

Summary
Background
Patients with non-alcoholic steatohepatitis (NASH)-related cirrhosis are at high risk of liver-related and all-cause morbidity and mortality. We investigated the efficacy and safety of the glucagon-like peptide-1 analogue semaglutide in patients with NASH and compensated cirrhosis.
Methods
This double-blind, placebo-controlled phase 2 trial enrolled patients from 38 centres in Europe and the USA. Adults with biopsy-confirmed NASH-related cirrhosis and body-mass index (BMI) of 27 kg/m2 or more were randomly assigned (2:1) to receive either once-weekly subcutaneous semaglutide 2·4 mg or visually matching placebo. Patients were randomly allocated via an interactive web response system, stratified by presence or absence of type 2 diabetes. Patients, investigators, and those assessing outcomes were masked to treatment assignment. The primary endpoint was the proportion of patients with an improvement in liver fibrosis of one stage or more without worsening of NASH after 48 weeks, assessed by biopsy in the intention-to-treat population. Safety was assessed in all patients who received at least one dose of study drug. The trial is closed and completed, and registered with ClinicalTrials.gov, number NCT03987451.
Findings
71 patients were enrolled between June 18, 2019, and April 22, 2021; 49 (69%) patients were female and 22 (31%) were male. Patients had a mean age of 59·5 years (SD 8·0) and mean BMI of 34·9 kg/m2 (SD 5·9); 53 (75%) patients had diabetes. 47 patients were randomly assigned to the semaglutide group and 24 to the placebo group. After 48 weeks, there was no statistically significant difference between the two groups in the proportion of patients with an improvement in liver fibrosis of one stage or more without worsening of NASH (five [11%] of 47 patients in the semaglutide group vs seven [29%] of 24 in the placebo group; odds ratio 0·28 [95% CI 0·06–1·24; p=0·087). There was also no significant difference between groups in the proportion of patients who achieved NASH resolution (p=0·29). Similar proportions of patients in each group reported adverse events (42 [89%] patients in the semaglutide group vs 19 [79%] in the placebo group) and serious adverse events (six [13%] vs two [8%]). The most common adverse events were nausea (21 [45%] vs four [17%]), diarrhoea (nine [19%] vs two [8%]), and vomiting (eight [17%] vs none). Hepatic and renal function remained stable. There were no decompensating events or deaths.
Interpretation
In patients with NASH and compensated cirrhosis, semaglutide did not significantly improve fibrosis or achievement of NASH resolution versus placebo. No new safety concerns were raised.
Introduction
Patients with non-alcoholic steatohepatitis (NASH)-related cirrhosis are at particularly high risk of developing potentially life-threatening liver-related morbidities, such as portal hypertension, hepatic decompensation, hepatocellular carcinoma, liver-related mortality, and cardiovascular events.1–4 By 2030, advanced liver disease amongst patients with NASH is expected to rise by 160% to nearly 8 million cases in the USA, leading to an estimated 3·5 million cases of cirrhosis and more than 100 000 cases of decompensated disease.5 This increase in prevalence is associated with an ageing population and increased incidence of metabolic syndrome related to the epidemic rise in obesity and type 2 diabetes.5 Indeed, approximately 71% of patients with NASH-related cirrhosis have type 2 diabetes,6 and suboptimal glycaemic control is a marker of advanced disease and adverse clinical outcomes.7,8
Patients with NASH-related cirrhosis have a high unmet need for effective pharmacotherapies to improve the natural history of this disease, including the associated increased risk for cardiovascular morbidity and mortality, yet there are currently no approved pharmacotherapies for the treatment of NASH.9 Currently, first-line treatment in patients with compensated cirrhosis and overweight or obesity involves lifestyle interventions to safely achieve weight loss and treat comorbidities (eg, hyperlipidaemia, hypertension, and diabetes).10,11 Despite this, there is limited evidence that lifestyle modification or treating comorbidities in patients with cirrhosis improves liver-related morbidity or mortality.12 From a liver perspective, the main aim of treatment is to prevent progression of cirrhosis to end-stage liver disease (hepatocellular carcinoma and liver failure, which are increasingly likely with increasing fibrosis13), for which liver transplantation remains the only curative treatment option.10,11 Unfortunately, due to obesity and metabolic and cardiovascular comorbidities, many patients may not be listed for liver transplantation.14
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) exert multiorgan effects and have been shown to lower HbA1c in people with type 2 diabetes and reduce bodyweight in individuals with overweight or obesity, and are associated with a reduced risk of adverse cardiovascular outcomes in patients with diabetes at high cardiovascular risk.15–18 It has been suggested that GLP-1RAs may have hepatoprotective effects.19 In a previous placebo-controlled trial, the GLP-1RA semaglutide improved metabolic parameters and NASH resolution and was well tolerated in patients with NASH without cirrhosis (fibrosis stage [F] 1–3).20 However, there are at present no data with semaglutide in patients with NASH-related cirrhosis. Therefore, we investigated the efficacy and safety of semaglutide in patients with NASH and compensated cirrhosis.
Methods
Study design and participants
This multinational, multicentre, randomised, double-blind, placebo-controlled, phase 2 trial was conducted in 38 centres in Europe and the USA. Before trial initiation, the protocol, consent form, and patient information sheet were reviewed and approved according to local regulations and by an independent ethics committee/review board. Patients provided written, informed consent before participating in the trial. The trial was conducted in accordance with the principles of the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice guidelines.
