

Abstract
A broad screening of phytochemicals has demonstrated that certain flavone and flavonol derivatives have a relatively high affinity at A3 adenosine receptors, with Ki values of ≥1 μM (Ji et al. J. Med. Chem. 1996, 39, 781–788). We have further modified the flavone structure to achieve a degree of selectivity for cloned human brain A3 receptors, determined in competitive binding assays versus [125I]AB-MECA [N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide)]. Affinity was determined in radioligand binding assays at rat brain A1 and A2A receptors using [3H]-N6-PIA ([3H]-(R)-N6-phenylisopropyladenosine) and [3H]CGS21680 [[3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine], respectively. The triethyl and tripropyl ether derivatives of the flavonol galangin, 4, had Ki values of 0.3–0.4 μM at human A3 receptors. The presence of a 5-hydroxyl group increased selectivity of flavonols for human A3 receptors. The 2′,3,4′,7-tetraethyl ether derivative of the flavonol morin, 7, displayed a Ki value of 4.8 μM at human A3 receptors and was inactive at rat A1/A2A receptors. 3,6-Dichloro-2′-(isopropyloxy)-4′-methylflavone, 11e, was both potent and highly selective (~200-fold) for human A3 receptors (Ki = 0.56 μM). Among dihydroflavonol analogues, the 2-styryl instead of the 2-aryl substituent, in 15, afforded selectivity for human A3 vs rat A1 or A2A receptors. The 2-styryl-6-propoxy derivative, 20, of the furanochromone visnagin was 30-fold selective for human A3 receptors vs either rat A1 or A2A receptors. Several of the more potent derivatives effectively antagonized the effects of an agonist in a functional A3 receptor assay, i.e. inhibition of adenylyl cyclase in CHO cells expressing cloned rat A3 receptors. In conclusion, these series of flavonoids provide leads for the development of novel potent and subtype selective A3 antagonists.
Introduction
The principal mechanism by which caffeine and other alkylxanthines act as physiological stimulants is by blocking the effects of the ubiquitous neuromodulator adenosine,1 which acts as a “local hormone” within a given organ. Adenosine is produced locally in response to increased activity or stress to the system. This feedback mechanism allows the organ to compensate for the stress by decreasing energy demand (depressant activity) and increasing oxygen supply (e.g. by vasodilation).2
Extracellular adenosine activates receptors of the A1 and A2A subtypes, which depresses the action of the heart, brain, kidneys, and the immune system, as well as other systems.3 Recently, the novel A3 subtype of adenosine receptors has been cloned,4,5 and its pharmacological6–9 and regulatory characteristics10 have been studied. Activation of A3 receptors requires relatively high, i.e. pathological, concentrations of adenosine; the Ki value of adenosine has been estimated as ~1 μM at A3 receptors versus 10 and 30 nM at A1 and A2A receptors, respectively.11 Thus the physiological role of A3 receptors may be very different from that of the A1 and A2A subtypes. A3 agonists induce hypotension and promote the release of inflammatory mediators from mast cells.8 We have introduced the first selective agonist ligands for the A3 subtype, including the agonist IB-MECA [N6-(3-iodobenzyl)adenosine-5′-(N-methyluronamide)].9,12 IB-MECA has been shown to elicit locomotor depression,9 and following chronic administration it acts as a cerebroprotectant in stroke and seizure models.13
In principle, a selective A3 adenosine receptor antagonist should serve as a cerebroprotective, antiasthmatic, or antiinflammatory agent.6,7,11,13 Previously, we have successfully designed specific antagonists for A1 and A2A receptors by chemically modifying the structures of caffeine and other xanthines.3 However, at A3 receptors xanthines are of particularly low affinity14 and have not yet provided satisfactory chemical leads for antagonists.15 Consequently, we have searched diverse chemical libraries for lead structures for developing A3 antagonists.16–19 A number of classes of non-xanthine adenosine antagonists have already been reported, including various nitrogen heterocycles and several classes of nonnitrogen heterocycles.16 We recently reported that tetrahydrobenzothiophenones, e.g. ethyl 3-(benzylthio)-4,5,6,7-tetrahydro-4-oxobenzo[c]thiophene-1-carboxylate (BTH4), bind to A1 and A2A adenosine receptors in the micromolar range.17 One member of this series, 4,5-dihydro-1-(methylthio)benzo[c]thiophene-3-carboxylic acid hydrazide, I (Figure 1), bound with slight selectivity for rat A3 receptors. Siddiqi et al.16 have shown that folic acid (II), a pyridopyrimidinone (III), cytochalasin H (IV), 11-hydroxytetracarbazolenine (V), the adenosine uptake inhibitor dipyridamole (VI), certain sulfonylpiperazines (e.g. the protein kinase C inhibitor HA-100, VII), and a number of other hetero-cyclic substances displace radioligand from adenosine receptors with low affinity but with apparent selectivity for rat A3 receptors. Recently, a broad screening of phytochemicals has demonstrated that certain flavones and flavonols, members of the larger class of phenolic natural products known as flavonoids, have a relatively high affinity at A3 adenosine receptors, with Ki values in the micromolar range, close to that of the natural ligand adenosine.19 The affinity of some flavones at A1 and A2A receptors was also noted by Ares et al.20 In the present study we have investigated the structure—activity relationships of flavonoids at adenosine receptors in an effort to develop novel A3 adenosine antagonists. Selected analogues have been tested in a functional A3 receptor assay, i.e. inhibition of adenylyl cyclase in Chinese hamster ovary (CHO) cells expressing cloned rat A3 receptors.4
Figure 1.

Substances that have selectivity in binding to rat A3 vs A1 and A2A adenosine receptors. Ki values shown are in competition for [125I]AB-MECA binding in membranes from CHO cells expressing the cloned rat A3 receptors.16,17
Results
Synthesis.
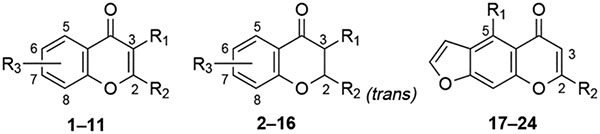
The structures of the flavonoid derivatives (including flavonols, 1–10; flavones, 11; flavanones, 12 and 13; dihydroflavonols, 14–16; and furylchromones, 17–24) examined in the present study are shown in Table 1, and the chemical characterization is summarized in Table 2. In our previous study,19 the flavonol galangin (3,5,7-trihydroxyflavone), 1, was shown to bind to adenosine receptors nonselectively, while its trimethyl ether derivative, 2, was shown to have enhanced affinity at A3 receptors. Thus, we synthesized and compared in receptor binding assays the series of alkyl ethers of galangin, 2–4, and other 3-substituted flavone derivatives.
Table 1.
|
|||||||
|---|---|---|---|---|---|---|---|
| Ki (μM) or % inhibitionc | rA1/hA3 | ||||||
| compound | R1 | R2 | R3 | rA1 | rA2A | hA3 | |
| 1f (galangin) | OH | Ph | 5,7-(OH)2 | 0.863 ± 0.092 | 0.966 ± 0.164 | 3.15 ± 0.85 | 0.27 |
| 2f (MRS928) | OMe | Ph | 5,7-(OMe)2 | 0.509 ± 0.049 | 6.45 ± 1.48 | 1.21 ± 0.30 | 0.42 |
| 3a (MRS1041) | OEt | Ph | 5,7-(OEt)2 | 0.603 ± 0.022 | 3.31 ± 1.24 | 0.364 ± 0.067 | 1.7 |
| 3b (MRS1093) | OEt | Ph | 5-OH-7-OEt | 1.89 ± 0.56 | 63.8 ± 18.8 | 0.748 ± 0.402 | 2.5 |
| 4 (MRS1042) | OPr | Ph | 5,7-(OPr)2 | 1.10 ± 0.20 | 3.22 ± 1.31 | 0.317 ± 0.089 | 3.5 |
| 5af (morin) | OH | 2′,4′-(OH)2Ph | 5,7-(OH)2 | 13.8 ± 3.1 | 17.3 ± 4.1 | 34 ± 4% (10−4) | ~0.1 |
| 5b | CH2CH=C(CH3)2 | 2′,4′-(OH)2Ph | 5-OH-6-CH=CHCH-(CH3)2-7-OMe | 9.09 ± 0.91 | d (10−4) | 4.59 ± 1.69 | 2.0 |
| 6f (MRS923) | OMe | 2′,4′-(OMe)2Ph | 5,7-(OMe)2 | 27.6 ± 7.5 | 46.7 ± 2.7 | 2.65 ± 0.72 | 10 |
| 7 (MRS1063) | OEt | 2′,4′-(OEt)2Ph | 5-OH-7-OEt | d (10−4) | d (10−4) | 4.83 ± 1.40 | >40 |
| 8 (MRS1086) | OEt | 2′,4′-(OEt)2Ph | 5,7-(OEt)2 | 32.3 ± 9.1 | 26.8 | 7.27 ± 1.88 | 4.4 |
| 9 | OH | 2′,4′,6′-(OMe)3Ph | H | 7.32 ± 1.36 | d (10−4) | 50.1 ± 7.8 | 0.15 |
| 10 (MRS1072) | OH | CtCPh | 6-OCH3 | 34 ± 9% (10−4) | d (10−4) | 24.0 ± 9.7 | ~8 |
| 11af (flavone) | H | Ph | H | 3.28 ± 0.92 | 3.45 ± 1.16 | 16.9 ± 3.8 | 0.19 |
| 11b (MRS1132) | Cl | Ph | H | 2.48 ± 0.72 | 25.1 ± 5.4 | 11.5 ± 3.7 | 0.22 |
| 11c (MRS1131) | H | Ph | 6-Cl | 28 ± 6% (10−5) | d (10−4) | 32 ± 5% (10−4) | |
| 11d (MRS1088) | Cl | Ph | 6-Cl | 15% (10−4) | 54.5 ± 26.3 | 0.741 ± 0.325 | >100 |
| 11e (MRS1067) | Cl | 2′-i-PrO-4′-MePh | 6-Cl | 36 ± 12% (10−4) | 19 ± 9% (10−4) | 0.561 ± 0.129 | ~200 |
| 11f (MRS1089) | Cl | 2′,4′,6′-Me3Ph | 6-Cl | 37 ± 5% (10−4) | d (10−4) | 5.24 ± 0.52 | ~20 |
| 12 (flavanone) | H | Ph | H | 32.0 ± 4.8 | nd | 50.1 ± 27.1 | 0.64 |
| 13a | H | 2′-OHPh | H | 2.64 ± 0.56 | 17.6 ± 2.1 | 6.07 ± 1.43 | 0.43 |
| 13b | H | 4′-OHPh | H | 11.5 ± 3.7 | 35% (10−4) | 42.8 ± 10.5 | 0.27 |
| 13cf (sakuranetin) | H | 4′-OHPh | 5-OH-7-OMe | 8.18 ± 2.53 | 35.6 ± 13.0 | 3.40 ± 0.18 | 2.4 |
| 14 e | OH | 2′-OHPh | H | 91.9 ± 0.3 | d (10−4) | 27 ± 3% (10−4) | <1 |
| 15e (MRS1061) | OH | CH=CHPh | 6-OMe | d (10−4) | d (10−4) | 21.1 ± 9.9 | >8 |
| 16e (MRS1062) | OH | C≡CPh | 6-OMe | 50.3 ± 17.0 | d (10−4) | 8.17 ± 0.43 | 6.2 |
| 17 (visnagin) | OCH3 | CH3 | 36 ± 3% (10−4) | 42.4 ± 11.9 | 60.0 ± 17.8 | ~2 | |
| 18 | OCH3 | CHO | d (10−4) | 25 ± 8% (10−4) | 88.9 ± 27.1 | >2 | |
| 19 (MRS1065) | OCH3 | CH=CHPh | 32.6 ± 10.5 | 11.5 ± 1.3 | 8.28 ± 2.69 | 0.37 | |
| 20 (MRS1066) | OC2H5 | CH=CHPh | 35.6 ± 12.7 | 33.8 ± 14.8 | 1.16 ± 0.45 | 31 | |
| 21 (MRS1084) | O(CH2)2CH3 | CH=CHPh | 40.0 ± 3.5 | 49.0 | 3.95 ± 1.98 | 10 | |
| 22 (MRS1070) | OCH3 | CH=CHCH=CHPh | d (10−4) | d (10−4) | d (10−4) | ||
| 23 (MRS1071) | OC2H5 | CH=CHCH=CHPh | d (10−4) | 167 ± 92 | 45.5 ± 10.3 | >4 | |
| 24 (MRS1078) | OCH3 | CH=NPh | d (10−4) | d (10−4) | 9.18 ± 2.56 | >20 | |
Displacement of specific [3H]PIA binding in rat brain membranes, expressed as Ki ± SEM in μM (n = 3–5), or as a percentage of specific binding displaced at 100 μM.
