

Abstract
Abdominal aortic aneurysms (AAAs) are prevalent with aging, and AAA rupture is associated with increased mortality. There is currently no effective medical therapy to prevent AAA rupture. The monocyte chemoattractant protein (MCP-1)/C–C chemokine receptor type 2 (CCR2) axis critically regulates AAA inflammation, matrix-metalloproteinase (MMP) production, and extracellular matrix (ECM) stability. We therefore hypothesized that a diet intervention that can modulate CCR2 axis may therapeutically impact AAA risk of rupture. Since ketone bodies (KBs) can trigger repair mechanisms in response to inflammation, we evaluated whether systemic ketosis in vivo could reduce CCR2 and AAA progression. Male Sprague–Dawley rats underwent surgical AAA formation using porcine pancreatic elastase and received daily β-aminopropionitrile to promote AAA rupture. Rats with AAAs received either a standard diet, ketogenic diet (KD), or exogenous KBs (EKB). Rats receiving KD and EKB reached a state of ketosis and had significant reduction in AAA expansion and incidence of rupture. Ketosis also led to significantly reduced aortic CCR2 content, improved MMP balance, and reduced ECM degradation. Consistent with these findings, we also observed that Ccr2−/− mice have significantly reduced AAA expansion and rupture. In summary, this study demonstrates that CCR2 is essential for AAA expansion, and that its modulation with ketosis can reduce AAA pathology. This provides an impetus for future clinical studies that will evaluate the impact of ketosis on human AAA disease.
Subject terms: Inflammation, Cardiovascular diseases, Vascular diseases, Aneurysm
Introduction
Abdominal aortic aneurysm (AAA) formation, expansion, and subsequent rupture results from a complex series of biomolecular processes1,2. AAAs are often asymptomatic during their formation and expansion stages, but lead to a high risk of morbidity and mortality when they spontaneously rupture3,4. Unfortunately, there is currently no effective medical therapy to alleviate AAA expansion and the risk of rupture. Invasive surgery is the only available management for AAAs that meet the traditional aortic diameter criteria or are rapidly expanding in diameter5. Given the limited medical treatment options for individuals with small AAAs that do not yet meet criteria for surgical repair, expectant management is usually the only remaining option6. Therefore, clinical stabilization of small AAAs remains a large unmet need, and a longer-term management strategy for individuals with AAA disease can be a tremendous value add7.
Inflammation plays an essential role in AAA disease progression and risk of rupture3. The release of inflammatory mediators within the aortic wall leads to a cascade of biomolecular signals that lead to the activation of matrix metalloproteinases (MMPs) that consequentially lead to extracellular matrix (ECM) degradation8–11. The C–C chemokine receptor type 2 (CCR2) mediates trafficking of leukocytes to site of aortic tissue inflammation following initial and repeated injury12. Our team previously demonstrated that CCR2 content in AAAs, as demonstrated by positron emission tomography (PET)/computed tomography (CT) imaging in a preclinical rodent model, highly correlates with the incidence of AAA expansion and the risk of rupture13,14. However, it remains unknown whether CCR2 content is essential for AAA rupture, and whether modulation of CCR2 can alleviate disease progression.
Various dietary interventions can impact immune function and inflammation15. Ketogenesis in particular can dramatically impact anti-inflammatory signaling, and is reported to promote vascular tissue repair15–17. As a natural physiologic process that leads to the production of ketone bodies (KBs) such as acetoacetate (AcAc), beta-hydroxybutyrate (βHB) and acetone, ketogenesis not only serves as an alternative fuel source for organ systems, but it also activates signaling cascades that can impact various cell functions. Although high fat diets are linked to increased AAA expansion and aortic plaque formation18,19, recent studies suggested that a high fat—low carbohydrate ketogenic diet, as well as exogenous ketone body supplementation, can reduce tissue inflammation and ameliorate the risk of vascular injury and atheroprogression due to increased catabolic metabolism20,21. It is unknown whether these potential benefits are limited to atherosclerosis, or whether ketosis can also have a broader impact on degenerative aortopathies such as AAAs. Therefore, we hypothesized that nutritional ketosis, either in the form of a ketogenic diet (KD) or exogenous ketone body (EKB) supplementation, can impact CCR2-mediated inflammation and improve MMP balance in aortic tissue to reduce the risk of AAA progression, aortic wall inflammation, ECM content, and the incidence of AAA rupture.
Results
Ketosis attenuates AAA formation and content of MMP9 in rat aortic tissue
We evaluated AAA pathology in rats maintained on either ketogenic diet (KD) or standard diet (SD). Rats maintained on a ketogenic diet prior to AAA induction (KDp; Fig. 1A) achieved a state of sustained ketosis from day 0–14 (Fig. 1B), and as previously reported22, had a moderate decrease in weight gain by week 1 (p < 0.001; Fig. 1C). By week 2 there was a substantial 42% decrease in AAA diameter in KDp rats (p = 0.008; Fig. 1D and Supplementary Fig. S1A). Aortic wall media demonstrated equivalent mason trichrome (MT)-stained collagen between rats maintained on SD and KDp (Fig. 1E–G). Zymography analysis of harvested aortic tissue at week 2 demonstrated a significant decrease in total-MMP9 in KDp rats whereas MMP2 was unchanged (Fig. 1H–J). Western blot of type 1 Collagen, α-smooth muscle actin (α-SMA) and TGF-β content demonstrated comparable values between KDp and SD rats (Fig. 1K–N). These data suggested that diet-induced ketosis can inhibit AAA expansion, and that this may in part be due to a decrease in aortic wall total MMP9 without substantial modification of the aortic architecture.
Figure 1.

Ketosis attenuates AAA formation and MMP9. (A) Rats underwent exposure to PPE for AAA creation. The experimental group was given a ketogenic diet that started one-week prior to PPE exposure (KDp; N = 6) while the control group was fed a standard chow diet (SD; N = 5). (B) Ketosis (βHB whole blood levels > 0.5 mM/L) was verified at week 0, 1 and 2 in SD (0.2 ± 0.1, 0.3 ± 0.1 and 0.4 ± 0.1) and KDp rats (3 ± 1, 3 ± 1 and 3 ± 0.5) respectively (p < 0.01). (C) Percent body weight difference in SD versus KDp rats at week 1 (13 ± 5 vs. 2 ± 1.3) and at week 2 (23 ± 5 vs. 8 ± 2) respectively (p < 0.001). (D) Percent aortic diameter in SD versus KDp rats at week 1 (154 ± 48 vs. 137 ± 42; p = ns) and at week 2 (332 ± 129 vs. 140 ± 152; p = 0.008) respectively (aneurysms were defined by a > 100% increase in the aortic diameter compared with pretreatment measurements). (E) AAA collagen staining quantification for SD and KDp at week 2 (33 ± 4 vs. 34 ± 2; p = ns) respectively. (F and G) Trichrome staining of abdominal aortas (cross-section of tissue slides) with 5× magnification for SD and KDp rats. Areas with blue staining signify areas with higher collagen deposition. (H) Zymogram demonstrating total MMP9 and MMP2 levels were measured by integrated optical density (IOD). (I) Total MMP-9 levels for SD and KDp at week 2 (4.6 ± 2 × 103 vs. 1.7 ± 0.8 × 103; p = 0.02) respectively. (J) Total MMP-2 levels for SD and KDp at week 2 (5.4 ± 3 × 102 vs. 1 ± 0.6 × 103; p = ns) respectively. (K) Representative western blots of collagen 1, α-SMA, TGFβ-1 and Caveolin 1 in AAA tissue. (L) Collagen I protein content expressed as a ratio to Caveolin 1 content in AAA tissue of SD and KDp rats (0.1 ± 0.1 vs. 0.1 ± 0.1 respectively; p = ns). (M) α-SMA protein content expressed as a ratio to Caveolin 1 content in AAA tissue of SD and KDp rats (2 ± 0.7 vs. 1.7 ± 0.6 respectively; p = ns). (N) TGFβ protein content expressed as a ratio to Caveolin 1 content in AAA tissue of SD and KDp rats (1.3 ± 0.3 vs. 1.2 ± 0.3 respectively; p = ns). Data presented as mean ± standard deviation. ns > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
Sustained ketosis reduces AAA expansion and CCR2 uptake in rupture-prone rats
The risk of AAA progression correlates with CCR2-mediated pro-inflammatory signaling14,23–25. We therefore evaluated whether dietary ketosis could impact CCR2 content using a AAA rupture model. For this we evaluated a cohort of rats that received either SD or KDp after AAA induction with PPE, as well as daily β aminopropionitrile (BAPN) administration to promote AAA rupture (Fig. 2A). Rats fed KDp, and received daily BAPN, remained in a state of ketosis from days 0–14 (Fig. 2B and Supplementary Fig. S2B), and weight gain was reduced at weeks 1 and 2 (p < 0.001 and p = 0.006, respectively; Fig. 2C and Supplementary Fig. S2C). Administration of BAPN did not significantly alter βHB levels between SD and KDp rats (Supplementary Fig. S3A). Notably, KDp rats also had significantly reduced AAA rupture rate (67% vs. 12%; p = 0.03; Fig. 2D and E), and aneurysm diameter at week 1 was significantly decreased in KDp rats (p = 0.002 and p = 0.01; Fig. 2F and Supplementary Fig. S1B). Although by week 2, AAA diameter was equivalent between groups, PET/CT with 64Cu-DOTA-ECL1i (selective CCR2-targeting PET radiotracer12,14,26) demonstrated significantly reduced CCR2 uptake in AAAs of KDp rats throughout study period (week 1, p = 0.05; week 2, p = < 0.0001; Fig. 2G and H). 18F-fluorodeoxyglucose PET/CT performed in KDp and SD rats at week 1 revealed comparable AAA signal uptake14,27,28, (p = ns; Supplementary Fig. S4A and B). These findings demonstrate that KDp rats developed smaller aneurysms, had a combined 54% absolute risk reduction in AAA rupture, and decreased CCR2 uptake in AAA tissue.
