

Abstract
Analogues of the P2 receptor antagonists pyridoxal-5′-phosphate and the 6-azophenyl-2′,4′-disulfonate derivative (PPADS), in which the phosphate group was cyclized by esterification to a CH2OH group at the 4-position, were synthesized. The cyclic pyridoxine-α4,5-monophosphate, compound 2 (MRS 2219), was found to be a selective potentiator of ATP-evoked responses at rat P2X1 receptors with an EC50 value of 5.9 ± 1.8 μM, while the corresponding 6-azophenyl-2′,5′-disulfonate derivative, compound 3 (MRS 2220), was a selective antagonist. The potency of compound 3 at the recombinant P2X1 receptor (IC50 10.2 ± 2.6 μM) was lower than PPADS (IC50 98.5 ± 5.5 nM) or iso-PPADS (IC50 42.5 ± 17.5 nM), although unlike PPADS its effect was reversible with washout and surmountable. Compound 3 showed weak antagonistic activity at the rat P2X3 receptor (IC50 58.3 ± 0.1 μM), while at recombinant rat P2X2 and P2X4 receptors no enhancing or antagonistic properties were evident. Compounds 2 and 3 were found to be inactive as either agonists or antagonists at the phospholipase C-coupled P2Y1 receptor of turkey erythrocytes, at recombinant human P2Y2 and P2Y4 receptors, and at recombinant rat P2Y6 receptors. Similarly, compounds 2 and 3 did not have measurable affinity at adenosine A1, A2A, or A3 receptors. The lack of an aldehyde group in these derivatives indicates that Schiff’s base formation with the P2X1 receptor is not necessarily required for recognition of pyridoxal phosphate derivatives. Thus, compounds 2 and 3 are relatively selective pharmacological probes of P2X1 receptors, filling a long-standing need in the P2 receptor field, and are also important lead compounds for future studies.
Extracellular adenine, and likely uracil, nucleotides have a physiological role to play in the central and peripheral nervous systems through their activation of P2 receptors.1,2 Two families of P2 receptors have been defined:3 ligand-gated cation channels (P2X subtype), activated by adenine nucleotides, and G-protein-coupled receptors (P2Y subtype), most of which are activated by both adenine and uracil nucleotides.4 P2X1–7 and P2Y1,2,4,6 designations have been unambiguously assigned to mammalian nucleotide receptors,5,6 although there is still uncertainty about the correspondence of these cloned sequences to the pharmacological phenotypes of native P2 receptors.
ATP acts as a cotransmitter with norepinephrine and other transmitters in sympathetic neurotransmission in mammals.7 In vas deferens, isolated blood vessels, intestine, kidney, and skin in a number of species, norepinephrine and ATP cause synergistic constriction via α1-adrenoceptors and P2X receptors (primarily the P2X1 subtype), respectively. In rabbit coronary vessels, guinea pig taenia coli, rat aorta, and rat mesenteric artery, the predominant effect of those transmitters is relaxation via β-adrenoceptors and P2Y receptors. Besides sympathetic neurotransmission, P2 receptors also function in parasympathetic, sensory-motor, nonadrenergic noncholinergic (NANC) inhibitory, and somatic neuromuscular neurotransmission. For example, it appears that activation of the P2X3 receptor subtype mediates nociception via the dorsal root ganglia; thus a selective antagonist may prove to be anti-nociceptive.8,9 The therapeutic potential and physiological role of P2 receptors in various central and peripheral biological systems has been reviewed.10
Progress in the field of P2 receptors has been impeded by the lack of stable, selective, and bioavailable ligands, especially antagonists.11 Synthetic polyanionic diazo derivatives of pyridoxal-5-phosphate (1a, Figure 1), PPADS (1b, pyridoxal-α5-phosphate-6-azophenyl-2′,4′-disulfonic acid), and iso-PPADS (1c, the 2,5-disulfonate isomer) were shown to be P2 receptor antagonists.12 Pyridoxal-5′-phosphate, itself, is a weak antagonist of P2 receptors.13 In smooth muscle assays PPADS irreversibly antagonized P2X receptors in rabbit vas deferens,14 urinary bladder,15 isolated blood vessels,16 guinea pig isolated vas deferens,17 and perfused rat mesenteric arterial bed.18 PPADS is a relatively non-selective antagonist at P2 receptors, since it also acts at P2Y1 receptors19 with a Ki value of approximately 1 μM. PPADS does not antagonize the action of ATP agonists at P2X4 and P2X6 receptors and has an IC50 of 45 μM at P2X7 receptors.20 Furthermore, PPADS has a low affinity at P2Y2 and P2Y6 receptors,21 at the P2YAC receptor in human platelets,21,22,40 and at the adenylate cyclase-coupled P2Y receptor in rat C6 glioma cells.21
Figure 1.

Structures of pyridoxal-5′-phosphate (1a) and its azo derivatives (1b, 1c) and the cyclic pyridoxine-α4,5-monophosphate derivatives (2, MRS 2219 and 3, MRS 2220) described in the present study.
