

Abstract
Background and Objectives
The GAA repeat expansion within the fibroblast growth factor 14 (FGF14) gene has been found to be associated with late‐onset cerebellar ataxia. This study aimed to investigate the genetic causes of cerebellar ataxia in patients in Japan.
Methods
We collected a case series of 940 index patients who presented with chronic cerebellar ataxia and remained genetically undiagnosed after our preliminary genetic screening. To investigate the FGF14 repeat locus, we employed an integrated diagnostic strategy that involved fluorescence amplicon length analysis polymerase chain reaction (PCR), repeat‐primed PCR, and long‐read sequencing.
Results
Pathogenic FGF14 GAA repeat expansions were detected in 12 patients from 11 unrelated families. The median size of the pathogenic GAA repeat was 309 repeats (range: 270–316 repeats). In these patients, the mean age of onset was 66.9 ± 9.6 years, with episodic symptoms observed in 56% of patients and parkinsonism in 30% of patients. We also detected FGF14 repeat expansions in a patient with a phenotype of multiple system atrophy, including cerebellar ataxia, parkinsonism, autonomic ataxia, and bilateral vocal cord paralysis. Brain magnetic resonance imaging (MRI) showed normal to mild cerebellar atrophy, and a follow‐up study conducted after a mean period of 6 years did not reveal any significant progression.
Discussion
This study highlights the importance of FGF14 GAA repeat analysis in patients with late‐onset cerebellar ataxia, particularly when they exhibit episodic symptoms, or their brain MRI shows no apparent cerebellar atrophy. Our findings contribute to a better understanding of the clinical variability of GAA‐FGF14‐related diseases.
Introduction
Late‐onset cerebellar ataxia belongs to a heterogeneous group of neurodegenerative disorders that are difficult to diagnose molecularly. Recent research has made groundbreaking discoveries in understanding the genetic basis of these ataxias. Notably, a monoallelic GAA repeat expansion within the first intron of the fibroblast growth factor 14 (FGF14) gene has emerged as a major genetic factor associated with late‐onset ataxia, alongside the biallelic repeat expansions in the RFC1 gene. 1 , 2 , 3 The GAA expansion within the FGF14 gene is responsible for the development of spinocerebellar ataxia 27B (SCA27B), which has been detected in a significant proportion (10–61%) of patients with late‐onset ataxia across diverse ethnic backgrounds. 1 , 2 , 4 , 5 , 6 , 7 The high prevalence of this genetic abnormality highlights the need to identify potential treatment options. One study demonstrated that 4‐aminopyridine results in symptomatic improvement and thus has therapeutic potential. 4 Furthermore, GAA repeats in FGF14 have been detected in patients who exhibit RFC1‐negative cerebellar ataxia accompanied by neuropathy and vestibular areflexia syndrome. 6 This finding expands the clinical spectrum of GAA‐FGF14‐related diseases and underscores the potential phenotypic similarities with RFC1‐related disorders.
In Japan, approximately 73% of patients with hereditary ataxia, including late‐onset cerebellar ataxia, remain undiagnosed. 8 Based on recent advancements, we conducted a thorough analysis of the FGF14 GAA repeat expansion using a diagnostic strategy integrating long‐range polymerase chain reaction (LR‐PCR), repeat‐primed PCR (RP‐PCR), and long‐read sequencing using Oxford Nanopore Technology platforms. We successfully identified FGF14 pathogenic repeat expansions from 12 patients who exhibited remarkable clinical variability.
Methods
Enrollment criteria
We conducted a comprehensive study involving 1288 unrelated Japanese index patients with chronic progressive cerebellar ataxia who were referred to our laboratory for genetic testing. Among these cases, 101 were clinically suspected to have multiple system atrophy, cerebellar type (MSA‐C). All patients underwent examinations by their respective neurologists, and their clinical data along with blood samples were submitted. Blood samples were collected from medical clinics and institutions situated in western Japan, primarily from the Kyushu region (including Kagoshima, Miyazaki, Oita, Fukuoka, and Okinawa Prefectures) as well as the Ehime Prefecture. Approval for this study was obtained from the Institutional Review Board of Kagoshima University (Application ID: 490). Prior to their involvement, all participants provided informed consent to participate in this study.
