

Abstract
1,4-Dihydropyridine and pyridine derivatives bound to three subtypes of adenosine receptors in the micromolar range. Affinity was determined in radioligand binding assays at rat brain A1 and A2A receptors using [3H]-(R)-PIA [[3H]-(R)-N6-(phenylisopropyl)adenosine] and [3H]CGS 21680 [[3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine], respectively. Affinity was determined at cloned human and rat A3 receptors using [125I]AB-MECA [N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine]. Structure–activity analysis at adenosine receptors indicated that sterically bulky groups at the 4-, 5-, and 6-positions are tolerated. (R,S)-Nicardipine, 12, displayed Ki values of 19.6 and 63.8 μM at rat A1 and A2A receptors, respectively, and 3.25 μM at human A3 receptors. Similarly, (R)-niguldipine, 14, displayed Ki values of 41.3 and 1.90 μM at A1 and A3 receptors, respectively, and was inactive at A2A receptors. A preference for the R- vs the S-enantiomer was observed for several dihydropyridines at adenosine receptors, in contrast with the selectivity at L-type Ca2+ channels. A 4-trans-β-styryl derivative, 24, with a Ki value of 0.670 μM at A3 receptors, was 24-fold selective vs A1 receptors (Ki) 16.1 μM) and 74-fold vs A2A receptors (Ki) 49.3 μM). The affinity of 24 at L-type Ca2+ channels, measured in rat brain membranes using [3H]isradipine, indicated a Ki value of 0.694 μM, and the compound is thus nonselective between A3 receptors and L-type Ca2+ channels. Inclusion of a 6-phenyl group enhanced A3 receptor selectivity: Compound 28 (MRS1097; 3,5-diethyl 2-methyl-6-phenyl-4-(trans-2-phenylvinyl)-1,4(R,S)-dihydro-pyridine-3,5-dicarboxylate) was 55-fold selective vs A1 receptors, 44-fold selective vs A2A receptors, and over 1000-fold selective vs L-type Ca2+ channels. In addition, compound 28 attenuated the A3 agonist-elicited inhibitory effect on adenylyl cyclase. Furthermore, whereas nicardipine, 12, displaced radioligand from the Na+-independent adenosine transporter with an apparent affinity of 5.36 ± 1.51 μM, compound 28 displaced less than 10% of total binding at a concentration of 100 μM. Pyridine derivatives, when bearing a 4-alkyl but not a 4-phenyl group, maintained affinity for adenosine receptors. These findings indicate that the dihydropyridines may provide leads for the development of novel, selective A3 adenosine antagonists.
The 1,4-dihydropyridines have been developed extensively as potent blockers and activators of L-type calcium channels.1 A number of these channel blockers, such as nifedipine (Figure 1, 9) and nicardipine (Figure 1, 12), are used therapeutically in the treatment of cardiovascular disorders, especially hypertension and coronary heart disease.2
Figure 1.

Structures of 1,4-dihydropyridines as potent calcium channel antagonists (nifedipine, 9, and nicardipine, 12) or as ligands at other receptor sites (SNAP 5089, an adrenoceptor antagonist, and 3-ethyl 5-[2-(phenylthio)ethyl] 2,4,6-trimethyl-1,4-dihydropyridine-3,5-dicarboxylate, 4, a PAF antagonist).
Dihydropyridines appear to be “privileged structures” in medicinal chemistry and pharmacology, i.e., they display affinity for many diverse binding sites.3 This adaptability of dihydropyridines has been utilized to optimize affinity in binding to α1a-adrenergic receptors (e.g., the antagonist SNAP 5089, Figure 1),4 to platelet activating factor (PAF, 1-O-hexadecyl/octadecyl-2-O-acetyl-sn-glycero-3-phosphorylcholine) receptors,5 and at other receptor targets.6 Thus, by careful structural modification, it has been possible to select for affinity at sites other than Ca2+ channels. For example, a dihydropyridine derivative, 3-ethyl 5-[2-(phenylthio)-ethyl] 2,4,6-trimethyl-1,4-dihydropyridine-3,5-dicarboxylate (Figure 1, 4), has been found to inhibit PAF binding with an affinity of 69 nM, while the same derivative was inactive as a calcium channel blocker.5 Several dihydropyridines were also found to bind to A1 adenosine receptors in rat brain.7,8 In the present study we have used the 1,4-dihydropyridine nucleus as a template for probing structure–activity relationships (SAR) at several subtypes of central adenosine receptors (A1, A2A, and A3), in an effort to design new selective antagonists. The classical7 (A1 and A2A) adenosine receptor antagonists are xanthines,9 but it would be desirable to expand the list of diverse chemical structures known to bind to adenosine receptors. There is already a large number of non-xanthine antagonists known for the A1 receptor.9–13 Although selective agents have been reported for both A1 and A2A subtypes of adenosine receptors,9 the development of ligands for the recently discovered A3 receptor is lagging behind the other subtypes.14 Activation of the A3 receptor results in hypotension and promotion of release of inflammatory mediators from mast cells.15 A3 adenosine receptor antagonists are being sought as potential antiasthmatic,15 antiinflammatory,15 and cerebroprotective agents.16 There may also be an involvement of A3 receptors in cancer17 and apoptosis.18 We have introduced the first selective agonists for the A3 adenosine receptor subtype, including N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine (IB-MECA),19,20 but studies of the SAR of xanthines at this subtype have thus far failed to identify principles of achieving selectivity.21 Furthermore, at A3 receptors the affinity of xanthines is generally much weaker than at the A1 or A2A subtypes,21,22 and considerable species variability in their affinity has been noted.23–25 A screen of >100 cyclic compounds of diverse structure has provided several unexplored leads for A3 selectivity, but potencies were generally low.11 A recent study has identified additional natural product leads having moderate affinity, but the degree of selectivity was still minimal.13 Thus, the design of potent and selective A3 antagonists has remained an unfulfilled challenge.14
Results
Synthesis.
The structures of the 1,4-dihydropyridines and related derivatives tested for affinity in radioligand binding assays at adenosine receptors are shown in Table 1. Some of the derivatives, i.e., both well-known Ca2+ channel ligands (nifedipine, 9; nitrendipine, 10; nicardipine, 12; nimodipine, 13; niguldipine, 14 and 15; Bay K 8644, 18 and 19) and a PAF antagonist (4),5 were obtained from commercial sources. Other compounds were synthesized using standard methodology (Scheme 1),26–28 in fair yield, as reported in Table 2. The synthesis consisted of the Hantzsch condensation of a 3-amino-2-butenoate ester, 33a,b, an aldehyde, 34a–o, and a 3-ketopropionate ester derivative, 35a–e, that were dissolved in ethanol and heated to 100 °C. In order to obtain substitution at the 4-position, the aldehyde component, 34a–o, was varied. Substitution at the 6-position was achieved by varying the 3-ketopropionate ester component, 35a–e (Scheme 1).
Table 1.
