

Pearls
“Leber hereditary optic neuropathy (LHON-Plus)” is a phenotype of LHON that is characterized by extraocular neurologic manifestations, which may be the first manifestations of the disease.
Extraocular manifestations of LHON-Plus can include movement disorders, peripheral neuropathy, seizures, and demyelinating-like syndromes.
Oy-sters
LHON-Plus can be highly difficult to distinguish from acquired demyelinating diseases based on radiologic findings alone, although a predilection for the dorsal columns in the spinal cord has been noted in LHON-Plus.
In rare cases, the co-occurrence of aquaporin-4 (AQP4+) NMOSD and LHON has been reported.
Case Report
A 2-year-old generally healthy White girl with 1 month of hyperphagia associated with weight gain from the 90th percentile to the 98th percentile for age, along with fatigue, incoordination, and hiccups, presented to the emergency department with a transient episode of unresponsiveness. Review of systems was negative for recent febrile illness, diarrhea, rash, or travel. Family history was notable for a strong maternal history of Leber hereditary optic neuropathy (LHON; Figure 1). Examination displayed encephalopathy and hypoxemia to 70% SpO2; chemistry panel was pertinent for hyponatremia to 123 mmol/L. Complete blood count, urinalysis, and urine drug screen were unremarkable. Head CT was normal. While in the emergency department, she had a second witnessed episode of unresponsiveness concerning for seizure, resulting in intubation and admission to the intensive care unit (ICU).
Figure 1. Genetic Pedigree With Age in Years and Designation of Individuals With LHON.
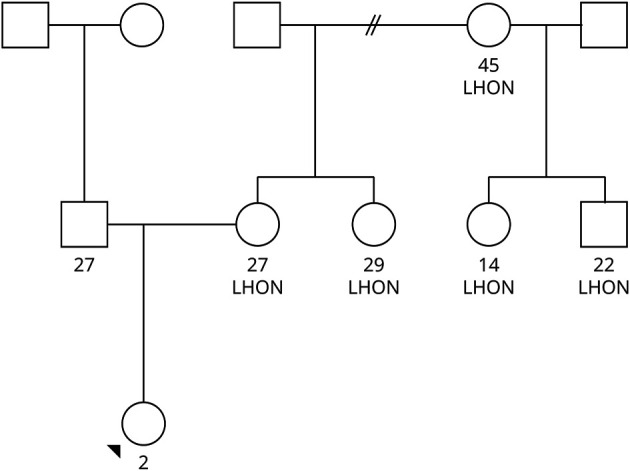
The patient is marked with a black arrow.
The patient's ICU course was complicated by ongoing respiratory failure and neurogenic bowel and bladder. She was noted to have ataxic respiration, cerebral salt wasting, and dysautonomia causing fevers and hypertension. Extended EEG monitoring showed diffuse slowing, but no epileptiform discharges. Extensive initial workup, including sepsis evaluation; routine CSF studies; CSF metabolites; rheumatologic laboratory tests; CSF meningitis-encephalitis panel; CSF herpes simplex virus; enterovirus PCR; metabolic screening laboratory tests; and endocrinologic testing of the thyroid, glucocorticoid, and gonadotropin axes, was unrevealing. Brain and spine MRI revealed T2 hyperintense lesions of the right substantia nigra, bilateral inferior colliculi, dorsal medulla, and most of the cervical and thoracic spinal cord, favored to represent demyelination (Figure 2). Chest, abdominal, and pelvic CT with contrast was normal. She was then transferred to another hospital for ongoing care.
Figure 2. Brain and Spine Imaging.

Brain MRI showing T2 fluid-attenuated inversion recovery signal abnormality (arrow) in the bilateral dorsal medulla in the axial (A), coronal (B), and sagittal (C) planes. Sagittal MRI of the spinal cord demonstrating a longitudinally extensive T2 hyperintense signal abnormality (arrow) in the cervical spinal cord (D).
