

Summary
Accurate measurement of the biological markers of the aging process could provide an “aging clock” measuring predicted longevity and enable the quantification of the effects of specific lifestyle choices on healthy aging. Using machine learning techniques, we demonstrate that chronological age can be predicted accurately from (1) the expression level of human genes in capillary blood and (2) the expression level of microbial genes in stool samples. The latter uses a very large metatranscriptomic dataset, stool samples from 90,303 individuals, which arguably results in a higher quality microbiome-aging model than prior work. Our analysis suggests associations between biological age and lifestyle/health factors, e.g., people on a paleo diet or with IBS tend to have higher model-predicted ages and people on a vegetarian diet tend to have lower model-predicted ages. We delineate the key pathways of systems-level biological decline based on the age-specific features of our model.
Subject areas: Microbiome, Omics
Graphical abstract
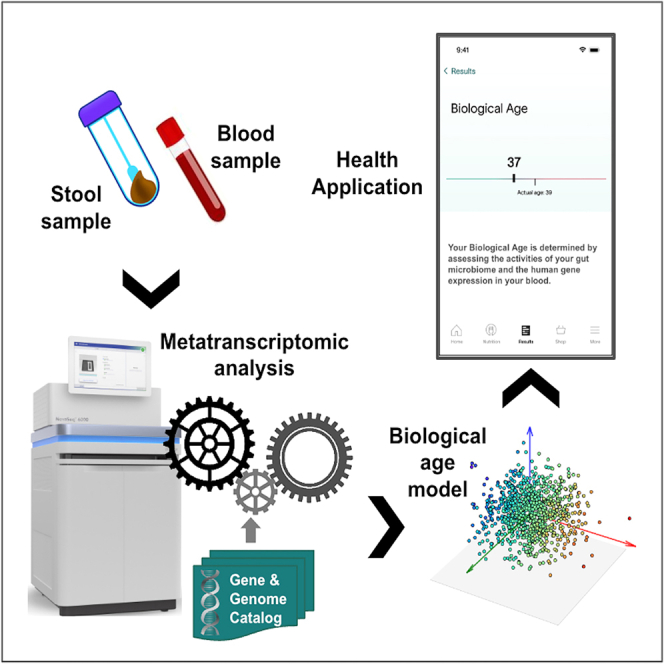
Highlights
-
•
Biological aging clocks developed from human gut microbiome and blood transcriptome
-
•
Microbiome dataset from metatranscriptomic analysis of stool from 90,303 individuals
-
•
Aging models account for around 46% and 53% of the variance in age by R2 respectively
-
•
Associations found between biological age and diet, drinking, diabetes, IBS, etc
Microbiome; Omics
Introduction
Biological age refers to biological markers of the aging process, and may be accelerated or slowed in some individuals relative to their chronological age. Recent research has proposed computational aging clocks based on various biomarkers including metabolites, blood cell count and other routine lab tests,1,2 DNA methylation,3,4,5,6 gene expression in tissue7 or blood,8,9 taxonomic composition of the gut microbiome,10,11 and others. Aging clocks propose to use a signal derived from these biomarkers as a health-related metric for aging. In this paper we present two biological age metrics, one derived from the metatranscriptome of the gut microbiome, and one from the transcriptome of capillary blood. These two metrics together arguably capture a very comprehensive view of human biology.10,12,13,14,15,16
Molecular markers from both microbial and human cells have been used to develop aging clocks. The composition and function of the gut microbiome changes with age, and may modulate healthy aging through multiple mechanisms. The increased dysbiosis associated with age can lead to innate proinflammatory immune responses, and the small molecules secreted by the gut microbiome affect host metabolism and signaling pathways that vary with age (see review in Kim & Jazwinski, 2018, i.a.).17 There is evidence that these microbiome changes over time are directly implicated in human healthspan. Maffei et al. (2017)18 show that certain properties of the gut microbiome, notably taxonomic diversity, are more predictive of a frailty index measuring mortality risk than chronological age. Similarly, on the human side, several molecular markers may modulate healthy aging. Perhaps the strongest aging clocks proposed so far have relied on biomarkers related to the epigenome such as DNA methylation. While these can act as an estimator of biological age, they are not comprehensive and they have limited ability to pinpoint the regulators of the biological clock. Both the gut microbiome and the human molecular mechanisms are known to participate in widespread epigenetic interactions (see survey in Watson & Søreide, 2017),19 so a biological clock based on both of these functions can potentially inform specific therapeutic avenues to slow down aging. These may include personalized diets, supplements (vitamins, minerals, prebiotics, probiotics, food extracts, etc.), pharmaceuticals, phages, immunotherapies (vaccines, antibodies), etc.
Here we briefly review related work using microbiome data to explore aging. (Lan et al. 2013)20 used metagenomic data from 5 studies but focused on one for their classification results. They repeatedly sample 110 samples and perform a binary classification using SVMs after feature selection using tf-idf and mRMR. The binary labels are created by separating the population in two groups based on an age threshold which they vary. Across all thresholds they report a mean AUC of 0.65. In (Odamaki et al. 2016),21 the authors analyze the 16s microbiome data of 367 healthy Japanese subjects to uncover age related changes to the microbiome. They do not perform any prediction of age based on the gut microbiome. In (Huang et al. 2020)11 the authors look at three sources of data, the oral microbiome, skin microbiome, and gut microbiome. They predict age from 1,975 skin microbiome samples with an MAE of 3.8 +- 0.45. With 2,550 saliva samples they report an MAE of 4.5 +- 0.14. With 4434 fecal samples, they achieve an MAE of 11.5 +- 0.12.
The work with the most similar objective to ours is that from Galkin et al. 2020. Galkin et al. report the results from applying a Deep Neural Network (DNN) model trained on ∼4000 aggregated metagenomic profiles from people from 18 to 90 years old. As our models are trained on metatranscriptomic data, a direct comparison of model performance to Galkin et al.’s DNN model is unfortunately not possible. Instead we evaluated our model performance by matching the age distribution of the cohort used in the DNN model reported by Galkin et al. This may give some indication of the utility of our approach and data on populations of similar age composition. More information about the comparison is provided in the STAR Methods section below.