Figure 2.

Sustained ketosis reduces AAA expansion and risk of rupture via CCR2 downregulation and Collagen 1 preservation. (A) Rats underwent exposure to PPE to develop AAAs and were also treated with β-aminoprionitrile (BAPN) to promote AAA rupture. (B) Ketosis (βHB whole blood levels > 0.5 mM/L) in SD and KDp rats at week 0 (0.2 ± 0.1 vs. 2 ± 0.5), week 1 (0.3 ± 0.1 vs. 1.8 ± 0.7) and week 2 (0.2 ± 0.1 vs. 3 ± 0.5; p < 0.01). (C) Percent body weight difference in SD and KDp rats at week 1 (12 ± 5 vs. 2 ± 1.3; p < 0.001) and week 2 (31 ± 6 vs. 4 ± 3; p = 0.006). (D) AAA rupture event rate with statistical analysis in SD and KDp rats (p = 0.03). (E) Kaplan–Meier curve demonstrating rate of survival following BAPN administration. 67% (8/12) of SD rats and 12% (1/8) KDp rats developed AAA rupture. (F) Percent aortic diameter in SD and KDp rats at week 1 (270 ± 93 vs. 154 ± 53; p = 0.002) and week 2 (485 ± 153 vs. 401 ± 246; p = ns). (G) Quantitative tracer uptake of CCR2 content in AAA tissue for SD and KDp rats at week 1 (0.9 ± 0.2 vs. 0.7 ± 0.2; p = 0.05) and week 2 (0.9 ± 0.2 vs. 0.4 ± 0.1; p < 0.001. (H) Representative PET/CT coronal images at day 14 post PPE exposure showed specific and intensive detection of AAA (yellow rectangle) in SD, compared with the low trace accumulations in the KDp group of rats. (I) Immunofluorescence staining of abdominal aortas (cross-sectional; 5× magnification and 10× magnification) marked with CCR2 (in green: CCR2 + cells) and CD68 marker (in red: CD68+ cells; macrophages) to visualize inflammatory cells infiltration within the AAA. (K) CCR2 concentration at week 1 in AAA tissue of SD and KDp rats (100 ± 22 vs. 67 ± 12; p = 0.02). (K) Macrophage marker CD68 content at week 1 in AAA tissue of SD and KDp rats (1.8 ± 0.4 vs. 1 ± 0.4; p = 0.002). (L) Chemokine MCP-1 content (3.7 ± 1.2 × 102 vs. 4.2 ± 2.2 × 102; p = ns). (M) Pro-inflammatory marker TNFα content (6.1 ± 0.6 vs. 2.1 ± 1; p = 0.001), (N) IL-10 content (5 ± 1.3 × 101 vs. 3.1 ± 0.9 × 101; p = 0.03), (O) IL-17A content (7.4 ± 1.1 × 101 vs. 0.8 ± 0.6 × 101; p < 0.001), (P) IFNγ content (7.3 ± 1.3 × 102 vs. 2.6 ± 0.7 × 102; p = 0.002), (Q) RANTES content (3 ± 0.7 × 102 vs. 1.1 ± 0.2 × 102; p < 0.001). Data presented as mean ± standard deviation. ns > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
Sustained ketosis inhibits cytokine profiles downstream to CCR2 in AAA tissue by week 1
We next evaluated whether sustained ketosis impacts CCR2 in AAA tissue and downstream cytokine profiles during AAA formation. We observed that immunostaining of AAA tissue in KDp rats demonstrated a marked decrease in CCR2 and CD68+ macrophages (Fig. 2I and J; Supplementary Figs. S5 and S6). KDp rats had reduced CD86+ pro-inflammatory macrophages (M1 phenotype; p = 0.04), whilst a modest increase in CD206+ pro-regenerative macrophages (M2 phenotype; p = ns), within the AAA wall (Supplementary Fig. S7A–D). Similarly, by using ELISA we confirmed that CCR2 and CD68 concentration was significantly decreased in AAAs of KDp rats at week 1 (p = 0.02 and p = 0.002, respectively; Fig. 2K and L). The CCR2 ligand, monocyte chemoattractant protein-1 (MCP-1) was unchanged between KDp and SD rats (Fig. 2M), but CCR2-dependent pro-inflammatory cytokines TNFα, IL-10, IL-17A, and IFN γ were significantly decreased in AAA tissue of KDp rats (p = 0.001, p = 0.03, p < 0.001, and p = 0.002 respectively; Fig. 2N–Q). Similarly, RANTES (the ligand for C–C motif chemokine receptor 5; CCR5) was significantly reduced in KDp rats (p < 0.001; Fig. 2R). These results indicates that KDp notably decreases AAA pro-inflammatory macrophage infiltration, CCR2-dependent cytokine production, and reduced AAA expansion and rupture.
Ketosis alters AAA collagen and elastin content and MMP balance
Previous work demonstrated that CCR2 can impact MMP balance and promote elastin degradation, while decreasing transforming growth factor beta (TGFβ) signaling that contributes to collagen formation29. Gelatin zymography of AAA tissue at week 1 demonstrated that although pro-MMP9 and total-MMP9 were equivalent between SD and KDp rats (Fig. 3A), active MMP9, known to promote AAA formation and rupture30, was significantly reduced in KDp rats (p = 0.04; Fig. 3B). Similarly, total MMP 2, known to promote AAA expansion31, was also significantly reduced in KDp rats (p < 0.001; Fig. 3C and D). Content of MMP9 and Tissue Inhibitor of Metalloproteinases 1 complex (MMP9/TIMP1; known to prevent MMP9 over-activation) was also significantly increased in KDp rats (p = 0.02; Fig. 3E; Supplementary Fig. S8A). Correspondingly, AAA tissue in KDp rats demonstrated equivalent levels of total MMP-9 (Fig. 3F), and significantly reduced TIMP1 compared to SD rats (p = 0.03; Fig. 3G). Overall, these data demonstrate that sustained ketosis with KDp decreases active MMP9 while increasing MMP9/TIMP1 stabilizing complex in AAA tissue.
Figure 3.

Sustained ketosis downregulates CCR2 content and inhibit its downstream signals. (A) Pro MMP9 levels at week 1 in AAA tissue of SD and KDp rats (11.3 ± 2 × 103 vs. 9.5 ± 2.1 × 103; p = ns). (B) Active MMP9 levels in AAA tissue of SD and KDp rats (2.1 ± 1.4 × 103 vs. 0.9 ± 0.7 × 103; p = 0.04). (C) Total MMP2 levels in AAA tissue of SD and KDp rats (7 ± 0.7 × 103 vs. 1.8 ± 1 × 103; p < 0.001). Pro and active MMP9 and total MMP2 levels were measured by integrated optical density (IOD). (D) Representative zymogram from AAA tissue homogenates at week 1 demonstrating pro and active MMP-9 and total MMP-2 levels in SD and KDp rats (E) MMP9/TIMP1 complex levels in AAA tissue of SD and KDp rats (6.3 ± 5 × 102 vs. 15 ± 8.7 × 102; p = 0.02). ELISA of AAA tissue homogenates in SD and KDp rats provided levels of (G) Total MMP9 (5 ± 1.1 × 10–1 vs. 6 ± 0.6 × 10–1; p = ns), (H) SD AAA tissue harvested at day 6 (week 1) stained with VVG at 10× and 5× magnification respectively. TIMP 1 (5.9 ± 1.6 × 103 vs. 3.4 ± 3 × 103; p = 0.03). (I) KD AAA tissue harvested at day 6 (week 1) stained with VVG at 10× and 5× magnification respectively. (J) AAA VVG-staining quantification for SD and KDp at week 1 (7 ± 2 vs. 26 ± 18 respectively; p = 0.03). (K) TGFβ-1 protein content expressed as a ratio to Caveolin 1 content in AAA tissue from SD and KDp rats at week 1 (2.2 ± 0.8 × 10–1 vs. 5.7 ± 3.4 × 10–1; p = 0.2). (L) TGFβ-1 protein content expressed as a ratio to Caveolin 1 content in AAA tissue from SD and KDp rats at week 2 (0.3 ± 0.1 vs. 0.5 ± 0.1; p = ns). (M and N) Trichrome staining of abdominal aortas (cross-sectional) with 5× magnification and 10× magnification in SD and KDp rats to visualize collagen deposition in animal aortic tissue. (O) AAA collagen staining quantification for SD and KDp at week 2 (20 ± 4 vs. 32 ± 6; p = 0.006). (P) Collagen 1 protein content expressed as a ratio to GAPDH content in AAA tissue at week 2 for SD and KDp rats (1.5 ± 1.0 × 10–2 vs. 10 ± 8.2 × 10–2, p = 0.03). (Q) α-SMA protein content expressed as a ratio to GAPDH content in AAA tissue of SD and KDp rats (3.4 ± 1 vs. 3.8 ± 0.7; p = ns). (R) NF Kappa β protein content expressed as a ratio to Caveolin I content in AAA tissue of SD and KDp rats (0.06 ± 0.03 vs. 0.03 ± 0.01; p = ns). (S) Representative Western blots of collagen 1, NF Kappa β, α-SMA, TGFβ-1, GAPDH and Caveolin 1 in AAA tissue. Data presented as mean ± standard deviation. ns > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
We also observed a significant positive correlation between active MMP9 and CCR2 content in the AAA tissue in both SD and KDp rats (p = 0.03 and p = 0.39 respectively; Supplementary Fig. S8B and C). These findings suggest that CCR2 content in AAA tissue may be responsible for activating MMPs, and therefore may be contributing to a higher incidence of AAA rupture. Along with MMP attenuation, VVG-staining demonstrated significantly increased elastin staining intensity within the AAA wall media in KDp rats by week 1 (p = 0.03; Fig. 3H-J), suggesting that KD protects against MMP-induced elastin degradation.
