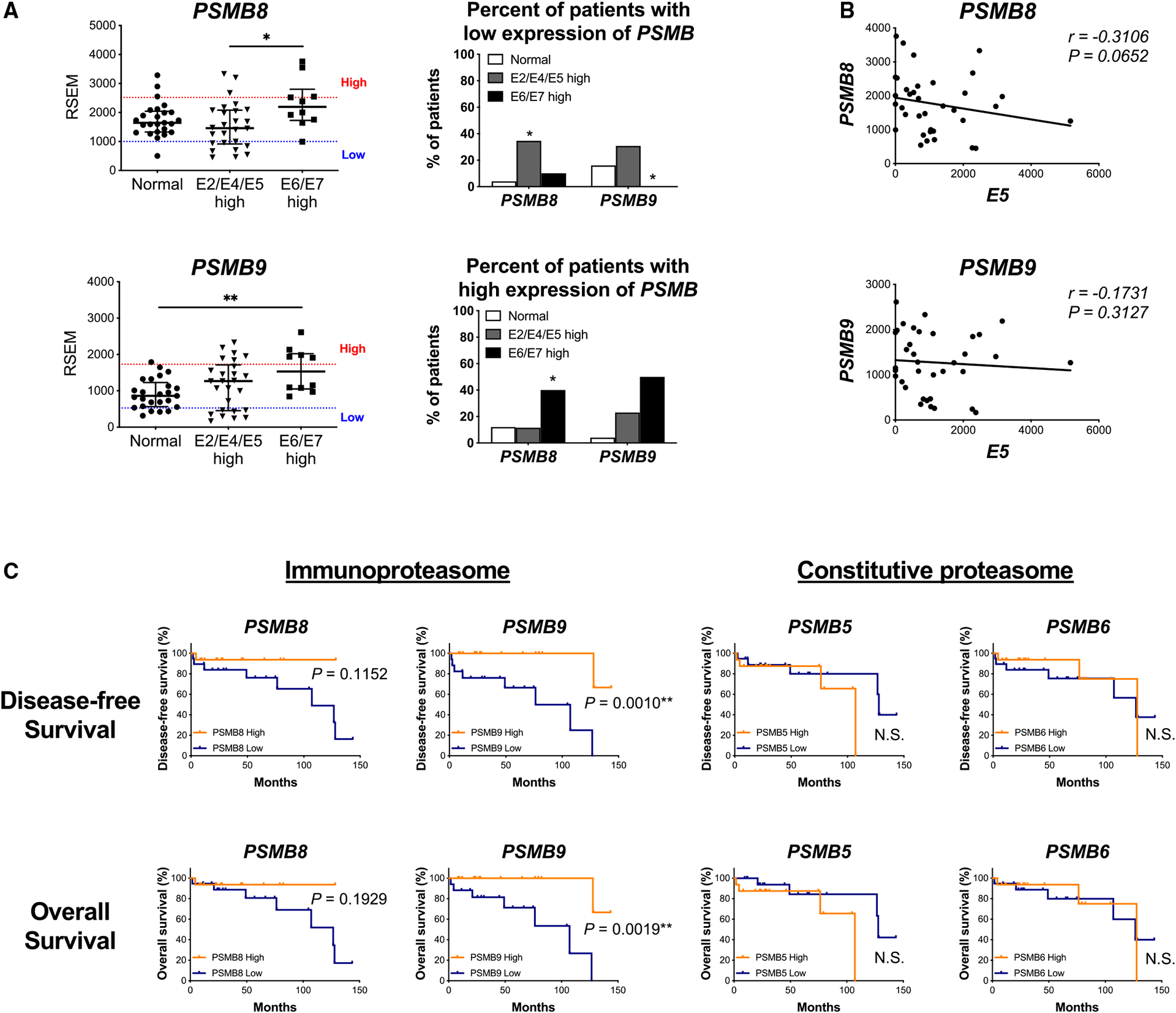
(A–C) Johns Hopkins University (JHU) cohort of HNSCC patients.
(A) Left: mRNA expression of immunoproteasome subunits analyzed by RNA-seq. (normal, n = 25; E2/E4/E5 high, n = 26; E6/E7 high, n = 10; median with interquartile range). Right: patients were assigned to one of three groups based on the mRNA expression level of immunoproteasome subunits (high, average, low). Patients with low and high expression are shown.
(B) Correlation analysis of HPV E5 expression and immunoproteasome subunits (n = 36, Spearman correlation coefficient).
(C) Disease-free survival and overall survival. Patients were assigned to groups based on HPV E5 and PSMB (immunoproteasome or constitutive proteasome subunits) expression level.
HPV E5 decreased the repertoire of antigens presented on MHC class I
Given the impact of HPV E5 on the immunoproteasome, we next examined the potential effects of HPV E5 on the repertoire of presented antigens in the MHC. To quantify changes in the repertoire of antigens presented in the MHC, we performed immunopeptidome analysis. HPV E5-expressing CAL-27 cells or parental empty vector-expressing control CAL-27 cells were lysed, and MHC class I was immunoprecipitated using a pan-HLA class I antibody (clone W6/32). Peptides on MHC class I were eluted and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Among all peptides detected, more than 60% were 9 amino acids long, and more than 95% were between 8-mer and 11-mer long, which is the typical length of peptides on MHC class I (Figure 4A). A total of 675 non-redundant peptides were detected in empty vector control cell lines, and 579 non-redundant peptides were detected in E5-positive cell lines. This indicates downregulation of approximately 15% in the repertoire of peptides presented on MHC class I in E5-positive cells (Figure 4B). Surprisingly, peptides generated from the E5 protein itself were not detected in E5-positive cells, suggesting that E5 blocked presentation of its own peptides and prevented their presentation on MHC class I as an antigen (Figure 4C). To investigate the types of proteins downregulated by E5, peptides were assigned to their source proteins and analyzed by Gene Ontology. One of the top altered peptidome pathways was viral process (Gene Ontology [GO]: 0016032), which was also downregulated in RNA-seq analysis (Figure 4D). We also observed alterations in the amino acid binding motifs of the 9-mer peptides. Namely, the relative frequencies of amino acids in positions 2, 3, and 4 were altered in E5-expressing cells (Figure 4E), raising the possibility that E5 regulates differential expression of HLA subclasses. Last, we analyzed the frequency of non-redundant source proteins because multiple different peptides can be generated from a single protein. The total number of non-redundant source proteins was also lower in E5-expressing cells, with 548 proteins presented on control cells and 482 proteins presented on E5-expressing cells, indicating that the repertoire of source proteins was also decreased in E5-expressing cells (Figure 4F). These data demonstrate that HPV E5 restricts the repertoire of antigens presented in MHC on the cell surface, highlighting a mechanism by which HPV mediates immune evasion.
Figure 4. HPV E5 decreased the repertoire of antigens presented on MHC class I.
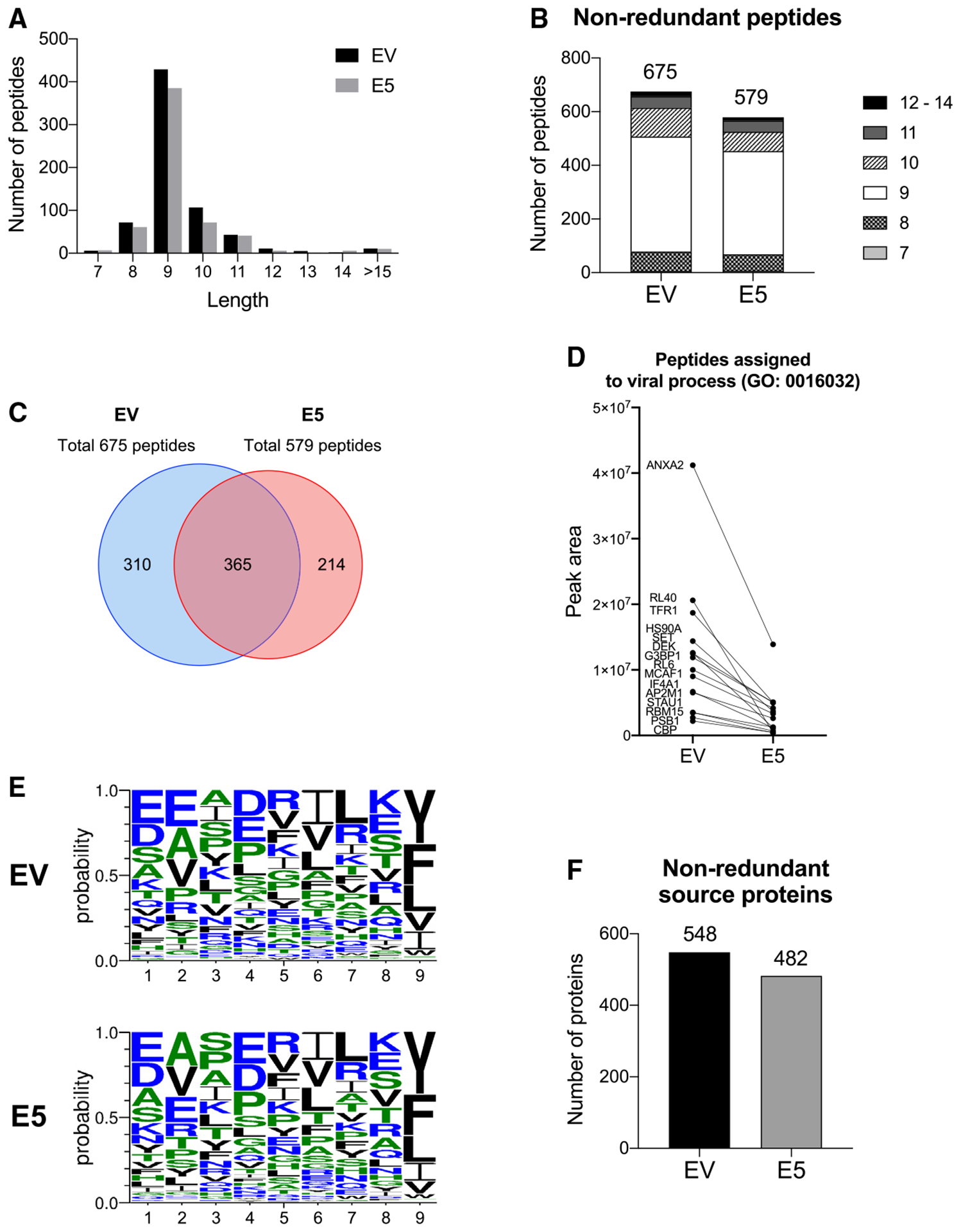
The immunopeptidome of HPV E5-expressing CAL-27 cells and its control were analyzed by LC-MS/MS.