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes, expressed as Ki ± SEM in μM (n = 3–6), or as a percentage of specific binding displaced at 100 μM.
Displacement of specific [125I]AB-MECA binding in the presence of 10 mM Mg2+ at human A3 receptors expressed in HEK293 cells, in membranes, expressed as Ki = SEM in μM (n = 2–5), or as a percentage of specific binding displaced at 100 μM.
Displacement of ≤10% of specific binding at the indicated concentrated (M).
2,3-trans.
Values from ref 19.
The systematic name for compounds 17–24 is different from the numbering scheme used in this table and in Results section.
nd = not determined.
Table 2.
Physical Characterization of Flavone, Flavanol, and Flavanone Derivatives
| compd no. | mp (°C) | MS | formula | anal. |
|---|---|---|---|---|
| 2 | 201 | 313 (CI) | C18H16O5·0.75H2O | C,H |
| 3a | 111–114 | 355 (FAB) | C21H22O5 | C,H |
| 3b | 100–101 | 337 (CI) | C19H18O5 | C,H |
| 4 | 90–93 | 397 (CI) | C24H28O5 | C,H |
| 6 | 153–156 | 373 (CI) | C20H20O7 | C,H |
| 7 | 122–123 | 415 (FAB) | C23H26O7 | C,H |
| 8 | 115–116 | 443 (CI) | C25H30O7 | C,H |
| 10 | oil | 293 (CI) | C18H12O4 | C,H |
| 11b | 121 | 256 (EI) | C15H9O2Cl·0.25EtOH | C,H |
| 11c | 170–172 | 257 (CI) | C15H9O2Cl·O.1H2O | C,H |
| 11d | 184–186 | 290 (EI) | C15H8O2Cl2 | a |
| 15 | 160–163 | 297 (CI) | C18H16O4·0.25H2O | C,H |
| 16 | 135–138 | 294 (EI) | C18H14O4 | a,b |
| 19 | 175–178 | 319 (CI) | C20H14O4·0.25H2O | C,H |
| 20 | 148–151 | 333 (CI) | C21H16O4 | C,H |
| 21 | 132 | 347 (CI) | C22H18O4 | C,H |
| 22 | 162–165 | 344 (EI) | C22H16O4·0.5H2O | C,H |
| 23 | 162–165 | 358 (EI) | C23H18O4 | C,H |
| 24 | 150–154 | 320 (CI) | C19H14NO4 | C,H |
High-resolution mass in FAB+ mode m/z determined to be within acceptable limits. 11d: calcd, 289.9901; found, 289.9893. 16: calcd, 294.0892; found, 294.0889.
C: calcd, 73.46; found, 74.30. H: calcd, 4.80; found, 5.27.
The alkylation of galangin was carried out using the appropriate alkyl iodide and potassium carbonate in refluxing acetone (Figure 2), providing the completely alkylated product, 3a or 4. Partial alkylation leaving the 5-position hydroxyl group unreacted, to provide 3b, was accomplished under less forceful conditions, e.g. alkyl bromide at room temperature. The O-alkylation of morin (2′,3,4′,5,7-pentahydroxyflavone) was complicated by an even lower reactivity of the 5-hydroxy group than in galangin, leading to tetra- rather than pentasubstitution (Figure 2). Thus, when ethyl sulfate was used the fully alkylated morin derivative, 8, was obtained, while reaction with ethyl iodide provided exclusively the tetraethyl derivative, 7. The position of the nonalkylated hydroxyl group of 3b and 7 was determined from the NMR chemical shift. The resonance for the 5-OH proton in flavone derivatives has been reported to appear at roughly 12 ppm.12 The downfield shift is apparently caused by the adjacent carbonyl to which it may form a hydrogen bond and thus a noncovalent six-membered ring. This proton signal is present in the spectra of 3b, 5a, and 7 but not 8.
Figure 2.
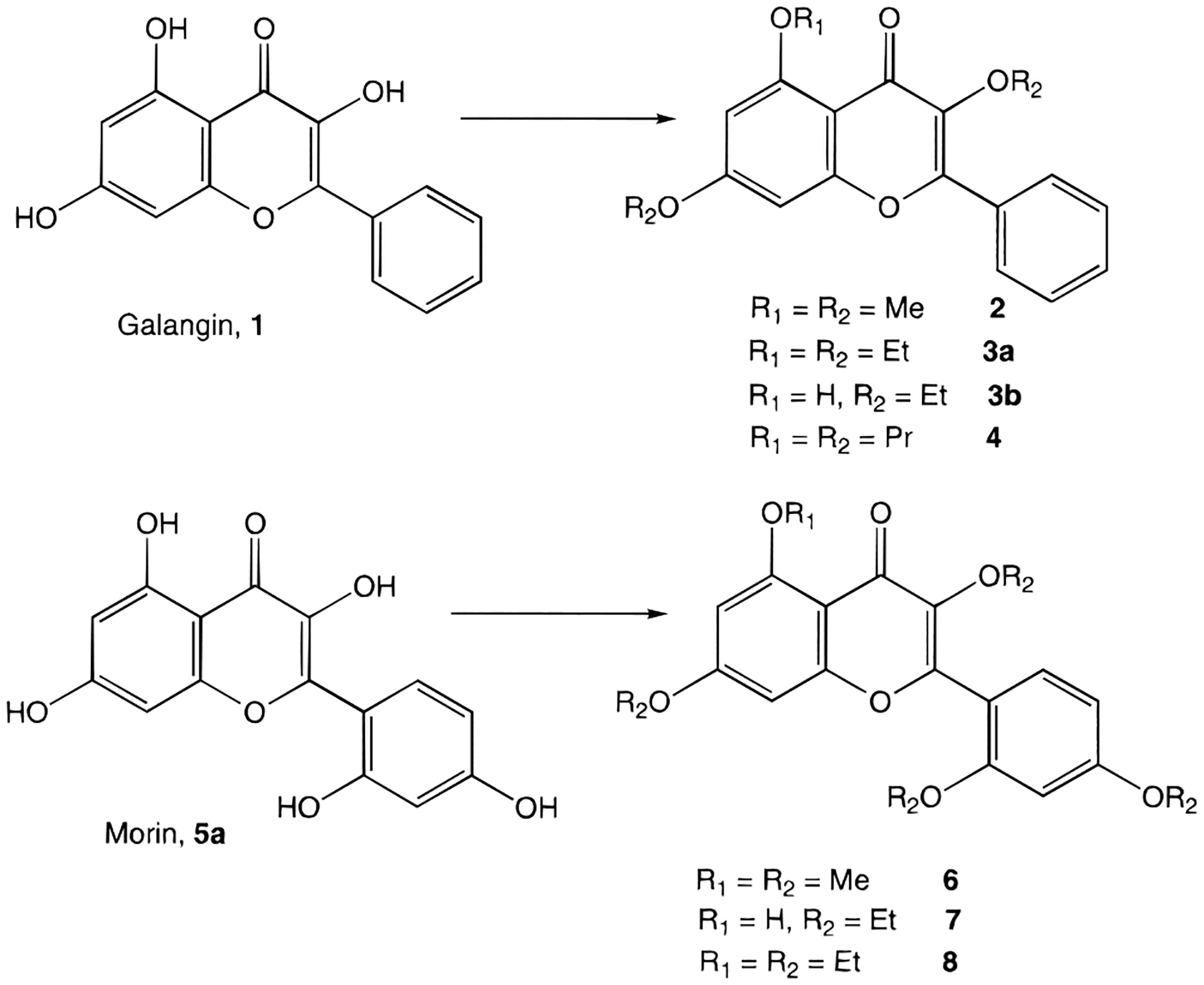
Synthesis of alkylated derivatives of galangin and morin. Reagents: 2, 3a, 4, and 7, R-I; 3b, Et-Br; 6 and 8, R2-SO4.