Affinities of Dihydropyridine and Pyridine Derivatives in Radioligand Binding Assays at A1, A2A, and A3 Receptorsa-d

|
||||||||
|---|---|---|---|---|---|---|---|---|
| Ki (mM) or % inhibitionc | ||||||||
| compd | R2 | R3 | R4 | R5 | R6 | rA1 | rA2A | hA3 |
| 1 | CH3 | CO2CH3 | CH3 | CO2CH2CH3 | CH3 | 32.6 ± 6.3 | 46.1 ± 6.8 | 32.3 ± 5.1 |
| 2 | CH3 | CO2CH3 | CH3 | CO2(CH2)2OCH3 | CH3 | 49.2 ± 0.7 | 37 ± 18%(10−4) | 62.3 ± 16.7 |
| 3 | CH3 | CO2CH3 | CH3 | CO2CH2Ph | CH3 | 6.45 ± 1.47 | 9.72 ± 0.63 | 2.78 ± 0.89 |
| 4 | CH3 | CO2CH2CH3 | CH3 | CO2(CH2)2SPh | CH3 | 6.50 ± 0.47 | 7.10 ± 2.46 | 5.56 ± 1.36 |
| 5 | CH3 | CO2CH3 | CH2CH3 | CO2CH2CH3 | CH3 | 7.52 ± 2.79 | 9.56 ± 2.69 | 13.6 ± 2.0 |
| 6 | CH3 | CO2CH3 | (CH2)2CH3 | CO2CH2CH3 | CH3 | 8.17 ± 1.58 | 11.5 ± 3.8 | 6.51 ± 0.74 |
| 7 | CH3 | CO2CH3 | CH2CHCH3(CH2)2-CH=C(CH3)2(R, S) | CO2CH2CH3 | CH3 | 9.10 ± 2.90 | 23.1 ± 8.6 | 7.90 ± 0.88 |
| 8 | CH3 | CO2CH3 | Ph | CO2CH2CH3 | CH3 | 11.0 ± 1.6 | 2.74 ± 0.85 | 12.0 ± 3.3 |
| 9(nifedipine) | CH3 | CO2CH3 | 2-NO2Ph | CO2CH3 | CH3 | 2.89 ± 0.23 | 18.2 ± 2.51 | 8.29 ± 2.41 |
| 10(nitrendipine) | CH3 | CO2CH3 | 3-NO2Ph | CO2CH2CH3 | CH3 | 8.96 ± 2.06 | 23.0 ± 3.7 | 8.30 ± 1.41 |
| 11 | CH3 | CO2CH2CH3 | 3-NO2Ph | CO2CH2CH3 | CH3 | 3.34 ± 2.17 | 18.2 ± 7.9 | 2.51 ± 0.15 |
| 12(nicardipine) | CH3 | CO2CH3 | 3-NO2Ph | CO2CH2CH2N(CH3)CH2Ph | CH3 | 19.6 ± 1.9 | 63.8 ± 4.2 | 3.25 ± 0.26 |
| 13(nimodipine) | CH3 | CO2CH(CH3)2 | 3-NO2Ph | CO2CH2CH2OCH3 | CH3 | 20.1 ± 1.7 | 44.3 ± 14.4 | 8.47 ± 2.75 |
| 14((R)-niguldipine) | CH3 | CO2CH3 | 3-NO2Ph |
|
CH3 | 41.3 ± 3.5 | d (10−4) | 1.90 ± 0.40 |
| 15((S)-niguldipine) | CH3 | CO2CH3 | 3-NO2Ph |
|
CH3 | d (10−4) | d (10−4) | 2.80 ± 0.35 |
| 16 | CH3 | CO2CH3 | 4-NO2Ph | CO2CH2CH3 | CH3 | 37 ± 14% (10−4) | 35.6 ± 1.9 | 5.90 ± 1.65 |
| 17 | CH3 | CO2CH3 | 2-CF3Ph | CO2CH2CH3 | CH3 | 6.68 ± 2.37 | 20.7 ± 2.8 | 11.6 ± 1.7 |
| 18((R)-BayK8644) | CH3 | CO2CH3 | 2-CF3Ph | NO2 | CH3 | 0.785 ± 0.113 | 35.1 ± 10.1 | 2.77 ± 0.34 |
| 19((S)-BayK8644) | CH3 | CO2CH3 | 2-CF3Ph | NO2 | CH3 | 6.66 ± 1.89 | 86.3 ± 23.4 | 23.5 ± 0.6 |
| 20 | CH3 | CO2CH3 | 4-CH3OPh | CO2CH2CH3 | CH3 | 2.75 ± 0.35 | 12.7 ± 3.8 | 4.10 ± 0.14 |
| 21 | CH3 | CO2CH3 | 3-CH3O-4-OHPh | CO2CH2CH3 | CH3 | 51.0 ± 3.7 | 56.8 ± 1.9 | 32.1 ± 9.2 |
| 22 | CH3 | CO2CH3 | 3,4-OCH2OPh | CO2CH2CH3 | CH3 | 3.66 ± 0.61 | 5.27 ± 1.97 | 4.58 ± 1.11 |
| 23 | CH3 | CO2CH3 | PhCH2CH2 | CO2CH2CH3 | CH3 | 8.81 ± 0.92 | 6.71 ± 2.06 | 2.30 ± 0.70 |
| 24 | CH3 | CO2CH3 | Ph-CH=CH-(trans) | CO2CH2CH3 | CH3 | 16.1 ± 0.5 | 49.3 ± 12.5 | 0.670 ± 0.195 |
| 25 | CH3 | CO2CH3 | Ph-C≡C- | CO2CH2CH3 | CH3 | 5.39 ± 0.33 | 38.3 ± 7.9 | 0.940 ± 0.070 |
| 26 | CH3 | CO2CH2CH3 | CH3 | CO2CH2CH3 | (CH2)3CH3 | 10.8 ± 3.52 | 38.0 ± 10.6 | 47.1 ± 10.8 |
| 27 | CH3 | CO2CH2CH3 | CH3 | CO2CH2CH3 | Ph | 25.9 ± 7.3 | 35.9 ± 15.3 | 7.24 ± 2.13 |
| 28(MRS1097) | CH3 | CO2CH2CH3 | Ph-CH=CH-(trans) | CO2CH2CH3 | Ph | 5.93 ± 0.27 | 4.77 ± 0.29 | 0.108 ± 0.012 |
| 29 | CH3 | CO2CH3 | CH3 | CO2CH2CH3 | CH3 | 6.95 ± 2.66 | 8.96 ± 0.93 | 29.5 ± 2.1 |
| 30 | CH3 | CO2CH2CH3 | CH3 | CO2CH2CH3 | Ph | 7.41 ± 1.29 | 28.4 ± 9.1 | 4.47 ± 0.46 |
| 31 | CH3 | CO2CH3 | 2-NO2Ph | CO2CH3 | CH3 | d (10−4) | d (10−4) | d (10−4) |
| 32 | H | H | 4-CH3OPh | H | H | 44.5 ± 1.0 | 71 ± 29 | d (10−4) |
Displacement of specific [3H]-(R)-PIA binding in rat brain membranes, expressed as Ki ± SEM in μM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes, expressed as Ki ± SEM in μM (n = 3–6).
Displacement of specific [125I]AB-MECA binding at human A3 receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM in μM (n = 2–3), or as a percentage of specific binding displaced at 10 μM.
Displacement of ≤ 10% of specific binding at the indicated concentration. nd, not determined.
Scheme 1.

General Procedure for the Synthesis of 1,4-Dihydropyridine Derivatives
Table 2.
Characterization of Dihydropyridine and Pyridine Derivatives
| no. | Tm (°C) | formula | MS | analysis | yield (%) | purification |
|---|---|---|---|---|---|---|
| 1 | 125–126 | C13H19NO4 | 253 (CI) | C,H,N | 98.2 | 1; MeOH |
| 2 | 88–89 | C14H21NO5·0.25H2O | 283 (CI) | C,H,N | 53.0 | 2; TLC |
| 3 | oil | C18H21NO4·0.50H2O | 315 (CI) | C,H,N | 70.5 | 2; TLC |
| 5 | 83–84 | C14H21NO4 | 267 (CI) | C,H,N | 50.5 | 1; MeOH |
| 6 | 95–96 | C15H23NO4 | 281 (CI) | H,Nb | 73.3 | 1; MeOH |
| 7 | glass | C21H33NO4 | 363 (CI) | c | 33.4 | 2; TLC |
| 8 | 121–122 | C18H21NO4 | 315 (CI) | C,H,N | 78.6 | 1; MeOH |
| 11 | 165–166 | C19H22N2O6 | 374 (EI) | C,H,N | 35.5 | 1; EtOAc |
| 16 | 151–152 | C18H20N2O6 | 360 (CI) | C,H,N | 68.1 | 1; MeOH |
| 17 | 114–115 | C19H20F3NO4 | 383 (CI) | C,Hd | 45.6 | 2; TLC |
| 20 | 112–113 | C19H23NO5·0.21EtOAc | 345 (CI) | C,H,N | 61.8 | 2; TLC |
| 21 | 190 | C19H23NO6 | 361 (CI) | C,H,N | 79.6 | 1; MeOH |
| 22 | oil | C19H21NO6·0.32EtOAc | 359 (CI) | C,H,N | 63.0 | 2; TLC |
| 23 | 87–88 | C20H25NO4 | 343 (CI) | e | 71.9 | 2; TLC |
| 24 | 135–136 | C20H23NO4 | 341 (CI) | C,H,N | 87.9 | 1; MeOH |
| 25 | 176–177 | C20H21NO4 | 339 (EI) | C,H,N | 43.3 | 1; EtOH |
| 26 | oil | C17H27NO4 | 309 (CI) | C,H,N | 58.8 | 2; TLC |
| 27 | 123–124 | C19H23NO4 | 329 (CI) | C,H,N | 53.0 | 1; pe 35–60 |
| 28 | oil | C26H27NO4 | 417 (CI) | C,H,N | 34.7 | 2; column |
| 29 | oil | C13H17NO4 | 251 (EI) | f | 57.9 | 2; TLC |
| 30 | oil | C19H21NO4·0.20EtOH | 327 (CI) | C,H,N | 48.9 | 2; TLC |
Purification was achieved by either (1) (re)crystallization from the solvent specified or (2) chromatography by the specified method, using EtOAc/pe, 20:80 (v/v), eluent. MeOH, methanol; EtOH, ethanol; EtOAc, ethyl acetate; pe, petroleum ether 35–60 °C fraction; TLC, preparative thin layer chromatography, silica 60, 1000 μm layer thickness; column, preparative column chromatography, silica 60, 220–440 mesh.
6 (C15H23NO4) H, N; C: calcd, 64.03; found, 64.75. EI: calcd, 281.1627; found, 281.1630.
7 (C21H33NO4). This compound decomposes upon prolonged standing. Biological data and mass spectrometry data were obtained with the freshly purified compound, but extensive drying and shipping proved detrimental for the elemental analysis. C, H, N: calcd, 69.39, 9.15, 3.85; found, 61.65, 7.27, 4.70. EI: calcd, 363.2410; found, 363.2398.
17 (C19H20F3NO4) C, H; N: calcd, 3.65; found, 4.17. EI: calcd, 383.1344; found, 383.1346.
23 (C20H25NO4) EI: calcd, 343.1784; found, 343.1791.
29 (C13H17NO4) EI: calcd, 251.1158; found, 251.1162.
For compound 26, the precursor butyl β-keto ester (35e) was prepared from the condensation of the enolate lithium salt of ethyl acetate and valeryl chloride.28
Good yields of the 6-phenyl-1,4-dihydropyridines were obtained using a 72 h reaction time.
Oxidation of 1,4-dihydropyridines (1 and 27) to the corresponding pyridine derivatives (29 and 30, respectively) was carried out using tetrachloro-1,4-benzoquinone (chloranil, 37) in tetrahydrofuran (Scheme 2).29
Scheme 2.
