

The stress-responsive transcription factor HSF1 reduces CD8+ T-cell infiltration in breast tumors to prevent immune-mediated killing, indicating that cellular stress responses affect tumor-immune interactions and that targeting HSF1 could improve immunotherapies.
Abstract
Heat shock factor 1 (HSF1) is a stress-responsive transcription factor that promotes cancer cell malignancy. To provide a better understanding of the biological processes regulated by HSF1, here we developed an HSF1 activity signature (HAS) and found that it was negatively associated with antitumor immune cells in breast tumors. Knockdown of HSF1 decreased breast tumor size and caused an influx of several antitumor immune cells, most notably CD8+ T cells. Depletion of CD8+ T cells rescued the reduction in growth of HSF1-deficient tumors, suggesting HSF1 prevents CD8+ T-cell influx to avoid immune-mediated tumor killing. HSF1 suppressed expression of CCL5, a chemokine for CD8+ T cells, and upregulation of CCL5 upon HSF1 loss significantly contributed to the recruitment of CD8+ T cells. These findings indicate that HSF1 suppresses antitumor immune activity by reducing CCL5 to limit CD8+ T-cell homing to breast tumors and prevent immune-mediated destruction, which has implications for the lack of success of immune modulatory therapies in breast cancer.
Significance:
The stress-responsive transcription factor HSF1 reduces CD8+ T-cell infiltration in breast tumors to prevent immune-mediated killing, indicating that cellular stress responses affect tumor-immune interactions and that targeting HSF1 could improve immunotherapies.
Graphical Abstract
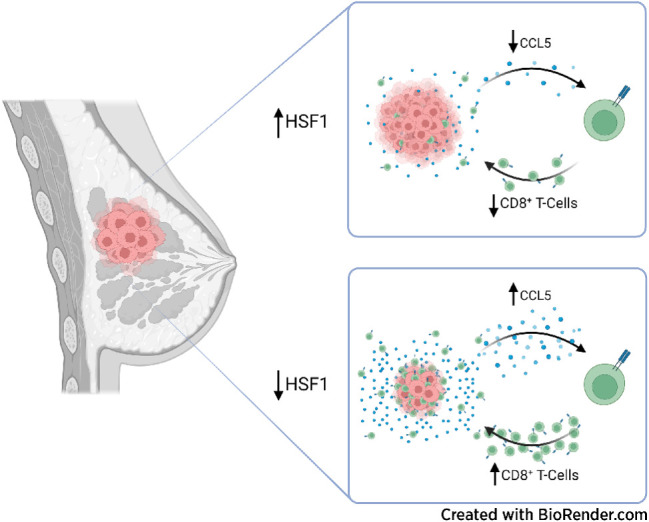
Introduction
Breast cancer is the second leading cause of cancer-related deaths in women with 1 in 8 developing invasive breast cancer over the course of their lifetime (1). Immune checkpoint therapy in patients with breast cancer has had mixed results with patients with triple-negative breast cancer (TNBC) primarily being the beneficiary of this therapeutic approach (2). Breast cancer has historically thought to be a low immunogenic tumor (3). TNBC has largely benefitted from immune checkpoint therapy due to it being the small subset of breast cancer that has shown to have PD-L1 expression and higher lymphocytic infiltration relative to other breast cancer subtypes (4, 5). While PD-L1-targeting checkpoint therapy was approved for high-risk TNBC, only a small percentage of these patients respond to immune checkpoint inhibition prompting the approval for anti–PD-L1 therapy to be removed for this patient population in 2021. Previous studies have identified that higher lymphocyte infiltration in breast cancers is associated with improved survival and improved response to treatments (6–9). Infiltration of CD8+ T cells, the primary cytotoxic lymphocyte, is an independent predictor of patient outcome and these immune cells are trafficked to tumors through chemotactic cytokines (10). While low tumor mutational burden (TMB) is a significant factor in the low lymphocyte infiltration in breast cancer, TMB does not fully explain low lymphocyte infiltration, indicating that additional mechanisms suppress lymphocyte attraction to breast tumors.
Heat shock factor 1 (HSF1) is a master transcription factor regulator of the heat shock response. The classical function of HSF1 is to regulate expression of chaperone genes in response to cellular stressors (11). HSF1 can be hyperactivated due to increased proteotoxic stress and upregulation of activating kinases including AKT1, mTORC1, and p38 among others (11–14). However, the seminal finding of a unique transcriptional program of HSF1 in cancer cells that is distinct from the heat stress response transcriptional program indicated that HSF1 has additional functions in cancer (15). It is now recognized that HSF1 plays key roles in cancer cells by promoting several protumor processes including epithelial-to-mesenchymal transition, promotion of the cancer stem-like state, promotion of cancer-associated metabolic changes, and several others that all support the malignancy of cancer cells (11–13, 15, 16). Furthermore, HSF1 levels and activity have been associated with worsened patient outcomes in many cancer types, including breast cancer (13, 15, 17). However, the role of HSF1 in tumor–immune interactions and avoiding immune destruction remains unclear.
Gene signatures, or sets of genes that represent biologic functions or features, have gained popularity since the introduction of gene set enrichment analysis (GSEA) and the molecular signatures database (mSigDB; ref. 18). Here we report a new gene signature termed “HSF1 Activity Signature,” or HAS, a 19-gene signature that reports HSF1 transcriptional activity generated using a novel approach. By using the HAS, we uncovered a novel mechanism underlying breast tumor evasion of immune destruction based on inhibition of CD8+ T-cell recruitment to tumors by HSF1-mediated suppression of CCL5, a key chemokine regulating CD8+ T cells.
Materials and Methods
Cell culture
MDA-MB-231 (DMEM, Gibco #10–013-CV) and 4T1 (RPMI, Gibco #10–040-CV) cells were obtained from ATCC (231: CRM-HTB-26, RRID: CVCL_0062; 4T1: CRL-2539, RRID: CVCL_0125) and were maintained at 37°C in 5% CO2 and culture media was supplemented with 10% FBS (Corning #35–011-CV) and 1% penicillin/streptomycin (Gibco #15140–122). Cells were tested monthly for Mycoplasma contamination using MycoAlert kit (Lonza #LT07–218). Lentiviral plasmids carrying control (5′-CCTAAGGTTAAGTCGCCCTCG-3′) or HSF1 (5′-GGAACAGCTTCCACGTGTTTG-3′) shRNA were synthesized by VectorBuilder using U6-driven promoter.
RNA sequencing and datasets used
Total RNA from MDA-MB-231 and 4T1 cells were collected (PureLink RNA Mini Kit, Thermo Fisher Scientific #12183018A) and subjected to mRNA-sequencing using an Illumina HiSeq 4000 and data were deposited in Gene Expression Omnibus at GSE236972. The sequencing data were first assessed using FastQC (Babraham Bioinfomatics) and then mapped to the mouse genome (mm10) using STAR RNA-seq aligner. Uniquely mapped sequencing reads were assigned to mm10 ref Gene genes using feature Counts (from subread). Genes with read count per million (CPM) >0.5 in more than 3 of the samples were kept. The data were further normalized using Reads Per Kilobase Million (RPKM). Differential gene expression analysis was conducted by using DESeq2 method with an FDR less than 0.05 as the significant cutoff. Microarray datasets from the public domain were accessed from NCBI GEO (Supplementary Table S1). The GEO datasets were all from the same platform, that is, Affymetrix Human Genome U133 Plus 2.0 Array. Renormalization was performed for each datasets from .CEL files using MAS5 method in R 4.1.2. The Cancer Genome Atlas (TCGA)-BRCA dataset was accessed through the TCGA data portal. The METABRIC dataset was accessed through the European Genome-Phenome Archive (EGA) under study ID EGAS00000000083.