The synthetic routes to flavone derivatives have been investigated extensively and optimized.22–25 As shown in Figure 3, a synthetic route used to form the flavone ring system was via a condensation of a 2-hydroxyacetophenone derivative, 25, with the appropriate aldehyde, 26, which gave rise to the 2-substituent. The condensation resulted first in a trans-olefin, 27, which could be cyclized under basic oxidizing conditions to give either the 2,3-saturated dihydroflavonol (e.g. 3-hydroxyflavanone) analogues, 15 and 16, or at higher temperature the corresponding dehydro derivative, 10, a flavonol. Cinnamaldehyde (26, R = PhCHd=CH−) used as the second component, led to styryl substitution at the 2-position (15). Similarly, use of phenylpropargyl aldehyde (26, R = PhC≡C−) provided 2-phenylethynyl substitution of the flavonol (10) or dihydroflavonol (16). Evidence that the dihydroflavonols obtained were of the trans configuration were deduced from NMR coupling constants for the 2- and 3-position protons in compounds 14-16 of ~12 Hz, characteristic of trans vicinal coupling.26 The 3-chloro substituent was introduced in flavone derivatives (as in the preparation of 11b and 11d) using sulfuryl chloride in carbon tetrachloride.37
Figure 3.
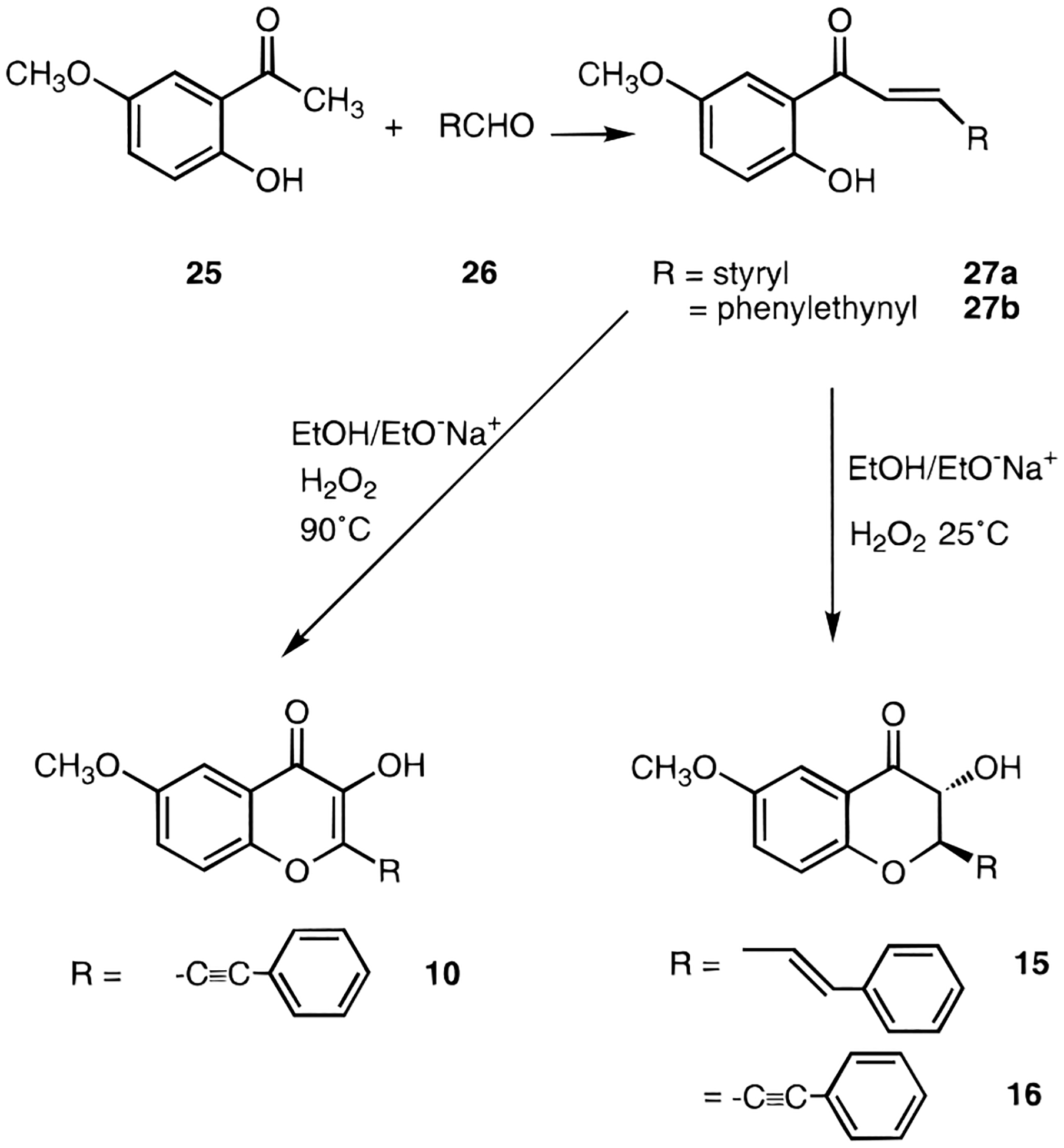
Synthesis of 2-phenylethynyl and 2-styryl derivatives of 4-methoxyfuranochromone.
An alternate approach to providing olefinic substitution at the 2-position (Figure 4) was to condense a 2-methylchromone (such as the natural product visnagin, 17)27 with an aldehyde, 28, under basic conditions. This method was used to prepare the 2-styryl derivative, 19. Evidence for a trans-styryl group of 19 was derived from the NMR spectra with an olefinic coupling constant of 15 Hz.26 In the presence of sodium ethoxide the 5-methoxy group readily exchanged, resulting in compound 20. The corresponding 5-propoxy derivative, 21, was obtained using an alternate approach (Figure 4), in which visnagin was demethylated to give 29, followed by condensation with benzaldehyde. The 2-methyl group of visnagin could also be oxidized to an aldehyde group, as in 18.28
Figure 4.
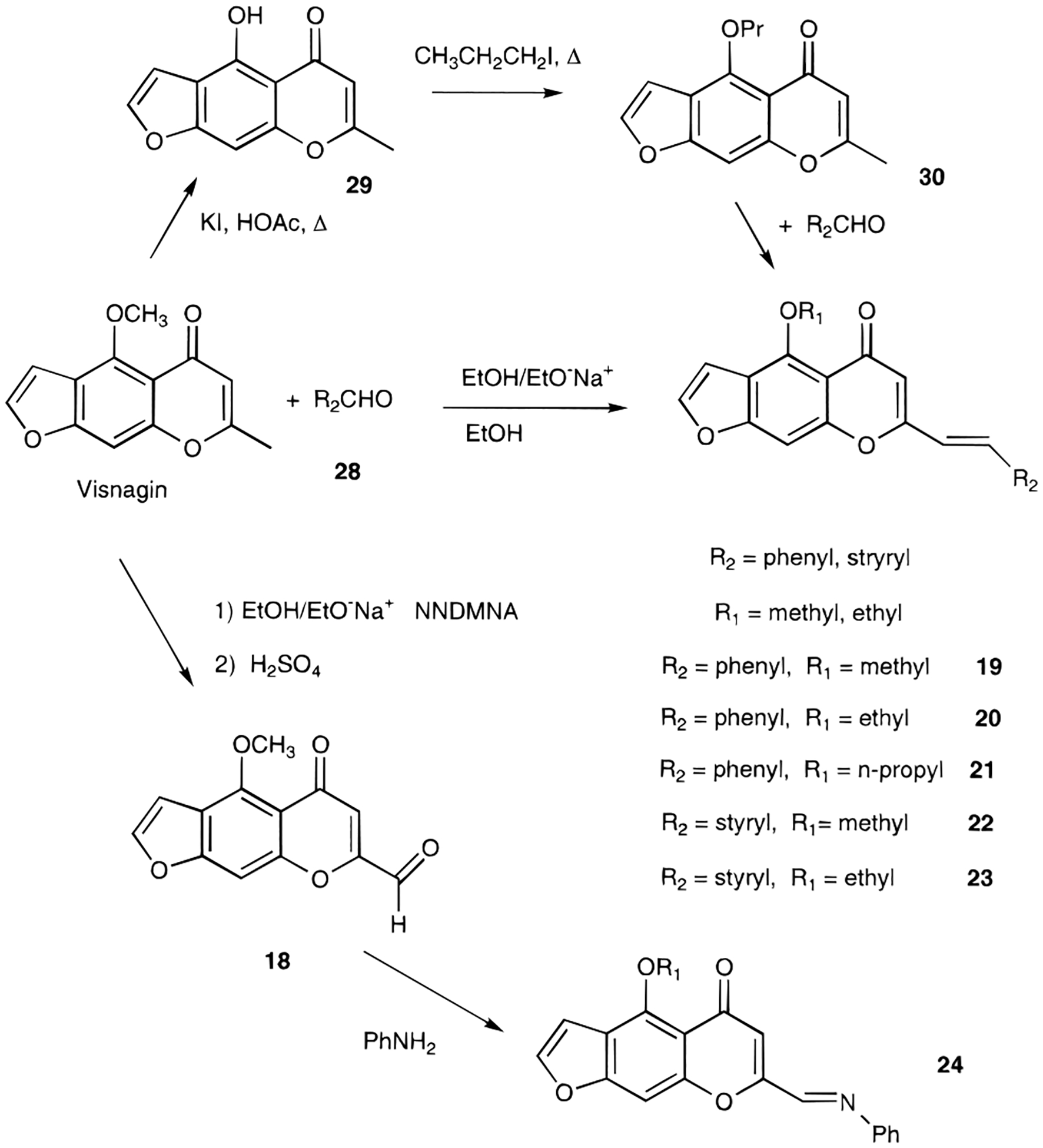
Chemical derivatization of visnagin and alkoxy substitution at the 5-position.
Affinity at Adenosine Receptors.
