

Abstract
BACKGROUND
To date, only a few cases of sellar and suprasellar glioblastomas have been reported even though high-grade glioma constitutes the most common adult brain tumor, commonly arising in the cerebral hemispheres. It arises de novo from astrocytes within the optic nerve, optic chiasm, or optic tracts and is quite challenging to diagnose and treat. To the authors’ knowledge, there are 72 cases (including this one) of optic glioma malignancies in the medical literature, 30 corresponding to glioblastomas.
OBSERVATIONS
The authors present the diagnostic considerations and challenges, management strategies, and clinical course of a very large sellar-suprasellar glioblastoma in a 19-year-female who had never received radiation therapy or prior surgery.
LESSONS
Sellar-suprasellar glioblastomas, although extremely rare, are known to occur and pose challenges in their diagnosis and preoperative treatment planning. The presence of diffusion restriction on diffusion-weighted magnetic resonance imaging in a mass lesion that has ring and nodular postcontrast enhancement in addition to absent calcification on computed tomography should be alert to the possibility of a high-grade mass. This is extremely important for preoperative patient counseling and planning for the multimodal treatments, because sellar-suprasellar glioblastomas carry a poorer prognosis than the common benign mass lesions in the region.
Keywords: glioblastoma, malignant optic pathway gliomas, sellar, suprasellar
ABBREVIATIONS: ACA = anterior cerebral artery, ADC = apparent diffusion coefficient, DWI = diffusion-weighted imaging, FLAIR = fluid-attenuated inversion recovery, GBM = glioblastoma, GRE = gradient echo, ICA = internal carotid artery, ICU = intensive care unit, MCA = middle cerebral artery, MRI = magnetic resonance imaging, NF1 = neurofibromatosis type 1, NF2 = neurofibromatosis type 2, OPG = optic pathway glioma, PMA = pituitary macroadenoma, WHO = World Health Organization
Malignant sellar gliomas are very rare phenomena even if they constitute the most common adult brain tumor, typically arising in the cerebral hemispheres. To date, only a few cases of sellar and suprasellar glioblastomas (GBMs) have been reported, most of which originated from the optic nerve or optic chiasm.1
Malignant optic pathway gliomas arise de novo from astrocytes within the optic nerve, optic chiasm, or optic tracts and are rare and very challenging to diagnose and treat.2,3 Malignant glioma involving the optic chiasm and hypothalamus usually occur only secondarily.4
Malignant gliomas involving the optic nerves and/or chiasm typically have a clinical presentation that is quite distinct from the more common benign optic pathway glioma (OPG) of the pediatric population.5 Suprasellar or hypothalamic/chiasmatic region GBMs are commonly interpreted radiographically as consistent with craniopharyngioma or other lesions and are a diagnostic challenge.1 Larger tumors involving the optic chiasm and hypothalamus often require surgery, both to distinguish them pathologically from other suprasellar neoplasms and to relieve mass effect.6,7
Gross-total resection is typically not performed due to potentially severe endocrine and visual morbidity. Depending on the age of the patient and the clinical and radiographic findings, chemotherapy, radiation therapy, or observation may be considered postoperatively.8 Here, we present the diagnostic considerations, management strategies, and clinical course of a very large sellar-suprasellar GBM in a patient who had never received radiation therapy or prior surgery.
Illustrative Case
A 19-year-old female college student, who claimed to have a 7-year history of global headache, presented with a new compliant of behavioral change and left visual deterioration of 10 months, as well as left-eye complete visual loss of 1 months’ duration. Otherwise, she claimed that she had experienced regular menses since the age of 14. She denied seizure, vomiting, body weakness, weight gain, increased urination, and fecal or urinary incontinence. Neither did she have known chronic medical illnesses or prior history of surgery or chemoradiation therapy.
