

SUMMARY
Nonoptimal synonymous codons repress gene expression, but the underlying mechanisms are poorly understood. We and others have previously shown that nonoptimal codons slow translation elongation speeds and thereby trigger messenger RNA (mRNA) degradation. Nevertheless, transcript levels are often insufficient to explain protein levels, suggesting additional mechanisms by which codon usage regulates gene expression. Using reporters in human and Drosophila cells, we find that transcript levels account for less than half of the variation in protein abundance due to codon usage. This discrepancy is explained by translational differences whereby nonoptimal codons repress translation initiation. Nonoptimal transcripts are also less bound by the translation initiation factors eIF4E and eIF4G1, providing a mechanistic explanation for their reduced initiation rates. Importantly, translational repression can occur without mRNA decay and deadenylation, and it does not depend on the known nonoptimality sensor, CNOT3. Our results reveal a potent mechanism of regulation by codon usage where nonoptimal codons repress further rounds of translation.
Graphical Abstract
In brief
Barrington et al. demonstrate that nonoptimal codon usage reduces protein expression post-transcriptionally by repressing translation initiation. These findings reconcile a long-standing contradiction in the field between slow elongation speed and reduced ribosome occupancy and suggest that elongation and initiation machinery cooperate to regulate gene expression.
INTRODUCTION
The open reading frame (ORF) does more than encode a protein: it also regulates gene expression. Our current understanding of the ORF as a major regulatory element represents a shift from the classical view that 5′ and 3′ untranslated regions (UTRs) are the predominant reservoirs of cis-elements for post-transcriptional regulation. Through structures and sequences bound by trans-regulatory factors, the UTRs can alter the translation, stability, and localization of messenger RNAs (mRNAs).1-6 However, we now know that sequences within the ORF can control many of these same processes.
Codon usage is a major way that the ORF regulates gene expression. The genetic code is redundant, meaning multiple synonymous codons can code for the same amino acid. However, synonymous codons are not used equally. When “nonoptimal codons” are used, they reduce gene expression compared to “optimized” variants. For this reason, optimal codons are often used more frequently in highly expressed genes.7 Thus, synonymous codon usage is an essential and conserved mechanism that controls gene expression, while the encoded primary sequence remains unchanged.8-10 This phenomenon underpins the common practice of codon optimization with heterologous expression of genes of interest.11-14 Despite the well-known importance of codon optimality on expression, understanding the underlying mechanisms remains an active area of study.
Codon usage can influence nearly every step of the gene expression pathway, but most of its impact occurs through translation. Due to various features, including tRNA abundance and charging, codon demand, and wobble position interactions, optimal codons are decoded more quickly than nonoptimal ones.8,15-21 Because decoding is often the rate-limiting step in translation elongation,15,22,23 nonoptimal decoding slows overall translation elongation. In some species (depending on the nature of the optimal codons), codon usage can change the GC content of the transcript, which can create additional secondary structural elements with the potential to slow ribosome progression.24-26 Slow translation elongation speed then affects other aspects of post-transcriptional regulation. One proposed mechanism is that translation elongation rates can alter protein folding.27-33 For instance, codons encoding linker regions between protein domains are frequently nonoptimal and are thought to slow ribosomes between structured elements, thus allowing enough time for proper co-translational folding.9,29-32,34 Indeed, complete optimization can limit ideal peptide folding kinetics, thereby reducing protein stability and overall yield.35,36 Another mechanism by which codon usage can affect gene expression is mediated within the so-called “translational ramp,” or the beginning of the ORF.37 This region is often disproportionately nonoptimal, which is thought to slow the ribosome, sterically block the start codon, and limit overall initiation rates.37,38 These observations have led to the model where the translational ramp acts to space out ribosomes on the ORF.37 Yet another pathway is through surveillance mechanisms; some substrates, including poly-lysine tracts, slow or stall elongating ribosomes and ultimately cause ribosomal collisions.39,40 Long-lived collisions activate the ZNF598-mediated ribosome quality control (RQC) pathway, resulting in translational repression, destruction of the nascent peptide, rescue of the stalled ribosomes, and degradation of the translated mRNA.41-47
Recent work has shown that slow elongation, specifically through nonoptimal codon usage and slow decoding, triggers mRNA decay by a mechanism distinct from the RQC pathway.10,48-50 Best described in yeast, this pathway is mediated by the CCR4-NOT deadenylase complex.16,21,48 Here, it is thought that a hallmark of slow decoding (i.e., a ribosome with empty A and E sites) leads to the binding of Not5p within the vacant E site.48,51 Because Not5p is a component of the CCR4-NOT deadenylase complex, its binding stimulates deadenylation of the mRNA, accelerating the eventual mRNA destruction by the Dhh1p decapping activator, the decapping enzyme, and 5′→3′ Xrn1-mediated decay of the mRNA body.16,48,52 The human ortholog of Not5p (CNOT3) has also been shown to sense slow translation in vitro, suggesting that this factor might have a conserved role in codon-optimality-mediated decay.51 In yeast, the decapping activator Dhh1p (DDX6 in humans) is also more associated with codon-poor transcripts, leading to decay.53,54 Indeed, orthologs of Dhh1, like human DDX6, have well-defined roles in translational silencing and mRNA decay, highlighting their ability to regulate gene expression post-transcriptionally.3,55-59 The connection between synonymous codon usage and transcript stability has now been expanded to numerous prokaryotic and eukaryotic organisms, including E. coli, S. pombe, Neurospora, Xenopus, zebrafish, Drosophila, mice, and humans, suggesting that this phenomenon is deeply conserved.60-72
Despite the substantial research into how codon usage impacts protein expression, these mechanisms can only partially explain the repressive effect of nonoptimal codons. This phenomenon has been most clearly seen in studies on Ras paralogs. Ras genes (HRas, NRas, and KRas) differ in their codon usage, and KRas, the least optimal paralog, produces substantially less protein than HRas, the most optimal one.33,60 Importantly, in studies where KRas has been optimized, more protein was produced than could be explained by changes in overall mRNA levels.33,60 Similarly, several studies have shown that more ribosomes are bound to optimal mRNAs than nonoptimal ones;33,62,73,74 however, this effect is counter to the expectation that, like slow traffic on a highway, slow elongation should result in more, not fewer, ribosomes on nonoptimal mRNAs. These results raise the possibility that codon usage may affect other post-transcriptional regulatory pathways, including translation. Consistent with this idea, experiments in Neurospora show that eIF2α (EIF2AK1) is phosphorylated in response to slow decoding, thus repressing translation initiation.75 However, whether this phenomenon is restricted to Neurospora or acts more generally in eukaryotes is unknown.
Here, we explored the mechanisms by which codon usage regulates gene expression. Using a series of reporters with large N-terminal tags (to exclude potential effects of the translational ramp), we found that optimal variants produce more protein than nonoptimal ones, beyond what can be explained by changes in mRNA abundance. The effect at the protein level can be substantially greater than that at the mRNA level, revealing a robust yet undescribed mechanism by which codon usage regulates gene expression. The reduction in protein abundance from nonoptimal reporters cannot be explained by differences in protein stability or folding, nor by premature ribosome termination. Instead, our results indicate that nonoptimal codons repress translation initiation. This translational repression appears to depend on reduced eIF4E binding but is independent of both EIF2AK1 phosphorylation and deadenylation. Finally, we show that more translational repression occurs in the absence of mRNA destabilization, revealing a contradictory relationship between two major drivers of codon-mediated regulation.
RESULTS
Codon optimality affects protein levels beyond what can be explained by mRNA changes
To determine how codon usage affects mRNA and overall protein expression levels, we made a series of optimality reporters using different metrics with various tags and lengths (Figure 1A). The first set of reporters contained an optimal or nonoptimal firefly luciferase coding region preceded by a large (~1 kb) N-terminal “Spaghetti Monster” (or “SM”) domain with interspaced FLAG tags.76,77 This large tag ensured that any regulation by codon optimality could not be explained by differences in the translational ramp.37,38 The firefly luciferase gene was either nonoptimized or optimized using the tRNA adaptation index (tAI) metric8,78 to obtain final optimality scores of 0.45 (6.2%) and 0.91 (83.7%) for the nonoptimal and optimal reporters, respectively. After transfecting these constructs into HEK293T cells, along with an optimized Renilla luciferase control, we measured transcript levels by RT-qPCR and protein activity using dual-luciferase assays. As expected, based on recent findings,68-71 the optimal reporter mRNA was more abundant than its nonoptimal counterpart (2.3− ± 0.7-fold, p < 10−5; Figure 1B, left).
Figure 1. Codon optimality affects protein levels beyond what can be explained by changes at the mRNA level.

(A) Schematic of optimality reporters used in this figure and throughout the article.
(B) SM-tagged firefly luciferase optimality reporters show differences in protein activity, even after controlling for mRNA abundance. HEK293T cells were transfected with plasmids expressing each reporter along with optimized Renilla luciferase. Shown are box-and-whisker plots for the relative fold change for mRNA abundance (RT-qPCR), protein activity (dual-luciferase assay), and normalized protein-per-mRNA abundance; n = 7.
(C) Nonoptimal firefly luciferase reporters have reduced protein levels. Western blotting was performed by probing for the SM tag and Renilla luciferase.
(D) Western blotting and luciferase assays show a comparable reduction in firefly luciferase levels due to nonoptimality. A scatterplot compares the fold change in the nonoptimal reporter firefly luciferase abundance as determined by luciferase activity or western blotting (p = 0.3; n = 3).
(E–G) As in (B)–(D) except for SM-tagged Renilla luciferase (n = 3).
(G) As in (D) except for Renilla luciferase (p = 0.2; n = 3).
(H) As in (B) except for γHA-tagged GFP. Protein levels were quantified by western blotting, with mCherry-Renilla as transfection control (n = 3).
(I) Nonoptimal GFP shows reduced protein levels. Shown is a representative western blot to quantify the reduction in γHA-GFP protein levels due to nonoptimal codons. Lysates were serially diluted for quantification, and fold change was calculated relative to Renilla luciferase.
(J) Translation of nonoptimal γHA firefly luciferase “ramp” reporter does not trigger mRNA decay nor reduce protein activity. HEK293T cells were transfected and analyzed as in (B) (n = 3)
However, differences in protein activity exceeded differences in mRNA abundance. The relative firefly luciferase activity of the optimal reporter was 4.2− ± 1.9-fold more than the nonoptimal one (p < 10−6; Figure 1B, middle). In control experiments, protein activity from the optimal firefly luciferase reporter changed linearly with mRNA levels (R2 = 0.994; Figure S1A), suggesting that our results with reporter variants may have revealed additional regulation. To confirm that these differences could not be explained by a potential impact on enzymatic activity (e.g., due to differences in protein folding, which has been reported to be affected by codon usage9,29-32), we analyzed protein abundance directly by western blotting and saw similar results (Figures 1C and 1D). Together, these data show an approximately 2-fold difference (1.86− ± 0.3-fold) in protein expression beyond that at the transcript level (p < 10−5; Figure 1B, right). These results were not cell-type specific, as the effect was recapitulated in U2OS cells (Figure S1B), nor did they depend on optimal Renilla luciferase for normalization, as results were unchanged when co-transfected with nonoptimal Renilla luciferase (Figure S1C).
To test the generality of this result, we repeated this assay with N-terminal FLAG SM Renilla luciferase reporters. The optimal Renilla coding sequence was optimized using the codon AI (CAI) metric,69,79 resulting in a score of 0.99 (99.6%), in contrast to the commonly used Renilla coding region, which has a score of 0.69 (33.4%). As before, optimal Renilla luciferase mRNA was more abundant than the nonoptimal reporter mRNA (1.37− ± 0.19-fold difference, p = 0.04; Figure 1E, left), but there was a larger difference at the protein level when measured by either luciferase assays or western blotting (2.44− ± 0.29-fold difference, p = 0.002; Figures 1E-1G).
To exclude possible artifacts due to the SM tag or plasmid-derived UTR sequences, we repeated the experiment using γHA-GFP optimality reporters; these reporters contained different 5′ and 3′ UTRs as well as a different N-terminal tag, which, while shorter than the SM tag, was still adequately long at 54 codons to exclude a possible translational ramp effect.37 These reporters were optimized using the tAI metric,78 like the firefly luciferase reporters, with a final score of 0.91 (83.5%) for the optimal and 0.51 (9.4%) for the nonoptimal. Again, the ~2.3-fold difference in mRNA levels was insufficient to account for the ~9.4-fold difference in protein abundance, revealing an almost 4-fold additional effect at the protein level (4.1− ± 0.7-fold; p < 10−3; Figures 1H and 1I). Thus, our results indicate that nonoptimal codons exert repressive effects beyond changes at the mRNA level in human cells.
We next asked if this additional protein effect from poor codon usage was conserved by performing similar assays in the Drosophila S2 cell line using γHA-firefly luciferase reporters. The reporters were optimized using the codon stability coefficient (CSC) metric previously determined for Drosophila,71 with a stabilizing score of −0.026/codon (0%) and +0.020/codon (100%) for nonoptimal and optimal reporters, respectively. Interestingly, although synonymous codon usage only had a marginal effect on mRNA abundance for reasons that are unclear (the optimal reporter was 1.06− ± 0.03-fold more abundant than the nonoptimal, p = 0.02; Figure S1D), the relative protein activity of the optimal reporter was nearly 4-fold higher than the nonoptimal counterpart (3.97− ± 0.89-fold; p < 10−4; Figure S1D).
We had included large N-terminal tags to minimize the impact of ribosomes slowly clearing the start codon (and blocking subsequent initiating ribosomes). However, it was still formally possible that the nonoptimal downstream sequence in the ORF could cause a pileup to decrease ribosome loading.37,38 Thus, we wanted to test whether the steric hindrance of translation initiation from slowed ribosomes was responsible for the repression we observed. To do so, we made a fourth set of reporters (Figure 1A) with either an optimal or nonoptimal N-terminal γHA tag (using the tAI metric78) upstream of our tAI-optimized firefly luciferase gene. However, mRNA levels were no longer significantly different (p = 0.09), and the additional repression at the protein level was no longer present (Figure 1J). Thus, four different sets of reporters in both vertebrates and invertebrates showed that poor codon usage in the body of the ORF represses protein expression more than can be explained by changes at the mRNA level.
Poor codon optimality does not affect protein stability
The observation that codon usage affects protein abundance beyond what can be explained by mRNA abundance could be due to effects on protein stability or translation. Given that the rate of translation elongation has been proposed to change the dynamics of co-translational folding and that incorrect folding can target peptides for degradation,31,35,36 we asked if each nonoptimal reporter was less stable than their optimal counterpart by measuring protein half-lives using cycloheximide translation shut-off assays. By measuring puromycin incorporation, we confirmed that cycloheximide effectively inhibited translation over a 12 h time course (Figure 2A). Optimal and nonoptimal firefly luciferase reporters had a functional half-life of 12 ± 3 and 15 ± 6 h, respectively; thus, if anything, the nonoptimal reporter was slightly more stable than the optimal one, which could be due to improper folding of the optimized firefly protein, although their half-lives did not differ significantly (p = 0.35; Figure 2B). Similar results were obtained with the Renilla luciferase reporters. Again, the nonoptimal variant had a longer, though not significantly different, functional half-life than its optimized counterpart (32 ± 17 and 18 ± 6 h, respectively, p = 0.18; Figure 2C). As expected, there was also no difference in the half-lives of either co-transfection control (p = 0.16 and 0.23; Figures S2A and S2B, respectively).