Eligible patients were male or female, aged 18–75 years (both inclusive) at the time of signing informed consent, and had histological evidence of NASH and Kleiner F4 according to the NASH Clinical Research Network (CRN) classification,21 based on single central pathologist evaluation of a liver biopsy obtained within 360 days before screening. In patients who had never had a liver biopsy showing NASH and F4, a liver stiffness of greater than 14 kPa by FibroScan at screening enabled selection for a trial-related liver biopsy. Further inclusion criteria were a histological non-alcoholic fatty liver disease (NAFLD) activity score (NAS) of 3 or more with a score of 1 or more for both lobular inflammation and hepatocyte ballooning, and a body-mass index (BMI) of 27 kg/m2 or greater.
Among the key exclusion criteria were presence or history of: hepatic decompensation (eg, ascites, variceal bleeding, hepatic encephalopathy, or spontaneous bacterial peritonitis) or liver transplantation; hepatocellular carcinoma; and gastro-oesophageal varices within the past 360 days before screening. Esophagogastroduodenoscopy was performed on patients with no known history of gastro-oesophageal varices but who had FibroScan of 20 kPa or more and thrombocyte count less than 150 000 per μL, in accordance with Baveno VI guidelines.22 Patients were also excluded if they were treated with: vitamin E (doses ≥800 IU/day) or pioglitazone, if not at a stable dose in the period from 90 days before screening; a GLP-1RA in the 90 days before screening; or other glucose-lowering agent(s) or weight loss medication not at a stable dose in the opinion of the investigator in the 28 days before screening. Full eligibility criteria are provided in the appendix (pp 14–15).
Randomisation and masking
Randomisation was done centrally using an interactive web response system and stratified for presence or absence of type 2 diabetes. Calyx (formerly Parexel) generated the randomisation list. Randomisation was done in a 2:1 ratio to the semaglutide or placebo group with a block size of six; patients were assigned to the next available treatment according to a randomisation schedule. Patients, investigators, trial site staff, and the sponsor (except for specific laboratory staff and individuals responsible for safety) remained blinded to treatment assignment throughout the trial. Semaglutide and placebo injections were visually identical—ie, used the same syringes and volume of injection, etc—to preserve blinding.
Procedures
Patients assigned to the semaglutide group received once-weekly subcutaneous semaglutide and those assigned to the placebo group received once-weekly, subcutaneous placebo for 48 weeks with a 7-week follow-up. Semaglutide was escalated from an initial dose of 0·24 mg to 0·5 mg after 4 weeks, and thereafter every 4 weeks to 1·0 mg, 1·7 mg, and finally 2·4 mg once weekly after 16 weeks’ treatment (appendix p 4). Dietary and lifestyle advice was given as standard of care according to local standards. During dose escalation, patients could remain at their existing level for up to 1 additional week for tolerability (eg, gastrointestinal events) or other reasons, as judged by the investigator. Patients were removed from the trial if any of the following criteria were met: simultaneous participation in another clinical trial, diagnosis of acute pancreatitis or medullary thyroid carcinoma, surgical treatment for obesity, or events of hepatic decompensation (eg ascites, variceal bleeding, hepatic encephalopathy, or spontaneous bacterial peritonitis).
A screening visit (6 weeks before randomisation) to assess patient eligibility was followed by visits or telephone contacts every second week during the first 12 weeks of the dose-escalation period. At the randomisation visit, patients attended in a fasting state and received instructions on trial product administration, completion of the study diary, and diet and lifestyle advice; patients with type 2 diabetes received a blood glucose meter. All patients underwent an MRI scan analysed by a central imaging supplier (blinded to treatment), and biosamples were collected from a selection of patients for exploratory biomarker analyses.
From week 12 until end of treatment, four visits were scheduled with an increasing time interval from 4 to 12 weeks between visits. At these visits, discontinuation criteria were evaluated; diaries were collected, reviewed, and transcribed; and diet and lifestyle advice was provided. All on-site visits (except for visits 4, 6, 9, 11, and 13) were attended in a fasting state. An end-of-trial follow-up visit for safety assessments was scheduled 7 weeks after end of treatment. Patients who prematurely discontinued treatment had a follow-up visit scheduled 48 weeks after randomisation.
The baseline liver biopsy was either retrospective (taken in the preceding 12 months) or performed de novo during the screening period, and all patients who completed treatment underwent a further biopsy 48 weeks after randomisation. Histology was assessed at a central site by one pathologist who was blinded to visit, patient characteristics, and treatment, but not time. Fibrosis was measured by MRI using magnetic resonance elastography (MRE) and steatosis was measured by MRI proton density fat fraction (MRI-PDFF).23 Pathologist evaluation included presence or absence of NASH, fibrosis stage, lobular inflammation, hepatocyte ballooning and steatosis, NAS, Ishak fibrosis score, steatosis-activity-fibrosis (SAF) score, and hepatic collagen content assessed via morphometry (collagen proportionate area).
Liver fat volume was calculated based on assessment of steatosis and liver volume assessed by MRI. Child–Pugh score was calculated,24 bodyweight and height recorded to calculate BMI, and waist circumference was measured. Blood samples were analysed for levels of the biomarkers pro-collagen 3 peptide (pro-C3), pro-C3 amino terminal peptide, total adiponectin, hyaluronic acid, tissue inhibitor of metalloproteinase-1 (TIMP-1), and high-sensitivity C-reactive protein (hsCRP). Levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma glutamyltransferase, as well as cardiometabolic parameters, were recorded.