Preliminary screening
In the initial screening phase, we examined the samples for repeat expansions associated with various hereditary ataxias, including SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, SCA17, SCA31, DRPLA, FXTAS, and PRNP gene point mutations. Additionally, whole‐exome sequencing was performed in a subset of undiagnosed patients using Ion Proton (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Subsequently, the remaining undiagnosed patients without a family history of autosomal dominant inheritance were screened for RFC1 repeat expansions. The detailed workflow has been described previously, 8 , 9 and the study flowchart is shown in Figure 1A. Age of onset distribution in our cerebellar ataxia cohort are summarized in Table S1.
Figure 1.
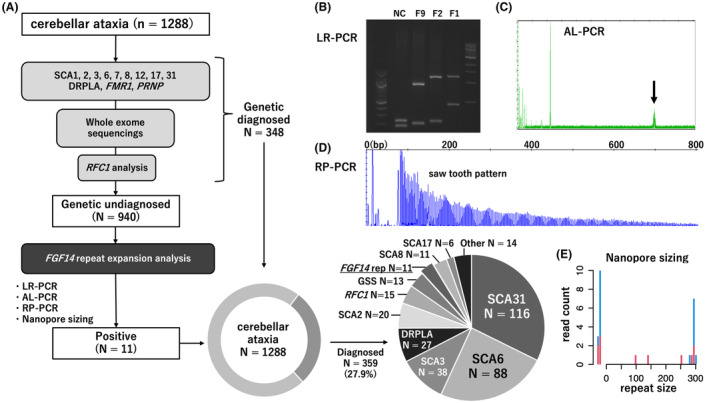
Flowchart, representative results of the FGF14 gene repeat analyses. (A) Comprehensive preliminary genetic screening of 1288 unrelated patients with cerebellar ataxia. The FGF14 repeat expansion was analyzed in 940 families/patients who tested negative after preliminary screening. FGF14 GAA repeat expansion was the eighth most frequent causative gene in our cohort. (B) PCR electrophoresis; NC, negative control. (C) Fluorescence amplicon length analysis PCR. (D) Saw‐tooth patterns obtained from repeat‐primed PCR. (E) Histogram of FGF14 repeat obtained from long‐read sequencing.
Molecular analysis of FGF14 GAA repeat expansion
For the 940 index patients who tested negative in the preliminary screening, we conducted analyses of FGF14 GAA repeat expansions. According to previous reports, we designed primers and performed RP‐PCR and fluorescence amplicon length analysis PCR (AL‐PCR). 1 , 2 , 5 All PCR products were subjected to capillary electrophoresis using the ABI PRISM 3130xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), and the results were visualized using the Peakscanner software (Applied Biosystems). Representative examples of PCR electrophoresis, AL‐PCR, and saw‐tooth patterns obtained from RP‐PCR are shown in Figure 1B–D. In accordance with previous studies, more than 250 GAA repeats were defined as pathogenic. 1 , 2
To accurately determine the repeat size in patients in whom the presence of GAA repeats was confirmed via the aforementioned analyses, we performed long‐read sequencing using the GridION platform (Oxford Nanopore Technologies, Oxford, UK) with the adaptive sampling option. The region of interest is listed in Data S1. Base calling of the acquired sequences was performed using Guppy in super accuracy mode. We used the tandem‐genotypes‐plot command to generate a histogram showing differences in the number of repeat units relative to the reference human genome (Fig. 1E). Repeat numbers were determined using consensus sequences generated using tandem‐genotypes‐merge and lamassemble 10 , 11 (Fig. 1F).
Statistical analysis
To compare frequencies or numerical variables, the Mann–Whitney U‐test and Pearson's correlation tests were utilized. A P < 0.05 was considered statistically significant. All statistical analyses were conducted using GraphPad Prism version 9.3.1 (GraphPad Software Inc., San Diego, CA, USA).