HSF1 gene module detection analysis
To identify potential genes that can act as reliable markers of HSF1 activity, we conducted an integrated coexpression analysis on a set of genes that have been demonstrated to be direct targets of HSF1 and are strongly linked to its activity. Our underlying premise is that HSF1 target genes are closely associated with the activation status of HSF1, making them indicative of its activity. By focusing on the core group of genes that exhibit simultaneous regulation by HSF1 and display robust coexpression patterns, we aim to pinpoint the most promising candidates for signifying HSF1 activity. First, we selected genes that were direct targets of HSF1 based on 49 chromatin immunoprecipitation sequencing (ChIP-seq) samples, including replicates, collected from three large ChIP-seq studies (15, 19, 20) on various cell types including colon cancer cell lines, breast cancer cell lines, lung cancer cell lines, normal epithelial cells with or without heat shock, among others. These direct targets were derived from the original analysis of these studies by the primary authors and we included all unique genes from any of the ChIP-seq samples (i.e., any gene identified as a target in these samples was included regardless of how many of the samples these genes were identified as targets) giving a total number of 3,415 genes. Eventual genes utilized in this study were analyzed to validate them as direct targets in multiple ChIP-seq datasets. Second, the genes were further filtered based on differential expression analysis on our bulk RNA-seq data, where only genes showing significantly decreased expression with HSF1 knockdown were retained. A total of 497 genes were significantly downregulated after HSF1 knockdown (fold change > 1.5, adjusted P < 0.05). There were a total of 60 genes common to the 3,415 genes detected as HSF1 direct targets and the 497 total genes downregulated after HSF1 knockdown. Finally, we performed our gene module detection analysis based on the retained 60 genes on 11 public breast cancer datasets (Supplementary Table S2), to identify a core set of genes consistently showing strong coexpression patterns across all 11 datasets. After doing MAS5 normalization on the raw .CEL files of the datasets, calculated the Spearman correlation matrix for the filtered genes. A binary matrix was constructed for each dataset where 1 indicated significant correlation and 0 indicated nonsignificant correlation. Note that here, a Bonferroni correction was conducted on the raw P value of correlation. An average ratio matrix, A, for the 11 binary matrices was further calculated, wherein each value denotes the total number of times for the pair of gene being significant divided by 11. Then we applied a greedy algorithm to identify the most interconnected modules. Basically, for a given α and β, we iteratively remove genes whose average significance ratio, shown in the average ratio matrix A, did not surpass α in at least 100*β percentage among all the genes. Here, α varies in a sequence of 0.01 to 0.9 with a step size of 0.09 and β varies from 0.1 to 0.9 with a step size of 0.01. For fixed α, and a given β, we run multiple iterations, and within each iteration, only a very small number of genes will be removed. This is because, in the beginning, we have a large number of genes, and if we use the given β, a large number of genes, which equates to the number of genes multiplied by β, will be removed, while the removed genes may contribute to a small but strongly coexpressed module. The iteration stops when all genes satisfy the constraint that their average significance ratio surpasses α in at least 100*β percentage among all the remaining genes. The gradual increase in β allows fine tuning of the coexpression module to ensure that only the most interconnected gene modules will be obtained. Note, we conducted robustness analysis for different α and β values, with smaller α, β resulting in relatively large co-expression modules, while large α, β resulting in small coexpression modules, but overall the resulting gene modules are highly overlapping. Here, considering that the HSF1 targets obtained from different ChIP-seq datasets collected by us are different, we performed the analysis described above for different combinations of the HSF1 target lists from different ChIP-seq datasets. In total, we identified nine different modules for different lists of HSF1 targets, and chose the modules obtained for only α > 0.36 and β > 0.3.
Heat map generation, principal component, and gene ontology analysis
Z-scores were used to generate heat maps using Morpheus. Gene expression values for the respective genes within each signature were subjected to PCA using GraphPad Prism 9. PC1 scores for samples from respective groups were used to rank order samples for survival analysis or used to compare signature scores between groups. Gene ontology for the HAS or genes associated with the HAS was performed using ShinyGO version 0.76. Genes associated with the HAS were selected through Pearson correlation of HAS score with all genes within the BRCA TCGA cohort.
Survival assessment
Patient survival was assessed using Kaplan–Meier plots within Prism 9. Log-rank tests were used for determination of statistical differences between groups. Age-adjusted Cox proportional hazard ratios were computed using PC1 scores from gene signatures with survival using SPSS 28.0 and calculated 95% confidence interval and computed P values. HRs were plotted in a forest plot using Prism 9. Forest plots have a dotted line at a HR of 1 with a box indicating the HR with red bars indicating the 95% confidence intervals.
GSEA
GSEA was done using GSEA software vs. 4.3.2 for Windows. Gene Cluster Text file (.gct) was generated from the TCGA BRCA, METABRIC, or GSE47561 breast cancer cohorts. Categorical class file (.cls) was generated by separating patients in these datasets based on high and low HAS score. The Gene Matrix file (.gmx) was generated using published gene signatures for immune cell types from the mSigDB and from TIMER2.0 portal (21–26). The number of permutations was set to 1,000 and the chip platform for TCGA gene lists was used. Heat maps were generated on Morpheus (https://software.broadinstitute.org/morpheus) using normalized enrichment scores.
Immune profile estimations
Immune proportion estimates were completed using deconvolution algorithms from TIMER, CIBERSORT, QuantiSeq, MCPCounter, and XCell for the TCGA-BRCA, METABRIC, and GSE47561 datasets using the TIMER2.0 portal (http://timer.cistrome.org; refs. 21–26). CD8+ T-cell proportions were compared between high and low HAS groups using Student t test.
IHC
IHC was performed on a breast cancer tumor microarray (BR1141A; tissuearray.com). Antibodies used for IHC include pHSF1 (S326; Abcam #76076; RRID: AB_1310328), CD3 (Santa Cruz Biotechnology #sc-20047; RRID: AB_627014), CD8A (CST #85336; RRID: AB_2800052), and CCL5 (Invitrogen #710001; RRID: AB_2532515). IHC was performed by deparaffinization using xylenes followed by rehydration with decreasing alcohol solutions. Slides were then incubated in Bloxall (Vector Laboratories #SP-6000–100) and subjected to antigen retrieval with 10 mmol/L citrate solution in a heated pressure cooker (2,100 Antigen Retriever; Aptum Biologics). Tissues were blocked in animal-free blocker (Vector Laboratories #SP-5035) and incubated in primary antibodies overnight in a humidified chamber. Primary antibodies were detected using Vectastain ABC-HRP kits (Rabbit: PK-4001; Mouse: PK-4010) and developed with DAB Immpact kit (Vector Laboratories #SK-4105) followed by dehydration with increasing alcohol solutions and mounted. Slides were imaged with Motic EasyScan scanner and analyzed with QuPath software (27).
