

Abstract
Background
Aminoacylase 1 (ACY1, EC 3.5.1.14) deficiency (ACY1D) is a very rare inherited metabolic disease (IMD) with autosomal recessive inheritance (OMIM #609924). Up to date, only 15 cases have been reported in the literature. It is diagnosed by detecting acetylated amino acids among the patient’s urine organic acids by gas chromatography-mass spectrometry. Its clinical manifestations are highly variable, ranging from severe neurological symptoms to being asymptomatic.
Case Description
We present a 14-year-old boy with mild intellectual disability, speech sound disorder and non-alcoholic fatty liver disease who exhibited increased urinary excretion of N-acetylalanine, N-acetylmethionine and N-acetylglutamine during testing for inherited metabolic disorders. A suspected ACY1D was subsequently confirmed by targeted next generation sequencing, which revealed the presence of a homozygous pathogenic missense mutation in the ACY1 gene, c.1057C>T (p.Arg353Cys). The proband underwent speech education with good outcome. The same homozygous mutation in ACY1 gene was found in the boy’s two brothers, who exhibited slightly varied intellectual abilities. Follow-up examinations of the siblings revealed no deterioration in their mental skills.
Conclusions
These results suggest that uneven mental abilities in pediatric patients with various disorders including autism spectrum disorder may be sufficient grounds to warrant metabolic testing for ACY1D. The acylglycines urine excretion could be a promising novel metabolic marker for ACY1D testing.
Keywords: Aminoacylase 1 deficiency (ACY1D), missense mutation, mental skills, case report
Highlight box.
Key findings
• Aminoacylase 1 deficiency (ACY1D) has variable clinical manifestation within one family.
What is known and what is new?
• ACY1D is rare inherited metabolic disease.
• ACY1D can be easily detected by high urinary excretion of acetylated amino acids and short-chain acylglycines using novel mass spectrometry methods.
What is the implication, and what should change now?
• Metabolic screening for ACY1D should be performed in children with neurodevelopmental disorders.
Introduction
Aminoacylase 1 (ACY1, EC 3.5.1.14) is a zinc-binding metalloenzyme belonging to the aminoacylase family, whose members catalyze the hydrolytic deacetylation of N-acetylated proteins. ACY1 is expressed most strongly in the kidney and brain, and catalyzes the cleavage of various N-acyl-L-amino acids (other than N-acetyl aspartate, which is hydrolyzed by aminoacylase 2) into the corresponding free amino acids and acetic acid. Deficiency of ACY1 (ACY1D, OMIM #609924) causes a very rare and potentially serious neurodegenerative disease known as Canavan disease, which has a highly variable phenotype. ACY1D individuals can be wholly asymptomatic but may also suffer from severe neurological and psychiatric disabilities. ACY1D is diagnosed on the basis of elevated levels of urinary N-acetylated amino acids, which can be detected by gas chromatography-mass spectrometry (GC/MS) or nuclear magnetic resonance (NMR) spectroscopy. Definitive confirmation of the diagnosis is obtained by enzyme activity determination or gene analysis. The disease exhibits autosomal recessive inheritance and the ACY1 gene is located on chromosome 3p21. This case report presents a 14-year-old boy with ACY1D who was referred for evaluation for obesity, hepatic steatosis, and incipient metabolic syndrome. The boy was screened for inherited metabolic disorders (IMD) due to the cognitive impairment characterized by below-average intelligence. We present this case in accordance with the CARE reporting checklist (available at https://acr.amegroups.com/article/view/10.21037/acr-23-46/rc).
