

Abstract
The adenosine antagonist 9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine (CGS15943) binds to human A3 receptors with high affinity (Ki = 14 nM), while it lacks affinity at rat A3 receptors. Acylated derivatives of the 5-amino group and other modifications were prepared in an effort to provide A3 subtype selectivity. Affinity was determined in radioligand binding assays at rat brain A1 and A2A receptors using [3H]-(R)-PIA ([3H]-(R)-N6-(phenylisopropyl)-adenosine) and [3H]CGS 21680 ([3H]-2-[[4-(2-carboxy ethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine), respectively. Affinity was determined at cloned human A3 receptors using [125I]AB-MECA (N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine). A series of straight chain alkyl amides demonstrated that the optimal chain length occurs with the 5-N-propionyl derivative, 3, which had a Ki value of 7.7 nM at human A3 receptors, and was 40- and 14-fold selective vs rat A1 and A2A receptors, respectively. The 5-N-benzoyl derivative, 10, displayed Ki values of 680 and 273 nM at rat A1 and A2A receptors, respectively, and 3.0 nM at human A3 receptors. A 5-N-phenylacetyl derivative, 12, was 470-fold selective for human A3 vs rat A1 receptors with a Ki value of 0.65 nM. A conjugate of Boc-γ-aminobutyric acid was also prepared but was nonselective. Conversion of the 5-amino group of CGS15943 to an oxo function resulted in lower affinity but 15-fold selectivity for human A3 receptors.
Introduction
Four subtypes of adenosine receptors (A1, A2A, A2B, and A3) have been cloned and studied pharmacologically.1 Since the recent discovery of the adenosine A3 receptor and the cloning of its species homologues,2 much effort has been made in order to characterize the receptor biochemically and pharmacologically. Principally, the development of selective agonist ligands3 has aided in this effort. The A3 receptor mediates processes of inflammation,2 hypotension,4 and mast cell degranulation.5 This receptor apparently also has a role in the central nervous system. The A3 selective agonist IB-MECA induces behavioral depression6 and upon chronic administration protects against cerebral ischemia.7 A3 selective agonists at high concentrations were also found to induce apoptosis in HL-60 human leukemia cells.8 These and other findings have made the A3 receptor a promising therapeutic target.3 Selective antagonists for the A3 receptor are sought as potential antiinflammatory or possibly antiischemic agents in the brain.9–11
Thousands of analogues of the classical adenosine antagonists, the xanthines, have been prepared, resulting in high degrees of selectivity for A1 or A2A receptor subtypes.12–14 However, the xanthines have not provided fruitful leads for A3 receptor antagonists, since they are generally much weaker in binding at the A3 subtype than at A1 or A2A subtypes.9,10 Thus, the search for A3 receptor antagonists has been directed toward the screening of libraries to identify structurally novel leads.11,15–18 Diverse classes of heterocycles, non-xanthine adenosine antagonists, have already been found to bind to A1 and A2A receptors and more recently to A3 adenosine receptors. We recently reported that two chemical classes, the flavonoids16 and dihydropyridines,18 are amenable to modification, resulting in ligands selective for the recently discovered A3 receptor. The flavonoid I (MRS 1067)17 and the dihydropyridine derivative II (MRS 1097)18 (Figure 1) are selective human A3 receptor antagonists.
Figure 1.
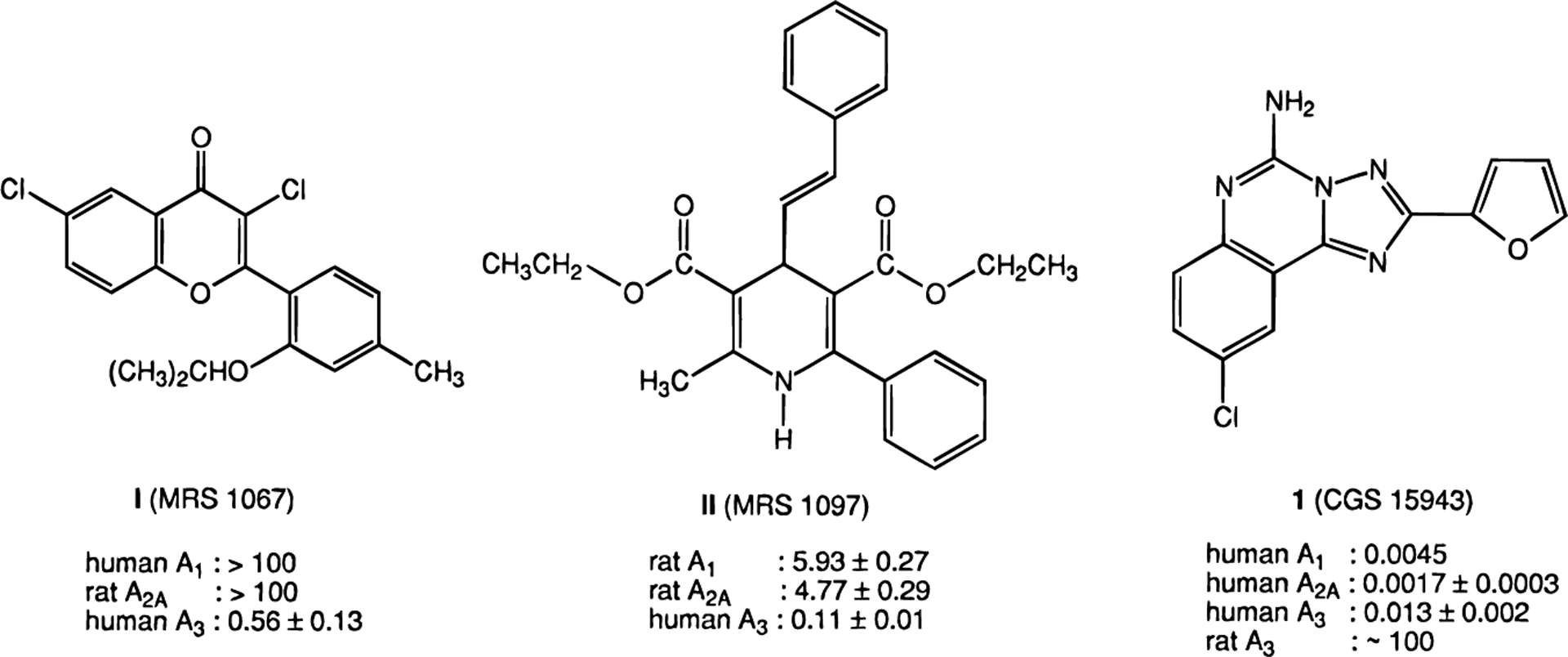
Structures and affinities (Ki value in μM) of novel A3 adenosine receptor antagonists the flavonoid I (MRS 1067),17 the dihydopyridine II (MRS 1097),18 and the nonselective antagonist triazoloquinazoline 1 (CGS15943).19,28,29
The non-xanthine adenosine antagonist 1 (CGS15943, 9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine)19,20 is very potent and slightly selective at human brain A2A receptors with a Ki value of 1.7 nM.21,29 It had been under development as an antiasthmatic agent, until unanticipated skin sensitivity was observed.12 This antagonist was reported to be inactive in binding at rat A3 receptors.10 Nevertheless, in light of the considerable species variability in affinity of non-adenosine derivatives at A3 receptors, the affinity of 1 (CGS15943) was determined at cloned human A3 receptors and was found to be relatively high. In this study we have prepared a series of derivatives of 1 and found that N-acylation greatly enhances affinity and selectivity for human A3 receptors.