Figure 2. Poor codon optimality does not affect protein stability.

(A) Cycloheximide (CHX) treatment reduces translation. HEK293T cells were treated with CHX or DMSO for 0, 6, and 12 h, and then puromycin was added 10 min before harvesting. Its incorporation was determined by western blotting.
(B) Codon optimality does not affect the stability of firefly luciferase protein. Cells were transfected with the SM-tagged firefly luciferase reporters and harvested at various time points after CHX addition. The amount of firefly luciferase was determined by luciferase assay and normalized to time = 0 h. Mean values ± SD are shown; p = 0.35, n = 3.
(C) As in (B) except for SM-tagged Renilla luciferase reporters; p = 0.18, n = 3.
(D) Codon optimality does not affect the proteasome-mediated degradation of firefly luciferase. After cells were transfected with SM-tagged firefly luciferase reporters, they were treated with MG132 (10 μM) and harvested at various time points before western blotting. An asterisk (*) refers to a cleavage product within the SM tag.
(E) As in (D) except for SM-tagged Renilla luciferase reporters. An asterisk (*) refers to a cleavage product within the SM tag.
(F) As in (D) except for γHA-GFP reporters.
To test whether optimality changed the likelihood of proteasome-mediated degradation, we treated the cells with the proteasome inhibitor MG132. Stabilized ubiquitinated proteins confirmed that MG132 treatment effectively inhibited proteasome activity (Figures 2D-2F). However, there was no detectable stabilization of the full-length or truncated protein products for either the optimal or the nonoptimal luciferase reporters, a result consistent with the relatively long half-lives of these reporters (Figures 2D and 2E) (note that the lower band results from a cleavage product within the SM tag [Figure S2C]). In fact, reporter band intensity was decreased upon MG132 treatment, perhaps because these proteins represented a smaller fraction of the now-stabilized total protein pool. Furthermore, no differential stabilization was seen when we examined the γHA-GFP optimality reporters (Figure 2F). Taken together, these data indicate that differences in protein stability cannot explain the differences in protein levels caused by codon usage.
Transcripts with nonoptimal codons are bound by fewer ribosomes
Because differences in protein stability could not explain how nonoptimal codons affected protein abundance, we turned to investigate potential translational regulation. We first measured the impact of codon usage on translation in bulk using polysome gradients. To minimize technical variation between the reporters, we co-transfected U2OS cells with the SM-tagged firefly luciferase nonoptimal and optimal reporters and separated ribosome fractions on a 10%–60% sucrose gradient (Figures 3A and 3B). Compared with the optimal reporter mRNA, a significantly higher proportion of nonoptimal reporter mRNA was present in the monosome fraction (1.38− ± 0.08-fold, p = 0.01; Figures 3B and 3C). These results suggest that the nonoptimal transcript may be associated with fewer ribosomes than its optimal counterpart.
Figure 3. Nonoptimal codons reduce ribosome association.

(A) Representative polysome fractionation. U2OS cells were transfected with both the nonoptimal and optimal SM-tagged firefly luciferase reporters before sucrose fractionation. Shown is an absorbance curve from a representative gradient.
(B) Nonoptimal codon usage affects ribosome association. Fractions from each gradient were pooled based on the number of ribosomes, and the amount of each reporter was quantified with RT-qPCR. Plotted is the percentage of each reporter per fraction for a representative biological replicate.
(C) Nonoptimal codon usage increases association with the 80S fraction. Plotted is the abundance of the nonoptimal firefly reporter mRNA in the 80S fraction relative to the optimal; n = 3.
(D) Schematic of the nascent chain tracking system.
(E) mRNA and protein signals colocalize. Fluorescent images of representative cells expressing optimal (left) and nonoptimal (right) firefly luciferase reporters. The white boxes highlight a single punctum with grayscale insets, showing both mRNA (MS2-coat proteins) and nascent chain (anti-FLAG Fab) signals. Scale bar, 5 μm.
(F) Codon usage does not affect the percentage of translating mRNAs. Shown is a box-and-whisker plot for the percent of reporter mRNAs translating per cell. Each point refers to a single cell. p value was determined by Mann-Whitney U test. Mean values ±SD are shown. N = 23 and 21 for optimal and nonoptimal reporters, respectively.
(G) Nonoptimal codons reduce the number of ribosomes bound to each translating mRNA. Plotted is a box-and-whisker plot for the number of nascent chains associated with each reporter mRNA. Each point refers to a single reporter molecule. p value was determined by Mann-Whitney U test. Mean values ± SD are shown. N = 347 and 190 for optimal and nonoptimal reporters, respectively.
This result is consistent with many observations in the field33,62,70,72-74,80 but is contrary to the a priori expectation of elongation dynamics. That is, this situation is analogous to the increased traffic when a highway lane is closed: with nonoptimal codons slowing elongating ribosomes,8,15,17,21,23 there should be more ribosomes on the nonoptimal reporter, not fewer.
To explore this finding, we turned to nascent chain tracking, where active translation on single transcripts can be visualized in living cells.81-85 In this system, individual reporter transcripts are visualized through 24× MS2 stem loops in their 3′ UTR, bound by Halo-tagged MS2 coat proteins (Figure 3D). At the same time, the large N-terminal SM tag contains 10× FLAG epitopes that are bound co-translationally by fluorescently conjugated α-FLAG Fabs. These reporters are the same as those used throughout this study (Figure 1A, top left). We first confirmed that the translation of both firefly luciferase reporters could be visualized in U2OS cells by bead loading the plasmid DNA of each optimality reporter along with purified α-FLAG Cy3-Fab and HaloTag-MCP probes. Reassuringly, for both reporters, we could detect untranslated mRNAs (labeled with the HaloTag alone) and mRNAs undergoing active translation (colabeled with both HaloTag and anti-FLAG Cy3 signals; Figure 3E). Puromycin treatment rapidly dissociated the anti-FLAG Cy3 signal from each mRNA punctum, confirming co-labeled spots as sites of active translation (Videos S1 and S2). Thus, the translation of both firefly luciferase reporter transcripts could be visualized in living cells at single-molecule resolution.
We next examined the impact of codon optimality on translation (Videos S3 and S4) and envisioned two, nonmutually exclusive possibilities explaining the impact of nonoptimal codons. First, nonoptimal codon usage might prevent translation initiation altogether, reducing the likelihood of translation occurring per mRNA. Thus, we measured the percentage of individual reporters in each cell, at steady state, that co-localized with the nascent chain signal and ultimately observed no difference in the fraction of mRNAs translated per cell (optimal reporter: 28.5% ± 12.9%; nonoptimal reporter: 23.8% ± 16.3%; p = 0.4; Figure 3F). These data suggest that codon usage does not change the fraction of mRNAs translated.
The second possibility is that the magnitude of translation itself is dampened on the nonoptimal mRNA (e.g., by reducing the number of initiating ribosomes per translation burst). We measured the intensity of the α-FLAG signal associated with each translating reporter (Figure 3G), which corresponds to the number of nascent peptides actively synthesized on each mRNA molecule. By normalizing intensity values with FLAG-tagged β-actin constructs,77 we found significantly reduced intensity (and thus the number of ribosomes translating) on the nonoptimal reporter (p < 10−5; Figure 3G). On the optimal reporter, the intensity corresponded to a median of 7.4 nascent chains, while the same measurements on the nonoptimal reporter showed a median of 5.4 nascent chains. Therefore, single-molecule imaging data support our previous polysome profiling results: nonoptimal codon usage reduces the number of translating ribosomes on an mRNA.
Poor codon usage does not cause incomplete translation nor does it trigger RQC
One possibility that could explain the reduced number of translating ribosomes on nonoptimal transcripts is that poor codon usage leads to ribosome drop-off. For instance, increased drop-off could occur through frameshifting, which has been proposed to take place on stretches of nonoptimal codons and to cause subsequent termination at out-of-frame stop codons.86 It could also occur through a mechanism analogous to what happens on RQC substrates, where coding regions stall the ribosome, cause collisions, and ultimately result in ribosome splitting and destruction of the nascent peptide.39-41,44-47 In theory, nonoptimal codons could act as an RQC substrate by slowing ribosome progression enough to cause stalls and trigger surveillance mechanisms.10,49,50 Two major signatures of these mechanisms would be (1) proteasomal degradation of truncated nascent peptides and (2) ribosome drop-off.
We first tested the extent to which nonoptimal codon usage leads to the production of truncated protein products. Because such products would likely be unstable, we transfected cells with either optimal or nonoptimal versions of each reporter (SM-tagged firefly luciferase, SM-tagged Renilla luciferase, and γHA-GFP; Figure 1A) and then treated the cells with MG132 to inhibit the proteasome. However, even when western blots were overexposed, there was no evidence of small fragments that accumulated explicitly with the nonoptimal reporters (Figures 4A, S3A, and S3B); therefore, truncated protein products are not produced in response to nonoptimal codon usage.
Figure 4. Nonoptimal codons do not lead to incomplete translation or RQC.

(A) Nonoptimal codons do not lead to truncated protein products. HEK239T cells were transfected with lHA-GFP optimality reporters and treated for 6 h with MG132 (10 μM) or DMSO before western blotting and probing for each reporter (α-GFP) and ubiquitin. Four times more nonoptimal γHA-GFP lysate was loaded, and the blot was overexposed.
(B) Schematic of the dual-tagged reporters used for the Ribo-seq experiment.
(C) Nonoptimal codons repress protein expression for the dual-tagged firefly luciferase reporters. HEK293T cells were transfected with the dual-tagged reporters alongside Renilla luciferase, and western blotting determined protein abundance (N-terminal SM tag [α-FLAG]), the C-terminal tag (α-GFP), Renilla luciferase, and hGAPDH. Lysates containing the optimized Ribo-seq reporter were serially diluted, and twice as much nonoptimal Ribo-seq reporter lysate was loaded.
(D) The decrease in FLAG and GFP signals from the nonoptimal dual-tagged Ribo-seq optimality reporter, compared to the optimal, is consistent. Scatterplot is a quantification of n = 3, one of which is shown in (C); p = 0.86.
(E) Nonoptimal codons do not lead to ribosome drop-off. Shown is the ratio of ribosome-protected fragments mapping to the C-terminal GFP tag relative to the N-terminal SM tag for both. The first 25 codons of the SM tag and the last 25 codons of the GFP tag were excluded from this analysis (n = 3).
Next, we looked directly at ribosome drop-off using ribosome profiling.87 We modified our SM-tagged firefly luciferase reporters such that they were followed by an invariant C-terminal optimized GFP (Figure 4B). Importantly, both reporters are identical except for the synonymous codon usage of the internal firefly luciferase region. Consistent with the previous single-tagged reporters (Figure 1), nonoptimal codon usage reduced mRNA abundance (2.6− ± 0.6-fold, p = 0.009; Figure S4A) and reduced protein expression even further (5.7− ± 0.8-fold, p = 0.0004), as determined by both western blotting and dual-luciferase assays (Figures 4C and S4A). Western blot immunofluo-rescent intensity (Figure 4C) from the SM (FLAG) tag compared to the downstream GFP tag was proportional, regardless of firefly luciferase optimality (p = 0.86; Figure 4D). Next, we performed RNA sequencing (RNA-seq) and Ribo-seq on cells transfected with these reporters in biological triplicate. As expected, RNA-seq measurements showed a decrease in nonoptimal mRNA abundance (3.1− ± 0.5-fold, p = 0.002; Figure S4B), indistinguishable from RT-qPCR measurements (p = 0.19; Figure S4B). To directly investigate drop-off, we determined the ratio of ribosome-associated reads mapping to the tags that preceded or followed the firefly luciferase coding region (SM and GFP, respectively). However, there was no difference in this ratio between the two reporters (p = 0.84; Figure 4E). Together, these results suggest that elongation through long stretches of nonoptimal codons does not result in ribosome drop-off.
Though ribosomes seem to not terminate prematurely due to the nonoptimal sequence, it was still formally possible that collisions were occurring, thereby initiating the RQC pathway. Although there are many known effectors of the RQC pathway,44-47 we chose to focus on ZNF598, a key protein that recognizes collided ribosomes.42,43 When we compared the repression of nonoptimal reporters in ZNF598 knockout cells with the parental HEK293T cell line,39 there was no difference in mRNA levels (p = 0.58; Figure S3C), protein activity (p = 0.81; Figure S3D), or in overall translational repression (p = 0.40; Figure S3E), indicating that ZNF598 is not required to repress translation due to nonoptimal codons. Taken together, these results are consistent with recent reports indicating a bifurcation in repression mediated by slow versus stalled ribosomes10,49,50 and suggest that the slow elongation on nonoptimal codons is not pronounced enough to cause the prolonged ribosome stalling and collisions needed for recognition by ZNF598.42,43
Poor codon usage inhibits translation initiation through reduced eIF4E and eIF4G1 binding
Another explanation for the reduced ribosome number on our nonoptimal reporters is repressed translation initiation. Reasoning that a decrease in translation initiation would lead to a reduction in translation before the nonoptimal region, we determined the translational efficiency of the upstream SM tag in our Ribo-seq datasets (Figures 4B and 5A). As a control, the translational efficiency of the co-transfected Renilla luciferase did not significantly differ between the conditions (p = 0.50; Figure S5A). In contrast, the translational efficiency of the upstream SM tag was reduced considerably in the context of the nonoptimal reporter relative to the optimal one (p = 0.02; Figure 5A). This effect continued further into the reporter, as there was reduced translational efficiency on the nonoptimal firefly luciferase that approached statistical significance (p = 0.06; Figure S5B, left), but this was no longer seen on the C-terminal GFP tag (p = 0.62; Figure S5B, right) for unclear reasons. Importantly, because the ribosome encounters the SM tag before the nonoptimal coding region, our results are consistent with a model of reduced translation initiation on the nonoptimal reporter.
Figure 5. Nonoptimal codons inhibit translation initiation through reduced eIF4E and eIF4G1 binding.

(A) Nonoptimal codons reduce ribosome association upstream. Shown is the normalized translational efficiency for the N-terminal SM tag, which is upstream of the optimal or nonoptimal firefly luciferase ORF, in the dual-tagged reporters (n = 3).
(B) ISRIB does not rescue the translational efficiency of the nonoptimal reporter. HEK293T cells were transfected with plasmids expressing each reporter alongside optimized Renilla luciferase. Shown are normalized protein-per-mRNA abundance, mRNA abundance (RT-qPCR), and protein activity (dual-luciferase assay); n = 3.
