

ABSTRACT
Chlamydia trachomatis and Neisseria gonorrhoeae are the most prevalent bacterial sexually transmitted infections (STIs) globally. Despite frequent co-infections in patients, few studies have investigated how mono-infections may differ from co-infections. We hypothesized that a symbiotic relationship between the pathogens could account for the high rates of clinical co-infection. During in vitro co-infection, we observed an unexpected phenotype where the C. trachomatis developmental cycle was impaired by N. gonorrhoeae. C. trachomatis is an obligate intracellular pathogen with a unique biphasic developmental cycle progressing from infectious elementary bodies (EB) to replicative reticulate bodies (RB), and back. After 12 hours of co-infection, we observed fewer EBs than in a mono-infection. Chlamydial genome copy number remained equivalent between mono- and co-infections. This is a hallmark of Chlamydial persistence. Chlamydial persistence alters inclusion morphology but varies depending on the stimulus/stress. We observed larger, but fewer, Chlamydia during co-infection. Tryptophan depletion can induce Chlamydial persistence, but tryptophan supplementation did not reverse the co-infection phenotype. Only viable and actively growing N. gonorrhoeae produced the inhibition phenotype in C. trachomatis. Piliated N. gonorrhoeae had the strongest effect on C. trachomatis, but hyperpiliated or non-piliated N. gonorrhoeae still produced the phenotype. EB development was modestly impaired when N. gonorrhoeae were grown in transwells above the infected monolayer. C. trachomatis serovar L2 was not impaired during co-infection. Chlamydial impairment could be due to cytoskeletal or osmotic stress caused by an as-yet-undefined mechanism. We conclude that N. gonorrhoeae induces a persistence-like state in C. trachomatis that is serovar dependent.
KEYWORDS: Chlamydia trachomatis, Neisseria gonorrhoeae, in vitro co-infection, persistence, serovar specificity
INTRODUCTION
Chlamydia trachomatis and Neisseria gonorrhoeae are, respectively, the number one and number two most prevalent bacterial sexually transmitted infections internationally (1, 2). In adults around the world, C. trachomatis is estimated to cause 130 million new infections and N. gonorrhoeae is estimated to cause 87 million new infections, annually. Both infections are frequently asymptomatic, especially in women, with primary infection beginning at the mucosal epithelium of the genital tract (3). Rarely, the infections can disseminate beyond the genital tract (4, 5). Women are particularly at risk of an upper genital tract infection leading to pelvic inflammatory disease (PID), ectopic pregnancy, and infertility due to chronic inflammation induced by both bacterial pathogens (6). While antibiotic resistance has not been reported for Chlamydia infections, N. gonorrhoeae first developed resistance to antibiotic therapy in the 1950s and continues to evolve resistance to newly introduced drugs (7). This makes treatment for co-infections more of a challenge as drugs effective against C. trachomatis may not always be effective against N. gonorrhoeae.
C. trachomatis is an obligate intracellular pathogen that undergoes a unique developmental cycle beginning with its infectious but not replicative form—the elementary body (EB) (8, 9). After invasion into a susceptible host cell and establishment of an inclusion within the host cell cytoplasm, the EB differentiates to become a reticulate body (RB) which is non-infectious. The RB divides by binary fission causing the inclusion to expand as the population of bacteria increases, until asynchronous differentiation begins and the RBs transition to EBs. At the end of the developmental cycle, the inclusion lyses or extrudes from the host cell. EBs can be detected as early as 18 hours post-infection (hpi) for C. trachomatis and the developmental cycle typically concludes at 48 hpi in in vitro settings (10). When the environment for the bacteria within the inclusion becomes sub-optimal, including but not limited to iron deprivation, nutrient deprivation, antibiotic exposure, or interferon gamma-induced tryptophan starvation, the RBs transition into aberrant bodies (ABs) and stop dividing while remaining viable (11). This is called “persistence” and enables long-term survival for C. trachomatis inside the parasitized host cell. This state is reversible, and the bacteria return to complete the developmental cycle once the stressing agent is gone. Recently, a comprehensive study characterized the persistent phenotypes and showed that different persistence-inducing conditions produce different Chlamydial phenotypes including loss of homotypic fusion (the ability of multiple inclusions within a single cell to fuse), peptidoglycan synthesis arrest, absence of secreted mid- and late-stage bacterial effectors and/or loss of cellular actin recruitment to the inclusion membrane (12).
N. gonorrhoeae is a facultative intracellular pathogen. The invasion of epithelial cells is dependent on the expression of both type 4 pili (T4P) and opacity-associated proteins (Opa) (13). Invasion leads to transcytosis through the epithelial cells (14). In symptomatic gonorrhea, gonococci are usually observed inside and attached to neutrophils which make up the characteristic purulent discharge. Extensive research has demonstrated the ability of N. gonorrhoeae to survive and replicate inside neutrophils and impair their antimicrobial activity (15). In N. gonorrhoeae, T4P are retractable extracellular structures, with most of the pilus structure comprised of PilE, and PilT the ATPase responsible for retracting the pilus. Alternating extrusion and retraction of T4P enables the characteristic gonococcal “twitching motility.” During retraction, the T4P exerts a measurable physical force on the plasma membrane of eucaryotic cells and is known to disrupt cell signaling of epithelial cells (16–18).
The extent of C. trachomatis–N. gonorrhoeae co-infections is difficult to accurately quantify due to the frequent asymptomatic nature of these infections. Incidence is generally estimated at 50%–70%. However, across seven studies, women with N. gonorrhoeae are most likely to be co-infected with C. trachomatis 17.6%–57.9%, whereas women with C. trachomatis are co-infected with N. gonorrhoeae in 2.1%–17.2% of cases (19–25). While both organisms are capable of ascension and causing PID, there does not seem to be evidence that co-infections specifically produce a higher proportion of acute PID (26).
Laboratory studies of C. trachomatis–N. gonorrhoeae co-infection in in vitro epithelial cells have only recently been reported (27). Onorini et al. examined the effect of gonococcal infection of HeLa cells prior to the C. trachomatis serovar E challenge. They observed that during co-infection, the C. trachomatis inclusions are reduced to 2% of their size when compared to a C. trachomatis mono-infection. N. gonorrhoeae pre-infection also reduced the quantity of inclusions observed, by up to 40%. The number of EBs produced during co-infective conditions decreased approximately 10-fold. This phenotype was not due to acidification of the medium nor nutrient deprivation from bacterial competition. The authors modified their experimental timeline and added N. gonorrhoeae after the C. trachomatis infection and allowed the bacteria to grow in parallel for 24 hours. As before, they observed reduced inclusion size and quantity when N. gonorrhoeae is added after C. trachomatis infection. Co-infection in human neutrophils has also been examined and found that C. trachomatis serovar L2 infection of neutrophils reduced their ability to kill N. gonorrhoeae without loss of neutrophil health (28).
To date, there are only two published reports that detail infection of mice with both Chlamydia and N. gonorrhoeae (29, 30). Both studies utilized the mouse pathogen C. muridarum as a surrogate for C. trachomatis. Vonck et al. pre-infected mice with C. muridarum and then challenged the mice with N. gonorrhoeae (29). In this study, co-infection did not increase Chlamydial titer but did increase N. gonorrhoeae shedding. In the same study, this in vivo result was not recapitulated by in vitro co-infection, as co-infection did not increase adhesion or invasion of N. gonorrhoeae to the infected cultured cells. While C. muridarum-specific antibody titers were not increased by co-infection, neutrophil influx was increased during co-infection. More recently, Onorini et al. established a latent C. muridarum infection in mice before challenge with N. gonorrhoeae at day 24 hpi (30). After 12 days of co-infective growth, C. muridarum titer was not increased by the gonococcal presence.
In this study, we aimed to identify whether a mid-cycle C. trachomatis infection is affected by a subsequent gonococcal challenge and, if so, how one or both bacterial species are affected. We found that while growth (as measured by genome copy number) of a urogenital serovar of C. trachomatis (serovar D) was unaffected, the titer of the infectious, fully differentiated bacteria was reduced 100-fold. We found that the phenotype was not produced when N. gonorrhoeae were killed by isopropanol or inhibited by the addition of gentamycin; could not be restored by tryptophan nor indole supplementation; that the phenotype was reversible; and that a partial phenotype was produced when gonococci were grown separated from the monolayer by transwells. When we challenged the Chlamydial infection with non-piliated or hyperpiliated N. gonorrhoeae, we saw a partial inhibition of EB development compared to a mono-infection. This suggested that while the action of T4P was responsible for the persistent-like phenotype in C. trachomatis, they are not the sole cause for the Chlamydial phenotype we observed during C. trachomatis–N. gonorrhoeae co-infection. Interestingly, the phenotype was biovar dependent as we did not observe the same inhibition when we used a lymphogranuloma venereum (LGV) serovar C. trachomatis L2. We considered that a major difference between C. trachomatis L2 and D serovars is the presence of the putative Chlamydia cytotoxin (31). We used C. muridarum (which has a complete cytotoxin locus) in the co-infection but did not observe a 100-fold or greater decrease in EB production as we anticipated. This result does not suggest that the cytotoxin alone is the cause behind the co-infection C. trachomatis phenotype as we hypothesized.
Co-infections are an important aspect of sexually transmitted infection, and it is critical that we know how they differ from mono-infections. Our observations provide insights into how co-infections of C. trachomatis and N. gonorrhoeae may be affected by current and future treatments.
RESULTS
N. gonorrhoeae reduces the number of chlamydial EBs after 12 hours of co-infection
To probe the interaction between C. trachomatis and N. gonorrhoeae in a cell culture model, we pre-infected sub-confluent HeLa monolayers with C. trachomatis serovar D and then challenged with N. gonorrhoeae at 12 hpi. The gonococcal challenge continued for 12 hours then all bacteria were enumerated. At this point in the Chlamydial developmental cycle, 24 hpi, we expect inclusions to contain mostly RBs and a small quantity of EBs, as EBs are detectable at 18 hpi (10). Figure 1A illustrates the timeline of the co-infection. When comparing the final number of N. gonorrhoeae attached to the monolayer, the colony forming units (CFU)/mL titer between mono-infection and co-infections showed no difference (Fig. 1B). We saw no difference between C. trachomatis genome copy (GC) number (used as a proxy for all C. trachomatis developmental forms) when comparing C. trachomatis quantity between mono- and co-infections (Fig. 1C). However, when we measured the titer of infectious EBs by counting the number of inclusions using immunofluorescence assay (IFA), there was a stark difference between mono- and co-infections and the EB titer was 100-fold fewer in a co-infection (Fig. 1D).