Animal studies
All animal studies were performed under an approved institutional animal care and use committee protocol on the Indiana University-Bloomington campus (Bloomington, IN). Control or HSF1 knockdown 4T1 cells (3e5) were injected into the mammary gland of 4-week-old female Balb/c mice and allowed to grow for 3–5 weeks. Tumor volume was measured with calipers. CD8+ T-cell depletion was accomplished by administration of 200 ug of CD8A (bioXcell #BE0061; RRID: AB_1125541) antibodies for 3 days and depletion was maintained throughout the study with 200 μg antibody injections (twice per week). After initial CD8+ T-cell depletion, mice were randomized to receive either 4T1 shCTL or 4T1 shHSF1 cells for injection. CD8+ T-cell depletion was confirmed at the end of the study by collecting the spleens and analyzing by flow cytometry.
Single-cell RNA sequencing
Tumors from 4T1 shCTL or shHSF1 were excised then dissociated using a tumor dissociation kit (Miltenyi Biotec #130–096–730). Approximately 10,000 cells per sample with greater than 70% viability were used as input to the 10X Genomics Chromium system using the Chromium Next GEM Single Cell 3′ Kit v3.1. Libraries were sequenced using a NovaSeq 6000 with a NovaSeq S2 reagent kit v1.0 (100 cycles) and raw data were deposited in Gene Expression Omnibus (GEO) at GSE236972. Count matrices were generated with 10X Genomics Cell Ranger (v4.0.0) with default settings and genome assembly with mm10 was used. Resulting matrices were processed and analyzed in Seurat (v.4.2.0). Quality control and filtering removed low-quality cells. Specifically, we kept only cells who has mitochondria percentage lower than 5%, expresses more than 100 but less than 5,000 genes, and has total counts less than 30,000. In addition, we noticed the presence of doublets, and removed them using the scDblFinder method (28). This left us with 25,039 (from 34,956) and 23,569 (from 38,134) cells for 4T1 shCTL or shHSF1 samples respectively. The two samples were integrated using function “IntegrateData” with 40 dimensions in the anchor weighting procedure in Seurat. We then selected variable genes and performed dimensionality reduction using principal component analysis and cell clustering on the integrated data with top 20 principal components using the “FindClusters” function in Seurat. Cluster-specific markers were identified by doing differential gene expression using Wilcoxin test with a log fold change threshold set up 0.25. The cluster markers were then overlapped to canonical cell type–defining signature genes. Ultimately, we recovered and annotated eight unique cell types from the two samples.
Flow cytometry
Single-cell suspensions were subjected to incubation with mouse-specific CD3-FITC (Miltenyi Biotec, #130–119–798; RRID: AB_2751851) and CD8A-APC (Miltenyi Biotec, #130–117–664; RRID: AB_2728016) antibodies in series followed by incubation with Zombie viability dye (Invitrogen #771847). After labelling, flow cytometry was performed using a MACS Quant (Miltenyi) system and data were analyzed with FlowJo 10.8.
Cytokine array
Control and HSF1 knockdown 4T1 cells were incubated in fresh media over 72 hours. Culture media was collected, centrifuged, and subjected to the Mouse XL Cytokine Array (R&D Systems #ARY028) according to manufacturer's instructions in triplicate.
RT-qPCR
Total RNA from was extracted using an RNA Extraction Kit (Zymo #R1050) and subjected to reverse transcription using RT Master Mix (Applied Biosystems #4368814). qPCR was performed with SYBR Green Master Mix (Applied Biosystems #4309155). qPCR primers used are listed in Supplementary Table S3.
Transwell migration of T cells
The assay was performed using a chemotaxis chamber (NeuroProbe) with 5-μm pore size filters. Briefly, the spleens were extracted from healthy Balb/c mice, dissociated using a gentleMACS and Spleen Isolation Kit (Miltenyi Biotec, #130–095–926), and T cells isolated using a pan-T Cell Isolation Kit (Miltenyi Biotec, #130–095–130). T cells (1 × 106) were placed in the top chamber in serum-free medium while the bottom chamber contained serum-free medium with or without exogenous CCL5 or conditioned medium from 4T1 cells with predesigned control siRNA (Bioneer #SN-1003), Hsf1 siRNA (Bioneer #15499), or Ccl5 siRNA (Bioneer #20304). Empty vector (pCMV-SPORT6) was obtained from Invitrogen and pCMV-SPORT6-CCL5 was obtained from Transomic Technologies (TCM1004). T-cells were incubated for 24 hours and subjected to flow cytometry with CD3/CD8A antibodies with five biological replicates.
Statistical analysis
All statistical tests were performed as two-tailed tests. For two-group comparisons, Student t test was used. For multiple group comparisons, ANOVA with Tukey post hoc test was used. All laboratory experiments were completed with a minimum of three biological replicates (e.g., qPCR, cytokine array) with flow cytometry experiments using a minimum of four biological replicates. Animal studies were completed with a minimum of five mice per group.
Data availability statement
Next-generation sequencing data generated in this study was deposited at Gene Expression Omnibus (GEO) at GSE236972. Publicly available datasets used to analyze and validate the HSF1 activity signature (HAS) included GSE38232 (MCF7/BPLER cells), GSE3697 (Hela cells), GSE83844 (A549 cells), GSE115973 (U2OS cells), GSE155248 (C4–2 cells; Supplementary Table S1). Publicly available expression datasets from patient samples used to analyze the intragene correlation for HSF1 gene module detection included GSE11121, GSE12093, GSE14020, GSE1456, GSE17705, GSE2034, GSE2603, GSE45255, GSE4922, GSE5327, GSE7390 (Supplementary Table S2). All other raw data generated in this study are available upon request from the corresponding author.
Results
Identification of an HAS that detects changes in HSF1 transcriptional activity
HSF1 was originally discovered for its role as the master regulator of the heat shock response (11, 13). The discovery of new functions of HSF1 has particularly been fruitful in the context of cancer where it has been found to regulate a target gene set distinct from the genes targeted by HSF1 in response to heat shock and, consequently, play a role in many processes that promote malignancy (11, 13, 15). In this study, we utilized a new computational approach to associate HSF1 transcriptional activity with cancer-related processes and patient outcomes. HSF1 has a complex posttranslational activation process whereby it must localize to the nucleus to undergo trimerization and DNA binding, in addition to phosphorylation prior to recruiting general transcription factors to initiate transcription at target genes. Because of this complex activation process at the protein level, RNA levels of the HSF1 gene are not highly predictive for HSF1 transcriptional activity and HSF1 RNA does not significantly increase expression in response to heat shock (Supplementary Fig. S1A–S1C). We reasoned that the most predictive gene set for HSF1 transcriptional activity will be RNA expression levels of genes that (i) are direct HSF1 target genes, (ii) have high intra-gene correlation within the gene set across multiple datasets, (iii) decrease expression when HSF1 is knocked down or inhibited, (iv) increase expression in response to heat shock, and (v) show increased expression in cancer samples compared with normal. Therefore, we attempted to identify a set of genes that meet these criteria utilizing the gene selection procedures outlined in Fig. 1A.
Figure 1.

Identification of the HAS. A, The schema for identifying an HSF1 activity signature, which included input data from ChIP-seq data to identify direct targets, followed by removing genes not dependent on HSF1 for their expression and finally generating gene sets that have high intragene correlation. B, Correlation matrix of the 19-gene HAS across 11 cancer datasets. C–E, Heat maps were generated from publicly available expression data from HeLa cells with HSF1 knockdown and/or heat stress (C), MDA-MB-231 cells with or without HSF1 knockdown (D), and A549 cells with or without heat stress (E).