Physical examination revealed well-appearing patient with normal range vital signs. On neurological examination, her Glasgow Coma Scale score was 15/15. Her visual acuity decreased in the right eye (counts from 3 m with temporal field cut), whereas there was no light perception on the left side and the pupil was dilated and fixed due to left-side partial oculomotor nerve injury. No other neurological or other pertinent physical system findings were noted. There were no stigmata suggesting neurocutaneous syndromes like neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2); neither was there a family history of NF1 and NF2. Baseline blood workup showed a normal complete blood count, organ function tests (renal and liver), and serum electrolytes. Serum hormone profiles showed low thyroid-stimulating hormone at −0.17 uIU (0.4–4), low free triiodothyronine at 2.38 pmol (4–8.3), and normal free thyroxine at 13.76 pmol (10.6–19.4). In addition, she had low serum cortisol at 80 nmol/L (140–700) and a normal level of prolactin at 4.09 ng/mL (5–35).
Imaging Findings
On preoperative brain magnetic resonance imaging (MRI), there was a sellar-suprasellar multicompartmental butterfly-shaped large mass 6 cm in size, and the tumor had interpeduncular fossa, hypothalamus, left subfrontal area, and paraventricular extension as well as secondary extension to the third ventricle, optic chiasm, and optic nerves, whereas the stalk was displaced posteroinferiorly to the tumor. The lesion was heterogeneously hyperintense and isointense on both T1- and T2-weighted sequences. The hyperintense part of the mass lesion (Fig. 1) was likely suggestive of intratumoral subacute bleeding. The lesion appeared predominantly hyperintense on fluid-attenuated inversion recovery (FLAIR) sequences, and there was no lesion that nulled out on FLAIR imaging, suggesting some sort of intratumoral bleeding. The mass had heterogeneous nodular and ring postcontrast enhancement (Fig. 2). Similarly, the T2* gradient echo (GRE) showed hypointense foci of the mass lesion (Fig. 1), suggesting either hemorrhage or calcification. In this case, the hypointense foci on T2* GRE was hemorrhage, as it appeared hypodense on noncontrast computed tomography (CT) scanning, and there was no calcification (Fig. 3D). In addition, the tumor had areas of restriction on diffusion-weighted imaging (DWI), suggesting a highly cellular or high-grade tumor (Fig. 2). The tumor had 360° encasement of major vessels, including internal carotid artery (ICA), middle cerebral artery (MCA), and anterior cerebral artery (ACA), especially on the left side (Fig. 3B). Altogether, the diagnosis of a sellar and suprasellar mass secondary to high-grade glioma plus R/O craniopharyngioma with or without malignant degeneration plus hypocortisolism was made.
FIG. 1.
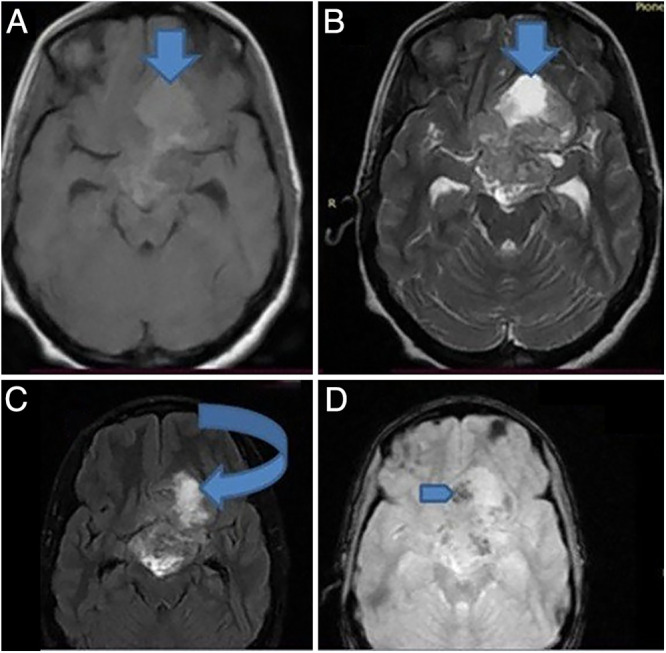
On T1-weighted (A), T2-weighted (B), and FLAIR (C) sequences, a sellar-suprasellar multicompartmental butterfly-shaped large mass 6 cm in size appeared hyperintense and isointense, and the tumor had interpeduncular fossa, hypothalamus, left subfrontal area, and paraventricular extension, as well as secondary extension to the third ventricle. The hyperintense part of the mass lesion (down arrows) was likely suggestive of intratumoral subacute bleeding. There was no lesion that nulled out on FLAIR imaging (curved arrow), suggesting some sort of intratumoral bleeding. The T2* GRE (D) sequence showed hypointense foci of the mass lesion (blue arrow), suggesting either hemorrhage or calcification (calcification was ruled out by CT scan, as seen in Fig. 3).