Fig 1.
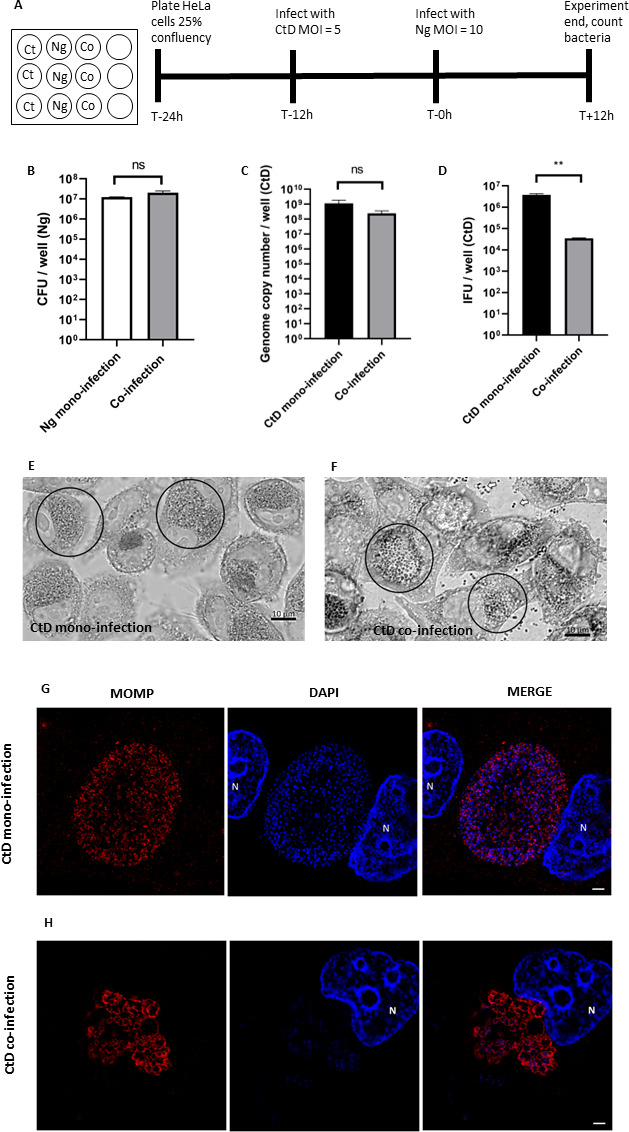
C. trachomatis EB titer is reduced after co-infection with N. gonorrhoeae. (A) Infections were carried out as described in Materials and Methods. (B) N. gonorrhoeae (Ng) CFU/mL after 12 hours of co-infection with or without C. trachomatis D. (C) Quantitative PCR measurement of genome copy number for all forms of C. trachomatis (CtD) (EBs, RBs, and ABs) after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae. (D) Quantitative immunofluorescence analysis of C. trachomatis D (CtD) EB titer after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae. Statistical analyses by Student’s t-test. Error bars are SEM. All experiments were performed in triplicate and repeated a total of three times. Differential interference contrast microscopy (DCIM) microscopy images of Giemsa stained (E) C. trachomatis mono-infection and (F) C. trachomatis–N. gonorrhoeae co-infection. Cells were fixed with 4% paraformaldehyde and stained with Giemsa stain then imaged at 1,000×. C. trachomatis inclusions are circled. In (F), N. gonorrhoeae cocci and diplococci can also be seen exterior to the cells (open arrows). Structured illumination microscopy (SIM) images of (G) C. trachomatis mono-infection and (H) C. trachomatis–N. gonorrhoeae co-infection. Chlamydial cells are larger after 12 hours of co-infection and aberrant bodies (ABs) are visible. Cells were fixed and permeabilized with methanol and 0.5% Triton X-100. Chlamydia appears red and DNA appears blue. Host cell nuclei are noted with “N.” White scale bars are 2 µm.
N. gonorrhoeae changes chlamydial inclusion morphology
We noticed the addition of N. gonorrhoeae impacted the appearance of the Chlamydial inclusions at 12 hpci. After fixing with 4% paraformaldehyde and staining with Giemsa, we were able to see the bacteria within the inclusions during co-infection. At 100×, the bacteria within the inclusions look enlarged and more sparsely distributed than the bacteria within a mono-infection (Fig. 1E and F). In addition, we used structured illumination microscopy to examine the morphology of the bacterial cells within the inclusions. Individual C. trachomatis cells are enlarged, consistent with an aberrant body (AB) phenotype (Fig. 1G and H)
Killed or inhibited N. gonorrhoeae do not affect chlamydial development
After our initial observations of a dramatic decrease in Chlamydial EB titer following C. trachomatis–N. gonorrhoeae co-infection, we asked whether this could be due to the presence of viable N. gonorrhoeae, or if it was because of nutrient depletion due to gonococcal growth. To answer this question, the co-infection experiment was repeated; however, before infection, the N. gonorrhoeae inocula were centrifuged and resuspended in 70% isopropanol or a medium containing gentamycin. After 10 minute of incubation in 70% isopropanol, the bacteria were re-centrifuged and resuspended in a fresh medium. Viability counts of the isopropanol-treated N. gonorrhoeae inoculum indicated that all the bacteria were effectively killed (Fig. 2A). Viability counts for gentamycin-treated N. gonorrhoeae inoculum indicated that the bacteria were alive when added to the co-infection but inhibited from growth during the duration of the experiment. The pre-treated inocula were added to C. trachomatis-infected cells as described above and the co-infection incubated for 12 hours. When pre-killed or growth-inhibited N. gonorrhoeae were used in the co-infection, the EB titer was restored to that of a mono-infection but the GC number for total C. trachomatis was not different between any of the conditions (Fig. 2B and C). This observation suggests that it is critical for N. gonorrhoeae to be actively growing with C. trachomatis-infected HeLa cells to decrease the Chlamydial EB titer.
Fig 2.
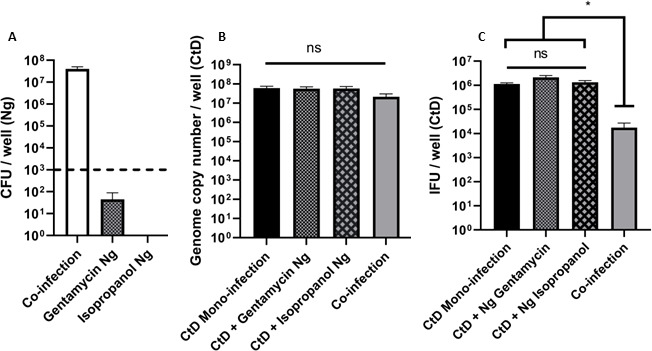
Dead or inactivated N. gonorrhoeae do not inhibit C. trachomatis EB titer during co-infection. (A) N. gonorrhoeae (Ng) CFU/mL after 12 hours of co-infection. The dotted line denotes the limit of detection for this experiment. (B) Quantitative PCR analysis of genome copy number for C. trachomatis (EBs, RBs, and ABs) after 24 hours of growth in HeLa cells including 12 hours of co-infection with treated or untreated N. gonorrhoeae. (C) Quantitative immunofluorescence analysis of C. trachomatis D (CtD) EB titer after 24 hours of growth in HeLa cells including 12 hours of co-infection with treated or untreated N. gonorrhoeae. (C). Statistical analysis for data (C) is by one-way ANOVA P = 0.0014, with Tukey’s repeated measures for the post hoc test, *P < .05. Error bars are SEM and each experiment was performed in triplicate, three or four times.
The inhibition phenotype is reversible
To fulfill all the requirements for a truly persistent phenotype, Chlamydial growth must be restored when the stressing agent is removed. We performed the standard co-infection but at 12 hours post-co-infection (hpci), the supplemented RPMI medium containing N. gonorrhoeae was removed, and the monolayer was carefully washed with PBS to detach loosely adherent N. gonorrhoeae without detaching any fragile aberrant inclusion-containing HeLa cells. The medium was replaced with fresh supplemented RPMI ± 20 µg/mL gentamycin. The co-infection was allowed to continue for an additional 12 hours and the experiment ended at 24 hpci. Figure S1 shows the modified co-infection timeline. All the bacteria were enumerated as for previous experiments. Figure 3A shows that the addition of gentamycin prevented regrowth of N. gonorrhoeae. The dotted line represents the limit of detection in this experiment. As with previous experiments, the GC number for C. trachomatis was unchanged between all samples (Fig. 3B). The EB titer for the C. trachomatis–N. gonorrhoeae gentamycin-treated co-infection was restored to an IFU/well count equivalent to the C. trachomatis mono-infection (Fig. 3C). We did not expect to see a partial recovery of EB titer in the absence of gentamycin (Fig. 3C, fourth bar). We hypothesize that this could be due to the medium change at 12 hpci which reduced the N. gonorrhoeae titer in the co-infection, replaced lost nutrients, and/or removed a secreted gonococcal molecule causing the chlamydial phenotype. During the following 12 hours of co-infection, C. trachomatis returned to its normal developmental cycle and produced EBs. The decrease between co-infections with and without gentamycin is not significantly different when analyzed by one-way ANOVA and Tukey’s post hoc test. The restoration of C. trachomatis EB titer in a co-infection with gentamycin indicated that when the non-adherent N. gonorrhoeae were removed and residual adherent N. gonorrhoeae prevented from regrowth, C. trachomatis re-entered its normal developmental cycle and started producing EBs again. This result reinforces the conclusion that this phenotype could be a persistent state.
Fig 3.