Further workup was initiated after hospital transfer. CSF oligoclonal bands (OCBs), serum anti-myelin oligodendrocyte (MOG), and anti-aquaporin 4 (AQP4) antibodies by a cell-based assay were negative. Dilated fundoscopic examination by ophthalmology was documented as normal and without disc pallor. She was found to have positive low-titer anti-NMDA receptor (NMDAR) serum antibodies at 1:10; however, CSF anti-NMDAR titers were negative. Given her recent history of hyperphagia and weight gain, Prader-Willi methylation studies, congenital hypoventilation, and obesity panels were sent, disclosing a variant of uncertain significance in the MAGE family member L2 (MAGEL2) gene concerning for Schaaf-Yang syndrome. Her case was discussed with several neuroimmunologists at various institutions. The temporal course of acute neurologic symptoms, combined with brain and spinal cord lesions, were suggestive of an acquired CNS demyelinating disease, including neuromyelitis optica spectrum disorder (NMOSD), acute disseminated encephalomyelitis, or myelin oligodendrocyte glycoprotein–associated disorder (MOGAD). Owing to the constellation of diencephalic and area postrema syndromes, longitudinally extensive transverse myelitis, and negative anti-AQP4 and anti-MOG antibodies, she was given a provisional diagnosis of antibody-negative NMOSD. Her treatment course consisted of daily intravenous methylprednisolone 30 mg/kg for 5 days, 10 exchanges of plasmapheresis, rituximab 375 mg/m2, and a 30-day prednisolone taper. Repeat brain MRI 2 weeks later demonstrated improvement of the aforementioned T2 hyperintense lesions, as well as new T2 hyperintense lesions of bilateral cranial nerves VII and VIII. Auditory brainstem response testing was normal. She was gradually weaned off respiratory support on ICU day 19 and was transferred to the floor. Her mental status gradually improved, and she was eventually discharged with neuroimmunology follow-up.
After discharge, the patient's symptoms continued to resolve, noting only residual fatigue and incoordination. Given the patient's family history of LHON, she was referred to genetics and ophthalmology, but due to the patient's mother's own poor vision attributable to LHON, transportation barriers did not allow family to make these appointments. Repeat brain and spine MRI demonstrated further improvement of the previously noted right substantia nigra, brainstem, and spinal cord lesions. Repeat anti-MOG and anti-AQP4 antibodies were negative. After extensive multi-institutional discussions, the variant of uncertain significance in the MAGEL2 gene was thought to be benign and her low serum NMDA antibody titer was considered incidental. She continued to undergo scheduled rituximab infusions with no recurrence of symptoms. After 2 years, the patient was able to undergo genetic mitochondrial testing, which revealed a pathogenic point sequence variant in m.14484T > C (MT-ND6,p.Met64Val). Repeat dilated ophthalmologic examination now revealed mild decreased visual acuity bilaterally and optic nerve pallor. Retinal nerve fiber layer testing showed severe bilateral thinning. Given her strong family history of LHON, bilateral optic nerve atrophy, and persistently negative anti-AQP4 antibodies, her diagnosis was revised to LHON-Plus. After shared decision making with the patient's family, rituximab treatment was discontinued with a plan to serially monitor anti-AQP4 antibodies for potential seroconversion.
Discussion
LHON is the most common mitochondrial disease. It characteristically causes sequential optic neuropathy, most commonly in young adults, which progresses over weeks to months and leads to blindness.1 A minority of cases have childhood onset, and it has been suggested that these patients may have slower progression of vision loss compared with teenagers and adults.2 Several genetic variants in mitochondrial genes encoding proteins in complex I of the electron transport chain have been implicated in the disease, with the most common being point sequence variants including m.3460G > A (MT-ND1, p.Ala52Thr), m.11778G > A (MT-ND4, p.Arg340His), m.14484T > C (MT-ND6,p.Met64Val), and m.3460G > A (MT-ND1, p.Ala52Thr). The disease is inherited in a matrilineal fashion, but displays incomplete penetrance due to heteroplasmy and has a male predominance.3,4 No FDA-approved therapy exists currently, although the coenzyme-Q10 analog idebenone is approved for the treatment of LHON in Europe, and gene therapy with adenovirus vectors is in development.5,6
“LHON-Plus” is a designation given to patients with LHON who display extraocular manifestations attributable to the disease. Neurologic manifestations in LHON-Plus include seizures, movement disorders, paraplegia, encephalopathy, peripheral neuropathy, and myopathy. In a case series of patients with LHON (n = 46), 59% (n = 27) had neurologic signs or extraocular symptoms.7 These protean manifestations are perhaps unsurprising, given the ubiquity of the electron transport chain, and likely reflect preferential involvement of metabolically active organs, such as the brain and muscle. It is interesting to note that there are reports of LHON-Plus with brain and spinal cord matter lesions mimicking acquired demyelinating diseases, such as multiple sclerosis (MS), NMOSD, and MOGAD.8,9 Such presentations can be highly difficult to distinguish from acquired demyelinating diseases based on radiologic findings alone, although a predilection for the dorsal columns in the spinal cord has been noted in LHON-Plus.10 In these patients, the diagnosis of LHON-Plus was made after pathogenic sequence variants consistent with LHON-Plus were revealed and disease markers such as CSF OCBs or serum anti-AQ4 and anti-MOG antibodies were negative.