Results
Analysis of repeat expansion in FGF14
Pathogenic GAA repeats (>250) in FGF14 were identified in 11 unrelated families among the 940 index patients with cerebellar ataxia included in this study. A subsequent analysis revealed GAA repeats from a sister (F7‐II) of patient F7 (F7‐I). Among all these 12 patients, their GAA repeat numbers were evaluated using both AL‐PCR and nanopore sequencing, and their GAA repeats were found ranging from 270 to 361, with a mean value of 309 repeats (Table 1). We further conducted a correlation analysis, and verified a linear relationship between the product length obtained through AL‐PCR and actual repeat number revealed by nanopore sizing (Fig. 2A). Similar to previous reports, 5 the repeat size determined with nanopore sequencing was larger than expected based on the PCR product size, and this discrepancy became more pronounced with increasing repeat size (Fig. 2A). Altogether, FGF14 GAA repeat expansions were detected in 1.2% (11 out of 940) of index patients with genetically undiagnosed cerebellar ataxia. Therein, in a subgroup of patients with late‐onset (>30 years) cerebellar ataxia, but without a diagnosis of MSA, we identified FGF14 GAA repeats in 0.9% (9 out of 1151) of cases.
Table 1.
Clinical manifestation of 12 patients with FGF14 GAA repeat expansions.
| Patient | F1 | F2 | F3 | F4 | F5 | F6 | F7‐I | F7‐II | F8 | F9 | F10 | F11 | All patients |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repeat size | 110/361 | 24/354 | 8/341 | 24/347 | 8/340 | 23/315 | 25/272 | 25/288 | 8/273 | 16/273 | 16/272 | 204/270 | |
| Onset age (years) | 59 | 69 | 59 | 58 | 67 | 77 | 50 | 66 | NA | 79 | 78 | 74 | 66.9 ± 9.6 |
| Exam age (years) | 64 | 76 | 67 | 61 | 73 | 77 | 70 | 67 | NA | 83 | 86 | 75 | 7.8 |
| Gender | F | F | M | M | F | M | F | F | F | F | F | F | Female 9 |
| Family history | − | − | − | − | − | − | + | NA | − | − | − | Sporadic 10 | |
| Cerebellar ataxia | + | + | + | + | + | + | + | + | NA | + | + | + | 11/11 (100%) |
| Nystagmus | + | + | + | + | + | + | + | + | NA | + | − | + | 10/11 (91%) |
| Dysarthria | + | + | − | + | + | + | + | + | NA | + | + | + | 10/11 (91%) |
| Limb ataxia | + | + | + | ± | + | + | + | + | NA | + | + | + | 11/11 (100%) |
| Trancal ataxia | + | − | + | + | + | + | + | + | NA | + | + | + | 10/11 (91%) |
| Episodic symptom | − | NA | − | + | + | NA | + | − | NA | + | + | − | 5/9 (56%) |
| Muscle weakness/atrophy | − | − | − | − | NA | NA | − | − | NA | − | − | − | 0/9 (0%) |
| Deep tendon reflex | Hypo | Normal | Normal | Normal | Slight hyper | Normal | Normal | Hyper | NA | Hyper | Hypo | Hypo | NA |
| Sensory disturbance | + | − | − | − | + | NA | − | − | NA | − | − | − | 2/10 (20%) |
| Pyramidal sign | − | − | − | − | − | Bilateral Babinski+ | − | − | NA | − | − | − | 1/11 (9%) |
| Parkinsonism | − | − | + | − | NA | − | − | − | NA | + | + | − | 3/10 (30%) |
| Cognitive impairment | − | NA | − | − | NA | − | − | − | NA | MCI | − | − | 1/9 (11%) |
| Dysautonomia | − | − | Erectile dysfunction | − | NA | − | − | − | NA | Hypohidrosis OH, constipation | − | − | 2/10 (20%) |
| Other | Diabetes mellitus | − | − | − | NA | NA | − | Diabetes Mellitus | NA | Bilateral vocal cord paralysis | − | − | NA |
| Brain MRI | |||||||||||||
| Cerebellar atrophy | ± | + | − | ± | + | + | + | + | NA | ± | ± | ± | + 5/11 (45%) ± 5/11 (45%) |
| Brain stem atrophy | − | − | − | − | − | − | − | − | NA | − | − | − | 0/11 (0%) |
| Cerebral atrophy | − | − | Frontal temporal | − | − | Frontal | − | − | NA | − | Frontal | − | 3/11 (27%) |
| SPECT | − | − | ECD‐SPECT No decrease in cerebellar blood flow | − | − | − | IMP‐SPECT reduced in cerebellar blood flow | ECD‐SPECT reduced in cerebellar blood flow | NA | DaTscan: reduced in striatal uptake | IMP‐SPECT No decrease in cerebellar blood flow | − | NA |
M, male; F, female; NA, not available; MCI, mild cognitive impairment; OH, orthostatic hypotension.