Removal of N. gonorrhoeae conditioned medium allows recovery of C. trachomatis EB titer. (A) N. gonorrhoeae (Ng) CFU/mL after 12 hours of co-infection and then 12 hours of ±gentamycin. The dotted line denotes the limit of detection for this experiment. (B) Quantitative PCR analysis of genome copy number for C. trachomatis D (EBs, RBs, and ABs) after 36 hours of growth in HeLa cells including 12 hours of co-infection ± N. gonorrhoeae and then 12 hours of ±gentamycin. (C) Quantitative immunofluorescence analysis of C. trachomatis D (CtD) EB titer after 36 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae and then 12 hours of ±gentamycin. Samples are not significantly different by one-way ANOVA or by Tukey’s modified post hoc test. Error bars are SEM. Experiments were performed three times in triplicate.
C. trachomatis EB titer is reduced during non-contact co-culture with N. gonorrhoeae
To further investigate the inhibitory effect of N. gonorrhoeae conditioned medium, we repeated the basic co-infection experiment (Fig. 1) but used 0.4 µm transwells to keep N. gonorrhoeae physically separated above the C. trachomatis-infected monolayer. N. gonorrhoeae are 0.5–1.0 µm in size due to their diplococcal morphology and cannot traverse these transwells. We hypothesized that we would see a decreased EB titer in the co-infected wells when C. trachomatis and N. gonorrhoeae shared the same culture medium. As seen previously, the gonococci grew to >108 CFU/mL in each of the three biological replicates (Fig. 4A) and the Chlamydial genome copy per well did not change significantly between mono- and co-cultures (Fig. 4B). We found that the EB titer for C. trachomatis was reduced approximately 10-fold which is a significant difference though not as pronounced as the 100-fold reduction when N. gonorrhoeae are grown in intimate contact with the eucaryotic cell plasma membrane (Fig. 4C). This indicated that merely sharing the nutrient pool was sufficient to produce a persistence-like response in C. trachomatis during C. trachomatis–N. gonorrhoeae co-culture.
Fig 4.
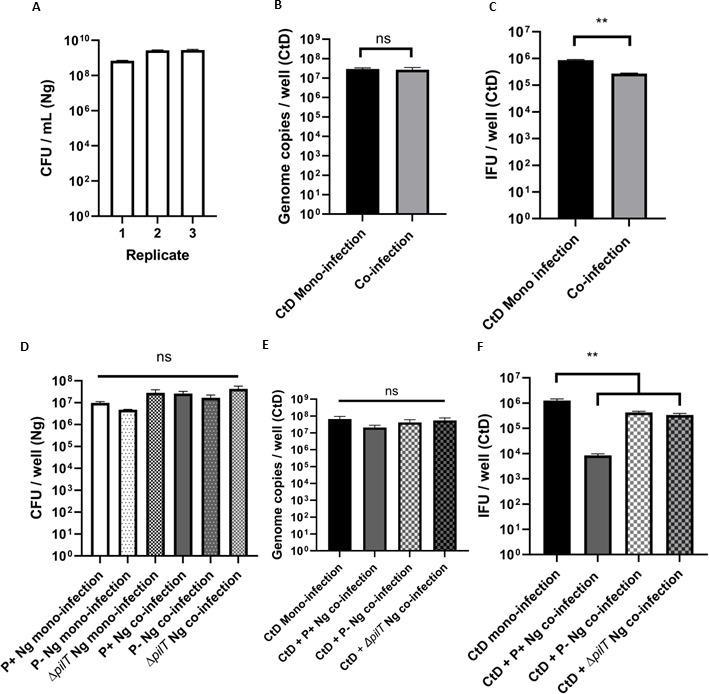
Aberrant Chlamydial phenotype is exacerbated by type 4 pili (T4P) but still observed during non-contact co-culture. HeLa cells were pre-infected with C. trachomatis D (CtD) and then challenged with N. gonorrhoeae (Ng) grown in the upper reservoir of 0.4 µm transwells for 12 hours. (A) N. gonorrhoeae grew to a stationary phase (108 CFU/mL) in all three biological replicates. (B) Genome copy number for C. trachomatis D remained equivalent between mono- and co-infected conditions. (C) Fewer IFU/well were produced during C. trachomatis–N. gonorrhoeae co-culture compared with a C. trachomatis mono-infection. Each experiment was performed in technical triplicate and three biological replicates were performed on separate days. IFU/well titers are significantly different by Student’s t-test, **P < .01. HeLa cells were pre-infected with C. trachomatis D (CtD) then challenged with piliated (P+), non-piliated (P-), or hyperpiliated (ΔpilT) N. gonorrhoeae (Ng) for 12 hours. (D) Co-infection did not significantly increase gonococcal attachment to monolayers for any of the piliated variants. (E) Chlamydial genome copy did not significantly change between mono-infection and any of the co-infection conditions. (F) Co-infections using non-piliated or hyperpiliated N. gonorrhoeae produced a decrease in Chlamydial EB titer. Error bars are the standard error of the mean (SEM) and ** represents P < 0.01 (Tukey’s multiple comparisons test). Results are significantly different by one-way ANOVA (P < 0.01).
Non-piliated N. gonorrhoeae decrease C. trachomatis EB titer
Following the results of our transwell experiments, we used phenotypically non-piliated (P-) N. gonorrhoeae or hyperpiliated (ΔpilT) N. gonorrhoeae in the co-infection experiment. Type 4 pili (T4P) are retractable and the PilT ATPase is responsible for retraction of the pilus. In our experiment, we utilized a gonococcal mutant MS11ΔpilT::KanR, which is constitutively hyperpiliated due to its inability to retract its pili. We hypothesized that we would see a moderate decrease in EB titer with non-piliated and ΔpilT N. gonorrhoeae, but not as great as observed with piliated N. gonorrhoeae. Figure 4D shows that there were slight, but not statistically significant differences between the quantity of cell-associated N. gonorrhoeae after the co-infection, and the C. trachomatis genome copy per well did not change significantly between the co-infection conditions (Fig. 4E). However, we observed a chlamydial EB titer decrease of just under one log when non-piliated or hyperpiliated N. gonorrhoeae were grown with C. trachomatis. This supported the observation from the transwell experiment that medium modification by N. gonorrhoeae was sufficient to alter the C. trachomatis EB production and while T4P exacerbates the persistence-like response, they are not solely responsible for it.
Inhibition phenotype is not due to tryptophan starvation
Given that the previous observations suggested that C. trachomatis was entering an aberrant state during co-infection with N. gonorrhoeae, we decided to investigate whether the phenotype could be rescued by tryptophan supplementation. One mechanism of Chlamydial persistence induction is tryptophan starvation (32). C. trachomatis is a tryptophan auxotroph and serovars D – L3 have only the genes for the final steps of the tryptophan biosynthetic pathway: trpAB which encodes the two subunits of the tryptophan synthase enzyme and permits synthesis of tryptophan from its precursor indole. C. trachomatis cannot synthesize indole but can scavenge it in the genital tract where it is produced by Prevotella spp (33). We hypothesized that gonococcal growth could potentially deplete tryptophan to a concentration too low to permit Chlamydial growth. To test this, we repeated the co-infection and supplemented the medium with 400 µM tryptophan or 10 µM indole in addition to NEAA and Fe(NO3)3. Previous research has shown that these concentrations are sufficient to rescue C. trachomatis from IFN-γ-induced persistence (33). However, co-infections supplemented with indole or tryptophan did not restore Chlamydial EB development and the EB titer remained equivalent to that of an un-supplemented co-infection while Chlamydial genome copy number and adherent gonococcal CFU/well remained equivalent (Fig. 5A through C).
Fig 5.

Supplementation of co-infection with 10 μM indole or 400 μM tryptophan does not restore the elementary body (EB) titer to mono-infection levels. HeLa cells were pre-infected with C. trachomatis CtD and then challenged with N. gonorrhoeae (Ng) with or without tryptophan or indole for 12 hours of growth. (A) N. gonorrhoeae (Ng) CFU/mL after 12 hours of co-infection with C. trachomatis D ± tryptophan or indole. (B) Quantitative PCR measurement of genome copy number for all forms of C. trachomatis (CtD) (EBs, RBs, and ABs) after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae ± tryptophan or indole. (C) Quantitative immunofluorescence analysis of C. trachomatis D (CtD) EB titer after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae ± tryptophan or indole. Each experiment was performed with three technical replicates and each experiment was performed three times. Error bars are the standard error of the mean (SEM) and * represents P < 0.05 (Tukey’s multiple comparisons test). Results are significantly different by one-way ANOVA (P < 0.01).
LGV biovar C. trachomatis is not sensitive to the effects of N. gonorrhoeae co-infection
We were curious whether this reduced EB titer effect was limited to a urogenital biovar of C. trachomatis. We repeated the basic co-infection experiment using an LGV serovar C. trachomatis serovar L2. L2 is reportedly more virulent than serovar D, so to account for this increased infectivity, we lowered the infection inoculum MOI from 5 to 1 for this experiment (34). All other parameters of the experiment remained unchanged. As with previous co-infections, the quantity of attached N. gonorrhoeae and the GC number for serovar L2 did not change between co- and mono-infections (Fig. 6A and B). We were surprised to find that serovar L2 EB titer did not change when C. trachomatis was challenged with N. gonorrhoeae (Fig. 6C). We examined the CtL2 inclusions using SIM and did not see aberrant bodies during the co-infection (Fig. 6E). These findings suggest that the C. trachomatis LGV serovar is resistant to the effect of N. gonorrhoeae during co-infection. As the genomes for both serovars D and L2 have been sequenced, this enables us to compare the two strains and hypothesize mechanisms for this observation. We tentatively explored the putative Chlamydial cytotoxin by repeating the co-infection with the mouse strain, C. muridarum. We hypothesized that whether serovar D’s putative partial cytotoxin was responsible for the inhibition phenotype, we would observe an increased effect with C. muridarum, which has a complete cytotoxin coding sequence (CDS) and has been previously demonstrated to exacerbate cytotoxin-dependent phenotypes (31). By contrast, the EB titer in N. gonorrhoeae–C. muridarum co-infection was only reduced by twofold (Fig. S2).