(A) Distribution of peptide length for all unique peptides detected.
(B) Number of each peptide length for non-redundant peptides in empty vector- and E5-expressing cells.
(C) Venn diagram of peptides detected in empty vector control and E5-expressing cells.
(D) Relative number of peptides from proteins assigned to the GO virus process pathway.
(E) Binding motif of 9-mer peptides.
(F) Number of non-redundant host proteins.
HPV E5 impairs the cGAS-STING and RIG-I/MDA5 signaling pathways
Our RNA-seq data (Figure 1) showed that basal expression of IFN-inducible genes was downregulated in E5-expressing cells without any stimulation. To identify the molecular mechanisms by which HPV E5 impacts IFN pathways, we treated cells with several stimulants for anti-viral and IFN pathways and then examined downstream signaling. When cells were stimulated directly with type I IFN (IFN-α2), there was no significant difference in downstream phosphorylation of STAT1 between empty vector and E5-expressing cell lines (Figure 5A). Similarly, there were no significant differences in mRNA levels of the IFN-inducible genes MX1, STAT1, OAS2, or IFIT1 after IFN treatment (Figure 5B). These results suggest that E5 does not directly inhibit signaling from the type I IFN receptor. HPV is a DNA virus and, as such, is recognized by cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS)-STING upon cell infection.23 DNA viruses are also recognized by retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) after transcription by RNA polymerase III.24 To determine whether E5 impacts expression of DNA virus detection pathways specifically, we performed qPCR for DDX58 (RIG-I), IFIH1 (MDA5), STING1 (STING), and CGAS (cGAS). As expected, while there were no changes in mRNA levels of STING or cGAS, there was a striking downregulation of RIG-I and MDA5 mRNA expression levels (Figure 5C). RIG-I and MDA5 are known type I IFN-inducible genes in E5-expressing cells. To investigate the independent effects of E5 relative to other HPV oncoproteins, we generated CAL-27 cells expressing different combinations of HPV E5, E6, and E7. Importantly, RIG-I and MDA5 were downregulated in the E5 and E5/E6/E7 lines, but not in the E6/E7-only-expressing line, demonstrating that E5 alone is sufficient for downregulation of RIG-I and MDA5 (Figure 5D). To investigate direct signaling downstream of the RIG-I/MDA5 and cGAS-STING pathways, E5-positive and control CAL-27 cells were treated by transfection with low- or high-molecular-weight (LMW or HMW, respectively) poly(I:C), which dominantly activates RIG-I and MDA5, respectively. We discovered a striking loss of IFNB1 induction in E5-positive cells upon LMW and HMW poly(I:C) treatment, demonstrating that signaling downstream of RIG-I/MDA5 is significantly impaired by E5 (Figure 5E). Furthermore, when cells were treated with the STING agonist 2′3′-c-di-AM(PS)2 (Rp,Rp) (ADU-S100), IFNB1 and IFIT1 mRNA expression remained significantly suppressed in E5-positive cells only (Figure 5F). Taken together, these data identify HPV E5 as a negative regulator of the RIG-I/MDA5 and cGAS-STING pathways.
Figure 5. HPV E5 inhibited the cGAS-STING and RIG-I/MDA5 pathways.
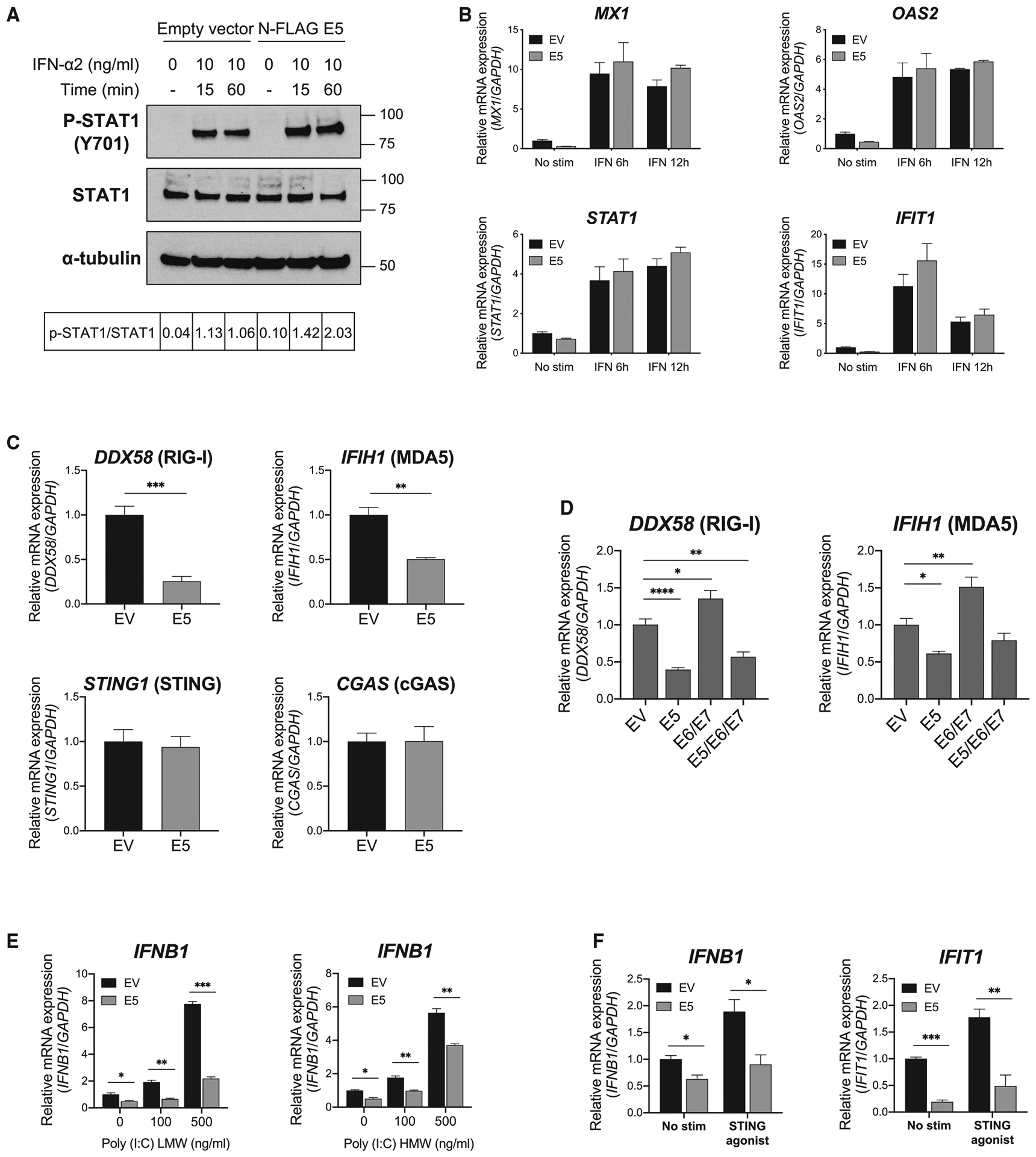
(A) Phosphorylation of STAT1 after IFN-α treatment in CAL-27 cells was analyzed by western blotting. The ratio of p-STAT1/total STAT1 is shown.
(B) mRNA expression in CAL-27 cells was analyzed by qPCR. Cells were treated with IFN-α at 10 ng/mL for 6 or 12 h
(C and D) Basal mRNA expression in CAL-27 cells was analyzed by qPCR.
(E) mRNA expression was analyzed after poly(I:C) treatment for 4 h at the indicated concentrations. LMW, low molecular weight; HMW, high molecular weight.
(F) mRNA expression after STING agonist treatment was analyzed. Cells were treated with 10 μg/mL of STING agonist for 4 h. Data are represented as mean ± SEM (n = 3).