Adverse events, either observed by the investigator or reported by patients, were recorded and evaluated for severity (mild, moderate, severe), seriousness, duration, outcome, and possible relationship to the study treatment. Data from physical examinations, vital signs, electrocardiograms, and clinical laboratory tests were recorded (appendix p 2).
Outcomes
The primary endpoint was the proportion of patients with an improvement in liver fibrosis of one stage or more on biopsy (using the NASH CRN classification) without worsening of NASH after 48 weeks. Worsening of NASH was a worsening of one grade or more of either lobular inflammation, hepatocyte ballooning, or steatosis, as defined by the NASH CRN. The primary endpoint was evaluated prospectively by an expert liver pathologist. In addition, a post-hoc exploratory analysis was undertaken using machine learning software developed by PathAI (Boston, MA, USA).
Primary endpoint assessment was changed from MRE to liver biopsy on Feb 21, 2020, after completion of the study protocol but before unblinding of trial data. This change was to align with new regulatory standards for NASH trials,25,26 introduced in 2018 during trial conduct, that specify endpoints deemed relevant clinical outcomes in NASH-related cirrhosis.
Secondary endpoints (all measured from baseline to week 48) were: relative change in liver fat content measured by MRI-PDFF and in liver stiffness measured by MRE; change in NASH resolution on biopsy (assessed by the liver pathologist and, post-hoc, by machine learning software), and in stage of fibrosis and NAS according to NASH CRN criteria; and number of treatment-emergent adverse events.
Exploratory endpoints were evaluation of the primary endpoint and NASH resolution by PathAI machine learning software, and changes from baseline in the biomarkers pro-C3, pro-C3 amino terminal peptide, total adiponectin, hyaluronic acid, TIMP-1, enhanced liver fibrosis (ELF) score, and hsCRP.
Statistical analysis
Although a sample size calculation was performed, there is currently no guidance on minimum treatment effect on histological endpoints that would be considered clinically relevant. Assuming 2:1 randomisation, a treatment ratio of 0·85 and a coefficient of variation of 0·17, with a 20% withdrawal rate in both groups, a total sample size of 69 participants was considered sufficient to provide 90% power to detect a difference between semaglutide and placebo at the 5% significance level for the initially defined primary endpoint. Following the change in the primary endpoint, there was no guidance on the minimum treatment effect on histological endpoints that would be considered clinically relevant. For the originally calculated sample size, assuming a semaglutide responder proportion of 35% and a placebo responder proportion of 10%, a power of 62% to observe a treatment in the binary histology endpoint was calculated.
Efficacy outcomes were assessed using intention-to-treat analysis, in all randomised patients. Safety outcomes were assessed in all patients exposed to at least one dose of randomised treatment. Patients were eligible for analysis based on the following: visit or measurement done; fasting status; screening or baseline measurements no later than first dose; visit scheduled; re-test rules followed if several observations for a given visit; and visit reallocation only for values collected at premature treatment discontinuation visits. The primary analysis was based on a Cochran–Mantel–Haenszel test based on all randomised patients for the in-trial period with missing data handled as non-responders. The common odds ratio between semaglutide and placebo, adjusting for baseline type 2 diabetes, was estimated along with exact 95% CI. To test for superiority, the exact two-sided p-value was calculated as the sum of probabilities of outcomes having equal or lower probability than the observed outcome under the null hypothesis.
A sensitivity analysis was performed in which missing data were handled by reference-based multiple imputation informed by data from placebo recipients. A supportive complete case on-treatment analysis, in which patients with missing week 48 data or for whom the data were collected after the on-treatment period were excluded, was also conducted.
Continuous endpoints were analysed using an analysis of covariance (ANCOVA) with missing outcomes handled by unconditional reference-based imputation. For each of 500 complete data sets, the treatment effect on change from baseline to week 48 was estimated with treatment and baseline diabetes status as factors, and baseline bodyweight and baseline biomarker as covariates. All parameters, except NAFLD and ELF scores, were logarithmically transformed and estimated treatment differences (ETDs) were back-transformed to the original scale as estimated treatment ratios (ETRs). The ETDs and SE were pooled and SE, 95% CIs for treatment difference, and associated two-sided p-values were calculated.
For the secondary endpoints, ordinal histological features were analysed by ordered logistic regression with the histological scores at week 48 as response; treatment, baseline diabetes status as factors; and baseline bodyweight and corresponding histological score at baseline as covariates. Change in hepatic collagen from baseline to week 48 was analysed using ANCOVA and a mixed model for repeated measures. Binary histological endpoints were analysed in the same way as the primary endpoint. Safety was analysed descriptively. See the appendix (p 3) for further information.
All statistical analyses were performed with SAS (version 9.4). The trial is closed and completed, and is registered with ClinicalTrials.gov, number NCT03987451.
Role of the funding source
The sponsor was responsible for the trial design, preparing the trial protocol and the statistical analysis plan, performing the statistical analyses, and analysis of the results.
Results
71 patients were enrolled between June 18, 2019, and April 22, 2021. The study was completed on June 10, 2021. 47 patients were randomly assigned to the semaglutide group and 24 to the placebo group. 64 (90%) patients completed treatment, of whom 63 had evaluable paired biopsies for primary endpoint assessment (61 on treatment; figure 1). Of the 71 patients, 47 had a screening biopsy with a collection date of more than 6 weeks before the randomisation date. All 71 patients were included in both the full and safety analysis sets. Patients were mainly female (49 [69%]) and white (62 [87%]), with a mean age of 59·5 years (SD 8·0), a mean BMI of 34·9 kg/m2 (SD 5·9), and mean NAS of 4·8 (SD 1·0); 53 (75%) patients had type 2 diabetes at baseline, with a mean HbA1c of 7·1% (SD 1·3; table 1). Histological parameters were generally balanced between treatment groups. More than three-quarters of patients had an Ishak score of 6/6, the mean baseline MELD score was 7·6 (SD 1·8), and mean albumin level was 4·2 g/dL (SD 0·3; table 1). Baseline liver parameters were also generally well balanced between treatments (table 1). Use of glucose-lowering medication was also generally well balanced between the groups and is shown in the appendix (p 16).