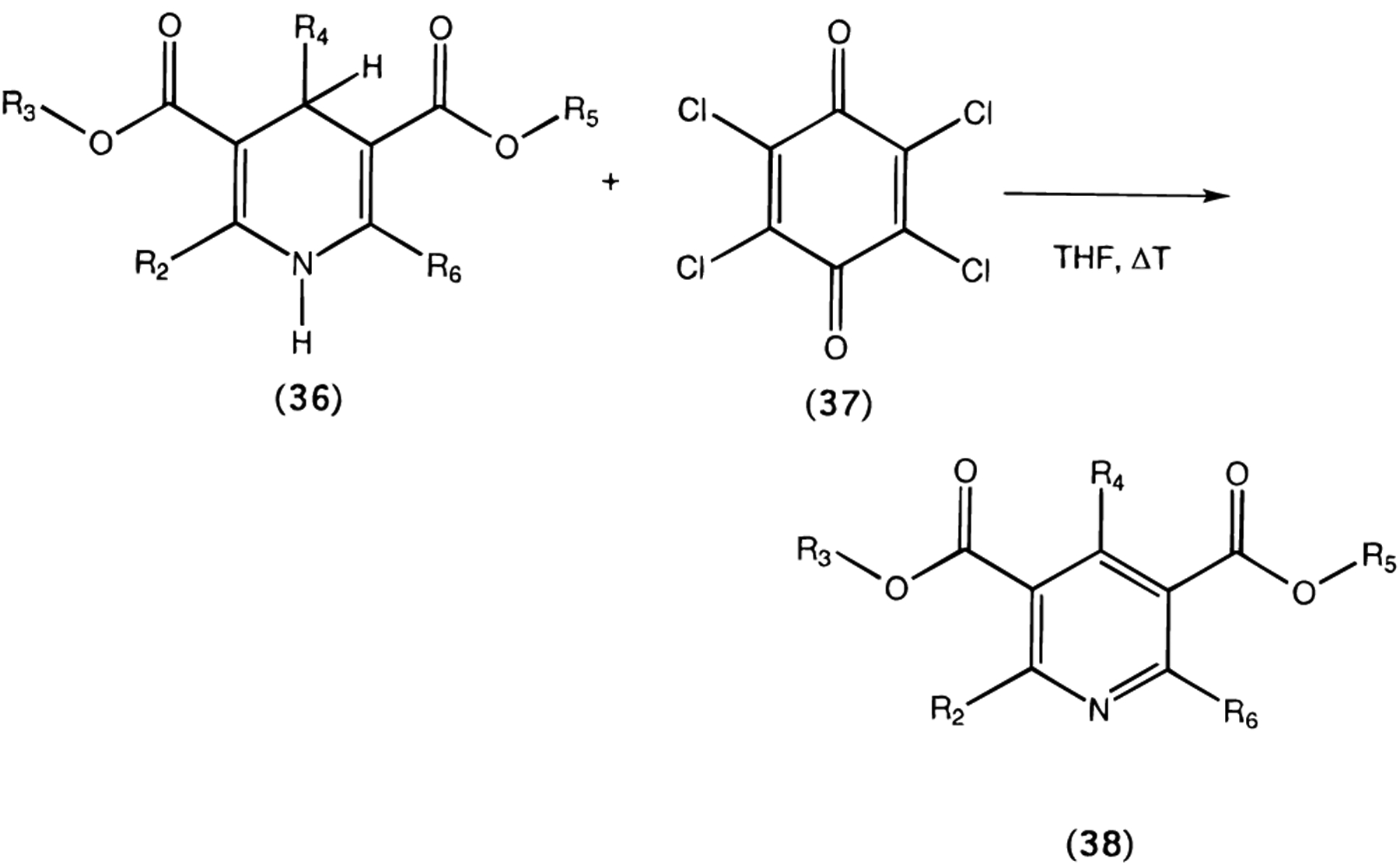
General Procedure for the Oxidation of 1,4-Dihydropyridine Derivatives (Compounds 29 and 30)
Most of the dihydropyridines examined, except for two pairs of pure enantiomers, 14 and 15, 18 and 19, and the nonchiral compounds 9 and 11, were racemic mixtures at position C-4.
Binding at Adenosine Receptors.
Ki values at A1 and A2A receptors were determined in radioligand binding assays in rat brain membranes vs [3H]-(R)-PIA [[3H]-(R)-N6-(phenylisopropyl)adenosine] or [3H]CGS 21680 [[3H]-2-[[4-[(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine], respectively.30,31 Affinity at cloned human A3 receptors expressed in HEK-293 cells23 was determined using [125I]AB-MECA [N6-(4-amino-3-[125I]iodobenzyl)-5′-(N-methylcarbamoyl)-adenosine].13,32
SAR analysis at adenosine receptors indicated that sterically bulky groups are tolerated at the 4-, 5-, and 6-positions (Table 1). R5 was varied from ethoxycarbonyl to larger ester groups in a homologous series (1 vs 2, 3, and 4), all having a methyl group at the 4-position, resulting in a general enhancement of affinity at all three adenosine receptor subtypes. Compound 4 is a potent PAF–acether antagonist5 and not a calcium channel antagonist; thus there is an uncoupling of the SAR in this series for interaction with both PAF and adenosine receptors on the one hand and L-type calcium channels on the other hand.
Another means of enhancing adenosine receptor affinity (relative to compound 1) was to enlarge the R4 substituent. In the series of ethyl, n-propyl, and even larger alkyl substituents at the 4-position (5, 6, and 7) it was demonstrated that steric bulk at this position is tolerated in adenosine receptor binding and favored at the A3 subtype and not detrimental at the A1 subtype. The affinity at the A2A subtype is generally decreased with increasing chain length of the alkyl substituent. Consecutively, 4-aryl substituents, typical of the potent Ca2+ channel blockers in clinical use, were also examined. In general, the affinity and receptor subtype selectivity of 4-aryl analogues was highly dependent on the substituents of the phenyl ring. For example, the unsubstituted 4-phenyl compound 8 was more potent (4-fold) at A2A receptors than at either A1 or A3 receptors. Of the nitrophenyl derivatives, compound 10, bearing an o-nitro group, was 3-fold selective for A1 vs A3, and 6-fold for A1 vs A2A receptors, whereas the m-nitro derivative 10 was about equipotent at A1 and A3 receptors, but 3-fold less potent at A2A receptors than at either A1 or A3 receptors. To complete the series, we also synthesized the p-nitro-substituted compound 16, which displayed a >17-fold selectivity for A3 receptors vs A1 receptors and 6-fold selectivity for A3 receptors vs A2A receptors. In addition, we prepared compounds with different substituents. Compound 17, bearing a o-trifluoromethyl group behaved similarly to the o-nitro-substituted compound 10 and was 2-fold selective for A1 vs A3 receptors. The p-methoxy derivative 20 was almost equipotent at A1 (Ki = 2.75 μM), and A3 receptors (Ki = 4.10 μM), but slightly less potent at A2A receptors (Ki = 12.7 μM). Affinity at A3 receptors did not appear to be a direct function of electron density of the phenyl substituents; i.e. compounds containing an electron-withdrawing p-nitro group (16) and an electron-donating p-methoxy group (20) were equipotent. Remarkably, the introduction of a hydroxy substituent on the para position of the 4-phenyl ring (in addition to a m-methoxy substituent) led to a general decrease in affinity (21; Ki = 51.0 μM at A1, 56.8 μM at A2A, and 32.1 μM at A3 receptors). Thus, steric factors surrounding the phenyl ring may be more important than electronic factors. The piperonal derivative 22 had a generally increased affinity at A1 and A3 adenosine receptor subtypes (vs the unsubstituted phenyl compound 8), but did not display subtype selectivity.
The relatively high affinity of a 4-aryl group larger than phenyl (22), an arylalkyl group (23), and dehydro analogues thereof (24, 25) further indicated a bulk tolerance at this position. The 4-trans-β-styryl derivative, 24, was particularly potent (Ki) 0.670 μM) and selective (24- and 74- fold vs rat A1 and A2A, respectively) at human A3 receptors. The phenylacetylenic analogue, 25, was nearly as potent at A3 receptors, but not as selective as 24 for A3 vs A1 receptors (only 6-fold).
Substitution at the 3- and 5-positions of the dihydropyridine moiety also had a large effect on affinity at adenosine receptors. Changing an asymmetrical mixed methyl and ethyl ester of nitrendipine, 10, to the corresponding symmetrical diethyl ester, 11, resulted in a 3.3-fold enhancement of A3 affinity. At A1 and A2A receptors a similar effect was observed, and subtype selectivity was therefore not affected. Thus, compound 11 displayed Ki values of 3.34 and 18.2 μM at rat A1 and A2A receptors, respectively, and 2.51 μM at human A3 receptors. Nicardipine,33 12, differing from nitrendipine,34 10, in the presence of a sterically bulky ester group at the 5-position, displayed a moderate enhancement of affinity at human A3 receptors of 2.6-fold, while at rat A1 and A2A receptors affinity was diminished 2–3 fold. Even larger ester substituents (i.e., 14, and 15) were well tolerated at human A3 receptors, while affinity at A1 and A2A receptors was drastically diminished.
Affinities of enantiomeric pairs, i.e., of Bay K 8644 (18 and 19, R and S, a Ca2+ channel blocker and activator, respectively) and the Ca2+ channel blocker niguldipine (14 and 15, R and S, respectively), indicated a general preference for the R- over the S-enantiomer at all of the receptor subtypes. This is in contrast to the affinity at L-type calcium channels [(R)-(−)-niguldipine, Ki) 8.1 nM; (S)-(+)-niguldipine, Ki) 0.18 nM; or 45-fold stereoselectivity]35 and at α1a adrenoceptors [(R)-(−)-niguldipine, Ki) 4.7 nM; (S)-(+)-niguldipine, Ki) 0.16 nM; or 29-fold stereoselectivity],4 at which the (S)-(+)-enantiomer is preferred. Both niguldipine enantiomers tended particularly towards selectivity for the A3 subtype (vs A1 and A2A receptors, but not vs Ca2+ channels). Thus, (R)-niguldipine, 14, displayed Ki values of 41.3 and >100 μM at rat A1 and A2A receptors, respectively, and 1.90 μM at human A3 receptors.