Case presentation
The proband (8/IV in Figure 1) was a boy and the 3rd child in his family, born from his mother’s 3rd pregnancy. He was born spontaneously at term by head delivery, birth weight was 3,800 g, birth length was 51 cm, and postpartum adaptation was uneventful. The boy’s psychomotor development was normal, but he attended speech therapy for a speech sound disorder. He was evaluated as socially immature at the beginning of his schooling and was diagnosed with dysgraphia at the age of 10 years during an examination by a special educator. He does not ride a bicycle because he reports that problems with coordination and balance make it impossible for him to do so. At 14 years of age, he was examined for elevated transaminase levels in obesity. His weight was 87.9 kg, height 174 cm, waist circumference 104 cm (Z score 2.5), body mass index (BMI) 29.0 kg/m2 (Z score 2.52), and blood pressure 132/83. Clinical examination revealed only marked gynecomastia and sonography showed diffuse hepatic hyperechogenicity consistent with steatosis and splenomegaly without sonographic evidence of portal hypertension. Biochemical results from his first outpatient examination are presented in Table 1. His condition was concluded to be non-alcoholic fatty liver disorder with obesity and incompletely expressed metabolic syndrome (1). Screening for IMD and psychological examinations were also performed because of the proband’s uneven mental skills. When tested with the Wechsler Intelligence Scale for Children (WISC-III), the boy exhibited unevenly distributed verbal and perceptual intellectual abilities. Verbal abilities were in the upper range of below average and perceptual abilities were in the average range. Overall cognitive ability was in the upper band of the below average range. The boy’s medical history was suspicious for attention deficit hyperactivity disorder (ADHD), he had a speech sound disorder, and he had been referred to the Special Education Center for Learning Disabilities since 5th grade (Table 2).
Figure 1.

Family pedigree tree. IM, myocardial infarction; Ca, carcinoma; GIT, gastrointestinal tract; DM, diabetes mellitus; AD, Alzheimer’s disease; FH, familial hypercholesterolemia; FoA, foramen ovale apertum.
Table 1. Proband’s laboratory test results at 14 and 18 years of age.
| Characteristic | Proband age | Normal range | |
|---|---|---|---|
| 14 years | 18 years | ||
| BMI (kg/m2) | 29.0 | 26.5 | 18.5–24.9 |
| Total cholesterol (mmol/L) | 5.7 | 4.6 | 2.9–5.0 |
| Triglycerides (mmol/L) | 1.53 | 0.95 | 0.42–1.7 |
| HDL-C (mmol/L) | 1.36 | 1.14 | 1.0–2.1 |
| Apo A1 (g/L) | 1.41 | ND | 1.0–1.7 |
| Apo B100 (g/L) | 1.09 | ND | 0.4–1.0 |
| Blood sugar (mmol/L) | 4.9 | 4.9 | 3.0–5.6 |
| HOMA-IR | 2.5 | 1.5 | ≤2.0 |
| ALT (μkat/L) | 0.6 | 0.45 | 0.1–0.78 |
| AST (μkat/L) | 0.54 | 0.72 | 0.22–0.67 |
| Uric acid (μmol/L) | 463 | 437 | 200–420 |
| Blood pressure | 132/83 | 121/73 | 115/70–120/80 |
BMI, body mass index; HDL, high density lipoprotein; Apo, apolipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance; ALT, alanine-aminotransferase, AST, aspartate-aminotransferase; ND, not determined.
Table 2. Psychological test results for the proband and his siblings.
| Psychological parameters | Proband (8/IV) | Sibling 1 (7/IV) | Sibling 2 (9/IV) |
|---|---|---|---|
| Verbal ability | Low | Average | Above |
| Perceptual ability | Average | Above | Above |
| Full scale IQ | Low | Above | Above |
| Attention deficit | Yes | No | Ambiguous |
| Hyperactivity | Yes | No | Ambiguous |
| SLD | Yes | No | Ambiguous |
| Speech disorder | Yes | No | Yes |
| Autism | No | No | No |
IQ, intelligence quotient; SLD, specific learning disabilities.
On first admission, the patient underwent analysis of organic acids, amino acids acylcarnitines, purines and pyrimidines, intermediates of creatine metabolism, acylglycines, and mucopolysaccharides in urine, serum and blood spot. Increased excretion of N-acetylalanine, N-acetylmethionine and N-acetylglutamine were detected by GC/MS. Interestingly, higher urinary excretions of acetyl, propionyl and butyrylglycine with calculated z-scores of 173.0; 154.1 and 40.3 were observed.