We first identified genes that are direct transcriptional targets of HSF1 utilizing 49 ChIP-seq samples in the public domain (15, 19, 20). All unique genes were included in the initial gene list as potential HSF1 target genes. We then removed genes whose expression was not decreased with the knockdown of HSF1 based on differential expression analysis. We then conducted an integrated coexpression analysis for the remaining genes using 11 different cancer expression datasets (Supplementary Table S2). This coexpression analysis resulted in identification of nine unique gene sets for consideration (Supplementary Table S4). One gene set, hereafter referred to as the HAS, was found to have the highest intra-gene correlation (Fig. 1B) compared with the other gene sets (Supplementary Fig. S2A-S2H) and outperformed all other gene sets in detecting changes in HSF1 activity after HSF1 knockdown or in response to heat stress (Fig. 1C; Supplementary Fig. S2I–S2P). The HAS gene set was consistently decreased when HSF1 was knocked down or when HSF1 was inhibited with DTHIB (Fig. 1C and D; Supplementary Fig. S3A–S3C). Inversely, the HAS increased after heat stress (Fig. 1C–E; Supplementary Fig. S3D and S3E). PCA showed that PC1 of the HAS accounted for 73%–92% of the variance across these experiments (Supplementary Fig. S4A–S4G). To statistically compare HAS across these groups, PC1 scores for each sample were compared and seen to have a significant decrease in HAS with HSF1 knockdown while a significant increase was observed in samples with heat shock (Supplementary Fig. S4A–S4G). Genes associated with the HAS were analyzed with gene ontology, which revealed several ontologies involved with protein folding, protein stabilization, heat shock proteins, unfolded proteins, chaperones, chaperonins, and chaperone complexes that were enriched (Supplementary Fig. S5A–S5C) that is indicative of the known function of HSF1 in proteostasis. There were also categories enriched indicative of the role of HSF1 in cancer cells such as metabolism-related categories and cell division. The HSF1 binding motif, heat shock elements (HSE), were also the most enriched motif among the genes within the HAS (Supplementary Fig. S5D). These data indicate the 19 gene set HAS can reliably detect changes in HSF1 transcriptional activity.
HSF1 activity is associated with breast cancer patient outcomes and molecular characteristics
HSF1 has previously been associated with several cancer phenotypes and increased expression and transcriptional activity in cancer cells (11, 13). The HAS genes showed a clear increase in expression in tumor samples compared with matched normal adjacent tissue (Fig. 2A), indicating the HAS was able to detect an increase in HSF1 activity in tumors. We next assessed whether the HAS could serve as a biomarker for outcomes of patients with breast cancer as previous studies show active HSF1 was associated with worse outcomes (15, 17, 19). As shown in Fig. 2B and C, high HAS was associated with worse overall survival (OS) in the TCGA breast cancer cohort and the METABRIC breast cancer cohort whereas expression of the HSF1 gene alone was not as consistent in reproducing this association (Supplementary Fig. S6A and S6B). In addition to Kaplan–Meier and log-rank tests, age-adjusted HR for HAS was also significantly associated with worse OS in the TCGA (HR = 2.42; 95% CI, 1.62–3.62; P < 0.001) and the METABRIC (HR = 1.58; 95% CI, 1.33–1.88; P < 0.001) cohorts. HAS was also associated with worse metastasis–free survival of patients with breast cancer (Fig. 2D) whereas expression of the HSF1 gene was weakly associated with metastasis-free survival (Supplementary Fig. S6C). These results further support previous studies suggesting a potential role for HSF1 in metastasis (15, 16, 29). Taken together, these results indicate that HAS can function as a marker of HSF1 activity and HAS predicted breast cancer patient outcomes consistent with previous assessments for HSF1 activity that include measurement of nuclear HSF1 levels or S326 phosphorylated HSF1 (15–17).
Figure 2.

HAS is associated with breast cancer outcomes and molecular subtypes. A, Heat map was generated using matched adjacent normal and tumor expression data from the TCGA-BRCA cohort (n = 100). B–D, Patients in the TCGA-BRCA (B), METABRIC (C), and GSE47561 (D) cohorts were sorted by their HAS scores and Kaplan–Meier plots were generated for overall survival (B and C) or metastasis-free survival (D). Patients in these analyses were separated into equal tertiles based on HAS scores. E and F, Heat map (E) was generated for the HAS of the METABRIC cohort delineated by PAM50 subtype and HAS PC1 scores (F) for each subtype were compared across subtypes via one-way ANOVA with Tukey post hoc test. *, significance compared with normal-like. G and H, Heat map (G) was generated for the HAS of the METABRIC cohort delineated by Integrated METABRIC Clusters and PC1 scores (H) were plotted across Integrated METABRIC Clusters. *, significance compared with the lowest cluster (#3). NL, normal-Like; LumA, luminal A; LumB, luminal B; HER2-E, HER2-enriched.
We next assessed the relationship of the HAS with molecular subtypes of breast cancer. HAS was increased within the luminal B (LumB), HER2-enriched (HER2-E), and basal subtypes in both the METABRIC and TCGA cohorts compared with the normal-like (NL) and luminal A (LumA) subtypes (Fig. 2E and F; Supplementary Fig. S6D and S6E). The HAS was also assessed in the METABRIC Integrated Clusters (IntClust) where the HAS was upregulated in several integrated clusters that appear to mirror HAS activation from PAM50 molecular subtypes. HAS was increased in IntClust 10, which is mostly associated with the basal molecular subtype, and IntClust 5, which is closely associated with the HER2-enriched molecular subtype (Fig. 2G and H). Interestingly, HAS was also elevated in clusters 1, 6, and 9 that are primarily ER-positive cancers but includes overlap with the LumB molecular subtype. Upregulation of the HAS in these clusters likely points toward a recently-identified interaction of HSF1 with ERα in breast cancer (30) and an interaction between HSF1 and HER2 signaling, which has been established (12, 29, 31). Furthermore, HAS was associated with both disease-free survival and overall survival across many of the TCGA cancer types (Fig. 3A and B). HSF1 has previously been associated with outcomes for many cancer types and the HAS reflected many of these known associations including liver (32), lung (33), and head/neck (34) cancers among others.
Figure 3.

Association of HAS with outcomes across TCGA cancer types. A and B, Cox proportional HRs were calculated for HAS across the TCGA cancer types for disease-free survival (A) and overall survival (B) and were controlled for age, sex, race, and histologic subtype. Forest plots were generated with HRs (black square) with 95% confidence intervals (red bars). Black dotted line indicates a HR of 1. HRs >1 indicate HAS is associated worse survival. *, significant (P < 0.05) HRs.