At the 6-position, n-butyl substitution (26) was tolerated, and phenyl substitution (27) enhanced A3 adenosine receptor binding (4.5-fold vs 1), and lead to slight subtype selectivity. Combination of 6-phenyl and 4-trans-β-styryl substituents greatly enhanced both affinity and selectivity. Consequently, compound 28 was 55-fold selective for A3 vs A1 receptors, and 44-fold selective for A3 vs A2A receptors. Two other blockers of L-type Ca2+ channels, verapamil and diltiazem, which are structurally unrelated to the dihydropyridines, failed to bind appreciably to adenosine receptors.
Pyridine derivatives maintained affinity for adenosine receptors, particularly when bearing a 4-alkyl group, e.g., the oxidized compound 29, displayed a Ki value of 29.5 μM at human A3 receptors, whereas the related dihydropyridine 1 displayed a Ki value of 32.3 μM at the same receptor subtype. Similarly, a comparison of the 6-phenyldihydropyridine 27 and the corresponding pyridine 30 shows only minor differences in affinity. In contrast, the nifedipine metabolite 31 no longer bound to adenosine receptors. The distantly related pyridine derivative 32 demonstrated that affinity of 4-arylpyridine analogues for adenosine receptors may be rescued to some extent by careful manipulation of the other substituents.
Binding at L-Type Calcium Channels.
Affinity at L-type Ca2+ channels, measured in rat brain membranes using [3H]isradipine36 (Table 3), indicated a Ki value of 0.694 ± 0.165 μM for the styryl derivative 24 (cf. 0.670 μM at human A3 receptors, and therefore not selective). In the same assay, (R)-niguldipine, 14, had a Ki value of 59.7 ± 0.1 nM (cf. 8.1 nM at guinea pig skeletal muscle35 and 1.90 μM at human A3 receptors, and therefore at least 32-fold selective for Ca2+ channels). Compound 30 at a concentration of 10 μM (Ki= 4.47 μM at A3 receptors) or 28 (Ki = 0.108 μM at human A3 receptors, and therefore at least 1000-fold A3 selective vs Ca2+ channels) at a concentration of 100 μM displaced <10% of specific binding. Slightly more potent in Ca2+ channel binding were 27 (17% displacement at 100 μM; Ki = 7.24 μM at human A3 receptors, and thus >14-fold A3 selective) and 25 (23% displacement at 100 μM; Ki = 0.94 μM at human A3 receptors, and thus >100-fold A3 selective). The 5-benzyl ester 3 (Ki = 1.17 ± 0.08 μM at Ca2+ channels and 2.78 ± 0.89 μM at A3 receptors) and the 4-(2-phenylethyl) analogue 23 (Ki = 0.910 ± 0.207 μM at Ca2+ channels and 2.30 ± 0.70 μM at A3 receptors) were about 2.5-fold selective for Ca2+ channels compared to human A3 receptors.
Table 3.
Inhibition of Specific Binding of [3H]Isradipine Binding at L-Type Calcium Channels by Various Dihydropyridine and Pyridine Derivatives in Rat Brain Membranes and the Selectivity Ratio vs Affinity at Cloned Human A3 Receptors
| compd | Ki (rCa2+)a | ratio of Ki (rCa2+)/Ki (hA3) |
|---|---|---|
| 3 | 1.17 ± 0.08 | 0.42 |
| 14 | 0.0597 ± 0.0001 | 0.031 |
| 23 | 0.910 ± 0.207 | 0.40 |
| 24 | 0.694 ± 0.165 | 1.04 |
| 25 | 23 ± 3% (10−4) | >106 |
| 27 | 17 ± 5% (10−4) | >14 |
| 28 | <10% (10−4) | >1000 |
| 30 | <10% (10−5) | >2.2 |
Expressed in μM as Ki ± SEM or percent displacement of specific binding at the indicated concentration (M) for three determinations, each performed in duplicate. Rat brain membranes (ca. 100 μg of protein/tube) were incubated for 1 h at 25 °C with 0.1 nM [3H]isradipine and varying concentrations of the dihydropyridine or pyridine derivative in a 50 mM Tris buffer, pH 7.4 in a total volume of 0.5 mL. Nonspecific binding was determined in the presence of 10 μM nitrendipine. Ki values were calculated using the Cheng-Prusoff equation43 assuming a Kd value of 0.13 nM for [3H]isradipine.
Functional Assays at A3-Adenosine Receptors.
We examined the antagonist properties of two dihydropyridine derivatives in a functional assay utilizing cloned rat A3 adenosine receptors expressed in CHO cells. Forskolin-stimulated adenylyl cyclase was inhibited by IB-MECA [N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine; 10−9–10−4 M) in CHO cells transfected with rat A3 adenosine receptors (maximum degree of inhibition was 40–50%).22 The concentration of IBMECA used to test prospective antagonists was between 10 nM and 1.0 μM, i.e., respectively 0.1- and 10-fold the IC50 of rat A3 receptor mediated inhibition of adenylyl cyclase. Both nicardipine, 12, and (R)-Bay K 8644, 18, at a concentration of 10 μM showed a significant (p < 0.02) effect on the agonist-mediated inhibition of adenylyl cyclase (Table 4). The inhibition of adenylyl cyclase induced by 10−6 M IB-MECA (40.2 ± 3.3%) was attenuated to 27.2 ± 8.5% by 10 μM nicardipine, 12, and the inhibition of adenylyl cyclase activity caused by 10−7 M IB-MECA was nearly completely reversed. A similar, but not as extensive, effect could be observed for (R)-Bay K 8644, 18, even though the affinity of these compounds at human A3 adenosine receptors was virtually identical. Compound 28 at a concentration of 50 μM attenuated the IB-MECA induced inhibition of adenylyl cyclase activity at all concentrations tested (Figure 2). This suggests that compound 28 is not only an effective displacer of the A3-selective radioligand [125I]AB-MECA but also an effective functional antagonist at this receptor subtype.
Table 4.
Effects of Dihydropyridine Derivatives at a Concentration of 10 μM on the Inhibition of Adenylyl Cyclase Elicited by the A3 Agonist IB-MECA
Figure 2.

Attenuation of A3 agonist elicited inhibition of forskolin stimulated adenylyl cyclase activity via cloned rat A3 adenosine receptors. Adenylyl cyclase activity was determined in membranes from CHO cells stably expressing the rat A3 receptor, as reported previously.28 Adenylyl cyclase activity was stimulated with 1 μM forskolin, and this stimulation was inhibited by the A3 selective agonist IB-MECA (shaded bars). Addition of 50 μM MRS1097, 28, attenuated the IB-MECA induced inhibition of adenylyl cyclase activity (closed bars). The asterisk (*) denotes a statistically significant effect (p < 0.02) vs the control experiment.
Interaction of 1,4-Dihydropyridines with the NBI-Sensitive Nucleoside Transporter Protein.
It was shown previously that the nucleoside uptake inhibitor NBI (S-(4-nitrobenzyl)-6-thioinosine) binds to 1,4-dihydropyridine ([3H]nimodipine) binding sites on human red blood cell membranes.37 We therefore investigated the possibility that 1,4-dihydropyridines may bind to the NBI-sensitive nucleoside transporter protein. Both the commercial Ca2+ channel blocker nicardipine, 12, and the A3 adenosine receptor selective ligand 24 displaced [3H]NBI from its binding site in rat forebrain membranes, with Ki values of 5.36 ± 1.51 and 1.20 ± 0.11 μM, respectively. In contrast, the A3-selective antagonist 28 displaced less than 10% of specific binding at a concentration of 100 μM. Compound 28 is therefore >900-fold selective for A3 receptors compared to the NBI-sensitive nucleoside transporter.
Affinity at Other Binding Sites.
Since compound 28 was the most potent and (A3 adenosine receptor) selective compound of the series, its affinity at non-adenosine receptor binding sites was examined in a battery of radioligand binding assays (NovaScreen, Division of Oceanix Biosciences, Hanover, MD).38 At a concentration of 10−5 M, there was no significant (0 ± 30%) displacement of radioligand from adrenergic (α1, α2, β), cholinergic (nicotinic and muscarinic M1, M2, M3), dopaminergic (D1 and D2), histaminergic (H1 and H2), serotoninergic (5-HT1, 5-HT2, and 5-HT3), C5a complement, central benzodiazepine (RO 151788), GABAA (muscimol), GABAB (baclofen), NMDA (kainate, quisqualate, and phencyclidine), glycine (strychnine sensitive and insensitive), σ (MK-801), angiotensin (AT-II), vasopressin V1, neuropeptide Y, cholecystokinin (CCK-B), neurotensin, somatostatin, ANF1, and EGF receptors. There was also no significant displacement of binding of radioligand from second messenger sites (forskolin, phorbol ester, and inositol trisphosphate) and ion channels (N-type calcium channels, chloride channels, and low conductance potassium channels). Significant displacement of radioligand was observed for the CCK-A (40%) and VIP (45%) binding sites, but the (estimated) IC50 for these compounds was still higher than 10 μM.