This finding was confirmed by liquid chromatography with tandem MS (Figure 2), where z-scores of 17.0, 148.2, 160.4 and 11.7 for acetylated acetyl-conjugates with glutamine, leucine, serine and valine were consistent with a ACY1D (2). The suspected ACY1D was confirmed by targeted next-generation sequencing (Ion Torrent S5, Thermo Fisher Scientific, Waltham, MA, USA), which revealed a homozygous pathogenic missense variant in the ACY1 gene c.1057C>T [p.Arg353Cys; rs21912698, minor allele frequency (MAF) 0.06%]. This missense mutation was subsequently detected in both parents in the heterozygous form (5/III and 6/III in Figure 1) using Sanger sequencing (ABI 3130, Thermo Fisher Scientific). IMD examination of the siblings (two brothers) also revealed similar increased urinary excretion of N-acetylated amino acids and selected acylglycines (Figure 2), and Sanger sequencing showed that both brothers were homozygous for the familial mutation (7/IV and 9/IV in Figure 1). The familial variant was not found in the eldest sister (6/IV in Figure 1). No abortions were reported in the family. Enzyme activity was not determined in any of the siblings or parents, and no brain magnetic resonance imagings (MRIs) were performed to evaluate neurological health. The boy was followed up on an outpatient basis, and it was found that his sonographic and biochemical status improved following compliance with a prescribed regimen and dietary modifications. The proband underwent speech education with good outcome and there was no deterioration in mental skills during this period.
Figure 2.
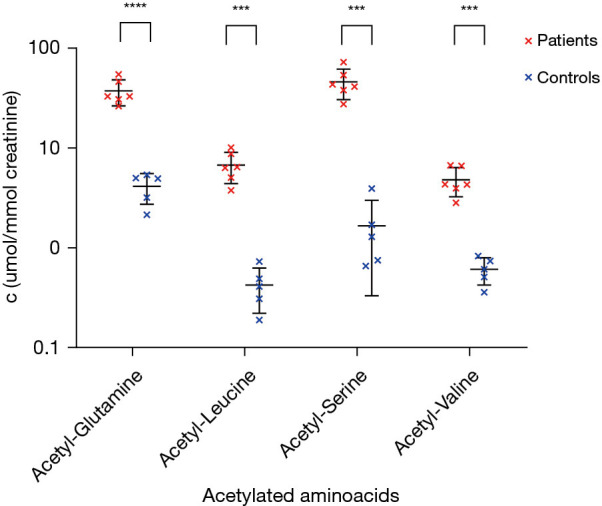
Concentrations of selected acetylated amino acids in siblings with ACY1 and healthy controls. ***, P<0.001; ****, P<0.0001.
The two brothers with familial homozygous variant c.1057C>T in ACY1 gene were also evaluated by a psychologist using the Wechsler Intelligence Scale. The adult scale (WAIS-III) was used in the case of the 25-year-old older brother, whose intellectual ability profile was unevenly distributed across the two components of intellect. His verbal abilities were in the average range and his perceptual abilities were in the higher average range. Overall, the cognitive abilities were in the higher average band.
Patient 3, the younger brother, was 13 years old and had a speech and behavioral disorder consistent with ADHD. He was examined using the WISC-III with the conclusion that the profile of rational abilities was balanced in the verbal part of the intellect, but unevenly distributed in the perceptual part. Verbal abilities were in the higher average range and the perceptual abilities were in the above average band. Overall, the cognitive abilities were in the above average band.
The cognition examinations confirmed that both the proband and his siblings exhibited better performance in perceptual abilities than in verbal abilities. Additionally, the proband and his younger brother had identical histories of ADHD symptomatology and speech impairment.
The proband’s father was further investigated for suspected familial hypercholesterolemia. Targeted sequencing revealed the ε4/ε4 isoform in the APOE gene associated with an increased risk of developing Alzheimer’s disease and with higher LDL cholesterol levels (3). No other pathogenic variants were found in genes associated with familial hypercholesterolemia, including the APOB and LDRL genes.
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient’s parents or legal guardian for the publication of this case report. A copy of the written consent is available for review by the editorial office of this journal.