Interestingly, at week 1 and 2, TGFβ content in AAA tissue in KDp rats trended higher (p = ns and p = 0.05, respectively; Fig. 3K and L), and MT-staining demonstrated significantly more collagen in the AAA media (aortic segment between internal and external elastic lamina; p = 0.006; Fig. 3M–O). In particular, type 1 Collagen content was increased in KDp rats compared to SD at week 2 (p = 0.03), while α-smooth muscle actin (αSMA) content remained unchanged (Fig. 3P and Q). Lastly, NF kappa β content in AAA tissue in KDp rats demonstrated a modest nonsignificant decrease (Fig. 3R and S).
Impact of ketosis that is initiated ‘therapeutically’ after AAA formation
Rats treated with an abbreviated course of KD, therapeutically initiated 3 days post-AAA formation with PPE (KDt; Fig. 4A), also led to a state of ketosis (Fig. 4B). Rats treated with supplemental exogenous ketone bodies by oral daily gavage (EKB; Supplementary Figs. S2 and 4A) also led to ketosis, but only for 8-h per day (Fig. 4B). Like KDp rats, KDt and EKB rats also had reduced weight gain at both week 1 and 2 when compared to SD rats (p < 0.001; Fig. 4C and Supplementary Fig. S2).
Figure 4.

Impact of therapeutic ketosis on AAA risk of rupture. (A) Rats underwent exposure to PPE to develop AAAs and were also treated with BAPN to promote AAA rupture. Following AAA induction, rats received a ketogenic ‘treatment’ via an oral diet (KDt) or exogenous supplement (EKB). (B) Ketosis (βHB whole blood levels > 0.5 mM/L) in SD, KDt, and EKB rats at week 1 (0.2 ± 0.1, 1.8 ± 0.9 and 1 ± 0.02, respectively; p < 0.05) and at week 2 (0.2 ± 0.1, 2.7 ± 1.1, and 1 ± 0.02 respectively; p < 0.01) analyzed using two-way ANOVA. (C) Percent body weight difference for SD, KDt, and EKB rats at week 1 (11 ± 4, 2.5 ± 1.4, and 2.4 ± 1.3, respectively; p < 0.01) and at week 2 (31 ± 6, 6 ± 3.5, and 5 ± 3.6, respectively; p < 0.01) analyzed using two-way ANOVA. (D) Free fatty Acids (FFA) serum levels in meq/L in rats fed SD (0.41 ± 0.05) versus KDp (0.86 ± 0.06; p = 0.001), KDt (0.9 ± 0.1 p = 0.005) and EKB (0.40 ± 0.1; p = ns) respectively. (E) Total Cholesterol serum levels in meq/L in rats fed SD (55.5 ± 6) versus KDp (57.5 ± 10; p = ns), KDt (58 ± 12; p = ns) and EKB (55.5 ± 7; p = ns) respectively. (F) Low-density lipoprotein (LDL) serum levels in meq/L in rats fed SD (11 ± 5) versus KDp (9 ± 2; p = ns), KDt (9 ± 3; p = ns) and EKB (9.5 ± 3; p = ns) respectively. (G) Triglycerides serum levels (meq/L) in rats fed SD (68 ± 32) versus KDp (112 ± 51; p = ns), KDt (206 ± 97; p = 0.01) and EKB (92 ± 37; p = ns) respectively. (H) High-density lipoprotein (HDL) serum levels (meq/L) in rats fed SD (21 ± 7) versus KDp (26 ± 6; p = ns), KDt (39 ± 6; p = 0.01) and EKB (27 ± 3; p = ns) respectively. (I) AAA rupture event rate in SD, KDt and EKB rats (p < 0.05 between SD and KDt) using survival analysis. (J) Kaplan–Meier curve demonstrating survival following BAPN administration. 67% (8/12) of SD rats, 40% (2/9) of KDt rats (p = 0.03), and 22% (4/10) of EKB rats (p = ns) developed AAA rupture by week 2. (K) Percent aortic diameter at week 1 in SD versus KDt rats (270 ± 94 and 155 ± 73; p = 0.06), and SD versus EKB rats (148 ± 94; p = 0.04). At week 2, in SD versus KDt rats (485 ± 153 and 234 ± 151; p < 0.01), and SD versus EKB rats (227 ± 147; p < 0.01 analyzed using two-way ANOVA. (L–O) Trichrome staining of AAA tissue at 5× and 10× magnifications to demonstrate collagen deposition in SD versus KDt rats (26 ± 3 and 34 ± 8; p = ns), and SD versus EKB rats (37 ± 5; p = 0.02) analyzed using one-way ANOVA. (P) CCR2 ELISA content in AAA tissue of SD versus KDt rats (5.7 ± 4 and 4.6 ± 3; p = ns), and SD versus EKB rats (4.7 ± 3; p = ns). (Q–S) Immunofluorescent staining of AAA tissue at 5× and 10× magnifications to demonstrate CD68, CCR2, and DAPI positive cells. (T) Immunofluorescent intensity of CD68 was analyzed using one-way ANOVA between SD, KDt, and EKB groups. Data presented as mean ± standard deviation. ns > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 using one-way ANOVA, or two-way ANOVA with multiple comparison.
Although KDt and EKB rats both achieved a state of ketosis throughout the study, resultant serum free fatty acids (FFA) were only significantly elevated in KDt rats when compared to EKB and SD rats (p = 0.005 and p < 0.001 respectively; Fig. 4D). While serum total cholesterol and LDL were observed to be unchanged among all rat groups (Fig. 4E and F), serum triglycerides and HDL levels were also notably elevated in KDt rats when compared to EKB and SD rats (p = ns and p < 0.05 respectively; Fig. 4G and H). These findings confirmed that KDt can lead to serum lipid changes that are expected with extended ketosis as previously demonstrated32.
Although, the AAA rupture rate was reduced in KDt and EKB rats compared to SD rats (40% reduction with KDt, p = 0.03, and 22% reduction EKB, p = 0.12: Fig. 4I and J), the relative decrease in rupture was not as low as that observed with KDp rats (Fig. 2D and E). AAA absolute diameter and percentage of aortic diameter increase were also significantly reduced at both week 1 and 2 in EKB rats while only significantly reduced at week 2 in KDt rats (Fig. 4K and Supplementary Fig. S1C). These findings demonstrate that KDt and EKB therapeutic regimens can lead to reduced AAA expansion and risk of rupture, albeit non-significant for EKB.
KDt and EKB rats also demonstrated increased AAA wall media Collagen content (p = 0.08 and p = 0.02, respectively; Fig. 4L–O), and reduced CCR2 content (p = 0.06 and p < 0.05, respectively; Fig. 4P–S). No difference was observed in CD68 content across groups (Fig. 4T). Equivalent levels of pro-MMP9 were observed among both treatment groups (Supplementary Fig. S9A and B), but active MMP9 was significantly decreased in KDt and EKB rats (p = 0.02 and p = 0.001, respectively; Supplementary Figs. S9A and C, S10). Total MMP2 was also notably attenuated in KDt rats (p < 0.001), but not in EKB rats (Supplementary Fig. S9A and D). These data suggest that even an abbreviated therapeutic course of ketosis following AAA formation can help stabilize AAAs, preserve aortic wall collagen content, reduce CCR2 tissue content, and promote MMP balance.