While there are multiple pediatric case reports of LHON-Plus, to our knowledge our patient is the youngest to present with neurologic symptoms consistent with the disease.11,12 Our patient's presentation was dominated by severe encephalopathy with an associated medullary lesion, which, combined with the patient's very young age and inability to participate in the visual examination, likely obfuscated the trademark vision loss of LHON. However, her monophasic course, presumed lack of ocular pain traditionally associated with optic neuritis, and family history of LHON were clues to her diagnosis. Although LHON and NMO are both quite rare, there have been case reports of patients with both pathogenic mtDNA sequence variants for LHON and AQP4+ NMOSD.11,13 It has been hypothesized that mtDNA sequence variants might be associated with AQP4 autoimmunity and predispose to NMO; however, this remains to be clarified.13 In our patient, anti-AQP4 antibodies will continue to be monitored for potential seroconversion.
This case illustrates several important points. First, while LHON is traditionally associated with optic neuropathy, it can also cause a myriad of neurologic symptoms involving the central and peripheral nervous systems. Young patients who will eventually be diagnosed with LHON-Plus may be too young to endorse visual symptoms or cooperate with formal visual acuity testing, and as a result will first present with other neurologic signs or symptoms. Second, LHON-Plus can mimic acquired demyelinating diseases, such as MS, NMOSD, and MOGAD, and should be kept in the differential for patients with progressive vision loss, especially if accompanied by demyelinating-like lesions in the brain and spinal cord. In such cases, a retrospective clue to a diagnosis of LHON-Plus may be the lack of recurrent attacks often seen in multiphasic acquired demyelinating diseases. Moreover, the rarity of each entity, NMOSD and LHON, does not preclude the co-occurrence of both entities, as has been described in rare adult cases.12 Finally, it is important to emphasize that LHON is a genetic diagnosis; accordingly, a diagnosis of LHON-Plus is contingent upon appropriate genetic testing and ruling out possible concomitant neurologic illnesses.
Acknowledgment
The authors thank care providers in the Nationwide Children's Hospital multidisciplinary neuroimmunology clinic, including Dr. Alana Leaver, PsyD, who provided direct patient care, as well as Paola Loreto Palacio, who participated in research enrollment.