Figure 1:
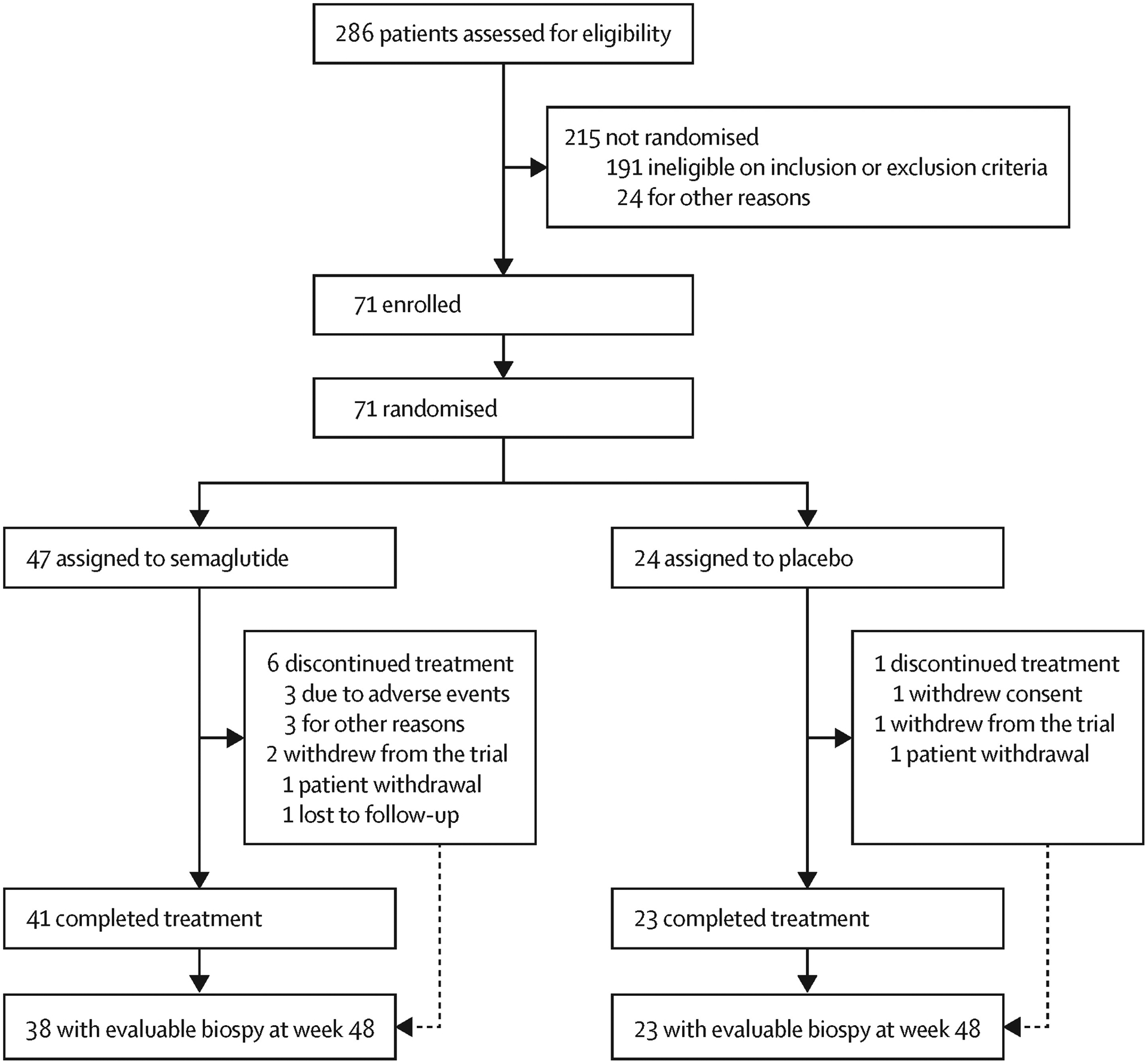
Trial profile
Table 1.