Ki values at A1 and A2A receptors were determined in radioligand binding assays in rat brain membranes vs [3H]PIA or [3H]CGS 21680, respectively.29,30 Affinity at human brain A3 receptors expressed in HEK-293 cells31 was determined using [125I]AB-MECA [N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide)].32 The complete set of compounds was tested at human A3 receptors, and only selected compounds at rat A3 receptors (stably expressed in CHO cells4,14–16), which would be preferred for comparison with the A1 and A2A data. We chose the human A3 receptor for our screen for the following reasons: (1) The affinity of most known adenosine receptor antagonists is minimal at rat A3 receptors.14,16 (2) The human A3, being more sensitive, allowed for a better comparison between compounds.31 (3) The human receptor is more relevant to therapeutic need. (4) The preliminary indications are that the A3 selectivity of flavones may be at least as great as indicated in this study when comparison is made between human receptor homologues. For example, the Ki value of galangin at human A2A receptors is 4.1 ± 0.7 μM (unpublished), compared to 0.97 μM at rat A2A receptors.19 At cloned human A1 receptors, the IC50 value for galangin in displacement 2 nM [125I]AB-MECA is 13.6 ± 1.5 μM (unpublished), i.e. weaker than at rat A1 receptors.
As we have previously reported,19 the flavonol galangin (2-phenyl-3,5,7-trihydroxy-4H-1-benzopyran-4-one), 1, inhibited binding at three adenosine receptor subtypes with Ki values of 1–3 μM. The trialkylation of galangin (compounds 2, 3a, and 4) caused an increase in affinity at human A3 receptors, in the order OPr = OEt > OMe > OH. At rat A1 and A2A receptors the affinity either was unchanged (A1) or varied in the order OH > OEt > OPr > OMe (A2A). The most potent and “A3 receptor selective” of the galangin ethers was the tripropyl derivative, 4, which had a Ki value of 0.32 μM at human A3 receptors and was 3-fold selective vs rat A1 receptors. The presence of a free 5-hydroxyl group, in 3b, greatly decreased A2A receptor affinity, while A3 receptor affinity was only 2-fold diminished. Morin, 5a, was considerably less potent than galangin at all three adenosine receptor subtypes.19 However, O-alkylation caused a gain in affinity, primarily at the human A3 receptors. As previously reported,19 pentamethylmorin, 6, appeared to bind with some selectivity (10- and 18-fold for human A3 vs rat A1 and A2A receptors, respectively), with a Ki value of 2.65 μM at human A3 receptors. The affinities of tetra- and pentaethyl ether derivatives of morin, 7 and 8, were compared. The presence of a free 5-hydroxyl group, in 2′,3,4′,7-tetraethylmorin, 7, greatly diminished affinity at A1 and A2A receptors and slightly enhanced affinity at A3 receptors, relative to the pentaethyl ether derivative, 8. Thus, compound 7 appeared to be selective for human A3 receptors vs rat A1 and A2A receptors. A derivative of 2′,4′,5,7-tetrahydroxyflavone, 5b, that was prenylated33 at the 3- and 6-positions and methylated at the 7-hydroxyl group had Ki values of 9.1 and 4.6 μM at rat A1 and human A3 receptors, respectively.
A trimethoxyphenyl derivative of flavonol, 9, only weakly displaced the binding of [125I]AB-MECA binding at human A3 receptors. Compound 9 also bound weakly at rat A2A receptors, but with moderate affinity at rat A1 receptors (Ki = 7.3 μM). The combination of an acetylenic substituent at the 2-position and a 6-methyl ether group (as in compound 10) failed to significantly enhance potency or selectivity.
The binding affinities of a number of flavones (non-hydroxylated at the 3-position) were compared. Flavone itself, 11a, was 5-fold selective for either rat A1 or A2A receptors vs human A3 receptors. Compound 11e, a 3,6-dichloro derivative, was particularly potent and selective for human A3 receptors. The affinities of compound 11a at cloned rat A3 receptors (Ki = 5.50 ± 2.221 μM) and at human A1 receptors (at 100 μM, only 35.0 ± 3.8% displacement of the binding of 2 nM [125I]AB-MECA) were also measured. Thus, this flavone derivative is ~20-fold selective for rat A3 vs rat A1 receptors, and ~200-fold selective for human A3 vs human A1 receptors. This compound, obtained from a chemical library of the National Cancer Institute (NCI), was found by Edwards et al.34 to lack potential as an anticancer agent. It was selected in this study for its combination of substituent groups at 3- and 2′-positions.19 The source of the selectivity apparently resided with 3,6-dichloro substitution, as indicated by the selectivity of compound 11d, which had a Ki value of 0.741 μM at human A3 receptors. This compound was >100-fold selective for human A3 receptors vs rat A1 receptors and 73-fold selective vs rat A2A receptors. The presence of trimethyl substitution of the phenyl ring in 11f did not eliminate selectivity, although the affinity at A3 receptors was somewhat diminished. Curiously, either 3-chloro or 6-chloro substitution alone (11b or 11c) did not result in increased potency or selectivity for human A3 receptors. 3-Chloroflavone, 11b, was more potent than the 6-chloro analogue, 11c; however, it did not exceed the affinity of unsubstituted flavone, 11a.
A number of derivatives of flavanone, 12, showed moderate affinity at adenosine receptors. Within a series of 2,3-saturated derivatives,35 a 2′-hydroxyl group enhanced the adenosine receptor affinity (e.g. the flavanone derivative 13a). Compound 13b, 4′-hydroxyflavanone, was less potent in binding to all three subtypes of adenosine receptors than the corresponding 2′-hydroxy analogue. Additional substitution of the A ring of 4′-hydroxyflavanone, as in sakuranetin, 13c, enhanced affinity at human A3 receptors. Previously we reported that reduction of the 2,3-olefinic bond of the flavonols, as in trans-(±)-dihydroquercetin,19 greatly diminished affinity at adenosine receptors. Consistently, the 3-hydroxyl group of 14 greatly diminished affinity relative to 13a.
Among dihydroflavonol analogues, the 2-styryl instead of the 2-aryl substituent, 15, afforded >8-fold selectivity for human A3 vs rat A1 and A2A receptors. The corresponding dihydroflavonol 2-phenylethynyl derivative, 16, was nearly as selective as 15 (6.2-fold for human A3 vs rat A1 receptors).
Visnagin, 17, a vasoactive furanochromone derivative27,36 isolated from Ammi visnaga, a fruit that has been used in folk medicine for its antispasmodic properties and in the treatment of angina pectoris,36 bound weakly to adenosine receptors. The corresponding 2-aldehyde, 18, was a similarly weak ligand at adenosine receptors. The presence of a trans-styryl group at the 2-styryl position, in 19, caused an 11-fold enhancement of A3 affinity, with less pronounced enhancements of affinity at A1 and A2A receptors. Elongation of the alkyl group at the 6-methyl ether of 19 caused a further affinity enhancement at A3 receptors, thus the corresponding 6-ethyl ether, 20, was 31-fold selective for human A3 receptors vs rat A1 receptors, with a Ki value of 1.16 μM. The selectivity of 20 for human A3 vs rat A2A receptors was 29-fold. Maximal selectivity was obtained with 20, since the corresponding 6-propyloxy derivative, 21, was less selective. The 6-ethoxy and 6-propyloxy derivatives, 20 and 21, were 7.1- and 2.1-fold more potent, respectively, than the 6-methoxy ether, 19. The 1-phenyl-1,3-butadiene derivative, 22, corresponding to an olefinic homologation of compound 19, was inactive in binding at all three adenosine receptor subtypes. The predicted enhancement of A3 receptor affinity upon ethyl substitution (23) of the 6-methyl ether, by analogy to compound 20, was insignificant.
Compound 24, the Schiff’s base equivalent of compound 19, i.e. in which the α-methyne group was formalistically replaced with nitrogen, bound selectively to human A3 (Ki 9.2 μM) vs rat A1 receptors.
At rat A3 receptors the affinity of flavones and flavonols, like that of many xanthines, has been shown to be weaker than at human A3 receptors.19 The Ki values of 1 and 2 have been shown to be 43.1 and 17.4 μM, respectively, at rat A3 receptors. This represents a human A3 vs rat A3 affinity ratio of 14-fold in both cases. The Ki values of several novel derivatives in this study (Table 3) have been measured in binding at cloned rat A3 receptors in membranes of transfected CHO cells.18,32 The Ki value of 11e at rat A3 receptors was 5.50 ± 2.21 μM; thus it was roughly 20-fold selective vs rat A1 receptors. In the rat compound 20 was only 3-fold selective for A3 receptors. The ratios between human and rat affinities were 50-, 9.8-, and 16-fold for 3a, 11e, and 20, respectively.
Table 3.
Inhibition of Binding of [125I]AB-MECA at Cloned Rat A3 Receptors and Effects of Flavone Derivatives (50 μM) on the A3 Agonist Elicited Inhibition of Adenylyl Cyclase
Functional Assay at Cloned Rat A3 Receptors.
We examined the antagonist properties of several flavonoid derivatives in a functional assay at A3 receptors, i.e. inhibition of adenylyl cyclase in CHO cells expressing cloned rat A3 receptors. Among the derivatives chosen, compounds 11e, 15, and 20 were subtype selective in binding, and compound 3a was very potent at human A3 receptors although nonselective. Adenylyl cyclase was inhibited by IB-MECA over the concentration range of 10−9−10−4 M in transfected CHO cells. The maximal extent of inhibition was 50–60% with an IC50 of approximately 0.1 μM, as reported.12 In Table 3 the concentration of IB-MECA used was either 0.1 or 1.0 μM, i.e. 1- or 10-fold the IC50 for the inhibition of rat A3 receptor mediated inhibition of adenylyl cyclase, respectively. Compound 3a at a concentration of 50 μM nearly completely abolished the inhibitory effect of 0.1 μM IB-MECA on adenylyl cyclase (Table 3). Compound 11e (Figure 5) also reversed the inhibition of adenylyl cyclase induced by IB-MECA at several concentrations. Compounds 15 or 16 at concentrations as high as 50 μM showed no significant effect on potency or maximal effect of agonist-induced inhibition of adenylyl cyclase.
Figure 5.