Results
Analogues of the P2 receptor antagonists pyridoxal-5′-phosphate and 6-azophenyl-2′,4′-disulfonate derivative (PPADS), in which the phosphate group was cyclized by esterification to a CH2OH group at the 4-position, were synthesized (Figure 1) with the aim of developing more potent and selective antagonists for P2 receptor subtypes. Cyclic pyridoxine-α4,5-monophosphate, 2 (MRS 2219), and cyclic pyridoxine-α4,5-monophosphate-6-azophenyl-2′,5′-disulfonic acid, 3 (MRS 2220), were prepared and characterized using NMR and high-resolution mass spectroscopy, and purity of >98% was demonstrated using high-pressure liquid chromatography (HPLC). The possibility of contamination of 3 with the acyclic pyridoxine-α5-phosphate-6-azophenyl-2′,5′-disulfonic acid was ruled out using HPLC. The latter substance was prepared through sodium borohydride reduction of 1c, and no corresponding peak was observed in the HPLC trace of 3, with a detection limit of 0.1%. Incubation of 3 in the buffer used for the P2X bioassay (see Materials and Methods) also failed to generate this acyclic compound.
The compounds were tested in a functional ion channel assay23,24 of ATP-induced current at recombinant rat P2X1, P2X2, P2X3, and P2X4 receptors, expressed in Xenopus oocytes, using the twin-electrode voltage-clamping technique. P1 and P2 receptors are present on the follicle cell layer of Xenopus oocytes,25 but our experiments were carried out on defolliculated oocytes to avoid activation of endogenous receptors. In the control uninjected, defolliculated oocytes, treatment with ATP (<100 μM) did not induce any current. Compounds 1a and 1b were previously reported to antagonize agonist-induced cation flux at recombinant P2X1 receptors, with the IC50 values of roughly 10 and 1 μM, respectively.26 Compounds 1a was not evaluated in the present study; however we have found compounds 1b and 1c to be considerably more potent than anticipated in inhibiting the inward current elicited by 3 μM ATP in defolliculated oocytes expressing recombinant P2X1 receptors, with IC50 values of 98.5 ± 5.5 and 42.5 ± 17.5 nM, respectively (Figure 2). In oocytes expressing rat P2X3 receptors, IC50 values were 240 ± 38 nM (1b) and 83.5 ± 3.6 nM (1c).
Figure 2.

Effects of compounds 1b (PPADS, squares) and 1c (iso-PPADS, triangles) on inward current induced by activation by 3 μM ATP of recombinant rat P2X1 receptors, expressed in Xenopus oocytes, using the twin-electrode voltage-clamping technique (pH 7.5, Ba2+ Ringer’s solution). IC50 values for compounds 1b and 1c (n = 4) were 98.5 ± 5.5 and 42.5 ± 17.5 nM, respectively. All data points were mean ± SEM of four observations. The apparent lack of error bars on some points is due to the size of symbols being greater than the size of error bars.
The novel cyclic phosphate structurally related to compound 1c, i.e., compound 3, also antagonized activation of recombinant P2X1 receptors by ATP (Figure 3B), while compound 2, related to compound 1a, selectively potentiated ATP-evoked responses at P2X1 receptors effectively doubling the current (Figure 3A) with an EC50 value of 5.9 ± 1.8 μM. Unlike PPADS, the effect of 1b at P2X1 receptors was readily reversible upon washout and surmountable. Furthermore, compound 3 showed weak antagonistic activity at rat P2X3 receptors (Figure 3B). The IC50 values for compound 3 at P2X1 and P2X3 receptors were 10.2 ± 2.6 and 58.3 ± 0.1 μM, respectively, in the presence of 3 and 1 μM ATP, respectively. Thus, compound 3 was 6-fold selective for P2X1 vs P2X3 receptors. Compounds 2 and 3 were both completely inactive as either potentiators or antagonists at P2X2 or P2X4 receptors (Figure 3). Therefore, although a less potent antagonist of P2X receptors than PPADS, the effect of 1b was selective for the P2X1 subtype.
Figure 3.