Patient baseline characteristics
| Empty Cell | Empty Cell | Semaglutide 2·4 mg group (n=47) | Placebo group (n=24) |
|---|---|---|---|
| Sex | |||
| Female | 31 (66%) | 18 (75%) | |
| Male | 16 (34%) | 6 (25%) | |
| Age, years | 59·9 (7·1) | 58·7 (9·7) | |
| Ethnicity | |||
| American Indian/Alaska Native | 1 (2%) | 0 | |
| Asian | 1 (2%) | 0 | |
| Black/African American | 0 | 2 (8%) | |
| White | 41 (87%) | 21 (88%) | |
| Other | 1 (2%) | 0 | |
| Not reported | 3 (6%) | 1 (4%) | |
| Bodyweight, kg | 95·2 (18·7) | 98·6 (22·2) | |
| BMI, kg/m2 | 34·6 (5·9) | 35·5 (6·0) | |
| Type 2 diabetes | 35 (75%) | 18 (75%) | |
| HbA1c, % | 7·1 (1·3) | 7·2 (1·2) | |
| Lipids, mg/dL | |||
| LDL cholesterol | 100·0 (34·4) | 88·1 (41·7) | |
| HDL cholesterol | 44·7 (10·0) | 45·8 (12·6) | |
| VLDL cholesterol | 32·5 (17·4) | 29·6 (11·0) | |
| Total cholesterol | 177·2 (34·9) | 163·4 (47·5) | |
| Free fatty acids | 15·6 (7·9) | 15·9 (8·1) | |
| Triglycerides | 168·9 (98·3) | 151·6 (56·2) | |
| Blood pressure, mm Hg | |||
| Diastolic | 78·7 (9·6) | 87·0 (6·6) | |
| Systolic | 132·6 (13·7) | 135·8 (14·7) | |
| Steatosis | |||
| 1 | 32 (68%) | 15 (63%) | |
| 2 | 12 (26%) | 7 (29%) | |
| 3 | 3 (6%) | 2 (8%) | |
| Lobular inflammation | |||
| 1 | 14 (30%) | 6 (25%) | |
| 2 | 31 (66%) | 17 (71%) | |
| 3 | 2 (4%) | 1 (4%) | |
| Hepatocyte ballooning | |||
| 1 | 18 (38%) | 8 (33%) | |
| 2 | 29 (62%) | 16 (67%) | |
| Ishak score* | |||
| 4 | 0 (0%) | 1 (4%) | |
| 5 | 9 (19%) | 6 (25%) | |
| 6 | 38 (81%) | 17 (71%) | |
| Total NAFLD activity score | 4·7 (1·0) | 4·9 (1·2) | |
| Hepatic collagen proportion | 11·5 (7·3) | 9·4 (4·8) | |
| Imaging, geometric mean (CV) | |||
| MRE, kPa | 6·4 (27·9) | 5·8 (30·7) | |
| MRI-PDFF, % | 10·0 (58·3) | 10·4 (54·7) | |
| Liver enzymes, U/L, geometric mean (CV) | |||
| ALT | 47·6 (59·0) | 36·4 (57·3) | |
| AST | 47·2 (45·7) | 39·0 (46·0) | |
| GGT | 94·3 (85·1) | 95·0 (133·5) | |
| Exploratory biomarkers, geometric mean unless stated | |||
| ELF | 10·7 (0·8) | 10·6 (0·7) | |
| Pro-C3, ng/mL (CV) | 20·4 (31·3) | 17·9 (26·1) | |
| Pro-C3 N-terminal peptide, ng/mL | 21·5 (8·6) | 18·5 (5·3) | |
| FIB-4, score (CV) | 2·4 (38·3) | 2·2 (54·2) | |
| Total adiponectin, μg/mL (CV) | 3·3 (69·0) | 4·2 (94·9) | |
| TIMP-1, ng/mL | 340·3 (83·0) | 350·8 (106·7) | |
| Hyaluronic acid, ng/mL | 188·9 (156·5) | 154·9 (92·9) | |
| Liver severity | |||
| MELD score | 7·6 (1·2) | 7·7 (2·6) | |
| Child–Pugh classification | 5·0 (0·1) | 5·0 (0·0) | |
| Albumin, g/dL | 4·2 (0·3) | 4·2 (0·3) | |
| Bilirubin, mg/dL | 0·3 (0·1) | 0·3 (0·1) | |
| INR | 1·1 (0·1) | 1·1 (0·4) | |
| Sodium, mmol/L | 140·2 (2·3) | 139·7 (2·5) | |
| Thrombocytes, 109/L | 178·4 (50·5) | 183·5 (63·1) | |
Data are n (%) or mean (SD) unless otherwise stated. Data based on full analysis set. ALT=alanine aminotransferase. AST=aspartate aminotransferase. BMI=body-mass index. CV=coefficient of variance. ELF=enhanced liver fibrosis. FIB-4=fibrosis-4 index. GGT=gamma glutamyltransferase. INR=international normalised ratio. MRE=magnetic resonance elastography. MRI-PDFF=MRI proton density fat fraction. NAFLD=non-alcoholic fatty liver disease. Pro-C3=pro-collagen 3 peptide. TIMP-1=tissue inhibitor of metalloproteinase-1.
Ishak score was not one of the inclusion criteria for this study.
There was no significant difference between groups in the proportion of patients with improvement in liver fibrosis and no worsening of NASH after 48 weeks (five [11%] of 47 patients in the semaglutide group vs seven [29%] of 24 patients in the placebo group; odds ratio 0·28 [95% CI 0·06–1·24]; p=0·087; figure 2). Outcomes were similar in the sensitivity and supportive analyses (appendix p 17). There was also no significant difference between treatments for the proportion of patients with NASH resolution (16 [34%] vs five [21%]; odds ratio 1·97 [95% CI 0·56–7·91]; p=0·29; figure 2). A lower proportion of patients achieved resolution of NASH and improvement in liver fibrosis at week 48 with semaglutide versus placebo, although this difference was not significant (three [6%] vs three [13%]; odds ratio 0·48 [95% CI 0·06–3·91]; p=0·40).
Figure 2: Improvement in liver fibrosis and no worsening of NASH (A) and resolution of NASH (B) at 48 weeks.