The Effect of Species Differences on Ligand Selectivity.
Ordinarily, a comparison of A1, A2A, and A3 affinities within one species would be preferred. We chose the human A3 receptor for our study instead of the rat A3 receptor for the following reasons: Whereas the affinity of many ligands varies slightly between rat and human A1 and A2A adenosine receptors, the affinity of most known adenosine receptor antagonists is only marginal at rat A3 receptors.11,12,21,25 The human A3 receptor, being more susceptible to displacement of a selective radioligand by competing compounds, therefore allowed for a better comparison between those compounds.25 The comparison was justified by the similarity of affinity of dihydropyridines at rat and human non-A3 adenosine receptors. For example, the calcium channel blocker nifedipine (9) displayed a Ki value of 5.6 ± 0.3 μM at human A2A adenosine receptors (unpublished data) and a Ki value of 18 ± 3 μM at rat A2A receptors (this manuscript), but the affinities of 1,4-dihydropyridines at rat A3 receptors differed considerably from their affinity at human A3 receptors (Table 5). Least affected by the species difference was (R)-Bay K 8644 (18) with Ki values of 2.77 ± 0.34 and 6.20 ± 2.12 μM at human and rat A3 receptors, respectively. Thus, the species selectivity ratio for (R)-Bay K 8644 (18) is only 2.2-fold, but this value increases to 38.6-fold for our most potent and selective compound (28), which displayed a Ki value of 4.16 ± 1.55 μM at rat A3 receptors. Consequently, the receptor subtype selectivity of the test compounds for A3 vs either A1 or A2A adenosine receptors was disproportionately affected (Table 5).
Table 5.
Affinity of and Selectivity Ratio for Selected Dihydropyridine Derivatives at Rat A3 vs Human A3, Rat A1, and Rat A2A Adenosine Receptors, Respectively
| compd | Ki (μM)a | selectivity ratio | ||
|---|---|---|---|---|
| rA3/hA3 | rA1/rA3 | rA2A/rA3 | ||
| 12 | 11.0 ± 7.3 | 3.38 | 1.78 | 5.80 |
| 18 | 6.20 ± 2.12 | 2.24 | 0.13 | 5.66 |
| 24 | 12.1 ± 2.6 | 18.1 | 1.33 | 4.07 |
| 28 | 4.16 ± 1.55 | 38.6 | 1.43 | 1.15 |
Discussion
In the present study, we have demonstrated that structural modification and careful examination of the SAR of 1,4-dihydropyridines resulted in the development of one of the first A3 adenosine receptor-selective non-xanthine ligands. Compound 28 [3,5-diethyl 2-methyl-6-phenyl-4-(trans-2-phenylvinyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate; MRS1097] displayed a good (submicromolar) affinity in a radioligand binding assay (Ki = 0.108 ± 0.012 μM). In addition, we have shown that dihydropyridines can be effective in attenuating the IB-MECA elicited inhibition of adenylyl cyclase in CHO cells expressing the cloned rat A3 adenosine receptor. Compound 28 was 55-fold selective vs A1 receptors, 44-fold selective vs A2A receptors, and over 1000-fold selective vs L-type Ca2+ channels.
It has been suggested that 1,4-dihydropyridines may bind to the nucleoside transporter protein of human red blood cells and human brain.37,43 We therefore measured the ability of some 1,4-dihydropyridines to displace [3H](nitrobenzyl)thioinosine ([3H]NBI) from the Na+-independent adenosine transporter in rat forebrain. Whereas the commercial compound nicardipine (12) and the novel compound MRS1045 (24) displaced [3H]NBI with affinities similar to their affinity at human A3 receptors (5.36 ± 1.51 and 1.20 ± 0.11 μM, respectively), the most selective compound of this series (MRS1097, 28) displaced less than 10% of total [3H]NBI binding at a concentration of 10−4 M.
In this study we have shown that the affinity of dihydropyridines can be optimized selectively for adenosine receptors vs L-type calcium channels and for A3 vs A1 and A2A receptors. Hu et al. previously found no effect of nifedipine binding on A1 receptor mediated adenylyl cyclase inhibition,7 but current work with the more recently cloned A3 receptor22 demonstrates that IB-MECA-induced inhibition of adenylyl cyclase was attenuated by several compounds of the 1,4-dihydropyridine class (Table 4). Preliminary tests show that both nicardipine, 12, and (R)-Bay K 8644, 18 (both Ca2+ channel blockers), functionally antagonize the inhibitory effects of an A3 selective agonist on adenylyl cyclase. Furthermore, we have shown that MRS1097, 28, significantly (p < 0.02) attenuates the effects of IB-MECA on adenylyl cyclase activity (Figure 2). Further pharmacological characterization will be required to determine if this is due to a competitive interaction with adenosine A3 receptors or at some other stage of the functional cascade. Thus, we have discovered dihydropyridines as a promising lead in the search for A3 adenosine receptor antagonists.
L-type calcium channel blockers such as nimodipine, 13, have been examined as cerebroprotective agents in experimental models of neurotoxicity in cell culture and in vivo.39 Adenosine agonists and antagonists have also been evaluated in models of brain ischemia. It has also been postulated that an adenosine A3 receptor antagonist would have cerebroprotective properties, based on the in vivo effects of the A3 receptor agonist IB-MECA administered to gerbils in a chronic regimen.16 Thus, a compound that would display a dual action in vivo might be even more useful in treating stroke than either an L-type calcium channel blocker or A3 receptor antagonist alone. Therefore, some of the compounds found in the present study to have affinity at both sites may be clinically useful as combined therapeutic agents.
Experimental Section
Materials.
(R)-(+)- and (S)-(−)-Bay K 8644, (R)-(−)- and (S)-(+)-niguldipine, nicardipine, nifedipine, nimodipine, and oxidized nifedipine were purchased from Research Biochemicals International (Natick, MA). Ethyl acetoacetate (35a), acetaldehyde (34a), propionaldehyde (34b), butyraldehyde (34c), benzaldehyde (34d), anisaldehyde (34e), vanillin (34f), methyl 3-aminocrotonate (33a), and trans-cinnamaldehyde (34g) were obtained from Fluka (Ronkonoma, NY). o-Nitrobenzaldehyde (34h), m-nitrobenzaldehyde (34i), p-nitrobenzaldehyde (34j), ethyl 3-aminocrotonate (33b), phenylpropargylaldehyde (34k), α,α,α -trifloro-o-tolualdehyde (34l), benzyl acetoacetate (35b), 2-methoxyethyl acetoacetate (35c), piperonal (34m), tetrachloro-1,4-benzoquinone (37), valeryl chloride, and ethyl benzoylacetate (35d) were from Aldrich (St. Louis, MO). Hydrocinnamaldehyde (34n) was from Eastman (Rochester, NY), and (±)-citronellal (34o) was from ICN (Plainview, NY). Compound 4 was from Tocris-Cookson (St. Louis, MO), and (S)-(4-nitrobenzyl)-6-thioguanosine was from Sigma (St. Louis, MO). [3H]PN200,110 (isradipine) was from DuPont NEN (Boston, MA). Compound 32 was prepared according to the literature.40 All other materials were obtained from commercial sources.
Synthesis.
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and spectra were taken in DMSO-d6 or CHCl3-d. Chemical-ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer, and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Atlantic Microlab Inc. (Norcross, GA). All melting points were determined with a Unimelt capillary melting point apparatus (Arthur H. Thomas Co., Philadelphia, PA) and were uncorrected.
General Procedure for Preparation of 1,4-Dihydropyridine-3,5-dicarboxylate Esters (36; 1–3, 5–8, 11, 16, 17, 20–28).
Equimolar amounts (0.5 mmol) of the appropriate 3-amino-2-butenoate ester (33a,b), aldehyde (34a–o), and 3-ketopropionate ester (35a–e) derivative were dissolved in 5 mL of absolute ethanol. The solution was sealed in a glass tube and heated to 100 °C for at least 24 h, and at most 72 h. The solvent was then evaporated, and products were purified either by crystallization, column chromatography (silica 60; 220–440 mesh; Fluka, CH; 20% ethyl acetate-80% petroleum ether 35–60) or preparative TLC (silica 60; 1000 μm; Analtech, DE; 20% ethyl acetate-80% petroleum ether 35–60). From the moment the reactants were sealed into the glass tube, all procedures were performed under nitrogen and low-light conditions to prevent oxidation of the products. The products were shown to be homogeneous by TLC.
3-Methyl 5-ethyl 2,4,6-trimethyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (1):
1H NMR (CHCl3-d) δ 0.98 (d, 3H, 4-CH3, J = 6.7 Hz), 1.31 (t, 3H, 5-methyl, J = 7.2 Hz), 2.28 (s, 6H, 2- and 6-CH3), 3.73 (s, 3H, 3-methyl), 3.83 (q, 1H, H-4, J = 6.4 Hz); 4.13–4.26 (m, 2H, 5-methylene), 5.51 (wide, 1H, H-1).
3-Methyl 5-(2-methoxyethyl) 2,4,6-trimethyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (2):
1H NMR (CHCl3-d) δ 0.99 (d, 3H, 4-CH3, J = 6.7 Hz), 2.28 (s, 6H, 2- and 6-CH3), 3.41 (s, 3H, 5-methoxy), 3.66 (t, 2H, 5-(2-methylene), J = 4.9 Hz), 3.73 (s, 3H, 3-methyl), 3.85 (q, 1H, H-4, J = 6.5 Hz), 4.22–4.38 (m, 2H, 5-(1-methylene)), 5.54 (wide, 1H, H-1).