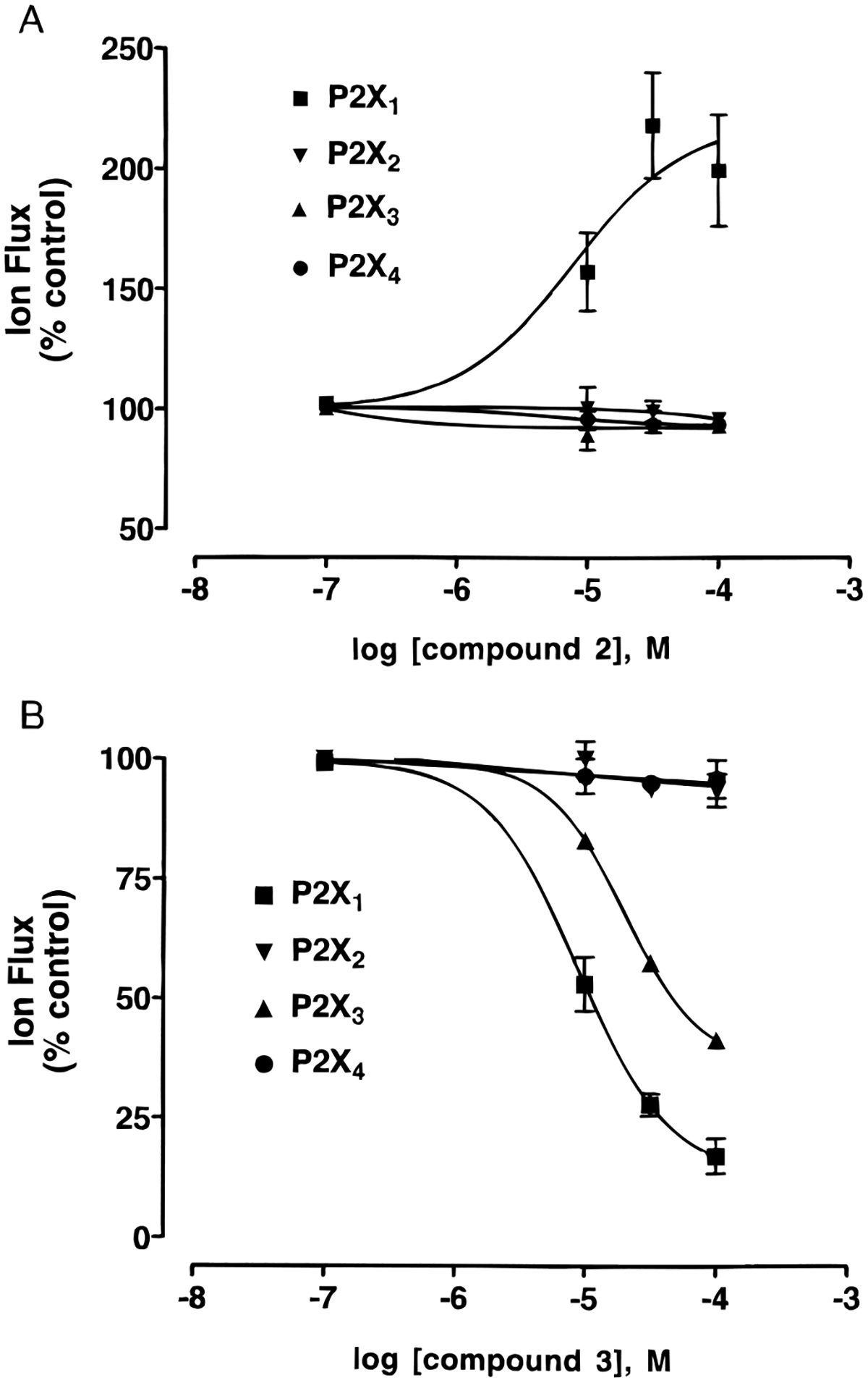
Effects of compounds 2 (A) and 3 (B) on inward current induced by activation by ATP, at the indicated concentrations, of recombinant rat P2X1 (3 μM), P2X2 (10 μM), P2X3 (1 μM), and P2X4 (30 μM) receptors, expressed in Xenopus oocytes, using the twin-electrode voltage-clamping technique. The agonist concentrations correspond approximately to the EC70 values. IC50 values for compounds 3 (n = 4) at P2X1 and P2X3 receptors were 10.2 ± 2.6 and 58.3 ± 0.1 μM, respectively. Slopes of the curves were 0.8 ± 0.1 and 0.9 ± 0.1, respectively. All data points were mean ± SEM of four observations. The apparent lack of error bars on some points is due to the size of symbols being greater than the size of error bars.
Antagonism of phospholipase C (PLC) activity27,28 induced by activation of single subtypes of P2Y receptors was studied. At the P2Y1 receptor of turkey erythrocytes, there was no significant effect on activation by 10 nM 2-MeSATP by compound 2 or 3 at concentrations up to 100 μM (Figure 4). Furthermore, compounds 2 and 3 were found to be essentially inactive in stimulating, potentiating, or blocking PLC at recombinant human P2Y2 and P2Y4 receptors and at rat P2Y6 receptors (Figure 4). Thus, compounds 2 and 3 were completely inactive as either agonists or antagonists of these four subtypes of P2Y receptors.
Figure 4.

Lack of either antagonism or potentiation (n = 3) by compounds 2 and 3 (100 μM) of activation of phospholipase C activity induced at turkey erythrocyte P2Y1 receptors (by 10 nM 2-MeSATP), recombinant human P2Y2 receptors (by 100 nM UTP), recombinant human P2Y4 receptors (by 100 nM UTP), and at recombinant rat P2Y6 receptors (by 100 nM UDP).
Affinity at adenosine (P1) receptors was examined in radioligand binding experiments at rat brain A1 and A2A adenosine receptors and at recombinant human A3 adenosine receptors expressed in HEK293 cells.29 Neither compound 2 nor 3 at 100 μM caused any significant displacement of [3H]R-N6-phenylisopropyladenosine at rat A1, [3H]-2-[4-[(2-carboxyethyl)phenyl]ethylamino]-5′-N-ethylcarbamoyladenosine at rat A2A, or [125I]N6-(4-amino-3-iodobenzyl)-5′-N-methylcarbamoyladenosine at human A3 receptors (data not shown). Thus, these two compounds display complete selectivity for the P2 over the P1 receptors.