CCR2 is essential for AAA formation and rupture in mice
We used whole body genetic knockdown of Ccr2 in C57BLK mice, to determine whether CCR2 is essential for AAA formation and rupture. Age-matched male wildtype (Ccr2+/+) and Ccr2−/− adult mice received angiotensin II osmotic pump administration to promote AAA formation13,33, and daily BAPN administration to promote AAA rupture27 (Supplementary Fig. S11A). Over the subsequent 2 weeks, Ccr2−/− mice had significantly reduced incidence of AAA rupture (p = 0.003; Supplementary Fig. S11B), and 47% higher survival compared to Ccr2+/+ (Supplementary Fig. S11C). Moreover, Ccr2−/− mice had significantly less AAA formation (p < 0.001; Supplementary Fig. S11D). PET/CT with 64Cu-DOTA-ECL1i demonstrated significantly reduced CCR2 content in the aorta of Ccr2−/− mice (p < 0.001; Supplementary Fig. S11E and F). These data confirm that CCR2 is essential for AAA formation and rupture.
Discussion
Our study demonstrates that Ccr2 is essential for AAA rupture, and that diet-induced ketosis can reduce CCR2 signaling and decrease AAA pathology progression. Using previously validated, pre-clinical murine mice and rat models for AAA13,14, and different ketogenic supplementation strategies, we provide a robust and comprehensive assessment of the impact of dietary ketosis on the AAA inflammatory milieu, aneurysm progression, and the risk of rupture. We also specifically demonstrate that administration of either a ketogenic diet (KDp or KDt) or an oral ketone body supplementation (EKB) can reliably induce systemic ketosis, significantly reduce aortic wall CCR2 and pro-inflammatory cytokines, increase collagen content in the AAA media, and promote an MMP balance that minimizes elastin degradation (Fig. 5).
Figure 5.
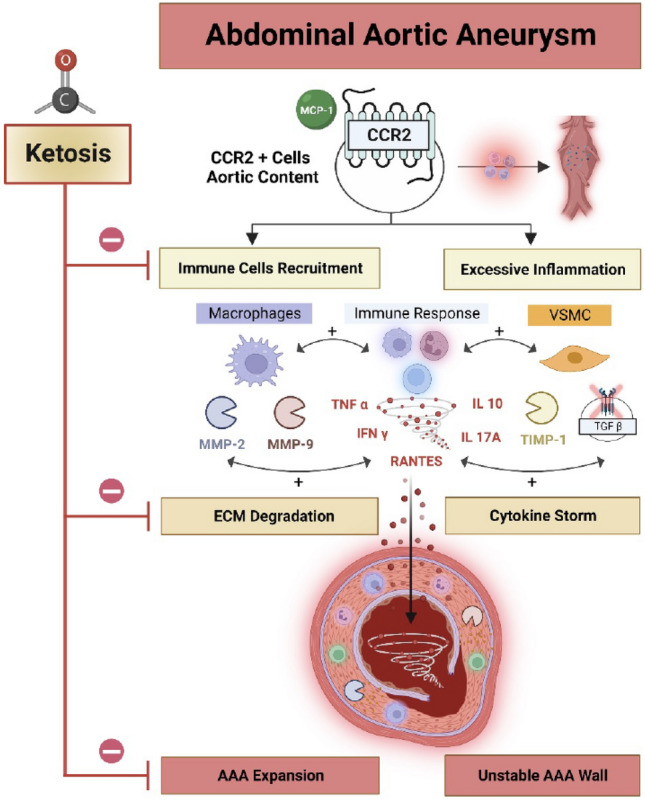
Ketosis impacts AAA expansion and risk of rupture. AAA expansion and risk rupture is influenced by CCR2, which in turn recruits CD68+ pro-inflammatory macrophages, and also leads to cytokine release, and MMP activation. Vascular smooth muscle cells (VSMCs) production of TIMP1 can complex with MMP9 to help balance out the rate of MMP-medicated ECM degradation. Decreased TIMP1/MMP9 complex can lead to higher ECM degradation and AAA expansion. CCR2-mediated release of TNFα, RANTES, IL-10, IL-17A, and IFNγ can further compound AAA tissue stability, and inhibition of TGFβ can progress AAA instability, which further escalates the risk of rupture. Ketosis inhibits inflammation and ECM degradation thereby stabilizes AAA tissue and reduces the risk of rupture. Figure was made using BioRender.com.
Rats that received KDp demonstrated the most notable decrease in AAA expansion and risk of rupture. Rats that received KDt and EKB supplements also demonstrated differences in AAA progression, but not to the same extent. There was also mild to moderate variability in the KDt and EKB values of CCR2, CD68, and MMP content in AAA tissue. Administration of BAPN reliably induced AAA rupture and did not appear to confound the impact of ketosis on AAA expansion and the risk of rupture. Additionally, our complementary studies demonstrated that ketosis can impact pro-inflammatory CCR2-mediated signaling mechanisms that can lead to AAA progression. Therefore, this pre-clinical study demonstrates that a low-risk, and relatively easy dietary intervention, can potentially alter the course of AAA disease progression, and provides important insights that can be easily translated to human patients with AAAs who lack an effective medical management strategy.
Ketogenic diets are thought to modify the host systemic energy metabolism to mimic the biochemical impact of starvation. This occurs with the significant increase in serum βHB levels, decrease in blood glucose, and increase in fatty acids34. These dietary regimens were originally introduced for the treatment of children with refractory epilepsy, and have now become popular remedies for weight loss, patients with diabetes, obesity, various types of cancer, and among high performance athletes22,35–38. Standard ketogenic diets that are devoid of carbohydrates can lead to elevated βHB serum levels that are consistently > 2 mM38. Recent studies demonstrate that βHB can serve as an important signaling mediator that can inhibit histone deacetylases39, blunt tissue oxidative stress40,41, activate G-protein-coupled receptors42,43, and regulate various inflammatory mediators44–47. In our study we similarly observed that rats with high serum βHB had blunted tissue inflammation and reduced CCR2 content in AAA tissue. This provides compelling evidence that a ketogenic diet may provide stability to AAAs and therefore prevent rupture. Future studies will help determine whether ketogenic diets can similarly stabilize small AAAs in human adults.
Uniquely, our study evaluated the impact of three different ketosis regimens: two types of ketogenic diets (KDp and KDt), and an oral supplement regimen (EKB). KDp included a 1-week priming period prior to AAA formation, that imitates the phenomenon of keto-adaptation that occurs in humans who are maintained longer-term on a ketogenic diet48. This regimen aided in determining whether a ketosis primer can have a ‘protective’ impact against AAA formation and expansion. On the other hand, KDt was designed to evaluate the potential ‘therapeutic’ impact of ketosis on expansion and rupture of AAA post-induction with PPE. This regimen would hypothetically be similar to how medical management would be prescribed in humans with small AAAs that do not yet meet the traditional size criteria for operative intervention. In the course of this study, we observed that rats tolerated both KDp and KDt, and that both were successful in inducing a sustained systemic state of ketosis. Interestingly, we observed that both regimens yielded significant reductions in both AAA expansion and the incidence of rupture relative to rats that received SD. However, the longer-term KDp regimen appeared to have a greater protective impact, and a more impressive reduction of CCR2 content in AAA tissue. These findings suggest that the length of diet-induced ketosis may be an important variable in the extent of AAA tissue inflammation reduction and the risk of AAA rupture.
Interestingly, rats that received KDt demonstrated higher levels of serum triglycerides and HDL, whereas rats that received KDp had lipid profiles that were similar to SD. These findings resemble human studies that showed that individuals on shorter regimens of KD tended to have elevated serum lipid profiles, while individuals who were maintained longer term on KD had normalized serum lipid profiles32. The variable serum lipid response to KDs has also led to differences in opinion regarding the relative risk assessments of arterial atherosclerosis. There is still an ongoing need for clinical trials to more clearly delineate the risks KD on atheroprogression32.
With the recent advent of EKB supplements, oral regimens have been increasingly utilized to manipulate human circulating blood ketone body concentrations for various health benefits49. While most studies involving EKB supplementation have traditionally focused on its impact among high-performance athletes50, these supplements are increasingly being studied as remedies for conditions such as epilepsy, heart failure, diabetes, and sepsis-related muscle atrophy51. Our study evaluated the impact of EKB on AAA expansion and risk of rupture. Interestingly, we observed that EKB not only decreased AAA tissue inflammation, but also reduced AAA expansion and incidence of rupture. The impact of EKB on CCR2 content and AAA rupture differed from the responses observed with KDp and KDt, and we suspect this is because EKB only induced intermittent ketosis (limited to 8 h per day). Similar to prior reports52, EKB supplementation did not alter serum lipid profiles. These findings suggest that oral supplementation with ketone bodies can serve as a minimally invasive method for the medical management of AAAs, and is a compelling topic for future human clinical trials—particularly in individuals with a genetic predisposition for impaired lipid metabolism53–55.