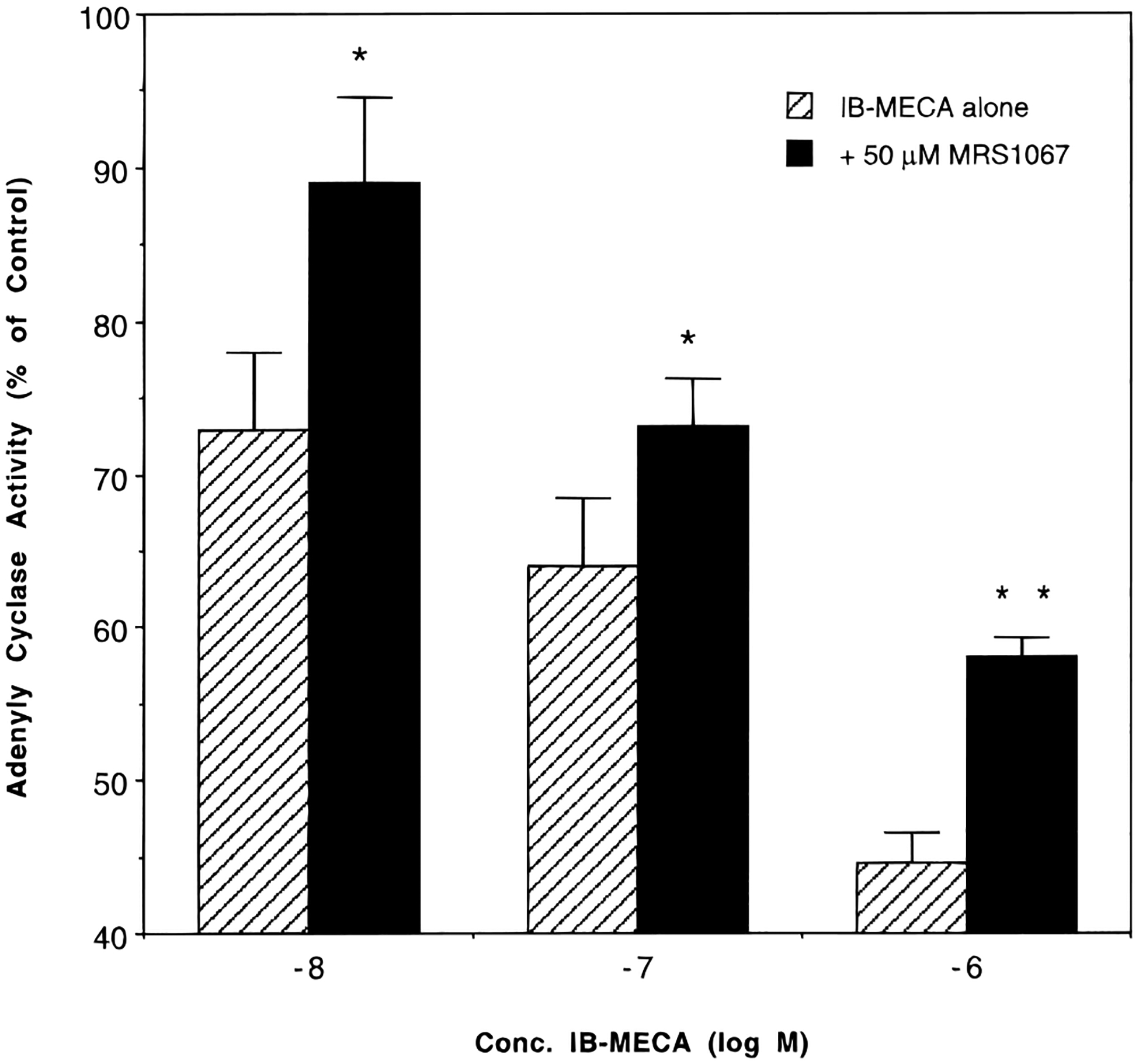
Inhibition of adenylate cyclase in membranes from CHO cells stably transfected with rat A3 receptors elicited by the agonist N6-(3-iodobenzyl)adenosine-5′-(N-methyluronamide) (IB-MECA), alone in the presence of 50 μM compound 11e (Ki = 5.50 ± 2.21 μM at rat A3 receptors). The assay was carried out as described in Experimental Section in the presence of 1 μM forskolin. Each data point is shown as mean ± S.E.M. for three determinations. *p < 0.05, **p < 0.001 (t test).
Discussion
The affinity of xanthines at A3 receptors, although species dependent, is generally extremely weak relative to their affinity at other subtypes.14 Thus, the naturally occurring xanthines do not act as antagonists at A3 receptors, as they do at the other subtypes. It is possible that certain members of another large family of natural products, the flavonoids, which are even more wide-spread in the human diet than the xanthines,38 may serve in this capacity as A3 receptor antagonists.19 More than 4000 flavonoids have already been isolated and identified from vascular plants,38 and they display a very wide range of biological activities. For example, flavonoids are used therapeutically to improve circulation and integrity of blood vessels40 and to suppress inflammation.41 They have been proposed as a potential treatment for diabetes, allergic reactions, infections, and other disorders.42 Even antineoplastic activities have been claimed for flavonoids.34 Many of these reported biological effects have not yet been fully explained mechanistically, although many mammalian enzymes and other proteins regulating intracellular biochemical processes, such as protein tyrosine kinases,23,24 are known to be affected by flavonoids.43,44 Flavones also inhibit phosphodiesterases and HIV integrase.48,49 Flavones have been studied in relation to the biological activity of adenosine45 and adenosine deaminase, which is inhibited at high concentrations of certain flavones.46
In several recent reports, flavones were found to bind to adenosine receptors,19,20 and certain derivatives have substantial affinity at the A3 receptor subtype.19 We identified flavones and flavonols as moderately potent adenosine receptor ligands as a result of screening a natural products library. Ares et al.20 independently discovered the affinity of flavones at A1 and A2A receptors in an investigation of the known gastroprotective effects of the substances,47 which yet lack a mechanistic explanation.
The most selective compounds for human A3 receptors identified in this study were compounds 7 and 11d-f, which were all essentially inactive at A1 and A2A receptors. The presence of a 5-hydroxyl group, as in 3b and 7, increased selectivity of flavonols for human A3 receptors. The most potent compounds in binding to A3 receptor (in order of increasing Ki value) were compounds 4, 3a > 11e, 11d ≥ 3b ≥ 20, 2 > 6. The degree of selectivity achieved was as high as 200-fold for compound 11e (Table 1). Since flavonoids are known to have affinity for many other proteins, it will be necessary to screen these derivatives in many other biochemical assays and at other receptors in order to establish true selectivity.
Conformational insights may be obtained from the present results. Compound 11e was highly selective for the human A3 receptor. In this derivative both the 3- and 2′-positions are highly substituted; thus there is likely steric hindrance to free rotation of the phenyl ring and assumption of a planar geometry. The potency and selectivity were similar to compound 11d, which was not as highly hindered sterically. Thus, we may conclude that a coplanar geometry between the two ring systems in the flavonols is not required.
Functional antagonism was indicated for derivatives, compounds 3a and 11e. It will be necessary to study the pharmacological effects in greater detail, in order to determine that other sites of interaction, for example at the level of G-proteins and second messengers, are not involved in the observed biological effect on adenylyl cyclase. It is likely that only flavonoids having Ki values of ≤1 μM at human A3 receptors will be useful as pharmacological probes.
In conclusion, these series of flavonoids provide leads for the development of novel potent and selective A3 adenosine receptor antagonists,14 although more detailed pharmacological characterization is required. The present findings indicate that the chemical modification of flavones at the 2- (with styryl groups), 3- (with hydroxyl or chloro), 6- (with chloro), and 2′-positions (with sterically bulky groups) may prove fruitful in this regard. The 2,3-position double bond appears to be required for recognition by human A3 receptors when both a 2-phenyl substituent and a 3-hydroxyl group are present. However the double bond is not an absolute requirement, since flavanone derivatives 13a–c, which lack the 3-hydroxyl group, displayed moderate affinity but not selectivity. Another means of restoring affinity in the reduced flavonol derivatives was to introduce a styryl or phenylethynyl group at the 2-position (compounds 15 and 16). Additional stucture–activity studies will be needed in order to design flavonoid analogues that are highly A3 receptor selective across species.
Experimental Section
Materials.
Compounds 1, 11a, 12, and 17 were obtained from Fluka, Ronkonoma, NY, or from Aldrich, St. Louis, MO. Compounds 13a, 13c, and 14 were obtained from Apin Chemicals, Ltd., Oxon, U.K. Compounds 5a and 13b were obtained from K+K Laboratories, Jamaica, NY. Compounds 5b (NSC #241010-z), 9 (NSC #78634-f), 11e (NSC #74899-t), and 11f (NSC #74931-f) were obtained from NCI (Bethesda, MD).
Synthesis.
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer and spectra were taken in DMSO-d6. Electron-impact mass spectrometry was performed with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Atlantic Microlabs, Inc. (Norcross, GA).
3,5,7-Trimethoxyflavone (2).
Galangin (27 mg, 0.1 mmol) was dissolved in dry acetone (stored over K2CO3, 20 mL), solid potassium carbonate (0.5 g) and dimethyl sulfate (1 mL) were added, and the mixture was refluxed for 4 h and then cooled to room temperature. The solution was filtered and evaporated, water (20 mL) and concentrated ammonium hydroxide (2 mL) were added, and the solution was extracted with ethyl acetate (15 mL × 2). The solvent was evaporated, and the residue was recrystallized from methanol/water to give 21 mg of product (67%): 1H-NMR (DMSO-d6) δ 3.73 (s, 3H, 3-OCH3), 3.86 (s, 3H, 7-OCH3), 3.86 (s, 3H, 5-OCH3), 6.48 (s, 1H, 8-H), 6.79 (s, 1H, 6-H), 7.99 (m, 3H, Ar), 8.02 (m, 2H, Ar); MS (CI/NH3) m/z 313 (MH+, base).
3,5,7-Triethoxyflavone (3a).
Galangin (30 mg, 0.11 mmol) was dissolved in dry acetone (45 mL), solid potassium carbonate (0.5 g) and iodoethane were added, and the mixture was refluxed overnight. The solution was filtered and evaporated, water (20 mL) and concentrated ammonia (1 mL) were added, and the solution was extracted with ethyl acetate. The ethyl acetate was evaporated, and the crude mass was purified on a preparative silica TLC plate to give 12 mg of 3,5,7-triethoxy-flavone: mp 111–114 °C; mass (FAB) m/z 355 (M+ + 1, base); 1H-NMR (CDCl3) δ 1.33 (t, 3H, CH3), 1.55 (t, 3H, J = 7 Hz, CH3), 1.62 (t, 3H, J = 7 Hz, CH3), 4.1–4.25 (m, 6H, CH2), 6.4 (s, 1H), 6.55 (s, 1H), 7.5–7.6 (m, 3H), 8.1–8.2.
3,7-Diethoxy-5-hydroxyflavone (3b).