Figure 2.
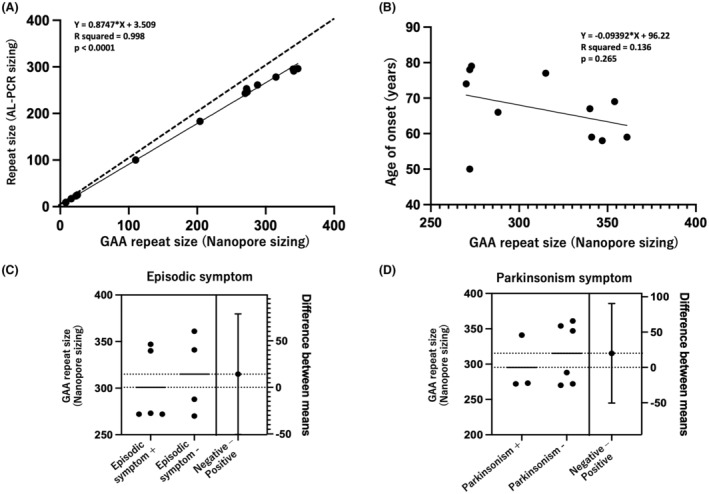
Statistical analysis of genetic findings and clinical findings. (A) Correlation analysis indicates a linear relationship between the product length in FGF14 obtained through AL‐PCR and actual repeat number revealed by nanopore sizing. (B) No significant correlation is identified between the onset age and repeat number (Pearson's correlation test, P > 0.05). (C, D) No significant difference is observed in repeat numbers between the groups with positive and negative cases of episodic ataxia or parkinsonism (Mann–Whitney U‐test, P > 0.05).
In three patients for whom a long product was obtained using LR‐PCR and AL‐PCR, but not RP‐PCR, long‐read sequencing revealed a unique repeat genotype in FGF14: (GAA)22/(GAA)22[(GAA)1–4(GCA)1–3]151(GAA)119, (GAA)8/(GAA)10[(GAA)1–5(GCA)1–2]107(GAA)71, (GAA)23/(GAA)22[(GAA)1–4(GCA)1–2]202(GAA)69, respectively. Previous reports observed (GAAGGA) n repeats and [(GAA)4(GCA)1] n repeats in the control group, suggesting that the non‐GAA expansions at the FGF14 locus are not associated with late‐onset cerebellar ataxia. 1 , 2 Therefore, we classified this repeat expansion as nonpathogenic.
Clinical summary
Table 1 summarizes the clinical findings of the 12 patients with FGF14 pathogenic repeat expansions. These expansions were sporadic in 10 of the 11 families (91%), and only 1 family (9%) had a positive family history (F7; Data S2). The mean age of disease onset was 66.9 ± 9.6 years (range: 50–79 years), and 75% of the patients were women. All patients exhibited cerebellar ataxia, of which 56% (5 out of 9) showed episodic symptoms, including episodic ataxia and episodic symptomatic fluctuation. Additionally, parkinsonism was observed in three patients: patient F3 presented with non‐levodopa‐responsive muscle rigidity, resting tremor, and a shuffling gait; patient F9 exhibited muscle rigidity and shuffling gait, but no administration of levodopa; patient F10 displayed non‐levodopa‐responsive muscle rigidity, bradykinesia, and a shuffling gait. Autonomic dysfunction in two patients, while motor neuropathic symptoms, such as muscle weakness/atrophy, were not observed.
Patient F10, who presented with cerebellar ataxia, parkinsonism, and autonomic failure, also developed bilateral vocal cord palsy. Although brain magnetic resonance imaging (MRI) showed only slight cerebellar atrophy, the diagnosis of MSA was made based on clinical features. The detailed clinical course of this patient is described in Data S3 and Figure S1.