HSF1 activity has a negative association with the presence of CD8+ T cells in breast tumors
Previous reports indicated a possible association between HSF1 and tumor–immune interactions as well as immune function but a clear role for HSF1 in these interactions has not been fully investigated (15, 35). To investigate the relationship between HSF1 activity and tumor–immune interactions, the HAS was used to sort tumors from breast cancer patient cohorts in the TCGA-BRCA (n = 1100), METABRIC (n = 1996), and GSE47561 (n = 1570) into high and low HSF1 activity groups. These groups were then subjected to GSEA to determine association of HSF1 activity with immune cell populations using multiple immune cell estimation algorithms (21–23, 36, 37). Interestingly, HSF1 activity was negatively associated with the presence of several immune cell types including CD8+ T cells, CD4+ T cells, and B cells across several different signatures for these cell types (Fig. 4A). Utilizing the immune cell estimation algorithms, it was consistently observed that patients with high HAS scores showed lower CD8+ T-cell proportions compared with patients with low HAS scores (Fig. 4B). To determine whether this relationship between HSF1 and CD8+ T-cell proportions is unique to any breast cancer subtype, GSEA was performed on subpopulations of patients with different receptor statuses. The relationship between the HAS score and CD8+ T cells appeared to be stronger in triple-negative breast cancer (TNBC) compared with tumors with other receptor statuses (Fig. 4C and D; Supplementary Fig. S7A–S7D). Patients with Breast cancer who had high HAS scores and low CD8+ T-cell proportions showed significantly worse overall survival or metastasis-free survival compared with patients with lower HAS and higher CD8+ T-cell proportions, which was consistent in the TNBC population (Fig. 4E and F; Supplementary Fig. S7E–S7H). While the relationship of the HAS to immune cell types were similar between TNBC and patients with ER−HER2+ (Fig. 4C; Supplementary Fig. S7D), the proportion of CD8+ T cells were more consistently significantly lower in patients with high HAS with TNBC compared with patients with ER−HER2+ (Fig. 4D; Supplementary Fig. S7D).
Figure 4.

HAS is negatively associated with presence of CD8+ T cells. A, GSEA was performed in TCGA-BRCA, METABRIC, and GSE47561 cohorts with patients separated into high or low HAS scores. Signatures for immune cell types were assessed for enrichment with high or low HAS patients. Normalized enrichment scores (NES) are plotted on a heat map. B, CD8+ T-cell proportions were estimated in the TCGA-BRCA cohort using the indicated deconvolution algorithms and plotted for high and low HAS patients. C, GSEA was performed as in A using only patients with TNBC in the TCGA-BRCA cohort. D, CD8+ T-cell proportions were estimated in patients with TNBC in the TCGA-BRCA cohort using the indicated deconvolution algorithms and plotted for patients with high and low HAS. E and F, Patients in the TCGA-BRCA cohort were separated by HAS scores and CD8+ T-cell proportions estimated by CIBERSORT and Kaplan–Meier graphs were plotted for patient outcomes using patients from all subtypes (E) or only patients with TNBC (F). Sample size for each group is indicated in the graph legend. Log-rank test was used to compute P values. NK, natural killer cells; Tregs, T regulatory cells.
These data suggest that tumors with high HSF1 activity have lower CD8+ T cells. To confirm these observations, tumor tissues from a cohort of 114 patients with breast cancer spanning all subtypes were subjected to IHC for the active mark of HSF1 (phosho-S326) that allowed us to separate tumors into high or low HSF1–active tumors based on nuclear presence of p-HSF1 (Fig. 5A and B). These same specimens were stained with CD8A antibodies to identify the CD8+ T cells. Similar to the computational analyses, these patient specimens had significantly less CD8+ cells in tumors with high levels of active HSF1 (Fig. 5C and D). This relationship was maintained in patients with TNBC tumors but was not significant in ER+ or HER2+ patient tumors (Fig. 5E and F; Supplementary Fig. S8A–S8D). Pearson correlation further support these conclusions as nuclear p-HSF1 was negatively correlated with CD8+ cells (r = −0.22, P = 0.038; Supplementary Fig. S8E) that was also consistent in a subanalysis of only patients with TNBC (r = −0.35, P = 0.048; Supplementary Fig. S8F). These data indicate a consistent relationship wherein tumors with high HSF1 activity have lower levels of CD8+ T cells that was strongest in patients with TNBC relative to other subgroups of patients.
Figure 5.

Active HSF1 in breast cancer tumor specimens coincides with low CD8+ T cells. A and B, A cohort of 114 breast tumors were subjected to IHC with antibodies for CD8A and pHSF1 (S326). C and D, All patients (n = 114) were separated into high (n = 46) or low (n = 68) HSF1-active tumors based on nuclear positivity percentage for pHSF1 (C) and CD8+ (D) cells were compared between these two groups based on active HSF1 levels using a Student t test. E and F, Only patients with TNBC; n = 38) were separated into high (n = 21) or low (n = 17) HSF1-active tumors based on nuclear positivity percentage for pHSF1 (E) and CD8+ (F) cells were compared between these two groups based on active HSF1 levels using a Student t test.
HSF1 functionally affects the level of CD8+ T cells in breast tumors
Having demonstrated a significant negative association between HSF1 activity and the presence of CD8+ T cells using computational approaches and assessment of patient specimens, it was important to examine whether a functional relationship also exists. First, to test for a functional effect of HSF1 on CD8+ T cells, mouse 4T1 breast cancer cells were engineered to express control or HSF1-directed shRNA (Supplementary Fig. S9A). These cells were orthotopically grown in the mammary glands of Balb/c mice for 3 weeks, after which, it was confirmed that HSF1 expression was lost and resulted in a significantly smaller tumor volume (Fig. 6A; Supplementary Fig. S9B). The number of CD8+ T cells in these tumors were assessed by IHC, which showed HSF1 knockdown tumors had significantly more CD8+ T cells compared with control tumors (Fig. 6B and C). To assess more fully the changes in immune cell types after HSF1 knockdown, a control and HSF1 knockdown tumor was digested and subjected to single-cell RNA sequencing (scRNA-seq). While shCTL 4T1 tumors were primarily comprised of tumor cells (71%) and macrophages (23%), the tumors expressing shHSF1 showed a more diverse population of cell types (Fig. 6D–F; Supplementary Fig. S9C). Specifically, HSF1 knockdown tumors showed increased proportions of several populations including CD8+ T cells (3.5 fold increase), B cells (14 fold increase), and neutrophils/granulocytes (13 fold increase; Fig. 6F; Supplementary Fig. S8C). Clustering of cell types did not specifically annotate any further subpopulations of CD8+ T cells. However, expression of markers for specific CD8+ T-cell subpopulations indicate various populations existed including exhausted CD8+ T cells (Cd274, Pdcd1, Tigit), active cytotoxic CD8+ T cells (Prf1, Gzmb), and naïve CD8+ T cells (Ccr7, Sell, Il7r; Supplementary Fig. S9D). These data confirm a role for HSF1 in regulating changes in proportions of CD8+ T cells within tumors but also points toward a potentially larger immunologic switch in breast tumors catalyzed by a loss of HSF1 in cancer cells that appears to promote a robust immunogenic response.
Figure 6.

HSF1 Functionally regulates the amount of CD8+ T cells in breast tumors. A, 4T1 cells (5 × 104 cells) with (n = 5) or without (n = 5) HSF1 knockdown were grown orthotopically in Balb/c mice for 3 weeks. Tumor volume at the conclusion of the study is graphed. B, IHC was performed on tumors from A detecting CD8A to identify CD8+ T cells. C, CD8+ T cells from B were quantified for control and HSF1 knockdown tumors by manual counting positive cells in >5 fields of the tumor tissue area. D–F, shCTL and shHSF1 tumors from A were subjected to scRNA-seq. Processed reads were used to map cell clusters for both samples using Seurat 4.2.0. The Uniform Manifold Approximation and Projection (UMAP) integrating both samples is shown in D. These cell types were annotated using expression of specific marker genes for each population, for which a sample of these marker genes is shown in E. The proportion of each cell population was also calculated and graphed in F.