(C) ISRIB does not rescue the translational efficiency throughout the human transcriptome in an optimality-dependent manner. Meta-analysis was performed from published Ribo-seq data. Shown is the change in translational efficiency of the HEK293T transcriptome when unstressed cells were treated with ISRIB, plotted against their tAI optimality score (Rep “A”).
(D) ISRIB does not rescue the number of ribosomes translating nonoptimal SM-tagged luciferase reporter transcripts. Plotted is the average change in translational intensity on individual reporter transcripts, tracked over time in U2OS cells, following DMSO or ISRIB treatment. For the nonoptimal reporter, 17 cells were imaged (338 total translation spots). For the optimal reporter, 13 cells were imaged (242 total translation spots). Mean values ± SD are shown.
(E) Nonoptimal codons reduce eIF4E binding. HEK293T cells were transfected with either optimal or nonoptimal SM-tagged firefly luciferase reporters, along with Renilla luciferase, and then eIF4E RNA immunoprecipitations (RIPs) were performed (n = 3).
(F) Nonoptimal codons reduce eIF4G1 binding. As in (E) except for eIF4G1 RIPs (n = 3).
(G) Schematic of reporter constructs with Renilla luciferase being expressed by an EMCV IRES.
(H) eIF4E is required to reduce translation initiation on nonoptimal transcripts. HEK293T cells were transfected with plasmids expressing each reporter, along with γHA-GFP as a transfection control. mRNA abundance was quantified by RT-qPCR, with values normalized to GFP. Protein activity was quantified by dual-luciferase activity. Shown are the fold changes in the nonoptimal reporter relative to the optimal: (1) (left) mRNA abundance, (2) (middle) firefly luciferase activity, and firefly luciferase activity relative to mRNA abundance, and (3) (right) Renilla luciferase activity, and Renilla luciferase activity relative to mRNA abundance; n = 3.
Previous reports in Neurospora indicated that in response to slow decoding, GCN2 phosphorylates eIF2α (EIF2AK1) and represses translation initiation,75 and so we asked whether this pathway might also play a role in the mammalian response. We made use of the small molecule ISRIB, which blocks the impact of EIF2AK1 phosphorylation, thereby increasing the available ternary complex and promoting translation initiation.88-91 The response of codon-mediated translational repression was tested in three ways. First, cells were transfected with the nonoptimal and optimal firefly luciferase reporters and treated with ISRIB or DMSO. ISRIB was active throughout the experiment, as it rescued overall translation after treatment with the oxidative stressor sodium arsenite (Figure S6A). Unlike the situation in Neurospora,75 in human cells, this treatment did not rescue protein levels, as measured by luciferase activity (p = 0.09; Figure 5B). Surprisingly, though, we saw a small but significant increase in repression of the optimal firefly luciferase reporter (p = 0.05; Figure S6B), which can be mainly attributed to increased optimal reporter mRNA abundance (Figures S6C and S6D). Second, we analyzed previously published Ribo-seq datasets,90 examining the impact of ISRIB on translation, and found no correlation between the effect of the small molecule on translational efficiency and optimality on a transcriptome-wide scale, regardless of the optimality metric (Figures 5C and S6E). Third, we examined the effect of ISRIB on individual transcripts in live cells using NCT. After validating that ISRIB rapidly dissolved stress granules in U2OS cells (Figure S6F; Video S5), we measured how the translational signal changed on individual reporter mRNAs, tracking translational intensity changes over time when treated with ISRIB or DMSO. Consistent with bulk measurements, ISRIB treatment did not increase the number of ribosomes associated with individual transcripts, regardless of optimality (Figures 5D, S6G, and S6H). These data demonstrate that, in humans, EIF2AK1 phosphorylation does not mediate translational repression in response to nonoptimal codons.
Next, we turned our investigation to two other core eukaryotic translation initiation factors: eIF4E, the cap-binding protein, and eIF4G1. These proteins are essential for cap-dependent translation,92-94 and reduced association of these key proteins has been implicated in translational repression in other contexts, such as microRNA (miRNA)-mediated repression.95-97 We immunoprecipitated eIF4E or eIF4G1 from cells transfected with either SM-tagged firefly luciferase optimality reporter along with a Renilla luciferase transfection control. We confirmed the pull-down using western blotting (Figures S5C and S5D) and used RT-qPCR to determine the extent to which each optimality reporter was associated with each initiation factor. We normalized each firefly luciferase reporter to the Renilla luciferase to control for technical differences in transfection and pull-down efficiency and then calculated the fraction of each reporter mRNA immunoprecipitated relative to input. Consistent with nonoptimal codons repressing translation initiation, the fraction of immunoprecipitated nonoptimal reporter mRNA was significantly reduced for both eIF4E and eIF4G1 (p = 0.03 and 0.01, respectively; Figures 5E and 5F).
To test whether reduced occupancy of eIF4E was required for reduced initiation, we modified our reporters to express a downstream Renilla luciferase driven by an EMCV IRES, or the internal ribosome entry site from the encephalomyocarditis virus (Figure 5G). Translation from this IRES is cap independent and does not require eIF4E but does use other downstream initiation factors like eIF3.98-100 γHA-tagged GFP was used as a transfection control. As expected, mRNA levels were significantly reduced for the nonoptimal reporter relative to the optimal one (p = 0.05; Figure 5H, left). Similarly, overall firefly luciferase activity was repressed with the nonoptimal reporter, and its expression was still reduced when the changes at the mRNA level were considered (p = 0.004; Figure 5H, middle). However, a different picture emerged when we analyzed Renilla luciferase activity (Figure 5H, right). Although overall activity was diminished, this reduction was entirely explained by the decreased mRNA abundance of the nonoptimal reporter. In other words, we observed no additional translational repression for the IRES-driven Renilla luciferase reporter (p = 0.34). Thus, these data indicate that nonoptimal codon usage limits translation by reducing eIF4E and eIF4G1 binding, and so the optimality-dependent inhibition of translation initiation is restricted to mRNAs that depend on eIF4E for ribosome loading.
Codon-mediated translational repression is independent of mRNA deadenylation and decay
To further explore how the translation of nonoptimal codons results in repression, we examined whether DDX6 played a role in repression. In yeast, this protein (Dhh1p) is preferentially associated with translating nonoptimal transcripts and is required for their targeted decay.53,54 DDX6 and its orthologs mediate translational repression in response to many post-transcriptional regulatory pathways and often link the CCR4-NOT deadenylase complex and decapping machinery.55-57 We depleted endogenously tagged DDX6 using the eDHFR degron system, where protein degradation is stimulated by the removal of trimethoprim (TMP) (Figure S7A).101 Due to reduced expression of HA-eDHFR-DDX6 even in the presence of TMP (Figure S7A; see α-hGAPDH immunoblot), the degron-tagged DDX6 serendipitously gave us a chance to see how codon-mediated regulation responded to different amounts of DDX6. However, regardless of DDX6 levels, the extent of mRNA destabilization or translational repression did not change (Figures S7B-S7D). As an orthogonal approach, we also performed an RNA immunoprecipitation against DDX6 and found no difference in its relative binding to either reporter (p = 0.77; Figure S7E). Thus, unlike in yeast, DDX6 does not significantly respond to codon optimality in human cells.
CNOT3 (Not5p in yeast) is a direct sensor of slow decoding and destabilizes nonoptimal transcripts by promoting deadenylation.48,51 Given the known links between translation and deadenylation, we next explored the role of CNOT3 as an upstream effector of mRNA decay and translational repression in human cells. We depleted CNOT3 using CRISPR-Cas9 with two independent guides targeting a ubiquitously expressed exon of CNOT3 and confirmed that the protein was efficiently depleted by western blotting (Figure 6A). (Note that because CNOT3 is an essential gene,102 we performed these experiments on pooled cells immediately following selection.) Consistent with previous studies,48,51 when CNOT3 was depleted, nonoptimal codons mediated less impact at the mRNA level (p = 0.0003 and 0.05 for guides #1 and #2, respectively; Figure 6B). However, we were surprised that relative protein activity was unchanged (p = 0.17 and 0.77; Figure 6C). In other words, there is more translational repression on the nonoptimal reporter without CNOT3-mediated destabilization (p = 0.006 and p < 10−3; Figure 6D).
Figure 6. Translation repression does not require mRNA decay or deadenylation.

(A) CRISPR guides can effectively knock down CNOT3. HEK293T lysates were collected 4 days following blasticidin selection and probed for CNOT3 (105 kDa expected; *, nonspecific band) and tubulin.
(B) Upon CNOT3 knockdown, nonoptimal mRNA levels are rescued relative to optimal. Following 4 days of blasticidin selection, cells were transfected with SM-firefly luciferase reporters and optimized Renilla luciferase. RT-qPCR quantified mRNA abundance. Box-and-whisker plots show relative fold change in transcript (guide #1: n = 7; guide #2: n = 5).
(C) Upon CNOT3 knockdown, nonoptimal protein activity is unchanged. Like in (B) but measured relative protein levels by dual-luciferase assay (guide #1: n = 7; guide #2: n = 5).
(D) Upon CNOT3 knockdown, there is more translational repression of the nonoptimal reporter. Values plotted are extrapolated from (B) and (C), calculating protein activity relative to transcript abundance.
(E) Neither deadenylation nor a poly(A) tail is required for translational repression. HEK293T cells were transfected with plasmids expressing optimality reporters, which, after processing, ended in a poly(A) tail or Malat1 triple helix. Shown is a dot plot depicting normalized protein-per-mRNA abundance (dual-luciferase assay and RT-qPCR; n = 3).
(F) Without a poly(A) tail, nonoptimal codons further reduce eIF4E binding. HEK293T cells were transfected with plasmids as in (E), and eIF4E RIPs were performed; n = 3.
Alternatively, we asked if the poly(A) tail (and thus deadenylation) is required to mediate translational repression in response to nonoptimal codons.103-107 We replaced the poly(A) signal in our firefly reporters with a Malat1 triple helix, which, after processing, results in a stable 3′ end lacking a poly(A) tail (Figure S7F).108,109 Consistent with our results with CNOT3 depletion, the Malat1 triple helix dramatically stabilized the nonoptimal reporter (Figure S7G), providing strong evidence that deadenylation is required for the mRNA decay of nonoptimal transcripts in humans. However, despite rescued transcript levels, there was a dramatic ~9.1-fold decrease in nonoptimal firefly protein levels, significantly more than the ~2.2-fold reduction from the “standard” poly(A)-tailed optimality reporters (p = 0.004; Figure 6E). These data demonstrate that translational repression mediated by codon optimality does not require deadenylation or mRNA decay.
Finally, considering the increased translational repression of the Malat1-ending reporters, we asked whether eIF4E binding was also reduced in that context. Indeed, eIF4E binding was decreased on the nonoptimal Malat1 reporter, an effect far greater than we observed with the poly(A)-tailed counterparts (p = 0.002; Figure 6F). These data also suggest that mRNA decapping is not a prerequisite for diminished eIF4E occupancy, as the Malat1-ending reporters are stable. Thus, nonoptimal codons lead to reduced eIF4E binding by a mechanism distinct from the impact on mRNA stability, and our results also show that translational repression is enhanced when mRNA decay is impaired.
DISCUSSION
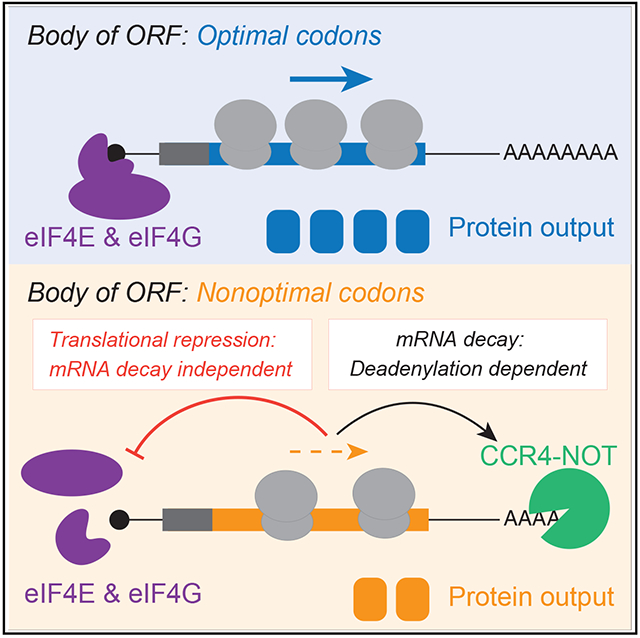
For over half a century, it has been known that synonymous codon usage regulates gene expression.7,11,110,111 This observation has provided a rationale for codon-optimizing transcripts in heterologous expression systems.12-14 However, the mechanisms by which synonymous codons modulate gene expression are still not fully understood. In addition to potentially affecting transcription and protein folding, we and others have shown that by slowing decoding rates of the elongating ribosome, poor codon usage leads to decreased mRNA stability; this mechanism appears to be deeply conserved and has been observed in bacteria and a wide variety of eukaryotic species.60-72 However, many studies suggest that mRNA abundance changes may be insufficient to explain the effects of nonoptimal codons.33,60,62,72-74,80,112,113 Here, we show that nonoptimal codons in the body of the ORF reduce ribosome association by repressing translation initiation in human cells (Figure 7). This observation is distinct from others’ that have linked nonoptimal codons at the beginning of the coding sequence with reduced initiation.37,38 Indeed, we identify decreased association of eIF4E and eIF4G1 as the proximal cause, and this is not dependent on phosphorylated EIF2AK1 like in Neurospora.75 These results are consistent with a recent study where dominant-negative mutations in the Gly-tRNA synthetase lead to global repression of translation, also independent of EIF2AK1 phosphorylation.113 Finally, despite the interconnected nature of mRNA degradation and translation, neither mRNA decay nor deadenylation is responsible for translational repression. Thus, our results reveal an additional, potent mechanism by which codon usage regulates gene expression.
Figure 7. Model: Nonoptimal codons repress gene expression by two post-transcriptional mechanisms.

Nonoptimal codons slow translation elongation and can lead to a ribosome state with empty A and E sites. CNOT3 can sense this state and thus recruits the CCR4-NOT deadenylase complex to trigger mRNA destabilization. Slow elongation also reduces eIF4E and eIF4G1 binding, thereby repressing translation initiation. It is unclear what ribosome state is sensed—and how this information is transmitted—to suppress translation. Nevertheless, neither mRNA deadenylation nor decay is required for translational repression, and in fact, they seem to limit the extent to which translation is repressed.
Our results resolve a contradiction in the field. Studies conducted in various model systems (including yeast, zebrafish, and human cells) have observed that nonoptimal codon usage decreases ribosome association.33,62,73,74,113 However, poor codon usage slows translation elongation, so the a priori expectation is that poor codon usage should increase, not decrease, ribosome association. This situation is analogous to a lane closure where cars are forced to slow down, leading to more, not less, cars on the highway. Our finding that nonoptimal codons repress translation initiation reconciles this apparent contradiction where slowdowns may lead to a response analogous to “on-ramp metering” that many motorists experience during rush hour.