While there are many ways to define biological age and operationalize the development of an aging clock,22 a common approach is to fit a machine-learned model to predict the chronological age of the human subject from the biomarker. This model’s predictions will deviate from chronological age to some extent: for example, if the subject’s biomarker profile is more similar to biomarker profiles of older people than to their peers, the model will overpredict age. The model’s predictions can be interpreted as a biological age in the sense that they approximate the age of a typical subject with the given biomarker profile. In this paper we show that the gut microbiome metatranscriptome as well as the blood transcriptome display strong associations with age to allow the creation of an aging clock.
Results
Table 1 gives an overview of the stool microbiome and blood transcriptome cohorts (All participants consented to participation, and the study protocol and informed consent forms were approved by the Viome IRB (VIRB), an IRB federally accredited in the USA by the Department of Health and Human Services. All data was de-identified for the purpose of the analyses reported here) used in this study. The stool microbiome cohorts consist of samples obtained from unique customers of Viome’s Gut Intelligence product. These samples were divided into a microbiome discovery cohort of 78,637 samples, and a microbiome validation cohort of 11,666 samples. The Galkin et al. matched microbiome cohorts are intended to allow comparison of this model to the one presented in that work, and were constructed by randomly choosing one Viome customer from our validation set with the same age as each person in the Galkin et al. datasets. The Galkin et al. CV (Cross Validation) Host matched cohort is equivalent to Galkin et al.’s training set for which they report cross-validation performance; and the Galkin et al. Independent HC (Healthy Cohort) matched cohort is equivalent to their independent healthy cohort. One person in the matched CV cohort could not be paired with a unique sample in our data, so our cohort has 1164 samples rather than Galkin et al.’s 1165. For the Independent HC cohort, we additionally matched on sex, which was impossible in the larger CV cohort. The human blood transcriptome cohort consists of samples obtained from 1494 unique customers of Viome’s Health intelligence product and associated research studies.
Table 1.
Model performance by cohort
| Stool microbiome |
Human blood transcriptome |
||||
|---|---|---|---|---|---|
| Microbiome discovery cohort (cross-validated) | Microbiome Validation cohort (prospective) | Galkin et al. CV Host matched cohort | Galkin et al. Independent HC matched cohort | Blood transcriptome discovery cohort (cross-validated) | |
| Cohort size | 78,637 | 11,666 | 1164 | 252 | 1494 |
| Sex (% female) | 64.10 | 65.37 | 64.95 | 48.41 | 61.29 |
| Age (y) mean s.d. | 46.79 15.9 | 43.22 15.75 | 49.00 15.35 | 48.06 11.36 | 47.24 14.22 |
| Baseline MAE | 12.98 | 12.90 | 13.03 | 9.36 | 11.75 |
| Prediction R2 | 0.42 0.00 | 0.46 | 0.46 | 0.31 | 0.53 0.02 |
| Prediction MAE | 9.49 0.02 | 9.21 | 9.14 | 7.64 | 7.63 0.25 |
Figure 1 presents descriptive statistics of the discovery cohort of Table 1. Ages of sample donors range from <1 year to 104 years, with 2686 donors below 18 years of age (included with parental consent). Study participants come from over 60 countries (86% US, 8% Canada, 3% Australia, 1.5% EU, 1% UK, rest from other countries). We do not observe any differences in taxonomic richness by age (Figures 1B and 1C); nor do we find differences in taxonomic diversity or active function richness. Neither richness nor diversity of functions or of taxa were found to increase predictive accuracy when included in our models. Figures 1D and 1E shows the taxa at the species level and KOs that vary the most with age. To identify these, we calculated the mean CLR for each feature in each decade of age in 70% of the discovery cohort, and chose those with the highest variance across ages. Then we plotted the trend in mean CLR by decade in the remaining 30% of the data, grouped by Viome Functional Category (VFC, see below). Notably, all of the KOs with the highest positive association with age are part of Methanogenesis Pathways resulting in production of methane gas.
Figure 1.

Descriptive statistics for the microbiome discovery cohort
(A) age distribution (B) richness and Pielou’s evenness index of active microbial richness by decade (C) richness and Pielou’s evenness index for active functions (D and E) mean CLR transformed expression levels of species/KOs by age for most variable species/KOs grouped by genus or Viome Functional Categories (see main text). In the boxplots shown in Figures 1B and 1C, the box denotes the quartiles and the whiskers extend 1.5 IQRs (Inter Quartile Range) above and below. See Table 1 for details regarding the discovery cohort.
Our biological age model’s performance is also presented in Table 1. For the independent validation cohort, the model predicts chronological age above the baseline MAE of the datasets, and accounts for around 46% of the variance in age by R2, the standard metric of quality of fit in regression tasks. Table 1 presents the performance of the model under 5-fold cross validation. The model accounts for around 53% of variance in ages in the dataset.
Figure 2 shows our biological age model’s predictions and most important predictors, for the model using the microbiome discovery cohort of Table 1. Figure 2B shows the features with highest absolute coefficients (above 0.3 for taxa and 0.15 for KOs) out of 2,507 non-zero features selected by the model. Those features are grouped by Viome Functional Categories (VFCs) and further grouped into themes, to facilitate interpretation and visual presentation of the models’ features. We introduce ‘Viome Functional Categories (VFC)’ as a novel annotation system that integrates both species level taxonomic activity and the functional expression profiles from KOs into higher order biological themes. VFCs are expert-curated themes that account for pathway directionality of feature association (activation/suppression, production/degradation, protective/harmful). Microbial taxonomic features are grouped into 11 biological themes covering 32 VFCs. Human gene expression features are grouped into 26 themes covering 65 VFCs. For example, the theme “ProInflammatory Activities in Aging” consists of 8 different VFCs. Within this theme, the “Ammonia Production Pathways” VFC contains 3 KOs, all with a positive association with aging in the model. On the other hand, the “Vaginal Commensal Microorganisms” VFC contains three known vaginal microorganisms that are negatively associated with aging. More details are provided in the Table S3.
Figure 2.

Biological aging model using the microbiome discovery cohort
(A) Predicted vs. actual age in held-out validation data (for clarity, only a random subset of points is shown), with the line of best fit superimposed (error bands represent a 95% confidence interval calculated through bootstrapping).
(B) Coefficients for the microbial taxonomic features (circles) and KO features (triangles) grouped into curated Viome Functional Categories (VFCs).