HPV E5 directly binds STING and MAVS and inhibits downstream signaling
To determine whether HPV E5 directly binds STING or MAVS, we performed co-immunoprecipitation assays. We first checked whether E5 binds to MAVS, a crucial molecule in the RIG-I and MDA5 pathways. We were able to confirm the interaction of E5 and MAVS in an overexpression system (Figure 6A) and at the endogenous level in CAL-27 cells (Figure 6B), consistent with a reported interaction by tandem affinity purification and MS.25 An interaction between HPV E5 and STING has not been reported previously, to our knowledge. Using multiple co-immunoprecipitation assays, we discovered that E5 directly binds STING (Figure 6C). The interaction of stably expressed E5 and endogenous STING was also observed in CAL-27 lines (Figure 6D). Upon activation of the STING pathway, IRF3 is phosphorylated and translocated to nuclei, leading to activation of gene promoters such as IFN-β. Importantly, STING-mediated phosphorylation of IRF3 was downregulated in the presence of E5 (Figure 6E), confirming that HPV E5 downregulates STING-mediated IFN responses. Additionally, phosphorylated IRF3 in the nuclear fraction was lower in E5-expressing cells (Figure 6F), indicating decreased nuclear translocation of IRF3. To further analyze the functional impact of HPV E5 on STING and MAVS, we used a dual-luciferase reporter assay to measure promoter activity in empty vector- or E5-expressing CAL-27 cells transfected with STING or MAVS. We observed that IFN-β promoter activation was not induced by STING transfection, and thus we were not able to evaluate the direct effect of E5 on STING-mediated IFN-β promoter activation. However, we observed that MAVS-mediated IFN-β promoter activity was downregulated in the presence of E5 (Figure 6G), indicating that E5 inhibits activation of the IFN-β promoter and subsequent transcription.
Figure 6. HPV E5 interacted with MAVS and STING and inhibited downstream pathways.
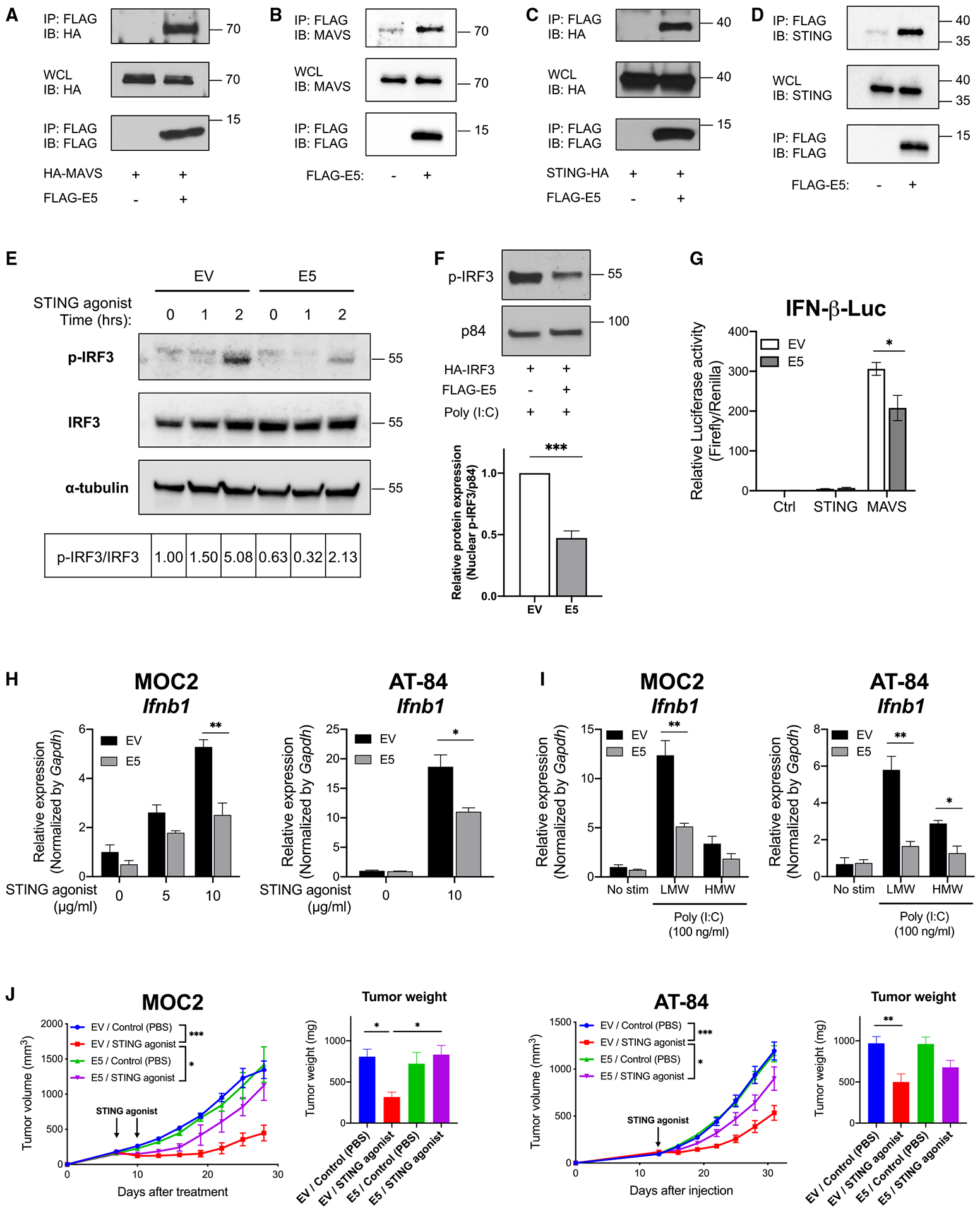
(A) The indicated constructs were transiently transfected in HEK293T cells, and co-immunoprecipitation was performed.
(B) Co-immunoprecipitation of stably expressed E5 and endogenous MAVS was performed with CAL-27.
(C) Indicated constructs were transiently transfected in HEK293T and co-immunoprecipitation was performed.
(D) Co-immunoprecipitation of stably expressed E5 and endogenous STING was performed with CAL-27 cells.
(E) Empty vector- or E5-expressing CAL-27 cells were treated with a STING agonist (10 μg/mL) for 1 or 2 h. Phosphorylation of IRF3 was analyzed by western blotting. The ratio of p-IRF3/total IRF3 is shown.
(F) The indicated constructs were transfected in HEK293T cells. Proteins from nuclear fractions were isolated and analyzed by western blotting (n = 3).
(G) Control, STING, or MAVS constructs were transfected in empty vector- or E5-expressing CAL-27 cells for 24 h. IFN-β promoter activity was measured by a dual-luciferase assay system (n = 3).
(H) mRNA expression after STING agonist treatment was analyzed by qPCR. Cells were treated with a STING agonist for 4 h at the indicated concentrations (n = 3).
(I) mRNA expression was analyzed by qPCR. Cells were treated with 100 ng/mL of poly(I:C) for 4 h (n = 3).
(J) Murine HNSCC lines (left: 1 × 105 MOC2 cells; right: 5 × 105 AT-84 cells) expressing empty vector or E5 were injected into mice (MOC2, n = 4–6 per group; AT-84, n = 6–7 per group). Tumors were treated with a STING agonist (ADU-S100) 20 μg two doses (MOC2) or 20 μg one dose (AT-84) when tumors reached 6–7 mm in diameter. Tumor weight at the time of sacrifice is also shown. Data are represented as mean ± SEM.
HPV E5 impairs anti-tumor activity of the STING agonist
Activating IFN responses with STING agonists is a promising anti-cancer treatment strategy. STING agonist treatment for HNSCC patients has been evaluated in clinical trials, and a recent study showed that STING is required for an effective response to radiotherapy in HNSCC.26 Despite these promising findings, STING agonists have had limited clinical success in HNSCC patients. Given our findings, we hypothesized that HPV E5 may be directly blocking the activity of STING agonists in HPV+ HNSCC. We evaluated the effect of E5 on the anti-tumor activity of STING agonists in two syngeneic mouse model strains: MOC2 in C57BL/6 mice and AT-84 in C3H/HeN mice. Consistent with our results from the human CAL-27 HNSCC line (Figures 5E and 5F), expression of the Ifnb1 gene was downregulated by HPV E5 upon treatment with the STING agonist or poly(I:C) in both cell lines (Figures 6H and 6I). Cell lines were implanted into the mice, and the STING agonist was administered intratumorally when tumors reached 6–7 mm in diameter. While STING agonist treatment controlled tumor growth in empty vector-expressing tumors, the anti-tumor response in E5-expressing tumors was significantly impaired (Figure 6J). Results were similar in both tumor models. We also tested the ability of E5 to inhibit the efficacy of RIG-1 agonism. The anti-tumor activity of the RIG-1 agonist was impaired in E5-expressing tumors (data not shown), although this effect did not reach statistical significance. Together, these experiments demonstrate that the anti-tumor effects of STING agonism were impaired by HPV E5 via inhibition of STING and subsequent gene transcription.
HPV E5 expression was inversely correlated with a type I IFN-inducible gene signature in HNSCC patients
To investigate the effect of HPV E5 on IFN-inducible gene expression in patients, we analyzed mRNA expression data from the JHU cohort. We analyzed type I IFN-inducible genes that were downregulated in E5-expressing CAL-27 cells (Figure 1A and Table S1). Compared with healthy tissue, expression of type I IFN-inducible genes was increased in tumors (E2/E4/E5 high and E6/E7 high groups), consistent with previous reports.27 Furthermore, IFN-inducible gene expression was lower in E2/E4/E5-high patient tumors compared with E6/E7-high patient tumors (Figures 7A and 7B). These clinical results are consistent with our CAL-27 cell findings (Figure 1). Correlation analysis revealed an inverse relationship between HPV E5, but not HPV E6 or E7, and IFN-inducible gene signatures (Figure 7C). These findings demonstrate a profound effect of HPV E5 on the majority of type I IFN-inducible genes. Notably, some genes, which may have other regulatory functions, show weak or no correlation with E5 (Figure 7D). Because we observed high variability in the expression pattern of IFN-inducible genes in the E2/E4/E5-high group, we conducted separate analyses on patients with clear patterns of high IFN-inducible gene expression (indicated by red arrows in Figure 7A) and patients with low expression (indicated by blue arrows). We found that low IFN-inducible expression was associated with decreased disease-free survival and overall survival (Figure 7E). These data indicate that HPV E5 modulates the expression of type I IFN-inducible genes in patients and that this E5-modulated gene expression impacts clinical outcomes.