We also investigated potential effectors of codon-mediated translational repression. Not5p (CNOT3) is a known sensor of slow decoding in yeast, recruiting the deadenylase complex to the transcript and expediting decay.48,49,51As an optimality sensor, we asked what effect(s) CNOT3 has in human cells. While CNOT3 regulates mRNA levels in response to nonoptimal codons, presumably in a similar way to budding yeast, its loss increased translational repression. In other words, CNOT3 is required for mRNA destabilization in response to nonoptimal codons but not for translational repression. In support of a model of bifurcating codon-mediated regulation, replacing the poly(A) tail on our reporters with the Malat1 triple helix (which is thus unable to be deadenylated) revealed a similar separation of mRNA decay and translational repression. In yeast, Dhh1p (DDX6 in humans) interacts with ribosomes translating nonoptimal sequences53,54 and has well-defined roles repressing translation in other contexts, such as miRNA-mediated silencing.3,55-59 Nevertheless, DDX6 did not impact codon-mediated regulation in humans. Therefore, no known sensor of poor codon usage appears to be a driver of the translational repression we observe.
Another possibility was that slowed ribosome progression might result in stalls and collisions.40-42,45,114 However, we found no evidence of this pathway acting in response to nonoptimal codons because the absence of ZNF598 does not change the relative mRNA or protein abundance from optimality reporters. The RQC pathway would also increase the likelihood of aberrant protein accumulation from nonoptimal transcripts while decreasing the probability that elongating ribosomes could associate with, and successfully translate, a gene immediately following a codon-poor sequence. Again, however, there was no evidence of these phenomena. Our results are consistent with other studies showing a distinction between no-go decay and codon-optimality-mediated decay, likely due to different ribosome elongation kinetics for slow decoding compared to persistent stalls.10,48-50
These data prompt several additional questions. What is the sensor of nonoptimal codons that results in translational repression, and what does it sense? In the case of CNOT3 and mRNA destabilization, the ribosome’s empty E and A sites are sensed, leading to eventual deadenylation.48 Given that eIF5A also interacts in the E site, there may be other, unknown factors that monitor this site.115,116 Additionally, what about other ways ribosomes are forced to slow, such as motifs that hinder peptide-bond formation or mRNA secondary structures that slow translocation?25,116-120 Is translation initiation also repressed in these instances, and if so, is the mechanism similar?
Our findings have revealed a potent mechanism by which nonoptimal codon usage represses gene expression: reduced translation initiation. This effect impacts gene expression as much, if not more, than the effect of nonoptimal codons on mRNA levels. The recent reliance on mRNA levels to investigate the impact of codon optimality on gene expression may explain why this effect has only been fully recognized now. Our findings underscore an emerging theme that elongation and initiation machinery tag team to regulate gene expression and raise important mechanistic questions about their cooperation for future studies.
Limitations of the study
Although we investigated several reporters in our study, one limitation is that we did not show how nonoptimal codons affect translational efficiency transcriptome-wide. Of course, to do so on a transcriptome-wide scale is inherently fraught with caveats because of the co-evolution of UTRs and coding regions.121,122 However, the discrepancy between ribosome association and slow elongation has been noted in endogenous genes in humans and other model organisms,62-72 strongly suggesting that this phenomenon is not unique to our constructs.
Additionally, though our data and reports from others indicate that nonoptimal codons do not act through ZNF598 or persistent collisions, we have not investigated other effectors of the RQC pathway, nor the presence of transient disomes or trisomes.44,46,123,124 Similarly, we still need to understand how nonoptimal codons reduce eIF4E association. Indeed, one formal possibility is that decapped intermediates accumulate for the nonoptimal reporter; because such RNAs would be unable to bind eIF4E, they would be unable to be translated. Given the known instability of such RNAs,125 this is unlikely but cannot be entirely excluded. Our data also suggest that eIF4E is required for the translational response to nonoptimal codons, but more research is needed to define the role of eIF4E. For instance, our nascent chain tracking results suggest that this mechanism involves fewer ribosomes being loaded per translational burst rather than an increase in a “translational off” state, in contrast to other mechanisms, such as that mediated by Ago2.126,127 Resolving these issues will be a critical next step.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Olivia Rissland (olivia.rissland@gmail.com).
Materials availability
Plasmids generated in this study are deposited on Addgene.
Data and code availability
Ribo-seq and RNA-seq data have been deposited at GEO: GSE202900, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The raw image data have been deposited at Mendeley Data (https://data.mendeley.com/datasets/tw2bjh857f/2) and are publicly available as of the date of publication. DOIs are also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-eIF4E | MBL Lifescience | Cat# RN001P, lot 004; RRID:AB_1570634 |
| Rabbit polyclonal anti-eIF4G1 | MBL Lifescience | Cat# RN002P, lot 005; RRID:AB_1570635 |
| Rabbit polyclonal anti-vinculin | Cell Signaling Technology | Cat# 4650, lot 5; RRID:AB_10559207 |
| Rabbit polyclonal anti-DDX6 | Abcam | Cat# ab151984, lot GR174405-10; RRID:AB_3073919 |
| Rabbit polyclonal anti-CNOT3 | Bethyl Laboratories | Cat# A302-156A; RRID:AB_1720348 |
| Mouse monoclonal anti-FLAG | Millipore Sigma | Cat# F1804, lot SLCK5688; RRID:AB_262044 |
| Mouse monoclonal anti-FLAG (for Flag fabs, imaging) | FUJIFILM Wako Shibayagi | Cat# 012-22384, clone 1E6; RRID:AB_10659717 |
| Mouse monoclonal anti-GFP | Roche | Cat# 11814460001, lot 42903200; RRID:AB_390913 |
| Rabbit polyclonal anti-Renilla luciferase | Abcam | Cat# ab187338, lot GR3382572-1; RRID: AB_3073920 |
| Mouse monoclonal anti-hGAPDH | Proteintech | Cat# 60004-1-Ig, lot 10020246; RRID:AB_2107436 |
| Mouse monoclonal anti-Ubiquitin | Thermo Fisher Scientific | Cat# 14-6078-82, lot 4342660; RRID:AB_837154 |
| Rabbit polyclonal anti-Firefly Luciferase | GeneTex | Cat# GTX125849, lot 42837; RRID:AB_11173184 |
| Mouse monoclonal anti-HA tag | Thermo Fisher Scientific | Cat# 26183, lot WA317137; RRID:AB_10978021 |
| Mouse monoclonal anti-Puromycin | Kerafast | Cat# EQ0001, lot 041918; RRID:AB_2620162 |
| Normal Rabbit Control IgG | SinoBiological | Cat# CR1; RRID: AB_3073921 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074S; RRID:AB_2099233 |
| Mouse monoclonal [SB62a] Anti-Rabbit IgG light chain (HRP) | Abcam | Cat# ab99697, lot GR254788-1; RRID:AB_10673897 |
| Rat monoclonal [187.1] Anti-Mouse kappa light chain (HRP) | Abcam | Cat# ab99617, lot GR256873-1; RRID:AB_10673885 |
| Bacterial and virus strains | ||
| One Shot™ TOP10 Chemically Competent E. coli | Invitrogen | C404010 |
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide | Millipore | C4859 |
| Salmon-sperm DNA | Sigma | D7656 |
| Puromycin | Gibco | A1113803 |
| ISRIB | Sigma Aldrich | SML0843 |
| Sodium (meta)arsenite | Sigma Aldrich | S7400 |
| Trimethoprim (TMP) | Sigma Aldrich | T7883 |
| P-32 | Perkin Elmer | Phosphorus-32, as monopotassium phosphate |
| Lipofectamine transfection reagent | Invitrogen | 11668019 |
| Effectene transfection reagent | Qiagen | 301425 |
| HaloTag-MCP protein | Protein Expression and Purification facility, CSU; H. Scherman | N/A |
| JF646-HaloTag ligand | HHMI Janelia; L. Lavis | N/A |
| TURBO™ DNase | Invitrogen | AM2388 |
| SUPERase•In™ RNase Inhibitor | Invitrogen | AM2696 |
| Random Hexamers | Invitrogen | N8080127 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | 18090050 |
| Critical commercial assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | E1960 |
| TRIzol™ Reagent | Invitrogen | 15596–018 |
| TRIzol™ LS Reagent | Invitrogen | 10296–010 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | 1725125 |
| EZview Red Protein G Affinity Gel beads | Millipore | E3403 |
| Amersham Hybond P 0.45 PVDF blotting membrane | Cytiva Life Sciences | 10600029 |
| 0.45 μM cellulose acetate Spin-X filters | Corning | CLS8162 |
| Micro Bio-Spin™ P-30 Gel Columns, Tris Buffer (RNase-free) | BioRad | 7326250 |
| ULTRAhyb-oligo hybridization buffer | Invitrogen | AM8663 |
| NuPAGE™ MES SDS Running Buffer (20X) | Invitrogen | NP0002 |
| NuPAGE™ Transfer Buffer (20X) | Invitrogen | NP00061 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 4693159001 |
| Qubit™ RNA Quantification, broad range kit | Invitrogen | Q10210 |
| Amersham MicroSpin S-400 HR Columns | Cytiva Life Sciences | 27514001 |
| Direct-zol RNA Micro-Prep Kit | Zymo Research | R2060 |
| Human riboPOOL for Ribosome Profiling samples | Galen Molecular | dp-K012-000042 |
| QIAseq miRNA Library Kit | Qiagen | 331502 |
| QIAseq miRNA NGS 96 Index IL plate kit | Qiagen | 331565 |
| Qubit™ dsDNA Quantitation, high sensitivity kit | Invitrogen | Q32851 |
| Hybridizing rRNA complementary DNA oligo mix | Baldwin et al., 2021132 | N/A |
| RNA Clean XP beads | Beckman Coulter | A63987 |
| KAPA RNA Hyperprep library kit | Roche | KK8540 |
| KAPA Dual-Indexed Adapter Kit | Roche | 08278555702 |
| Tapestation D1000 HS Screentape | Agilent | 5067–5584 |
| Tapestation D1000 HS Screentape reagents | Agilent | 5067–5585 |
| Deposited data | ||
| Paired Ribo-seq and RNA-seq data | This study | GEO Database: GSE202900 |
| Mendeley image dataset | This study | Mendeley Data: https://data.mendeley.com/datasets/tw2bjh857f/2 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| Drosophila S2 | ThermoFisher | R69007 |
| ZNF598 KO Flp-In T-REx 293 dox inducible GFP-P2A-(KAAA)0-P2A-RFP | Juszkiewicz and Hegde, 201739 | N/A |
| ZNF598 KO Flp-In T-REx 293 dox inducible GFP-P2A-(KAAA)21-P2A-RFP | Juszkiewicz and Hegde, 201739 | N/A |
| eDHFR-degron tagged DDX6 HEK293T | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 for full list of RT-qPCR primers, CRISPR guides, and probes | N/A | N/A |
| DNA oligonucleotide pool complementary to human rRNA transcripts | Baldwin et al., 2021132 | N/A |
| Recombinant DNA | ||
| pUB_SMFLuc0—MS2 | This paper | Addgene #210568 |
| pUB_SMFLuc100—MS2 | This paper | Addgene #210570 |
| pIS1_Opt_Renilla | This paper | Addgene 210571 |
| Nonopt_WT_Renilla_pIS1 | pIS1 | Addgene #12179 |
| pUB_SM-RLuc0—MS2 | This paper | Addgene #210572 |
| pUB_SM-RLuc100—MS2 | This paper | Addgene # 210573 |
| pUB_FLuc100—MS2 | This paper | Addgene # 210574 |
| pcDNA3.1_γHA-GFPopt | This paper | Addgene # 210575 |
| pcDNA3.1_γHA-GFPnonopt | This paper | Addgene # 210576 |
| pUB_OptγHAramp-FLuc100 | This paper | Addgene #210577 |
| pUB_NonoptγHAramp-FLuc100 | This paper | Addgene # 210579 |
| pcDNA3.1_SM-FLuc0-malat1 | This paper | Addgene # 210580 |
| pcDNA3.1_SM-FLuc100-malat1 | This paper | Addgene # 210581 |
| pcDNA3.1_SM-FLuc0-pA | This paper | Addgene # 210583 |
| pcDNA3.1_SM-FLuc100-pA | This paper | Addgene # 210584 |
| pUB_SM-FLuc0_EMCV-Ren | This paper | Addgene # 210590 |
| pUB_SM-FLuc100_EMCV-Ren | This paper | Addgene # 210591 |
| Dm_γHA-FLuc0_CSC | This paper | Addgene # 210589 |
| Dm_γHA-FLuc100_CSC | This paper | Addgene #210592 |
| Dm_Opt_Renilla_CSC | This paper | Addgene # 210593 |
| pUB_SM-FLuc0-GFPOPT | This paper | Addgene # 210594 |
| pUB_SM-FLuc100-GFPOPT | This paper | Addgene #210596 |
| pUB_mCherry-Rluc100 | This paper | Addgene # 210600 |
| lentiCRISPR v2-blast | Mohan Babu; Addgene | Addgene #83480 |
| psPax2 | Mikko Taipale | N/A |
| pCMV-VSV-G | Mikko Taipale | N/A |
| Software and algorithms | ||
| UMI-tools | Smith et al., 2017133 | N/A |
| Bowtie version 1.2.2. | Langmead et al., 2009135 | N/A |
| SAMtools version 1.12 | Danecek et al., 2021136 | N/A |
| STAR version 2.6.0a | Dobin et al., 2013137 | N/A |
| BEDtools | Quinlan and Hall, 2010141 | N/A |
| Salmon version 1.6.0. | Patro et al., 2017138 | N/A |
| Cutadapt | Martin et al., 2011134 | N/A |
| deepTools (Bamcoverage) | Ramírez et al., 2014139 | N/A |
| ribosomeProfilingQC R package | Ou and Hoye, 2022140 | N/A |
| Fiji ImageJ | Schindelin et al., 2012130 | N/A |
| Adobe Photoshop | https://www.adobe.com/products/photoshop.html | N/A |
| Adobe Illustrator | https://www.adobe.com/products/illustrator.html | N/A |
| Other | ||
| 35 mm glass bottom MatTek dishes | MatTek Corporation | P35G-1.5-14-C |
| Open-Top Polyclear Centrifuge tubes | Seton Scientific | 7030 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T (ATCC) and U2OS (ATCC) cells were grown in DMEM with high glucose and pyruvate (Gibco), supplemented with 10% FBS (Gibco) in a humidified incubator maintained at 37°C with 5% CO2. Unless otherwise stated, cells were transfected with plasmids expressing reporters using Lipofectamine 2000 (Invitrogen) and harvested 24 h after transfection.