Fig 6.

C. trachomatis serovar L2 is resistant to the EB titer reduction during co-infection with N. gonorrhoeae. C. trachomatis (CtL2) pre-infected HeLa cells were challenged with live N. gonorrhoeae (Ng) for 12 hours of growth. (A) N. gonorrhoeae (Ng) CFU/mL after 12 hours of co-infection with or without C. trachomatis L2. (B) Quantitative PCR measurement of genome copy number for all forms of C. trachomatis (CtL2) (EBs, RBs, and ABs) after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae. (C) Quantitative immunofluorescence analysis of C. trachomatis L2 (CtL2) EB titer after 24 hours of growth in HeLa cells including 12 hours of co-infection with or without N. gonorrhoeae. Statistical analyses by Student’s t-test. Error bars are SEM. All experiments were performed in triplicate and repeated a total of three times. There is no significant difference between the mono- and co-infections when measured by Student’s t-test. Super illumination microscopy (SIM) fluorescence microscopy representative images of (D) CtL2 mono-infection and (E) CtL2 - N. gonorrhoeae co-infection. Chlamydial cells are the same size as mono-infected cells after 12 hours of co-infection. Cells were fixed and permeabilized with methanol and 0.5% Triton X-100. Chlamydia appears red and DNA appears blue. Host cell nuclei are noted with “N.” White scale bars are 2 µm.
Co-infection with N. gonorrhoeae alters C. trachomatis gene expression
Several studies into Chlamydial persistence in epithelial cells have attempted to identify a profile of genes that are upregulated or downregulated because of persistence (34–37). However, there is no clear single transcriptome pattern associated with all the methods used to induce persistence.
We selected four genes to probe and normalized the transcripts to chlamydial 16S. All the primers used for these genes are listed in Table S1. We chose to probe hctB as this is a late gene associated with the transition from RB to EB; ftsK as this gene has previously been observed unchanged in some transcript studies but upregulated in others (35–37). Lastly, we chose trpB and recA as these are two genes upregulated in IFN-γ-induced persistence. Table 1 shows the log2 fold change (log2 FC) for both C. trachomatis serovar D and C. trachomatis serovar L2 co-infections with N. gonorrhoeae when compared to mono-infections, and how these results compare to four transcript analyses in the literature. C. trachomatis serovar L2 showed no significant differences between mono- and co-infections. C. trachomatis serovar D showed a significant 4.6 log2 FC decrease in hctB transcripts. The other genes did not show significant differences between mono- and co-infection.
TABLE 1.
Late gene hctB is downregulated during C. trachomatis serovar D co-infectiona
| Gene | CtD FC | P value | CtL2 FC | P value | IFN (37) | HSV (35) |
|---|---|---|---|---|---|---|
| HctB | −4.6489 | 0.0285 | −0.7593 | 0.1398 | Down | N/A |
| FtsK | −0.9555 | 0.2788 | 0.3418 | 0.7236 | No change | No change |
| TrpB | −1.8177 | 0.1442 | −2.0526 | 0.3107 | Up | N/A |
| RecA | −0.2669 | 0.7850 | 0.3114 | 0.7564 | Up | N/A |
cDNA was prepared from RNA from C. trachomatis (CtD or CtL2) and N. gonorrhoeae co-infected HeLa cells. Real-time quantitative PCR was performed for C. trachomatis genes hctB, ftsK, trpB, and 16S. Transcripts were normalized to 16S for each experiment. FC equals log2 fold change for each averaged transcript. The IFN column shows the transcript change measured during IFN-induced persistence, and the HSV column shows the transcript change measured during herpes simplex virus (HSV)–C. trachomatis co-infection. Each experiment was performed three times with three technical replicates. Statistical analysis was performed by Student’s t-test and P ≤ 0.05 is considered significant.
DISCUSSION
The findings from this study have revealed an outcome of C. trachomatis–N. gonorrhoeae co-infection that we did not anticipate but is fascinating, nonetheless. There is no apparent cooperation between these two bacterial pathogens, at least when investigated in the narrow context of epithelial cell co-infection. Instead, we found that when an existing mid-cycle Chlamydia infection (12 hpi) is challenged by extracellular N. gonorrhoeae brought into contact with the host cell membrane, C. trachomatis continues growing and dividing but does not transition to the infectious EB state as readily as they do in a mono-infection. The Chlamydial response is not immediate, otherwise we would see a lack of EB development as EBs are not detectable until 18 hours in the developmental cycle. While this is not an apparent benefit for Chlamydia, an innate ability to delay a complete developmental cycle culminating with the release of progeny bacteria, until an environment becomes less hostile can be interpreted as a “benefit.” Persistence has been observed in C. trachomatis experiments for decades and shown to be induced by a variety of stimuli (12). These stimuli include but are not limited to nutrient deprivation, antibiotic use, and iron deprivation. As N. gonorrhoeae is known to be highly inflammatory and recruit large numbers of immune cells to the site of infection, this would present a challenge for EBs upon cell lysis as increased risk of phagocytosis by neutrophils would reduce the likelihood of re-infection into a new cell. C. trachomatis serovar D EBs can infect neutrophils but this is an abortive infection as the transition from EB to RB leads to cytotoxic death before the developmental cycle can be completed (38). As such, entering a viable but non-replicative state would further prolong the longevity of C. trachomatis within epithelial cells in the genital tract until the N. gonorrhoeae infection had been cleared.
When we added gonococci treated in a manner where they were dead (isopropanol) or unable to grow (gentamycin), we did not see a decrease in Chlamydial EB titer. We hypothesized that our previous observation could be due to receptor engagement between gonococci and the HeLa cell resulting in a detrimental cell signaling event causing stress on the developing Chlamydia which was sufficient to produce persistence. N. gonorrhoeae possesses several pathogenesis factors used for colonization and survival in the human host (39). Outer membrane structures include porin, T4P, opacity-associated proteins (Opa), and lipooligosaccharide (LOS). Porin (PorB) is a voltage-gated pore involved in small nutrient acquisition and ion exchange (40). PorB is capable of translocation into mammalian cell membranes via outer membrane vesicles (OMV) causing calcium influx leading to apoptosis (41, 42). PorB has also been implicated in the gonococcal invasion of epithelial cells (43, 44) and contributes to serum resistance through complement component binding (45).
There are 11 Opa proteins in N. gonorrhoeae and, in theory, any combination from all or none of them can be expressed by the bacteria (46). These proteins bind to cellular carcinoembryonic antigen-related cell adhesion molecules (CEACAMs), some of which are found on epithelial cells and are involved in the mucosal immune response (47). However, in our study, we used Opa- bacteria to prevent confounding results which could be Opa dependent. Therefore, we did not consider Opa involvement in the C. trachomatis response to co-infection.
LOS in N. gonorrhoeae is present in the outer leaflet of the outer membrane and is similar to lipopolysaccharide (LPS; endotoxin) in other Gram-negative bacteria but lacking the O-antigen polymer (48). Like the other outer membrane structures discussed above, it is important for adherence, invasion, and immune system evasion (49–51). Lipid A is a danger signal to the immune system and is recognized by toll-like receptor 4 (TLR4), which leads to activation of inflammatory transcription factors and chemokine and cytokine release (52).
T4P are dynamic structures that have been previously demonstrated to exert tremendous force on the eucaryotic plasma cell membrane. When T4P retracts, they produce forces of 80–100 pN on HeLa cells which leads to enhanced expression of infection-regulated genes, most of which are under the control of mitogen-activated protein kinases (MAPKs) (16, 18). During intracellular growth, the Chlamydial inclusion is encased in actin, intermediate filaments, and microtubules which stabilize the inclusion and aid in the transport of essential nutrients (53, 54). Loss of this stability could result in the enlargement of the inclusion. It is difficult to hypothesize if this could be the case during C. trachomatis–N. gonorrhoeae co-infection as Howie et al. showed that when magnetic beads are used to exert forces on eucaryotic cells equivalent to that of pilus retraction, actin recruitment is increased not decreased (16). Actin is recruited toward the magnetic beads; therefore, it could theoretically be recruited away from the inclusion during co-infection.
Our gentamycin assay showed that when 12 hours of gonococcal conditioned medium is removed and replaced with fresh medium with or without gentamycin, the Chlamydial EB titer recovers to levels similar to a mono-infection. As gentamycin is a plasma membrane impermeable antibiotic, we did not expect the gentamycin to have a strong detrimental effect on EB titer. However, as shown in Fig. 3, we did not expect the gentamycin-free medium replacement replicates to have a C. trachomatis EB titer recovery not significantly different from the gentamycin-containing wells. We hypothesize that this is due to an unidentified factor in the N. gonorrhoeae conditioned medium which inhibits C. trachomatis development. In addition, most N. gonorrhoeae in the co-infection are non-adherent and are removed with the medium. This creates a window of opportunity for C. trachomatis to transition back to an actively growing state before the gonococci can multiply to high CFU/mL again. Further experiments are required to identify the factor within the medium causing this phenotype. N. gonorrhoeae secretes a variety of molecules that contribute to virulence including peptidoglycan fragments, LOS, DNA, and OMV all of which are pathogen-associated molecular patterns recognized by HeLa cells.
The finding that C. trachomatis serovar L2 is resistant to the inhibitory effect generated by co-infection is especially interesting. A recent study on C. trachomatis–N. gonorrhoeae co-infection also saw decreased C. trachomatis EB titer and inclusion size (27). In this study, HeLa cells were incubated with N. gonorrhoeae for 24 hours before being infected with C. trachomatis and cycloheximide was added throughout the experiment. Cycloheximide prevents the translation of new proteins in eucaryotic cells, meaning that the host cells are restricted from responding to Chlamydial triggers except with proteins that are already present. A different urogenital C. trachomatis serovar (serovar E) and strain of N. gonorrhoeae (FA1090) were also used in this study. The authors also noted that they saw a similar effect with C. muridarum. In our study, we saw a similar but diminished effect with C. muridarum. When comparing the genomes of the two urogenital serovars (D and E), C. trachomatis serovar L2, and C. muridarum, one notable observation is the presence of genes in the Plasticity Zone (PZ). Of the pathogenic Chlamydia, C. muridarum has the largest PZ with several large CDS for putative Chlamydial cytotoxins. Serovar L2 does not have any CDS which resemble the putative cytotoxin genes, but Serovar D and E both have genes with some cytotoxin gene homology. However, when we used C. muridarum in co-infection with N. gonorrhoeae, we did not see a 100-fold decrease in EB titer as we observed with serovar D. This suggests that the Chlamydial cytotoxin is not playing a role in the phenotype we have observed.