Results
Synthesis.
The structures of the triazoloquinazoline derivatives tested for affinity in radioligand binding assays at adenosine receptors are shown in Table 1. Several of the derivatives, e.g. the bromo derivative, 13, and the 5-oxo derivative, 15, had been reported previously by Francis et al.19 to bind to A1 and A2A receptors. The 5-oxo derivative19 was prepared in the present study by an alternate route using an acid treatment of commercially available 1 (Figure 2). Various N-acyl derivatives were synthesized from 1 using standard acylation methodologies (Figure 2). Compound 1319,20 was prepared by first protecting the amino group as the tert-butyloxycarbonyl derivative, followed by bromination using N-bromosuccinimide and acidic deprotection. The yields and chemical characterization of these compounds are reported in Table 2.
Table 1.
Affinities of Triazoloquinazoline Derivatives in Radioligand Binding Assays at A1, A2A, and A3 Receptors
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Ki (nM), IC50 (nM), or % inhibition | ||||||||
| compd | R1 | R2 | R3 | rA1a | rA2Ab | hA3c | rA1/hA3 | rA2A/hA3 |
| 1 (CGS15943) | H | H | 21 ± 3d | 3.3 ± 1.7d | 13.8 ± 2.4 | |||
| 2 | COCH3 | H | 52.2 ± 2.6 | 36.3 ± 3.2 | 13.9 ± 2.5 | 3.8 | 2.6 | |
| 3 (MRS1186) | COCH2CH3 | H | 283 ± 42 | 106 ± 30 | 7.66 ± 3.03 | 40 | 14 | |
| 4 | CO(CH2)2CH3 | H | 32.5 ± 9.4 | 37.6 ± 5.6 | 14.6 ± 2.8 | 2.2 | 2.6 | |
| 5 | CO(CH2)3CH3 | H | 28.9 ± 3.7 | 48.8 ± 10.5 | 21.5 ± 6.2 | 1.3 | 2.3 | |
| 6 | COC(CH3)3 | H | 205 ± 20 | 88.8 ± 20.5 | 244 ± 6 | 0.84 | 0.36 | |
| 7 | COOC(CH3)3 | H | 190 ± 16 | 92.0 ± 8.0 | 82.5 ± 23.3 | 2.3 | 1.1 | |
| 8 | CO(CH2)3NH-Boc | H | 91.3 ± 13.2 | 135 ± 38 | 32.9 ± 1.7 | 2.8 | 4.1 | |
| 9 | CO(CH2)3NH2 | H | 11.3 ± 2.4 | 14.4 | 80.8 ± 7.4 | 0.14 | 0.18 | |
| 10 (MRS1177) | COPh | H | 680 ± 82 | 273 ± 42 | 3.03 ± 1.73 | 220 | 90 | |
| 11 | CO(3-I-Ph) | H | 200 | 310 ± 151 | 23.8 ± 5.2 | 8.4 | 13 | |
| 12 (MRS1220) | COCH2Ph | H | 305 ± 51 | 52.0 ± 8.8 | 0.65 ± 0.25 | 470 | 80 | |
| 13 | H | Br | 1570d | 531d | 64.0 ± 13.1 | |||
| 14 | COPh | Br | <10%e | <10%e | 856 ± 156 | |||
| 15 | H | 3950 ± 250 | 4380 | 260 ± 87 | 15 | 17 | ||
| 16 | (CH2)2CH3 | 1850 ± 420 | 6960 | 1813 ± 720 | 1.0 | 3.8 | ||
Displacement of specific [3H]-(R)-PIA binding in rat brain membranes, expressed as Ki ± SEM in nM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes, expressed as Ki ± SEM in nM (n = 3–6).
Displacement of specific [125I]AB-MECA binding at human A3 receptors expressed in HEK-293 cells, in membranes, expressed as Ki ±SEM in nM (n = 3–4).
IC50 values from Francis et al.19
Percent displacement of specific binding at concentration 1 μM.
Figure 2.
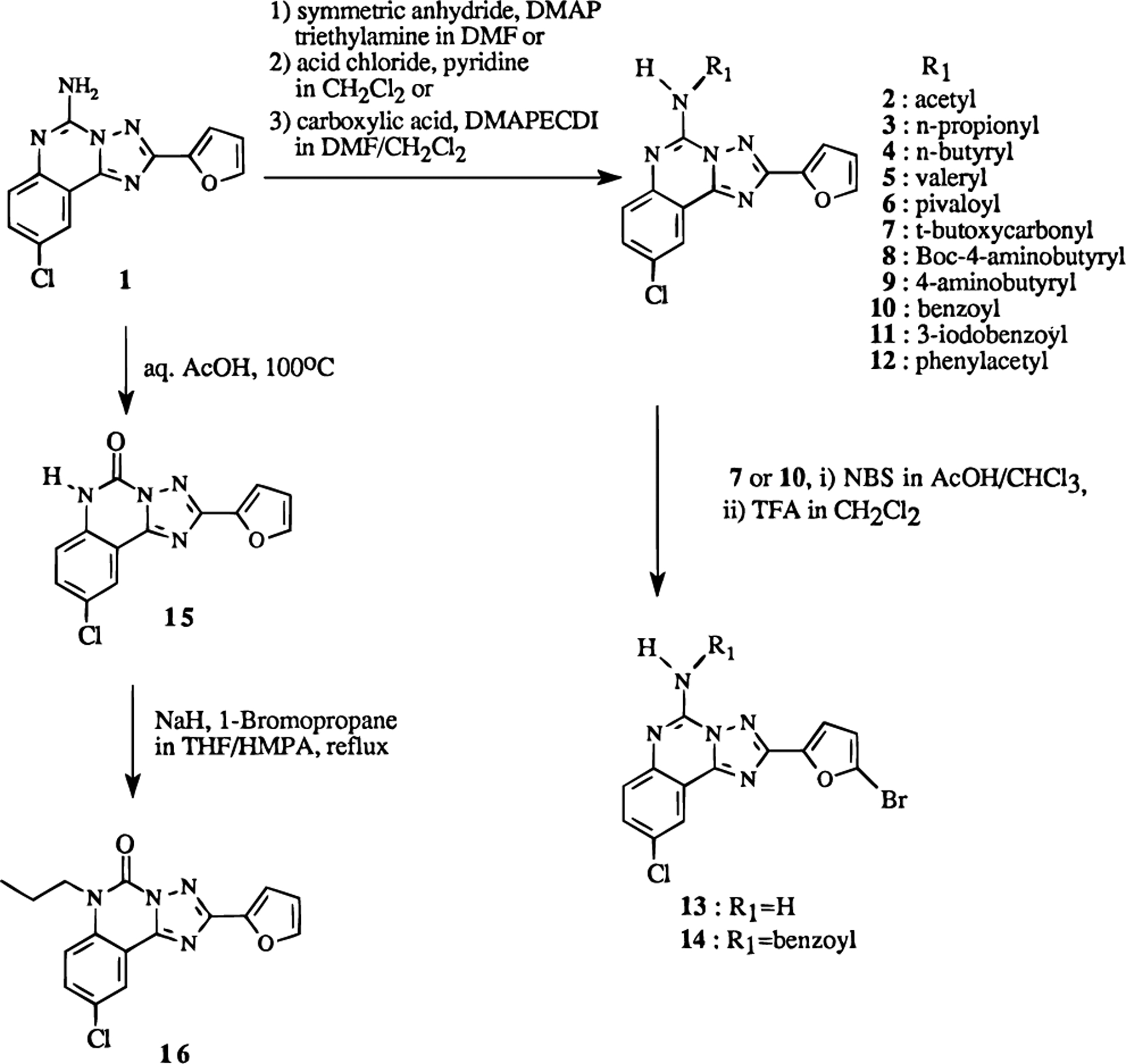
Synthesis of N-acylated and various other derivatives of 1 (CGS15943).