FIG. 2.

Tumor had bright and dark areas of restriction on DWI (A) and ADC (B). The restricting foci of tumor is indicative of a highly cellular or high-grade tumor. The mass had heterogeneous nodular and ring postcontrast enhancement (C–E).
FIG. 3.

Different CT angiography views (A–C). The red double arrow (B) shows a 360° encasement of major vessels, including the ICA, MCA, and ACA, especially on the left side by the tumor. The hypodensity (blue triangle, D) on a noncontrast CT scan suggests hemorrhage and no calcification. Immediate postoperative control MRI (E and F) showed a well-decompressed sella and suprasellar region with a rim of residual tumor (red arrow). The major vessels and the stalk were also well preserved (arrow, G).
Surgery
A left pterional craniotomy was made, and the dura mater was opened in C-shaped manner and tented. Proximal sylvian dissection was done, and a combined transsylvian plus subfrontal approach was used to excise the tumor once the critical neurovascular structures were identified. The hemorrhagic part of the tumor was decompressed first to relax the brain, and it was a subacute hemorrhage. The tumor was reddish and fleshy with a soft consistency. The tumor was highly vascular and easily aspirated; hence, the encased ICA, MCA, and ACA were circumferentially freed by gently aspirating the tumor along the course of the vessels. In resecting the tumor, the oculomotor carotid, opticocarotid, and translamina terminalis corridors were used. The tumor was debulked internally to stay safe and protect the neurovascular structures. The optic chiasm was not identified intraoperatively, and clear cleavage between the optic nerve and tumor was not identified. The intraoperative gross pathology findings were more suggestive of a high-grade tumor like GBM over craniopharyngioma, because there were no cholesterol crystals and the tumor was highly vascular. Hence, after achieving significant safe decompression of the tumor, hemostasis was achieved, and the dura was closed in watertight fashion. Galea and skin were closed in layers. The patient then was extubated and transferred to intensive care unit (ICU) with a stable neurological status.
Postoperative Course
The patient was transferred to the ward after she stayed 1 night in the neurocritical ICU. There was no neurological deficit postoperatively, and she was able to count from 4 m by her right eye with improvement of the temporal field cut, and her headache improved. Postoperative investigations were normal except for low serum cortisol at <80 (140–700). Immediate postoperative control MRI showed safe maximal resection of the tumor with some residual tumors along the interpeduncular and hypothalamic region (Fig. 3E–F). In addition, the sella and suprasellar region was well decompressed and the major vessels and the stalk were also well preserved (Fig. 3G). She was discharged on her sixth postoperative day and linked to the oncology clinic for initiation of radiation and chemotherapy (Stupp regimen). Her follow-up at the first and third month postoperatively showed marked relief of her complaints, and she was able to count from 4 m by her right eye.
Histopathology Result
Histopathology examination revealed highly cellular oval to polygonal hyperchromatic cells, mitosis, and proliferation of pleomorphic bizarre cells (Fig. 4 right) that was accompanied by extensive tumor necrosis (depicted by downward-facing arrows of Fig. 5) and vascular proliferation (Fig. 4 left) with a focus of keratin suggesting a World Health Organization (WHO) grade 4 brain tumor (GBM). The histopathological appearance of the tumor under higher and lower magnifications is presented in Figs. 4 and 5. Immunohistochemistry evaluation of the biopsied tissue showed strong and diffuse positivity to GFAP and S100 but negative to CD34 and CK7 with a Ki-67 index of 50%. These immunohistochemical findings confirmed once again that the tumor was indeed a high-grade glioma.