When comparing C. trachomatis serovars D (urogenital) and L2 (LGV), an important difference is the site of infection. A typical urogenital infection may ascend to the upper reproductive tract (in women), but LGV strains invade from the genital tract into the regional lymph nodes. An infected individual first develops a small painless ulcer or papule which may resolve or progress to a secondary stage with lymphadenopathy. If untreated, the disease can result in necrosis and fibrosis. Disseminated gonorrhea leading to complications such as endocarditis, dermatitis, or arthritis results from transit via the bloodstream. In a clinical situation, an LGV C. trachomatis serovar is unlikely to encounter N. gonorrhoeae as it does not invade lymph nodes. The LGV serovars may have undergone rounds of natural selection and lost the ability to respond to gonococcal threat as it was simply not needed. To probe this hypothesis further, the co-infection model would need to be repeated with other LGV serovars: L1, L2b, and L3. Repeating co-infections with other serovars in the urogenital and LGV biovars would start to disentangle the potential avenues for exploration. For example, LGV strains L1, L2, L2b, and L3 are still genetically different and if the phenotype observed here with serovar L2 is not reproducible with L1, L2b, and/or L3, that would tell us that the observation is not as binary as LGV vs urogenital biovar specificity.551313,5614 There are many future directions arising from these initial observations. These can be broken down into three distinct questions: (1) What is C. trachomatis modulating in its developmental cycle to generate this phenotype? (2) What is the gonococcus doing to produce the C. trachomatis persistence-like phenotype? And (3) What is the role of the host cell in this phenotype? A future direction we have already undertaken is a dual RNA-seq analysis of the C. trachomatis and N. gonorrhoeae transcriptomes during in vitro co-infection (data in analysis). We observed extensive transcriptome changes in C. trachomatis serovar D with generally more downregulation than upregulation of genes. When considering the role of the gonococcus, there are many secreted virulence factors produced by N. gonorrhoeae which would accumulate in the shared culture medium These include peptidoglycan fragments, OMV and DNA.
In this study, we limited N. gonorrhoeae to extracellular growth only by using phenotypically Opa- bacteria. Clinical isolates typically express some combination of Opas meaning they are potentially invasive to epithelial cells. With that in mind, it would be important to investigate how cellular invasion by N. gonorrhoeae affects C. trachomatis inclusion development.
Lastly, the question of the effect on the host cell is an avenue we did not explore. Previous transcriptome analysis of C. trachomatis infection of HeLa cells has revealed that the number of host cell transcripts is reduced 10-fold (from approximately 100 million reads to approximately 10 million) at 24 hpi (55). In the same study, Humphrys et al. identified that upregulated eucaryotic transcripts included pro-fibrotic genes as well as “immune dampening” genes for antimicrobial peptides and extracellular matrix mucin expression. Overall, we have identified a distinct phenotype for urogenital C. trachomatis during co-habitation with N. gonorrhoeae which may translate over to clinical infections. This phenotype is defined by decreased EB production, but an absence of an effect upon N. gonorrhoeae. It refutes our initial hypothesis of a symbiotic effect on these two pathogens during co-infection. We developed a reliable in vitro model using immortalized cervical epithelial cells and have shown that the impact of co-infection on extracellular N. gonorrhoeae is negligible. Previous co-infection studies have focused on the observations of chlamydial development when epithelial cells are pre-infected with N. gonorrhoeae, or both C. trachomatis and N. gonorrhoeae at the same time (27, 56). Therefore, this work contributes to the fledgling field of C. trachomatis–N. gonorrhoeae in vitro co-infection by revealing how a productive mid-cycle chlamydial infection responds to the new challenge of N. gonorrhoeae entering its environmental niche.
MATERIALS AND METHODS
Bacterial strains
C. trachomatis serovar D/UW-3/CX (ACE022) and C. trachomatis serovar L2434/Bu (ACE004) were originally provided by Catherine O’ Connell (University of North Carolina, Chapel Hill, NC) and Harlan Caldwell (National Institutes of Health, Bethesda, MD), respectively. C. muridarum Nigg M9 (ACE132) was originally provided by Roger Rank (University of Arkansas for Medical Sciences, Little Rock, AR). Each serovar was expanded and stored as multiple single-use aliquots. The inclusion forming unit (IFU) per mL was calculated using an immunofluorescence assay (57) for each serovar batch. N. gonorrhoeae strain MS11 was provided by Joseph Dillard (University of Wisconsin, Madison, WI) and freshly streaked from a frozen stock onto gonococcal medium base agar (GCB) with Kellogg’s supplements (58). Phenotypically piliated (P+) and Opa-negative (Opa-) colonies were visualized using a stereo microscope (Leica), and approximately 10 colonies were passaged to a fresh GCB plate and allowed to grow for 12 to 13 hours (h). This bacterial lawn was swabbed using Dacron swabs (Puritan) and resuspended in 1× phosphate-buffered saline (PBS) to prepare inocula for experiments. P- MS11 N. gonorrhoeae was isolated by colony morphology and passaged to give a P-stock. Hyperpiliated MS11 was prepared by transforming MS11 with gDNA extracted from FA1090 “Opaless” ΔpilT:KanR N. gonorrhoeae provided by Dr. Alison Criss (University of Virginia). Kanamycin-resistant colonies were passaged and confirmed by PCR using primers pilT F and pilT R, also provided by Dr. Alison Criss. Phenotypically, these clones were visibly hyperpiliated. MS11 ΔpilT:KanR was stocked and used in subsequent experiments.
Eucaryotic cell lines
HeLa-USU, a derivative of HeLa-CCL2 endocervical epithelial cells originally sourced from the American Type Culture Collection but passaged at the Uniformed Services University (USU) (59), was used in all co-infection and other tissue culture assays. The HeLa cells were maintained in Roswell Park Memorial Institute medium (RPMI 1640, Gibco) supplemented with heat-inactivated 10% fetal bovine serum (FBS, Gibco). When used in IFU titer assays, HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS. Cells expanded from a single frozen HeLa aliquot were not passaged more than 30 times and were tested for mycoplasma contamination at passages 1, 12, and at the end of use. C. trachomatis L2 434/Bu was originally expanded in L2 mouse fibroblast cells before the start of this study. L2 cells were routinely grown in DMEM supplemented with 10% FBS.
C. trachomatis–N. gonorrhoeae co-infection
Two microliters of 6.2 × 104 cells/mL HeLa cells was plated per well in a 12-well plate (Corning) 24 hours before infection with C. trachomatis serovar D. Estimating that confluency is approximately 5 × 105 cells/well, and with the assumption that HeLa cells divide every 24 hours, this approach considers the physical requirements of the growing eucaryotic cells throughout the co-infection experiment and minimizes the impact of overcrowding on the experiment outcome. Twenty-four hours after seeding the HeLa cells, the medium was removed, monolayers were washed once with PBS, and the medium was replaced with 1.5 mL RPMI plus 10% FBS. C. trachomatis serovar D was diluted to 1.2 × 107 IFU/mL in sucrose phosphate glutamic acid buffer (SPG), and 100 µL added to each well, that is, a MOI ≈ 5. HeLa cells that would receive N. gonorrhoeae only (see below) were mock infected with 1.5 mL RPMI plus 10% FBS and 100 µL SPG. Cells plus bacteria were rocked for 30 minutes (min) at 37°C in 5% CO2, then centrifuged at 500× g at room temperature for another 30 minutes. Media and inocula were removed by aspiration and monolayers were washed twice with PBS. One microliter of RPMI plus 10% FBS was added to each well and the infection was allowed to progress for 12 hours. At the start of the co-infection, N. gonorrhoeae were swabbed from a 12- to 13-hour-old lawn into 3–5 mL of PBS and vigorously vortexed to resuspend the bacteria. Bacterial CFU/mL was estimated by measuring the optical density (OD) at wavelength 550 nanometers (OD550) using a Biomate S3 spectrophotometer (Thermofisher) and using the standard equivalent of 0.2 OD550 ≈1 × 108 CFU/mL. Bacteria were diluted to approximately 1.7 × 106 CFU/mL in RPMI plus 10% FBS. The C. trachomatis-infected monolayers were washed twice with PBS, and 1.5 mL (2.5 × 106 CFU, MOI ≈ 10) N. gonorrhoeae was added to each well. C. trachomatis only wells received 1.5 mL RPMI. To facilitate N. gonorrhoeae attachment to the HeLa cells, the plates were centrifuged at 600× g for 4 minutes at room temperature. Immediately after centrifugation, the inoculum was removed and each well was washed twice with PBS. Cell monolayers were incubated in 1 mL of supplemented medium [RPMI plus 10% FBS, plus non-essential amino acids (NEAA) and 12 µM iron (III) nitrate (Fe(NO3)3] at 37°C in 5% CO2 for 12 hours. To harvest bacteria from the wells at 12 hpci, the media was removed, and monolayers were gently washed with PBS twice. HeLa cells were detached and lysed by vigorously rolling five-5 mm glass beads (Fisher Scientific) across each monolayer. Lysate was collected in 1 mL of PBS, and the well surface was washed with 1 mL of PBS twice. The wash was added to the lysate to bring the sample total to 3 mL. The samples were vortexed well, and 100 µL was stored in SPG at −80°C for Chlamydial EB enumeration. One microliter was collected separately for Chlamydial genome copy (GC) number calculation. Finally, 100 µL was serially diluted 1:3 in gonococcal medium base liquid (GCBL) for N. gonorrhoeae enumeration (see below). Figure 1A shows the timeline for the co-infection. When C. trachomatis serovar L2 was used in the co-infection, the MOI was reduced to 1.