Discussion
In the present study we describe a structural modification, a reduction of the aldehyde group and cyclization of the pyridoxal-5-phosphate moiety of pyridoxal phosphate-related P2 receptor antagonists, that apparently results in molecules that exhibit complete selectivity for P2X over P2Y receptors, and further selectivity for the P2X1 subtype within the P2X class of signaling proteins. By virtue of this modification, the analogue corresponding to iso-PPADS, 3, retains antagonist properties at this subtype, while the analogue corresponding to pyridoxal-5-phosphate, 2, becomes a potentiator of activation of this subtype. Compound 3 also weakly antagonized ATP action at recombinant P2X3 receptors. Evans et al.20 reported that PPADS blocks ATP action at recombinant P2X3 receptors expressed in HEK293 cells with an IC50 of 2 μM.
PPADS also potentiated UTP responses in cell lines possessing P2U receptors and pyrimidinoceptors, but by an action considered to involve ectoATPase inhibition.30 This action may explain the modest increase by compound 3 of uridine nucleotide activity at recombinant hP2Y2 and hP2Y4 receptors (see Figure 4).
While selective antagonists have been reported for P2YAC and P2Y1 receptors,22,29 this is the first example of a highly selective antagonist (compound 3) for any P2X receptor subtype. Furthermore, although potentiation by various antagonists of the effects of P2X receptor agonists have been reported previously (e.g., suramin at P2X receptors in enteric neurons and PPADS at recombinant P2X4 receptors),31,32 compound 2 displays unique selectivity as a potentiator of the P2X1 subtype. Defolliculated oocytes contain no ectoATPases;33 therefore the possibility that compound 2 could be acting as a potentiator through enzymatic inhibition can be ruled out.
The dramatic qualitative difference in activity between the structurally related compounds 2 and 3 may provide clues in the molecular modeling of the receptors34 to distinguish native and activated conformations of this ion channel. It will be useful to study the effects of these compounds at mutant P2X receptors35,36 in order to form a hypothesis for the amino acids responsible for antagonist/potentiator binding.
Antagonism by PPADS at native P2X receptors in whole tissue bioassays is nonsurmountable and often reversed slowly,14–18 if at all, consistent with irreversible antagonism. However, blockade by iso-PPADS at P2X receptors on rat vagus nerve37 and blockade by PPADS at recombinant P2X2 receptors expressed in oocytes23 are fully reversible within 1–2 h. The possibility of Schiff’s base formation between the aldehyde group on the ligand and a P2X receptor to explain the slow reversibility was raised by Buell et al.35 and Collo et al.38 PPADS blockade was slowly reversible, but not fully reversible, at recombinant P2X1,2,5 receptors (expressed in HEK293 cells). PPADS blockade reversed rapidly at recombinant P2X3 receptors (expressed in HEK293 cells), whereas PPADS only showed irreversible blockade at P2X4,6 receptors at 100-fold higher concentrations. It was hypothesized that the aldehyde of PPADS might form a Schiff’s base with lysine residues of P2X1 and P2X2 receptors. Scrutiny of the structures of P2X1–6 receptors drew attention to a lysine on P2X1,2,5 receptors at a site equivalent to position 249 on P2X4 receptors and position 251 on P2X6 receptors. A threonine is present at the equivalent position on P2X3 receptors. Substitution of glutamate with lysine (E249K) at P2X4 receptors enhanced the antagonist potency of PPADS by 30-fold, although blockade was slowly reversible. The same substitution at the equivalent position (L251K) at P2X6 receptors also enhanced PPADS sensitivity with slow reversibility. Substitution of lysine with glutamate (K246E) at P2X2 receptors increased the rate of recovery of PPADS blockade. None of these procedures affected the blocking activity of the reversible antagonist suramin at these recombinant P2X receptors. At P2X1 receptors, the conserved lysine would be unreactive with compounds 2 and 3, since the aldehyde group is absent. Thus, Schiff’s base formation between the ligand and receptor is not necessarily required for recognition of pyridoxal phosphate derivatives at P2X1 receptors. The absence of an aldehyde group in compound 3 may be responsible for the complete reversibility of its antagonism.
The pharmacological properties of compounds 2 and 3 at putative heterooligomers of the P2X1 receptors, such as have already been demonstrated for P2X2/P2X3 subtypes,39 should be examined.
Further structure activity studies are in progress to improve the affinity of compounds 2 and 3 at the P2X1 receptor. Such a selective P2X1 receptor antagonist derived from 3 may have potential utility in controlling receptor-mediated contraction of visceral and vascular smooth muscle (e.g., vascular hypertension and instability of the urinary bladder detrusor muscle). A selective enhancer of P2X1 receptor activity derived from 2 may have potential utility in enhancing contractions at P2X1 receptor-controlled muscle sphincter tone (e.g., treatment of urinary incontinence) and sympathetic vascular tone. In conclusion, these novel derivatives are useful pharmacological probes, filling a long-standing need in the P2 receptor field, and are also important lead compounds for future studies.