S2 cells (ThermoFisher) were maintained in serum-free ExpressFive media (Gibco) supplemented with 20 mM L-glutamine (Gibco) and kept in a humidified incubator at 28°C. S2 cells were transfected plasmids expressing reporters using Effectene reagent (Qiagen) and harvested 48 h after transfection.
CNOT3 KO cells were made with CRISPR guides (Table S1) integrated into lentiCRISPRv2-blast vector (Addgene: #83480). HEK293T cells were co-transfected with guides and plasmids expressing psPax2 and pCMV-VSV-G (Mikko Taipale) to package the lentivirus. Viral titers were then used to infect HEK293T cells. Media was changed the day after infection, then selection begin (5 μg/μL blasticidin) the following day. Since CNOT3 is an essential gene,102 we pooled the cells after selection. After four days of blasticidin treatment, CNOT3 was strongly knocked down.
The eDHFR-degron tagged DDX6 HEK293T cell line was made using eDHFR cassettes as described.101 The eDHFR cassette was PCR amplified with linear, double-stranded HDR donors, including overhangs containing homology to either side of the Cas9 cleavage site positioned close to the stop codon of endogenous DDX6. The cassette was designed to be in-frame with the rest of the gene (see Table S1). The HDR donor and pX330 plasmid was co-transfected 1:1; then puromycin and neomycin selection begin 72 h later. Single colonies were isolated, and PCR screening and western blotting against the endogenous protein were performed on clones to ensure eDHFR cassette incorporation into all copies of DDX6. The degron-DDX6 protein was stabilized with 100 μM antibiotic trimethoprim (TMP) (Sigma) and could be quickly depleted following TMP washout. (−) TMP cell samples were collected 24 h post-washout.
Flp-In 293 T-Rex cells were maintained in DMEM with 10% tetracycline-free FBS in the presence of 15 mg/mL blasticidin and 100 mg/mL hygromycin.
METHOD DETAILS
RT-qPCR
Cells were washed three times with cold PBS before adding 1 mL TRIzol Reagent (Invitrogen), and RNA was extracted according to the manufacturer’s instructions. 1 μg RNA was treated with TuRBO DNase (Invitrogen) and reverse transcribed with random hexamer priming (Invitrogen) and Superscript IV Reverse Transcriptase (Invitrogen). RT-qPCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) on a LightCycler 480 (Roche) machine. Cycling conditions were as follows: 15 min at 95°C, and then cycled 35x at 95°C for 15 s, 65°C for 30 s, and 72°C for 30 s. Signal acquisition was performed after each cycle. Samples were measured in technical triplicate, and at least three biological replicates were performed per experiment.
Dual luciferase assays
According to the manufacturer’s instructions, cells were lysed with 1x passive lysis buffer from the Dual-Luciferase Reporter Assay System kit (Promega). 20 μL lysate was added to a 96-well flat bottom plate (Greiner Bio-One) in technical triplicate, before performing the dual luciferase assay on a Glomax Navigator plate reader (Promega). Relative luciferase enzymatic activity was quantified by taking the ratio of luciferase values, and the geometric mean was calculated between technical triplicates.
Puromycin incorporation assay
Cells were treated with 80 μg/mL cycloheximide (Millipore) for a variable amount of time before replacing the media to contain 2 μg/mL puromycin for 10 min. Puromycin incorporation into nascent peptides was detected using western blotting (anti-Puromycin, Kerafast). Puromycin signal is a readout of active translation, while the lack of puromycin incorporation indicates translational shut-off.
RNA immunoprecipitation
EZview Red Protein G Affinity Gel beads (Millipore) were prepared by washing twice with lysis buffer A (100 mM KCl, 0.1 mM EDTA, 20 mM HEPES-KOH, pH 7.6, 0.4% NP-40, and 10% glycerol, along with freshly added SUPERase•In RNase Inhibitor [Invitrogen], 1 mM DTT, and cOmplete mini EDTA-free protease inhibitor cocktail tablets [Roche]). Beads were resuspended in lysis buffer A and blocked with salmon-sperm DNA (Sigma), rotating overnight at 4°C. The next day, cells were washed three times with cold PBS pH 7.2 (Gibco) and lysed in cold lysis buffer A, clarified by spinning 21,000 x g at 4°C for 5 min, and 50 μL was removed as input. The remaining lysate was incubated with the appropriate antibody for 1 h, rotating at 4°C. Blocked EZview Protein G beads (Millipore) were washed three times with lysis buffer A before the slurry was added to each IP. Lysates were further incubated for 1 h, rotating at 4°C. The beads were washed three times with lysis buffer A, transferred to new tubes after the first wash, and split for future analysis: TRIzol reagent (Invitrogen) was added to 90% and processed for RT-qPCR, while the remaining 10% was prepared for western blotting to confirm immunoprecipitation efficiency. Both eIF4E and eIF4G1 antibodies were purchased from MBL International (2.5 μL and 6 μL used per IP, respectfully), and normal rabbit polyclonal IgG control antibody was purchased from SinoBiological (7.5 μL per IP).
Western blotting
Lysates were prepared with NuPage 4x LDS Sample buffer (Invitrogen) and 10x Bolt Sample Reducing Agent (Invitrogen) and boiled at 95°C for 5 min. Samples were run on NuPAGE 4–12%, Bis-Tris, 1.0 mM, Mini Protean precast gels (Invitrogen) with 1x NuPAGE MES SDS Western Running Buffer (Invitrogen). The protein was transferred to an Amersham Hybond 0.4 PVDF blotting membrane (Cytiva Life Sciences), with NuPAGE Transfer Buffer (Invitrogen), according to the manufacturer’s instructions. The membrane was blocked with PBST (1x PBS and 0.1% Tween) with 5% nonfat powdered milk and rocked at room temperature for 30 min. Antibodies were diluted in PBST with 5% milk, and blots were incubated rocking overnight at 4°C. The following day, the membranes were washed four times with PBST and incubated with light-chain specific HRP-linked rabbit and mouse (Abcam) secondary antibodies (except for eIF4E, where HRP-linked rabbit [Cell Signaling] secondary was used). Membranes were washed four times with PBST. Protein signal was detected with ECL (Cytiva) and imaged on Sapphire Western blot imager (Azure). Western intensity was quantified using Fiji ImageJ.128 Western blotting was performed in biological triplicate.
Northern blotting
3.5 μg of purified total RNA per sample was loaded on a 6% denaturing polyacrylamide TBE gel, then transferred to a positively charged nylon membrane. The membrane was crosslinked before blocking with prewarmed ULTRAhyb-oligo hybridization buffer (Invitrogen) for 1 h with rotation at 42°C. 32P-radiolabeled probes (PerkinElmer) were added (previously filtered through Micro Bio-Spin P-30 Gel Columns, Tris buffer, RNase-free [BioRad]) and incubated overnight. The membrane was washed 4x with warm 2x SSC buffer containing 0.1% SDS at 42°C, before being exposed to a phosphor screen and imaged.
Sucrose gradients
U2OS cells were co-transfected with both SM-firefly luciferase optimality reporters. Just before harvesting, cycloheximide (CHX) was added to 100 μg/mL final concentration (Millipore), and the cells were incubated at 37°C for 10 min. All subsequent harvesting steps were done on ice with cold buffers. The cell monolayer was washed twice with PBS + CHX (100 μg/mL), and 500 μL polysome lysis buffer (10 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 100 mM KCl, and 1% Triton X-100, along with freshly added 2 mM DTT, 500 U/mL SUPERase•In RNase inhibitor [Invitrogen], 100 μg/mL cycloheximide, and cOmplete mini EDTA-free protease inhibitor cocktail tablets [Roche]), was added per 10-cm plate. Lysed cells were collected by scraping, sheared four times with a 26-gauge needle, and centrifuged at 1300 x g for 10 min at 4°C to clarify. Lysates were flash-frozen in liquid nitrogen and stored at −80°C.
10% and 60% sucrose gradient solutions were made with gradient buffer base (20 mM HEPES-KOH, pH 7.4, 5 mM MgCl2, and 100 mM KCl) and filter-sterilized using 0.22-μm PES filter (Millipore). Just prior to pouring, 2 mM DTT, 500 U/mLSUPERase•In RNase Inhibitor (Invitrogen), and 100 mg/mL cycloheximide was added. 10%–60% gradients were made using the Biocomp Gradient Station per pre-programmed instructions into Open-Top Polyclear centrifuge tubes (Seton Scientific) and stored at 4°C for at least an hour. Lysate was loaded onto the gradient and spun at 36,000 rpm for 2 h at 4°C with SW41 TI rotor. 60 fractions were collected per sample using Piston Gradient fractionator with Triax Full Spectrum Flow Cell. Those fractions were pooled based on ribosome density and brought to 750 μL with RNase-free water. RNA was isolated using phenol-chloroform extraction and precipitated at −80°C overnight with ethanol and sodium acetate. The extracted RNA was processed for RT-qPCR. The percentage of both optimality reporter transcripts in each fraction was calculated. Sucrose gradients were performed in biological triplicate.
Single-molecule imaging
U2OS cells were plated on 35 mm glass-bottom MatTek dishes at ~80% confluency in DMEM (Thermo Fisher Scientific) with 10% (v/v) FBS and 1mM L-Glutamine (DMEM+) the day prior to imaging. On the day of imaging, cells were bead loaded with the nascent chain tracking components: 1.5 μg reporter plasmid DNA (either spaghetti-monster-tagged firefly luciferase nonoptimal and optimal reporters, with 24x MS2 stem loops in the 3′UTR), 100 μg/mL Cy3-labeled α-Flag Fab, and 33 μg/L purified HaloTag-MCP protein. Cells were washed three times with phenol red free DMEM and the media was changed with phenol red free DMEM+ 1 h after bead-loading to remove beads. Cells were stained with 200 nM JF646-HaloTag ligand in one mL phenol red free DMEM+ 30 min prior to imaging. After ligand incubation, cells were washed three times with phenol red free DMEM and the media was replaced with two mL phenol red free DMEM+. Imaging was done 5–7 h after bead-loading.
Images were taken on a custom-built microscope.81 Briefly, the microscope is equipped with 488, 561 and 637 nm solid-state lasers (Vortran), all coupled and focused on the back focal plane of the objective (60×, NA 1.49 oil immersion objective, Olympus). Fluorescent emission signals are split using an imaging grade, ultra-flat dichroic mirror (T660lpxr, Chroma) and the emission beams are focused with a 300 mm tube lens onto two aligned EM-CCD cameras (iXon Ultra 888, Andor). The combination of the imaging objective, 300 mm tube lens, and EM-CCD camera sensors produce 100× images with 130 nm/pixel. One camera detects far-red fluorescence (in this case corresponding to mRNA marked by JF646 HaloTag-MCP), while the other detects red fluorescence (in this case corresponding to translation marked by Cy3-conjugated Fab). The far-red signal is excited with the 637 nm laser using a 731/137 nm emission filter (FF01-731/137/25, Semrock), while the red signal is excited with the 561 nm laser using a 593/46 nm emission filter (FF01-593/46-25, Semrock). Signal-to-noise is enhanced by operating the microscopy in HILO (highly inclined and laminated optical sheet) mode.129
The lasers, filter wheel, cameras, and piezoelectric stage were all synchronized by an Arduino Mega board (Arduino). The exposure time of the cameras was 53.64 msec throughout the experiments. The camera’s readout time from the combination of imaging size, readout mode, and the vertical shift speed was 23.36 msec, resulting in an imaging rate of 13 Hz (77 msec per image). The excitation laser lines were digitally synched to ensure they only illuminated cells when the camera was exposing to avoid excessive photo-bleaching. Laser powers for all images were 20 mW for 637 nm and 5 mW for 561 nm with an ND10 neutral density filter at the beam expander.
Before imaging, live cells were placed into an incubation chamber (Okolab) at 37°C with 5% CO2 on a piezoelectric stage (PZU-2150, Applied Scientific Instrumentation). The focus was maintained with the CRISP Autofocus System (CRISP-890, Applied Scientific Instrumentation). Image acquisition was performed using opensource Micro-Manager.128 Cell volumes (comprising 13 z-stacks with 0.5 μm steps between each stack) were acquired at full speed, with an imaging delay set so that consecutive volume acquisitions were separated by a 6-s interval. Cells were selected for imaging if they exhibited at least ten translating mRNA.
For image analysis, collected images were first pre-processed with Fiji.130 First, a 2D maximum intensity projection was generated from the original 3D movies, followed by background subtraction (the background was chosen from the dimmest cell- and debris-free regions of images). Post-processed images were then analyzed using a custom Mathematica (Wolfram Research) routine to detect and track particles in the RNA channel (red color). In this code, images are band-pass filtered so the positions of the intensity centroid of single mRNA spots can be easily detected using the built-in Mathematica routine ComponentMeasurements. Particles detected were then linked through time if they were within 5 pixels (650 nm) of one another in consecutive frames. Particle tracks lasting at least five frames were selected for further analysis.
For each frame of each track, crops (15 pixels × 15 pixels) centered on the registered mRNA coordinate were made and averaged through time. Again, using Mathematica’s built-in bandpass filter and ComponentMeasurements command, the time-averaged crops corresponding to each track were categorized based on their signals in the red (mRNA) and green (translation/nascent chains) channels. If particles were detected in the red channel only, the spot was categorized as a ‘non-translating’ spot. If particles were detected in both the red and green channels, the spot was categorized as a ‘translating’ spot. After categorizing spots in this automated fashion, all spots were again hand-checked to minimize error. Finally, the original 2D maximum intensity projected images corresponding to each hand-checked track were fit to a 2D Gaussian to determine their precise XY-coordinates and signal intensities (using the built-in Mathematica routine NonlinearModelFit). The 2D Gaussian used for fitting had the following form:
where is the background signal intensity, the particle peak intensity, (, ) the spread of the particle signal, and (, ) the particle location. From these data, the intensity, position through time, and number of spots over time in each track were quantified.
To correct for any positional offset between the two cameras, Mathematica’s built-in command FindGeometricTransform was used to transform detected spot coordinates. The correct transformation function was determined by aligning the positions of 100 nm diameter Tetraspeck beads (fluorescent in all channels) imaged on the same day of each experiment. The transformation function was only used to correct for spot positions so that spots could be properly categorized, as described above. It was not used to correct the raw images themselves, as this could lead to artifactual image distortions when reassigning pixel values. Therefore, a slight offset between channels can be observed in the raw and 2D max-projected images and movies.
To calibrate the intensity of single-mRNA translation signals to the number of mature proteins (a proxy for the number of ribosomes translating the mRNA), the average fluorescence signals of the spaghetti-monster-tagged firefly luciferase nonoptimal and optimal reporters were compared to that of spaghetti monster-β-Actin-MS2, a reporter that was previously shown to have an average fluorescence equivalent to 11.4 ± 2.0 SM-tagged proteins (or ribosomes) in U2OS cells.77 To make the comparison as fair as possible, cells were imaged in two chambers on the same day with the same imaging conditions (50 mW for 561 nm and 15 mW for 637 nm laser). In the first chamber, cells were bead loaded with α-FLAG Fab (Cy3) and spaghetti monster-β-Actin-MS2. In the second chamber, cells were bead loaded with α-Flag Fab (Cy3) and either of the spaghetti-monster-tagged firefly luciferase optimality reporters.