FIG. 4.
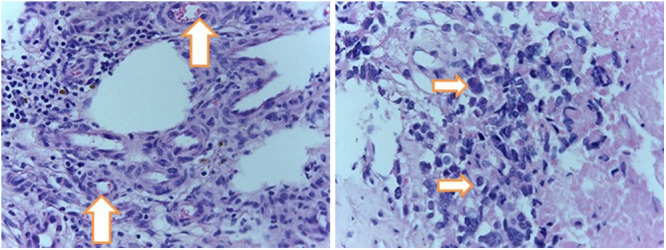
Histopathology examination under higher magnifications revealed highly cellular oval to polygonal hyperchromatic cells, mitosis, and proliferation of pleomorphic bizarre cells (arrows, right) that was accompanied by and vascular proliferation (arrows, left) with a focus of keratin suggesting WHO grade 4 brain tumor (GBM). Hematoxylin and eosin, original magnification ×20.
FIG. 5.
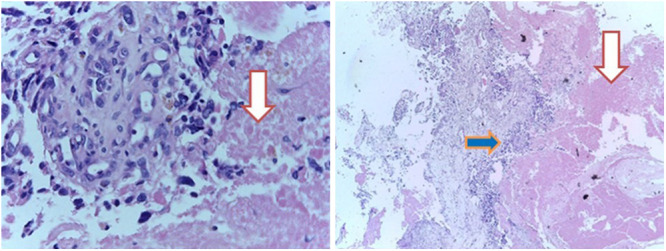
The histopathological appearance of the tumor under higher (left, original magnification ×20) and lower (right, original magnification ×4) magnifications showing extensive tumor necrosis (downward-facing arrows) seen in GBM. The right-facing blue arrow signifies a highly cellular sheet of hyperchromatic malignant glial cells with extensive mitosis.
Patient Informed Consent
The necessary patient informed consent was obtained in this study.
Discussion
Observations
Optic gliomas represent 0.6% to 1.2% of all brain tumors detected in general.9 The tumor can originate in the optic chiasm, the optic tract, or the optic nerves and can occur as multifocal neoplasia. Most gliomas are benign, occur in the pediatric population before age 15 (85%), and are strongly associated with NF1.6,10 Malignant optic gliomas rarely occur in adults; this entity was defined for the first time in 1973 by Hoyt et al.11 To our knowledge, there are at least 71 cases of optic glioma malignancies in the medical literature, 30 corresponding to GBM.12
Clinically, patients with OPGs present with rapidly progressive vision loss, either unilateral or bilateral, in conjunction with headache or periorbital pain.6 Although the presentation symptoms of our patient and most patients in the literature were mainly visual complaints, the rapidity of visual deterioration was extremely faster in reported cases with an average time to complete vision loss of 5 to 6 weeks in contrast to our patient who had a 7-year history of headaches and whose complete visual loss took nearly 10 months.
The tumor’s site of origin determines whether the initial vision loss is unilateral or bilateral and the presence or absence of ophthalmic symptoms. Tumors originating in the optic nerve initially cause unilateral vision loss and bilateral changes in fundoscopy (disc edema and venous stasis). On the other hand, tumors in the optic chiasm present bilateral visual alterations and normal fundoscopy. The latter is more likely to infiltrate the hypothalamus and the walls of the third ventricle. If the tumor is in the optic tract, there will be a visual field deficit contralateral to the side of the lesion.6,12 Our patient had bilateral visual complaints (left-sided complete visual loss and right-sided significant field cut and decreased visual acuity), which is consistent with an optic chiasm origin. A hypothalamic origin is unlikely, because she did not have any one of the hypothalamic syndrome manifestations.