Materials and Methods
Synthesis.
Pyridoxine and the reagents for azo coupling reactions were purchased from Aldrich (St. Louis, MO). Aniline-2,5-disulfonic acid was obtained from K & K Laboratories, Inc. (Hollywood, CA).
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and spectra were taken in D2O. The chemical shifts are expressed in ppm relative to the HOD peak at 4.78 ppm. 31P NMR spectra were recorded without proton decoupling mode, at room temperature using Varian XL-300 spectrometer (121.419 MHz); orthophosphoric acid (85%) was used as an external standard. High-resolution FAB (fast atom bombardment) mass spectra were determined with a JEOL SX102 spectrometer, and electron spray mass spectra were obtained using a Hewlett-Packard 1100 LC-ESPRAY system. Determination of purity were performed with a Hewlett-Packard 1090 HPLC system using an OD-5–60 C18 analytical column (250 mm × 4.6 mm, Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M triethylammonium acetate buffer:CH3CN = 95:5 to 40:60 for 20 min with flow rate 1 mL/min. The other (B) was 5 mM tetrabutylammonium phosphate buffer:CH3CN = 80:20 to 40:60, in 20 min with flow rate 1 mL/min. Peaks were detected by UV absorption using a diode array detector.
Cyclic Pyridoxine-α4,5-monophosphate (2).
To a suspension of 0.1 g (0.59 mmol) of pyridoxine in 2 mL of anhydrous benzene was added 1 mL of trimethylsilyl polyphosphate under nitrogen atmosphere. The mixture was stirred at 40 °C for 2 days and poured into 10 mL of anhydrous ether. The white precipitate was collected by filtration, washed with anhydrous ether, and dissolved in 2 mL of water. The water solution was stirred at 40 °C for 30 min to remove the trimethylsilyl group and passed through Amberlite CG-50 resin (H+ form, weakly acidic) with the elution of water (flow rate 0.5 mL/min). The pure fractions were combined and lyophilized. The solid residue was washed with a minimum of water, and 0.05 g of white powdered product was obtained (yield 37%). 1H NMR (D2O): δ 2.62 (3H, s, CH3), 5.14 (2H, d, J = 15.6 Hz, CH2O), 5.34 (2H, d, J = 15.6 Hz, CH2O), 8.08 (1H, s, H-6). 31P NMR (D2O): 4.62 (pen, J = 15.9 Hz). MS: negative FAB, 230 (M – H), positive FAB, 232 (M + H); negative API-ES, 230 (M – H). HRMS (FAB–): calcd 230.0218, found 230.0216. HPLC retention time: 4.0 min using solvent system A, 5.4 min using solvent system B (purity >98%).
Cyclic Pyridoxine-α4,5-monophosphate-6-azophenyl-2′,5′-disulfonic Acid (3).
To a solution of 0.055 g (0.216 mmol) of aniline-2,5-disulfonic acid in 2 mL of water and 0.22 mL of 1 N HCl was added 0.015 g (0.216 mmol) of solid sodium nitrite at 0 °C. This solution was stirred for 5 min, and the pH was adjusted to ~10 with 1 N NaOH. To the mixture was added dropwise a solution of 0.05 g (0.216 mmol) of 2, previously dissolved in aqueous NaOH (pH ~10). The pH was adjusted to ~9, and the yellow color changed to red. After 30 min of stirring 0 °C, the mixture was purified by ion-exchange column chromatography using Amberlite CG-50 resin (H+ form, weakly acidic) with the elution of water (flow rate 0.5 mL/min). The red fraction showing a single peak in HPLC was collected and lyophilized to give 0.08 g of the disodium salt form of the desired compound (yield 69%). 1H NMR (D2O): δ 2.56 (3H, s, CH3), 5.24 (2H, d, J = 15.6 Hz, CH2O), 5.70 (2H, d, J = 15.6 Hz, CH2O), 7.89 (1H, bs, phenyl), 8.08–8.14 (2H, m, phenyl). 31P NMR (D2O): 5.56 (pen, J = 15.9 Hz). HRMS (FAB–): calcd 493.9729, found 493.9721. HPLC: 7.7 min using solvent system A, 12.4 min using solvent system B (purity >98%).
Pyridoxine-α5-phosphate-6-azophenyl-2′,5′-disulfonic Acid (HPLC Standard).
To a stirred solution of 0.027 g of 1c (0.05 mmol, Tocris Cookson Inc., St. Louis, MO) in 2 mL of H2O was added 0.004 g of solid NaBH4 (0.1 mmol) at room temperature. The mixture was stirred for 30 min and passed through Amberlite CG-50 resin (H+ form, weakly acidic) with water elution (flow rate 0.5 mL/min). The pure fractions of the major component, indicated by single peak in HPLC, were collected and lyophilized (0.015 g, 58%). 1H NMR (D2O): δ 2.70 (3H, s, CH3), 5.21 (2H, s, CH2OH), 5.61 (2H, d, J = 6.8 Hz, CH2OP), 7.88–8.23 (3H, m, phenyl). 31P NMR (D2O) 0.05 (t, J = 6.8 Hz). HPLC: 5.9 min using solvent system A. HRMS (FAB–): calcd 511.9835, found 511.9828.