Figure 3 presents the predictions of our aging model based on human blood transcriptome cohort of Table 1. Figure 3B shows the features with highest absolute coefficients out of 2293 features selected by the model on average, grouped by VFC.
Figure 3.

Biological aging model using the human blood transcriptome discovery cohort
(A) Actual vs. predicted age, with the line of best fit superimposed (error bands represent a 95% confidence interval calculated through bootstrapping).
(B) Top coefficients grouped by Viome Functional Categories (VFCs).
Cohort comparisons
As an additional exploration of our stool microbiome model, we compare the biological age predicted for a number of specific subsets of the full microbiome dataset (discovery plus validation) corresponding to populations of interest. In each case we select all available samples from the population of interest (e.g., vegetarians), and create an appropriate control cohort (e.g., omnivores) where each member of the control cohort is matched on age to one member of the reference cohort. For each member in the population of interest, one control is randomly sampled from the pool of users with the same age in the control population. All diet information was self-reported and collected through a questionnaire (we interpret an organic diet as favoring organically grown produce, although no definition is given beyond this label). We perform Wilcoxon signed-rank tests to determine whether there is a significant difference in biological age between the cohorts. The cohorts consist of: people reporting the special diets vegan, vegetarian, organic, paleo, ketogenic (contrasted with people following no special diet); people with self-reported IBS and Diabetes (contrasted with people reporting no health issues); and heavy drinkers (contrasted with non-drinkers), where heavy drinkers were defined following Mayo Clinic guidelines as consuming 15 or more drinks per week for males and 8 or more for females.
To explore the biological age of specific populations of interest, we present summaries of Wilcoxon signed-rank tests in Figure 4A, which depicts the difference in mean biological ages for specific cohort comparisons of interest, together with p values from corresponding tests. Figure 4B shows the difference in chronological age for these same populations within the discovery cohort. We note that the model picks up several interesting differences between these populations and their age-matched controls. Vegetarians and vegans both tend to have a lower biological age than omnivores, while those following the ketogenic or paleo diets are biologically older than omnivores. Heavy drinkers are biologically older than non-drinkers. People with diabetes or IBS appear older than healthy controls. Some of these results may reflect chance patterns in the training data, as discussed below.
Figure 4.

Cohort comparisons
(A) Mean and standard error of biological age differences between cohorts and age-matched controls where p values <0.01 from Wilcoxon signed-rank tests.
(B) Mean and standard error of chronological age differences between cohorts and controls in the discovery cohort. Standard errors are small enough to appear as zero width bars.
Discussion
Model performance
The models presented here are capable of predicting chronological age above the baseline MAE of the datasets, and account for around 46% (stool) and 53% (blood) of the variance in age by R2. We note that some discrepancy between predicted and actual age is expected in a useful biological age candidate. If age was perfectly predicted, it would indicate either that the aspects of health captured by the biomarker decline in lockstep with chronological age, or that the biomarker is statistically associated with properties that vary systematically with age but are irrelevant to health.
Previously published attempts to predict age from the gut microbiome have used 16S (e.g., Huang et al., 2020, Odamaki et al., 2016)11,21 or metagenomic data.20 We present a comparison of our model with the gut metagenomic aging clock of Galkin et al. (2020),10 which is to our knowledge the most accurate microbiome-based clock published to date. Galkin et al. report MAE of 10.60 and R2 of 0.21 (vs. our 9.49 and 0.42) in a dataset (CV Host) with a baseline MAE of 13.03 (vs. our 12.98). In a secondary validation exercise, their model obtains MAE of 6.81 and R2 of 0.134 when applied to a separate dataset (Independent HC) with a lower baseline MAE of 9.27 (Galkin et al. also report MAE of 5.91 for this dataset but note that the data contains multiple samples from many of the subjects, and after merging duplicates into averaged samples, performance falls to 6.85. As performance on averaged samples is not representative of performance on individual samples, we report metrics for the Galkin et al. model after randomly excluding all but one sample from each donor. The metrics we report here are calculated from the predictions for individual samples shared as part of that paper’s supplementary data). Since metatranscriptomic data are unavailable for that cohort, we created an additional validation cohort with exactly the same age distribution shown in Table 1. In this cohort, our model attains MAE of 7.64 (vs. their 6.81) and R2 of 0.31 (vs. their 0.13). R2 is the standard metric used for quality of fit in a regression task, and these numbers suggest that our metatranscriptomic model provides a better overall fit to this distribution of ages.
Interestingly, Galkin et al. report that an Elastic Net model was unable to extract significant signal from the data, and achieved their best performance using a deep neural net (DNN). In contrast, we found similar performance across model types, including when using a neural network architecture modeled after the one they report. Further details of these attempts are given in the supplemental information. Using a linear model is advantageous in terms of interpretability and actionability: the influence of each biological feature on the model’s age prediction is transparent, making it straightforward to determine a set of candidate targets to act on.
Although we report separate models for the microbiome and human gene cases, these models could be combined to give a single prediction, which we have performed since the publication of this paper and will be publishing separately in the future.
The resolution of the data supplied by our clinical grade and fully automated lab analysis method allows identification of microorganisms at the strain level, although for this analysis we aggregate data to the species level. This contrasts with 16S gene sequencing, which does not discriminate between species within most genera. This additional resolution appears to be important to capture age-related variation. In several cases, some species of a genus are associated with older age, and others associated with younger age (shown in Figure S1 of supplemental information).
Contrary to much published literature23,24,25,26,27(cf. Bian et al., 2017),28 chronological age was not associated with significant changes in alpha diversity (richness or evenness) of taxa or KOs across decades (Figures 1B and 1C) despite significant changes in individual taxa and KOs over time (Figures 1D and 1E). This difference may be due to our RNA-based approach, whereas previous studies have used DNA-based approaches (amplicon or metagenomics). It is intriguing that the richness of active gene expression captured in this RNA-based data remains steady throughout life. Focusing on functional activity (RNA) and not potential (DNA) suggest that the number of actual genes active doesn’t change much throughout life.