3-Methyl 5-benzyl 2,4,6-trimethyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (3):
1H NMR (CHCl3-d) δ 0.98 (d, 3H, 4-CH3, J = 5.0 Hz), 2.28 (s, 6H, 2- and 6-CH3), 3.72 (s, 3H, 3-methyl), 3.89 (q, 1H, H-4, J = 6.5 Hz), 5.21 (q, 2H, 5-methylene, J = 14.8 Hz), 5.54 (wide, 1H, H-1), 7.30–7.41 (m, 5H, 5-phenyl).
3-Methyl 5-ethyl 2,6-dimethyl-4-ethyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (5):
1H NMR (DMSO-d6) δ 0.63 (t, 3H, 4-methyl, J = 7.4 Hz), 1.18 (t, 3H, 5-methyl, J = 7.1 Hz), 2.19 (s, 6H, 2- and 6-CH3), 3.58 (s, 3H, 3-methyl), 3.73 (t, 1H, H-4, J = 5.4 Hz), 4.00–4.13 (m, 4H, 4- and 5-methylene).
3-Methyl 5-ethyl 2,6-dimethyl-4-propyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (6):
1H NMR (DMSO-d6) δ 0.77 (t, 3H, 4-methyl, J = 6.7 Hz), 1.12 (wide, 4H, 4a- and 4b-methylene), 1.19 (t, 3H, 5-methyl, J = 7.2 Hz), 2.19 (s, 6H, 2- and 6-CH3), 3.59 (s, 3H, 3-methyl), 3.76 (t, 1H, H-4, J = 5.2 Hz), 4.07 (q, 2H, 5-methylene, J = 6.6 Hz).
3-Methyl 5-ethyl 2,6-dimethyl-4-(2(R,S),6-dimethylhexen-5-yl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (7):
1H NMR (CHCl3-d) δ 0.91 (double d, 3H, 4-(2-CH3), J1 = 5.8 Hz, J2 = 24.0 Hz), 1.30 (m, 7H), 1.63 (d, 6H, 4-(6- and 7-CH3), J = 25.4 Hz), 2.28 (d, 6H, 2- and 6- CH3, J = 7.8 Hz), 3.71 (s, 3H, 3-methyl), 3.97 (t, 1H, H-4, J = 7.2 Hz), 4.11–4.24 (m, 2H, 5-methylene), 5.07 (t, 1H, 4-(2-methyne), J = 6.5 Hz), 5.71 (wide, 1H, H-1).
3-Methyl 5-ethyl 2,6-dimethyl-4-phenyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (8):
1H NMR (DMSO-d6) δ 1.11 (t, 3H, 5-methyl, J = 6.4 Hz), 2.23 (s, 6H, 2- and 6-CH3), 3.51 (s, 3H, 3-methyl), 3.97 (m, 2H, 5-methylene), 4.84 (s, 1H, H-4), 7.08–7.20 (m, 5H, 4-phenyl).
3,5-Diethyl 2,6-dimethyl-4-(3-nitrophenyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (11):
1H NMR (CHCl3-d) δ 1.23 (t, 6H, 3- and 5-methyl, J = 6.9 Hz), 2.38 (s, 6H, 2- and 6-CH3), 4.02–4.18 (m, 4H, 3- and 5-methylene), 5.10 (s, 1H, H-4), 5.68 (wide, 1H, H-1), 7.38 (t, 1H, H-5′, J = 8.0 Hz), 7.65 (d, 1H, H-6′, J = 7.8 Hz), 8.02 (d, 1H, H-4′, J = 7.4 Hz), 8.14 (s, 1H, H-2′).
3-Methyl 5-ethyl 2,6-dimethyl-4-(4-nitrophenyl)-1,4-(R,S)-dihydropyridine-3,5-dicarboxylate (16):
1H NMR (DMSO-d6) δ 1.11 (t, 3H, 5-methyl, J = 7.2 Hz), 2.26 (s, 6H, 2- and 6-CH3), 3.52 (s, 3H, 3-methyl), 4.00 (m, 2H, 5-methylene), 4.96 (s, 1H, H-1), 7.39 (d, 2H, H-2′ and H-6′, J = 8.7 Hz), 8.09 (d, 2H, H-3′ and H-5′, J = 8.7 Hz).
3-Methyl 5-ethyl 2,6-dimethyl-4-(2-α,α,α-trifluoromethylphenyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (17):
1H NMR (DMSO-d6) δ 1.05 (m, 3H, 5-methyl), 2.21 (s, 6H, 2- and 6-CH3), 3.44 (s, 3H, 3-methyl), 3.84–4.10 (m, 2H, 5-methylene), 5.38 (s, 1H, H-4), 7.31 (t, 1H, J = 7.3 Hz), 7.51 (m, 3H, 4-phenyl).
3-Methyl 5-ethyl 2,6-dimethyl-4-(4-methoxyphenyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (20):
1H NMR (CHCl3-d) δ 1.23 (t, 3H, 5-methyl, J = 7.8 Hz), 2.34 (s, 6H, 2- and 6-CH3), 3.65 (s, 3H, 4′-OCH3), 3.76 (s, 3H, 3-methyl), 4.07–4.14 (m, 2H, 5-methylene), 4.94 (s, 1H, H-4), 5.57 (wide, 1H, H-1), 6.76 (d, 2H, H-2′ and H-6′, J = 8.5 Hz), 7.20 (d, 2H, H-3′ and H-5′, J = 8.6 Hz).
3-Methyl 5-ethyl 2,6-dimethyl-4-(4-hydroxy-3-methoxyphenyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate
(21): 1H NMR (DMSO-d6) δ 1.13 (t, 3H, 5-methyl, J = 7.4 Hz), 2.21 (s, 6H, 2- and 6-CH3), 3.52 (s, 3H, 3′-OCH3), 3.66 (s, 3H, 3-methyl), 3.96–4.01 (m, 2H, 5-methylene), 4.74 (s, 1H, H-4), 6.49 (t, 1H, H-6′, J = 3.9 Hz), 6.58 (d, 1H, H-5′, J = 7.9 Hz), 6.66 (s, 1H, H-2′).
3-Methyl 5-ethyl 2,6-dimethyl-4-[3,4-(methylenedioxy)-phenyl]-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (22):
1H NMR (CHCl3-d) δ 1.24 (t, 3H, 5-methyl, J = 7.3 Hz), 2.33 (s, 6H, 2- and 6-CH3), 3.66 (s, 3H, 3-methyl), 3.73 (s, 1H, H-4), 4.09–4.22 (m, 2H, 5-methylene), 4.92 (s, 2H, 3′,4′-methylenedioxy), 5.57 (wide, 1H, H-1), 5.89 (s, 1H, H-2′), 6.66 (d, 1H, H-5′, J = 8.1 Hz), 6.75 (d, 1H, H-6′, J = 7.9 Hz).
3-Methyl 5-ethyl 2,6-dimethyl-4-(2-phenyl)ethyl-1,4-(R,S)-dihydropyridine-3,5-dicarboxylate (23):
1H NMR (CHCl3-d) δ 1.25–1.55 (m, 3H, 5-methyl), 1.63–1.71 (m, 2H, 4-(α-methylene)), 2.31 (s, 6H, 2- and 6-CH3), 2.51–2.57 (m, 2H, 4-(β-methylene)), 3.73 (s, 3H, 3-methyl), 4.05 (t, 1H, H-4, J = 5.6 Hz), 4.12–4.27 (m, 2H, 5-methylene), 5.58 (wide, 1H, H-1), 7.15 (d, 2H, H-2′ and H-6′, J = 6.4 Hz), 7.24 (t, 3H, H-3′, and H-4′ and H-5′, J = 6.8 Hz).
3-Methyl 5-ethyl 2,6-dimethyl-4-(trans-2-phenylvinyl)-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (24):
1H NMR (CHCl3-d) δ 1.31 (t, 3H, 5-methyl, J = 7.1 Hz), 2.34 (s, 6H, 2- and 6-CH3), 3.74 (s, 3H, 3-methyl), 4.14–4.28 (m, 2H, 5-methylene), 4.63 (d, 1H, H-4, J = 5.4 Hz), 5.60 (wide, 1H, H-1), 6.19 (t, 1H, 4-(H-1 vinylidene), J = 6.0 Hz), 7.18 (d, 1H, 4-(H-2 vinylidene), J = 6.6 Hz), 7.24–7.34 (m, 5H, 4-phenyl).
3-Methyl 5-ethyl 2,6-dimethyl-4-(phenylethynyl)-1,4-(R,S)-dihydropyridine-3,5-dicarboxylate (25):
1H NMR (CHCl3-d) δ 1.35 (t, 3H, 5-methyl, J = 7.1 Hz), 2.36 (s, 6H, 2- and 6-CH3), 3.80 (s, 3H, 3-methyl), 4.23–4.31 (m, 2H, 5-methylene), 4.99 (s, 1H, H-4), 5.71 (wide, 1H, H-1), 7.24 (t, 3H, H-3′, and H-4′, and H-5′, J = 3.2 Hz), 7.36 (d, 2H, H-2′ and H-6′, J = 3.6 Hz).
3,5-Diethyl 2,4-dimethyl-6-butyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (26):
1H NMR (CHCl3-d) δ 0.92–0.98 (m, overlap, 6H, 6-(4-CH3) and 4-CH3, J = 7.0 Hz), 1.27 (t, 6H, 3- and 5-methyl, J = 7.0 Hz), 1.51 (m, 4H, 6-(2- and 3-CH2)), 2.30 (s, 3H, 2-CH3), 2.52–2.76 (m, 2H, 6-(1-CH2)), 3.85 (q, 1H, H-4, J = 7.0 Hz), 4.18 (m, 4H, 3- and 5-methylene), 5.53 (wide, 1H, H-1).