Table 2.
Yields and Chemical Characterization of Triazoloquinazoline Derivatives
| compd no. | methoda | % yield | mp (°C) | MS | formula | analysis |
|---|---|---|---|---|---|---|
| 2 | A | 96 | 245 | CI: 328 | C15H10cN5O2C1·0.16CH2C12 | C,H,N |
| 3 | A | 84 | 224 | CI: 342 | C16H12N5O2Cl | C,H,N |
| 4 | A | 96 | 232 | CI: 356 | C17H14N5O2Cl·0.2MeOH | C,H,N |
| 5 | A | 73 | 220 | FAB: 370 | C18H16N5O2Cl·0.1EtOAc | C,H,N |
| 6 | B | 70 | 200 | CI: 370 | C18H16N5O2Cl | C,H,N |
| 7 | A | 98 | 285 | CI: 386 | C18H16N5O3Cl | C,H,N |
| 8 | C | 49 | 209 | CI: 471 | C22H23N6O4Cl·0.11CH2Cl2 | C,H,N |
| 9 | 83 | 252 | FAB: 371 | C17H15N6O2Cl·C2H1O2F3·0.15CH2Cl2 | C,H,N | |
| 10 | A | 73 | 239 | CI: 390 | C20H12N5O2Cl·0.6CH2Cl2 | C,H,N |
| 11 | C | 50 | 243 | CI: 515 | C20cH11N5O2ClI·0.75MeOH | C,H,N |
| 12 | B | 35 | 233 | EI: 403 | C21H14N5O2Cl·0.38EtOAc | C,H,N |
| 14 | 28 | 266 | FAB: 469 | C20H11N5O2BrCl | b | |
| 16 | 50 | 178 | EI: 328 | C16H13N502C1 | b |
A, symmetric anhydride method; B, acid chloride method; C, carbodiimide method.
High-resolution mass in EI or FAB+ mode (m/z) determined to be within acceptable limits. 14: calcd, 467.9863; found, 467.9871. 16: calcd, 328.0727; found, 328.0726.
Binding at Adenosine Receptors.
Ki values were determined in radioligand binding assays at rat cortical A1 receptors vs [3H]-(R)-PIA ([3H]-(R)-N6-(phenylisopropyl)adenosine) or rat striatal A2A receptors vs [3H]CGS 21680 ([3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine.22,23 Affinity at cloned human A3 receptors expressed in HEK-293 cells24 was determined using [125I]AB-MECA (N6-(4-amino-3-[125I]iodobenzyl)-5′-(N-methyl carbamoyl)adenosine).25 In this study, affinities at adenosine receptors from different species are compared. The species differences in ligand affinity for triazoloquinazolines at A1 and A2A receptors are minor, whereas those at A3 receptors are significant (Figure 1).2,26 Thus, our ratios of affinity are indicative of selectivities in human, but not rat tissue.
The primary amine and 2-furanyl moieties in 1 have been considered most important for binding to rat A1 and rat A2A receptors.19 In an effort to investigate the effects on binding at human A3 receptors, various structural modifications of these groups were made. The structure–activity relationships (SAR) for binding at adenosine receptors indicated that a variety of N-acyl groups are tolerated at the 5-amino position (Table 1). As with the parent compound 1,19 the affinity of the N-acylated derivatives (2–12) at A2A receptors tended to equal or surpass the affinity at A1 receptors. The substituent R1 was varied from acetyl to valeryl in a homologous series (2–5). Also several acyl congeners containing either bulky tert-butyl or derivatized aromatic moieties were synthesized to investigate steric and electrostatic effects on the competitive binding at each of the adenosine receptors.
Among the straight acyl chain derivatives, the N-propionyl derivative, 3, had the highest binding affinity (Ki = 7.7 nM) and selectivity (40-fold vs rat A1, 14-fold vs rat A2A) at human A3 receptors. Within this series, the affinity at A3 receptors varied only 3-fold with varying chain lengths, while at A2A receptors a greater variation was observed. The affinities of compounds 6 and 7 indicated that bulky groups at position R1 were less well tolerated than straight alkyl chains in human A3 receptor binding. The pivaloyl derivative, 6, was 32-fold less potent at A3 receptors than the propionyl derivative, 3, from which it differed only in the presence of two methyl groups at a branched carbon. The change from an amide, 6, to the corresponding urethane, 7, had no effect on affinity at A1 and A2A receptors, while the affinity at A3 receptors was enhanced ~3-fold. The competitive binding curves of compounds 3, 6, and 7 at human A3 receptors are shown in Figure 3A.
Figure 3.