Discussion
The clinical manifestation of ACY1D is highly variable. Patients have been described with severe neurological findings including generalized dystonia (4) and convulsions (2,5,6), but cases involving only motor clumsiness and sluggishness have also been reported (7,8). Patients with documented ACY1D show varying mental deficits including speech delay (9) and behavioral disorders with autistic features (9,10). However, patients with no loss of mental skills, no behavioral disturbances, and normal neurological findings have also been diagnosed (7). Several sets of brain MRI data on patients with ACY1D have been published, with results ranging from normal brain imaging (6,8,11) to cortical laminar necrosis, delayed myelination with hypoplasia of the corpus callosum and cerebellar vermis, and cortical dysplasia with gray matter heterotopia (2,5,7,9). Syringomyelia in the lumbar spinal cord has also been described in one patient (7). Patients exhibiting different clinical symptoms and brain MRI results also differ in terms of genetic analysis results, and there is no correlation between phenotype and genotype.
The proband in our study exhibited delayed development of verbal and opinion mental abilities, with discordant patterns of perception, memory, and intellectual abilities together with attention problems similar to those described by Alessandrì et al. (11). A secondary finding, unrelated to the diagnosed ACY1D, was obesity with hepatic steatosis and hyperuricemia with a less physically active lifestyle. The boy did not satisfy the diagnostic criteria for metabolic syndrome, although weight above the 92nd percentile, a systolic blood pressure of 130 mmHg, and homeostatic model assessment of insulin resistance (HOMA-IR >2) were all present. These symptoms were corrected by lifestyle improvements upon follow-up (1).
Discrepancies in verbal and perceptual abilities are also present in the proband’s brothers, who have genetically proven ACY1 deficiencies. The brothers’ mental abilities range from above normal to above average. Interestingly, neither the proband nor his brothers were diagnosed with autism spectrum disorders, unlike the patient with the same homozygous c.1057C>T mutation in ACY1 gene described by Tylki-Szymanska et al. (10). In accordance with Alessandrì et al. (11), we do not anticipate progression or more rapid deterioration of intellectual abilities based on the follow-up of our ACY1D proband.
The homozygous c.1057C>T missense variant in ACY1 gene found in the studied siblings is the most common pathogenic mutation in ACY1D patients. It has been described in 7 of 15 probands diagnosed with ACY1D based on molecular genetic analysis (11). In four cases, the mutation was homozygous (c.1057C>T/c.1057C>T). Reduced intellectual ability and susceptibility to convulsions are most frequently described in patients with this mutation (2,4,6,7,10). The pathogenic significance of the c.1057C>T missense variant was evaluated based on the most recent ClinVar database entry, and then based on the PhyloP score, predictive software, and low population frequency. The variant’s clinical significance was demonstrated by the loss of ACY1 catalytic activity in patient cells compared with controls (2,5). Five carriers bearing the heterozygous form of this mutation were found among 161 controls (2), suggesting that the prevalence of ACY1D may be underestimated. To our knowledge, only 15 patients with ACY1D who were diagnosed before the age of 11 years have been described in the literature to date. Another patient with first dystonic symptoms at age 12 years, which gradually progressed to generalized dystonia, was diagnosed at age 63 years. The symptoms described in the literature vary widely, and it is not yet clear whether there is any relationship between clinical symptomatology and ACY1D. The family history of our patients does not support the hypothesis of an increased number of miscarriages associated with mutations in the ACY1 gene (11).
All three patients showed a very similar abnormal urinary profile of acetylated amino acids, a typical finding in ACY1D. Moreover, the increased excretion of short-chain acylglycines, which has not been previously described in literature, appears to be interesting. The selected acylglycines may help in refining the diagnostic process in patients with ACY1D.
Conclusions
The low frequency of ACY1D diagnoses may be because it frequently causes only minor intellectual disabilities in childhood that are not indicated for selective screening including GC-MS analysis of urinary organic acids. It is therefore possible that selective IMD screening including tests for urinary acetylated amino acids should be indicated not only for equivocal neurological findings but also for cognitive impairment and in children with various disorders consistent with the autism spectrum.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was funded by Ministry of Education, Youth and Sport of the Czech Republic (NCMG - LM2023067 and EATRIS-CZ – LM2018133 to J.S.), Palacky University Olomouc (LF 2023_006 to J.S.) and Czech Health Research Council AZV CR (No. NU20-08-00367 to D.F.).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from from the patient’s parents or legal guardian for publication of this case report. A copy of the written consent is available for review by the editorial office of this journal.