Galangin (27 mg, 0.1 mmol) was dissolved in dry acetone (20 mL), solid potassium carbonate (0.5 g) and bromoethane (1 mL) were added, and the mixture was stirred overnight, at room temperature. The solution was filtered and evaporated, water (20 mL) and concentrated ammonium hydroxide (2 mL) were added, and the solution was extracted with ethyl acetate (20 mL × 2). The solution was dried, and the solvent was evaporated. The residue was purified by preparative TLC plate (silica, ethyl acetate/petroleum ether (2:8) to give 21 mg of product (64%): 1H-NMR (CDCl3) δ 1.34 (t, 3H, J = 7.3 Hz, 3-CH3), 1.48 (t, 3H, J = 7.1 Hz, 7-CH3), 4.14 (m, 4H, 2 × OCH2), 6.38 (s, 1H, 6-H), 6.47 (s, 1H, 8-H), 12.64 (s, 1H, 5-OH); MS (CI/NH3) m/z 327 (MH+, base).
3,5,7-Tripropyloxyflavone (4).
Galangin (30 mg, 0.11 mmol) was dissolved in dry acetone (45 mL), solid potassium carbonate (0.5 g) and 1-iodopropane were added, and the mixture was refluxed overnight. The solution was filtered and evaporated, water (20 mL) and concentrated ammonia (1 mL) were added, and the solution was extracted with ethyl acetate. The ethyl acetate was evaporated, and the crude mass was purified on a preparative TLC plate (silica) to give 25 mg (63%) of 3,5,7-tripropyloxyflavone: mp 90–93 °C; mass spectra (CI/NH3) m/z 397 (M+ + 1, base); 1H-NMR (CDCl3) δ 0.85–1.2 (m, 9H), 1.6–2.1 (m, 6H), 3.9–4.1 (m, 6H), 6.32 (s, 1H), 6.5 (s, 1H), 7.4–7.5 (m, 3H), 8.04–8.12 (m, 2H).
2′,3,4′,5,7-Pentakis(methyloxy)flavone (6).
Morin (120 mg, 0.4 mmol) was dissolved in dry acetone (80 mL). Solid potassium carbonate (2 g) and iodomethane (2 mL) were added, and the mixture was refluxed overnight and then cooled to room temperature. The solution was filtered and evaporated, and the residue was separated by preparative TLC plates (silica, ethyl acetate) to give 80 mg of product (55%); 1H-NMR (CDCl3) δ 3.86, 3.88, 3.91, 3.93, 4.02 (5s, 15H, 5 × OCH3), 6.79 (3m, 3H, phenyl), 6.65 (m, 1H, 8-H), 7.42 (m, 1H, 6-H); MS (EI) m/z 372 (M+), 371 (M − H)+, 341 (M − OCH3)+.
2′,4′,3,7-Tetraethoxy-5-hydroxymorin (7).
Compound 7 was synthesized according to the above procedure for 6, except using iodoethane. The product, 7, was separated on a preparative TLC plate with petroleum ether/ethyl acetate and displayed a mass (FAB) m/z 415 (M+ + 1, base): 1H-NMR (CDCl3) δ 1.15 (t, 3H, CH3), 1.35 (t, 3H, CH3), 1.45 (dt, 6H, CH3), 3.9–4.2 (m, 8H, CH2), 6.33 (d, 1H), 6.6 (m, 3H), 7.4 (d, 1H).
2′,3,4′,5,7-Pentaethoxyflavone (8).
Morin (302 mg, 1 mmol) was dissolved in dry acetone (80 mL), solid potassium carbonate (5 g) and diethyl sulfate (5 mL) were added, and the mixture was refluxed overnight and then cooled to room temperature. The solution was filtered and evaporated, water (50 mL) and concentrated ammonium hydroxide (10 mL) were added, and the solution was extracted with ethyl acetate (40 mL × 2). The solution was dried, and the solvent was evaporated. The residue was crystallized from ethyl acetate and petroleum ether (1:9) to give 335 mg of product (76%): 1H-NMR (CDCl3) δ 1.12 (t, 3H, J = 7.1 Hz, CH3), 1.32 (t, 3H, J = 7.1 Hz, CH3), 1.45 (2t, overlap, 6H, 2 × CH3), 1.56 (t, 3H, J = 7.1 Hz, CH3), 4.10 (m, 10 H, 5 × OCH2),6.32 (d, 1H, J = 1.2 Hz, 2-phenyl), 6.39 (s, 1H, 2-phenyl), 6.53 (s, 1H, 2-phenyl), 6.58 (s, 1H, 6-H), 7.37 (d, 1H, 8-H); MS (CI/NH3) m/z 443 (MH+, base).
2-(Phenylethynyl)-3-hydroxy-6-methoxyflavone (10).
1-(2-Hydroxy-5-methylphenyl)-5-phenylpenta-2-en-4-yn-1-one (compound 27b, 60 mg, 0.2 mmol) was dissolved in ethanol (1.5 mL). Sodium hydroxide (1 N, 0.4 mL) and hydrogen peroxide (0.5 g) were added, and the mixture was heated at 90 °C for 35 min. The cooled mixture was diluted with water (5 mL) and acidified with hydrochloric acid (1 N). A precipitate was collected and recrystallized from methanol: mass (CI/NH3) m/z 293 (M+ + 1, base); 1H-NMR (CDCl3) δ 3.95 (s, 3H, CH3), 6.92 (s, 1H), 7.25–8.0 (m, 7H).
3-Chloroflavone (11b).
Sulfuryl chloride (85 mg, 0.55 mL) in 2 mL of carbon tetrachloride was added dropwise to a solution of flavone (111 mg, 0.5 mmol) in 5 mL of carbon tetrachloride. The mixture was stirred overnight. The solution was diluted with CH2Cl2 (5 mL), washed with water (10 mL × 2), saturated sodium bicarbonate solution (10 mL × 2), and brine (10 mL × 2), and dried over sodium sulfate. The solvent was evaporated, and the residue was purified on a preparative TLC plate (silica, ethyl acetate/petroleum ether, 2:8) to give 80 mg of product (63%): 1H-NMR (CDCl3) δ 7.46–7.58 (m, 5H, 2-C6H5), 7.74 (m, 1H, 7-H), 7.94 (m, 2H, 5,6-H), 8.33 (dd, 1H, J = 7.8, 2.0 Hz, 8-H); MS (EI) m/z 256 (M+, base), 228 (M − CO)+, 221 (M − Cl)+; mp 121 °C.
6-Chloroflavone (11c).
Bromine (185 mg, 1.15 mmol) was added a solution of 2-hydroxy-5-chlorochalcone (258 mg, 1.0 mmol) in acetic acid (10 mL). After the reaction was stirred at ambient temperature for 3 h, 1% aqueous sodium bisulfite solution (20 mL) was added slowly. The resulting precipitate was filtered off, washed with water, and suspended in ethanol (10 mL); potassium hydroxide (200 mg, 3.5 mmol) dissolved in water (3.5 mL) was added; and stirring was continued overnight. The reaction mixture was acidified with 2 N HCl. The precipitate formed was filtered off and washed with water to give 105 mg of residue, which was recrystallized from EtOH/H2O to give 85 mg of product (33%): 1H-NMR (CDCl3) δ 6.85 (s, 1H, 3-H), 7.54–7.69 (m, 5H, 2-C6H5), 7.93 (m, 2H, 5, 7-H), 8.22 (d, 1H, J = 2.7 Hz, 8-H); MS (CI/NH3) m/z 257 (MH+, base) and 259 (M + 2H+) in 3:1 ratio; mp 170–172 °C.
3,6-Dichloroflavone (11d).
Sulfuryl chloride (28 μL, 0.28 mmol) in 2 mL of carbon tetrachloride was added dropwise to a solution of 6-chloroflavone (65 mg, 0.25 mmol) in 5 mL of carbon tetrachloride. The mixture was stirred overnight. The solution was diluted with CH2Cl2 (5 mL), washed with water (5 mL × 2), saturated sodium bicarbonate solution (5 mL × 2), and brine (5 mL × 2), and dried over sodium sulfate. The solvent was evaporated, and the residue was purified on a preparative TLC plate (silica, ethyl acetate/petroleum ether, 2:8) to give 8 mg of product (10%): 1H-NMR (CDCl3) δ 7.49–7.70 (m, 5H, 2-C6H5), 7.91 (m, 2H, 5,7-H), 8.28 (d, 1H, J = 2.6 Hz, 8-H); MS (EI) m/z 290, 292, 294 (M+:(M + 2+):(M + 4+) = 9:6:1); mp 184–186 °C.
trans-2-Styryl-3-hydroxy-6-methoxyflavanone (15).
2′-Hydroxy-5′-methoxyacetophenone (25, 4.1 g, 22 mmol) and cinnamaldehyde (3.35 g, 25 mmol) were dissolved in minimal methanol (2.5 mL). Concentrated sodium hydroxide (12.5 mL, 50%) was added, and the mixture was kept on ice for 8 h. The resulted solid was suspended in water and acidified using HCl (4 N). The oil that separated was dissolved in ethanol and crystallized from ethanol/water to give 1-(2-hydroxy-5-methylphenyl)-5-phenylpenta-2,4-dien-1-one (1.2 g) as a red-brown powder: mass (CI/NH3) m/z 281 (M+ + 1, base); 1H-NMR (CDCl3) δ 3.9 (s, 3H, CH3), 6.95–7.8 (m, 12H). The above compound (170 mg, 0.6 mmol) was dissolved in an mixture of ethanol (3.5 mL) and acetone (4 mL). Sodium hydroxide (1 N, 1 mL) and hydrogen peroxide (1 mL, 35%) were added, and the solution was stirred for 6 h at room temperature. The mixture was precipitated by adding water and HCl and purified on prep TLC (petroleum ether/ethyl acetate) to give compound 15: mp 160–163 °C; mass (CI/NH3) m/z 297 (M+ + 1,base); 1H-NMR (CDCl3) δ 3.9 (s, 3H, CH3), 4.45 (d, J = 12 Hz, 1H), 4.8 (m, 1H), 6.55 (dd, 1H), 6.9–7.6 (m, 9H).
trans-2-(Phenylethynyl)-3-hydroxy-6-methoxyflavanone (16).