Electrophysiological analyses were performed on two patients, which revealed a slight decrease in median sensory nerve conduction velocity and tibial motor nerve conduction velocity (Table S2).
Genotype–phenotype correlation
No significant correlation was observed between the age of onset and repeat size (P = 0.27, Pearson's correlation, Fig. 2C). Furthermore, no significant differences in repeat size were observed in patients with or without episodic symptoms (P = 0.62) or parkinsonism (P = 0.52) (Mann–Whitney U‐test; Fig. 2D, E).
Radiological findings
Brain MRI data were available for 11 of the 12 patients and showed mild cerebellar atrophy (n = 5), slight cerebellar atrophy or almost normal (n = 5), and no cerebellar atrophy (n = 1) (Table 1, Fig. 3A, Figure S2). None of the patients exhibited brainstem atrophy. Single‐photon emission computed tomography of patients F7‐I and II who had mild cerebellar atrophy showed hypoperfusion confined to the cerebellum (Fig. 3B, C, Figure S3A,B). However, two patients did not show cerebellar hypoperfusion (F3 and F10, data not shown). DaTscan (123I‐ioflupane single‐photon emission computed tomography) of patient F9 with parkinsonism showed reduced striatal uptake of dopamine transporter (Fig. 3D, Figure S3C). These radiological characteristics are summarized in Table 1.
Figure 3.
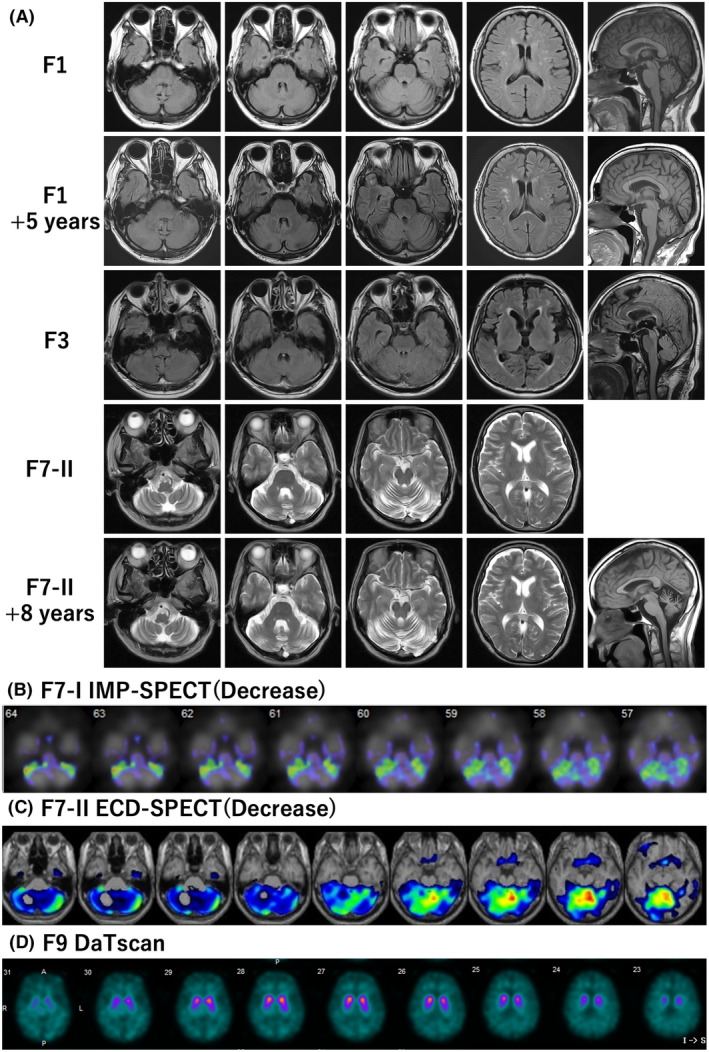
Brain MRI and scintigraphy of patients with FGF14 GAA repeat expansions. (A) Brain MRI of F1, 3, and 7‐II. F1 and F7‐II exhibit slight and mild cerebellar atrophy, respectively. While F3 shows no signs of cerebellar atrophy. Brain MRI follow‐up indicates minimal longitudinal progression of cerebellar atrophy. Brain MRI follow‐up showed minimal longitudinal progression of cerebellar atrophy. (B, C) Isopropyl‐p‐iodoamphetamine (IMP)‐single‐photon emission computed tomography (SPECT) of patient F7‐I and ethylene cysteinate dimer (ECD)‐SPECT of F7‐II. In both patients, hypoperfusion can be observed, specifically limited to the cerebellum. (D) DaTscan (123I‐Ioflupane SPECT) of patient F9. A decrease of striatal uptake is evident. All images depicting these radiological findings are available in Figure S1 and S2.