Puromycin imaging experiments
Five time points were taken prior to the addition of 0.5 mL of DMEM+ containing puromycin to achieve a final concentration of 50 μg/mL puromycin.
Assessing stress granule disassembly with ISRIB
U2OS cells were transfected with a plasmid DNA encoding GFP-G3BP1, a stress granule marker. After 6 h, live cell imaging confirmed the formation of stress granules in response to sodium (meta)arsenite (NaAs) stress. The cells were exposed to 0.5 mM NaAs (Sigma) for 30 min. Following the NaAs exposure, cells were imaged at 5-min intervals to assess the disassembly of stress granules with ISRIB (Sigma). Immediately after the first time point (t = 0 min), 200 nM ISRIB was added to the cell culture chamber.
Ribo-seq library preparation
Libraries were prepared according to Calviello et al., 2016.131 Briefly, cells were coated with ice-cold PBS supplemented with 100 μg/mL cycloheximide, before immediately aspirating wash and dipping the bottom of each plate in LN2. Ice-cold lysis buffer B (LyB: 20 mM Tris HCL, pH 7.4, 150 mM NaCl, 5 mM MgCl2, with freshly added 1 mM DTT, 100 μg/mL cycloheximide [Sigma], 1% (v/v) Triton X-100, and 25U/mL TuRBO DNase [Invitrogen]) was dripped onto frozen dishes. Plates were thawed on ice, and the lysate was collected by scrapping and transferred to a pre-cooled 1.5 tube. Lysates were incubated on ice for 10 min, triturated ten times with a 26-gauge needle, and centrifuged 10 min, 20,000 x g at 4°C. Clarified lysate was transferred to pre-cooled tubes, flash-frozen in LN2, and stored at −80°C. Ribo-seq experiments were performed in biological triplicate.
RNA lysate concentration was measured using the Qubit RNA BR kit (Invitrogen). 85 mg lysate was footprinted for ribosomal fragments with 750 U RNase I (Ambion) and brought to 300 μL with 1x lysis buffer B. Samples were incubated for 45 min, shaking at 300 rpm at room temperature. 10 μL SUPERase•In RNase Inhibitor (Invitrogen) was added to stop digestion, and samples were placed on ice. Amersham MicroSpin S-400 HR columns (Cytiva Life Sciences) were equilibrated by removing storage buffer and washing with PB buffer (20 mM Tris HCl, pH 7.4, 150 mM NaCl, 5 mM MgCl2, and 1 mM DTT and 100 μg/mL cycloheximide [Sigma]). 100 μL digested lysate was loaded per spin, and flowthrough was transferred to a pre-chilled five mL tube. Three volumes of TRIzol LS (Invitrogen) were added before purification using Direct-zol RNA Micro-Prep Kit (with in-column DNase treatment; Zymo Research), and RNA was eluted with 11 μL RNase-free water. Five μg of RNA was ribo-depleted using siTOOLs Human RiboPOOL for Ribosome Profiling (Galen Molecular), following manufacturer’s instructions. Ribo-depleted RNA was precipitated in ethanol overnight at −80°C and resuspended in RNase-free water. Next, 27–30 nt fragments were recovered from 15% TBE-Urea PAGE gel, using 27 nt and 30 nt ribomarkers as guides (Rib’27: 5′ rArUrG rUrArC rArCrG rGrArG rUrCrG rArGrC rUrCrA rArCrC rCrGrC/3Phos/; Ribo’0: 5′ rArUrG rUrArC rArCrG rGrArG rUrCrG rArGrC rUrCrA rArCrC rCrGrC rArArC/3Phos/). Bands were visualized using 1x SybrGold (Invitrogen). The extracted gel was shredded by centrifugation and incubated in RNA extraction buffer (400mM NaCl, 1mM EDTA, and 0.25% (wt/v) SDS) at room temperature, rotating, for 2.5 h, before being filtered through 0.45 μm cellulose acetate Spin-X filter (Corning) at top speed. RNA was precipitated with isopropanol overnight at −80°C and resuspended in RNase-free water. Samples were treated with PNK, precipitated with isopropanol overnight at −80°C, and resuspended in RNase-free water.
Libraries were prepared from fragments using QIASeq miRNA Library preparation kit (Qiagen), with indexes supplied in QIAseq miRNA NGS 96 Index IL plate kit (Qiagen). Pilot PCRs using HotStarTaq 2X Mastermix (Qiagen) were performed and run out on 6% TBE-PAGE gels with 186bp Qiagen marker PCR product to best determine cycle number. After amplification, samples were purified according to manufacturer instructions, and libraries were eluted in RNase-free water. The quality of the libraries was determined with Qubit dsDNA HS assay and Tapestation D1000 HS Screentape (Invitrogen). Ribo-seq libraries were sequenced on NovaSeq6000 Illumina sequencer at the Shared Genomics Core at the University of Colorado Anschutz.
RNA-seq
10% of each lysate from the Ribo-seq sample was stored at −80°C with three volumes of TRIzol LS (Invitrogen). Purified RNA input sample was ribo-depleted using hybridizing rRNA complementary DNA oligo mix (final conc 16.5 μM) and thermostable RNaseH (10 U), as previously described.132 Samples were purified with 2.2X RNA Clean XP beads (Beckman Coulter) and eluted with RNase-free water. Cleaned samples were DNase-treated with TuRBO DNase (Invitrogen), incubating at 37°C for 30 min. The samples were cleaned again using 2.2X RNA Clean XP beads and eluted in 22 μL 1x Elute Prime Fragment Buffer included in KAPA RNA Hyperprep library kit (Roche). Samples were fragmented by heating at 85°C for 6 min. RNA-seq libraries were generated per KAPA RNA Hyperprep library kit protocol, with Illumina compatible IDT-UBI adapters (KAPA Dual-Indexed Adapter Kit [Roche]) cDNA was amplified for ten cycles. PCR products were purified with 0.8X bead-based cleanup (KAPA Pure beads) and eluted in 10 mM Tris-HCl (pH 8.0–8.5). Samples were checked for quality control with Qubit dsDNA HS kit (Invitrogen) and Tapestation HS D1000 Screentape (Agilent). Libraries were sequenced on NovaSeq6000 Illumina sequencer at the Shared Genomics Core at the University of Colorado Anschutz.
Ribo-seq data processing
UMI-containing reads (Read 2 only) were extracted from the Fastq files using UMI-tools.133 Next, adapters were trimmed, and reads shorter than 18 nts were filtered with Cutadapt.134 The trimmed fastq files were reverse complemented, and any remaining rRNA-mapped reads were filtered out using Bowtie (version 1.2.2) and SAMtools (version 1.12).135,136 The remaining reads were aligned and mapped to the human genome (hg38) with STAR (version 2.6.0a).137 The output.bam files were de-duplicated using UMI-tools and transcriptome-wide transcript expression was calculated with Salmon (version 1.6.0).138 The transcriptome used was Gencode v26. Bigwig files were also generated using bamCoverage139 to visualize RNA-seq and Ribo-seq coverage across the genome. The libraries were further checked for quality control with ribosomeProfilingQC R package,140 where the following were analyzed: 1) average read length (27–30 nts for ribosome-associated fragments expected), 2) percentage of reads mapping to CDS, 3) the mean of coverage across the CDS, and 4) proper phasing across the ORFs generated with Ribo-seq fragments of 27, 28, and 29 nts in length.
To analyze the translational efficiency of our reporters, we extracted the reads that did not map to the genome from the output.bam files. These reads were de-duplicated using UMI-tools (ribo-seq only) and mapped to the reporter sequences using Bowtie (version 1.2.2). Each condition included the Fasta sequence from the transfected 1) Renilla luciferase plasmid and 2) optimal or nonoptimal dual-tagged firefly luciferase reporter plasmids. Bedtools coverage (version v2.26.0–129-gc8b58bc)141 and dplyr tools in R were used to quantify reads across each reporter. RPKM was measured using the equation “RPKM = numReads/(geneLength/1000 * totalNumReads/1,000,000)” in R and Excel. The P-site was estimated to be 13 nucleotides downstream of’the 5′ end of each read. Additional analysis was performed using in-house scripts in R.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using built-in R Studio Student’s t test or Mann-Whitney U tests to calculate statistical significance. Statistical comparisons were made using Paired Student’s t tests to calculate p values unless specified otherwise in the figure legend. Unless otherwise stated, n refers to the number of biological replicates. A p value of < or = 0.05 was considered significant.
Supplementary Material
Highlights.
Synonymous codon usage impacts protein expression beyond mRNA levels
Nonoptimal transcripts are engaged by fewer ribosomes
Reduced eIF4E binding is required to repress translation initiation
Translational repression is not a consequence of mRNA decay nor deadenylation
ACKNOWLEDGMENTS
We thank Dr. Sujatha Jagannathan, Dr. Matt Taliaferro, Dr. David Bentley, Dr. Srinivas Ramachandran, and other members of the Rissland lab for thoughtful and helpful discussions. We thank Dr. Néel Mukherjee for sharing several Ribo-seq reagents and helping with Ribo-seq data processing. We also thank Amber Baldwin and Kathryn Walters for their expertise when helping with the Ribo-seq protocol and analysis. We thank Kent Riemondy for his bioinformatics skills. We thank the Hegde lab for their ZNF598 knockout (KO) Flp-In TRex cells. This work was supported by NIH grant R35GM128680 (O.S.R.) and the RNA Bioscience Initiative, University of Colorado School of Medicine (O.S.R.), NIH training grant T32-GM136444 (C.L.B.), and NSF grant MCB-1845761 (T.J.S.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113413.
DECLARATION OF INTERESTS
O.S.R. is a member of the Molecular Cell advisory board and the Cell Reports advisory board.
REFERENCES
- 1.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, and Bartel DP (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105. 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinnebusch AG, Ivanov IP, and Sonenberg N (2016). Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rissland OS, Subtelny AO, Wang M, Lugowski A, Nicholson B, Laver JD, Sidhu SS, Smibert CA, Lipshitz HD, and Bartel DP (2017). The influence of microRNAs and poly(A) tail length on endogenous mRNA-protein complexes. Genome Biol 18, 211. 10.1186/s13059-017-1330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leppek K, Das R, and Barna M (2018). Author Correction: Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol 19, 673. 10.1038/s41580-018-0055-5. [DOI] [PubMed] [Google Scholar]
- 5.Mayr C (2019). What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol 11. 10.1101/cshperspect.a034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong D, and Jeong S (2023). 3′UTR Diversity: Expanding Repertoire of RNA Alterations in Human mRNAs. Mol. Cells 46, 48–56. 10.14348/molcells.2023.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikemura T (1985). Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol 2, 13–34. 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 8.dos Reis M, Savva R, and Wernisch L (2004). Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32, 5036–5044. 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y (2020). A code within the genetic code: codon usage regulates cotranslational protein folding. Cell Commun. Signal 18, 145. 10.1186/s12964-020-00642-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bae H, and Coller J (2022). Codon optimality-mediated mRNA degradation: Linking translational elongation to mRNA stability. Mol. Cell 82, 1467–1476. 10.1016/j.molcel.2022.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikemura T (1981). Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. Journal of Molecular Biology 146, 1–21. 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- 12.Gustafsson C, Govindarajan S, and Minshull J (2004). Codon bias and heterologous protein expression. Trends Biotechnol 22, 346–353. 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Mauro VP, and Chappell SA (2014). A critical analysis of codon optimization in human therapeutics. Trends Mol. Med 20, 604–613. 10.1016/j.molmed.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain R, Jain A, Mauro E, LeShane K, and Densmore D (2023). ICOR: improving codon optimization with recurrent neural networks. BMC Bioinformatics 24, 132. 10.1186/s12859-023-05246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardin J, Yeasmin R, Yurovsky A, Cai Y, Skiena S, and Futcher B (2014). Measurement of average decoding rates of the 61 sense codons in vivo. Elife 3, e03735. 10.7554/eLife.03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Presnyak V, Alhusaini N, Chen Y-H, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, et al. (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamble CE, Brule CE, Dean KM, Fields S, and Grayhack EJ (2016). Adjacent Codons Act in Concert to Modulate Translation Efficiency in Yeast. Cell 166, 679–690. 10.1016/j.cell.2016.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep 14, 1787–1799. 10.1016/j.celrep.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brule CE, and Grayhack EJ (2017). Synonymous Codons: Choose Wisely for Expression. Trends Genet 33, 283–297. 10.1016/j.tig.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao F, Yu C-H, and Liu Y (2017). Codon usage regulates protein structure and function by affecting translation elongation speed in Drosophila cells. Nucleic Acids Res 45, 8484–8492. 10.1093/nar/gkx501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanson G, Alhusaini N, Morris N, Sweet T, and Coller J (2018). Translation elongation and mRNA stability are coupled through the ribosomal A-site. RNA 24, 1377–1389. 10.1261/rna.066787.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dana A, and Tuller T (2014). Mean of the typical decoding rates: a new translation efficiency index based on the analysis of ribosome profiling data. G3 5, 73–80. 10.1534/g3.114.015099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dana A, and Tuller T (2014). The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res 42, 9171–9181. 10.1093/nar/gku646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pop C, Rouskin S, Ingolia NT, Han L, Phizicky EM, Weissman JS, and Koller D (2014). Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol. Syst. Biol 10, 770. 10.15252/msb.20145524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauger DM, Cabral BJ, Presnyak V, Su SV, Reid DW, Goodman B, Link K, Khatwani N, Reynders J, Moore MJ, et al. (2019). mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. U. S. A 116, 24075–24083. 10.1073/pnas.1908052116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrzejewska A, Zawadzka M, and Pachulska-Wieczorek K (2020). On the Way to Understanding the Interplay between the RNA Structure and Functions in Cells: A Genome-Wide Perspective. Int. J. Mol. Sci 21. 10.3390/ijms21186770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komar AA, Lesnik T, and Reiss C (1999). Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett 462, 387–391. 10.1016/s0014-5793(99)01566-5. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Hubalewska M, and Ignatova Z (2009). Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol 16, 274–280. 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 29.Pechmann S, and Frydman J (2013). Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol 20, 237–243. 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu C-H, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, and Liu Y (2015). Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 59, 744–754. 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou M, Wang T, Fu J, Xiao G, and Liu Y (2015). Nonoptimal codon usage influences protein structure in intrinsically disordered regions. Mol. Microbiol 97, 974–987. 