Figure 7. HPV E5 expression was inversely correlated with an IFN-inducible gene signature in the HNSCC patient cohort.
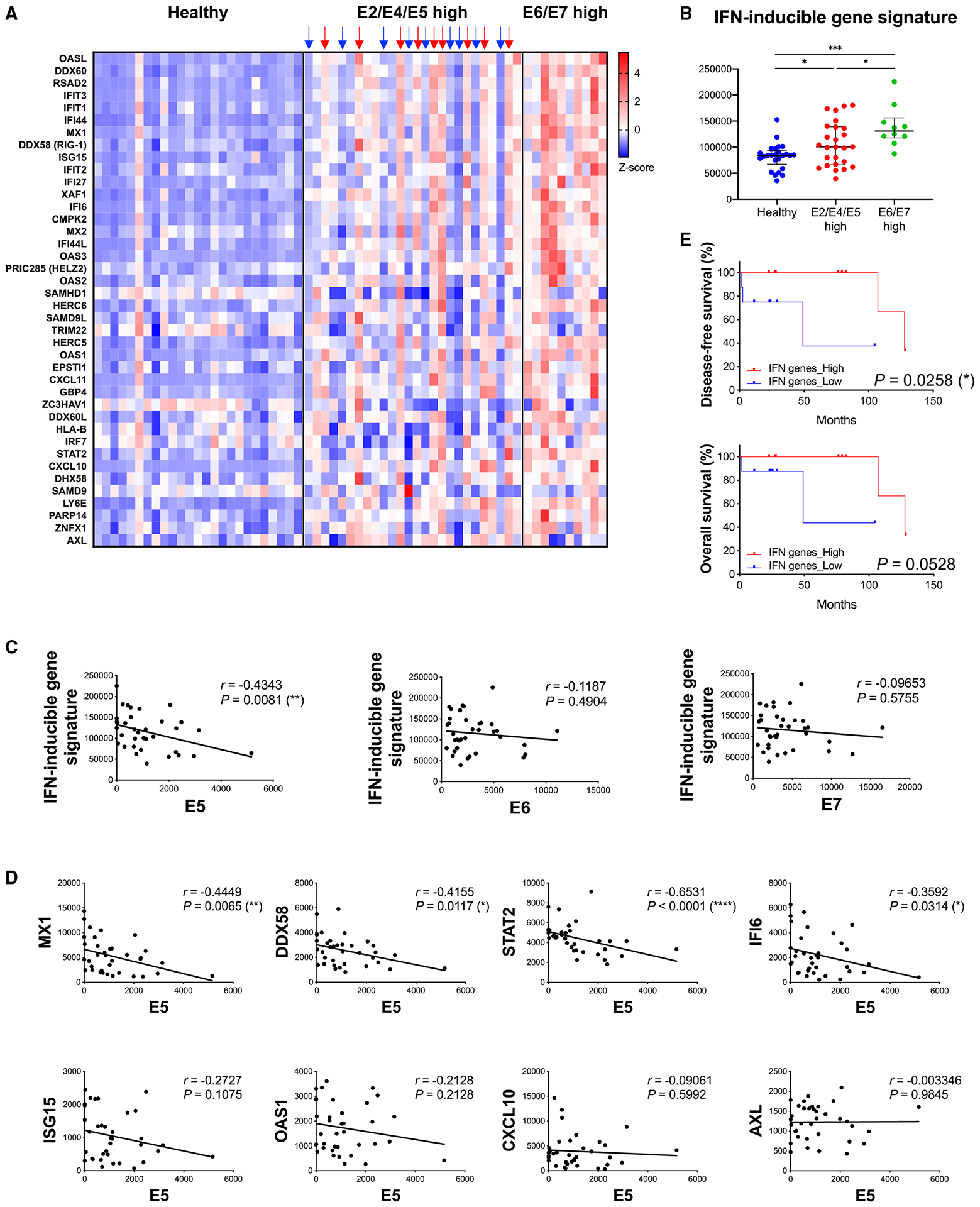
(A–E) JHU cohort of HNSCC patients.
(A) Heatmap of IFN-inducible gene expression analyzed by RNA-seq (normal, n = 25; E2/E4/E5 high, n = 26; E6/E7 high, n = 10).
(B) Expression of the IFN-inducible gene signature in each group. Median with interquartile range.
(C) Correlation analysis of HPV E5, E6, or E7 expression and IFN-inducible gene signature (n = 36, Spearman correlation coefficient).
(D) Correlation analysis of HPV E5 and selected genes from the IFN-inducible gene signature.
(E) Overall survival and disease-free survival of patients with high IFN-inducible gene expression (indicated by red arrows in Figure 7A) and low expression (indicated by blue arrows) from the E2/E4/E5 high group (n = 9 per group).
DISCUSSION
HPV infection is linked to development of cervical cancer, HNSCC, anal cancers, and other gynecologic and genitourinary malignancies. The incidence of these cancers has increased dramatically over the past two decades. Previous work has shown that HPV-associated cancers are more responsive to standard therapies, including chemotherapy and radiation therapy, compared with non-HPV-associated cancers. Moreover, patients with HPV-associated HNSCCs have better survival outcomes compared with patients with non-HPV-associated HNSCCs.28 HPV-positive tumors are associated with increased immune cell infiltration compared with HPV-negative tumors and have been thought to be more immunogenic because of the presence of foreign viral epitopes. However, a 2-year long-term follow-up of a randomized phase III trial (CheckMate 141) found almost identical response rates to checkpoint blockade immunotherapy (CBI) among HPV-positive and HPV-negative HNSCC, the latter of which harbors a greater number of carcinogen-induced mutations.29,30 These results highlight the dual roles that viral oncogenes play in suppressing host immune responses. Namely, viral oncogenes support viral replication and life cycle while also suppressing the immune system and potential responses to immunotherapies.
Among the multiple oncoproteins expressed by HPV, E6 and E7 have been extensively studied. Notably, these oncoproteins inhibit critical tumor suppressors. E6 and E7 bind to p53 and pRb, respectively, resulting in inhibition of apoptosis, loss of cell-cycle arrest, and promotion of cell proliferation.31–33 Far less is known about the functions of oncoprotein E5, perhaps because of its small molecular weight and hydrophobic features.
In the present study, we investigated the effects of E5 on the host immune response. The RNA-seq data with HPV E5-positive cells showed clear downregulation in anti-virus and type I IFN pathways. Surprisingly, most of the highly downregulated genes were IFN-inducible genes. Previous work has demonstrated immunosuppressive effects of E5 via downregulation of MHC class I and inhibition of endosomal acidification.8–10 However, our data indicate much broader immunosuppressive functions of E5. We observed impaired immunoproteasome activity in E5-positive lines, which we attributed to a downstream effect of downregulation in the anti-virus response and type I IFN pathways rather than direct inhibition of the immunoproteasome by E5. Transcription factors regulating expression of immunoproteasome subunits are induced by immune stimulation. E5 suppresses immunoproteasome activity by downregulating these anti-viral immune response pathways. Recognition of antigens by T lymphocytes is necessary for adaptive immunity, and the immunoproteasome and TAP play essential roles in generating robust immunogenic antigens. Immunopeptidomics is a powerful tool for identifying and understanding the repertoire of peptides presented on MHCs. We found that HPV E5 downregulates immunoproteasome activity, resulting in a smaller repertoire of peptides presented on the cell surface. This is one of the mechanisms by which HPV escapes host immune surveillance. Indeed, in the clinical cohort, expression of immunoproteasome subunits, but not the constitutive proteasome subunits, predicted survival outcomes, suggesting that activation of the immunoproteasome may improve outcomes in patients with HNSCC and other HPV-associated malignancies. Importantly, HPV may selectively impair the immunoproteasome, but not the constitutive proteasome, to allow the cell to present common antigens and prevent natural killer (NK) cell activation. Furthermore, some responses to IFN signaling remain intact, while the virus-specific detection systems are impaired. Effectively, we hypothesize that this selective impairment allows an HPV-infected cell to respond physiologically to certain stimuli to go undetected by the immune system, ultimately facilitating viral latency and survival of the HPV virus.