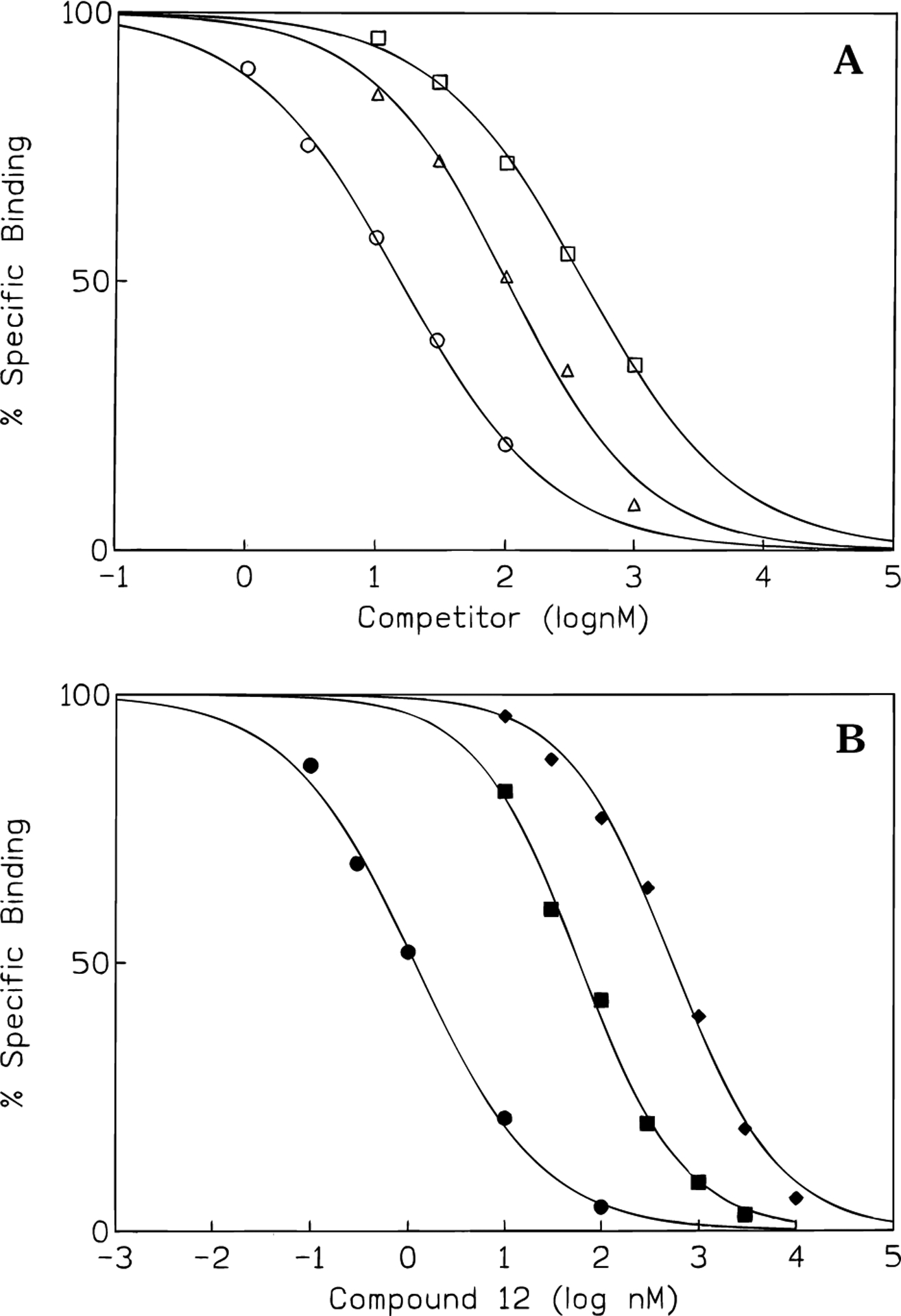
Representative competition curves comparing inhibition of binding at adenosine receptors. (A) Inhibition by compounds 3 (circles), 6 (squares), and 7 (triangles) of [125I]AB-MECA binding at human A3 receptors, expressed in HEK-293 cells. (B) Inhibition by compound 12 of [125I]AB-MECA binding at human A3 receptors (filled circles, transfected HEK-293 cell membranes, nH = 0.75), [3H]-(R)-PIA binding at rat brain A1 (filled diamonds, nH) = 0.89), and [3H]CGS 21680 binding at A2A (filled squares, nH = 0.84) receptors (in brain membranes).
A Boc-γ-amino butyric acid conjugate, 8, of 1 was slightly selective for human A3 receptors. The removal of the Boc group resulting in an acyl chain containing the primary amine functionality, 9, which is positively charged at pH 7.4, reduced potency at human A3 receptors, while increasing potency at rat A1 and rat A2A receptors.
Among aromatic acyl derivatives, the N-benzoyl derivative, 10, showed higher binding affinity (Ki = 3.0 nM) at the human A3 receptor than 1, while affinities at rat A1 and rat A2A receptors were significantly decreased. At rat A3 receptors,9 compound 10 was much less potent (data not shown), with only 23% of radioligand binding displaced at 3 μM. Substitution of the phenyl ring of the N-benzoyl group with a 3-iodo substituent, 11, caused a marked reduction in affinity only at human A3 receptors. However, the less sterically hindered N-phenylacetyl compound, 12, showed yet higher affinity (Ki = 0.65 nM) and selectivity (470-fold vs rat A1 receptors) for human A3 receptors than 10. The competitive binding curves of compound 12 at rat A1 and A2A receptors and at human A3 receptors are shown in Figure 3B.
Two 5-bromofuranyl derivatives were prepared. Compound 13, which was used as the synthetic precursor in the preparation of tritiated 120 and reported to be much less potent at rat A1 and rat A2A receptors,19 showed appreciable potencies at human A3 receptors. Compound 14 was prepared with an expectation of improving selectivity and affinity with respect to compounds 10 and 13. Unfortunately, this combination of functionalities displayed a dramatic loss of binding activity at all three adenosine receptor subtypes.
Finally, two 5-oxo compounds were also prepared and tested. Compound 15 was previously reported to be much less potent than 1 at A1 and A2A receptors,19 and we found it to be 15–17-fold A3 receptor selective. However, compound 16, the N6-propyl derivative of 15 displaced radioligand binding with lower affinity at the A3 adenosine receptor subtype.
Discussion
Until the present there have been few reports of leads for the development of selective antagonists for the A3 receptor,17,18 especially having high potency. We have screened chemical libraries11 and known adenosine receptor ligands10 for potential A3 receptor antagonists. Initially we reported that the triazoloquinazoline 1, which was reported as the first potent, non-xanthine, but nonselective adenosine A1 and A2A receptor antagonist, did not have appreciable binding activity at rat A3 receptors.10 Nevertheless, in this study, it proved to be highly potent at human A3 receptors (Ki = 13.8 nM), suggesting that it may serve as a lead compound in this species. This finding encouraged us to investigate structure–activity relationships of 1 derivatives for the development of highly potent and selective antagonists at human but not rat A3 receptors. There are dramatic species differences2,26 in the affinity of antagonists binding at A3 receptors. The overall identity of protein sequences between the A3 adenosine receptors of rat and human was reported as 72%,1,2 which is relatively low, consistent with the high variability in antagonist affinity.