Our study also suggest that ketosis has a multifaceted impact on aortic wall architecture and function. Inflammation is the major molecular mediator of AAA disease progression (Fig. 5). Previous studies demonstrated that excessive aortic wall inflammation can inhibit reparative signaling, wall fibrosis, and collagen deposition, which in turn can accelerate AAA expansion and lead to a higher risk of rupture56. Tissue macrophages are known to promote AAA disease, in particular subsets that highly express CCR211. We as well as others, also previously demonstrated that genetic or molecular targeting of CCR2 can reduce AAA progression12–14. Here we provide further completing evidence that CCR2 content indeed correlates with AAA disease progression, and that systemic ketosis in vivo can significantly reduce CCR2 content as well as downstream pro-inflammatory cytokines in AAA tissue.
Previous studies investigating the inflammasome in AAAs demonstrated that TNFα and RANTES are both up-regulated in expanding AAA tissue57,58. Inhibition of TNFα appears to decrease aortic wall MMP activity, reduce ECM disruption, and decrease aortic diameter in a murine AAA model59. In another study, Empagliflozin, a sodium-glucose cotransporter 2 inhibitor that increases plasma ketone bodies60,61, was found to reduce aortic aneurysm diameter and aortic wall RANTES in Apoe−/− mice62. Similarly, our study observed that diet-induced ketosis can significantly decrease aortic wall pro-inflammatory cytokines TNFα and RANTES, as well as increase aortic wall Collagen content. Although the direct mechanism of action for this is yet to be fully elucidated, we suspect that the molecular interplay between macrophages and other pro-inflammatory cell types contribute to the immune modulation in AAA tissue63,64.
A central pathological feature of AAA disease progression is excessive and aberrant extracellular matrix (ECM) remodeling. This results from reduced inactive pro-MMP form, increased MMP activity, and higher rapid ECM breakdown and disruption of the integrity of the aortic wall65,66. Previous work demonstrates that MMP2 plays a central role in the formation and early expansion of AAAs, while MMP9 is more related to late AAA expansion and risk of aneurysm rupture27,67,68. Synergistic activation of both MMP2 and MMP9 provides an unfavorable environment that can accelerate AAA dilation and lead to a higher risk of aneurysm rupture69. Previous studies also demonstrate that ketosis, high serum βHB, and signaling via NF-Kβ, play key roles in suppressing MMP-9 expression in colonic tissue70. Our studies build upon this, and demonstrate that along with blunted CCR2 content, ketosis resulted in attenuated active MMP9, reduced total MMP2 content, and increased TIMP1/MMP9 stabilizing complex in AAA tissue (Fig. 5). This is consistent with a recent study that demonstrated CCR2 antagonism similarly downregulated MMP-9 in lung cancer cells, resulting in reduced cellular motility71.
Our study also demonstrated a mild-moderate, but non-significant, increase in AAA tissue TGFβ content in rats treated with ketogenic diets (Fig. 5). TGFβ belongs to a superfamily of growth factors that regulate many cellular functions such as cell growth, adhesion, migration, differentiation, and apoptosis72. In aortic tissue, TGFβ appears to have a beneficial role. For example, administration of TGFβ neutralizing antibodies appeared to promote excessive monocyte-macrophage infiltration within murine and rat AAA tissue29,73, while overexpression or administration of TGFβ1 significantly increased aortic wall collagen deposition74, and collagen synthesis in normal arteries75.Recent studies demonstrated that TGFβ content is reduced in human AAA tissue76, and ketosis promoted TGFβ-induced myocardial fibrosis and Collagen 1 and 3 deposition in spontaneously hypertensive rats77. In this context, our study suggests that KDs lead to increased AAA TGFβ content and increase aortic wall Collagen 1 deposition.
We acknowledge that a dietary regimen that mimics a fasting state can be physiologically challenging to evaluate mainly due to its significant impact on energy metabolism. None the less, ad libitum intake of a precisely formulated KD (Supplementary Fig. S12) appears to be the best approach to promote satiety and evaluate the impact of ketogenesis in the most naturally occurring way78. For example, similar to prior studies22,79, our results demonstrate that an ad libitum ketogenic diet can lead to reduced weight gain. We observed that this reduction mostly occurred during week 1, but by week 2 rats started to gain more weight (Figs. 1C, 2C, and 4C). A similar trend was also observed in sham rats that did not have AAAs (Supplementary Fig. S13). Moreover, we confirmed in all ketogenic diet groups had the hypothesized rise in serum βHB, which is a highly sensitive predictor of ketosis80.
Given the ad libitum study design it was nearly impossible to precisely track oral food intake quantities among our different diet groups. We acknowledge that this may have introduced confounding variables into the study, as satiety may have been variably impacted among the diet intervention groups. Nevertheless, we observed that rats that received either KD or EKB had no difference in total cholesterol or LDL (Fig. 4E and F). On the other hand, in addition to elevated serum βHB, rats that received KD had elevated serum FFAs, which is typically released from adipose tissue to serve as an alternative fuel and is converted into acetoacetate, βHB, and acetone81,82. These observations provide supporting evidence that KD had its intended effect, despite the potential limitations of an ad libitum study design.
Furthermore, we acknowledge that all our data was derived from male pre-clinical rodent models that may not be necessarily representative of human AAA pathophysiology. Our male rat AAA rupture model was previously validated and shown to be the most reliable and consistent AAA rupture model to date. Given the known impact of estrogen on AAAs1,3, there is currently no reliable AAA rupture model in female rodents and is an area that requires further investigation and development. In the future, we hope to conduct similar studies using female rodents to determine the impact of sex on our study findings. Additionally, our studies did not systematically evaluate arterial blood pressure. This would have required more sophisticated monitoring apparatuses, and possibly indwelling sensors that evaluate continuous telemetry. While such monitoring systems are feasible for shorter experimental protocols, our 2–3 week experimental protocol would have greatly complicated the experimental design and led to several confounding variables. Instead, we serially monitored AAA endpoints via ultrasound, which provided reliable and reproducible data. Lastly, our study used a single composition for the ketogenic diet intervention. We acknowledge that this is not fully representative of the variety of human lipid and oil-based ketogenic diets, but this was selected to maintain consistency and adherence within all rodent study groups.
In conclusion, this study demonstrates that KDs and EKB supplementation can significantly reduce AAA expansion and reduce the incidence of AAA rupture. Importantly, a ketogenic priming period appears to be further protective, while EKB appears to be less effective than other dietary regimens. KDs reduced CCR2 content, promoted MMP balance, and attenuated ECM degradation in AAA tissue. These findings provide the impetus for future pre-clinical and clinical studies geared to determine the role of ketosis as a medical management tool for human patients with AAAs that do not yet meet the criteria for surgical intervention.
Methods
Animals
Male Sprague–Dawley rats (200–300 g) were obtained from Charles River Laboratories (Wilmington, MA; see supplemental methods). Adult 12-week-old, male, Ccr2−/− and Ccr2+/+ mice on a C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME; see supplementary methods). All animals were housed at 21 °C in a 12/12 h light/dark cycle and had access to food and water ad libitum. Anesthesia was administered with a mixture of ~ 1.5% isoflurane and oxygen for all procedures. The core body temperature was continuously monitored and maintained with a heating pad at 37 °C. Use of all animal experiments were performed in accordance with relevant guidelines and regulations, were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine in St. Louis and reported in accordance with ARRIVE guidelines. At the conclusion of studies, live animals were sacrificed appropriately using anesthetic agents and cervical dislocation.
Induction of AAA and sham-control models
AAA rats were induced to develop infrarenal AAAs via an established model using porcine pancreatic elastase (PPE; 12 U/mL), while sham-control rats (n = 8) were exposed to heat-inactivated PPE (12 U/mL, heated at 90 °C for 45 min; Supplementary Fig. S13) as previously described28. Ventral abdominal wall laparotomy is performed, and the infrarenal abdominal aorta was exposed (Supplementary Fig. S14). A customized polyethylene catheter (Braintree Scientific, Braintree, MA) was introduced through an infrarenal aortotomy, and elastase was infused into the isolated aortic segment for 30 min. The exposed aortic segment was dilated to a maximal diameter, and constant pressure was maintained with the use of a syringe pump. Using a video micrometer, the baseline maximum aortic diameter was measured. After 14 days, all rat aortas were re-exposed via ventral abdominal laparotomy, maximal aortic diameters were measured, and aortic tissue was harvested for further analysis (Supplementary Fig. S14).
Promoting AAA rupture model
As previously described, BAPN is reported to promote AAA tissue inflammation by day 6, and AAA rupture between days 7 and 14, but unlikely to cause rupture after day 14 if it has not already occurred14. We therefore promoted AAA rupture with daily administration of BAPN on a specific cohort of rats (RAAA) starting 3 days before PPE exposure. Through drinking water, 300 mg BAPN was administered daily (0.3% BAPN in 25 mL water consumed per day by a 250 g rat)83. AAA growth was monitored for 1 week (6–7 days) or 2 weeks (14 days). At the 1 or 2-week timepoints, rats were sacrificed, AAA diameters were evaluated, and whole blood and aortic tissue were collected for further analysis. Rats that developed ruptured AAAs (RAAA) during the study period promptly underwent necropsy to confirm and analyze the pathology (Supplementary Fig. S14). By day 14, rats that survived (non-ruptured AAA; NRAAA) were evaluated via open surgical laparotomy. Tissue harvested at week 1, prior to rupture, was mainly used to assess AAA tissue inflammation (Supplementary Fig. S2).