Brain MRI follow‐up studies were conducted in seven patients, with a mean interval of 6 years (range: 4–8 years). Throughout this observation period, none of the patients showed significant progression of cerebellar atrophy (Fig. 3, Figure S2).
Discussion
In our study, which involved a case series of 940 index patients with undiagnosed cerebellar ataxia, we identified pathogenic FGF14 repeat expansions in 11 unrelated families. This finding represents advancements in our diagnostic rate and expands the genetic spectrum within our case series of cerebellar ataxia.
FGF14 point mutations have been reported to cause SCA27 and episodic ataxia type 9, respectively. 12 , 13 More recently, an autosomal dominant expansion of GAA repeats in FGF14 emerged as a common cause of hereditary ataxia, particularly late‐onset cerebellar ataxia (SCA27B, MIM 620174). 1 , 2 The initial report uncovered a complete penetrance of (GAA)>300 expansions and an incomplete penetrance of (GAA)250–300 expansions in FGF14. 1 SCA27B due to FGF14 GAA repeats has been identified in ataxia cohorts from various ethnic backgrounds, including French–Canadian, German, Australian, Indian, and French populations. 1 , 2 , 4 , 5 , 6 , 7 Additionally, a family lineage with a monoallelic FGF14 GAA repeat and biallelic RFC1 AAGGG repeats was reported from Chile. 14 Taken together, these findings suggest that FGF14 is implicated in cerebellar ataxia across diverse ethnic groups. However, FGF14 repeat expansions have not been documented so far in the Japanese cerebellar ataxia cohort whose genetic characteristics remained unclear.
In this study, we detected FGF14 GAA repeat expansions in 1.2% (11 out of 940) of index patients with undiagnosed cerebellar ataxia. This frequency is lower than that reported in previous studies. Pellerin et al. reported frequencies of 61%, 18%, 15%, and 10% in French‐Canadian, German, Australian, and Indian cohorts, respectively. 1 The occurrence of GAA‐FGF14‐related disease may be relatively low in Asian countries, including Japan. However, it is important to acknowledge the impact of potential patient enrollment bias as a limitation of our study. Specifically, our case series included approximately 100 patients with MSA, and it is possible that it also included patients with nonhereditary ataxia, such as immune‐mediated cerebellar ataxia. These factors might have contributed to the observed low frequency of GAA‐FGF14‐related disease in our study.
An examination of the clinical records of the 12 patients with FGF14 GAA repeat expansions revealed that most patients exhibited a late‐onset pure cerebellar form of the disease, with episodic symptom fluctuations observed in 56% of patients. Although the age of onset has been reported to have an inverse correlation with the size of FGF14 GAA repeats in certain studies, 1 , 2 , 5 , 6 other study has reported no correlation. 5 In our statistical analysis, we observed a trend suggesting decreasing age at onset with increasing repeat size; however, this trend did not reach statistical significance. Episodic symptoms have been previously observed in 13–59% of patients with GAA‐FGF14‐related disease, representing a characteristic feature of the condition. 1 , 4 , 6
Additionally, we identified Parkinsonism in 30% (3/10) of our patients. Whole exome analysis was conducted on these patients and no known gene mutations associated with parkinsonism were detected. Parkinsonism is considered a rare symptom in GAA‐FGF14‐related disorders, 7 and thus far, only one patient with bradykinesia has been reported. 6 Furthermore, we identified the FGF14 pathogenic repeat expansion in a patient who was clinically diagnosed with MSA‐C, characterized by cerebellar ataxia, parkinsonism, autonomic neuropathy, and bilateral vocal cord palsy. Despite the penetrance of (GAA) expansions spanning between 250 and 300 may not be complete, this patient carrying 273 (GAA) expansions, exhibited clinical and radiological characteristics consistent with SCA27B, in contrast to her initial clinical diagnosis of MSA‐C. Therefore, it is advisable to consider genetic screening for FGF14 repeat expansions in patients with such clinical symptoms.