When C. muridarum was used in the co-infection, the incubation periods were shortened to account for its relatively faster developmental cycle. C. muridarum was infected at an MOI of 1 onto sub-confluent HeLa cells as described above. Infected cells were incubated at 37°C in 5% CO2 for 8 hours. The medium was removed and N. gonorrhoeae was infected by centrifugation at an MOI of 10 as described above. The co-infection was incubated for an additional 10 hours and then harvested as described above. We performed a growth curve using C. muridarum and identified 18 hpi as a feasible timepoint because EBs were above the limit of detection (data not shown).
Colony counts for N. gonorrhoeae titer
One hundred microliters of co-infection harvest sample was serially diluted 1:3 (100 µL sample plus 200 µL GCBL) and 3–10 μL spots were added onto pre-dried GCB agar plates. Plates were incubated at 37°C with 5% CO2 overnight. Triplicate spots with 20–200 colonies were counted using a stereomicroscope and used to calculate CFU/well. Samples with <20 colonies counted per spot at 100 were considered below the limit of detection.
Immunofluorescence for EB titer
HeLa cells were pre-seeded onto acid-washed coverslips in 24-well plates (Corning) at 1.3–1.5 × 105 cells/well in DMEM plus 10% FBS. One or two days later, media were removed from the monolayers and 1:10 serial dilutions of the previously harvested EBs were prepared in SPG. The medium was removed from the 24-well plates and replaced with 100 µL of diluted sample. Plates were gently rocked at 37°C with 5% CO2 for 2 hours to facilitate the adsorption of bacteria to the HeLa cell membrane. Inoculum was removed and replaced with 1 mL DMEM plus 10% FBS supplemented with 1 µg/mL cycloheximide. When this method was used to count EBs produced during co-infection experiments, 20 µg/mL gentamycin was included to prevent N. gonorrhoeae growth. Plates were incubated at 37°C in 5% CO2 for 42–48 hpi. Cells were fixed and permeabilized by removing the growth medium and adding ice-cold 100% methanol for 20–30 minutes. Methanol was promptly removed, and cells were washed in IFA wash buffer (1× PBS, 0.1% bovine serum albumin, 0.05% Tween-20). C. trachomatis inclusions were stained using Pathfinder anti-Chlamydial LOS antibody (BioRad, 30701) for 30–60 minutes. Coverslips were individually washed by dipping 10–20 times in 50–100 mL of IFA wash buffer, blotting on a paper towel, and mounting on glass slides with Fluoromount-G (Southern Biotech). Stained inclusions across duplicate coverslips were counted at 200× magnification using an inverted epifluorescence microscope (Zeiss). The limit of detection is considered 20 inclusions/coverslip, and, wherever possible, coverslips with ≥20 inclusions were used for EB titer calculation. Data are presented as average IFU/well ± standard error of the mean (SEM).
DNA extraction by phenol-chloroform
One microliter of co-infection harvest sample was centrifuged at 20,000 g for 5 minutes at room temperature and then the pellet was resuspended in 400 µL Lysis buffer (Tris-EDTA pH 8.0, 1% sodium dodecyl sulfate) and incubated at room temperature overnight. Total DNA was extracted using a standard phenol:chloroform protocol (60). Briefly, 200 µL of phenol and then 200 µL chloroform were added to each sample and vortexed well. Samples were centrifuged at 16,000 g for 5 minutes to separate phases and 300 µL of the aqueous phase transferred to a new microfuge tube. An equal volume of chloroform was added and the sample vortexed and centrifuged at 16,000 g for 5 minutes. Two hundred microliters of the aqueous phase was transferred to a new microfuge tube and 20 µL 3M sodium acetate pH 5.2 and 400 µL of ice-cold 100% ethanol was added to precipitate DNA. Samples were vortexed and incubated at −20°C for 1 hours overnight. DNA was pelleted by centrifuging at 19,000× g in a microfuge at 4°C for 30 minutes. The supernatant was carefully removed and replaced with ice-cold 70% ethanol. Samples were centrifuged at maximum speed at 4°C for 15 minutes and the supernatant was carefully removed. DNA pellets were allowed to air dry, and dissolved in 50 µL molecular biology grade water, and the DNA concentration was measured on a Nanodrop 2000 spectrophotometer (Fisher Scientific). Samples were stored at −20°C until used in a quantitative polymerase chain reaction (qPCR).
qPCR for C. trachomatis genome copy number
The C. trachomatis 16S rRNA gene was amplified via qPCR using primers: u_16 s F TAGTGTGTGAGGGGATAAATTGAGAG and u_16s R GTTTAGCATCTATACTGGCCTGCATTCT (61). The C. muridarum 16S rRNA gene was amplified via qPCR using primers Cm 16S F CGCCTGAGGAGTACACTCGCAAGG and Cm 16S R CCAACACCTCACGGCACGAG (62). For each qPCR run, a fresh standard curve was prepared to compare measured threshold values (Cq) from a known concentration of C. trachomatis or C. muridarum genomic DNA (gDNA.). A 1:10 dilution series standard curve from 4 × 102 to 4 × 108 genome copies was amplified in triplicate for each qPCR run. A water template negative control was included each time. Before routine use in quantification assays, both an absence of amplification of HeLa or N. gonorrhoeae gDNA and good amplification efficiency were confirmed for these primer pairs. Each technical replicate was amplified once in a final volume of 10 µL using Luna Universal qPCR Mastermix (Qiagen), 0.25 µM of each primer, and with cycling conditions: 95°C/1 minute [95°C/15 s, 60°C/30 s, plate read] × 45 cycles. A melt curve was also performed after the amplification to ensure a single target had been amplified.
Giemsa staining and microscopy
Co-infections were performed as described above, using the same MOIs but in 24-well plates with covered glass bottoms (Nunc). Medium was removed from monolayers, and the cells were fixed at room temperature using 4% paraformaldehyde for 10 minutes. Cells were washed with 1× PBS and stained with a freshly prepared 1:40 dilution of Giemsa stock solution (7.6 mg/mL) for 2 hours at room temperature. Excess dye was removed and replaced with 1× PBS for imaging. Cells were viewed at 1,000× with oil immersion using the brightfield setting on an inverted epifluorescence microscope (Zeiss) and imaged with Zen Blue software (Zeiss).
Fluorescence microscopy and imaging
Co-infections were performed as described above, using the same MOIs. HeLa cells were cultured on acid-washed coverslips and after the co-infection, the monolayer was gently washed twice with PBS and then fixed with room temperature methanol for 5 minutes. Monolayers were washed with PBS three times and then incubated with 0.5% Triton-X100 for 5 minutes. Monolayers were washed with PBS and then blocked with 3% BSA for 1 hour. Coverslips were incubated with α-MOMP primary antibody (Lifespan Biosciences) for 1 hour and then washed with 3% BSA. Coverslips were incubated with a secondary antibody conjugated to Alexfluor 594 (Invitrogen) for 1 hour and then washed with 3% BSA. Coverslips were mounted on slides with Fluoromount-G and sealed with nail polish. Fluorescent images were obtained using super-resolution microscopy via a Zeiss ELYRA PS.1 set to SIM-imaging mode.
Gentamycin recovery assays
The co-infection protocol as described above was performed, but at 12 hpci, the media were removed from all wells, the monolayers were washed twice with 1X PBS, and fresh medium supplemented with 10% FBS, 1× NEAA, and 12 µM Fe3+ with or without 20 µg/mL gentamycin was added to the wells. The co-infection continued for an additional 12 hours at 37°C, 5% CO2. Figure 3 shows the timeline for this modification of the co-infection procedure. At 24 hpci, the media were removed, and the monolayers were washed twice with 1× PBS. HeLa cells were detached and lysed by vigorously rolling five- 5 mm glass beads across each monolayer. The lysate was collected in 1 mL of PBS and the well surface was washed twice with 1 mL of PBS. The wash was added to the collected lysate to bring the sample total to 3 mL. The samples were vortexed well and 100 µL was stored in SPG at −80°C for Chlamydial EB enumeration. One microliter was collected separately for Chlamydial GC number enumeration. Finally, 100 µL was serially diluted 1:3 in GCBL for N. gonorrhoeae enumeration as described above.
Isopropanol and gentamycin killing assays
The co-infection was performed as described above. Before inoculating the wells with N. gonorrhoeae, 108 bacteria were centrifuged at 10,000× g for 3 minutes and resuspended in 1 mL 70% isopropanol for 10 minutes at room temperature, then centrifuged again and resuspended in supplemented medium. This inoculum was further diluted in the medium and added to the appropriate wells for infection. At the same time, another 108 N. gonorrhoeae were centrifuged at 10,000× g for 3 minutes and resuspended in 1 mL supplemented medium with 20 µg/mL gentamycin. This inoculum was also further diluted in the supplemented medium with 20 µg/mL gentamycin and centrifuged into contact with the Chlamydia-infected monolayer as described above. After infection, all monolayers were washed with PBS twice and incubated for 12 hours in a supplemented medium. For the gentamycin N. gonorrhoeae wells, the supplemented medium also contained 20 µg/mL gentamycin. The co-infection experiment was harvested and enumerated as previously described.
Tryptophan and indole recovery assay
The co-infection experiment was set up as previously described. After centrifuging N. gonorrhoeae into contact with the monolayer and washing non-adherent bacteria away, the medium was replaced with a supplemented medium containing 10 µM indole or 400 µM L-tryptophan. C. trachomatis mono-infections were also incubated in a supplemented medium containing 10 µM indole or 400 µM L-tryptophan. At 12 hpci, all wells were harvested and enumerated as previously described.
Co-infection in transwell plates
The co-infection assay was performed as previously described with the exception that 0.4 µm transwells were placed above C. trachomatis-infected monolayers. The transwells contained 150 µL of N. gonorrhoeae at approximately 6.7 × 109 CFU/mL or 150 µL of supplemented medium. At 12 hpci, the transwells were carefully removed and all bacteria enumerated as described above.