3,5-Diethyl 2,4-dimethyl-6-phenyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (27):
1H NMR (CHCl3-d) δ 0.89 (t, 3H, 3-methyl, J = 7.4 Hz), 1.14 (d, 3H, 5-methyl, J = 7.4 Hz), 1.31 (t, 3H, J = 7.0 Hz), 2.3 (s, 3H, CH3), 3.94 (m, 3H), 4.23 (m, 2H), 7.28–7.41 (m, 5H, C6H5).
3,5-Diethyl 2-methyl-4-(trans-2-phenylvinyl)-6-phenyl-1,4(R,S)-dihydropyridine-3,5-dicarboxylate (28):
1H NMR (CHCl3-d) δ 0.92 (t, 3H, 5-methyl, J = 7.0 Hz), 1.32 (t, 3H, 3-methyl, J = 7.0 Hz), 2.37 (s, 3H, 2-CH3), 3.94 (q, 3H, 5-methylene, J = 6.7 Hz), 4.22, (q, 3H, 3-methylene, J = 6.7 Hz), 4.76 (d, 1H, H-4, J = 6.2 Hz), 5.78 (wide, 1H, H-1), 6.30–6.38 (m, 2H, CH=CH), 7.20–7.44 (m, 10H, 2 x C6H5).
General Procedure for Oxidation of 1,4-Dihydropyri-dine-3,5-dicarboxylate Esters (38; 29, 30).
Equimolar amounts (0.25 mmol) of the 1,4-dihydropyridine-3,5-dicarboxylate ester (36; 1, 27) and tetrachloro-1,4-benzoquinone (37) in tetrahydrofuran (2 mL) were mixed and refluxed for up to 4 h. The solvent was then evaporated, and products were purified by preparative TLC (silica 60; 1000 μm; Analtech, DE; 20% ethyl acetate–80% petroleum ether 35–60).
3-Methyl 5-ethyl 2,4,6-trimethylpyridine-3,5-dicarboxylate (29):
1H NMR (DMSO-d6) δ 1.27 (t, 3H, 5-methyl, J 6.9 Hz), 2.16 (s, 3H, 4-CH3), 2.39 (s, 6H, 2- and 6-CH3), 3.87 (s, 3H, 3-methyl), 4.36 (q, 2H, 5-methylene, J = 7.4 Hz).
3,5-Diethyl 2,4-dimethyl-6-phenyl-pyridine-3,5-dicarboxylate (30):
1H NMR (CHCl3-d) δ 1.00 (t, 3H, 3-methyl, J = 7.4 Hz), 1.43 (t, 3H, 5-methyl, J = 7.5 Hz), 2.37 (s, 3H,4-CH3), 2.62 (s, 3H, 2-CH3), 4.10 (q, 2H, 3-methylene, J = 7.4 Hz), 4.47 (q, 2H, 5-methylene, J = 7.4 Hz), 7.40–7.58 (m, 5H, C6H5).
Ethyl Valerylacetate (35e).
A solution of n-butyllithium (6.3 mL, 1.6 M in hexane) was slowly added to a solution of N-isopropylcyclohexylamine (1.41 g, 10 mmol) in dry THF (20 mL) at −5 °C. The mixture was stirred for 15 min and then cooled to −78 °C. Ethyl acetate (440 mg, 5 mmol) was added dropwise over a period of 5 min, followed by valeryl chloride (600 mg, 5 mmol). The reaction mixture was allowed to stir an additional 10 min, at which point it was quenched with 5 mL of 20% HCl in water. The organic layer was separated off, and the aqueous layer was extracted with 10 mL of diethyl ether twice. The combined organic phases were washed with a saturated sodium bicarbonate solution (10 mL × 2) and brine (5 mL × 2) and dried over anhydrous sodium sulfate. The solvent was evaporated and the residue purified by column chromatography (silica gel 60, eluted with ethyl acetate/petroleum ether, 1:9) to yield ethyl valerylacetate (772 mg, 90%): 1H NMR (CHCl3-d) δ 0.90 (t, 3H, CH3, J = 7.0 Hz), 1.20–1.65 (m, 7H, CH3 and 2 × CH2), 2.30 (t, 2H, COCH2, J = 7.0 Hz), 2.65 (s, 2H, COCH2CO), 4.15 (q, 2H, COOCH2, J = 7.0 Hz); MS (EI) 85 [CH3(CH2)3CO+], 172 (base), 57 [CH3-(CH2)3]+. The ethyl valerylacetate (35e) was then used in the procedure described above for the preparation of compound 26.
Pharmacology: Radioligand Binding Studies.
Binding of [3H]-(R)-N6-(phenylisopropyl)adenosine ([3H]-(R)-PIA) to A1 receptors from rat cerebral cortex membranes and of [3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-N-(ethylcarbamoyl)-adenosine ([3H]CGS 21680) to A2A receptors from rat striatal membranes was performed as described previously.30,31 Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in a preincubation of 30 min at 30 °C, and during the incubation with the radioligands.
Binding of [125I]-N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine ([125I]AB-MECA) to membranes prepared from HEK-293 cells stably expressing the human A3 receptor (Receptor Biology, Inc., Baltimore, MD) or to membranes prepared from CHO cells stably expressing the rat A3 receptor was performed as described.13,32 The assay medium consisted of buffer containing 50 mM Tris, 10 mM MgCl2, and 1 mM EDTA at pH 8.0. The glass incubation tubes contained 100 μL of the membrane suspension (0.3 mg of protein/mL, stored at −80 °C in the same buffer), 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of a solution of the proposed antagonist. Nonspecific binding was determined in the presence of 100 μM N6-(phenylisopropyl)adenosine (R-PIA).
Binding of [3H]isradipine to rat cerebral cortex membranes was performed essentially as described in Moody et al.36 Briefly, an aliquot of a membrane suspension corresponding to 100 μg of protein was incubated in a total volume of 0.5 mL of Tris buffer (50 mM, pH 7.4, 25 °C) for 1 h in the presence of 0.1 nM [3H]isradipine and varying concentrations of the dihydropyridine or pyridine derivatives. Nonspecific binding was determined in the presence of 10 μM nitrendipine.
All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%.
Incubations were terminated by rapid filtration over What-man GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each.
At least five different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in the InPlot program (Graph-PAD, San Diego, CA), were converted to apparent Ki values using the Cheng–Prusoff equation41 and Kd values of 1.0, 14, and 0.13 nM for [3H]-(R)-PIA, [3H]CGS 21680, and [3H]isradipine, respectively, and 0.59 nM for binding of [125I]-AB-MECA at human A3 receptors, respectively.
Inhibition of Adenylyl Cyclase Activity.
Adenylyl cyclase activity was determined in membranes from CHO cells stably expressing the rat A3 receptor, prepared as reported previously.32 [α−32P]ATP was added to a membrane suspension containing 1 μM forskolin to stimulate adenylyl cyclase. The reaction was terminated by addition of a stop buffer containing 20 000 cpm/mL [3H]cyclic AMP. Cyclic AMP was isolated by consecutive chromatography over columns of Dowex 50 ion exchange resin and alumina. Maximal stimulation of adenylyl cyclase activity was obtained in the presence of 1 μM forskolin (circa 6–8-fold), and maximal inhibition of forskolin-stimulated adenylyl cyclase activity (40–50% residual activity from the stimulated level) was obtained in the presence of 10 μM IB-MECA. Statistical significance was calculated in a t-test, with 4 degrees of freedom at a significance level of 98%.
Interaction of 1,4-Dihydropyridines with the NBI-Sensitive Nucleoside Transporter Protein.
Competition of 1,4-dihydropyridines for binding of [3H]NBI to the NBI-sensitive nucleoside transporter protein was carried out by the slightly modified procedure of Marangos et al.42 Rat forebrain membranes were incubated for 30 min at 23 °C with 0.7 nM [3H]NBI and varying concentrations of competitor in Tris buffer (50 mM, pH 7.4) in a total of 500 μL. Then 5 μM (S)-(4-nitrobenzyl)-6-thioguanosine was added to determine non-specific binding. Nonspecific binding did not exceed 15% of total binding. A Kd value of 0.15 nM was used for the calculation of the Kivalues.42
Acknowledgment.
We thank Gilead Sciences (Foster City, CA) and the Cystic Fibrosis Foundation (Silver Spring, MD) for financial support and Dr. Bernhard Witkop (NIH) for helpful discussions. We thank Dr. Pat Towers of Amersham (Cardiff, U.K.) and Dr. Garth Brown of DuPont NEN (North Billerica, MA) for providing samples of [125I]AB-MECA.