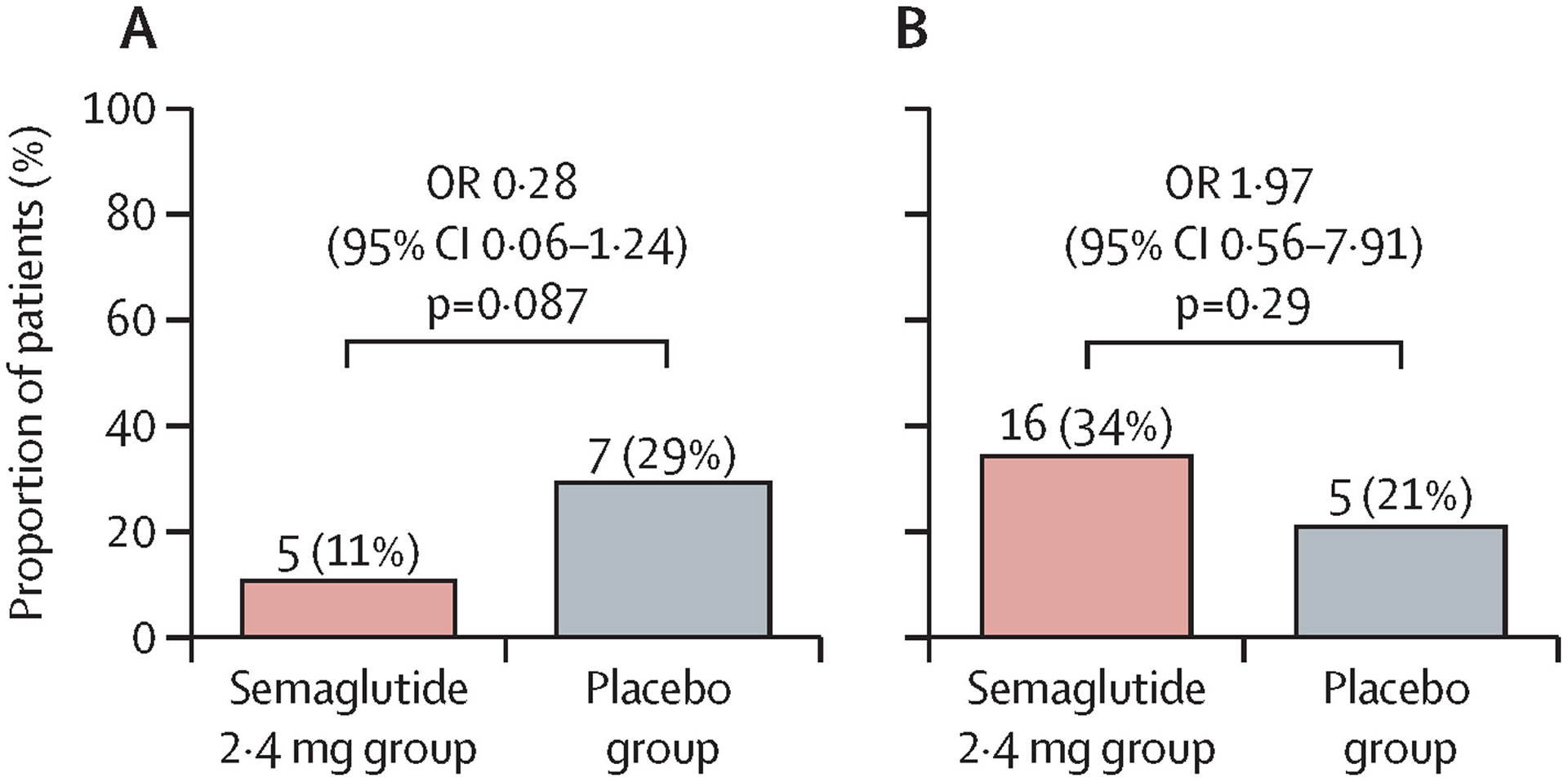
p-values are two-sided and taken from a Cochran–Mantel–Haenszel test stratified by baseline diabetes status. Patients with missing outcomes were imputed as non-responders. NASH=non-alcoholic steatohepatitis. OR=odds ratio.
Exploratory assessment by PathAI machine learning software also showed no significant differences between treatments for improvement in liver fibrosis and no worsening of NASH after 48 weeks (three [7%] vs two [8%]; p=1·0) or NASH resolution (eight [17%] vs two [8%]; p=0·50; appendix p 5).
Outcomes for ordinal histological endpoints are shown in the appendix (pp 6–8). A lower proportion of patients had evaluable biopsies in the semaglutide group versus the placebo group (38 [81%] vs 23 [96%]) but all patients were included in the evaluation of histological endpoints. A lower proportion of patients had an improvement in liver fibrosis stage (NASH CRN and Ishak scores) with semaglutide versus placebo (appendix pp 6–8).
There was no significant difference between treatments for components of NASH. 21 (45%) patients in the semaglutide group had an improvement in steatosis grade compared with eight (33%) in the placebo group (p=0·45), and a lower proportion experienced worsening of steatosis (one [2%] vs four [17%]). In the semaglutide group, 20 (43%) patients had an improvement in lobular inflammation compared with nine (38%) in the placebo group (p=0·80); similar proportions of patients in both groups had worsening of lobular inflammation (six [13%] vs three [13%]). A higher proportion of patients (26 [55%]) in the semaglutide group had an improvement in hepatocyte ballooning compared with the placebo group (eight [33%]; p=0·088); one (2%) patients in the semaglutide group and two (8%) in the placebo group experienced worsening. NAS improvement was achieved by 29 (62%) patients who received semaglutide versus 14 (58%) in the placebo group (p=0·80), whereas one (2%) in the semaglutide group and four (17%) in the placebo group had a worsening of NAS. Finally, in the semaglutide group, 27 (57%) patients had an improvement in SAF score versus 12 (50%) in the placebo group (p=0·61); four (9%) patients in the semaglutide group and three (13%) in the placebo group had a worsening of SAF.