Animal diets
We evaluated four different dietary interventions. First, control groups in the AAA (n = 5) and RAAA (n = 12) cohorts were fed with a standard chow diet (SD). Second, experimental groups in the AAA (n = 6) and RAAA (n = 8) cohorts were fed a very high fat diet with almost no carbohydrate, also known as a classic ketogenic diet one week prior to PPE exposure to induce a ‘priming’ keto-adapted status84, before AAA induction (Supplementary Fig. S12). Ketogenic diet was then maintained prospectively in these groups following AAA formation. Third, experimental groups in the RAAA (n = 9) cohorts were separately started on ketogenic diets as a ‘treatment’ intervention 3 days after AAA induction. Lastly, experimental RAAA rats were administered a SD along with exogenous ketone body (EKB) supplementation starting 3 days after PPE exposure: RAAA + EKB (n = 10). As previously described52, this EKB supplementation was performed with daily intragastric gavage of 1,3-Butanediol85 (BD; 5 g per kg dose; Prod # B84785-100ML; Sigma-Aldrich, St. Louis, MO) and rats achieved a ketosis state only for 8 h per day (Supplementary Fig. S12).
Synthesis and radiolabeling of DOTA-ECL1i
The ECL1i peptide (LGTFLKC) was synthesized from D-form amino acids by CPC Scientific (Sunnyvale, CA). DOTA-ECL1i was prepared following our previous report. Copper-64 (64Cu, t1/2 = 12.7 h) was produced by the Washington University Cyclotron Facility. The DOTA-ECL1i conjugate was radiolabeled with 64CuCl2 (64Cu-DOTA-ECL1) as described, and the radiochemical purity was determined by radio-HPLC14.
Animal PET/CT imaging analysis
Dynamic PET scan and corresponding CT images were obtained using Inveon MM PET/CT (Siemens, Malvern, PA) at 45–60 min after a tail vein injection of 64Cu-DOTA-ECL1i (selective CCR2-targeting radiotracer; 11.1 MBq per rat) to minimize the effect of blood retention on AAA uptake. To localize tracer uptake, a CT contrast agent (1.0 mL, eXIA 160XL, Binitio, Canada) was administrated via tail vein after PET imaging. Contrast-enhanced CT (Bin of 2, 90 mm axial FOV, 60 kV, 500 μA, 500 ms exposure time, 10 ms settle time, no magnification, pixel size: 80–100 μm) was performed. The AAA uptake was calculated as standardized uptake value (SUV) in 3-dimensional regions of interest from PET images without correction for partial volume effect using Inveon Research Workplace software (Siemens). Imaging specificity of CCR2 tracer binding detection within the AAA wall was previously identified by CT contrast agent using the same rat AAA model14. Dynamic (0–90 min) 18F-fluorodeoxyglucose (41.1 MBq per rat) PET was also performed in AAA rats at week 1 and 2 post-PPE exposure to identify areas with high glucose metabolism. Only a specific number of each group of rats received PET/CT imaging.
Ultrasound aortic assessments
Noninvasive ultrasound (GE, 12 MHz Zonare, Mountain View, CA), was used to evaluate serial aortic maximum diameter measurements. Relative to baseline aortic diameter prior to PPE exposure, the percentage increase in aortic diameter was evaluated at 1- and 2-weeks post-PPE exposure. As previously described, aortic aneurysms were defined as > 100% increase in the aortic maximum diameter relative to baseline diameter28,86.
Blood βHB assessments
Animal state of ketosis was evaluated via whole blood D-βHB (Keto-MoJo blood ketone meter; Keto-Mojo, Napa, CA, USA) concentrations79. Tail vein puncture was used for blood sample, which was tested on day 0 pre-PPE exposure, and then 1- and 2-weeks following AAA induction. βHB values > 0.5 mmol/L were indicative of ketosis.
Serum free fatty acids and lipids measurements
Whole blood samples (1 mL) were collected from Inferior Vena Cava (IVC) puncture at 2-week timepoint (14 days), and centrifugated for serum isolation. Serum samples were sent to Washington University Core Lab for analysis of free fatty acids (FFA), total cholesterol (TC), direct low-density lipoprotein (LDL), triglycerides (TG) and high-density lipoprotein (HDL).
Animal weight
Animal whole body weights were evaluated at day 0 pre-PPE exposure, and 1 and 2 weeks followed AAA induction. Body weight was evaluated by the difference between the values at the baseline (Day 0) and the values at week 1 and 2 respectively and then divided by the baseline to assess difference. All these absolute numbers were then multiplied by 100 to present it as the percentage of difference in weight throughout the time of the study.
Histology and immunostaining
Aortic tissue was harvested from all animals. AAA tissue was fixed in Histochoice (VWR), and paraffin embedded. Paraffin blocks were sectioned at 5 μm, and deparaffinized. Processing for antigen retrieval was performed with Sodium Citrate solution, pH 6.0, for 10 min. Tissue sections were blocked with 10% serum, and sections were incubated with primary antibody anti-CD68, 1:100 (Bio-Rad, MCA341GA), CCR2, 1:200 (Novus-Bio, NBP1-48338), CD86 (Thermo-Fisher, #942-MSM-P0) and CD206 (Thermo-Fisher, #187004-1-AP). For Immunofluorescence, sections were incubated with donkey anti mouse (Alexa Fluor 647), and donkey anti rabbit (Cy3) (Jackson ImmunoResearch Laboratories). All sections were imaged using a Leica THUNDER Imager 3D tissue microscope system. Sections were then incubated with anti-mouse secondary antibodies conjugated with HRP (Cell Signaling), DAB peroxidase substrate kit (Vector Laboratories), and counter stained with hematoxylin, imaged using an Olympus fluorescent microscope system. To evaluate AAA tissue morphology and pathology, tissue sections were also evaluated using Hematoxylin and Eosin (H&E), Mason Trichrome (MT) and Verhoeff-Van Gieson (VVG), imaged using NanoZoomer, Supported by the Alafi Neuroimaging Laboratory. All AAA wall staining intensity was analyzed in 3 random ROIs, quantified with Image J, and normalized to the total area of the aortic wall media.
ELISA and cytokine array assessments
AAA tissue protein was extracted using RIPA buffer with proteinase inhibitor (Sigma #MCL1). Protein quantification was done by Bradford assay. For each AAA tissue samples 25ug of protein was analyzed for Tissue Inhibitor of Metalloproteinases 1 (TIMP1)-specific ELISA (RayBiotech, ELR-TIMP1-CL-2), MMP-9 (MyBioSource, MBS722532), CD68 (MyBioSource, MBS705029) and Cytokine multiplex assay (Millipore, RECYTMAG-65 K) using manufacturer instructions.
MMP activity zymography
For each AAA tissue sample, 25ug of protein was loaded on wells of 10% Gelatin Zymogram electrophoresis gels. Gels were then incubated in Zymogram renature buffer for 30 min, followed by 36 h of Zymogram development buffer at 37 °C. Gels were then stained with Coomassie Brilliant Blue R-25 solution from BioRad for 30 min, followed by de-staining buffer (20% Methanol, 20% Acetic acid, 60% DI water) until MMP bands were visualized. Gels were scanned on BioRad Chemi doc and analyzed using ImageJ software.
Immunoblotting
From each AAA sample 25 μg of denatured protein was separated on 4–12% polyacrylamide by electrophoresis and then transferred to PVDF membrane. The membranes were then incubated with collagen type-1 (Millipore #ABT123, 1:2000), TGF-β1 (Sigma #AV44268, 1:1000), α-SMA (Sigma #A2547, 1:1000) and KFkB (Thermo-Fisher #PA516545). GAPDH (Sigma # G9545, 0.1 mg/ml) and Caveolin-1 (Santa Cruz # sc-53564, 1:1000) were used as loading controls. Membranes were treated with HRP-conjugated secondary antibody at room temperature for 1 h and evaluated with chemiluminescence. Blot band intensities were analyzed using ImageJ software.
Statistics
All data are presented as the mean ± SD. Most group comparisons were performed using unpaired t test. For comparisons that included one endpoint in more than one animal/diet groups, an ordinary one-way ANOVA with multiple comparison was performed. For comparisons that included more than one endpoint in more than one animal/diet group, we utilized a two-way ANOVA with multiple comparison. Data was considered statistically significant with p ≤ 0.05. Kaplan–Meier curve was generated to assess the survival of BAPN-exposed animals. GraphPad Prism 9 (La Jolla, CA) was used for all statistical analyses and graphical data representations. MT and VVG cross section staining’s were analyzed using ImageJ software by color deconvolution, adjust threshold and region of interest assessment of the AAA to analyze collagen deposition and elastin degradation. Immunofluorescence in AAA wall tissue-stained with CD-68 (red channel), CCR2 (green channel), and DAPI (blue channel) were also analyzed by ImageJ software using split channels, inversion and threshold adjustment and shown as mean intensity (content) for each staining in the specific regions of interest (ROI).