The present findings indicated that certain less polar derivatives of 1 displayed increased affinity at the human A3 receptor, and this enhanced binding affinity could be distinguished from effects at rat A1 and rat A2A receptors. Compound 13 and 15, which were previously reported to have poor binding affinities at rat A1 and A2A receptors, showed binding affinities to human A3 receptors in the sub-micromolar range; thus some degree of selectivity was present. The most significant findings in the present study are that compounds 10 and 12, containing the N-benzoyl and N-phenylacetyl substituents, respectively, are highly potent and selective for human A3 receptors. A competitive binding assay of 10 showed high affinity at human A3 receptors (Ki = 3.0 nM), with 220-fold selectivity vs rat A1 receptors and 90-fold selectivity vs rat A2A receptors. Compound 12 displayed a Ki value of 0.65 nM, with 470-fold selectivity vs rat A1 receptor and 80-fold selectivity vs rat A2A receptors. The displacement of radioligand binding at rat A3 receptors by 10 and 12 was only 23% (3 μM) and 12% (1 μM), respectively, showing that there are marked differences of affinities between human A3 and rat A3 receptors.
Compound 14, the benzoyl derivative of compound 13, however, had greatly diminished affinity at adenosine receptors; thus the A3 receptor selectivity enhancing effects induced by N-acylation and by 5-bromination are not additive.
N-Acylations with various alkanecarboxylic acids resulted in less selective ligands than the aryl carboxylic acid derivatives. A structure–activity relationship analysis of various length of acyl chain substituents (2–7) resulted in the interesting finding that compound 3 displayed the optimum acyl chain length for A3 receptor binding.
There appears to be a hydrophobic pocket in the receptor in the vicinity of the N5-amino group, since hydrophobic N-acyl and N-aryl substituents could enhance potency. In contrast, removal of the hydrophobic Boc group from 8 resulted in the positively charged congener 9 that displayed a lower affinity at A3 receptors. According to molecular modeling,15 in the receptor bound states the N6 region of adenosine corresponds to the position of N5 of 1. In the present study, the introduction of an aryl ring in that region enhanced selectivity, as was found in the case of N6-benzylad-enosine derivatives.3 Compound 11 was synthesized to test further the hypothesis of overlap with the N6 region of adenosine, since the N6-3-iodobenzyl substituent, vs unsubstituted N6-3-benzyl, has been shown to enhance A3 receptor affinity and selectivity. Nevertheless, in the present series, the iodo substituent offered no advantage. N5 is also proposed to correspond to the N3-position of xanthines,15 at which large hydrophobic substituents are tolerated,10 consistent with our findings.
The selective ligands introduced here should be useful as antagonists in characterizing and probing the physiological role of human A3 receptors. They may also provide affinity probes such as radioligands for human A3 receptors. These selective agents must now be studied in functional assays.
Experimental Section
Materials.
Compound 1, (R)-PIA, and 2-chloroadenosine were purchased from Research Biochemicals International (Natick, MA). All acylating agents were obtained from Aldrich (St. Louis, MO).
Synthesis.
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer and spectra were taken in DMSO-d6 or CDCl3. Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Chemical-ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Atlantic Microlab Inc. (Norcross, GA) or Galbraith Laboratories, Inc. (Knoxville, TN). All melting points were determined with a Unimelt capillary melting point apparatus (Arthur H. Thomas Co., PA) and were uncorrected. All triazoloquinone derivatives showed one spot on TLC (MK6F silica, 0.25 mm, glass backed, Whatman Inc., Clifton, NJ). Where needed evaluation of purity was done on a Hewlett-Packard 1090 HPLC system using OD-5–60 C18 analytical column (150 mm × 4.6 mm, Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M TEAA/CH3CN, 30:70 to 10:90, in 20 min with flow rate 1 mL/min. The other (B) was H2O/MeOH, 40:60 to 10:90, in 20 min with flow rate 1 mL/min. Peaks were detected by UV absorption using a diode array detector.
General Procedure for Preparation of 5-N-Acyl Derivatives of 1.
Method A (Symmetrical Anhydride).
To a stirred solution of 1 (10 mg, 0.035 mmol), anhydride (0.105 mmol) and (dimethylamino)pyridine (0.5 mg, 0.004 mmol) in 1.5 mL of anhydrous DMF was added triethylamine (73 μL, 0.525 mmol) at room temperature. The mixture was stirred for 48 h and then evaporated to dryness under reduced pressure. The residue was purified by preparative silica gel TLC (CH2Cl2/MeOH, 50:1 ~ 75:1) to afford the desired compounds (2–5, 7, and 10).
Method B (Acid Chloride).
To a stirred solution of 1 (10 mg, 0.035 mmol) and anhydrous pyridine (40 μL, 0.5 mmol) in 1.5 mL of anhydrous CH2Cl2 was added acyl chloride (0.105 mmol) at 0 °C. The mixture was stirred at room temperature for 24–48 h and then treated with same procedure as method A for purification of the desired compounds (6 and 12).
Method C (Carbodiimide).
A solution of 1 (10 mg, 0.035 mmol), the carboxylic acid component (0.210 mmol), 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide (41 mg, 0.210 mmol), 1-hydroxybenzotriazole (28 mg, 0.210 mmol), 4-(dimethylamino)pyridine (0.5 mg, 0.004 mmol), and triethylamine (74 μL, 0.530 mmol) in 2 mL of anhydrous DMF/CH2Cl2 (1:1 v/v) was stirred at room temperature for 48 h. The mixture was treated with same procedure as method A for purification of desired compounds (8 and 11).
5-(Acetylamino)-9-chloro-2-(2-furanyl)[1,2,4]triazolo-[1,5-c]quinazoline (2):
1H NMR (CDCl3) δ 2.78 (3H, s, CH3CO), 6.63–6.65 (1H, m, H-4′), 7.30 (1H, d, J = 2.9, H-3′),7.68 (1H, broad s, H-5′), 7.73 (1H, dd, J = 1.9, 8.8, H-8), 7.86 (1H, d, J = 8.8, H-7), 8.48 (1H, d, J = 2.9, H-10), 8.99 (NH, broad s).
9-Chloro-2-(2-furanyl)-5-(n-propionylamino)[1,2,4]-triazolo[1,5-c]quinazoline (3):
1H NMR (CDCl3) δ 1.34 (3H, t, J = 7.8, CH3CH2CO), 3.10 (2H, q, J = 7.8, CH3CH2CO), 6.63–6.65 (1H, m, H-4′), 7.31 (1H, d, J = 3.9, H-3′), 7.68 (1H, d, J = 1.9, H-5′), 7.73 (1H, dd, J = 2.0, 8.8, H-8), 7.88 (1H, d, J = 8.8, H-7), 8.48 (1H, d, J = 2.0, H-10), 9.01 (NH, broad s).