Interestingly, age related discriminatory taxon previously identified using DNA based approaches are similar to the ones we found to be correlated with age. Species belonging to the Ruminococcaceae, Bifidobacteriaceae, Lachnospiraceae, and Clostridiaceae families have previously been shown to be important at predicting age from the gut microbial composition29 were also important predictors in our model. Similar to Hopkins et al.23 we also found a Bifidobacterium to be negatively correlated with age, while a Bacteroides was positively associated with age (in our case we identified a specific species). In addition, Yatsuneko et al. also showed that methanogenesis related genes are higher in adult microbiomes compared to children.26
Cohort comparisons
Figure 4A shows that populations following certain lifestyle choices or suffering from specific conditions are systematically assigned a different biological age from their age-matched controls. For instance, those on plant-based diets have lower model-predicted ages (see review in Medawar et al., 2019). On the other hand, heavy alcohol drinkers and IBS and diabetes sufferers have higher model-predicted ages. Those on a ketogenic or paleo diet tend to have a higher model-predicted age than controls. For some of these cohorts (e.g., drinking, diabetes) the age difference in the training data has the same sign as the predicted age (Figure 4B). In these cases, it is possible that age-based trends in the training data may have influenced the model’s estimation of the aging signal. However, for other cohorts (i.e., vegetarians, paleo, IBS) the opposite pattern is seen, which shows that the microbiome features associated with these populations include features associated with aging in the general population. Overall, these results are consistent with an interpretation of our biological age metric as reflecting an accumulation of lifestyle choices and disease status that contribute to healthy aging.
Molecular feature interpretation
The primary goal of this paper is to show that it is possible to estimate age with high accuracy from microbiome and human gene expression data. However, in this section we offer some possible interpretation of the features selected by the models. This is intended to highlight parallels to previous literature and potential avenues for future investigation.
The VFC are expert curated functional themes that provide mechanistic insights into the aging process from the features predictive of the biological age model. For instance, the Pro-Inflammatory Activities in Aging theme captures several VFCs like Oral Pathobionts, Ammonia Production Pathways and Methanogenesis Pathways that could lead to increased systemic inflammation and aging. The presence and activity of oral taxa in the gut is an established marker of hypochlorhydria (low stomach acid), which is known to develop with age as stomach acid levels and digestive efficiency progressively decline.30,31,32 It is worthy to highlight that both KO and Taxa features of our model collectively contribute to microbial Proinflammatory Activities in Aging. On the other hand, Cell Protective Activities in Aging theme captures Anti-Inflammatory and Antioxidant Production Pathways, such as Glutathione Production Pathways, which are known to be diminished with aging.33 Other than microbial Proinflammatory Activities, the model also predicts an association of Human Inflammatory Pathways with aging. Our model predicts increased demands on activity of pathways involved in B-cell differentiation, T cell proliferation, T cell differentiation, Eosinophil Migration, Cytokine Secretion, which can be viewed as the human response to activation of TLR4 and other signaling pathways with aging from microbial or environmental origins. While some of the proinflammatory activities increase with age, however, many of the crucial T cell response elements actually decrease in expression, which can result in insufficient ability of the immune system to respond to the sources of inflammation with age.
The role of the gut microbiome in neuro-generative processes is increasingly evident, and the perturbation of the microbiome and microbial products has been demonstrated to affect behavior34 through the well-known gut-brain axis.35 In the Neuronal Activities in Aging theme, the VFCs Glutamate and Gamma Amino Butyric Acid (GABA) Pathways, Serotonin Metabolism Pathways and Pro-apoptotic Pathways in neuronal cells show association with aging from the model, in line with current knowledge of Neuroinflammation and cognitive aging.
Some prior work has attempted to validate the relevance of biological age metrics to health by relating them to measures of health or mortality,1,36 and we are investigating a similar approach in ongoing work. We note that biological age may in general serve as a proxy for longevity or healthspan. Additionally, our results uncover novel machine-learned associations between age and specific metatranscriptomic features that could guide the design of nutritional interventions to reduce biological age and increase the human healthspan.37,38 We will continue to evaluate and improve the models as we obtain more data.
Conclusions
A major contribution of this paper is the development and validation of two models for biological age. Our stool metatranscriptome model uses what is to date the largest published cohort of stool microbiome samples, 90,303 in total. Although our human transcriptome model is based on a smaller dataset, it performs very well. Both models are capable of predicting chronological age well beyond the baseline MAE of the datasets, and account for 46% and 53% of the variance in age by R2 respectively.
Another contribution of this paper is the biological age characterization of populations following specific dietary and lifestyle choices. While not all of these initial results are readily interpretable, the trends in some populations indicate a relationship between health and biological age - for example, those on a vegetarian diet have lower model-predicted ages, while those on a paleo diet or suffering from IBS have higher model-predicted ages. These results are consistent with an interpretation of our biological age metric as reflecting an accumulation of lifestyle choices and disease status, and suggest that the microbiome features associated with these populations include features associated with aging in the general population.
Moreover, pathway analysis of the features and interpretation of functional themes suggests mechanistic insights predictive of aging, and further connects age-related microbial activities with human cellular expression patterns on a molecular level. This predictive signature not only offers additional clues to the role of immune function in progressive systems-level decline, but also provides possibilities for future nutritional or pharmacological anti-aging interventions.
This is the first report of using functional (i.e., gene expression) microbiome features and human gene expression data to build accurate aging models. We have incorporated these models into a health application showing a variety of insights into the biology of individuals. Investigating the causal relationship between these microbial and human gene expression features with age could potentially lead to fruitful interventions into human aging. These include therapeutic modalities such as diet, supplements, and lifestyle as well as pharmaceuticals such as small molecules, phages, engineered probiotics and immunotherapy.
Limitations of the study
We report results from models trained separately on stool microbiome and blood transcriptome data. Using both types of data together might better model the interactions between the host and gut microbiome, this is an area that we have actively worked on since the completion of this manuscript.
Though we get the best performance from linear models, non-linear models would be expected to more accurately model the complex relationships between the features (microbial and host). The lack of success of non-linear models might be due to the amount of data used, the diversity of available data, or the need for better modeling assumptions.
When developing the cohorts used to train the models reported in this study, we do not take into account information like health status, demographics, medication use or other relevant metadata. There are many ways to use this metadata to inform modeling, downstream task development, evaluation, and biological/clinical interpretation. These would all be fruitful areas for improvement and future work.