While these data suggest the loss of HSF1 affects the proportions of CD8+ T cells in breast tumors, loss of HSF1 can also intrinsically impact cancer cells. HSF1 has been shown to be important to several aspects of cancer cell function, including maintaining proteostasis, regulating cancer cell metabolism, cell cycle, and many others (11, 15, 20). As such, to determine the importance of CD8+ T cells to the loss of tumor volume after HSF1 knockdown, both control and HSF1 knockdown 4T1 cells were grown orthotopically in Balb/c mice with or without CD8+ T-cell depletion. If the loss in tumor volume with HSF1 knockdown is due to the direct intrinsic effects on the cancer cells, depletion of CD8+ T cells should have no effect on tumor volume. Splenic T-cell counts confirmed that CD8+ T cells were indeed depleted at the end of the 3-week tumor growth period (Fig. 7A). Depletion of CD8+ T cells led to an expected increased volume of control tumors as CD8+ T cell depletion removes a tumor suppressor (Fig. 7B). In addition, the depletion of CD8+ T cells rescued tumor growth with HSF1 knockdown, indicated by a greater increase in tumor growth after CD8+ T-cell depletion (∼50 fold increase) compared with the increase in control tumors (∼9 fold increase). The decrease in tumor volume with HSF1 knockdown was accompanied by an increase in CD8+ T cells while mice with CD8+ T-cell depletion were largely void of these cells within the tumors (Fig. 7C). These results suggest that CD8+ T cells were a significant contributor to the decreased tumor volume with HSF1 knockdown and that HSF1 can protect breast tumors from immune-mediated killing through suppression of CD8+ T cells.
Figure 7.

Depletion of CD8+ T cells rescues tumor growth after HSF1 knockdown. A, Balb/c mice were given either PBS control or CD8A antibodies to deplete CD8+ T cells in vivo. Control and HSF1 knockdown cells were then grown orthotopically for 3 weeks. Spleens were collected at the conclusion of the study, dissociated, and cells were subjected to flow cytometry to confirm the depletion of CD8+ T cells. B, Tumor volume at the conclusion of the study from A is plotted. C, Tumor tissue from B was subjected to IHC for CD8A to assess the CD8+ T cells.
HSF1 suppresses expression and secretion of CCL5, which is necessary to attract CD8+ T cells after HSF1 knockdown
To understand how HSF1 activity could affect proportions of CD8+ T cells in breast tumors, we tested whether HSF1 regulates expression or secretion of cytokines that attract CD8+ T cells. We compared expression of cytokines from RNA-seq of 4T1 control and HSF1 knockdown cells with a cytokine array detecting over 100 cytokines that was performed on conditioned media from 4T1 control and HSF1 knockdown cells. The only cytokine that showed a significant increase with HSF1 knockdown at the RNA level and secreted protein was CCL5/RANTES (Fig. 8A; Supplementary Fig. S10A and S10B), which is also a canonical chemoattractant cytokine for CD8+ T cells (38). The effect of HSF1 knockdown on CCL5 expression was confirmed with qPCR in both mouse and human breast cancer cells (Fig. 8B and C) while overexpression of HSF1 decreased CCL5 transcript levels (Supplementary Fig. S10C). HSF1 knockdown 4T1 tumors also showed an increase in CCL5 protein by IHC compared with control tumors (Fig. 8D). In addition, assessing CCL5 levels by IHC in the same cohort of breast cancer specimens from Fig. 5, CCL5 levels were significantly lower in tumors with high active HSF1, which remained significant when analyzing only patients with TNBC (Fig. 8E–G).
Figure 8.

HSF1 suppresses CCL5 to prevent attraction of CD8+ T cells. A, Conditioned media was grown for 72 hours on control or HSF1 knockdown 4T1 cells. Conditioned media was then subjected to a cytokine array detecting over 100 cytokines. Cytokines are ordered by P value and fold change (FC) is calculated as HSF1 knockdown (KD) divided by control (CTL) cells. B and C,CCL5 mRNA levels in control and HSF1 knockdown 4T1 (B) and MDA-MB-231 (C) cells assessed by RT-qPCR. D, IHC of CCL5 in 4T1 control and HSF1 knockdown tumors from Fig. 6A. E, Tumor specimens from Fig. 5 were subjected to IHC for CCL5. CCL5 levels were quantified by QuPath. F, CCL5 levels are plotted in high (n = 46) or low (n = 68) active HSF1 tumors from all patients (n = 114). G, CCL5 levels are plotted in high (n = 21) or low (n = 17) active HSF1 tumors from patients with TNBC (n = 38). H, Conditioned media from 4T1 cells expressing either control, HSF1, or HSF1+CCL5 siRNA were placed in the bottom chamber for the T-cell transwell migration assay. CD8+ T-cell proportions are plotted for each group (n = 5) and statistically compared using one-way ANOVA with Tukey post hoc test. I, Conditioned media from 4T1 cells expressing either empty vector or CCL5-expressing construct were placed in the bottom chamber for the T-cell transwell migration assay. CD8+ T-cell proportions are plotted for each group (n = 3) and statistically compared using one-way ANOVA with Tukey post hoc test. J, Model indicating HSF1 suppresses CCL5 expression and secretion, leading to decreased attraction of CD8+ T cells toward breast cancer cells.
To test the importance of increased expression and secretion of CCL5 after HSF1 knockdown, transwell migration of T cells was tested. In this assay, CD3+ T cells were isolated from Balb/c mice spleens and T cells were placed in the top chamber while the bottom chamber contained chemokines or conditioned media and were incubated for 24 hours followed with assessment of the bottom chamber by flow cytometry for CD3+/CD8+ T cells (Supplementary Fig. S10D). Adding exogenous CCL5 resulted in a directed migration of CD8+ T cells to the bottom chamber, indicating the assay is responding to a positive control (Supplementary Fig. S10E). To test the effect of HSF1 knockdown, conditioned media from 4T1 cells with control or HSF1 siRNA were added the bottom chamber of the T-cell migration assay, which resulted in a significant increase in CD8+ T-cell migration with HSF1 knockdown (Fig. 8H). Knockdown of both HSF1 and CCL5 significantly reduced the CD8+ T-cell migration observed with only HSF1 knockdown (Fig. 8H), suggesting the increased expression and secretion of CCL5 after HSF1 knockdown is necessary to attract CD8+ T-cells. In addition, conditioned media used in this assay from 4T1 cells with forced expression of CCL5 also led to a significant increase in CD8+ T-cell migration (Fig. 8I) without any alteration in HSF1, indicating CCL5 is both necessary and sufficient in this system to attract CD8+ T cells. Taken together, these data suggest a model whereby tumors with high activity of HSF1 leads to suppression of CCL5 transcript and secreted protein that ultimately prevents attraction of CD8+ T cells toward breast tumors allowing the tumors to evade immune-mediated destruction (Fig. 8J).