Glossary
- AQP4
aquaporin-4
- ICU
intensive care unit
- LHON
Leber hereditary optic neuropathy
- MAGEL-2
MAGE Family Member L2
- MOG
myelin oligodendrocyte glycoprotein
- MOGAD
myelin oligodendrocyte glycoprotein antibody–associated disease
- MS
multiple sclerosis
- NMDAR
NMDA receptor
- NMOSD
neuromyelitis optica spectrum disorder
- OCB
oligoclonal band
Appendix. Authors
| Name | Location | Contribution |
| Alex Sunshine, MD, PhD | Neurology, Nationwide Children's Hospital | Drafting/revision of the manuscript for content, including medical writing for content |
| Quinton J Mandle, MD | Neurology, Nationwide Children's Hospital | Drafting/revision of the manuscript for content, including medical writing for content |
| Ana M Cabal Herrera, MD | Pediatrics, Nationwide Children's Hospital | Drafting/revision of the manuscript for content, including medical writing for content |
| Bianca Zapanta, DO | Genetics, Nationwide Children's Hospital | Major role in the acquisition of data |
| Hersh Varma, MD | Ophthalmology, Nationwide Children's Hospital | Major role in the acquisition of data |
| Setty Magaña, MD, PhD | Neurology, Nationwide Children's Hospital | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the NIH under award number K12NS098482 (S. Magaña).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Manickam AH, Michael MJ, Ramasamy S. Mitochondrial genetics and therapeutic overview of Leber's hereditary optic neuropathy. Indian J Ophthalmol. 2017;65(11):1087-1092. doi: 10.4103/ijo.IJO_358_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barboni P, La Morgia C, Cascavilla ML, et al. Childhood-onset leber hereditary optic neuropathy–clinical and prognostic insights. Am J Ophthalmol. 2023;249:99-107. doi: 10.1016/j.ajo.2022.12.014 [DOI] [PubMed] [Google Scholar]
- 3.Sundaramurthy S, SelvaKumar A, Ching J, Dharani V, Sarangapani S, Yu-Wai-Man P. Leber hereditary optic neuropathy—new insights and old challenges. Graefes Arch Clin Exp Ophthalmol. 2020;259(9):2461-2472. doi: 10.1007/s00417-020-04993-1 [DOI] [PubMed] [Google Scholar]
- 4.Berardo A, Emmanuele V, Vargas W, Tanji K, Naini A, Hirano M. Leber hereditary optic neuropathy plus dystonia, and transverse myelitis due to double mutations in MT-ND4 and MT-ND6. J Neurol. 2020;267(3):823-829. doi: 10.1007/s00415-019-09619-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guy J, Qi X, Pallotti F, Schon E, Manfredi G. Rescue of a Mitochondrial Deficiency Causing Leber Hereditary Optic Neuropathy. Wiley Online Library; 2002. Accessed March 10, 2023. [DOI] [PubMed] [Google Scholar]
- 6.Carelli V, Carbonelli M, de Coo IF, et al. International consensus statement on the clinical and therapeutic management of leber hereditary optic neuropathy. J Neuro Ophthalmol. 2017;37(4):371-381. doi: 10.1097/WNO.0000000000000570 [DOI] [PubMed] [Google Scholar]
- 7.Nikoskelainen EK, Marttila RJ, Huoponen K, et al. Leber's “plus”: neurological abnormalities in patients with Leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 1995;59(2):160-164. doi: 10.1136/jnnp.59.2.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birnbaum FA, Gospe SM. Severe leber hereditary optic neuropathy plus disease in a middle-aged man. J Neuro Ophthalmol. 2021;41(4):e715-e717. doi: 10.1097/WNO.0000000000001151 [DOI] [PubMed] [Google Scholar]
- 9.Mansukhani SA, Mehta DG, Renaud DL, Whealy MA, Chen JJ, Bhatti MT. Nuclear DNA mutation causing a phenotypic leber hereditary optic neuropathy plus. Ophthalmology. 2021;128(4):628-631. doi: 10.1016/j.ophtha.2020.09.011 [DOI] [PubMed] [Google Scholar]
- 10.McClelland CM, Van Stavern GP, Tselis AC. Leber hereditary optic neuropathy mimicking neuromyelitis optica. J Neuro Ophthalmol. 2011;31(3):265-268. doi: 10.1097/WNO.0b013e318225247b [DOI] [PubMed] [Google Scholar]
- 11.Shiraishi W, Hayashi S, Kira J. A Case of Neuromyelitis Optica Harboring Both Anti-aquaporin-4 Antibodies and a Pathogenic Mitochondrial DNA Mutation for Leber's Hereditary Optic Neuropathy. Sage Journals; 2013. Accessed March 10, 2023. journals.sagepub.com/doi/10.1177/1352458513513057 [DOI] [PubMed] [Google Scholar]
- 12.Simão LM. Neuromyelitis optica antibody in Leber hereditary optic neuropathy: case report. Arq Bras Oftalmol. 2012;75(4):280-282. doi: 10.1590/s0004-27492012000400013 [DOI] [PubMed] [Google Scholar]
- 13.Dujmovic I, Jancic J, Dobricic V, et al. Are Leber's mitochondrial DNA mutations associated with aquaporin-4 autoimmunity? Mult Scler. 2016;22(3):393-394. doi: 10.1177/1352458515590649 [DOI] [PubMed] [Google Scholar]