Our cohort comparisons show interesting differences between the predicted ages of different groups of interest but could be improved to better interrogate the nature of the difference. Though we report differences in predicted age, where there are none in actual age, the explanation for why those differences exist is unexplored, both from a statistical and clinical perspective. More systematic, and clinically informed, probing of this question is necessary to determine the degree to which the reported associations are clinically relevant.
Comparisons with prior work is important to contextualize results, but due to the lack of proliferation of metatranscriptomic microbiome data, we found this difficult to do. We try to compare our results with the results reported by Galkin 202010 but are limited in our ability to do so because of the differences between the types of data used in both studies. It would be fruitful to better connect the results we report in this study using metatranscriptomics data to the existing literature.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Quant-iT RiboGreen RNA Kit | ThermoFisher | R11490 |
| Quant-iT PicoGreen dsDNA Assay Kit | ThermoFisher | P7589 |
| HS NGS Fragment Kit (1-6000bp) | Agilent | DNF-474-0500 |
| NextSeq 500 High Output kit, 300 cycles | Illumina | FC-404-1004 |
| NovaSeq 6000 S4 Reagent Kit v1.5 (300 cycles) | Illumina | 20028312 |
| Deposited data | ||
| Summary data from the figures and additional summary statistics for the features relevant to the discussion. | This paper. | Figshare: https://doi.org/10.6084/m9.figshare.22635025.v2 |
| Software and algorithms | ||
| The code used for the model used in the paper | This paper. | Figshare: https://doi.org/10.6084/m9.figshare.22266436.v1 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Guruduth Banavar (guru@viome.com).
Materials availability
This study did not generate new unique reagents (over and above prior published studies).
Data and code availability
-
•
A summary dataset containing data from the figures and additional summary statistics for the features relevant to the discussion is available at the DOI: Figshare: https://doi.org/10.6084/m9.figshare.22635025.v2. The raw data used in this study cannot be shared publicly due to privacy and legal reasons. However, if data is specifically requested, we may be able to share a summary and/or portions of the data. Researchers requiring more data for non-commercial purposes can request via: https://www.viomelifesciences.com/data-access. Viome provides access to full summary statistics through a Data Transfer Agreement that protects the privacy of participants' data.
-
•
The code used for the model used in the paper is at Figshare: https://doi.org/10.6084/m9.figshare.22266436.v1.
Method details
Sample processing and bioinformatics
Stool samples were collected, preserved and processed using the metatranscriptomic method described in (Hatch 2019).39 See that article for data supporting the validity and reliability of our methods. Paired-end reads were mapped to genomes40 and to a catalog of microbial genes with KEGG ortholog (KO) annotations,41 and quantified using the expectation maximization algorithm.42 This yields two views of the relative activity of each gut microbiome sample, one taxonomic and one functional. The taxonomic view aggregates reads to the species level, while the functional view aggregates the same reads to KOs. A total of 5,010 taxa and 6,576 KO features are detected in the data for model development. On average, a taxa is detected in 5,202.64 of 78,637 samples (s.d. 1,4829.80, s.e. 209.52); a KO is detected in 20,723.21 of the samples (s.d. 28395.90, s.e. 350.17).
Blood samples were collected, preserved and processed using the whole blood transcriptomic method described in Toma et al.43 This method is selective for polyadenylated RNA. Paired-end reads were mapped to the human genome. Gene expression levels were computed by aggregating transcripts per million estimates per gene using an approach based on Salmon version 1.1.0,44 as described in (Toma et al.).43 A total of 18,457 unique genes are detected in the data. On average, a gene is detected in 985.41 of the samples (s.d. 615.33, s.e. 4.53).
Lab analysis of stool and blood samples was performed in a CLIA-licensed clinical laboratory. All samples were collected, preserved, and analyzed as described in (Hatch et al., Toma et al.). Briefly, samples were collected at home and immediately preserved with the Viome preservation solution, then shipped to the laboratory. After highly efficient sample lysis and RNA extraction, total RNA was quantified with RiboGreen (ThermoFisher). Messenger RNAs were either selected (in blood) or enriched (in stool) and converted to directional sequencing libraries, which were quality controlled with PicoGreen (ThermoFisher) and Fragment Analyzer (Advanced Analytical) assays, to accurately measure the library concentration and average fragment size. After sequencing on Illumina NextSeq 500 or NovaSeq 6000, raw sequencing reads were aligned to human transcriptome, microbial genomes (RefSeq), and/or microbial gene functions (Integrated Gene Catalog, IGC, and Kyoto Encyclopedia of Genes and Genomes, KEGG) databases. On average, 8.5 million paired-end sequencing reads were generated for each stool sample, and 8 million paired-end reads were generated for blood samples. For stool samples, quality control passing criteria included a minimum of 1 million reads and 50 strain-level taxa per sample. For blood samples, a minimum of 1 million paired-end reads were required to align to the human transcriptome. The bioinformatic methods (described below), as well as the laboratory ones, have been validated for accuracy, precision, sensitivity, specificity, and dynamic range, to ensure highly accurate data are generated for machine learning.
Stool samples passed internal quality assurance if they met all of the following criteria: a) sample sequencing depth greater than 5e3, b) the number of detected species must be greater than or equal to 50, c) the number of detected distinct KOs must be greater than or equal to 200, and d) the average paired read length must be greater than 90 bps. Furthermore, our quality control method involves internal controls to quantify sample-to-sample cross-talk and background contamination by microbial taxa allowing us to re-run samples that do not meet our stringent criteria.
Blood samples passed internal quality assurance if they met all of the following criteria: a) read depth of annotated protein-coding genes and not genes considered to form an integral part of the hemoglobin complex (ENSG00000086506, ENSG00000130656, ENSG00000169877, ENSG00000188536, ENSG00000196565, ENSG00000206172, ENSG00000206177, ENSG00000206178, ENSG00000213931, ENSG00000213934, ENSG00000223609, ENSG00000229988, and ENSG00000244734) must be greater than or equal to 5e5 reads b) the number of detected genes with at least one read mapped must have counts greater than or equal to 5e3.
Quantification and statistical analysis
Definitions for all statistical terms can be found in Methods S1.
We offer a detailed description of the ML approaches used in the paper below, full results can be found in Methods S1. Methods S1 also contains the exact values of N (number of samples) for all the ML experiments.