To evade host immune surveillance, many viruses manipulate the host immune system. Specifically, HPV may suppress IFN responses.34,35 Mechanistically, HPV proteins inhibit anti-virus responses and downstream type I IFN production in several ways. For example, HPV16/18 E2 downregulates mRNA expression of STING,36 HPV18 E7 binds and antagonizes STING,37 HPV16 E7 enhances STING protein destabilization,38 and HPV16 E6 binds to IRF3 and impairs its transcriptional activity.39 Indeed, cGAS-STING responses are dampened in HPV16-positive HNSCC cells,40 and loss of HPV16 E7 restores cGAS-STING responses.41 Two recent studies demonstrated that E5 is also involved in suppression of the IFN response.42,43 Notably, Scott et al.43 reported that IFN response suppression was mediated by IFN-κ, a type I IFN produced by keratinocytes. In our model, expression of IFN-κ was undetectable in CAL-27 cells. Moreover, downregulation of the IFN pathway was observed in a murine dendritic cell line that does not usually express IFN-κ, implicating mechanisms not involving IFN-κ. To our knowledge, ours is the first study to investigate the role of E5 and its interacting protein in anti-virus response pathways. We found that E5 interacted with MAVS and STING, both critical components in virus recognition. The interaction of E5 and MAVS, initially reported in a study using tandem affinity purification and MS,25 was verified by our co-immunoprecipitation assay. After RIG-I and MDA5 sense viral RNA, MAVS-dependent signaling is initiated, resulting in phosphorylation of IRF3 and transcriptional activation of antiviral signaling.44,45 Importantly, we found an interaction between E5 and STING, which has not been reported previously, to our knowledge. STING and cGAS are key molecules in cytosolic DNA sensing.46,47 Upon DNA sensing, cGAS generates cyclic-GMP-AMP (2′3′-cGAMP) which works as a second messenger, binding to STING and activating downstream signaling and transcription. Interestingly, we observed that E5 inhibited cGAS-STING and the RIG-I/MDA5 axis, two of the major virus-recognizing pathways. This suggests that E5 almost completely shuts down virus recognition machinery. As we reported previously,11 HPV E5 also mediates resistance to PD-L1 CBI, suggesting that targeting E5 may overcome resistance to PD-L1 CBI in HNSCC. While we also observed a trend toward a similar blunting of anti-tumor activity in E5-expressing cell lines after RIG-1 agonism, this difference did not reach statistical significance. The differences in efficacy between STING and RIG-1 agonists on tumors expressing E5 may have resulted from differential binding of E5 to effector proteins within these two different IFN response pathways. Importantly, in our models, expression of HPV E5 blunted the anti-tumor activity of the STING agonist, suggesting that inhibition of the HPV E5 oncogene may enhance responses to immunotherapies dependent on IFN or STING activity.
In conclusion, we elucidated the role of HPV E5 as a key negative regulator of anti-viral IFN responses via inhibition of the RIG-I/MAVS and cGAS-STING pathways. We identified that expression of immunoproteasome subunits correlates with improved survival in HNSCC patients and that HPV E5 inhibits the immunoproteasome. These effects culminate in a diminished repertoire of antigens presented on the tumor cell surface and resistance to immunotherapies, including STING agonists in HPV E5-expressing tumors. These findings advance our understanding of the mechanisms underlying virus-mediated resistance to immunotherapy and provide a rationale for novel treatment strategies to improve outcomes for patients with HPV-associated malignancies.
Limitations of the study
Unlike E6 and E7, the E5 oncogene generally presents and replicates episomally. However, we used E5 stably expressed lines, in which E5 is integrated into the genome, to ensure that E5 expression levels were adequate for experimental requirements. Moreover, because of the low expression nature of E5, analysis of endogenous E5 in HPV-positive HNSCC cells was difficult. Finally, while we focused on E5 in the present study, it should be noted that E5 is expressed in the same HPV infection phase as other HPV proteins, including E2 and E4. Interactions with these other proteins remain to be determined. Future studies using an infection model with HPV harboring mutant E5 may reveal additional mechanisms of action and more faithfully recapitulate viral oncogene expression and function.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andrew B. Sharabi (sharabi@ucsd.edu).
Materials availability
The following plasmids were generated in this study: MIP-FLAG-HPV16 E5 (codon-optimized), pcDNA-FLAG-HPV16 E5 (codon-optimized), pcDNA-HA-HPV16 E5 (codon-optimized), pcDNA-HA-hMAVS, pcDNA-hSTING-HA, pcDNA-hSTING-Myc, pcDNA-HA-hIRF3, pMSCV-Blasticidin-HPV16 E6, pMSCV-Zeocin-HPV16 E7, and pGL3-IFN-β gene promoter. All materials generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Bulk RNAseq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LMP2 (clone G-3) | Santa Cruz Biotechnology | Cat#: sc-373996; RRID: AB_10918476 |
| Mouse monoclonal anti-LMP7 (clone D-2) | Santa Cruz Biotechnology | Cat#: sc-374089; RRID: AB_10947104 |
| Mouse monoclonal anti-α-tubulin | Developmental Studies Hybridoma Bank (DSHB) | Cat#: 12G10; RRID: AB_2315509 |
| Rabbit polyclonal anti-STAT1 | Cell Signaling Technology | Cat#: 9172; RRID: AB_2198300 |
| Rabbit monoclonal anti-phospho-STAT1 (Tyr701) (clone 58D6) | Cell Signaling Technology | Cat#: 9167; RRID: AB_561284 |
| Rabbit polyclonal anti-MAVS | Cell Signaling Technology | Cat#: 3993; RRID: AB_823565 |
| Rabbit monoclonal anti-STING (clone D2P2F) | Cell Signaling Technology | Cat#: 13647; RRID: AB_2732796 |
| Rabbit monoclonal anti-IRF3 (clone D83B9) | Cell Signaling Technology | Cat#: 4302; RRID: AB_1904036 |
| Rabbit monoclonal anti-phospho-IRF3 (Ser396) (clone 4D4G) | Cell Signaling Technology | Cat#: 4947; RRID: AB_823547 |
| Mouse monoclonal anti-Nuclear Matrix Protein p84 (clone 5E10) | GeneTex | Cat#: GTX70220; RRID: AB_372637 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat#: F3165; RRID: AB_259529 |
| Mouse monoclonal anti-HA.11 Epitope Tag (clone 16B12) | BioLegend | Cat#: 901501; RRID: AB_2565006 |
| Mouse monoclonal anti-c-Myc (Clone 9E10) | BioLegend | Cat#: 626801; RRID: AB_2235686 |
| Mouse monoclonal anti-HLA-ABC (clone W6/32) | BioLegend | Cat#: 311441; RRID: AB_2800814 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#: 31430; RRID: AB_228307 |
| Goat anti-Rabbit IgG (H + L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#: 31460; RRID: AB_228341 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human IFN-α2 (carrier-free) | BioLegend | Cat#: 592702 |
| Poly(I:C) (LMW)/LyoVec™ | InvivoGen | Cat#: tlrl-picwlv |
| Poly(I:C) (HMW)/LyoVec™ | InvivoGen | Cat#: tlrl-piclv |
| ADU-S100 [2′3′-c-di-AM(PS)2 (Rp,Rp)] | InvivoGen | Cat#: tlrl-nacda2r |
| TRIzol™ Reagent | Thermo Fisher Scientific (Invitrogen) | Cat#: 15596018 |
| qScript cDNA Synthesis Kit | Quantabio | Cat#: 95047 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725124 |
| PEI (Polyethylenimine, branched) | Sigma-Aldrich | Cat#: 408727 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich (Roche) | Cat#: 11873580001 |
| PhosSTOP™ | Sigma-Aldrich (Roche) | Cat#: 4906837001 |
| Protein G Plus/Protein A Agarose Suspension | EMD Millipore | Cat#: IP05 |
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat#: A2220 |
| Critical commercial assays | ||
| RNeasy Plus Mini Kit | QIAGEN | Cat#: 74136 |
| Immunoproteasome Activity Fluorometric Assay Kit II | UBPBio | Cat#: J4170 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#: E1980 |
| Deposited data | ||
| RNAseq data of cell lines | This paper | GEO: GSE226754 |
| RNAseq data of HPV-positive OPSCCs from Johns Hopkins University cohort | Guo et al.21 | GEO: GSE112027 |
| Experimental models: Cell lines | ||
| Human: CAL-27 | Dr. J. Silvio Gutkind (University of California, San Diego) | RRID: CVCL_1107 |
| Human: HEK293T | Dr. Dong-Er Zhang (University of California, San Diego) | RRID: CVCL_0063 |
| Mouse: DC2.4 | Dr. Dong-Er Zhang (University of California, San Diego) | RRID: CVCL_J409 |
| Mouse: MOC2 | Dr. J. Silvio Gutkind (University of California, San Diego) | RRID: CVCL_ZD33 |
| Mouse: AT-84 | Dr. Aldo Venuti (Regina Elena National Cancer Institute, Italy) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#: 000664; RRID: IMSR_JAX:000664 |
| Mouse: C3H/HeNCrl | Charles River Laboratories | Strain Code: 025; RRID: IMSR_CRL:025 |
| Oligonucleotides | ||
| See Table S5 for qPCR primers | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pLXSN-AU1-HPV16 E5 (codon-optimized) | Dr. Frank Suprynowicz (Georgetown University Medical School) | N/A |
| Plasmid: MIP (MSCV-IRES-Puro) | Dr. Dong-Er Zhang (University of California, San Diego) | N/A |
| Plasmid: pMSCV-Blasticidin | Addgene | Plasmid: #75085; RRID: Addgene_75085 |
| Plasmid: pMSCV-Zeo | Addgene | Plasmid: #75088; RRID: Addgene_75088 |
| Plasmid: pCL-10A1 | Novus | Cat#: NBP2–29542 |
| Plasmid: pCL-Eco | Novus | Cat#: NBP2–29540 |
| Plasmid: pcDNA™3.1 (+) Mammalian Expression Vector | Thermo Fisher Scientific (Invitrogen) | Cat#: V79020 |
| Plasmid: MIP-FLAG-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pMSCV-Blasticidin-HPV16 E6 | This paper | N/A |
| Plasmid: pMSCV-Zeo-HPV16 E7 | This paper | N/A |
| Plasmid: pcDNA-FLAG-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pcDNA-HA-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pcDNA-HA-human MAVS | This paper | N/A |
| Plasmid: pcDNA-human STING-HA | This paper | N/A |
| Plasmid: pcDNA-human STING-Myc | This paper | N/A |
| Plasmid: pcDNA-HA-human IRF3 | This paper | N/A |
| Plasmid: pGL3-Basic | Promega | Cat#: E1751 |
| Plasmid: pGL3-IFN-β gene promoter | This paper | N/A |
| Plasmid: pRL-TK | Promega | Cat#: E2241 |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | N/A |
| ImageJ | Schneider et al.48 | https://imagej.nih.gov/ij/ |
| STAR (v2.6.1) | Dobin et al.49 | N/A |
| DESeq2 (v2_1.6.3) | Love et al.50 | N/A |
| Gene Ontology | Ashburner et al.51; Gene Ontology Consortium52; Mi et al.53 |
http://geneontology.org/ |
| Reactome | Fabregat et al.54 | https://reactome.org/ |
| KEGG | Kanehisa et al.55–57 | http://www.kegg.jp/ |
| PEAKS | Bioinformatics Solutions Inc. | N/A |
| Immune Epitope Database and Analysis Resource (IEDB) | Vita et al.58 | https://www.iedb.org |
| WebLogo 3 | Schneider et al.59; Crooks et al.60 |
https://weblogo.threeplusone.com |
| Other | ||
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat#: LT07–710 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse studies
All experimental protocols were approved by the Institutional Animal Care and Use Committee of UCSD (#S15281). Animal experiments were performed in specific pathogen-free facilities at Moores Cancer Center accredited by the American Association for the Accreditation of Laboratory Animal Care. Wild-type female 6-week-old mice were used for experiments. C3H/HeN mice were purchased from Charles River. C57BL/6 mice were purchased from The Jackson Laboratory. Mice were injected subcutaneously with 1.0 × 105 MOC2 or 5.0 × 105 AT-84 cells resuspended in 100 μL of PBS in the right flank. Tumor diameter was measured every 3 days with an electronic caliper and reported as volume using the formula; tumor volume (mm3) = (length × width2)/2. Once tumors reached 6–7 mm in diameter, mice were treated with STING agonist (ADU-S100) 20 μg two doses (MOC2) or 20 μg one dose (AT-84).