5-(n-Butyrylamino)-9-chloro-2-(2-furanyl)[1,2,4]triazolo-[1,5-c]quinazoline (4):
1H NMR (CDCl3) δ 1.10 (3H, t, J = 7.4, 7.3, CH3CH2CH2CO), 1.84–1.91 (2H, m, CH3CH2CH2CO), 3.03 (2H, t, J = 7.4, 7.3, CH3CH2CH2CO), 6.63–6.65 (1H, m, H-4′), 7.31 (1H, d, J = 3.4, H-3′), 7.68 (1H, d, J = 1.8, H-5′), 7.73 (1H, dd, J = 2.3, 8.8, H-8), 7.88 (1H, d, J = 8.8, H-7), 8.48 (1H, d, J = 2.3, H-10), 8.97 (NH, broad s).
9-Chloro-2-(2-furanyl)-5-(n-pentanoylamino)[1,2,4]-triazolo[1,5-c]quinazoline (5):
1H NMR (CDCl3) δ 1.01 (3H, t, J = 6.8, 7.8, CH3CH2CH2CH2CO), 1.48–1.55 (2H, m, CH3CH2CH2CH2CO), 1.77–1.85 (2H, m, CH3CH2CH2CH2CO), 3.05 (2H, t, J = 7.82, 6.8, CH3CH2CH2CH2CO), 6.63–6.65 (1H, m, H-4′), 7.31 (1H, d, J = 2.9, H-3′), 7.68 (1H, broad s, H-5′), 7.73 (1H, dd, J = 2.0, 8.8, H-8), 7.88 (1H, d, J = 8.8, 7-H), 8.48 (1H, d, J = 2.9, 10-H), 8.98 (NH, broad s).
9-Chloro-2-(2-furanyl)-5-[(trimethylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline (6):
1H NMR (CDCl3) δ 1.47 (9H, s, (CH3)3CCO), 6.63–6.65 (1H, m, H-4′), 7.31 (1H, d, J = 3.4, H-3′), 7.68–7.69 (1H, m, H-5′), 7.74 (1H, dd, J = 2.5, 8.9, H-8), 8.01 (1H, d, J = 8.9, H-7), 8.49 (1H, d, J = 2.5, H-10), 9.39 (NH, broad s).
5-[(tert-Butoxycarbonyl)amino]-9-chloro-2-(2-furanyl)-[1,2,4]triazolo[1,5-c]quinazoline (7):
1H NMR (CDCl3) δ 1.63 (9H, s, (CH3)3COCO), 6.65–6.66 (1H, m, H-4′), 7.35 (1H, d, J = 3.5, H-3′), 7.69 (1H, broad s, H-5′), 7.74 (1H, dd, J = 2.6, 8.9, H-8), 8.01 (1H, d, J = 8.9, H-7), 8.49 (1H, d, J = 2.6, H-10), 8.56 (NH, broad s).
5-[[4-[(tert-Butoxycarbonyl)amino]butyryl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (8):
1H NMR (CDCl3) δ 1.44 (9H, s, (CH3)3COCONH), 2.03 (2H, pen, J = 6.8, NHCH2CH2CH2CO), 3.13 (2H, t, J = 6.8, 7.3, NHCH2CH2CH2CO), 3.32 (2H, q, J = 6.4, NHCH2CH2CH2-CO), 4.81 (NH, broad s), 6.63–6.65 (1H, m, H-4′), 7.27–7.32 (1H, m, H-3′), 7.67–7.68 (1H, m, H-5′), 7.71–7.75 (1H, m, H-8), 7.91 (1H, d, J = 8.8, H-7), 8.48 (1H, d, J = 2.0, H-10), 9.15 (NH, broad s).
5-[(4-aminobutyryl)amino]-9-chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazoline (9).
A solution of 8 (3 mg, 6.4 μmol) in 1 mL of 50% TFA in CH2Cl2 was left to stand for 10 min and evaporated under reduced pressure. Crystallization of the residue with MeOH/CH2Cl2 gave 3.2 mg of 9 (83% as TFA salt) as a white solid: 1H NMR (DMSO-d6) δ 1.87–1.97 (2H, m, H2NCH2CH2CH2CO), 2.82 (2H, t, J = 7.32, H2NCH2-CH2CH2CO), 2.88–2.94 (2H, m, H2NCH2CH2CH2CO), 6.78–6.80 (1H, m, H-4′), 7.34 (1H, d, J = 3.05, H-3′), 7.77 (NH2, broad s), 7.93–8.02 (3H, m), 8.40 (1H, d, J = 1.2, H-10), 11.17 (NH, s).
5-(Benzoylamino)-9-chloro-2-(2-furanyl)[1,2,4]triazolo-[1,5-c]quinazoline (10):
1H NMR (CDCl3) δ 6.64–6.66 (1H, m, H-4′), 7.35 (1H, d, J = 2.9, H-3′), 7.61–7.68 (3H, m), 7.68–7.70 (1H, m, H-5), 7.77 (1H, dd, J = 2.0, 8.8, H-8), 8.04–8.10 (3H, m), 8.52 (1H, d, J = 2.0), 9.75 (NH, broad s).
9-Chloro-2-(2-furanyl)-5-[(3-iodobenzoyl)amino][1,2,4]-triazolo[1,5-c]quinazoline (11):
1H NMR (CDCl3) δ 6.64–6.66 (1H, m, H-4′), 7.30–7.36 (3H, m), 7.70 (1H, broad s, H-5′), 7.77 (1H, dd, J = 2.0, 10.7, H-8), 7.92–8.10 (2H, broad m), 8.39 (1H, m), 8.52 (1H, d, J = 2.0, hH-10), 9.62 (NH, broad s).
9-Chloro-2-(2-furanyl)-5-[(phenylacetyl)amino][1,2,4]-triazolo[1,5-c]quinazoline (12):
1H NMR (CDCl3) δ 4.38 (2H, s, CH2CO), 6.62–6.65 (1H, m, H-4′), 7.24–7.26 (1H, m, H-3′), 7.35–7.44 (5H, m), 7.67–7.68 (1H, m, H-5′), 7.74 (1H, dd, J = 2.2, 9.0, H-8), 7.93 (1H, d, J = 8.8, H-7), 8.49 (1H, m, H-10), 9.10 (NH, broad s).