Hepatic collagen decreased from baseline to week 48 in both the semaglutide and placebo groups, albeit from a higher baseline level in the semaglutide group (appendix p 18).
At week 48, change in liver stiffness (assessed by MRE) from baseline was not significantly different between groups (ETR 0·93 [95% CI 0·80–1·07]; p=0·30; figure 3; appendix p 18). At week 48, improvement in liver steatosis (assessed by MRI-PDFF) from baseline was significantly greater in the semaglutide group than in the placebo group (ETR 0·67 [95% CI 0·51–0·88]; p=0·0042; figure 3; appendix p 18). In the semaglutide group, 23 (49%) patients had a 30% or greater reduction in steatosis compared with three (13%) patients in the placebo group (odds ratio 6·58 [95% CI 1·63–39·31]; p=0·0037). At week 48, reduction in liver fat volume and thus total liver volume was significantly greater in the semaglutide group than in the placebo group (appendix pp 9, 18).
Figure 3: Change in imaging parameters and liver enzymes from baseline to week 48.
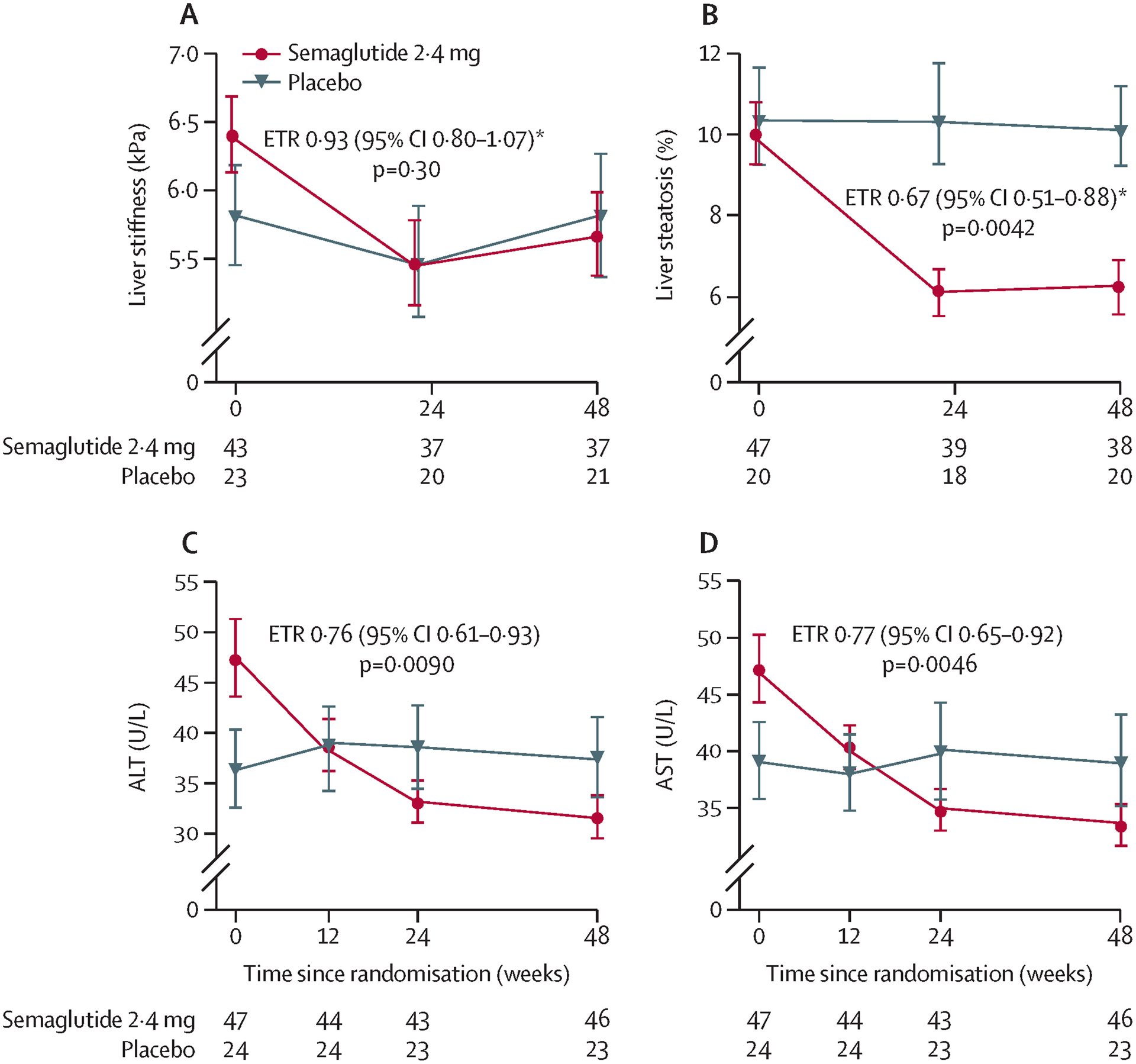
Liver stiffness assessed by MRE (A), liver steatosis assessed by MRI-PDFF (B), ALT (C), and AST (D). Number of observations per treatment group and visit is presented in the lower part of each plot. Error bars show the SE of the mean for observed values. ALT=alanine aminotransferase. ANCOVA=analysis of covariance. AST=aspartate aminotransferase. ETR=estimated treatment ratio. MRE=magnetic resonance elastography. MRI-PDFF=MRI proton density fat fraction. *ETRs with 95% CI and two-sided p-values were calculated using the same ANCOVA analysis. Missing data were imputed from the observed data in the placebo group using the same ANCOVA model but without treatment as factor.