Clinical patient cohorts
HPV-positive OPSCCs from Johns Hopkins University (JHU) patient cohort were analyzed as previously described.21 Patients were recruited with written informed consent under a protocol approved by the institutional review board of JHU (#NA_00–36235). Detailed patient demographic and clinical data have been previously published.11 The data include normal (n = 25), E2/E4/E5 high (n = 26), and E6/E7 high (n = 10). HPV status was classified based on the expression of high-risk HPV16, 33, and 35. mRNA expression was analyzed by RNA-seq and assessed as RSEM.
Cell lines
Human and murine HNSCC lines CAL-27 (tongue origin) and MOC2 were kindly provided by Dr. J. Silvio Gutkind (UCSD) in March 2018. Murine dendritic cell line DC2.4 and HEK293T were a kind gift from Dr. Dong-Er Zhang (UCSD) in September 2017. Murine HNSCC line AT-84 was kindly provided by Dr. Aldo Venuti (Regina Elena National Cancer Institute, Italy) in February 2021. CAL-27 and HEK293T were grown in DMEM containing 10% FBS, 1% L-glutamine, and 1% Penicillin/streptomycin. DC2.4 and AT-84 were grown in RPMI 1640 containing 10% FBS, 1% L-glutamine, and 1% Penicillin/streptomycin. MOC2 was cultured in the media previously described.61 All cell lines were cultured in an incubator with 5% CO2 at 37°C. Routine monitoring for Mycoplasma contamination was performed using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza).
HPV16 E5-expressing stable cell lines were generated as previously described.11 Briefly, codon-optimized HPV16 E562 (from Dr. Frank Suprynowicz, Georgetown University Medical School) with FLAG tag on N-terminus was amplified and cloned into MIP (MSCV-IRES-Puro) vector (from Dr. Dong-Er Zhang, UCSD). HPV16 E6 and HPV16 E7 were amplified and cloned into pMSCV-Blasticidin (Addgene) or pMSCV-Zeo (Addgene), respectively. Retroviruses were generated by co-transfection of HPV oncogene-coding vector (MIP-FLAG-HPV16 E5, pMSCV-Blasticidin-HPV16 E6, or pMSCV-Zeo-HPV16 E7) and packaging vector (pCL-10A1 or pCL-Eco from Novus) into HEK293T and infected CAL-27 and DC2.4 cell lines. The infected cells were then selected by using appropriate selection drugs. The following reagents were used for in vitro treatment; recombinant human IFN-α2 (BioLegend), LMW and HMW Poly(I:C) (InvivoGen), and 2′3′-c-di-AM(PS)2 (Rp,Rp) (ADU-S100) (InvivoGen).
METHOD DETAILS
Reverse transcription and quantitative PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) and reverse transcribed with qScript cDNA Synthesis Kit (Quantabio) according to the manufacturer’s instructions. Quantitative PCR analysis was conducted using iTaq Universal SYBR Green Supermix (Bio-Rad) on the CFX96 Touch (Bio-Rad). Primer sequences were listed in Table S5.
Plasmid construction and transfection
FLAG-, HA-, or Myc-tagged codon-optimized HPV16 E5,62 MAVS, and STING were cloned into pcDNA3.1(+) vector (Invitrogen). HEK293T cells were transfected with plasmids using PEI reagent (Sigma-Aldrich) for 24 h.
Immunoprecipitation and immunoblotting
Cells were lysed in lysis buffer composed of 25 mmol/L Tris-HCl, pH8.0, 150 mmol/L NaCl, 1 mmol/L EDTA, 0.5% IgepalCA-630, and protease and phosphatase inhibitors (Roche). The cell lysates were centrifuged (15,000 × g), at 4°C for 5 min. For co-immunoprecipitation assay, cell lysates were immunoprecipitated for 1 to 2 h with FLAG M2 Affinity Gel for FLAG-tagged proteins or anti-HA antibody followed by protein G/A-Agarose Suspension (EMD Millipore) for HA-tagged proteins. Immunocomplexes were then washed three times. All samples were denatured in 1x sample buffer (50 mmol/L Tris-HCl, pH6.8, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.01% bromophenol blue) for 5 min at 100°C. Proteins were electroblotted onto nitrocellulose membranes (GE Healthcare). The following primary antibodies were used: anti-LMP2 and anti-LMP7 from Santa Cruz Biotechnology, anti-α-tubulin from Developmental Studies Hybridoma Bank, anti-STAT1, anti-p-STAT1, anti-MAVS, anti-STING, anti-IRF3, and anti-p-IRF3 from Cell Signaling Technology, anti-p84 from GeneTex, anti-FLAG M2 from Sigma, anti-HA and anti-Myc from BioLegend. HRP-conjugated secondary antibodies (Thermo Fisher Scientific) were used for detection with Western ECL substrates (Thermo). Chemiluminescent signals were detected by film or ChemiDoc (Bio-Rad). Quantification was performed with ImageJ software.48
RNAseq analysis
The total RNA from empty vector or E5-expressing CAL-27 and DC2.4 was extracted with RNeasy Plus Mini Kit (QIAGEN). Library preparation and sequencing with Illumina sequencer (PE150) were performed at Novogene. STAR (v2.6.1)49 was used for alignment. Differential gene expression analysis was performed using the DESeq2 (v2_1.6.3).50 Genes with an adjusted p-value < 0.05 were considered as differentially expressed. Gene Ontology,51–53 Reactome,54 and KEGG55–57 were used for pathway enrichment analysis of differentially expressed genes.
Immunoproteasome activity assay
Immunoproteasome activity was measured by using Immunoproteasome Activity Fluorometric Assay Kit (UBPBio). Briefly, cells were lysed with cell lysis buffer (40 mM Tris, pH 7.2, 50 mM NaCl, 2 mM 2-mercaptoethanol, 2 mM ATP, 5 mM MgCl2, 10% glycerol) and sonicated. Lysates were then centrifuged at 17,000 × g for 20 min at 4°C, and the supernatant was collected and diluted if needed. The supernatant was applied to a 96-well plate and warmed-up substrates were added. Fluorescence was monitored at 37°C on a TECAN infinite M200 microplate with excitation and emission filters at 360/40 and 460/30 nm, respectively.