Abbreviations:
- [125I]AB-MECA
[125I]-N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- Bay K 8644
3-methyl 2,6-dimethyl-5-nitro-4-(2-(α,α,α-trifluoromethyl)phenyl)-1,4-dihydropyridine-3-carboxylate
- CGS 21680
2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)-adenosine
- CHA
N6-cyclohexyladenosine
- CHO cells
Chinese hamster ovary cells
- DMSO
dimethyl sulfoxide
- HEK cells
human embryonic kidney cells
- IB-MECA
N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine
- K i
equilibrium inhibition constant
- NBI
S-(4-nitrobenzyl)-6-thioinosine
- NECA
(N-ethylcarbamoyl)adenosine
- PAF
1-O-hexadecyl/octadecyl-2-O-acetyl-sn-glycero-3-phosphorylcholine, platelet activating factor
- (R)-PIA
(R)-N6-(phenylisopropyl)adenosine
- SAR
structure–activity relationship
- Tris
tris(hydroxymethyl)aminomethane
- XAC
8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine, xanthine amine congener
References
- (1).Triggle DJ Drugs acting on ion channels and membranes. In Comprehensive Medicinal Chemistry; Emmett JC, Ed.; Pergamon Press: London, 1985; Vol. 3, pp 1047–1099. [Google Scholar]
- (2).Borchard U Calcium antagonists in comparison: view of the pharmacologist. J. Cardiovasc. Pharmacol 1994, 24, Suppl 2, S85–91. [PubMed] [Google Scholar]
- (3).Coburn RA; Wierzba M; Suto MJ; Solo AJ; Triggle AM; Triggle DJ. 1,4-Dihydropyridine antagonist activities at the calcium channel: a quantitative structure-activity relationship approach. J. Med. Chem 1988, 31, 2103–2107. [DOI] [PubMed] [Google Scholar]
- (4).Wetzel JM; Miao SW; Forray C; Borden LA; Branchek TA; Gluchowski C Discovery of alpha 1a-adrenergic receptor antagonists based on the L-type Ca2+ channel antagonist niguldipine. J. Med. Chem 1995, 38, 1579–1581. [DOI] [PubMed] [Google Scholar]
- (5).Sunkel CE; de Casa-Juana MF; Santos L; Gomez MM; Villarroya M; Gonzalez-Morales MA; Priego JG; Ortega MP 4-Alkyl-1,4-dihydropyridines derivatives as specific PAF-acether antagonists. J. Med. Chem 1990, 33, 3205–3210. [DOI] [PubMed] [Google Scholar]
- (6).Lopez MG; Fonteriz RI; Gandia L; de la Fuente M; Villarroya M; Garcia-Sancho J; Garcia AG The nicotinic acetylcholine receptor of the bovine chromaffin cell, a new target for dihydropyridines. Eur. J. Pharmacol 1993, 247, 199–207. [DOI] [PubMed] [Google Scholar]
- (7).Hu PS; Lindgren E; Jacobson KA; Fredholm BB Interaction of dihydropyridine calcium channel agonists and antagonists with adenosine receptors. Pharmacol. Toxicol 1987, 61, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ismail NA; Shaheen AA; El-Sawalhi MM; Megahed YM Effect of calcium channel antagonists in modifying the inhibitory influence of adenosine on insulin secretion. Arzneim.- Forsch./Drug Res 1995, 45, 865–868. [PubMed] [Google Scholar]
- (9).Jacobson KA; van Galen PJM; Williams M Adenosine receptors - pharmacology, structure activity relationships, and therapeutic potential. J. Med. Chem 1992, 35, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Daly JW; Hong O; Padgett WL; Shamim MT; Jacobson KA; Ukena D Non-xanthine heterocycles: activity as antagonists of A1- and A2-adenosine receptors. Biochem. Pharmacol 1988, 37, 655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Siddiqi SM; Ji XD; Melman N; Olah ME; Jain R; Evans P; Glashofer M; Padgett WL; Cohen LA; Daly JW; Stiles GL; Jacobson KA A survey of non-xanthine derivatives as adenosine receptor ligands. Nucleosides Nucleotides 1996, 15, 693–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).van Rhee AM; Siddiqi SM; Melman N; Shi D; Padgett WL; Daly JW; Jacobson KA Tetrahydrobenzothiophenone derivatives as a novel class of adenosine receptor antagonists. J. Med. Chem 1996, 39, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ji XD; Melman N; Jacobson KA Interactions of flavonoids and other phytochemicals with adenosine receptors. J. Med. Chem 1996, 39, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jacobson KA; Kim HO; Siddiqi SM; Olah ME; Stiles G; von Lubitz DKJE A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future 1995, 20, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Beaven MA; Ramkumar V; Ali H Adenosine-A3 receptors in mast-cells. Trends Pharmacol. Sci 1994, 15, 13–14. [DOI] [PubMed] [Google Scholar]
- (16).von Lubitz DKJE; Lin RCS; Popik P; Carter MF; Jacobson KA Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol 1994, 263, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).MacKenzie WM; Hoskin DW; Blay J Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adeno-carcinoma cells through an A3 receptor. Cancer Res. 1994, 54, 3521–3526. [PubMed] [Google Scholar]
- (18).Kohno Y; Sei Y; Koshiba M; Kim HO; Jacobson KA Induction of apoptosis in HL-60 human promyelocytic leukemia cells by selective adenosine A3 receptor agonists. Biochem. Biophys. Res. Commun 1996, 219, 904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jacobson KA; Nikodijevic O; Shi D; Gallo-Rodriguez C; Olah ME; Stiles GL; Daly JW A role for central A3-adenosine receptors: Mediation of behavioral depressant effects. FEBS Lett. 1993, 336, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim HO; Ji XD; Siddiqi SM; Stiles GL; Jacobson KA 2-Substitution of N6-benzyl adenosine-5′-uronamides enhances selectivity for A3 receptors, J. Med. Chem 1994, 37, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim HO; Ji XD; Melman N; Olah ME; Stiles GL; Jacobson KA Structure-activity relationships of 1,3-dialkylxanthine derivatives at rat A3 adenosine receptors. J. Med. Chem 1994, 37, 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhou QY; Li CY; Olah ME; Johnson RA; Stiles GL; Civelli O Molecular cloning and characterization of an adeno-sine receptor - The A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Linden J; Taylor HE; Robeva AS; Tucker AL; Stehle JH; Rivkees SA; Fink JS; Reppert SM Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol. Pharmacol 1993, 44, 524–532. [PubMed] [Google Scholar]
- (25).Ji XD; von Lubitz DKJE; Olah ME; Stiles GL; Jacobson KA Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev. Res 1994, 33, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stout DM; Myers AI Recent advances in the chemistry of dihydropyridines. Chem. Rev 1982, 82, 223–243. [Google Scholar]
- (27).Singer A; McElvain SM 2,6-Dimethylpyridine. Org. Synth 1934, 14, 30–33. [Google Scholar]
- (28).Rathke MW; Deitch J The reaction of lithium ester enolates with acid chlorides. A convenient procedure for the preparation of β-keto esters. Tetrahedron Lett. 1971, 31, 2953–2956. [Google Scholar]
- (29).Braude EA; Hannah J; Linstead R Hydrogen Transfer. Part XVI. Dihydrides of nitrogenous heterocycles as hydrogen donors. J. Chem. Soc 1960, 3249–3257. [Google Scholar]
- (30).Schwabe U; Trost T Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol 1980, 313, 179–187. [DOI] [PubMed] [Google Scholar]
- (31).Jarvis MF; Schutz R; Hutchison AJ; Do E; Sills MA; Williams M [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther 1989, 251, 888–893. [PubMed] [Google Scholar]
- (32).Olah ME; Gallo-Rodriguez C; Jacobson KA; Stiles GL [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- (33).Triggle DJ 1,4-Dihydropyridine activators and antagonists: structural and functional distinctions. Trends Pharmacol. Sci 1989, 10, 507–511. [DOI] [PubMed] [Google Scholar]
- (34).Meyer H; Bossert F; Wehinger E; Stoepel K; Vater W Synthesis and comparative pharmacological studies of 1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)pyridine-3,5-dicarboxylates with non-identical ester functions. Arzneim.-Forsch 1981, 31, 407–9. [PubMed] [Google Scholar]
- (35).Höllt V; Kouba M; Dietel M; Vogt G Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem. Pharmacol 1992, 43, 2601–2608. [DOI] [PubMed] [Google Scholar]
- (36).Moody EJ; Harris B; Hoehner P; Skolnick P Inhibition of [3H]isradipine binding to L-type calcium channels by the optical isomers of isoflurane. Lack of stereospecificity. Anesthesiology 1994, 81, 124–128. [DOI] [PubMed] [Google Scholar]
- (37).Striessnig J; Zernig G; Glossmann H Ca2+ antagonist receptor sites on human red blood cell membranes. Eur. J. Pharmacol 1985, 108, 329–330. [DOI] [PubMed] [Google Scholar]
- (38).Sweetnam PM; Caldwell L; Lancaster J; Bauer C; Mc-Millan B; Kinnier WJ; Price CH The role of receptor binding in drug discovery. J. Nat. Prod 1993, 56, 441–455. [DOI] [PubMed] [Google Scholar]
- (39).Gerlach M; Riederer P; Youdim M Neuroprotective therapeutic strategies: Comparison of experimental and clinical results. Biochem. Pharmacol 1995, 50, 1–16. [DOI] [PubMed] [Google Scholar]
- (40).Gessner W; Brossi A; Shen R-S; Abell CW Synthesis and dihydropteridine reductase inhibitory effects of potential metabolites of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Med. Chem 1985, 28, 311–317. [DOI] [PubMed] [Google Scholar]
- (41).Cheng YC; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (42).Marangos PJ; Patel J; Clark-Rosenberg R; Martino AM Nitrobenzylthio-inosine binding as a probe for the study of adenosine uptake sites in brain. J. Neurochem 1982, 39, 184–191. [DOI] [PubMed] [Google Scholar]
- (43).Deckert J; Bereznai B; Hennemann A; Gsell W; Götz M; Fritze J; Riederer P Nimodipine inhibits [3H]nitrobenzyl-thioinosine binding to the adenosine transporter in human brain. Eur. J. Pharmacol 1993, 238, 131–133. [DOI] [PubMed] [Google Scholar]