2′-Hydroxy-5′-methoxyacetophenone and phenylpropargyl aldehyde were condensed according to the procedure for the preparation of compound 15 to give 1-(2-hydroxy-5-methylphenyl)-5-phenylpenta-2-en-4-yn-1-one: mass (CI/NH3) m/z 279 (M+ + 1, base); 1H-NMR (CDCl3) δ 3.9 (s, 3H, CH3), 7.0 (d, 1H), 7.1–7.6 (m, 9H). The above compound was allowed to react with hydrogen peroxide to give compound 16 as a white powder: mp 135–138 °C; mass (EI) m/z 295 (M+1), 150 (base); 1H-NMR (CDCl3) δ 3.85 (s, 3H, OCH3), 4.6 (d, J = 12 Hz, 1H), 5.09 (d, J = 12 Hz, 1H), 7.0–7.6 (m, 8H).
4-Methoxy-7-formyl-5H-furo[3,2-g][1]benzopyran-5-one (18)
4-Methoxy-7-formyl-5H-furo[3,2-g][1]benzopyran-5-one (18) was synthesized from visnagin (4-methoxy-7-methyl-5H-furo[3,2-g][1]benzopyran-5-one) according to ref 28.
4-(Methyloxy)-7-trans-styrylvisnagin (19) and 4-(Ethyloxy)-7-trans-styrylvisnagin (20).
Visnagin (160 mg, 0.7 mmol) and benzaldehyde (120 mg, 1.1 mmol) were dissolved in ethanol (4 mL). Sodium ethoxide (20% in ethanol, 0.5 mL) was added, and the mixture was stirred for 10 min at 80 °C. Disappearance of the starting material was accompanied by formation of two products, Rf 0.7 and 0.8 in ethyl acetate. Both were separated on preparative TLC plates (ethyl acetate) to give compound 19: mp 175–178 °C; mass (CI/NH3) m/z 319 (M+ + 1, base); 1H-NMR (CDCl3) δ 4.2 (s, 3H, OCH3), 6.23 (s, 1H), 6.75 (d, J = 15 Hz, 1H), 7.05 (s, 1H), 7.3–7.8 (m, 8H).
Compound 20: mp 148–151 °C; mass (CI/NH3) m/z 333 (M+ + 1, base); 1H-NMR (CDCl3) δ 1.55 (t, 3H, CH3), 4.4 (q, 2H, OCH2), 6.23 (s, 1H), 6.78 (d, J = 15 Hz, 1H), 7.0 (s, 1H), 7.35–7.7 (m, 8H).
4-(Propyloxy)-7-trans-styrylvisnagin (21).
A mixture of visnagin (2 g, 8.7 mmol), potassium iodide (10 g), and acetic acid (50 mL) was refluxed for 7 h. After cooling, the precipitate was removed by filtration, and the filtrate was evaporated under reduced pressure, and coevaporated with toluene (10 mL × 2). The residue was crystallized from ethanol to give 1.62 g (81%) of demethylation product of visnagin: 1H-NMR (CDCl3) δ 2.40 (s, 3H, 7-CH3), 6.12 (s, 1H, 6-H), 7.00 and 7.62 (2d, 2H, J = 2.9 Hz, 3-H and 2H), 7.05 (s, 1H, 9-H), 13.6 (s, 1H, 4-OH). The mixture of demethylated visnagin (430 mg, 2 mmol), iodopropane (3 mL), and potassium carbonate (5 g) in dry acetone (80 mL) was refluxed overnight. The solid was removed by filtration, the solvent was evaporated, water (50 mL) and concentrated ammonium hydroxide (15 mL) were added, and the solution was extracted with ethyl acetate (40 mL × 2). The organic layer was dried over sodium sulfate, and the solvent was evaporated. The residue was purified by preparative TLC plates (silica, ethyl acetate/petroleum ether, 4:6) to give 465 mg (90%) of 4-(propyloxy)visnagin (30): 1H-NMR (CDCl3) δ 1.10 (t, 3H, J = 7.5 Hz, CH3), 1.94 (m, 2H), 2.33 (s, 3H, 7-CH3), 4.23 (t, 2H, J = 6.7 Hz, 4-OCH2), 6.03 (s, 1H, 6-H), 6.97 and 7.60 (2d, 2H, J = 2.7 Hz, 3-H and 2-H), 7.27 (s, 1H, 9-H). 4-(Propyloxy)visnagin (86 mg, 0.3 mmol) and benzaldehyde (50 mg, 0.5 mmol) were dissolved in ethanol (2 mL). While stirring, sodium ethoxide (20% in ethanol, 0.3 mL) was added, and the mixture was stirred for 15 min at 80 °C. The solvent was evaporated, the residue was partitioned between ice water (20 mL) and ethyl acetate (20 mL), the aqueous layer was extracted with ethyl acetate (20 mL), and the combined organic layer was dried over sodium sulfate. The solvent was evaporated, and the residue was separated by preparative TLC plate (silica, ethyl acetate/petroleum ether, 4:6) to give 26 mg of the desired product (25%): 1H-NMR (CDCl3) δ 1.07 (t, 3H, J = 7.5 Hz, CH3), 1.97 (m, 2H), 4.25 (t, 2H, J = 6.7 Hz, 4-OCH2), 6.21 (s, 1H, 6-H), 6.78 (d, 2H, J = 14 Hz, 3-H), 7.00 (s, 1H, 9-H), 7.33 (m, 8H); MS (CI/NH3) m/z 347 (MH+, base).
4-Methoxy-7-(4-phenyl-1,3-pentadienyl)-5H-furo[3,2-g][1]benzopyran-5-one (22)and 4-Ethoxy-7-(4-phenyl-1,3-pentadienyl)-5H-furo[3,2-g][1]benzopyran-5-one (23).
Compound 22 was prepared by dissolving visnagin and cinnamaldehyde in ethanol in the presence of sodium ethoxide according to the above procedure for 19 and 20: mp 162–165 °C; mass (EI) m/z 344 (M+, base); 1H-NMR (CDCl3) δ 4.2 (s, 3H, OCH3), 6.15 (s, 1H), 6.33 (d, J = 15 Hz, 1H), 6.95 (s, 1H), 7.0–7.6 (m, 10H).
Compound 23 was also isolated from the above reaction: mass (EI) m/z 358 (M+), 343 (M – 15, base); 1H-NMR (CDCl3) δ 1.55 (t, 3H, CH3), 4.4 (q, 2H, OCH2), 6.15 (s, 1H), 6.35 (d, J = 15 Hz, 1H), 7.4 (d, J = 15 Hz, 1H), 6.9–7.6 (m, 10H).
4-Methoxy-7-formyl-5H-furo[3,2-g][1]benzopyran-5-one Aniline Schiff base (24).
Compound 18 (400 mg, prepared according to ref 28) and aniline (0.5 mL) were dissolved in toluene and stirred overnight at 25 °C. The course of the reaction was followed using analytical TLC (silica, CHCl3/MeOH, 20:1). The solvent was evaporated, and the excess aniline was removed under high vacuum. The product (Rf ~ 0.7) was purified using preparative TLC (silica, ethyl acetate): mass (CI/NH3) m/z 320 (M+ + 1, base).
Pharmacology. Radioligand Binding Studies.
The radioligand binding data for the novel compounds were determined as described previously.17,18 Membranes prepared from HEK-293 cells stably expressing the human A3 receptor were obtained from Receptor Biology, Inc. (Baltimore, MD). CHO cells stably expressing the rat A3 adenosine receptor were grown in F-12 medium containing 10% fetal bovine serum and penicillin/streptomycin (100 units/mL and 100 μg/mL, respectively) at 37 °C in a 5% CO2 atmosphere, and membrane homogenates were prepared as reported.18 Binding of [125I]-N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide) ([125I]AB-MECA) to rat A3 receptors in stably transfected CHO cell membranes was performed as described.32 Assays at human A3 receptors were performed in a buffer containing 50 mM Tris, 10 mM Mg2+, and 1 mM EDTA at pH 8.0. The glass tubes contained 100 μL of the membrane suspension (0.3 mg of protein/mL, stored at −80 °C in the same buffer), 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of a solution of the proposed antagonist. All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 1%. Duplicate incubations were carried out for 1 h at 25 °C19 and were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each. The radioactivity on the filters was determined with a Beckman 5500B γ-counter. Nonspecific binding was determined in the presence of 100 μM N6-phenylisopropyladenosine [(R)-PIA].
Binding of [3H]-(R)-PIA to A1 receptors from rat cortical membranes and of [3H]CGS 21680 to A2A receptors from rat striatal membranes was performed as described previously.29,30
Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in which an incubation of 30 min at 30 °C is carried out, and during the incubation with the radioligands. At least five different concentrations spanning 3 orders of magnitude, adjusted appropriately for the IC50 of each compound, were used. IC50 values, computer-generated using the nonlinear regression method implemented in the InPlot program (Graph-PAD, San Diego, CA), were converted to apparent Ki values using Kd values of 1.0 and 14 nM for [3H]PIA and [3H]CGS 21680 binding, respectively, applying the Cheng–Prusoff equation.39 A Kd value of 0.59 nM for [125I]AB-MECA binding at human A3 receptors was assumed.19
Adenylyl cyclase activity was measured in membranes from CHO cells stably expressing the rat A3 receptor, prepared as above, using a previously reported method.4,12 The method involved addition of [α-32P]ATP to membranes in the presence of forskolin to stimulate adenylyl cyclase and papaverine as a phosphodiesterase inhibitor. The reaction was terminated by addition of a stop solution containing 20 000 cpm/mL [3H]cyclic AMP. The total radiolabeled cyclic AMP was isolated on columns of Dowex 50 ion exchange resin and alumina. Maximal inhibition of adenylyl cyclase activity corresponded to 40–50% of total activity under conditions of stimulation (typically 6–8-fold) in the presence of 1 μM forskolin. IC50 values were calculated using InPlot (GraphPad, San Diego, CA).
Acknowledgment.
We thank Marc Glashofer for technical assistance and helpful discussions and Dr. George W. Milne of the National Cancer Institute (Bethesda, MD) for providing samples of several of the compounds used in this study. We thank Gilead Sciences (Foster City, CA) and the Cystic Fibrosis Foundation for financial support. We thank Dr. Pat Towers of Amersham (Cardif, U.K.) and Dr. Garth Brown of DuPont NEN (Billerica, MA) for providing samples of [125I]AB-MECA.