Immunopeptidome analysis
Immunopeptidome analysis was performed following a published protocol.63 Briefly, 5 × 108 cells of CAL-27/Empty vector or CAL-27/N-FLAG-E5 were lysed and MHC immunoaffinity purification was performed by using pan-HLA antibody (clone W6/32, BioLegend). MHC class I-bound peptides were eluted with 10% acetic acid and the elute was analyzed at Biomolecular & Proteomics Mass Spectrometry Facility at UCSD. The MHC elute was separated by RP-HPLC and then run by LC-MS/MS. The raw data was quality controlled with an FDR cutoff of 5%. Peptides smaller than 14-mer were further analyzed. Detected peptides were mapped to protein by using PEAKS (Bioinformatics Solutions Inc.) and the quality was evaluated with Immune Epitope Database and Analysis Resource (IEDB).58 The peptide-binding motif was constructed by using WebLogo 3.59,60
Luciferase assay
HEK293T or CAL-27 stably expressing empty vector or E5 were transfected with the following constructs by using PEI reagent: the pGL3-Basic vector encoding the IFN-β gene promoter cloned upstream of the Firefly luciferase reporter gene, the pRL-TK control vector with Renilla luciferase reporter gene (Promega), and pcDNA3.1 encoding either MAVS or STING. HEK293T cells were also transfected with pcDNA3.1 empty vector or pcDNA3.1/FLAG-HPV16 E5 construct. Twenty-four hours after the transfection, Dual-Luciferase Reporter Assay System (Promega) was used to measure luciferase activity. Firefly luciferase activity was normalized by Renilla luciferase activity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analysis was performed using Prism 8 (GraphPad). Unpaired-two-tailed t test or ordinary one-way ANOVA multiple comparison test with post hoc Tukey were conducted for comparisons of two groups and comparisons of more than three groups, respectively. For the JHU cohort, Chi-square test and residual analysis were used for mRNA expression analysis. Spearman’s correlation coefficient was used for correlation analysis. p value < 0.05 was considered to be statistically significant: *; p < 0.05, **; p < 0.01, ***; p < 0.001.
ADDITIONAL RESOURCES
Data from the previously reported HPV-positive OPSCC cohort from Johns Hopkins University (#NA_00–36235) were utilized in this study.21 RNAseq data are available at GEO (accession GSE112027).
Supplementary Material
Highlights.
HPV E5 downregulates anti-viral type I interferon response pathways
HPV E5 binds STING and MAVS to inhibit downstream type I IFN signaling
HPV E5 limits the MHC class I antigen repertoire by suppressing the immunoproteasome
ACKNOWLEDGMENTS
We thank Dr. Majid Ghassemian (Biomolecular and Proteomics Mass Spectrometry Facility, University of California, San Diego) for mass spectrometry analysis. The 12G10 anti-α-tubulin monoclonal antibody deposited by Frankel, J./Nelsen, E.M. was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology (Iowa City, IA, USA). We would also like to thank and acknowledge Nathalie Boutros for her assistance and editorial support. This work was supported in part by NIH grants (1KL2TR001444, 1R01DE028563, and 1U01DE028227) and the San Diego Center for Precision Immunotherapy (to A.B.S.).
DECLARATION OF INTERESTS
E.E.W.C. has received compensation from MSD and Roche for attending advisory boards and is on the scientific advisory board for Toragen Inc. A.B.S. reports research funding and honoraria from Pfizer and Varian Medical Systems, consultant fees from AstraZeneca and Primmune, and other fees from Merck. A.B.S. is the scientific founder and has an equity interest in Toragen Inc., which could potentially benefit from the research results. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112508.
REFERENCES
- 1.de Martel C, Plummer M, Vignat J, and Franceschi S (2017). Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 141, 664–670. 10.1002/ijc.30716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamani M, Grønhøj C, Jensen DH, Carlander AF, Agander T, Kiss K, Olsen C, Baandrup L, Nielsen FC, Andersen E, et al. (2020). The current epidemic of HPV-associated oropharyngeal cancer: an 18-year Danish population-based study with 2,169 patients. Eur. J. Cancer 134, 52–59. 10.1016/j.ejca.2020.04.027. [DOI] [PubMed] [Google Scholar]
- 3.Mahal BA, Catalano PJ, Haddad RI, Hanna GJ, Kass JI, Schoenfeld JD, Tishler RB, and Margalit DN (2019). Incidence and demographic burden of HPV-associated oropharyngeal head and neck cancers in the United States. Cancer Epidemiol. Biomarkers Prev 28, 1660–1667. 10.1158/1055-9965.EPI-19-0038. [DOI] [PubMed] [Google Scholar]
- 4.Gravitt PE, and Winer RL (2017). Natural history of HPV infection across the lifespan: role of viral latency. Viruses 9, 267. 10.3390/v9100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoppe-Seyler K, Bossler F, Braun JA, Herrmann AL, and Hoppe-Seyler F (2018). The HPV E6/E7 oncogenes: key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 26, 158–168. 10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Tsao YP, Li LY, Tsai TC, and Chen SL (1996). Human papillomavirus type 11 and 16 E5 represses p21(WafI/SdiI/CipI) gene expression in fibroblasts and keratinocytes. J. Virol 70, 7535–7539. 10.1128/JVI.70.11.7535-7539.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedroza-Saavedra A, Lam EWF, Esquivel-Guadarrama F, and Gutierrez-Xicotencatl L (2010). The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 400, 44–52. 10.1016/j.virol.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Campo MS, Graham SV, Cortese MS, Ashrafi GH, Araibi EH, Dornan ES, Miners K, Nunes C, and Man S (2010). HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 407, 137–142. 10.1016/j.virol.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 9.Cortese MS, Ashrafi GH, and Campo MS (2010). All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 126, 1675–1682. 10.1002/ijc.25004. [DOI] [PubMed] [Google Scholar]
- 10.Straight SW, Herman B, and McCance DJ (1995). The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol 69, 3185–3192. 10.1128/JVI.69.5.3185-3192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyauchi S, Sanders PD, Guram K, Kim SS, Paolini F, Venuti A, Cohen EEW, Gutkind JS, Califano JA, and Sharabi AB (2020). HPV16 E5 mediates resistance to PD-L1 blockade and can Be targeted with rimantadine in head and neck cancer. Cancer Res. 80, 732–746. 10.1158/0008-5472.CAN-19-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka K (2009). The proteasome: overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 85, 12–36. 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloetzel PM (2001). Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol 2, 179–187. 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 14.Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, Tanahashi N, Yoshimura T, Tanaka K, and Ichihara A (1994). Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem 115, 257–269. 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]
- 15.Hallermalm K, Seki K, Wei C, Castelli C, Rivoltini L, Kiessling R, and Levitskaya J (2001). Tumor necrosis factor-alpha induces coordinated changes in major histocompatibility class I presentation pathway, resulting in increased stability of class I complexes at the cell surface. Blood 98, 1108–1115. 10.1182/blood.v98.4.1108. [DOI] [PubMed] [Google Scholar]
- 16.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ, Valenzuela DM, et al. (2011). Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol 13, 129–135. 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V (2012). Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol 12, 253–268. 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mantovani A, Marchesi F, Malesci A, Laghi L, and Allavena P (2017). Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14, 399–416. 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masucci MT, Minopoli M, and Carriero MV (2019). Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol 9, 1146. 10.3389/fonc.2019.01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright KL, White LC, Kelly A, Beck S, Trowsdale J, and Ting JP (1995). Coordinate regulation of the human TAP1 and LMP2 genes from a shared bidirectional promoter. J. Exp. Med 181, 1459–1471. 10.1084/jem.181.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T, Gaykalova DA, Considine M, Wheelan S, Pallavajjala A, Bishop JA, Westra WH, Ideker T, Koch WM, Khan Z, et al. (2016). Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. Int. J. Cancer 139, 373–382. 10.1002/ijc.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren S, Gaykalova DA, Guo T, Favorov AV, Fertig EJ, Tamayo P, Callejas-Valera JL, Allevato M, Gilardi M, Santos J, et al. (2020). HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 39, 6327–6339. 10.1038/s41388-020-01431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Z, and Damania B (2016). The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe 19, 150–158. 10.1016/j.chom.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, and Hornung V (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol 10, 1065–1072. 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M, et al. (2012). Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495. 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayman TJ, Baro M, MacNeil T, Phoomak C, Aung TN, Cui W, Leach K, Iyer R, Challa S, Sandoval-Schaefer T, et al. (2021). STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat. Commun 12, 2327. 10.1038/s41467-021-22572-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borden EC (2019). Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov 18, 219–234. 10.1038/s41573-018-0011-2. [DOI] [PubMed] [Google Scholar]