A description of the “Cohort comparison” results can be found in the section “Cohort comparisons.” The size of N for each of the cohorts can be found in Figures 4A and 4B.
For the “Cohort comparison” we used a non-parametric test (Wilcoxon signed-rank tests) because our test data did not meet the assumptions of normality required for a t-test.
Machine learning
The microbiome data was transformed using the centered log ratio transformation (CLR)45,46 after imputation of zero values using multiplicative replacement47 Both taxonomic and functional features were included in the model. The human gene expression data was transformed using a Yeo-Johnson power transformation.
Machine-learned models for both stool microbiome and blood transcriptome are Elastic Nets (EN: linear regression with tunable L1 and L2 regularization). We also tried other approaches including deep neural networks (DNN), random forest and gradient boosting machines (GBM). As results were similar across all approaches, we report the simplest and most interpretable model class, ElasticNet.
For the stool microbiome model, hyperparameter optimization was done using a 5-fold cross-validation on the discovery cohort. A final model using the optimal hyperparameter setting was then trained on the full discovery cohort and applied to the validation cohort to test generalization. Hyperparameter settings were scored using R2 and the selected model was evaluated using Mean Absolute Error (MAE) and R2.
For the human blood transcriptome model, model evaluation was performed using a nested CV due to the smaller dataset size. An inner 3-fold CV was used for hyperparameter selection while an outer 5-fold CV was used for model evaluation. Hyperparameter settings in each were scored using R2 and evaluated using MAE and R2.
Baseline MAE was computed as MAE from the median age of the cohort. The discovery cohort was used to tune model hyperparameters using cross validation, then a final model was trained on the full discovery cohort. The final model was applied to the validation cohort (drawn from the same population but not used in training), cohorts matched to two datasets of healthy individuals analyzed in Galkin et al. (2020),10 and additional cohorts described in Cohort Comparisons below.
Modeling details
The model class used for both the blood based and stool based data is Elastic net, which is linear regression trading off L1 and L2 regularization. The hyperparameters for this model class are alpha, which controls the penalty strength, and l1_ratio which specifies the mixing parameter. An l1_ratio of 0 is equivalent to Ridge regression and an l1_ratio of 1 is equivalent to LASSO. The following hyperparameter space was explored for both models: (1) 25 logarithmically spaced values between −3 and 3 for alpha (2) l1_ratios of: [0.25, 0.5, 0.75, 0.9, 0.95, 1].
For the stool based model, model selection is done using Cross validation on the discovery cohort (training set) where the optimal hyperparameter setting was alpha=0.1 and l1_ratio=0.5.
For the blood based model, due to the smaller data set size, a nested cross validation is used to evaluate model performance. In each outer fold, hyperparameter search occurs on the train data using an inner CV and the selected model is evaluated on the outer test set. In our case, the same hyperparameters were optimal in every outer fold, which reassures us that model selection does not depend heavily on any particular choice of validation or train set. These values were: alpha = 0.25 and l1_ratio=0.56.
In terms of hardware and software, a variety of Amazon EC2 instances were used to train the models and for analysis. A representative example: m5.16xlarge. The python programming language and ecosystem was used for analysis and experiments, specific packages are given in Methods S2.
Methods for Alternate models
Our primary motivation is to gauge the strength and nature of the relationship between the gut microbiome, blood metatranscriptome and chronological aging. From this vantage point, we are sensitive to trading off simplicity and interpretability for marginal gains in performance. In Methods S3 we show alternative model classes that we experimented with for the task of age prediction. Listed are the model class, the hyperparameter search space, and the method of model selection. Model selection is done with either random search or grid search, and models are evaluated with either cross validation or a held out validation set (separate from test set). The selected models can be found in Methods S4. These choices are made depending on the computational costs involved in training. Future directions in this area would be to more rigorously attempt to improve model performance by expanding the relevant search spaces, trying many different model classes, trying different architectures for the neural net model, and imparting domain knowledge through appropriate inductive biases. We would also like to explore modeling approaches that can relate the predicted age to quantifiable metrics of wellness and health.
Model Performance Comparison to Galkin et al.
Galkin et al. reported a DNN model trained on ∼4000 aggregated metagenomic profiles from people from 18 to 90 years old. As our models are trained on metatranscriptomic data, a direct comparison of model performance to Galkin’s DNN model is not possible.. Instead we evaluated our model performance by matching the age distribution of the cohort used in the DNN model reported by Galkin et al. We provide below the detailed steps.
Comparing model performance on “CV Host” data set from Galkin et al.
-
•
Galkin et al. reported cross-validation performance from their DNN model on a cohort containing 1,165 healthy individuals where samples from the same donor were collapsed into one average profile (denoted “CV Host” in Table 1 of Galkin et al. and “CV Host” in our main text). The actual age and predicted age from their DNN model for this cohort is publicly available.
-
•
For each donor in the Galkin’s “CV Host” cohort, we randomly matched one samples from users with exactly the same age (rounded) in our validation cohort. We were able to match 1,164 samples out of 1,165 samples with exactly the same age distribution. This matched cohort is denoted as “Galkin et al. CV Host matched cohort ” in Table 1 or “matched CV cohort” in the main text. We were not able to match one sample with age of 90. Dropping one sample barely changed the baseline MAE and the performance evaluation of the Galkin’s model.
-
•
Ages were predicted by our EN model. R2 and MAE of this matched cohort is reported under column “Galkin et al. CV Host matched cohort” in Table 1 along with age, sex and baseline MAE.
Comparing model performance on Independent HC data set from Galkin et al.
-
•
Galkin et al. reported DNN performance on an independent HC cohort containing 402 profiles from 252 healthy individuals. (denoted “Independent HC” in Table 1 of Galkin et al. and “HC cohort” in our main text). The actual age, sex and predicted age from the DNN model for this cohort is publicly available. We dropped replicated samples from the same donor and reported the performance for performance comparison.
-
•
For each donor in Galkin’s “Independent HC”, we randomly matched one sample from users with exactly the same age (rounded) and sex in our validation cohort. This matched cohort is denoted as “Galkin et al. Independent HC matched cohort” in Table 1.