10.1111/mmi.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaney JL, Steele A, Carmichael R, Rodriguez A, Specht AT, Ngo K, Li J, Emrich S, and Clark PL (2017). Widespread position-specific conservation of synonymous rare codons within coding sequences. PLoS Comput. Biol 13, e1005531. 10.1371/journal.pcbi.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu J, Dang Y, Counter C, and Liu Y (2018). Codon usage regulates human KRAS expression at both transcriptional and translational levels. J. Biol. Chem 293, 17929–17940. 10.1074/jbc.RA118.004908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou T, Weems M, and Wilke CO (2009). Translationally optimal codons associate with structurally sensitive sites in proteins. Mol. Biol. Evol 26, 1571–1580. 10.1093/molbev/msp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, and Komar AA (2016). Synonymous Codons Direct Co-translational Folding toward Different Protein Conformations. Mol. Cell 61, 341–351. 10.1016/j.molcel.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson GN, and Clark PL (2016). Quality over quantity: optimizing co-translational protein folding with non-’optimal’ synonymous codons. Current Opinion in Structural Biology 38, 102–110. 10.1016/j.sbi.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, and Pilpel Y (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354. 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 38.Chu D, Kazana E, Bellanger N, Singh T, Tuite MF, and von der Haar T (2014). Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J 33, 21–34. 10.1002/embj.201385651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juszkiewicz S, and Hegde RS (2017). Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol. Cell 65, 743–750.e4. 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arthur LL, and Djuranovic S (2018). PolyA tracks, polybasic peptides, poly-translational hurdles. Wiley Interdiscip. Rev. RNA 9, e1486. 10.1002/wrna.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandman O, and Hegde RS (2016). Ribosome-associated protein quality control. Nat. Struct. Mol. Biol 23, 7–15. 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, and Hegde RS (2018). ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 72, 469–481.e7. 10.1016/j.molcel.2018.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikeuchi K, Tesina P, Matsuo Y, and Sugiyama T (2019). Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J 38, e100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hickey KL, Dickson K, Cogan JZ, Replogle JM, Schoof M, D’Orazio KN, Sinha NK, Hussmann JA, Jost M, Frost A, et al. (2020). GIGYF2 and 4EHP Inhibit Translation Initiation of Defective Messenger RNAs to Assist Ribosome-Associated Quality Control. Mol. Cell 79, 950–962.e6. 10.1016/j.molcel.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juszkiewicz S, Slodkowicz G, Lin Z, Freire-Pritchett P, Peak-Chew S-Y, and Hegde RS (2020). Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. Elife 9, e60038. 10.7554/eLife.60038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinha NK, Ordureau A, Best K, Saba JA, Zinshteyn B, Sundaramoorthy E, Fulzele A, Garshott DM, Denk T, Thoms M, et al. (2020). EDF1 coordinates cellular responses to ribosome collisions. Elife 9, e58828. 10.7554/eLife.58828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu CC-C, Peterson A, Zinshteyn B, Regot S, and Green R (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416.e14. 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buschauer R, Matsuo Y, Sugiyama T, Chen Y-H, Alhusaini N, Sweet T, Ikeuchi K, Cheng J, Matsuki Y, Nobuta R, et al. (2020). The Ccr4-Not complex monitors the translating ribosome for codon optimality. Science 368. 10.1126/science.aay6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veltri AJ, D’Orazio KN, Lessen LN, Loll-Krippleber R, Brown GW, and Green R (2022). Distinct elongation stalls during translation are linked with distinct pathways for mRNA degradation. Elife 11, e76038. 10.7554/eLife.76038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishima Y, Han P, Ishibashi K, Kimura S, and Iwasaki S (2022). Ribosome slowdown triggers codon-mediated mRNA decay independently of ribosome quality control. EMBO J.e109256 10.15252/embj.2021109256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Absmeier E, Chandrasekaran V, O’Reilly FJ, Stowell JAW, Rappsilber J, and Passmore LA (2022). Specific recognition and ubiquitination of slow-moving ribosomes by human CCR4-NOT. bioRxiv, 2022. 07.24.501325. 10.1101/2022.07.24.501325. [DOI] [Google Scholar]
- 52.Webster MW, Chen Y-H, Stowell JAW, Alhusaini N, Sweet T, Graveley BR, Coller J, and Passmore LA (2018). mRNA Deadenylation Is Coupled to Translation Rates by the Differential Activities of Ccr4-Not Nucleases. Mol. Cell 70, 1089–1100.e8. 10.1016/j.molcel.2018.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sweet T, Kovalak C, and Coller J (2012). The DEAD-box protein Dhh1 promotes decapping by slowing ribosome movement. PLoS Biol 10, e1001342. 10.1371/journal.pbio.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radhakrishnan A, Chen Y-H, Martin S, Alhusaini N, Green R, and Coller J (2016). The DEAD-Box Protein Dhh1p Couples mRNA Decay and Translation by Monitoring Codon Optimality. Cell 167, 122–132.e9. 10.1016/j.cell.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Y, Boland A, Kuzuoğlu-Öztürk D, Bawankar P, Loh B, Chang C-T, Weichenrieder O, and Izaurralde E (2014). A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol. Cell 54, 737–750. 10.1016/j.molcel.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 56.Mathys H, Basquin J, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, and Filipowicz W (2014). Structural and biochemical insights to the role of the CCR4-NOT complex and DDX6 ATPase in microRNA repression. Mol. Cell 54, 751–765. 10.1016/j.molcel.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 57.Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, and Sonenberg N (2014). Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 20, 1398–1409. 10.1261/rna.045302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamenska A, Simpson C, Vindry C, Broomhead H, Bénard M, Ernoult-Lange M, Lee BP, Harries LW, Weil D, and Standart N (2016). The DDX6–4E-T interaction mediates translational repression and P-body assembly. Nucleic Acids Res 44, 6318–6334. 10.1093/nar/gkw565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J, Muraoka M, Okada H, Toyoda A, Ajima R, and Saga Y (2022). The RNA helicase DDX6 controls early mouse embryogenesis by repressing aberrant inhibition of BMP signaling through miRNA-mediated gene silencing. PLoS Genet 18, e1009967. 10.1371/journal.pgen.1009967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lampson BL, Pershing NLK, Prinz JA, Lacsina JR, Marzluff WF, Nicchitta CV, MacAlpine DM, and Counter CM (2013). Rare codons regulate KRas oncogenesis. Curr. Biol 23, 70–75. 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, and Liu Y (2013). Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115. 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bazzini AA, Del Viso F, Moreno-Mateos MA, Johnstone TG, Vejnar CE, Qin Y, Yao J, Khokha MK, and Giraldez AJ (2016). Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J 35, 2087–2103. 10.15252/embj.201694699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boël G, Letso R, Neely H, Price WN, Wong K-H, Su M, Luff J, Valecha M, Everett JK, Acton TB, et al. (2016). Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529, 358–363. 10.1038/nature16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harigaya Y, and Parker R (2016). Analysis of the association between codon optimality and mRNA stability in Schizosaccharomyces pombe. BMC Genomics 17, 895. 10.1186/s12864-016-3237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mishima Y, and Tomari Y (2016). Codon Usage and 3′ UTR Length Determine Maternal mRNA Stability in Zebrafish. Mol. Cell 61, 874–885. 10.1016/j.molcel.2016.02.027. [DOI] [PubMed] [Google Scholar]
- 66.Zhou Z, Dang Y, Zhou M, Li L, Yu C-H, Fu J, Chen S, and Liu Y (2016). Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U. S. A 113, E6117–E6125. 10.1073/pnas.1606724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burow DA, Martin S, Quail JF, Alhusaini N, Coller J, and Cleary MD (2018). Attenuated Codon Optimality Contributes to Neural-Specific mRNA Decay in Drosophila. Cell Rep 24, 1704–1712. 10.1016/j.celrep.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hia F, Yang SF, Shichino Y, Yoshinaga M, Murakawa Y, Vandenbon A, Fukao A, Fujiwara T, Landthaler M, Natsume T, et al. (2019). Codon bias confers stability to human mRNA s. EMBO Rep. 20, e48220. 10.15252/embr.201948220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Narula A, Ellis J, Taliaferro JM, and Rissland OS (2019). Coding regions affect mRNA stability in human cells. RNA 25, 1751–1764. 10.1261/rna.073239.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Q, Medina SG, Kushawah G, DeVore ML, Castellano LA, Hand JM, Wright M, and Bazzini AA (2019). Translation affects mRNA stability in a codon-dependent manner in human cells. Elife 8, e45396. 10.7554/eLife.45396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Forrest ME, Pinkard O, Martin S, Sweet TJ, Hanson G, and Coller J (2020). Codon and amino acid content are associated with mRNA stability in mammalian cells. PLoS One 15, e0228730. 10.1371/journal.pone.0228730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen SR, Stewart RK, Rogers M, Ruiz IJ, Cohen E, Laederach A, Counter CM, Sawyer JK, and Fox DT (2022). Distinct responses to rare codons in select Drosophila tissues. Elife 11, e76893. 10.7554/eLife.76893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heyer EE, and Moore MJ (2016). Redefining the Translational Status of 80S Monosomes. Cell 164, 757–769. 10.1016/j.cell.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 74.Lima SA, Chipman LB, Nicholson AL, Chen Y-H, Yee BA, Yeo GW, Coller J, and Pasquinelli AE (2017). Short poly(A) tails are a conserved feature of highly expressed genes. Nature Structural & Molecular Biology 24, 1057–1063. 10.1038/nsmb.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lyu X, Yang Q, Zhao F, and Liu Y (2021). Codon usage and protein length-dependent feedback from translation elongation regulates translation initiation and elongation speed. Nucleic Acids Res. 10.1093/nar/gkab729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Viswanathan S, Williams ME, Bloss EB, Stasevich TJ, Speer CM, Nern A, Pfeiffer BD, Hooks BM, Li W-P, English BP, et al. (2015). High-performance probes for light and electron microscopy. Nat. Methods 12, 568–576. 10.1038/nmeth.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koch A, Aguilera L, Morisaki T, Munsky B, and Stasevich TJ (2020). Quantifying the dynamics of IRES and cap translation with single-molecule resolution in live cells. Nat. Struct. Mol. Biol 27, 1095–1104. 10.1038/s41594-020-0504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, and Pan T (2015). Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837. 10.1038/nmeth.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sharp PM, and Li WH (1987). The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15, 1281–1295. 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tunney R, McGlincy NJ, Graham ME, Naddaf N, Pachter L, and Lareau LF (2018). Accurate design of translational output by a neural network model of ribosome distribution. Nat. Struct. Mol. Biol 25, 577–582. 10.1038/s41594-018-0080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, Lavis LD, Grimm JB, Viswanathan S, Looger LL, et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science 352, 1425–1429. 10.1126/science.aaf0899. [DOI] [PubMed] [Google Scholar]
- 82.Wu B, Eliscovich C, Yoon YJ, and Singer RH (2016). Translation dynamics of single mRNAs in live cells and neurons. Science 352, 1430–1435. 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang C, Han B, Zhou R, and Zhuang X (2016). Real-Time Imaging of Translation on Single mRNA Transcripts in Live Cells. Cell 165, 990–1001. 10.1016/j.cell.2016.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yan X, Hoek TA, Vale RD, and Tanenbaum ME (2016). Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 165, 976–989. 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pichon X, Bastide A, Safieddine A, Chouaib R, Samacoits A, Basyuk E, Peter M, Mueller F, and Bertrand E (2016). Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J. Cell Biol 214, 769–781. 10.1083/jcb.201605024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Celik A, Baker R, He F, and Jacobson A (2017). High-resolution profiling of NMD targets in yeast reveals translational fidelity as a basis for substrate selection. RNA 23, 735–748. 10.1261/rna.060541.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001). Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol 21, 5018–5030. 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, and Ron D (2015). Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030. 10.1126/science.aaa6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sidrauski C, McGeachy AM, Ingolia NT, and Walter P (2015). The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 4. 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA, Sakamoto A, Nishimoto M, Yonemochi M, Shirouzu M, et al. (2021). ISRIB Blunts the Integrated Stress Response by Allosterically Antagonising the Inhibitory Effect of Phosphorylated eIF2 on eIF2B. Mol. Cell 81, 88–103.e6. 10.1016/j.molcel.2020.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marcotrigiano J, Gingras AC, Sonenberg N, and Burley SK (1997). Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89, 951–961. 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 93.Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras A-C, Sonenberg N, and Wagner G (1997). Structure of translation factor elF4E bound to m7GDP and interaction with 4E-binding protein. Nat. Struct. Biol 4, 717–724. 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- 94.Gingras AC, Raught B, and Sonenberg N (1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem 68, 913–963. 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 95.Humphreys DT, Westman BJ, Martin DIK, and Preiss T (2005). MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proceedings of the National Academy of Sciences 102, 16961–16966. 10.1073/pnas.0506482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, and Filipowicz W (2005). Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309, 1573–1576. 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 97.Mathonnet G, Fabian MR, Svitkin YV, Parsyan A, Huck L, Murata T, Biffo S, Merrick WC, Darzynkiewicz E, Pillai RS, et al. (2007). MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 317, 1764–1767. 10.1126/science.1146067. [DOI] [PubMed] [Google Scholar]