5-Amino-2-[2-(5-bromofuranyl)]-9-chloro[1,2,4]triazolo-[1,5-c]quinazoline (13):
A solution of 7 (0.01 g, 0.026 mmol) and N-bromosuccinimide (0.005 g, 0.028 mmol) in 2 mL of AcOH/CHCl3 (1:1) was stirred for 1 h at room temperature. The mixture was poured into 10 mL of saturated NaHCO3 solution, and the product was extracted with 10 mL of CHCl3 three times. The combined CHCl3 solution was washed with brine, dried over anhydrous Na2SO4, and evaporated to dryness under reduced pressure. The residue was purified by preparative silica gel TLC (CHCl3/MeOH, 80:1) to afford 2-[2-(5-bromofuranyl)]-5-[(tert-butoxycarbonyl)amino]-9-chloro[1,2,4]-triazolo[1,5-c]quinazoline (0.012 g, 99%) as a white solid: MS (CI, NH3) 466 (M+ + 1); 1 H-NMR (CDCl3) δ 1.63 (9H, s, (CH3)3-OCO), 6.58 (1H, d, J = 3.6, H-4′), 7.29 (1H, d, J = 3.6, H-3′), 7.73 (1H, dd, J = 2.3, 8.8, H-8), 7.98 (1H, d, J = 9.0, H-7), 8.46 (1H, d, J = 2.3, H-10), 8.53 (NH, broad s). To a solution of this compound in 2 mL of CH2Cl2 was added TFA (0.05 mL, 0.67 mmol), and the mixture was stirred for 2 h at room temperature. The reaction mixture was treated with same workup procedure above. A preparative silica gel TLC (n-hexane/CHCl3/MeOH, 1:1:0.1) of the crude product gave 13 (4.5 mg, 48%) as a white solid: MS (CI, NH3) 366 (M+ + 1), 383 (M+ + 18); 1H-NMR (CDCl3) δ 5.94 (NH2, broad s), 6.56 (1H, d, J = 3.8, H-4′), 7.25 (1H, d, J = 3.8, H-3′), 7.63–7.65 (2H, m, H-8 + H-7), 8.41 (1H, d, J = 2.1, H-10).
5-(Benzoylamino)-2-[2-(5-bromofuranyl)]-9-chloro[1,2,4]-triazolo[1,5-c]quinazoline (14).
The furanyl group in compound 10 was brominated by the same method as above: 1H-NMR (CDCl3) δ 6.64–6.66 (1H, m, H-4′), 7.35 (1H, d, J = 2.9, H-3′), 7.61–7.68 (2H, m), 7.68–7.70 (1H, m), 7.77 (1H, dd, J = 2.0, 8.8, H-8), 8.04–8.10 (3H, m), 8.52 (1H, d, J = 2.0, H-10), 9.75 (NH, broad s); HPLC retention time: 4.6 min (>95% purity) using solvent system A, 12.5 min (>95% purity) using solvent system B.
9-Chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5(6H)-one (15).
A solution of 1 (0.075 g, 0.263 mmol) in 8.0 mL of AcOH and 2.0 mL of H2O in a sealed tube was heated for 72 h at 100 °C. The solution was coevaporated with toluene under reduced pressure, and the residue was purified by preparative silica gel TLC (CHCl3/MeOH, 15:1) to afford 15 (0.065 g, 86%) as a white solid: mp >310 °C; MS (CI NH3) 287 (M+ + 1), 304 (M+ + 18); 1H-NMR (DMSO-d6) δ 6.73–6.74 (1H, m, H-4′), 7.26 (1H, d, J = 3.5, H-3′), 7.46 (1H, d, J = 8.8, H-7), 7.77 (1H, dd, J = 2.5, 8.9, H-8), 7.97 (1H, s, H-5′), 8.16 (1H, d, J = 2.5, H-10), 12.47 (NH, s).
9-Chloro-2-(2-furanyl) 6-n-propyl[1,2,4]triazolo[1,5-c]-quinazolin-5-one (16).
To a suspension of 15 (0.021 g, 0.073 mmol) in 2 mL of anhydrous THF was added a suspension of NaH (6 mg, 60% in mineral oil, prewashed with n-hexane, 0.15 mmol) in 2 mL of anhydrous THF followed by HMPA (0.21 mL, 12 mmol) under N2 atmosphere at room temperature. The mixture was stirred vigorously for 30 min (H2 gas evolved). 1-Bromopropane (28 μL, 0.3 mmol) was added, and the reaction mixture was refluxed for 6 h. After cooling, the precipitate was removed by filtration through a small volume of silica gel bed and the filtrate was evaporated. The residue was purified by preparative silica gel TLC (n-hexane/EtOAc, 2:1) to afford 16 (0.012 g, 50%) as a white solid: 1H-NMR (CDCl3) δ 1.11 (3H, t, J = 7.5, 7.5, CH3CH2CH2N), 1.85–1.93 (2H, m, CH3CH2-CH2N), 4.35 (2H, t, J = 8.0, 7.5, CH3CH2CH2N), 6.60–6.61 (1H, m, H-4′), 7.32 (1H, d, J = 3.4, H-3′), 7.37 (1H, d, J = 9.3, H-7), 7.66 (1H, broad s, H-5′), 7.68 (1H, dd, J = 2.39, 9.0, H-8), 8.52 (1H, d, J = 2.5, H-10); HPLC retention time 3.5 min (>95% purity) using solvent system A, 9.1 min (>95% purity) using solvent system B.
Pharmacology: Radioligand Binding Studies.
Binding of [3H]-(R)-PIA to A1 receptors from rat cerebral cortex membranes and of [3H]CGS 21680 to A2A receptors from rat striatal membranes was performed as described previously.6,8 Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in a preincubation of 30 min at 30 °C and during the incubation with the radioligands.
Binding of [125I]AB-MECA in membranes prepared from HEK-293 cells stably expressing the human A3 receptor (Receptor Biology, Inc., Baltimore, MD), or from CHO cells stably expressing the rat A3 receptor, was as described.17,25 The assay medium consisted of a buffer containing 50 mM Tris, 10 mM Mg2+, and 1 mM EDTA, at pH 8.0. The glass incubation tubes contained 100 μL of the membrane suspension (0.3 mg of protein/mL, stored at −80 °C in the same buffer), 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of a solution of the proposed antagonist. Nonspecific binding was determined in the presence of 200 μM NECA.
All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%.
Incubations were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL buffer each.
At least five different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in the InPlot program (Graph-PAD, San Diego, CA), were converted to apparent Ki values using the Cheng-Prusoff equation27 and Kd values of 1.0 nM, 14 nM for [3H]-(R)-PIA and [3H]CGS 21680, and 0.59 nM for binding of [125I]AB-MECA at human A3 receptors, respectively. Most Hill coefficients of tested compounds were in the range 0.8–1.1.
Acknowledgment.
We thank Dr. Mark E. Olah and Prof. Gary L. Stiles (Duke University Medical Center, Durham, NC) and Dr. Garth Brown of DuPont NEN (North Billerica, MA) for providing samples of [125I]AB-MECA.