-
•
Ages were predicted by our EN model. R2 and MAE of this matched cohort is reported under column “Galkin et al. Independent HC matched cohort” in Table 1 along with age, sex and baseline MAE.
Acknowledgments
This work was funded by Viome Life Sciences Inc.
Author contributions
V.G. contributed to Conceptualization, Data curation, Investigation, Formal Analysis, Methodology, Software, Visualization, Writing – original draft, and Writing – review and editing. F.C. and P.T. contributed to Bioinformatics software. R.T. contributed to Lab processing. Y.C. contributed to Formal Analysis, Validation, and Writing – review and editing. S.K. and S.R. contributed to Interpretation, Writing – original draft, and Writing – review and editing. HT contributed to Formal Analysis, Investigation, and Writing – review and editing. M.V. contributed to Funding acquisition, Investigation, Resources, and Writing – review and editing. G.B. contributed to Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, and Writing – review and editing.
Declaration of interests
All authors of this manuscript were employees of Viome Life Sciences Inc at the time of their contributions, and held stock options in the company. M.V. and G.B. hold management positions within the company. The authors have submitted a patent application but have not been granted a patent.
Published: December 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108538.
Supplemental information
References
- 1.Earls J.C., Rappaport N., Heath L., Wilmanski T., Magis A.T., Schork N.J., Omenn G.S., Lovejoy J., Hood L., Price N.D. Multi-Omic Biological Age Estimation and Its Correlation With Wellness and Disease Phenotypes: A Longitudinal Study of 3,558 Individuals. J. Gerontol. 2019;74:S52–S60. doi: 10.1093/gerona/glz220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mamoshina P., Kochetov K., Putin E., Cortese F., Aliper A., Lee W.S., Ahn S.M., Uhn L., Skjodt N., Kovalchuk O., et al. Population specific biomarkers of human aging: A big data study using South Korean, Canadian, and Eastern European patient populations. J. Gerontol. A Biol. Sci. Med. Sci. 2018;73:1482–1490. doi: 10.1093/gerona/gly005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fraga M.F., Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Horvath S., Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018;19:371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 5.Bell C.G., Lowe R., Adams P.D., Baccarelli A.A., Beck S., Bell J.T., Christensen B.C., Gladyshev V.N., Heijmans B.T., Horvath S., et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 2019;20:249. doi: 10.1186/s13059-019-1824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M., Lemos B. Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 2019;29:325–333. doi: 10.1101/gr.241745.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mamoshina P., Volosnikova M., Ozerov I.V., Putin E., Skibina E., Cortese F., Zhavoronkov A. Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front. Genet. 2018;9:242. doi: 10.3389/fgene.2018.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harries L.W., Hernandez D., Henley W., Wood A.R., Holly A.C., Bradley-Smith R.M., Yaghootkar H., Dutta A., Murray A., Frayling T.M., et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell. 2011;10:868–878. doi: 10.1111/j.1474-9726.2011.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin H., Lunetta K.L., Zhao Q., Mandaviya P.R., Rong J., Benjamin E.J., Joehanes R., Levy D., van Meurs J.B.J., Larson M.G., Murabito J.M. Whole Blood Gene Expression Associated With Clinical Biological Age. J. Gerontol. A Biol. Sci. Med. Sci. 2019;74:81–88. doi: 10.1093/gerona/gly164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galkin F., Mamoshina P., Aliper A., Putin E., Moskalev V., Gladyshev V.N., Zhavoronkov A. Human gut microbiome aging clock based on taxonomic profiling and deep learning. iScience. 2020;23:101199. doi: 10.1016/j.isci.2020.101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang S., Haiminen N., Carrieri A.P., Hu R., Jiang L., Parida L., Russell B., Allaband C., Zarrinpar A., Vázquez-Baeza Y., et al. Human Skin. mSystems. 2020;5 doi: 10.1128/mSystems.00630-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogunrinola G.A., Oyewale J.O., Oshamika O.O., Olasehinde G.I. The Human Microbiome and Its Impacts on Health. Int. J. Microbiol. 2020;2020:8045646. doi: 10.1155/2020/8045646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan Y., Pedersen O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021;19:55–71. doi: 10.1038/s41579-020-0433-9. [DOI] [PubMed] [Google Scholar]
- 14.Askarova S., Umbayev B., Masoud A.R., Kaiyrlykyzy A., Safarova Y., Tsoy A., Olzhayev F., Kushugulova A. The Links Between the Gut Microbiome, Aging, Modern Lifestyle and Alzheimer’s Disease. Front. Cell. Infect. Microbiol. 2020;10:104. doi: 10.3389/fcimb.2020.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renson A., Mullan Harris K., Dowd J.B., Gaydosh L., McQueen M.B., Krauter K.S., Shannahan M., AE A., Shannahan M., Aiello A.E. Early Signs of Gut Microbiome Aging: Biomarkers of Inflammation, Metabolism, and Macromolecular Damage in Young Adulthood. J. Gerontol. A Biol. Sci. Med. Sci. 2020;75:1258–1266. doi: 10.1093/gerona/glaa122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry S.E., Valdes A.M., Drew D.A., Asnicar F., Mazidi M., Wolf J., Capdevila J., Hadjigeorgiou G., Davies R., Al Khatib H., et al. Human postprandial responses to food and potential for precision nutrition. Nat. Med. 2020;26:964–973. doi: 10.1038/s41591-020-0934-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S., Jazwinski S.M. The gut microbiota and healthy aging: A mini-review. Gerontology. 2018;64:513–520. doi: 10.1159/000490615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maffei V.J., Kim S., Blanchard E., 4th, Luo M., Jazwinski S.M., Taylor C.M., Welsh D.A. Biological aging and the human gut microbiota. J. Gerontol. A Biol. Sci. Med. Sci. 2017;72:1474–1482. doi: 10.1093/gerona/glx042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watson M.M., Søreide K. The Gut Microbiota influence on human epigenetics, health, and disease. Handb. Epigenet. 2017;32:495–510. [Google Scholar]