- 98.Jang SK, Kräusslich HG, Nicklin MJ, Duke GM, Palmenberg AC, and Wimmer E (1988). A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol 62, 2636–2643. 10.1128/JVI.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pelletier J, and Sonenberg N (1988). Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334, 320–325. 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 100.Kieft JS (2008). Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci 33, 274–283. 10.1016/j.tibs.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sheridan RM, and Bentley DL (2016). Selectable one-step PCR-mediated integration of a degron for rapid depletion of endogenous human proteins. Biotechniques 60, 69–74. 10.2144/000114378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neely GG, Kuba K, Cammarato A, Isobe K, Amann S, Zhang L, Murata M, Elmén L, Gupta V, Arora S, et al. (2010). A global in vivo Drosophila RNAi screen identifiesNOT3 as a conserved regulator of heart function. Cell 141, 142–153. 10.1016/j.cell.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amrani N, Ghosh S, Mangus DA, and Jacobson A (2008). Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453, 1276–1280. 10.1038/nature06974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gallie DR (1991). The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 5, 2108–2116. 10.1101/gad.5.11.2108. [DOI] [PubMed] [Google Scholar]
- 105.Gebauer F, Xu W, Cooper GM, and Richter JD (1994). Translational control by cytoplasmic polyadenylation of c-mos mRNA is necessary for oocyte maturation in the mouse. EMBO J 13, 5712–5720. 10.1002/j.1460-2075.1994.tb06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jacobson A, and Peltz SW (1996). Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem 65, 693–739. 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- 107.Kahvejian A, Svitkin YV, Sukarieh R, M’Boutchou M-N, and Sonenberg N (2005). Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev 19, 104–113. 10.1101/gad.1262905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilusz JE, Freier SM, and Spector DL (2008). 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135, 919–932. 10.1016/j.cell.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilusz JE, JnBaptiste CK, Lu LY, Kuhn C-D, Joshua-Tor L, and Sharp PA (2012). A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26, 2392–2407. 10.1101/gad.204438.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grosjean H, and Fiers W (1982). Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene 18, 199–209. 10.1016/0378-1119(82)90157-3. [DOI] [PubMed] [Google Scholar]
- 111.Sharp PM, Tuohy TM, and Mosurski KR (1986). Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14, 5125–5143. 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Biever A, Glock C, Tushev G, Ciirdaeva E, Dalmay T, Langer JD, and Schuman EM (2020). Monosomes actively translate synaptic mRNAs in neuronal processes. Science 367. 10.1126/science.aay4991. [DOI] [PubMed] [Google Scholar]
- 113.Mendonsa S, von Kuegelgen N, Bujanic L, and Chekulaeva M (2021). Charcot–Marie–Tooth mutation in glycyl-tRNA synthetase stalls ribosomes in a pre-accommodation state and activates integrated stress response. Nucleic Acids Res 49, 10007–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Filbeck S, Cerullo F, Pfeffer S, and Joazeiro CAP (2022). Ribosome-associated quality-control mechanisms from bacteria to humans. Mol. Cell 82, 1451–1466. 10.1016/j.molcel.2022.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schmidt C, Becker T, Heuer A, Braunger K, Shanmuganathan V, Pech M, Berninghausen O, Wilson DN, and Beckmann R (2016). Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res 44, 1944–1951. 10.1093/nar/gkv1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pelechano V, and Alepuz P (2017). eIF5A facilitates translation termination globally and promotes the elongation of many non polyproline-specific tripeptide sequences. Nucleic Acids Res 45, 7326–7338. 10.1093/nar/gkx479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gutierrez E, Shin B-S, Woolstenhulme CJ, Kim J-R, Saini P, Buskirk AR, and Dever TE (2013). eIF5A promotes translation of polyproline motifs. Mol. Cell 51, 35–45. 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, and Jung K (2013). Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85. 10.1126/science.1228985. [DOI] [PubMed] [Google Scholar]
- 119.Mao Y, Liu H, Liu Y, and Tao S (2014). Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res 42, 4813–4822. 10.1093/nar/gku159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barba-Aliaga M, Mena A, Espinoza V, Apostolova N, Costell M, and Alepuz P (2021). Hypusinated eIF5A is required for the translation of collagen. J. Cell Sci 134. 10.1242/jcs.258643. [DOI] [PubMed] [Google Scholar]
- 121.Zrimec J, Börlin CS, Buric F, Muhammad AS, Chen R, Siewers V, Verendel V, Nielsen J, Töpel M, and Zelezniak A (2020). Deep learning suggests that gene expression is encoded in all parts of a coevolving interacting gene regulatory structure. Nat. Commun 11, 6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Medina-Muñoz SG, Kushawah G, Castellano LA, Diez M, DeVore ML, Salazar MJB, and Bazzini AA (2021). Crosstalk between codon optimality and cis-regulatory elements dictates mRNA stability. Genome Biol 22, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peter D, Weber R, Sandmeir F, Wohlbold L, Helms S, Bawankar P, Valkov E, Igreja C, and Izaurralde E (2017). GIGYF1/2 proteins use auxiliary sequences to selectively bind to 4EHP and repress target mRNA expression. Genes Dev 31, 1147–1161. 10.1101/gad.299420.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Farooq Z, Kusuma F, Burke P, Dufour CR, Lee D, Tabatabaei N, Toboz P, Radovani E, Greenblatt JF, Rehman J, et al. (2022). The amino acid sensor GCN2 suppresses terminal oligopyrimidine (TOP) mRNA translation via La-related protein 1 (LARP1). J. Biol. Chem 298, 102277. 10.1016/j.jbc.2022.102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parker R, and Song H (2004). The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol 11, 121–127. 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- 126.Cialek CA, Galindo G, Morisaki T, Zhao N, Montgomery TA, and Stasevich TJ (2022). Imaging translational control by Argonaute with single-molecule resolution in live cells. Nat. Commun 13, 3345. 10.1038/s41467-022-30976-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Livingston NM, Kwon J, Valera O, Saba JA, Sinha NK, Reddy P, Nelson B, Wolfe C, Ha T, Green R, et al. (2022). Bursting Translation on Single mRNAs in Live Cells. bioRxiv, 2022.11.07.515520. 10.1101/2022.11.07.515520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J Biol Methods 1. 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tokunaga M, Imamoto N, and Sakata-Sogawa K (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161. 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 130.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Calviello L, Mukherjee N, Wyler E, Zauber H, Hirsekorn A, Selbach M, Landthaler M, Obermayer B, and Ohler U (2016). Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 13, 165–170. 10.1038/nmeth.3688. [DOI] [PubMed] [Google Scholar]
- 132.Baldwin A, Morris AR, and Mukherjee N (2021). An Easy, Cost-Effective, and Scalable Method to Deplete Human Ribosomal RNA for RNA-seq. Current Protocols 1. 10.1002/cpz1.176. [DOI] [PubMed] [Google Scholar]
- 133.Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 135.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ou J, and Hoye M (2023). ribosomeProfilingQC Ribosome Profiling Quality. Control. 10.18129/B9.bioc.ribosomeProfilingQC. [DOI] [Google Scholar]
- 141.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Ribo-seq and RNA-seq data have been deposited at GEO: GSE202900, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The raw image data have been deposited at Mendeley Data (https://data.mendeley.com/datasets/tw2bjh857f/2) and are publicly available as of the date of publication. DOIs are also listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-eIF4E | MBL Lifescience | Cat# RN001P, lot 004; RRID:AB_1570634 |
| Rabbit polyclonal anti-eIF4G1 | MBL Lifescience | Cat# RN002P, lot 005; RRID:AB_1570635 |
| Rabbit polyclonal anti-vinculin | Cell Signaling Technology | Cat# 4650, lot 5; RRID:AB_10559207 |
| Rabbit polyclonal anti-DDX6 | Abcam | Cat# ab151984, lot GR174405-10; RRID:AB_3073919 |
| Rabbit polyclonal anti-CNOT3 | Bethyl Laboratories | Cat# A302-156A; RRID:AB_1720348 |
| Mouse monoclonal anti-FLAG | Millipore Sigma | Cat# F1804, lot SLCK5688; RRID:AB_262044 |
| Mouse monoclonal anti-FLAG (for Flag fabs, imaging) | FUJIFILM Wako Shibayagi | Cat# 012-22384, clone 1E6; RRID:AB_10659717 |
| Mouse monoclonal anti-GFP | Roche | Cat# 11814460001, lot 42903200; RRID:AB_390913 |
| Rabbit polyclonal anti-Renilla luciferase | Abcam | Cat# ab187338, lot GR3382572-1; RRID: AB_3073920 |
| Mouse monoclonal anti-hGAPDH | Proteintech | Cat# 60004-1-Ig, lot 10020246; RRID:AB_2107436 |
| Mouse monoclonal anti-Ubiquitin | Thermo Fisher Scientific | Cat# 14-6078-82, lot 4342660; RRID:AB_837154 |
| Rabbit polyclonal anti-Firefly Luciferase | GeneTex | Cat# GTX125849, lot 42837; RRID:AB_11173184 |
| Mouse monoclonal anti-HA tag | Thermo Fisher Scientific | Cat# 26183, lot WA317137; RRID:AB_10978021 |
| Mouse monoclonal anti-Puromycin | Kerafast | Cat# EQ0001, lot 041918; RRID:AB_2620162 |
| Normal Rabbit Control IgG | SinoBiological | Cat# CR1; RRID: AB_3073921 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074S; RRID:AB_2099233 |
| Mouse monoclonal [SB62a] Anti-Rabbit IgG light chain (HRP) | Abcam | Cat# ab99697, lot GR254788-1; RRID:AB_10673897 |
| Rat monoclonal [187.1] Anti-Mouse kappa light chain (HRP) | Abcam | Cat# ab99617, lot GR256873-1; RRID:AB_10673885 |
| Bacterial and virus strains | ||
| One Shot™ TOP10 Chemically Competent E. coli | Invitrogen | C404010 |
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | C2987H |
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide | Millipore | C4859 |
| Salmon-sperm DNA | Sigma | D7656 |
| Puromycin | Gibco | A1113803 |
| ISRIB | Sigma Aldrich | SML0843 |
| Sodium (meta)arsenite | Sigma Aldrich | S7400 |
| Trimethoprim (TMP) | Sigma Aldrich | T7883 |
| P-32 | Perkin Elmer | Phosphorus-32, as monopotassium phosphate |
| Lipofectamine transfection reagent | Invitrogen | 11668019 |
| Effectene transfection reagent | Qiagen | 301425 |
| HaloTag-MCP protein | Protein Expression and Purification facility, CSU; H. Scherman | N/A |
| JF646-HaloTag ligand | HHMI Janelia; L. Lavis | N/A |
| TURBO™ DNase | Invitrogen | AM2388 |
| SUPERase•In™ RNase Inhibitor | Invitrogen | AM2696 |
| Random Hexamers | Invitrogen | N8080127 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | 18090050 |
| Critical commercial assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | E1960 |
| TRIzol™ Reagent | Invitrogen | 15596–018 |
| TRIzol™ LS Reagent | Invitrogen | 10296–010 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | 1725125 |
| EZview Red Protein G Affinity Gel beads | Millipore | E3403 |
| Amersham Hybond P 0.45 PVDF blotting membrane | Cytiva Life Sciences | 10600029 |
| 0.45 μM cellulose acetate Spin-X filters | Corning | CLS8162 |
| Micro Bio-Spin™ P-30 Gel Columns, Tris Buffer (RNase-free) | BioRad | 7326250 |
| ULTRAhyb-oligo hybridization buffer | Invitrogen | AM8663 |
| NuPAGE™ MES SDS Running Buffer (20X) | Invitrogen | NP0002 |
| NuPAGE™ Transfer Buffer (20X) | Invitrogen | NP00061 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 4693159001 |
| Qubit™ RNA Quantification, broad range kit | Invitrogen | Q10210 |
| Amersham MicroSpin S-400 HR Columns | Cytiva Life Sciences | 27514001 |
| Direct-zol RNA Micro-Prep Kit | Zymo Research | R2060 |
| Human riboPOOL for Ribosome Profiling samples | Galen Molecular | dp-K012-000042 |
| QIAseq miRNA Library Kit | Qiagen | 331502 |
| QIAseq miRNA NGS 96 Index IL plate kit | Qiagen | 331565 |
| Qubit™ dsDNA Quantitation, high sensitivity kit | Invitrogen | Q32851 |
| Hybridizing rRNA complementary DNA oligo mix | Baldwin et al., 2021132 | N/A |
| RNA Clean XP beads | Beckman Coulter | A63987 |
| KAPA RNA Hyperprep library kit | Roche | KK8540 |
| KAPA Dual-Indexed Adapter Kit | Roche | 08278555702 |
| Tapestation D1000 HS Screentape | Agilent | 5067–5584 |
| Tapestation D1000 HS Screentape reagents | Agilent | 5067–5585 |
| Deposited data | ||
| Paired Ribo-seq and RNA-seq data | This study | GEO Database: GSE202900 |
| Mendeley image dataset | This study | Mendeley Data: https://data.mendeley.com/datasets/tw2bjh857f/2 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| Drosophila S2 | ThermoFisher | R69007 |
| ZNF598 KO Flp-In T-REx 293 dox inducible GFP-P2A-(KAAA)0-P2A-RFP | Juszkiewicz and Hegde, 201739 | N/A |
| ZNF598 KO Flp-In T-REx 293 dox inducible GFP-P2A-(KAAA)21-P2A-RFP | Juszkiewicz and Hegde, 201739 | N/A |
| eDHFR-degron tagged DDX6 HEK293T | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 for full list of RT-qPCR primers, CRISPR guides, and probes | N/A | N/A |
| DNA oligonucleotide pool complementary to human rRNA transcripts | Baldwin et al., 2021132 | N/A |
| Recombinant DNA | ||
| pUB_SMFLuc0—MS2 | This paper | Addgene #210568 |
| pUB_SMFLuc100—MS2 | This paper | Addgene #210570 |
| pIS1_Opt_Renilla | This paper | Addgene 210571 |
| Nonopt_WT_Renilla_pIS1 | pIS1 | Addgene #12179 |
| pUB_SM-RLuc0—MS2 | This paper | Addgene #210572 |
| pUB_SM-RLuc100—MS2 | This paper | Addgene # 210573 |
| pUB_FLuc100—MS2 | This paper | Addgene # 210574 |
| pcDNA3.1_γHA-GFPopt | This paper | Addgene # 210575 |
| pcDNA3.1_γHA-GFPnonopt | This paper | Addgene # 210576 |
| pUB_OptγHAramp-FLuc100 | This paper | Addgene #210577 |
| pUB_NonoptγHAramp-FLuc100 | This paper | Addgene # 210579 |
| pcDNA3.1_SM-FLuc0-malat1 | This paper | Addgene # 210580 |
| pcDNA3.1_SM-FLuc100-malat1 | This paper | Addgene # 210581 |
| pcDNA3.1_SM-FLuc0-pA | This paper | Addgene # 210583 |
| pcDNA3.1_SM-FLuc100-pA | This paper | Addgene # 210584 |
| pUB_SM-FLuc0_EMCV-Ren | This paper | Addgene # 210590 |
| pUB_SM-FLuc100_EMCV-Ren | This paper | Addgene # 210591 |
| Dm_γHA-FLuc0_CSC | This paper | Addgene # 210589 |
| Dm_γHA-FLuc100_CSC | This paper | Addgene #210592 |
| Dm_Opt_Renilla_CSC | This paper | Addgene # 210593 |
| pUB_SM-FLuc0-GFPOPT | This paper | Addgene # 210594 |
| pUB_SM-FLuc100-GFPOPT | This paper | Addgene #210596 |
| pUB_mCherry-Rluc100 | This paper | Addgene # 210600 |
| lentiCRISPR v2-blast | Mohan Babu; Addgene | Addgene #83480 |
| psPax2 | Mikko Taipale | N/A |
| pCMV-VSV-G | Mikko Taipale | N/A |
| Software and algorithms | ||
| UMI-tools | Smith et al., 2017133 | N/A |
| Bowtie version 1.2.2. | Langmead et al., 2009135 | N/A |
| SAMtools version 1.12 | Danecek et al., 2021136 | N/A |
| STAR version 2.6.0a | Dobin et al., 2013137 | N/A |
| BEDtools | Quinlan and Hall, 2010141 | N/A |
| Salmon version 1.6.0. | Patro et al., 2017138 | N/A |
| Cutadapt | Martin et al., 2011134 | N/A |
| deepTools (Bamcoverage) | Ramírez et al., 2014139 | N/A |
| ribosomeProfilingQC R package | Ou and Hoye, 2022140 | N/A |
| Fiji ImageJ | Schindelin et al., 2012130 | N/A |
| Adobe Photoshop | https://www.adobe.com/products/photoshop.html | N/A |
| Adobe Illustrator | https://www.adobe.com/products/illustrator.html | N/A |
| Other | ||
| 35 mm glass bottom MatTek dishes | MatTek Corporation | P35G-1.5-14-C |
| Open-Top Polyclear Centrifuge tubes | Seton Scientific | 7030 |