We provided detailed radiographic findings for 11 patients with GAA‐FGF14‐related diseases. Brain MRI revealed variable extents of cerebellar atrophy, which were classified as mild (n = 5), slight or nearly normal (n = 5), and completely normal (n = 1). None of these patients displayed brainstem atrophy. Furthermore, after a mean follow‐up period of 6 years, brain MRI did not reveal any notable progression of cerebellar atrophy. Cerebellar atrophy in GAA‐FGF14‐related disease has been reported to be mild. 7 Previous MRI follow‐up results in one case indicated only mild progression of cerebellar atrophy. 4 Our longitudinal MRI analysis further supports and reinforces these findings. Cerebellar neuropathology in GAA‐FGF14‐related disease typically manifests as cerebellar cortical atrophy, predominantly affecting the vermis, with less involvement of the hemispheres. 4 Therefore, attention should be directed toward the cerebellar vermis when examining brain MRI to detect cerebellar atrophy in these patients. However, it is important to note that there might be patients in whom cerebellar atrophy is minimal or absent over prolonged periods of time.
Our diagnostic strategy, which included LR‐PCR, AL‐PCR, RP‐PCR, and long‐read sequencings, has shown promising results. The utilization of adaptive sampling with GridION has enabled us to rule out the possibilities of other known repeat expansion disorders simultaneously. Limitations of the current study comprise incomplete clinical data by local hospitals and insufficient segregation analysis, particularly in patients without a family history.
In conclusion, our study identified FGF14 GAA repeat expansions in 1.2% of a large Japanese case series of patients with genetically undiagnosed cerebellar ataxia. This is the first report of GAA‐FGF14‐related disease in Japan. Repeat expansion analysis of FGF14 is recommended for patients presenting with late‐onset cerebellar symptoms, especially when accompanied by episodic symptoms or when brain MRI reveals only mild cerebellar atrophy or normal findings. The clinical variability observed in our patients, particularly the undescribed phenotype of MSA, has expanded the clinical spectrum of GAA‐FGF14‐related diseases.
Author contributions
Masahiro Ando, Yujiro Higuchi, and Hiroshi Takashima conceived the project and designed the study. Masahiro Ando, Yujiro Higuchi, Junhui Yuan, Akiko Yoshimura, and Fumikazu Kojima contributed to the analysis and interpretation of data. Yuki Yamanishi, Yasuhiro Aso, Kotaro Izumi, Minako Imada, Yoshimitsu Maki, Hiroto Nakagawa, Takahiro Hobara, Yutaka Noguchi, Jun Takei, Yujiro Higuchi, Satoshi Nozuma, Yusuke Sakiyama, Akihiro Hashiguchi, Eiji Matsuura, and Yuji Okamoto participated in analysis of clinical data. Masahiro Ando produced the original manuscript and all authors approved the final version.
Funding information
This work was supported by Grants‐in‐Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (201610002B). This research was also supported by JSPS KAKENHI Grant Numbers JP18H02742, JP20K16604, JP21K15702, JP21H02842, JP22K15713, JP22K07495, JP22K07519, and JP23K06931.
Conflict of interest
All authors declare that there is no conflict of interest.
Supporting information
Table S1. Age of onset distribution in our cerebellar ataxia cohort.
Data S1. The region of interest used for adaptive sampling option.
Data S2. A family tree (F7) with a family history.
Data S3. The clinical course of F9 (FGF14 16/273 repeats).
Data S4. Laryngeal fiberscope findings in F9.
Table S2. Electrophysiological findings of F1 and F9.
Figure S1. Brain MRI of all patients with FGF14 GAA repeat expansions and longitudinal follow‐up.
Figure S2. Full scintigraphic image of patients with FGF14 GAA repeat expansions.