References
- (1).Daly JW Mechanism of action of caffeine. In Caffeine, Coffee and Health; Garattini S, Ed.; Raven Press: New York, 1993; Chapter 4, pp 97–150. [Google Scholar]
- (2).Bruns RF The role of adenosine in energy supply/demand balance. Nucleosides Nucleotides 1991, 10, 931–944. [Google Scholar]
- (3).Jacobson KA; van Galen PJM; Williams M Perspective. Adenosine receptors: pharmacology, structure-activity relationships and therapeutic potential. J. Med. Chem 1992, 35, 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhou QY; Li CY; Olah ME; Johnson RA; Stiles GL; Civelli O Molecular cloning and characterization of an adenosine receptor - The A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 7432–7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Jacobson MA Cloning and expression of human adenosine receptor subtypes. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology; Bellardinelli L, Pelleg A, Eds.; Kluwer: Norwell, 1995; pp 5–13. [Google Scholar]
- (6).Ramkumar V; Stiles GL; Beaven MA; Ali H The A3AR is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem 1993, 268, 16887–16890. [PubMed] [Google Scholar]
- (7).Linden J Cloned adenosine A3 receptors - pharmacological properties, species-differences and receptor functions. Trends Pharmacol. Sci 1994, 15, 298–306. [DOI] [PubMed] [Google Scholar]
- (8).Hannon JP; Pfannkuche HJ; Fozard JR A role for mast-cells in adenosine A(3) receptor-mediated hypotension in the rat. Br. J. Pharmacol 1995, 115, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jacobson KA; Nikodijević O; Shi D; Gallo-Rodriguez C; Olah ME; Stiles GL; Daly JW A role for central A3-adenosine receptors: Mediation of behavioral depressant effects. FEBS Lett. 1993, 336, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Palmer TM; Gettys TW; Stiles GL Differential interaction with and regulation of multiple G-proteins by the rat A(3) adenosine receptor. J. Biol. Chem 1995, 270, 16895–16902. [DOI] [PubMed] [Google Scholar]
- (11).Jacobson KA; Kim HO; Siddiqi SM; Olah ME; Stiles G; von Lubitz DKJE A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future 1995, 20, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kim HO; Ji X.-d.; Siddiqi SM; Olah ME; Stiles GL; Jacobson KA 2-Substitution of N6-benzyladenosine-5′-urona-mides enhances selectivity for A3-adenosine receptors. J. Med. Chem 1994, 37, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).von Lubitz DKJE; Deutsch SI; Carter MF; Lin RC-S; Mastropaolo J; Jacobson KA The effects of adenosine A3 receptor stimulation on seizures in mice. Eur. J. Pharmacol 1995, 275, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ji X.-d.; von Lubitz D; Olah ME; Stiles GL; Jacobson KA Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev. Res 1994, 33, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kim HO; Ji X.-d.; Melman N; Olah ME; Stiles GL; Jacobson KA Structure-activity relationships of 1,3-dialkyl-xanthine derivatives at rat A3 adenosine receptors. J. Med. Chem 1994, 37, 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Siddiqi SM; Ji X.-d.; Melman; Olah ME; Jain R; Evans P; Glashofer M; Padgett WL; Cohen LA; Daly JW; Stiles GL; Jacobson KA A survey of non-xanthine derivatives as adenosine receptor ligands. Nucleosides Nucleotides 1995, 15, 693–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).van Rhee AM; Siddiqi SM; Melman N; Shi D; Padgett WL; Daly JW; Jacobson KA Tetrahydrobenzothiophenone derivatives as a novel class of adenosine receptor antagonists. J. Med. Chem 1996, 39, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).van Galen PJM; van Bergen AH; Gallo-Rodriguez C; Olah ME; IJzerman AP; Stiles GL; Jacobson KA A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol. Pharmacol 1994, 45, 1101–1111. [PMC free article] [PubMed] [Google Scholar]
- (19).Ji X.-d.; Melman N; Jacobson KA Interactions of flavonoids and other phytochemicals with adenosine receptors. J. Med. Chem 1996, 39, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ares JJ; Pong SF; Outt PE; Blank MA; Murray PD; Portlock DE The binding of flavonoids to adenosine receptors; 209th ACS National Meeting, Chicago, IL, April 1995; Abstract MEDI190. [Google Scholar]
- (21).Markham KR; Geiger H 1H Nuclear magnetic resonance spectroscopy of flavonoids and their glycosides in hexadeu-terodimethylsulfoxide. In The Flavonoids, Advances in Research Since 1986; Harborne JB, Ed.; Chapman and Hall: London, 1993; Chapter 10, pp 441–497. [Google Scholar]
- (22).Wollenweber E Flavones and flavonols. In The Flavonoids, Advances in Research Since 1986; Harborne JB, Ed.; Chapman and Hall: London, 1993; Chapter 10, pp 259–335. [Google Scholar]
- (23).Cushman M; Zhu H; Gaehlen RL; Kraker A Synthesis and biochemical evaluation of a series of aminoflavones as potential inhibitors of protein-tyrosine kinases p56lck, EGFr, and p60v-src. J. Med. Chem 1994, 37, 3373–3382. [DOI] [PubMed] [Google Scholar]
- (24).Cunningham BDM; Threadgill MD; Groundwater PW; Dale IL; Hickman JA Synthesis and biological activity of a series of flavones as inhibitors of protein tyrosine kinases. Anti-Cancer Drug Des. 1992, 7, 365–384. [PubMed] [Google Scholar]
- (25).Lee YR; Morehead AT A new route for the synthesis of furanoflavone furanochalcone natural products. Tetrahedron 1995, 51, 4909–4922. [Google Scholar]
- (26).Williams DH; Fleming I Spectroscopic Methods in Organic Chemistry, 4th ed.; McGraw Hill: London, 1989. [Google Scholar]
- (27).Duarte J; Perez-Viscaino F; Torres AI; Zarzuelo A; Jimenez J; Tamargo J Vasodilator effects of visnagin in isolated rat vascular smooth muscle. Eur. J. Pharmacol 1995, 286, 115–122. [DOI] [PubMed] [Google Scholar]
- (28).El-Shaaer HM; Zahradník P; Lácová M; Matulová M Study of substituted formylchromones. Collect. Czech. Chem. Commun 1994, 59, 1673–1681. [Google Scholar]
- (29).Schwabe U; Trost T Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol 1980, 313, 179–187. [DOI] [PubMed] [Google Scholar]
- (30).Jarvis MF; Schutz R; Hutchison AJ; Do E; Sills MA; Williams M [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther 1989, 251, 888–893. [PubMed] [Google Scholar]
- (31).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Olah ME; Gallo-Rodriguez C; Jacobson KA; Stiles GL [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- (33).Ikuta J; Hano Y; Nomura T; Kawakami Y; Sato T Chem. Pharm. Bull 1986, 35, 602-. [Google Scholar]
- (34).Edwards JM; Rauffauf RF; LeQuesne PW Antineoplastic activity and cytotoxicity of flavones, isoflavones, and flavanones. J. Nat. Prod 1979, 42, 85–91. [DOI] [PubMed] [Google Scholar]
- (35).Portert LJ Flavans and proanthocyanidins. In The Flavonoids, Advances in Research Since 1986; Harborne JB, Ed.; Chapman and Hall: London, 1993; Chapter 2, pp 23–55. [Google Scholar]
- (36).Rauwald HW; Brehm O; Odenthal K-P The involvement of a Ca2+ channel blocking mode of action in the pharmacology of Ammi visnaga fruits. Planta Med. 1994, 60, 101–105. [DOI] [PubMed] [Google Scholar]
- (37).Merchant JR; Martyres G Reactions of benzopyrones with sulfuryl chloride. Chem. Ind. (London) 1980, 937. [Google Scholar]
- (38).Harborne J Flavonoids in the environment. In Plant Flavonoids in Biology and Medicine II: Biochemical, Cellular and Medicinal Properties; Alan R. Liss: New York, 1988; pp 17–27. [Google Scholar]
- (39).Cheng YC; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (40).Beretz A; Cazenave JP The effect of flavonoids on blood-vessel wall interactions. Prog. Clin. Biol. Res 1988, 280, 187–200. [PubMed] [Google Scholar]
- (41).Gabor M Anti-inflammatory and anti-allergic properties of flavonoids. Prog. Clin. Biol. Res 1986, 213, 471–80. [PubMed] [Google Scholar]
- (42).Middleton E Jr. Some biological properties of plant flavonoids. Ann. Allergy 1988, 61, 53–57. [PubMed] [Google Scholar]
- (43).Roger CR The nutritional incidence of flavonoids: some physiological and metabolic considerations. Experientia 1988, 44, 725–733. [DOI] [PubMed] [Google Scholar]
- (44).Beretz A; Anton R; Cazenave JP The effect of flavonoids on cyclic nucleotide phosphodiesterases. In Plant Flavonoids in Biology and Medicine; Cody V, Middleton E, Harborne J, Eds.; Alan R. Liss: New York, 1986; pp 281–296. [PubMed] [Google Scholar]
- (45).Melzig MF; Franke S Effect of flavonoids and phenocarboxylic acids on adenosine activity in endothelial cells. Pharmazie 1995, 50, 510–511. [PubMed] [Google Scholar]
- (46).Koch HP; Jager W; Groh U; Plank G In vitro inhibition of adenosine deaminase by flavonoids and related compounds. New insight into the mechanism of action of plant phenolics. Methods Find. Exp. Clin. Pharmacol 1992, 14, 413–417. [PubMed] [Google Scholar]
- (47).Pfister JR; Wyman WE; Schuler ME; Roszkowski AP Inhibition of histamine-induced gastric secretion by flavone-6-carboxylic acids. J. Med. Chem 1980, 23, 335–338. [DOI] [PubMed] [Google Scholar]
- (48).Kuppusamy UR; Das NP Potentiation of beta-adrenoceptor agonist-mediated lipolysis by quercetin and fisetin in isolated rat adipocytes. Biochem. Pharmacol 1994, 47, 521–529. [DOI] [PubMed] [Google Scholar]
- (49).Raghavan K; Buolamwini JK; Fesen MR; Pommier Y; Kohn KW; Weinstein JN Three-dimensional quantitative structure-activity relationship (QSAR) of HIV integrase inhibitors: a comparative molecular field analysis (CoMFA) study. J. Med. Chem 1995, 38, 890–897. [DOI] [PubMed] [Google Scholar]