Supplementary Information
Author contributions
Conceptualization: S.S.D., S.E.B., S.J.E., M.A.Z.; Methodology: B.A., S.S.D., S.S.B., R.M., M.S.Z.; Investigation: S.S.D., S.E.B., B.A., G.S.H., H.L.; Data Collection: S.E.B., G.S.H., H.L., Y.L.; Supervision: S.J.E., Y.L., M.A.Z.; Writing—original draft: S.E.B., M.A.Z.; Writing—review & editing: S.E.B., S.S.D., M.A.Z.
Funding
This work was supported by grants from National Institute of Health, National Heart Lung and Blood Institute R01HL153436 (Mohamed A. Zayed), R01HL150891 (Mohamed A. Zayed), R01HL153262 (Mohamed A. Zayed).
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Sergio Sastriques-Dunlop and Santiago Elizondo-Benedetto.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-51996-7.
References
- 1.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 2.Moll FL, et al. Management of abdominal aortic aneurysms clinical practice guidelines of the European Society for Vascular Surgery. Eur. J. Vasc. Endovasc. Surg. 2011;41:S1–S58. doi: 10.1016/j.ejvs.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Carino D, Sarac TP, Ziganshin BA, Elefteriades JA. Abdominal aortic aneurysm: evolving controversies and uncertainties. Int. J. Angiol. 2018;27:58–80. doi: 10.1055/s-0038-1657771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: Basic mechanisms and clinical implications. Curr. Probl. Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421. [DOI] [PubMed] [Google Scholar]
- 5.Golledge J, et al. Lack of an effective drug therapy for abdominal aortic aneurysm. J. Intern. Med. 2020;288:6–22. doi: 10.1111/joim.12958. [DOI] [PubMed] [Google Scholar]
- 6.Brewster DC, et al. Guidelines for the treatment of abdominal aortic aneurysms: Report of a subcommittee of the Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. J. Vasc. Surg. 2003;37:1106–1117. doi: 10.1067/mva.2003.363. [DOI] [PubMed] [Google Scholar]
- 7.Lindeman JH, Matsumura JS. Pharmacologic management of aneurysms. Circ. Res. 2019;124:631–646. doi: 10.1161/CIRCRESAHA.118.312439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potteaux S, Tedgui A. Monocytes, macrophages and other inflammatory mediators of abdominal aortic aneurysm. Curr. Pharm. Des. 2015;21:4007–4015. doi: 10.2174/1381612821666150826093855. [DOI] [PubMed] [Google Scholar]
- 9.Li H, et al. Modulation of immune-inflammatory responses in abdominal aortic aneurysm: Emerging molecular targets. J. Immunol. Res. 2018 doi: 10.1155/2018/7213760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ijaz T, Tilton RG, Brasier AR. Cytokine amplification and macrophage effector functions in aortic inflammation and abdominal aortic aneurysm formation. J. Thorac. Dis. 2016;8:E746–E754. doi: 10.21037/jtd.2016.06.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs; their role and potential as targets for therapy. Arterioscler. Thromb. Vasc. Biol. 2015 doi: 10.1161/ATVBAHA.115.305269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, et al. Chemokine (C-C motif) receptor 2 mediates mast cell migration to abdominal aortic aneurysm lesions in mice. Cardiovasc. Res. 2012;96(3):543–551. doi: 10.1093/cvr/cvs262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daugherty A, et al. Angiotensin II infusion promotes ascending aortic aneurysms: Attenuation by CCR2 deficiency in apoE-/- mice. Clin. Sci. 2010;118:681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.English SJ, et al. CCR2 positron emission tomography for the assessment of abdominal aortic aneurysm inflammation and rupture prediction. Circ. Cardiovasc. Imaging. 2020 doi: 10.1161/CIRCIMAGING.119.009889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe M, et al. Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: A comprehensive review of the literature. Obes. Rev. 2020;21:1–11. doi: 10.1111/obr.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayadi JJ, Sayadi L, Satteson E, Chopan M. Nerve injury and repair in a ketogenic milieu: A systematic review of traumatic injuries to the spinal cord and peripheral nervous tissue. PLoS One. 2021;16:1–16. doi: 10.1371/journal.pone.0244244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradshaw, P. C., Seeds, W. A., Miller, A. C., Mahajan, V. R. & Curtis, W. M. COVID-19: Proposing a ketone-based metabolic therapy as a treatment to blunt the cytokine storm. Oxid. Med. Cell. Longev.2020 (2020). [DOI] [PMC free article] [PubMed]
- 18.Hashimoto K, et al. The effect of a high-fat diet on the development of abdominal aortic aneurysm in a vascular hypoperfusion-induced animal model. J. Vasc. Res. 2018;55:63–74. doi: 10.1159/000481780. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, et al. Small rodent models of atherosclerosis. Biomed. Pharmacother. 2020;129:110426. doi: 10.1016/j.biopha.2020.110426. [DOI] [PubMed] [Google Scholar]
- 20.Castro R, et al. A hypomethylating ketogenic diet in apolipoprotein E-deficient mice: A pilot study on vascular effects and specific epigenetic changes. Nutrients. 2021 doi: 10.3390/nu13103576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Jie S, et al. Ketone body 3-hydroxybutyrate ameliorates atherosclerosis via receptor Gpr109a-mediated calcium influx. Adv. Sci. 2021;8:1–12. doi: 10.1002/advs.202003410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno B, et al. Obesity treatment by very low-calorie-ketogenic diet at two years: Reduction in visceral fat and on the burden of disease. Endocrine. 2020;54:681–690. doi: 10.1007/s12020-016-1050-2. [DOI] [PubMed] [Google Scholar]
- 23.Middleton RK, et al. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: A protein array study. J. Vasc. Surg. 2007;45:574–580. doi: 10.1016/j.jvs.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 24.Wang SK, Green LA, Gutwein AR, Drucker NA, Motaganahalli RL, Gupta AK, Fajardo A, Murphy MP. Description of human AAA by cytokine and immune cell aberrations compared to risk-factor matched controls. Surgery. 2019;72:2964–2979. doi: 10.1016/j.surg.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puchenkova OA, et al. Cytokines in abdominal aortic aneurysm: master regulators with clinical application. Biomark. Insights. 2022;17:11772719221095676. doi: 10.1177/11772719221095676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brody SL, et al. Chemokine Receptor 2–targeted Molecular Imaging in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021;203:78–89. doi: 10.1164/rccm.202004-1132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.English SJ, et al. Increased 18f-fdg uptake is predictive of rupture in a novel rat abdominal aortic aneurysm rupture model. Ann. Surg. 2015;261:395–404. doi: 10.1097/SLA.0000000000000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.English SJ, et al. Utility of 18 F-FDG and 11C-PBR28 microPET for the assessment of rat aortic aneurysm inflammation. EJNMMI Res. 2014;4:1–14. doi: 10.1186/s13550-014-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, et al. TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Invest. 2010 doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandooren J, Van Den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013;48:222–272. doi: 10.3109/10409238.2013.770819. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, et al. Angiotensin II induces an increase in matrix metalloproteinase 2 expression in aortic smooth muscle cells of ascending thoracic aortic aneurysms through JNK, ERK1/2, and p38 MAPK activation. J. Cardiovasc. Pharmacol. 2015;66:285–293. doi: 10.1097/FJC.0000000000000276. [DOI] [PubMed] [Google Scholar]
- 32.Zaghloul MS, Elizondo-Benedetto S, Zayed MA. Impact of ketogenic diet on cardiovascular disease. Nutr. Rev. J. 2023 doi: 10.1093/nutrit/nuad152. [DOI] [PubMed] [Google Scholar]
- 33.Fashandi AZ, et al. A novel reproducible model of aortic aneurysm rupture. Surgery. 2018;163:397–403. doi: 10.1016/j.surg.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selvaraj S, Kelly DP, Margulies KB. Implications of altered ketone metabolism and therapeutic ketosis in heart failure. Circulation. 2020;141:1800–1812. doi: 10.1161/CIRCULATIONAHA.119.045033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrero JR, et al. Safety and effectiveness of the prolonged treatment of children with a ketogenic diet. Nutrients. 2020;12:306. doi: 10.3390/nu12020306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Augustin K, et al. Review mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Oncol. 2018;17:84–93. doi: 10.1016/S1474-4422(17)30408-8. [DOI] [PubMed] [Google Scholar]
- 37.Ludwig DS, Willett WC, Volek JS, Neuhouser ML. Dietary fat: From foe to friend? Science. 2018;362:764–770. doi: 10.1126/science.aau2096. [DOI] [PubMed] [Google Scholar]
- 38.Peterman MG. The ketogenic diet in epilepsy. J. Am. Med. Assoc. 1925;84:1979–1983. doi: 10.1001/jama.1925.02660520007003. [DOI] [Google Scholar]
- 39.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Nagao M, et al. b-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016 doi: 10.1016/j.bbrc.2016.05.097. [DOI] [PubMed] [Google Scholar]
- 41.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor NIH Public access. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug. Discov. 2012 doi: 10.1038/nrd3777. [DOI] [PubMed] [Google Scholar]