The mean age at onset for 66 reported cases was 57 years (standard deviation ± 15; range, 22–83 years), with females and males almost equally affected (30 females and 36 males). It rarely occurs in pediatric populations, either as a primary high-grade glioma or as malignant transformation of a low-grade glioma.6,12 This finding contrasts with our case, because our patient presented at 19 years of age; it is an extremely rare event for a GBM to develop elsewhere in the brain let alone the sellar-suprasellar area.
The differential diagnosis includes pituitary adenomas, the most common tumors in the sellar region, followed by craniopharyngiomas and Rathke cleft cysts and, less commonly, meningiomas, germinomas, hamartomas, and metastatic tumors. On the contrary, malignant sellar gliomas are very rare, and only a few cases of suprasellar malignant gliomas have been reported.1,12–17
Endosuprasellar malignant gliomas, originating from the hypothalamic/pituitary axis, also contribute to the differential diagnosis of sellar-suprasellar masses even if only five cases have been reported.1,12–15
Overall, considering the high malignancy of GBMs, it is important to inform clinicians and radiologists about these tumors in the chiasmatic-sellar region. Besides, it is essential to discuss the possibility of the differential diagnosis during the preoperative stage, which can be useful for early selection of a treatment plan.12
MRI of the brain is the primary diagnostic tool. However, MRI examination of pituitary tumors does not clearly differentiate the most common pituitary adenomas from gliomas and GBMs of the pituitary gland and sellar-suprasellar region, as both types of lesions can be hyperintense on T2-weighted images and iso- or hypointense on T1-weighted images, with features of central necrosis in the tumors.12–17 The presence of signs of restricted diffusion on the sellar-suprasellar GBMs could not help us establish an accurate diagnosis because it is well known that macroadenomas with a soft consistency have high signal intensity on DWI low apparent diffusion coefficient (ADC).18 Given that the pituitary and the stalk were partly pushed posteroinferiorly and that the restricting component of the tumor was the suprasellar, interpeduncular, and paraventricular area of the tumor, it was clear that the part restricting in our patient was likely to be from another high-grade tumor rather than a soft pituitary macroadenoma (PMA), and the consideration of PMA as a diagnosis was unlikely in our patient.
Rapid tumor growth, unrelated to hormonal abnormalities (in the absence of hormonal activity of the tumor), may lead to a suspicion of GBM in the sellar region. That means that a malignant endosuprasellar mass originating from the pituitary gland can also be suspected when there is a significant decrease in the prolactin level while the tumor fails to decrease in size or even increase rapidly. The initial higher level of prolactin in these cases is likely from the Hook effect that developed from the mass effect rather than a prolactin-secreting PMA; hence, the diagnosis of a prolactin-secreting PMA should be doubted.
Moreover, squamous papillary craniopharyngiomas, which are more common in adults, may have similar MRI characteristics of sellar-suprasellar GBM, complicating the differential diagnosis of these masses.18,19
The more common craniopharyngioma, especially those that have cystic and nodular postcontrast enhancement, may mimic these tumors; however, the presence of calcification on CT scanning, the commonly absent diffusion restriction on DWI, and a more common pediatric patient favor craniopharyngioma over GBM. The fact that our patient had diffuse restriction, no calcification, and narrowed calibers of the major vessels, the diagnosis of craniopharyngioma was questioned even though the location was highly suggestive of it. But given that our patient claimed that she had 7 years of long-standing headaches, the possibility of malignant craniopharyngioma was pondered, taking into consideration the highly cellular nature of the tumor on DWI and the absence of a classic imaging appearance even if it is said to be rare and tends to occur in adults.
Similarly, apart from the exceedingly rare sellar suprasellar location for GBM, the heterogeneous T1 and T2 isointense to hyperintense signal with areas of internal hemorrhage, as evidenced by blooming on T2* GRE and ring and nodular postcontrast enhancement, the absence of calcification was highly suggestive of a higher-grade glioma like GBM. Hence, a high index of suspicion for malignant degeneration of a more common benign OPG was considered, and it was the most likely diagnosis made preoperatively even if malignant craniopharyngioma was difficult to rule out.