RNA purification, reverse transcription, and qPCR for gene expression
The co-infection was performed as previously described. Infected monolayers were resuspended into 150 µL of PBS and 750 µL RNA. Later, reagents were added for preservation. Total RNA was extracted using the Direct-Zol RNA miniprep kit (Zymo) including the on column DNaseI treatment step, according to the manufacturer’s instructions. A second DNA removal step was performed using TURBO DNase (Invitrogen). RNA concentration was measured using a Nanodrop spectrophotometer (Fisher Scientific) and gDNA removal was validated using qPCR (primers u_16S F and u_16S R). cDNA was synthesized using the High Capacity cDNA Reverse Transcriptase kit (Applied Biosystems) and a no reverse transcriptase (NRT) control was included for each sample. cDNA was diluted 1:5 in molecular biology water before real-time amplification on a CFX 96 thermocycler (BioRad) using Luna Universal qPCR Mastermix (New England Biolabs). Primer combinations for each gene target are listed in Table S1. Chlamydial genomic DNA was used to prepare a standard curve for calculating transcript copy number and all target genes were normalized to 16S transcript levels.
Statistical analysis
Data sets with three or more groups were analyzed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons for the post hoc test. Data sets with less than three groups were analyzed using an unpaired Student’s t-test. P values of ≤ 0.05 are considered statistically significant. Graphpad Prism was used for all calculations.
ACKNOWLEDGMENTS
The authors would like to acknowledge the members of the Maurelli and Liechti labs for their helpful discussions. We also thank Drs. Alison Criss, Joseph Dillard, and Cory Leonard for their advice, discussions, and reagents with growing Neisseria gonorrhoeae.
Funding support to L.M.B. was provided by the Graduate Program for Biomedical Sciences at the University of Florida, and the Department of Environmental and Global Health at the University of Florida. Funding support to G.W.L. was provided via a MIRA ESI award from NIGMS (GM138202).
G.W.L. and E.B. The opinions or assertions contained herein are ours and are not to be construed as official or as reflecting the views of the Department of Defense or the Uniformed Services University.
Contributor Information
Anthony T. Maurelli, Email: amaurelli@phhp.ufl.edu.
Kimberly A. Kline, Universite de Geneve, Geneva, Switzerland
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/iai.00179-23.
Modified co-infection timeline.
Chlamydia muridarum co-infection with N. gonorrhoeae.
Legends for Fig. S1 and S2 and Table S1.
Primers used in this study.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. HHS CDC, NCSSHTP . 2019. CDC FACT SHEET: reported STDs in the United States. Available from: www.cdc.gov/nchhstp/newsroom
- 2. WHO . https://www.who.int/health-topics/sexually-transmitted-infection. (2016).
- 3. Wiesenfeld HC, Sweet RL, Ness RB, Krohn MA, Amortegui AJ, Hillier SL. 2005. Comparison of acute and subclinical pelvic inflammatory disease. Sex Transm Dis 32:400–405. doi: 10.1097/01.olq.0000154508.26532.6a [DOI] [PubMed] [Google Scholar]
- 4. Holmes KK, Counts GW, Beaty HN. 1971. Disseminated gonococcal infection. Ann Intern Med 74:979–993. doi: 10.7326/0003-4819-74-6-979 [DOI] [PubMed] [Google Scholar]
- 5. Richardson D, Goldmeier D. 2007. Lymphogranuloma venereum: an emerging cause of proctitis in men who have sex with men. Int J STD AIDS 18:11–14; doi: 10.1258/095646207779949916 [DOI] [PubMed] [Google Scholar]
- 6. den Heijer CDJ, Hoebe CJPA, Driessen JHM, Wolffs P, van den Broek IVF, Hoenderboom BM, Williams R, de Vries F, Dukers-Muijrers NHTM. 2019. Chlamydia trachomatis and the risk of pelvic inflammatory disease, ectopic pregnancy, and female infertility: a retrospective cohort study among primary care patients. Clin Infect Dis 69:1517–1525. doi: 10.1093/cid/ciz429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willcox RR. 1970. A survey of problems in the antibiotic treatment of gonorrhoea. with special reference to South-East Asia. Br J Vener Dis 46:217–242. doi: 10.1136/sti.46.3.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moulder JW. 1991. Interaction of chlamydiae and host cells in vitro. Microbiol Rev 55:143–190. doi: 10.1128/mr.55.1.143-190.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tamura A, Matsumoto A, Higashi N. 1967. Purification and chemical composition of reticulate bodies of the meningopneumoniitis organisms. J Bacteriol 93:2003–2008. doi: 10.1128/jb.93.6.2003-2008.1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LaBrie SD, Dimond ZE, Harrison KS, Baid S, Wickstrum J, Suchland RJ, Hefty PS. 2019. Transposon mutagenesis in Chlamydia trachomatis identifies CT339 as a ComEC homolog important for DNA uptake and lateral gene transfer. mBio 10:e01343-19. doi: 10.1128/mBio.01343-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beatty WL, Byrne GI, Morrison RP. 1993. Morphologic and antigenic characterization of interferon γ-mediated persistent Chlamydia trachomatis infection In vitro. Proc Natl Acad Sci U S A 90:3998–4002. doi: 10.1073/pnas.90.9.3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brockett MR, Liechti GW. 2021. Persistence alters the interaction between Chlamydia trachomatis and its host cell. Infect Immun 89:e0068520. doi: 10.1128/IAI.00685-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griffiss JM, Lammel CJ, Wang J, Dekker NP, Brooks GF. 1999. Neisseria gonorrhoeae coordinately uses Pili and OPA to activate HEC-1- B cell Microvilli, which causes engulfment of the gonococci. Infect Immun 67:3469–3480. doi: 10.1128/IAI.67.7.3469-3480.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang JA, Meyer TF, Rudel T. 2008. Cytoskeleton and motor proteins are required for the transcytosis of Neisseria gonorrhoeae through polarized epithelial cells. Int J Med Microbiol 298:209–221. doi: 10.1016/j.ijmm.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 15. Johnson MB, Criss AK. 2011. Resistance of Neisseria gonorrhoeae to neutrophils. Front Microbiol 2:77. doi: 10.3389/fmicb.2011.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Howie HL, Glogauer M, So MTN. 2005. Gonorrhoeae type IV pilus stimulates mechanosensitive pathways and cytoprotection through a pilT-dependent mechanism. PLoS Biol 3:e100. doi: 10.1371/journal.pbio.0030100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maier B, Potter L, So M, Long CD, Seifert HS, Sheetz MP. 2002. Single pilus motor forces exceed 100 pN. Proc Natl Acad Sci U S A 99:16012–16017. doi: 10.1073/pnas.242523299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Biais N, Ladoux B, Higashi D, So M, Sheetz M. 2008. Cooperative retraction of bundled type IV pili enables nanonewton force generation. PLoS Biol 6:e87. doi: 10.1371/journal.pbio.0060087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forward KR. 2010. Risk of coinfection with Chlamydia trachomatis and Neisseria gonorrhoeae in nova scotia. Can J Infect Dis Med Microbiol 21:e84–6. doi: 10.1155/2010/760218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Creighton S, Tenant-Flowers M, Taylor CB, Miller R, Low N. 2003. Co-infection with Gonorrhoea and Chlamydia: how much is there and what does it mean? Int J STD AIDS 14:109–113. doi: 10.1258/095646203321156872 [DOI] [PubMed] [Google Scholar]
- 21. McKenna JG, Young H, Moyes A, Smith IW. 1990. Is coexisting chlamydial infection more common in gonococcal infections with serogroup WI Int J STD AIDS 1:340–342. doi: 10.1177/095646249000100507 [DOI] [PubMed] [Google Scholar]
- 22. Mårdh PA, Lind I, From E, Andersen AL. 1980. Prevalence of Chlamydia trachomatis and Neisseria gonorrhoeae infections in greenland. a seroepidemiological study. Br J Vener Dis 56:327–331. doi: 10.1136/sti.56.5.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das S, Allan PS, Wade AAH. 2002. A retrospective study of the clinical effectiveness of the treatment of genital co-infection with N. gonorrhoeae and C. trachomatis in coventry. Int J STD AIDS 13:178–180. doi: 10.1258/0956462021924875 [DOI] [PubMed] [Google Scholar]
- 24. Biro FM, Rosenthal SL, Kiniyalocts M. 1995. Gonococcal and chlamydial genitourinary infections in symptomatic and asymptomatic adolescent women. Clin Pediatr (Phila) 34:419–423. doi: 10.1177/000992289503400804 [DOI] [PubMed] [Google Scholar]
- 25. Smith TF, Weed LA, Pettersen GR, O’Brien PC. 1978. A comparison of genital infections caused by Chlamydia trachomatis and by Neisseria gonorrhoeae. Am J Clin Pathol 70:333–336. doi: 10.1093/ajcp/70.3.333 [DOI] [PubMed] [Google Scholar]
- 26. Eschenbach DA. 2008. Acute pelvic inflammatory disease. GLOWM:1–28. doi: 10.3843/GLOWM.10029 [DOI] [PubMed] [Google Scholar]
- 27. Onorini D, Borel N, Schoborg RV, Leonard CA. 2022. Neisseria gonorrhoeae limits Chlamydia trachomatis inclusion development and infectivity in a novel In vitro co-infection model. Front. Cell. Infect. Microbiol 12:1–15. doi: 10.3389/fcimb.2022.911818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajeeve K, Das S, Prusty BK, Rudel T. 2018. Chlamydia trachomatis paralyses neutrophils to evade the host innate immune response. Nat Microbiol 3:824–835. doi: 10.1038/s41564-018-0182-y [DOI] [PubMed] [Google Scholar]