Pharmacology: Antagonist Activity at Recombinant P2X Receptors.
Xenopus oocytes were harvested and prepared as previously described.23 Defolliculated oocytes were injected cytosolically with rat P2X1, P2X2, P2X3, or P2X4 receptor cRNA (40 nL, 1 μg/mL), incubated for 24 h at 18 °C in Barth’s solution, and kept for up to 12 days at 4 °C until used in electrophysiological experiments.
ATP-activated membrane currents (Vh = −90 mV) were recorded from cRNA-injected oocytes using the twin-electrode voltage-clamp technique (Axoclamp 2B amplifier). Voltage recording (1–2 MΩ tip resistance) and current-recording microelectrodes (5 MΩ tip resistance) were filled with 3.0 M KCl. Oocytes were held in an electrophysiological chamber and superfused with Ringer’s solution (5 mM/min, at 18 °C) containing (mM) NaCl, 110; KCl, 2.5; HEPES, 5; BaCl2, 1.8, adjusted to pH 7.5.
ATP (at the EC70 values in μM for respective subtypes: P2X1 3, P2X2 10, P2X3 1, and P2X4 30) was superfused over oocytes for 60–120 s and then washed out for a period of 20 min. For inhibition curves, data were normalized to the current evoked by ATP at pH 7.5. Test substances were added for 20 min prior to ATP exposure; all compounds were tested for reversibility of their effects. The concentration required to inhibit the ATP-response by 50% (IC50) was taken from Hill plots constructed using the formula log(I/Imax – I), where I is the current evoked by ATP in the presence of an antagonist. Data are presented as Mean ± SEM (n = 4) for data from different batches of oocytes.
Phospholipase C Assay at P2Y Receptors.
P2Y1 receptor-promoted stimulation of inositol phosphate formation by 2-MeSATP (10 nM) was measured in turkey erythrocyte membranes as previously described.25,26 The values were averaged from three to eight independent determinations. Briefly, 1 mL of washed turkey erythrocytes was incubated in inositol-free medium (DMEM; Gibco) with 0.5 mCi of 2-[3H]myo-inositol (20 Ci/mmol; American Radiolabelled Chemicals Inc., St. Louis, MO) for 18–24 h in a humidified atmosphere of 95% air 5% CO2 at 37 °C. Erythrocyte ghosts were prepared by rapid lysis in hypotonic buffer (5 mM sodium phosphate, pH 7.4, 5 mM MgCl2, 1 mM EGTA) as described.26 PLC activity was measured in 25 μL of [3H]inositol-labeled ghosts (~175 μg of protein, 200–500000 cpm/assay) in a medium containing 424 μM CaCl2, 0.91 mM MgSO4, 2 mM EGTA, 115 mM KCl, 5 mM KH2PO4, and 10 mM Hepes, pH 7.0. Assays (200 μL final volume) contained 1 μM GTPγS and the indicated concentrations of nucleotide analogues. Ghosts were incubated at 30 °C for 5 min, and total [3H]inositol phosphates were quantitated by anion exchange chromatography as previously described.25,26
Stimulation of inositol phosphate formation in 1321N1 human astrocytoma cells stably expressing recombinant human P2Y2 receptors (activated by 100 nM UTP), recombinant human P2Y4 receptors (activated by 100 nM UTP), and recombinant rat P2Y6 receptors (activated by 100 nM UDP) was measured in a similar fashion.
Acknowledgment.
We thank to Dr. Lewis Pannell and Wesley White for determination of high-resolution mass spectroscopy and NMR. This work was supported by USPHS Grants GM38213, HL34322, and HL54889.
Abbreviations.