Unique to our case was the extremely large size of the tumor at presentation compared to those in most reports, in which they presented with a smaller size as well as an earlier presentation. The late presentation would have contributed to the large size. However, our patient’s long-standing headache as a presentation symptom and 10-month visual deterioration could mark the time for malignant degeneration of a preexisting benign pathology even if a de novo origin is still possible.
The treatment of GBM regardless of its location is more or less similar and involves surgery with the goal of achieving gross-total resection if possible and safe maximal resection or biopsy in a critical location, followed by multimodal chemoradiation therapy including the Stupp regimen. Our patient received prednisolone for her hypocortisolism together with surgical and chemotherapy treatment. However, the prognosis is poor, with an average survival time of 8 to 9 months among all 27 cases found in the literature. Alabiad et al.12 reported the case with the most extended survival (7 years, and still alive). In addition, because patients with OPGs who have NF1 tend to have tumors with a more benign course than those without NF1, an effort to avoid radiation therapy is made to decrease malignant degeneration in this population.4
Last, a limitation of this illustrative case was that molecular and genetic studies (that could have told us more information about prognostic, diagnostic, and targeted therapies) were not done because of a lack of affordability. This precluded the initiation of targeted therapy other than the standard Stupp regimen.
Lessons
Sellar-suprasellar GBMs, although extremely rare, are known to occur and pose challenges in the diagnosis and preoperative treatment planning. The presence of diffusion restriction on DWI in a mass lesion that has ring and nodular postcontrast enhancement in addition to absent calcification on CT should alert one to the possibility of a high-grade mass. This is extremely important for preoperative patient counseling and planning for the multimodal treatments, as sellar-suprasellar GBMs carry a poorer prognosis than the common benign mass lesions in the region.
Acknowledgments
We would like to thank our patient for her permission to publish this case and use her diagnostic image.
Author Contributions
Conception and design: Shiferaw, Teklemariam, Aklilu, Gebrewahd, Mekuria, Robele. Acquisition of data: Shiferaw, Gebrewahd, Mekuria, Robele. Analysis and interpretation of data: Shiferaw, Gebrewahd, Robele. Drafting the article: Shiferaw, Gebrewahd, Mekuria, Yesuf. Critically revising the article: Shiferaw, Mekuria, Yesuf. Reviewed submitted version of manuscript: Shiferaw, Teklemariam, Aklilu, Gebrewahd, Yesuf, Robele. Approved the final version of the manuscript on behalf of all authors: Shiferaw. Statistical analysis: Shiferaw. Administrative/technical/material support: Shiferaw, Gebrewahd. Study supervision: Shiferaw, Aklilu.