Discussion
The major finding from these studies is activity of the stress response transcription factor HSF1 in breast cancer cells, known to support malignancy, prevents CD8+ T cells from attacking breast tumors. Furthermore, depletion of HSF1 resulted in a recruitment of CD8+ T cells that was found to be critical to reducing tumor volume after HSF1 depletion, thereby directly connecting HSF1 function to CD8+ T cells within breast tumors. The recruitment of CD8+ T cells appears to be part of an entire reprogramming of the tumor microenvironment after loss of HSF1 in cancer cells that was accompanied by increases in several immune populations such as neutrophils/granulocytes, B cells, and CD4+ T cells. While a direct connection between HSF1 function within tumors and immune cell presence or infiltration has not yet been established, a negative association between CD8+ T-cell proportion estimates and HSF1 gene expression has been observed in breast cancer (35). This study clearly indicates loss of HSF1 decreases tumor volume in an immune-competent system, consistent with previous results (29) and point to HSF1 functioning as an immune-suppressive factor in the context of cancer. These data indicate for the first time that CD8+ T cells, specifically, are critical to the tumor-suppressive effect of HSF1 depletion. We suggest that therapeutic targeting of HSF1 in breast tumors could lead to a reprogramming of the tumor microenvironment to be more immunogenic, which would enhance therapeutic response to several standard-of-care breast cancer therapies including taxanes and anthracyclines (7, 39).
HSF1 has been known as the master regulator of the heat shock response since the mid 1980s. It was first observed to be altered in metastatic prostate cancer and has since been found to play a pleiotropic role in cancer regulating many functions in cancer cells from metabolism to proliferation. Because of the complex activation of HSF1 protein activity, the transcript levels have poor utility in predicting or assessing HSF1 activity. We identified a gene signature, named HAS, comprised of 19 genes that are direct HSF1 gene targets and depend on HSF1 for their expression. Increased HSF1 activity largely results in increased expression of the majority of the 19 genes, indicated by the high intra-gene correlation among these 19 genes providing for the first time an accurate and sensitive readout of HSF1 activity using transcript data. Studies wherein HSF1 gene was knocked down or exposed to heat shock, both of which have predictable effects on HSF1 activity, confirmed this observation. There is one previously reported HSF1 gene signature, the CaSig, developed from elegant studies identifying the many roles for HSF1 in cancer cells (15). The performance of the HAS in our studies is possibly due to the intra-gene correlation as the CaSig (456 genes) had a lower intra-gene correlation. Including intra-gene correlation as a criterion is an additional novel aspect to the HAS and the development of gene signatures. Finally, while we have applied the use of the HAS in this study primarily in breast cancers, we did utilize direct target genes from multiple cell types. As such, the HAS likely has utility that extends further than just breast cancer, which can be evidenced by the use of the HAS to calculate HRs across the TCGA cancer study spectrum (Fig. 3). The application of the HAS will allow for future analysis of HSF1 transcriptional activity with many cancer-related studies that cannot be done based on HSF1 expression alone.
Our results indicate the influx of CD8+ T cells after HSF1 depletion is because of an increase in CCL5 expression and secretion, suggesting CCL5 would serve a tumor-suppressive function. However, the role of CCL5 in breast cancer is controversial. A recent report indicates CCL5 is associated with breast cancer metastasis (40), while another study indicated therapeutic response to HDAC inhibitors in lung cancer is accompanied by a decrease in MYC function and an increase in CCL5 that supported therapeutic efficacy (41), suggesting tumor-suppressive effects of CCL5. The disparity in results for the function of CCL5 in cancer is puzzling but is possibly related to expression of the CCL5 receptor, CCR5, on cancer cells. There is also precedent for an increase in cytokine-related signaling with HSF1 inhibition in other contexts, such as LPS exposure (42, 43). Because these results saw a partial loss of CD8+ T-cell attraction by silencing CCL5, it is likely that attraction of T cells after HSF1 depletion is regulated by additional mechanisms, including antigen presentation (44). Future studies will address other possible mechanisms by which HSF1 regulates the composition of the tumor microenvironment and the regulation of the CCL5 gene as it was not identified as a direct HSF1 target when generating the HAS.
Further investigation is needed to understand if immune suppression is a general feature of HSF1 biology. Inversely to cancer cells, neurodegenerative diseases share etiologies with an imbalance of proteostasis leading to protein aggregate formation in neurons. Opposite to cancer cells, these neurons lose HSF1 expression and activity with aging that ultimately escalates these diseases (45). Furthermore, several neurodegenerative diseases have been linked to immune-mediated mechanisms for neuronal death in recent years (46). Consequently, it is possible HSF1 serves to protect over-stressed cells from immune-mediated destruction and our findings point to how cancer cells have coopted this function for their benefit and survival.
These studies indicate a novel mechanism regulating CD8+ T-cell presence in breast tumors wherein hyperactive HSF1 suppresses CCL5, leading to decreased attraction of CD8+ T cells. Breast cancer is a low immunogenic tumor, which could partially be because of high basal activity of HSF1. Further studies will need to identify other mechanisms contributing to the effect of HSF1 on CD8+ T cells and other immune cells in the tumor microenvironment. Studies are underway to test whether inhibition of HSF1, with compounds such as DTHIB (47), can result in an influx of CD8+ T cells that would support targeting HSF1 as a therapeutic approach to making breast tumors more immune cell-rich. The role of HSF1 in CD8+ T-cell activity or exhaustion warrants further investigation considering HSF1 has been reported to upregulate PD-L1 expression on cancer cells (48). These results generate several new lines of investigation for the role of HSF1 in new aspects of tumor biology and tumor–immune interactions.