Acknowledgments
The authors thank all the patients and their families for participating in this study. The authors are supported by Enago (www.enago.jp) for reviewing the English in this report. The authors extend their appreciation to the Division of Gene Research, Research Support Centre, Kagoshima University, for the use of their facilities.
First Author: Masahiro Ando
Funding Statement
This work was funded by JSPS KAKENHI grants JP18H02742, JP20K16604, JP21H02842, JP21K15702, JP22K07495, JP22K07519, JP22K15713, and JP23K06931; Ministry of Health, Labour and Welfare, Japan grant 201610002B; Research Committee of Ataxia, Health Labour Sciences Research.
Data availability statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Pellerin D, Danzi MC, Wilke C, et al. Deep intronic FGF14 GAA repeat expansion in late‐onset cerebellar ataxia. N Engl J Med. 2023;388(2):128‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rafehi H, Read J, Szmulewicz DJ, et al. An intronic GAA repeat expansion in FGF14 causes the autosomal‐dominant adult‐onset ataxia SCA50/ATX‐FGF14. Am J Hum Genet. 2023;110(1):105‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late‐onset ataxia. Nat Genet. 2019;51(4):649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilke C, Pellerin D, Mengel D, et al. GAA‐FGF14 ataxia (SCA27B): phenotypic profile, natural history progression and 4‐aminopyridine treatment response. Brain. 2023;146:4144‐4157. doi: 10.1093/brain/awad157 [DOI] [PubMed] [Google Scholar]
- 5. Bonnet C, Pellerin D, Roth V, et al. Optimized testing strategy for the diagnosis of GAA‐FGF14 ataxia/spinocerebellar ataxia 27B. Sci Rep. 2023;13(1):9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pellerin D, Wilke C, Traschütz A, et al. Intronic FGF14 GAA repeat expansions are a common cause of ataxia syndromes with neuropathy and bilateral vestibulopathy. J Neurol Neurosurg Psychiatry. 2023:30:jnnp‐2023‐331490. doi: 10.1136/jnnp-2023-331490. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wirth T, Clément G, Delvallée C, et al. Natural history and phenotypic spectrum of GAA‐FGF14 sporadic late‐onset cerebellar ataxia (SCA27B). Mov Disord. 2023;38(10):1950‐1956. doi: 10.1002/mds.29560 [DOI] [PubMed] [Google Scholar]
- 8. Ando M, Higuchi Y, Yuan JH, et al. Genetic and clinical features of cerebellar ataxia with RFC1 biallelic repeat expansions in Japan. Front Neurol. 2022;13:952493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higuchi Y, Ando M, Yoshimura A, et al. Prevalence of fragile X‐associated tremor/ataxia syndrome in patients with cerebellar ataxia in Japan. Cerebellum. 2022;21(5):851‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitsuhashi S, Frith MC, Mizuguchi T, et al. Tandem‐genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 2019;20(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miyatake S, Koshimizu E, Fujita A, et al. Rapid and comprehensive diagnostic method for repeat expansion diseases using nanopore sequencing. NPJ Genom Med. 2022;7(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Swieten JC, Brusse E, de Graaf BM, et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia. Am J Hum Genet. 2003;72(1):191‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piarroux J, Riant F, Humbertclaude V, et al. FGF14‐related episodic ataxia: delineating the phenotype of Episodic Ataxia type 9. Ann Clin Transl Neurol. 2020;7(4):565‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saffie Awad P, Lohmann K, Hirmas Y, et al. Shaking up ataxia: FGF14 and RFC1 repeat expansions in affected and unaffected members of a Chilean family. Mov Disord. 2023;38:1107‐1109. doi: 10.1002/mds.29390 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Age of onset distribution in our cerebellar ataxia cohort.
Data S1. The region of interest used for adaptive sampling option.
Data S2. A family tree (F7) with a family history.
Data S3. The clinical course of F9 (FGF14 16/273 repeats).
Data S4. Laryngeal fiberscope findings in F9.
Table S2. Electrophysiological findings of F1 and F9.
Figure S1. Brain MRI of all patients with FGF14 GAA repeat expansions and longitudinal follow‐up.
Figure S2. Full scintigraphic image of patients with FGF14 GAA repeat expansions.
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.