- ATP
adenosine 5′-triphosphate
- HEK
human embryonic kidney
- Hepes
N-[2-hydroxyethyl]piperazine-N′-[3-propanesulfonic acid]
- HPLC
high-pressure liquid chromatography
- HRMS
high-resolution mass spectroscopy
- K i
equilibrium inhibition constant
- iso-PPADS
pyridoxal-α5-phosphate-6-azophenyl-2′,5′-disulfonic acid
- MeATP
adenosine-5′-methylenetri-phosphate, (α,β) or (β,γ) isomers
- 2-MeSATP
2-methylthioadenosine-5′-triphosphate
- MRS 2219
cyclic pyridoxine-α4,5-monophosphate
- MRS 2220
cyclic pyridoxine-α4,5-monophosphate-6-azophenyl-2′,5′-disulfonic acid
- MS
mass spectrum
- PLC
phospholipase C
- PPADS
pyridoxal-α5-phosphate-6-azophenyl-2′,4′-disulfonic acid
- SAR
structure–activity relationship
- TLC
thin-layer chromatography
References
- (1).North RA; Barnard EA Nucleotide receptors. Curr. Opin. Neurobiol 1997, 7, 346–357. [DOI] [PubMed] [Google Scholar]
- (2).Burnstock G P2 purinoceptors – historical-perspective and classification. Ciba Found. Symp 1996, 198, 1–34. [DOI] [PubMed] [Google Scholar]
- (3).Abbracchio MP; Burnstock G Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol. Ther 1994, 64, 445–475. [DOI] [PubMed] [Google Scholar]
- (4).Communi D; Boeynaems JM Receptors responsive to extracellular pyrimidine nucleotides. Trends Pharmacol. Sci 1997, 18, 83–86. [DOI] [PubMed] [Google Scholar]
- (5).Burnstock G; King BF Numbering of cloned P2 purinoceptors. Drug Dev. Res 1996, 38, 67–71. [Google Scholar]
- (6).Fredholm BB; Abbracchio MP; Burnstock G; Dubyak GR; Harden TK; Jacobson KA; Schwabe U; Williams M Towards a revised nomenclature for P1 and P2 receptors. Trends Pharmacol. Sci 1997, 18, 79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Burnstock G Physiological and pathological roles of purines: an update. Drug Dev. Res 1993, 28, 195–206. [Google Scholar]
- (8).Burnstock G; Wood JN Purinergic receptors – their role in nociception and primary afferent neurotransmission. Curr. Opin. Neurobiol 1996, 6, 526–532. [DOI] [PubMed] [Google Scholar]
- (9).Gu JG; MacDermott AB Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 1997, 389, 749–753. [DOI] [PubMed] [Google Scholar]
- (10).Williams M; Jacobson KA P2 purinoceptors: Advances and therapeutic opportunities. Exp. Opin. Invest. Drugs, 1995, 4, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jacobson KA; Kim Y-C; Camaioni E; van Rhee AM Structure activity relationships of P2 receptor agonists and antagonists. Chapter II.4 in The P2 Nucleotide Receptors, in the series The Receptors; Turner JT, Weisman G, Fedan J, Eds.; Humana Press: Clifton, NJ, 1997; pp 81–107. [Google Scholar]
- (12).Lambrecht G; Ardanuy U; Bäumert HG; Bo X; Hoyle CHV; Nickel P; Pfaff O; Ralevic V; Windschief U; Ziganshin AU; Ziyal R; Mutscheler E; Burnstock G Design and pharmacological characterization of selective P2-purinoceptor antagonists. In Perspectives in Receptor Research; Elsevier Science Publishers: Amsterdam, 1996; Pharmaco Chemistry Library, Giardinà D, Piergentili S, and Pigini M Eds., pp 337–350. [Google Scholar]
- (13).Tresize DJ; Bell NJ; Khakh BS; Michel AD; Humphrey PPA P2 purinoceptor antagonist properties of pyridoxal-5-phosphate Eur. J. Pharmacol 1994, 259, 295–300. [DOI] [PubMed] [Google Scholar]
- (14).Lambrecht G; Friebe T; Grimm U; Windscheif U; Bungardt E; Hildebrandt C; Baumert HG; Spatzkumbel G; Mutschler E PPADS, a Novel Functionally Selective Antagonist of P2 Purinoceptor-Mediated Responses. Eur. J. Pharmacol 1992, 217, 217–219. [DOI] [PubMed] [Google Scholar]
- (15).Ziganshin AU; Hoyle C; Bo XN; Lambrecht G; Mutschler E; Bäumert HG; Burnstock G PPADS selectively antagonizes P2X purinoceptor-mediated responses in the rabbit urinary-bladder. Br. J. Pharmacol 1993, 110, 1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ziganshin AU; Hoyle CHV; Lambrecht G; Mutschler E; Baumert HG; Burnstock G Selective antagonism by PPADS at P2X-purinoceptors in rabbit isolated blood vessels. Br. J. Pharmacol 1994, 111, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McLaren GJ; Lambrecht G; Mutschler E; Baumert HG; Sneddon P; Kennedy C Investigation of the actions of PPADS, a novel of P2X-purinoceptor antagonist, in the guinea-pig isolated vas deferens. Br. J. Pharmacol 1994, 111, 913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Windscheif U; Ralevic V; Bäumert HG; Mutschler E; Lambrecht G; Burnstock G Vasoconstrictor and vasodilator responses to various agonists in the rat perfused mesenteric arterial bed – selective-inhibition by PPADS of contractions mediated via P2X-purinoceptors. Br. J. Pharmacol 1994, 113, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Boyer JL; Zohn IE; Jacobson KA; Harden TK Differential effects of P2-purinergic receptor antagonists on phospholipase C- and adenylyl cyclase-coupled P2Y purinergic receptors. Br. J. Pharmacol 1994, 113, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Evans RJ; Surprenant A; North RA P2X receptors. Chapter 2 in The P2 Nucleotide Receptors, in the series The Receptors; Turner JT, Weisman G, Fedan J, Eds.; Humana Press: Clifton, NJ, 1997; pp 43–61. [Google Scholar]