- 29. Vonck RA, Darville T, O’Connell CM, Jerse AE. 2011. Chlamydial infection increases gonococcal colonization in a novel murine coinfection model. Infect Immun 79:1566–1577. doi: 10.1128/IAI.01155-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Onorini D. 2023. Neisseria gonorrhoeae coinfection during Chlamydia muridarum shedding genital latency does not modulate murine vaginal bacterial shedding Delia. Microbiol Spectr. doi: 10.1128/spectrum.04500-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belland RJ, Scidmore MA, Crane DD, Hogan DM, Whitmire W, McClarty G, Caldwell HD. 2001. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc Natl Acad Sci U S A 98:13984–13989. doi: 10.1073/pnas.241377698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Byrne GI, Lehmann LK, Landry GJ. 1986. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun 53:347–351. doi: 10.1128/iai.53.2.347-351.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziklo N, Huston WM, Taing K, Katouli M, Timms P. 2016. In vitro rescue of genital strains of Chlamydia trachomatis from interferon-γ and tryptophan depletion with indole-positive, but not indole-negative prevotella spp. BMC Microbiol 16:286. doi: 10.1186/s12866-016-0903-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edwards VL, Smith SB, McComb EJ, Tamarelle J, Ma B, Humphrys MS, Gajer P, Gwilliam K, Schaefer AM, Lai SK, Terplan M, Mark KS, Brotman RM, Forney LJ, Bavoil PM, Ravel J. 2019. The cervicovaginal microbiota-host interaction modulates Chlamydia trachomatis infection. mBio 10:e01548-19. doi: 10.1128/mBio.01548-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deka S, Vanover J, Dessus-Babus S, Whittimore J, Howett MK, Wyrick PB, Schoborg RV. 2006. Chlamydia trachomatis enters a viable but non-cultivable (persistent) state within herpes simplex virus type 2 (HSV-2) co-infected host cells. Cell Microbiol 8:149–162. doi: 10.1111/j.1462-5822.2005.00608.x [DOI] [PubMed] [Google Scholar]
- 36. Gérard HC, Krausse-Opatz B, Wang Z, Rudy D, Rao JP, Zeidler H, Schumacher HR, Whittum-Hudson JA, Köhler L, Hudson AP. 2001. Expression of Chlamydia trachomatis genes encoding products required for DNA synthesis and cell division during active versus persistent infection. Mol Microbiol 41:731–741. doi: 10.1046/j.1365-2958.2001.02550.x [DOI] [PubMed] [Google Scholar]
- 37. Belland RJ, Nelson DE, Virok D, Crane DD, Hogan D, Sturdevant D, Beatty WL, Caldwell HD. 2003. Transcriptome analysis of chlamydial growth during IFN-γ-mediated persistence and reactivation. Proc Natl Acad Sci U S A 100:15971–15976. doi: 10.1073/pnas.2535394100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang C, Lei L, Collins JWM, Briones M, Ma L, Sturdevant GL, Su H, Kashyap AK, Dorward D, Bock KW, Moore IN, Bonner C, Chen C-Y, Martens CA, Ricklefs S, Yamamoto M, Takeda K, Iwakura Y, McClarty G, Caldwell HD. 2021. Chlamydia evasion of neutrophil host defense results in NLRP3 dependent myeloid-mediated sterile inflammation through the purinergic P2X7 receptor. Nat Commun 12:1–16. doi: 10.1038/s41467-021-25749-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quillin SJ, Seifert HS. 2018. Neisseria gonorrhoeae host adaptation and pathogenesis. Nat Rev Microbiol 16:226–240. doi: 10.1038/nrmicro.2017.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen A, Seifert HS. 2014. Saturating mutagenesis of an essential gene: a majority of the Neisseria gonorrhoeae major outer membrane Porin (PorB) is mutable. J Bacteriol 196:540–547. doi: 10.1128/JB.01073-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Müller A, Günther D, Düx F, Naumann M, Meyer TF, Rudel T. 1999. Neisserial porin (PorB) causes rapid calcium influx in target cells and induces apoptosis by the activation of cysteine proteases. EMBO J. 18:339–352. doi: 10.1093/emboj/18.2.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deo P, Chow SH, Hay ID, Kleifeld O, Costin A, Elgass KD, Jiang J-H, Ramm G, Gabriel K, Dougan G, Lithgow T, Heinz E, Naderer T. 2018. Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathog 14:e1006945. doi: 10.1371/journal.ppat.1006945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Putten JP, Duensing TD, Carlson J. 1998. Gonococcal invasion of epithelial cells driven by P.IA, a bacterial ion channel with GTP binding properties. J Exp Med 188:941–952. doi: 10.1084/jem.188.5.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bauer FJ, Rudel T, Stein M, Meyer TF. 1999. Mutagenesis of the Neisseria gonorrhoeae porin reduces invasion in epithelial cells and enhances phagocyte responsiveness. Mol Microbiol 31:903–913. doi: 10.1046/j.1365-2958.1999.01230.x [DOI] [PubMed] [Google Scholar]
- 45. Ram S, Cullinane M, Blom AM, Gulati S, McQuillen DP, Monks BG, O’Connell C, Boden R, Elkins C, Pangburn MK, Dahlbäck B, Rice PA. 2001. Binding of C4B-binding protein to porin: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J Exp Med 193:281–295. doi: 10.1084/jem.193.3.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhat KS, Gibbs CP, Barrera O, Morrison SG, Jähnig F, Stern A, Kupsch EM, Meyer TF, Swanson J. 1991. The opacity proteins of Neisseria gonorrhoeae strain MS11 are encoded by a family of 11 complete genes. Mol Microbiol 5:1889–1901. doi: 10.1111/j.1365-2958.1991.tb00813.x [DOI] [PubMed] [Google Scholar]
- 47. Klaile E, Müller MM, Schäfer MR, Clauder A-K, Feer S, Heyl KA, Stock M, Klassert TE, Zipfel PF, Singer BB, Slevogt H. 2017. Binding of Candida albicans to human CEACAM1 and CEACAM6 modulates the inflammatory response of intestinal epithelial cells. mBio 8:1–16. doi: 10.1128/mBio.02142-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Preston A, Mandrell RE, Gibson BW, Apicella MA. 1996. The lipooligosaccharides of pathogenic gram-negative bacteria. Crit Rev Microbiol 22:139–180. doi: 10.3109/10408419609106458 [DOI] [PubMed] [Google Scholar]
- 49. Song W, Ma L, Chen R, Stein DC. 2000. Role of lipooligosaccharide in OPA-independent invasion of Neisseria gonorhoeae into human epithelial cells. J Exp Med 191:949–960. doi: 10.1084/jem.191.6.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Vliet SJ, Steeghs L, Bruijns SCM, Vaezirad MM, Snijders Blok C, Arenas Busto JA, Deken M, van Putten JPM, van Kooyk Y. 2009. Variation of Neisseria gonorrhoeae lipooligosaccharide directs dendritic cell-induced T helper responses. PLoS Pathog 5:e1000625. doi: 10.1371/journal.ppat.1000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wetzler LM, Barry K, Blake MS, Gotschlich EC. 1992. Gonococcal lipooligosaccharide sialylation prevents complement-dependent killing by immune sera. Infect Immun 60:39–43. doi: 10.1128/iai.60.1.39-43.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Murphy K, Weaver C. 2017. Janeway’s immunobiology. 9th ed. Garland Science. [Google Scholar]
- 53. Al-Zeer MA, Al-Younes HM, Kerr M, Abu-Lubad M, Gonzalez E, Brinkmann V, Meyer TF. 2014. Chlamydia trachomatis remodels stable microtubules to coordinate golgi stack recruitment to the chlamydial inclusion surface. Mol Microbiol 94:1285–1297. doi: 10.1111/mmi.12829 [DOI] [PubMed] [Google Scholar]
- 54. Kumar Y, Valdivia RH. 2008. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds yadunanda. Cell Host Microbe 4:159–169. doi: 10.1016/j.chom.2008.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Humphrys MS, Creasy T, Sun Y, Shetty AC, Chibucos MC, Drabek EF, Fraser CM, Farooq U, Sengamalay N, Ott S, Shou H, Bavoil PM, Mahurkar A, Myers GSA. 2013. Simultaneous transcriptional profiling of bacteria and their host cells. PLoS One 8:e80597. doi: 10.1371/journal.pone.0080597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vonck RA, Darville T, O’Connell CM, Jerse AE. 2011. Chlamydial infection increases gonococcal colonization in a novel murine coinfection model. Infect Immun 79:1566–1577. doi: 10.1128/IAI.01155-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Slade JA, Brockett M, Singh R, Liechti GW, Maurelli AT. 2019. Fosmidomycin, an inhibitor of isoprenoid synthesis, induces persistence in chlamydia by inhibiting peptidoglycan assembly. PLoS Pathog 15:e1008078. doi: 10.1371/journal.ppat.1008078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kellogg DS, Peacock WL, Deacon WE, Brown L, PIrkle DI. 1963. Neisseria gonorrhoeae. I. virulence genetically linked to clonal variation. J Bacteriol 85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang L-F, Eaton BT, Broder CC. 2005. Ephrin-B2 ligand is a functional receptor for hendra virus and nipah virus. Proc Natl Acad Sci U S A 102:10652–10657. doi: 10.1073/pnas.0504887102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Green MR, Sambrook J. 2017. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb Protoc 2017:pdb.prot093450. doi: 10.1101/pdb.prot093450 [DOI] [PubMed] [Google Scholar]
- 61. Turingan RS, Kaplun L, Krautz-Peterson G, Norsworthy S, Zolotova A, Joseph SJ, Read TD, Dean D, Tan E, Selden RF. 2017. Rapid detection and strain typing of Chlamydia trachomatis using a highly multiplexed microfluidic PCR assay. PLoS One 12:e0178653. doi: 10.1371/journal.pone.0178653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mishkin N, Ricart Arbona RJ, Carrasco SE, Lawton S, Henderson KS, Momtsios P, Sigar IM, Ramsey KH, Cheleuitte-Nieves C, Monette S, Lipman NS. 2022. Reemergence of the murine bacterial pathogen Chlamydia muridarum in research mouse colonies. Comp Med 72:230–242. doi: 10.30802/AALAS-CM-22-000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Modified co-infection timeline.
Chlamydia muridarum co-infection with N. gonorrhoeae.
Legends for Fig. S1 and S2 and Table S1.
Primers used in this study.