References
- 1. Lemm D, de Oliveira FH, Bernays RL, et al. Rare suprasellar glioblastoma: report of two cases and review of the literature. Brain Tumor Pathol. 2012;29(4):216–220. doi: 10.1007/s10014-012-0086-0. [DOI] [PubMed] [Google Scholar]
- 2. Miller NR. Primary tumours of the optic nerve and its sheath. Eye (Lond) 2004;18(11):1026–1037. doi: 10.1038/sj.eye.6701592. [DOI] [PubMed] [Google Scholar]
- 3. Alabiad CR, Shah VS, Eatz TA, Sternau LL, Lam BL. Malignant optic nerve glioma in a young woman with 7 year follow up without recurrence. Am J Ophthalmol Case Rep. 2020;19:100862. doi: 10.1016/j.ajoc.2020.100862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zoeller GK, Brathwaite CD, Sandberg DI. Malignant transformation of an optic pathway glioma without prior radiation therapy. J Neurosurg Pediatr. 2010;5(5):507–510. doi: 10.3171/2009.12.PEDS09173. [DOI] [PubMed] [Google Scholar]
- 5. Cirak B, Unal O, Arslan H, Cinal A. Chiasmatic glioblastoma of childhood. A case report. Acta Radiol. 2000;41(4):375–376. doi: 10.1080/028418500127345505. [DOI] [PubMed] [Google Scholar]
- 6. Dinh TT, Wang YY, Rosenfeld JV, Cherny M. Glioblastoma of the optic chiasm. J Clin Neurosci. 2007;14(5):502–505. doi: 10.1016/j.jocn.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 7. Tow SL, Chandela S, Miller NR, Avellino AM. Long-term outcome in children with gliomas of the anterior visual pathway. Pediatr Neurol. 2003;28(4):262–270. doi: 10.1016/s0887-8994(02)00628-8. [DOI] [PubMed] [Google Scholar]
- 8. Janny P, Cure H, Mohr M, et al. Low grade supratentorial astrocytomas. Management and prognostic factors. Cancer. 1994;73(7):1937–1945. doi: 10.1002/1097-0142(19940401)73:7<1937::aid-cncr2820730727>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 9. Nagaishi M, Sugiura Y, Takano I, et al. Clinicopathological and molecular features of malignant optic pathway glioma in an adult. J Clin Neurosci. 2015;22(1):207–209. doi: 10.1016/j.jocn.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 10. Kornreich L, Blaser S, Schwarz M, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol. 2001;22(10):1963–1969. [PMC free article] [PubMed] [Google Scholar]
- 11. Hoyt WF, Meshel LG, Lessell S, Schatz NJ, Suckling RD. Malignant optic glioma of adulthood. Brain. 1973;96(1):121–132. doi: 10.1093/brain/96.1.121. [DOI] [PubMed] [Google Scholar]
- 12. Traber GL, Pangalu A, Neumann M, et al. Malignant optic glioma—the spectrum of disease in a case series. Graefes Arch Clin Exp Ophthalmol. 2015;253(7):1187–1194. doi: 10.1007/s00417-015-3045-8. [DOI] [PubMed] [Google Scholar]
- 13. Anvari K, Samini F, Faraji M, Khooei A, Ghiasi T, Dehghan P. Pituitary glioblastoma: a case report. Iran J Cancer Prev. 2015;8(4):e3436. doi: 10.17795/ijcp-3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahta A, Buhl R, Huang H, Jansen O, Kesari S, Ulmer S. Sellar and supra-sellar glioblastoma masquerading as a pituitary macroadenoma. Neurol Sci. 2013;34(4):605–607. doi: 10.1007/s10072-012-1110-1. [DOI] [PubMed] [Google Scholar]
- 15. Deng S, Liu L, Wang D, Tong D, Zhao G. Small cell glioblastoma of the sella turcica region: case report and review of the literature. World Neurosurg. 2018;110:174–179. doi: 10.1016/j.wneu.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 16. Barbaro NM, Rosenblum ML, Maitland CG, Hoyt WF, Davis RL. Malignant optic glioma presenting radiologically as a “cystic” suprasellar mass: case report and review of the literature. Neurosurgery. 1982;11(6):787–789. doi: 10.1227/00006123-198212000-00011. [DOI] [PubMed] [Google Scholar]
- 17. Tekkök IH, Tahta K, Saglam S. Optic nerve glioma presenting as a huge intrasellar mass. Case report. J Neurosurg Sci. 1994;38(2):137–140. [PubMed] [Google Scholar]
- 18. Mohamed FF, Abouhashem S. Diagnostic value of apparent diffusion coefficient (ADC) in assessment of pituitary macroadenoma consistency. Egypt J Radiol Nucl Med. 2013;44(3):617–624. [Google Scholar]
- 19. Kumar J, Kumar A, Sharma R, Vashisht S. Magnetic resonance imaging of sellar and suprasellar pathology: a pictorial review. Curr Probl Diagn Radiol. 2007;36(6):227–236. doi: 10.1067/j.cpradiol.2007.04.004. [DOI] [PubMed] [Google Scholar]