- 28.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tân PF, Westra WH, Chung CH, Jordan RC, Lu C, et al. (2010). Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med 363, 24–35. 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferris RL, Licitra L, Fayette J, Even C, Blumenschein G Jr., Harrington KJ, Guigay J, Vokes EE, Saba NF, Haddad R, et al. (2019). Nivolumab in patients with recurrent or metastatic squamous cell carcinoma of the head and neck: efficacy and safety in CheckMate 141 by prior cetuximab use. Clin. Cancer Res 25, 5221–5230. 10.1158/1078-0432.CCR-18-3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington KJ, Kasper S, Vokes EE, Even C, et al. (2018). Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 81, 45–51. 10.1016/j.oraloncology.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Werness BA, Levine AJ, and Howley PM (1990). Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248, 76–79. 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 32.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, and Howley PM (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136. 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 33.Dyson N, Howley PM, Münger K, and Harlow E (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243, 934–937. 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 34.Chang YE, and Laimins LA (2000). Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol 74, 4174–4182. 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuel CE (2001). Antiviral actions of interferons. Clin. Microbiol. Rev 14, 778–809. 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, Sangkomkamhang U, and Ekalaksananan T (2014). E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS One 9, e91473. 10.1371/journal.pone.0091473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau L, Gray EE, Brunette RL, and Stetson DB (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 38.Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, Moghbeli T, Tan YS, Lin X, Bellile E, et al. (2020). HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Invest 130, 1635–1652. 10.1172/JCI129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronco LV, Karpova AY, Vidal M, and Howley PM (1998). Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 12, 2061–2072. 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaikh MH, Bortnik V, McMillan NA, and Idris A (2019). cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog 132, 162–165. 10.1016/j.micpath.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Bortnik V, Wu M, Julcher B, Salinas A, Nikolic I, Simpson KJ, McMillan NA, and Idris A (2021). Loss of HPV type 16 E7 restores cGAS-STING responses in human papilloma virus-positive oropharyngeal squamous cell carcinomas cells. J. Microbiol. Immunol. Infect 54, 733–739. 10.1016/j.jmii.2020.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Raikhy G, Woodby BL, Scott ML, Shin G, Myers JE, Scott RS, and Bodily JM (2019). Suppression of stromal interferon signaling by human papillomavirus 16. J. Virol 93, 00458–19. 10.1128/JVI.00458-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott ML, Woodby BL, Ulicny J, Raikhy G, Orr AW, Songock WK, and Bodily JM (2020). Human papillomavirus 16 E5 inhibits interferon signaling and supports episomal viral maintenance. J. Virol 94, 01582–19. 10.1128/JVI.01582-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, and Chen ZJ (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobs JL, and Coyne CB (2013). Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol 425, 5009–5019. 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gene Ontology Consortium (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, D’Eustachio P, Stein L, and Hermjakob H (2017). Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics 18, 142. 10.1186/s12859-017-1559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanehisa M (2019). Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanehisa M, Furumichi M, Sato Y, Kawashima M, and Ishiguro-Watanabe M (2023). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, and Peters B (2019). The immune epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343. 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider TD, and Stephens RM (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100. 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Judd NP, Allen CT, Winkler AE, and Uppaluri R (2012). Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol. Head Neck Surg 147, 493–500. 10.1177/0194599812442037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Disbrow GL, Sunitha I, Baker CC, Hanover J, and Schlegel R (2003). Codon optimization of the HPV-16 E5 gene enhances protein expression. Virology 311, 105–114. 10.1016/s0042-6822(03)00129-6. [DOI] [PubMed] [Google Scholar]
- 63.Purcell AW, Ramarathinam SH, and Ternette N (2019). Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc 14, 1687–1707. 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNAseq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LMP2 (clone G-3) | Santa Cruz Biotechnology | Cat#: sc-373996; RRID: AB_10918476 |
| Mouse monoclonal anti-LMP7 (clone D-2) | Santa Cruz Biotechnology | Cat#: sc-374089; RRID: AB_10947104 |
| Mouse monoclonal anti-α-tubulin | Developmental Studies Hybridoma Bank (DSHB) | Cat#: 12G10; RRID: AB_2315509 |
| Rabbit polyclonal anti-STAT1 | Cell Signaling Technology | Cat#: 9172; RRID: AB_2198300 |
| Rabbit monoclonal anti-phospho-STAT1 (Tyr701) (clone 58D6) | Cell Signaling Technology | Cat#: 9167; RRID: AB_561284 |
| Rabbit polyclonal anti-MAVS | Cell Signaling Technology | Cat#: 3993; RRID: AB_823565 |
| Rabbit monoclonal anti-STING (clone D2P2F) | Cell Signaling Technology | Cat#: 13647; RRID: AB_2732796 |
| Rabbit monoclonal anti-IRF3 (clone D83B9) | Cell Signaling Technology | Cat#: 4302; RRID: AB_1904036 |
| Rabbit monoclonal anti-phospho-IRF3 (Ser396) (clone 4D4G) | Cell Signaling Technology | Cat#: 4947; RRID: AB_823547 |
| Mouse monoclonal anti-Nuclear Matrix Protein p84 (clone 5E10) | GeneTex | Cat#: GTX70220; RRID: AB_372637 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat#: F3165; RRID: AB_259529 |
| Mouse monoclonal anti-HA.11 Epitope Tag (clone 16B12) | BioLegend | Cat#: 901501; RRID: AB_2565006 |
| Mouse monoclonal anti-c-Myc (Clone 9E10) | BioLegend | Cat#: 626801; RRID: AB_2235686 |
| Mouse monoclonal anti-HLA-ABC (clone W6/32) | BioLegend | Cat#: 311441; RRID: AB_2800814 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#: 31430; RRID: AB_228307 |
| Goat anti-Rabbit IgG (H + L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#: 31460; RRID: AB_228341 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human IFN-α2 (carrier-free) | BioLegend | Cat#: 592702 |
| Poly(I:C) (LMW)/LyoVec™ | InvivoGen | Cat#: tlrl-picwlv |
| Poly(I:C) (HMW)/LyoVec™ | InvivoGen | Cat#: tlrl-piclv |
| ADU-S100 [2′3′-c-di-AM(PS)2 (Rp,Rp)] | InvivoGen | Cat#: tlrl-nacda2r |
| TRIzol™ Reagent | Thermo Fisher Scientific (Invitrogen) | Cat#: 15596018 |
| qScript cDNA Synthesis Kit | Quantabio | Cat#: 95047 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725124 |
| PEI (Polyethylenimine, branched) | Sigma-Aldrich | Cat#: 408727 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich (Roche) | Cat#: 11873580001 |
| PhosSTOP™ | Sigma-Aldrich (Roche) | Cat#: 4906837001 |
| Protein G Plus/Protein A Agarose Suspension | EMD Millipore | Cat#: IP05 |
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat#: A2220 |
| Critical commercial assays | ||
| RNeasy Plus Mini Kit | QIAGEN | Cat#: 74136 |
| Immunoproteasome Activity Fluorometric Assay Kit II | UBPBio | Cat#: J4170 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#: E1980 |
| Deposited data | ||
| RNAseq data of cell lines | This paper | GEO: GSE226754 |
| RNAseq data of HPV-positive OPSCCs from Johns Hopkins University cohort | Guo et al.21 | GEO: GSE112027 |
| Experimental models: Cell lines | ||
| Human: CAL-27 | Dr. J. Silvio Gutkind (University of California, San Diego) | RRID: CVCL_1107 |
| Human: HEK293T | Dr. Dong-Er Zhang (University of California, San Diego) | RRID: CVCL_0063 |
| Mouse: DC2.4 | Dr. Dong-Er Zhang (University of California, San Diego) | RRID: CVCL_J409 |
| Mouse: MOC2 | Dr. J. Silvio Gutkind (University of California, San Diego) | RRID: CVCL_ZD33 |
| Mouse: AT-84 | Dr. Aldo Venuti (Regina Elena National Cancer Institute, Italy) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#: 000664; RRID: IMSR_JAX:000664 |
| Mouse: C3H/HeNCrl | Charles River Laboratories | Strain Code: 025; RRID: IMSR_CRL:025 |
| Oligonucleotides | ||
| See Table S5 for qPCR primers | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pLXSN-AU1-HPV16 E5 (codon-optimized) | Dr. Frank Suprynowicz (Georgetown University Medical School) | N/A |
| Plasmid: MIP (MSCV-IRES-Puro) | Dr. Dong-Er Zhang (University of California, San Diego) | N/A |
| Plasmid: pMSCV-Blasticidin | Addgene | Plasmid: #75085; RRID: Addgene_75085 |
| Plasmid: pMSCV-Zeo | Addgene | Plasmid: #75088; RRID: Addgene_75088 |
| Plasmid: pCL-10A1 | Novus | Cat#: NBP2–29542 |
| Plasmid: pCL-Eco | Novus | Cat#: NBP2–29540 |
| Plasmid: pcDNA™3.1 (+) Mammalian Expression Vector | Thermo Fisher Scientific (Invitrogen) | Cat#: V79020 |
| Plasmid: MIP-FLAG-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pMSCV-Blasticidin-HPV16 E6 | This paper | N/A |
| Plasmid: pMSCV-Zeo-HPV16 E7 | This paper | N/A |
| Plasmid: pcDNA-FLAG-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pcDNA-HA-HPV16 E5 (codon-optimized) | This paper | N/A |
| Plasmid: pcDNA-HA-human MAVS | This paper | N/A |
| Plasmid: pcDNA-human STING-HA | This paper | N/A |
| Plasmid: pcDNA-human STING-Myc | This paper | N/A |
| Plasmid: pcDNA-HA-human IRF3 | This paper | N/A |
| Plasmid: pGL3-Basic | Promega | Cat#: E1751 |
| Plasmid: pGL3-IFN-β gene promoter | This paper | N/A |
| Plasmid: pRL-TK | Promega | Cat#: E2241 |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | N/A |
| ImageJ | Schneider et al.48 | https://imagej.nih.gov/ij/ |
| STAR (v2.6.1) | Dobin et al.49 | N/A |
| DESeq2 (v2_1.6.3) | Love et al.50 | N/A |
| Gene Ontology | Ashburner et al.51; Gene Ontology Consortium52; Mi et al.53 |
http://geneontology.org/ |
| Reactome | Fabregat et al.54 | https://reactome.org/ |
| KEGG | Kanehisa et al.55–57 | http://www.kegg.jp/ |
| PEAKS | Bioinformatics Solutions Inc. | N/A |
| Immune Epitope Database and Analysis Resource (IEDB) | Vita et al.58 | https://www.iedb.org |
| WebLogo 3 | Schneider et al.59; Crooks et al.60 |
https://weblogo.threeplusone.com |
| Other | ||
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat#: LT07–710 |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.