Footnotes
Reporting Checklist: The authors have completed the CARE reporting checklist. Available at https://acr.amegroups.com/article/view/10.21037/acr-23-46/rc
Peer Review File: Available at https://acr.amegroups.com/article/view/10.21037/acr-23-46/prf
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://acr.amegroups.com/article/view/10.21037/acr-23-46/coif). J.S. reports he obtained institutional funding from Ministry of Education, Youth and Sport of the Czech Republic (NCMG - LM2023067, EATRIS-CZ – LM2018133) and Palacky University Olomouc (LF 2023_006) and he is the co-founder of spin-off company Intellmed, Ltd. and Cancer Research Czech Republic Foundation. D.F. obtained institutional funding from Czech Health Research Council AZV CR (NU20-08-00367). The other authors have no conflicts of interest to declare.
References
- 1.Tock L, Prado WL, Caranti DA, et al. Nonalcoholic fatty liver disease decrease in obese adolescents after multidisciplinary therapy. Eur J Gastroenterol Hepatol 2006;18:1241-5. 10.1097/01.meg.0000243872.86949.95 [DOI] [PubMed] [Google Scholar]
- 2.Van Coster RN, Gerlo EA, Giardina TG, et al. Aminoacylase I deficiency: a novel inborn error of metabolism. Biochem Biophys Res Commun 2005;338:1322-6. 10.1016/j.bbrc.2005.10.126 [DOI] [PubMed] [Google Scholar]
- 3.Khalil YA, Rabès JP, Boileau C, et al. APOE gene variants in primary dyslipidemia. Atherosclerosis 2021;328:11-22. 10.1016/j.atherosclerosis.2021.05.007 [DOI] [PubMed] [Google Scholar]
- 4.Sass JO, Vaithilingam J, Gemperle-Britschgi C, et al. Expanding the phenotype in aminoacylase 1 (ACY1) deficiency: characterization of the molecular defect in a 63-year-old woman with generalized dystonia. Metab Brain Dis 2016;31:587-92. 10.1007/s11011-015-9778-6 [DOI] [PubMed] [Google Scholar]
- 5.Sommer A, Christensen E, Schwenger S, et al. The molecular basis of aminoacylase 1 deficiency. Biochim Biophys Acta 2011;1812:685-90. 10.1016/j.bbadis.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 6.Sass JO, Olbrich H, Mohr V, et al. Neurological findings in aminoacylase 1 deficiency. Neurology 2007;68:2151-3. 10.1212/01.wnl.0000264933.56204.e8 [DOI] [PubMed] [Google Scholar]
- 7.Sass JO, Mohr V, Olbrich H, et al. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am J Hum Genet 2006;78:401-9. 10.1086/500563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alessandrì MG, Casarano M, Pezzini I, et al. Isolated Mild Intellectual Disability Expands the Aminoacylase 1 Phenotype Spectrum. In: Zschocke J, Gibson K, Brown G, et al. editors. JIMD Reports Volume 16. Berlin, Heidelberg: Springer; 2014:81-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferri L, Funghini S, Fioravanti A, et al. Aminoacylase I deficiency due to ACY1 mRNA exon skipping. Clin Genet 2014;86:367-72. 10.1111/cge.12297 [DOI] [PubMed] [Google Scholar]
- 10.Tylki-Szymanska A, Gradowska W, Sommer A, et al. Aminoacylase 1 deficiency associated with autistic behavior. J Inherit Metab Dis 2010;33 Suppl 3:S211-4. 10.1007/s10545-010-9089-3 [DOI] [PubMed] [Google Scholar]
- 11.Alessandrì MG, Milone R, Casalini C, et al. Four years follow up of ACY1 deficient patient and pedigree study. Brain Dev 2018;40:570-5. 10.1016/j.braindev.2018.03.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as