Supplementary Material
Supplemental Figure 1
{kind=link}
Supplemental Figure 2
{kind=link}
Supplemental Figure 3
{kind=link}
Supplemental Figure 4
{kind=link}
Supplemental Figure 5
{kind=link}
Supplemental Figure 6
{kind=link}
Supplemental Figure 7
{kind=link}
Supplemental Figure 8
{kind=link}
Supplemental Figure 9
{kind=link}
Supplemental Figure 10
{kind=link}
Supplementary Legends and Tables
Acknowledgments
This publication was made possible, in part, with support from the NCI (K22CA207575 to R.L. Carpenter; R01CA248033 to X. Lu), the Catherine Peachey Fund (to R.L. Carpenter), and the Indiana Clinical and Translational Sciences Institute (to R.L. Carpenter) funded, in part by grant number UL1TR002529 from the NIH, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. Research funding was also provided by P30CA82709–20 (Tumor Microenvironment and Metastasis Program) and Van Andel Institute through the Van Andel Institute – Stand Up To Cancer Epigenetics Dream Team (to R.L. Carpenter and K.P. Nephew). The indicated Stand Up To Cancer Grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C. We would also like to acknowledge the Light Microscopy Imaging Center, the Flow Cytometry Facility, the Laboratory Animal Resources facility, and the Center for Medical Genomics at Indiana University for use of their core facilities.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
No disclosures were reported.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authors' Contributions
C. Jacobs: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. S. Shah: Formal analysis, investigation, methodology. W.-C. Lu: Formal analysis, validation, investigation, methodology. H. Ray: Formal analysis, validation, investigation, methodology. J. Wang: Formal analysis, validation, investigation, methodology. N. Hockaden: Resources, data curation, software, formal analysis, methodology. G. Sandusky: Conceptualization, resources, software, formal analysis, supervision, methodology, writing–original draft, writing–review and editing. K.P. Nephew: Conceptualization, resources, data curation, formal analysis, supervision, visualization, writing–original draft, writing–review and editing. X. Lu: Conceptualization, Resources, data curation, software, formal analysis, visualization, writing–review and editing. S. Cao: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. R.L. Carpenter: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. American Cancer Society. Cancer Facts & Figures 2023. Atlanta, GA; 2023. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf. [Google Scholar]
- 2. Gaynor N, Crown J, Collins DM. Immune checkpoint inhibitors: key trials and an emerging role in breast cancer. Semin Cancer Biol 2022;79:44–57. [DOI] [PubMed] [Google Scholar]
- 3. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jamiyan T, Kuroda H, Yamaguchi R, Nakazato Y, Noda S, Onozaki M, et al. Prognostic impact of a tumor-infiltrating lymphocyte subtype in triple negative cancer of the breast. Breast Cancer 2020;27:880–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loi S, Drubay D, Adams S, Pruneri G, Francis PA, Lacroix-Triki M, et al. Tumor-infiltrating lymphocytes and prognosis: a pooled individual patient analysis of early-stage triple-negative breast cancers. J Clin Oncol 2019;37:559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ali HR, Provenzano E, Dawson SJ, Blows FM, Liu B, Shah M, et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann Oncol 2014;25:1536–43. [DOI] [PubMed] [Google Scholar]
- 7. Denkert C, Loibl S, Noske A, Roller M, Müller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010;28:105–13. [DOI] [PubMed] [Google Scholar]
- 8. Lee HJ, Seo JY, Ahn JH, Ahn SH, Gong G. Tumor-associated lymphocytes predict response to neoadjuvant chemotherapy in breast cancer patients. J Breast Cancer 2013;16:32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seo AN, Lee HJ, Kim EJ, Kim HJ, Jang MH, Lee HE, et al. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br J Cancer 2013;109:2705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ogiya R, Niikura N, Kumaki N, Bianchini G, Kitano S, Iwamoto T, et al. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci 2016;107:1730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol 2018;19:4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carpenter RL, Paw I, Dewhirst MW, Lo HW. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 2015;34:546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carpenter RL, Gokmen-Polar Y. HSF1 as a cancer biomarker and therapeutic target. Curr Cancer Drug Targets 2019;19:515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu WC, Omari R, Ray H, Wang J, Williams I, Jacobs C, et al. AKT1 mediates multiple phosphorylation events that functionally promote HSF1 activation. FEBS J 2022;289:3876–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012;150:549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carpenter RL, Sirkisoon S, Zhu D, Rimkus T, Harrison A, Anderson A, et al. Combined inhibition of AKT and HSF1 suppresses breast cancer stem cells and tumor growth. Oncotarget 2017;8:73947–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 2011;108:18378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- 19. Kourtis N, Moubarak RS, Aranda-Orgilles B, Lui K, Aydin IT, Trimarchi T, et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol 2015;17:322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santagata S, Mendillo ML, Tang Y-c, Subramanian A, Perley CC, Roche SP, et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 2013;341:1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Medicine 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, et al. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019;35:i436–i45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Germain PL, Lun A, Garcia Meixide C, Macnair W, Robinson MD. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 2021;10:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J Biol Chem 2012;287:35646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vydra N, Janus P, Kus P, Stokowy T, Mrowiec K, Toma-Jonik A, et al. Heat shock factor 1 (HSF1) cooperates with estrogen receptor α (ERα) in the regulation of estrogen action in breast cancer cells. eLife 2021;10:e69843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meng L, Gabai VL, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010;29:5204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab 2011;14:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S, Jung J, Lee YJ, Kim SK, Kim JA, Kim BK, et al. Targeting HSF1 as a therapeutic strategy for multiple mechanisms of EGFR inhibitor resistance in EGFR mutant non-small-cell lung cancer. Cancers (Basel) 2021;13:2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishiwata J, Kasamatsu A, Sakuma K, Iyoda M, Yamatoji M, Usukura K, et al. State of heat shock factor 1 expression as a putative diagnostic marker for oral squamous cell carcinoma. Int J Oncol 2012;40:47–52. [DOI] [PubMed] [Google Scholar]
- 35. Chen F, Fan Y, Cao P, Liu B, Hou J, Zhang B, et al. Pan-cancer analysis of the prognostic and immunological role of HSF1: a potential target for survival and immunotherapy. Oxid Med Cell Longev 2021;2021:5551036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 2020;48:W509–W14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roussot N, Ghiringhelli F, Rébé C. Tumor immunogenic cell death as a mediator of intratumor CD8 T-cell recruitment. Cells 2022;11:3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018;19:40–50. [DOI] [PubMed] [Google Scholar]
- 40. Qiu J, Xu L, Zeng X, Wu H, Liang F, Lv Q, et al. CCL5 mediates breast cancer metastasis and prognosis through CCR5/Treg cells. Front Oncol 2022;12:972383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 2017;171:1284–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murapa P, Ward MR, Gandhapudi SK, Woodward JG, D'Orazio SE. Heat shock factor 1 protects mice from rapid death during Listeria monocytogenes infection by regulating expression of tumor necrosis factor alpha during fever. Infect Immun 2011;79:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan H, Huang F, Huang M, Wu X, Tong Z. HSF1 Attenuates the release of inflammatory cytokines induced by lipopolysaccharide through transcriptional regulation of Atg10. Microbiol Spectr 2023;11:e0305922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng H, Li Z. Cutting edge: cross-presentation of cell-associated antigens to MHC class I molecule is regulated by a major transcription factor for heat shock proteins. J Immunol 2004;173:5929–33. [DOI] [PubMed] [Google Scholar]
- 45. Gomez-Pastor R, Burchfiel ET, Neef DW, Jaeger AM, Cabiscol E, McKinstry SU, et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease. Nat Commun 2017;8:14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 2022;22:657–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dong B, Jaeger AM, Hughes PF, Loiselle DR, Hauck JS, Fu Y, et al. Targeting therapy-resistant prostate cancer via a direct inhibitor of the human heat shock transcription factor 1. Sci Transl Med 2020;12:eabb5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang T, Ren C, Lu C, Qiao P, Han X, Wang L, et al. Phosphorylation of HSF1 by PIM2 induces PD-L1 expression and promotes tumor growth in breast cancer. Cancer Res 2019;79:5233–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Figure 5
Supplemental Figure 6
Supplemental Figure 7
Supplemental Figure 8
Supplemental Figure 9
Supplemental Figure 10
Supplementary Legends and Tables
Data Availability Statement
Next-generation sequencing data generated in this study was deposited at Gene Expression Omnibus (GEO) at GSE236972. Publicly available datasets used to analyze and validate the HSF1 activity signature (HAS) included GSE38232 (MCF7/BPLER cells), GSE3697 (Hela cells), GSE83844 (A549 cells), GSE115973 (U2OS cells), GSE155248 (C4–2 cells; Supplementary Table S1). Publicly available expression datasets from patient samples used to analyze the intragene correlation for HSF1 gene module detection included GSE11121, GSE12093, GSE14020, GSE1456, GSE17705, GSE2034, GSE2603, GSE45255, GSE4922, GSE5327, GSE7390 (Supplementary Table S2). All other raw data generated in this study are available upon request from the corresponding author.