- 20.Lan Y., Kriete A., Rosen G.L. Selecting age-related functional characteristics in the human gut microbiome. Microbiome. 2013;1:2. doi: 10.1186/2049-2618-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odamaki T., Kato K., Sugahara H., Hashikura N., Takahashi S., Xiao J.Z., Abe F., Osawa R. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90. doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia L., Zhang W., Chen X. Common methods of biological age estimation. Clin. Interv. Aging. 2017;12:759–772. doi: 10.2147/CIA.S134921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopkins M.J., Sharp R., Macfarlane G.T. Variation in human intestinal microbiota with age. Dig. Liver Dis. 2002;34:S12–S18. doi: 10.1016/s1590-8658(02)80157-8. [DOI] [PubMed] [Google Scholar]
- 24.Mariat D., Firmesse O., Levenez F., Guimarăes V., Sokol H., Doré J., Corthier G., Furet J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koenig J.E., Spor A., Scalfone N., Fricker A.D., Stombaugh J., Knight R., Angenent L.T., Ley R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA. 2011;108:4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P., et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de la Cuesta-Zuluaga J., Kelley S.T., Chen Y., Escobar J.S., Mueller N.T., Ley R.E., McDonald D., Huang S., Swafford A.D., Knight R., et al. Age and sex-dependent patterns of gut microbial diversity in human adults. mSystems. 2019;4 doi: 10.1128/mSystems.00261-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bian G., Gloor G.B., Gong A., Jia C., Zhang W., Hu J., Zhang H., Zhang Y., Zhou Z., Zhang J., et al. The Gut Microbiota of Healthy Aged Chinese Is Similar to That of the Healthy Young. mSphere. 2017;2 doi: 10.1128/mSphere.00327-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de la Cuesta-Zuluaga J., Kelley S.T., Chen Y., Escobar J.S., Mueller N.T., Ley R.E., McDonald D., Huang S., Swafford A.D., Knight R., Thackray V.G. Age- and sex-dependent patterns of gut microbial diversity in human adults. mSystems. 2019;4 doi: 10.1128/mSystems.00261-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinsen T.C., Bergh K., Waldum H.L. Gastric Juice: A Barrier Against Infectious Diseases. Basic Clin. Pharmacol. Toxicol. 2005;96:94–102. doi: 10.1111/j.1742-7843.2005.pto960202.x. [DOI] [PubMed] [Google Scholar]
- 31.Banoo H., Nusrat N. Implications of Low Stomach Acid: An Update. RAMA Univ. J. Med Sci. 2016;2:16–26. [Google Scholar]
- 32.D’Souza A.L. Ageing and the gut. Postgrad. Med. J. 2007;83:44–53. doi: 10.1136/pgmj.2006.049361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Homma T., Fujii J. Application of Glutathione as Anti-Oxidative and Anti-Aging Drugs. Curr. Drug Metab. 2015;16:560–571. doi: 10.2174/1389200216666151015114515. [DOI] [PubMed] [Google Scholar]
- 34.Hsiao E.Y., McBride S.W., Hsien S., Sharon G., Hyde E.R., McCue T., Codelli J.A., Chow J., Reisman S.E., Petrosino J.F., et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dumitrescu L., Popescu-Olaru I., Cozma L., Tulbă D., Hinescu M.E., Ceafalan L.C., Gherghiceanu M., Popescu B.O. Oxidative Stress and the Microbiota-Gut-Brain Axis. Oxid. Med. Cell. Longev. 2018;2018 doi: 10.1155/2018/2406594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holly A.C., Melzer D., Pilling L.C., Henley W., Hernandez D.G., Singleton A.B., Bandinelli S., Guralnik J.M., Ferrucci L., Harries L.W. Towards a gene expression biomarker set for human biological age. Aging Cell. 2013;12:324–326. doi: 10.1111/acel.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sae-Lee C., Corsi S., Barrow T.M., Kuhnle G.G.C., Bollati V., Mathers J.C., Byun H.M. Dietary Intervention Modifies DNA Methylation Age Assessed by the Epigenetic Clock. Mol. Nutr. Food Res. 2018;62 doi: 10.1002/mnfr.201800092. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh T.S., Rampelli S., Jeffery I.B., Santoro A., Neto M., Capri M., Giampieri E., Jennings A., Candela M., Turroni S., et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: the NU-AGE 1-year dietary intervention across five European countries. Gut. 2020;69:1218–1228. doi: 10.1136/gutjnl-2019-319654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatch A., Horne J., Toma R., Twibell B.L., Somerville K.M., Pelle B., Canfield K.P., Genkin M., Banavar G., Perlina A., et al. A Robust Metatranscriptomic Technology for Population-Scale Studies of Diet, Gut Microbiome, and Human Health. Int. J. Genom. 2019;2019 doi: 10.1155/2019/1718741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breitwieser F.P., Lu J., Salzberg S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019;20:1125–1136. doi: 10.1093/bib/bbx120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanehisa M., Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dempster A.P., Laird N.M., Rubin D.B. Maximum Likelihood from Incomplete Data Via the EM Algorithm. J. Roy. Stat. Soc. Ser. B Methodol. 1977;39:1–22. [Google Scholar]
- 43.Toma R., Duval N., Pelle B., Parks M.M., Gopu V., Torres P.J., Camacho F.R., Shen N., Krishnan S., Hatch A., et al. A clinically validated human capillary blood transcriptome test for global systems biology studies. BioTechniques. 2020;69 doi: 10.2144/btn-2020-0088. [DOI] [PubMed] [Google Scholar]
- 44.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aitchison J. The Statistical Analysis of Compositional Data. J. Roy. Stat. Soc. Ser. B Methodol. 1982;44:139–160. [Google Scholar]
- 46.Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martín-Fernández J.A., Barceló-Vidal C., Pawlowsky-Glahn V. Dealing with zeros and missing values in compositional data sets using nonparametric imputation. Math. Geol. 2003;35:253–278. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
A summary dataset containing data from the figures and additional summary statistics for the features relevant to the discussion is available at the DOI: Figshare: https://doi.org/10.6084/m9.figshare.22635025.v2. The raw data used in this study cannot be shared publicly due to privacy and legal reasons. However, if data is specifically requested, we may be able to share a summary and/or portions of the data. Researchers requiring more data for non-commercial purposes can request via: https://www.viomelifesciences.com/data-access. Viome provides access to full summary statistics through a Data Transfer Agreement that protects the privacy of participants' data.
-
•
The code used for the model used in the paper is at Figshare: https://doi.org/10.6084/m9.figshare.22266436.v1.