Abbreviations:
- AcOH
acetic acid
- Boc
tert-butoxycarbonyl
- CGS 21680
2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-N-(ethylcarbamoyl)adenosine
- CGS15943
9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- CHO cells
Chinese hamster ovary cells
- CI
chemical ionization
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacatate
- HEK cells
human embryonic kidney cells
- HMPA
hexamethylphosphotriamide
- [125I]AB-MECA
[125I]-N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- K i
equilibrium inhibition constant
- MS
mass spectrum
- NECA
(N-ethylcarbamoyl)adenosine
- (R)-PIA
(R)-N6-(phenylisopropyl)adenosine
- SAR
structure-activity relationship
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- Tris
tris(hydroxymethyl)amino-methane
References
- (1).Olah ME; Stiles GL Adenosine receptor subtypes : characterization and therapeutic regulation. Annu. Rev. Pharmacol. Toxicol 1995, 35, 581–606. [DOI] [PubMed] [Google Scholar]
- (2).Linden J Cloned adenosine A3 receptors - pharmacological properties, species -differences and receptor functions. Trends Pharmacol. Sci 1994, 15, 298–306. [DOI] [PubMed] [Google Scholar]
- (3).Jacobson KA; Kim HO; Siddiqi SM; Olah ME; Stiles G; von Lubitz DKJE A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future 1995, 20, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hannon JP; Pfannkuche HJ; Fozard JR A role for mast-cells in adenosine A3 receptor-mediated hypotension in the rat. Br. J. Pharmacol 1995, 115, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fozard JR; Pfannkuche H-J; Schuurman H-J Mast cell degranulation following adenosine A3 receptor activation in rats. Eur. J. Pharmacol 1996, 298, 293–297. [DOI] [PubMed] [Google Scholar]
- (6).Jacobson KA; Nikodijević O; Shi D; Gallo-Rodriguez C; Olah ME; Stiles GL; Daly JW A role for central A3-adenosine receptors: Mediation of behavioral depressant effects. FEBS Lett. 1993, 336, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).von Lubitz DKJE; Lin RCS; Popik P; Carter MF; Jacobson KA Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol 1994, 263, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kohno Y; Sei Y; Koshiba M; Kim HO; Jacobson KA Induction of apoptosis in HL-60 human promyelocytic leukemia cells by selective adenosine A3 receptor agonists. Biochem. Biophys. Res. Commun 1996, 219, 904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim HO; Ji XD; Melman N; Olah ME; Stiles GL; Jacobson KA Structure-activity relationships of 1,3-dialkyl-xanthine derivatives at rat A3 adenosine receptors. J. Med. Chem 1994, 37, 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).van Galen PJM; van Bergen AH; Gallo-Rodriquez C; Melman N; Olah ME; IJzerman AP; Stiles GL; Jacobson KA A binding-site model and structure-activity-relationships for the rat A3-adenosine receptor. Mol. Pharmacol 1994, 45, 1101–1111. [PMC free article] [PubMed] [Google Scholar]
- (11).Siddiqi SM; Ji XD; Melman N; Olah ME; Jain R; Evans P; Glashofer M; Padgett WL; Cohen LA; Daly JW; Stiles GL; Jacobson KA A survey of non-xanthine derivatives as adenosine receptor ligands. Nucleoside Nucleotide 1996, 15, 693–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jacobson KA; van Galen PJM; Williams M Adenosine receptorsspharmacology, structure activity relationships, and therapeutic potential. J. Med. Chem 1992, 35, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Belardinelli L; Shryock JC; Zhang Y; Scammells PJ; Olsson R; Dennis D; Milner P; Pfister J; Baker SP 1,3-dipropyl-8-[2-(5,6-epoxy)norbornyl] xanthine, a potent, specific and selective A1 adenosine receptor antagonist in the guinea-pig heart and brain and in DDT(1)MF-2 cells. J. Pharmacol. Exp. Ther 1995, 275, 1167–1176. [PubMed] [Google Scholar]
- (14).Baraldi PG; Cacciari B; Spalluto G; Borioni A; Viziano M; Dionisotti S; Ongini E Current developments of A2a adenosine receptor antagonists. Curr. Med. Chem 1995, 2, 707–722. [Google Scholar]
- (15).van Rhee AM; Siddiqi SM; Melman N; Shi D; Padgett WL; Daly JW; Jacobson KA Tetrahydrobenzothiophenone derivatives as a novel class of adenosine receptor antagonists. J. Med. Chem 1996, 39, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ji XD; Melman N; Jacobson KA Interactions of flavonoids and other phytochemicals with adenosine receptors. J. Med. Chem 1996, 39, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Karton Y; Jiang J.-l.; Ji XD; Melman N; Olah ME; Stiles GL; Jacobson KA Synthesis and biological activities of flavonoid derivatives as A3 adenosine receptor antagonists. J. Med. Chem 1996, 39, 2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).van Rhee AM; Jiang J -l.; Melman, N.; Olah, M. E.; Stiles, G. L.; Jacobson, K. A. Interaction of 1,4-dihydropyridine and pyridine derivatives with adenosine receptors: selectivity for A3 receptors. J. Med. Chem 1996, 39, 2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Francis JE; Cash WD; Psychoyos S; Ghai G; Wenk P; Friedmann RC; Atkins C; Warren V; Furness P; Hyun JL Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J. Med. Chem 1988, 31, 1014–1020. [DOI] [PubMed] [Google Scholar]
- (20).Jarvis MF; Williams M; Do UH; Sills MA Characterization of the binding of a novel non-xanthine adenosine antagonist radioligand, [3H]CGS 15943, to multiple affinity states of the adenosine A1 receptor in the rat cortex. Mol. Pharmacol 1991, 39, 49–54. [PubMed] [Google Scholar]
- (21).Ji X-D; Stiles GL; Galen P. J. M. v.; Jacobson KA Characterization of human striatal A2-adenosine receptors using radioligand binding and photoaffinity labeling. J. Recept. Res 1992, 12, 149–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schwabe U; Trost T Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol 1980, 313, 179–187. [DOI] [PubMed] [Google Scholar]
- (23).Jarvis MF; Schutz R; Hutchison AJ; Do E; Sills MA; Williams M [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther 1989, 251, 888–893. [PubMed] [Google Scholar]
- (24).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Olah ME; Gallo-Rodriguez C; Jacobson KA; Stiles GL [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- (26).Ji XD; von Lubitz DKJE; Olah ME; Stiles GL; Jacobson KA Species differences in ligand affinity at central A3-adenosine receptors, Drug Dev. Res 1994, 33, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cheng YC; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (28).Rivkees SA; Lasbury ME; Barbhaiya H Identification of domains of human A1 adenosine receptor that are important for binding receptor subtype-selective ligands using chimeric A1/A2A adenosine receptors. J. Biol. Chem 1995, 270, 20485–20490. [DOI] [PubMed] [Google Scholar]
- (29).Kim J; Jiang Q; Glashofer M; Yehle S; Wess J; Jacobson KA Glutamate residues in the second extracellular loop of the human A2A adenosine receptor are required for ligand recognition. Mol. Pharmacol 1996, 49, 683–691. [PMC free article] [PubMed] [Google Scholar]