- 43.Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL. Short chain fatty acids and their receptors: New metabolic targets. Transl. Res. 2013;161:131–140. doi: 10.1016/j.trsl.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 44.Rahman M, et al. ARTICLE The b-hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014 doi: 10.1038/ncomms4944. [DOI] [PubMed] [Google Scholar]
- 45.Fu S-P, et al. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-κ B activation. Mediat. Inflamm. 2014 doi: 10.1155/2014/983401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu S-P, et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2012;12:1–14. doi: 10.1186/s12974-014-0230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youm YH, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherrier M, Li H. The impact of keto-adaptation on exercise performance and the role of metabolic-regulating cytokines. Am. J. Clin. Nutr. 2019;110:562–573. doi: 10.1093/ajcn/nqz145. [DOI] [PubMed] [Google Scholar]
- 49.Newport MT, Vanitallie TB, Kashiwaya Y, King MT, Veech RL. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 2015;11:99–103. doi: 10.1016/j.jalz.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dearlove DJ, Faull OK, Clarke K. Context is key: Exogenous ketosis and athletic performance. Curr. Opin. Physiol. 2019;10:81–89. doi: 10.1016/j.cophys.2019.04.010. [DOI] [Google Scholar]
- 51.Soto-Mota A, Norwitz NG, Clarke K. Why a D-β-hydroxybutyrate monoester? Biochem. Soc. Trans. 2020 doi: 10.1042/BST20190240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kesl SL, et al. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr. Metab. 2016;13:1–15. doi: 10.1186/s12986-016-0069-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koeslag JH. Post-exercise ketosis and the hormone response to exercise. Med. Sci. Sports Exerc. 1982;14:327–334. doi: 10.1249/00005768-198205000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Tew GA, et al. Randomized feasibility trial of high-intensity interval training before elective abdominal aortic aneurysm repair. Br. J. Surg. 2017;104:1791–1801. doi: 10.1002/bjs.10669. [DOI] [PubMed] [Google Scholar]
- 55.Perissiou M, et al. Aortic and systemic arterial stiffness responses to acute exercise in patients with small abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2019;58:708–718. doi: 10.1016/j.ejvs.2019.02.021. [DOI] [PubMed] [Google Scholar]
- 56.Kaneko H, et al. Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc. Res. 2011;91:358–367. doi: 10.1093/cvr/cvr080. [DOI] [PubMed] [Google Scholar]
- 57.Kaneko H, et al. Tumor necrosis factor-α converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218:470–478. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 58.Jones GT, Phillips LV, Williams MJA, van Rij AM, Kabir TD. Two C-C family chemokines, eotaxin and RANTES, are novel independent plasma biomarkers for abdominal aortic aneurysm. J. Am. Heart Assoc. 2016;5:1–14. doi: 10.1161/JAHA.115.002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Worth J, et al. Formation in a murine model attenuates aneurysm α blocking TNF. J. Immunol. Ref. 2022;183:2741–2746. doi: 10.4049/jimmunol.0803164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrannini E, et al. Clinical medicine Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014;124:499. doi: 10.1172/JCI72227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferrannini E, et al. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65:1190–1195. doi: 10.2337/db15-1356. [DOI] [PubMed] [Google Scholar]
- 62.Ortega R, et al. Arteriosclerosis, thrombosis, and vascular biology SGLT-2 (sodium-glucose cotransporter 2) inhibition reduces Ang II (angiotensin II)-induced dissecting abdominal aortic aneurysm in ApoE (apolipoprotein E) knockout mice. Arter. Thromb. Vasc. Biol. 2019;39:1614–1628. doi: 10.1161/ATVBAHA.119.312659. [DOI] [PubMed] [Google Scholar]
- 63.University, S. Limiting AAA with Metformin (LIMIT) Trial (LIMIT). https://clinicaltrials.gov/ct2/show/NCT04500156 (2020).
- 64.Guangdong Provincial People’s Hospital. Low-dose Colchicine Inhibit Abdominal Aortic Aneurysm Growth Trial (COIN). https://clinicaltrials.gov/ct2/show/NCT05361772 (2022).
- 65.Zhang G, Miyake M, Lawton A, Goodison S, Rosser CJ. Matrix metalloproteinase-10 promotes tumor progression through regulation of angiogenic and apoptotic pathways in cervical tumors. BMC Cancer. 2014;14:1–14. doi: 10.1186/1471-2407-14-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.García-Irigoyen O, et al. Matrix metalloproteinase 10 contributes to hepatocarcinogenesis in a novel crosstalk with the stromal derived factor 1/C-X-C chemokine receptor 4 axis. Hepatology. 2015;62:166–178. doi: 10.1002/hep.27798. [DOI] [PubMed] [Google Scholar]
- 67.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 68.Koole D, et al. Intraluminal abdominal aortic aneurysm thrombus is associated with disruption of wall integrity. J. Vasc. Surg. 2013;57:77–83. doi: 10.1016/j.jvs.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Maleux G, et al. Incidence, etiology, and management of type III endoleak after endovascular aortic repair. J. Vasc. Surg. 2017;66:1056–1064. doi: 10.1016/j.jvs.2017.01.056. [DOI] [PubMed] [Google Scholar]
- 70.Zhang R, et al. Ketogenic diet elicits antitumor properties through inducing oxidative stress, inhibiting MMP-9 expression, and rebalancing M1/M2 tumor-associated macrophage phenotype in a mouse model of colon cancer. J. Agric. Food Chem. 2020;68:11182–11196. doi: 10.1021/acs.jafc.0c04041. [DOI] [PubMed] [Google Scholar]
- 71.An J, et al. Targeting CCR2 with its antagonist suppresses viability, motility and invasion by downregulating MMP-9 expression in non-small cell lung cancer cells. Oncotarget. 2017;8:39230–39240. doi: 10.18632/oncotarget.16837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmierer B, Hill CS. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 73.Dai J, Michineau S, Franck G, Desgranges P, Becquemin J-P. Long term stabilization of expanding aortic aneurysms by a short course of cyclosporine A through transforming growth factor-beta induction. PLoS One. 2011;6:28903. doi: 10.1371/journal.pone.0028903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roberts AB, et al. Transforming growth factor type,8: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. U. S. A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nabel EG, et al. Direct transfer of transforming growth factor j81 gene into arteries stimulates fibrocellular hyperplasia (gene transfer/gene expression/extraceliular matrix/ceilular proliferation) Proc. Natl. Acad. Sci. U. S. A. 1993;90:10759–10763. doi: 10.1073/pnas.90.22.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Biros E, Walker PJ, Nataatmadja M, West M, Golledge J. Downregulation of transforming growth factor, beta receptor 2 and Notch signaling pathway in human abdominal aortic aneurysm. Atherosclerosis. 2012;221:383–386. doi: 10.1016/j.atherosclerosis.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 77.You Y, et al. Ketogenic diet aggravates cardiac remodeling in adult spontaneously hypertensive rats. Nutr. Metab. 2020;17:1–11. doi: 10.1186/s12986-020-00510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taylor MK, Swerdlow RH, Sullivan DK. Dietary neuroketotherapeutics for Alzheimer’s disease: An evidence update and the potential role for diet quality. Nutrients. 2019;11:1910. doi: 10.3390/nu11081910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deemer SE, et al. Exogenous dietary ketone ester decreases body weight and adiposity in mice housed at thermoneutrality. Obesity. 2020;28:1447–1455. doi: 10.1002/oby.22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anderson JC, Mattar SG, Greenway FL, Lindquist RJ. Measuring ketone bodies for the monitoring of pathologic and therapeutic ketosis. Obes. Sci. Pract. 2021;7:646–656. doi: 10.1002/osp4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.George F, Cahil J. Starvation in man. N. Engl. J. Med. 1970;282:668–675. doi: 10.1056/NEJM197003192821209. [DOI] [PubMed] [Google Scholar]
- 82.Masood W, Annamaraju P, Khan Suheb MZ, Uppaluri KR. Stat Pearls. Content is Key. StatPearls Publishing; 2023. Ketogenic diet. Stat Pearls. [PubMed] [Google Scholar]
- 83.Lu G, et al. A novel chronic advanced stage abdominal aortic aneurysm murine model. J. Vasc. Surg. 2017;66:232–242.e4. doi: 10.1016/j.jvs.2016.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hyde PN, Lustberg MB, Miller VJ, LaFountain RA, Volek JS. Pleiotropic effects of nutritional ketosis: Conceptual framework for keto-adaptation as a breast cancer therapy. Cancer Treat. Res. Commun. 2017;12:32–39. doi: 10.1016/j.ctarc.2017.06.001. [DOI] [Google Scholar]
- 85.Shaw DM, et al. The effect of 1, 3-butanediol on cycling time-trial performance. Int. J. Sport Nutr. Exerc. Metabol. 2019;29:466–473. doi: 10.1123/ijsnem.2018-0284. [DOI] [PubMed] [Google Scholar]
- 86.Knipp BS, et al. Ultrasound measurement of aortic diameters in rodent models of aneurysm disease. J. Surg. Res. 2003 doi: 10.1016/S0022-4804(03)00114-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.