- (21).Harden TK; Nicholas RA; Schachter JB; Lazarowski E; Boyer JL Pharmacological selectivities of molecularly defined subtypes of P2Y receptors. Chapter 5 in The P2 Nucleotide Receptors, in the series The Receptors; Turner JT, Weisman G, Fedan J, Eds.; Humana Press: Clifton, NJ, 1997, pp 109–134. [Google Scholar]
- (22).Leff P; Robertson MJ; Humphries RG The role of ADP in thrombosis and the therapeutic potential of P2T-receptor antagonists as novel antithrombotic agents, Chapter 11, in Purinergic Approaches in Experimental Therapeutics; Jacobson KA, Jarvis MF, Eds.; Wiley: New York, 1997; pp 203–216. [Google Scholar]
- (23).King BF; Wildman SS; Ziganshina LE; Pintor J; Burnstock G Effects of extracellular pH on agonism and antagonism at recombinant P2X2 receptors. Br. J. Pharmacol 1997, 121, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brake AJ; Wagenbach MJ; Julius D New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature 1994, 371, 519–523. [DOI] [PubMed] [Google Scholar]
- (25).King BF; Wang S; Burnstock G P2 purinoceptor activated inward currents in Xenopus oocytes. J. Physiol. (London) 1996, 494, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Evans RJ; Lewis C; Buell G; Valera S; North RA; Surprenant A Pharmacological characterization of heterologously expressed ATP-gated cation channels (P2X purinoceptors). Mol. Pharmacol 1995, 48, 178–183. [PubMed] [Google Scholar]
- (27).Harden TK; Hawkins PT; Stephens L; Boyer JL; Downes P Phosphoinositide hydrolysis by guanosine 5′-(gammathio)triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem. J 1988, 252, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Boyer JL; Downes CP; Harden TK Kinetics of activation of phospholipase C by P2Y purinergic receptor agonists and guanine nucleotides. J. Biol. Chem 1989, 264, 884–90. [PubMed] [Google Scholar]
- (29).Camaioni E; Boyer JL; Mohanram A; Harden TK; Jacobson KA Deoxyadenosine-bisphosphate derivatives as potent antagonists at P2Y1 receptors. J. Med. Chem 1998, 41, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen BC; Lee CM; Lin WW Inhibition of ecto-ATPase by PPADS, suramin and reactive blue in endothelial cells, C6 glioma cells and RAW 264.7 macrophages. Br. J. Pharmacol 1996, 119, 1628–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Barajas-Lopez C; Huizinga JD; Collins SM; Gerzanich V; Espinosa-Luna R; Peres AL P2x-purinoceptors of myenteric neurones from the guinea-pig ileum and their unusual pharmacological properties. Br. J. Pharmacol 1995, 119, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Buell G; Lewis C; Collo G; North RA; Surprenant A An antagonist-insensitive P2X receptor expressed in epithelia and brain. EMBO J 1996, 15, 55–62. [PMC free article] [PubMed] [Google Scholar]
- (33).Ziganshin AU; Ziganshina LE; King BF; Burnstock G Characteristics of ecto-ATPase of Xenopus oocytes and the inhibitory actions of suramin on ATP breakdown. Pflugers Arch 1995, 429, 412–418. [DOI] [PubMed] [Google Scholar]
- (34).van Rhee AM; Jacobson KA; Garrad R; Weisman GA; Erb L P2 receptor modeling and identification of ligand binding sites. Chapter 6 in The P2 Nucleotide Receptors, in the series The Receptors; Turner JT, Weisman G, Fedan J, Eds.; Humana Press: Clifton, NJ, 1997; pp 135–166. [Google Scholar]
- (35).Buell G; Collo G; Rassendren F P2X receptors: an emerging channel family. Eur. J. Neurosci 1996, 8, 2221–2228. [DOI] [PubMed] [Google Scholar]
- (36).Rassendren F; Buell G; Newbolt A; North RA; Suprenant A Identification of amino acid residues contributing to the pore of a P2X receptor. EMBO J. 1997, 16, 3446–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Trezise DJ; Kennedy I; Humphrey PPA The use of antagonists to characterize the receptors mediating depolarization of the rat isolated vagus nerve by alpha,beta- methylene adenosine 5′-triphosphate. Br. J. Pharmacol 1994, 112, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Collo G; North RA; Kawashima E; Merlo-Pich E; Neidhart S; Surprenant A; Buell G Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci 1996, 16, 2495–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Radford KM; Virginio C; Surprenant A; North RA; Kawashima E Baculovirus expression provides direct evidence for heteromeric assembly of P2X2 and P2X3 receptors. J. Neurosci 1997, 17, 6529–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Daniel JL; Dangelmaier C; Jin J; Ashby B; Smith JB; Kunapuli SP Molecular basis for ADP-induced platelet activation. I. Evidence for three distinct ADP receptors on human platelets. J. Biol. Chem 1998, 273, 2024–2029. [DOI] [PubMed] [Google Scholar]