At week 48, change in ALT concentration from baseline was significantly greater in the semaglutide group than in the placebo group (ETR 0·76 [95% CI 0·61–0·93]; p=0·0090; figure 3; appendix p 18). In a post-hoc analysis, a significantly greater proportion of patients receiving semaglutide had a clinically significant decrease of 17 units in ALT27 compared with placebo (19 [40%] vs two [8%]; p=0·0057), and had both a 17-unit ALT plus a 30% or greater MRI-PDFF decrease (14 [30%] vs one [4%]; p=0·013). Similar improvements were seen with semaglutide compared with placebo for both AST concentrations (ETR 0·77 [95% CI 0·65–0·92]; p=0·0046; figure 3; appendix p 18) and gamma glutamyltransferase (ETR 0·74 [95% CI 0·62–0·88]; p=0·0007; appendix p 18).
Pro-C3 (ETR 0·84 [95% CI 0·73 to 0·98]; p=0·027) and hsCRP (ETR 0·59 [95% CI 0·40 to 0·87]; p=0·0072) levels were significantly reduced after treatment with semaglutide versus placebo (appendix p 10). ELF decreased from baseline to week 48 in the semaglutide group by −0·44 and in the placebo group by −0·13; the difference between groups was not significant (ETD −0·31 [95% CI −0·69 to 0·07; p=0·12).
Bodyweight decreased from baseline by a greater extent in patients treated with semaglutide (relative change from baseline −8·83% in the semaglutide group vs −0·09% in the placebo group); the difference between groups was significant (ETD −8·75 [95% CI −12·41 to −5·09]; p<0·0001; figure 4). The proportion of patients who achieved a 5% or greater (29 [62%] vs six [25%]; p=0·0047) and 10% or greater (19 [40%] vs none; p=0·016) weight reduction at week 48 was significantly higher with semaglutide than placebo. BMI and waist circumference were also significantly lower with semaglutide versus placebo at week 48 (appendix pp 19–20).
Figure 4: Change in (A) bodyweight and (B) HbA1c (in patients with type 2 diabetes) from baseline to week 48.
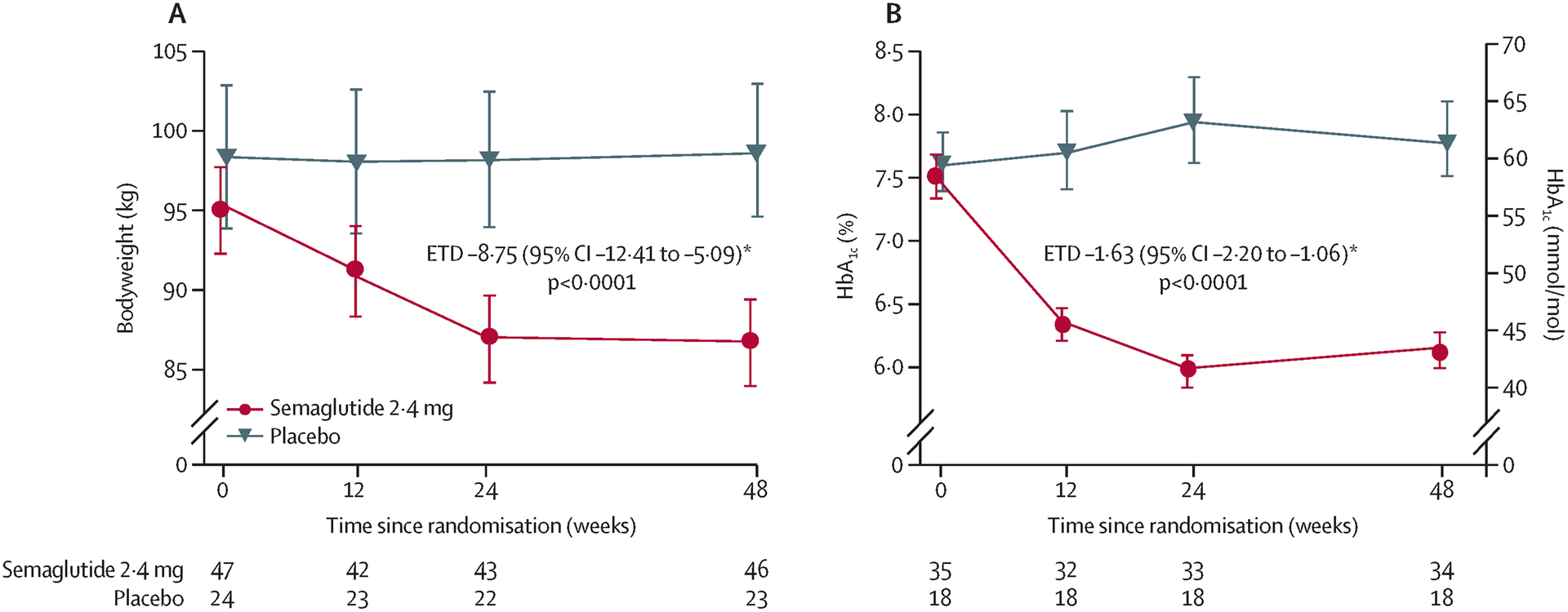
Number of observations per treatment group and visit is presented in the lower part of each plot. Error bars show the SE of the mean for observed values. ANCOVA=analysis of covariance. ETD=estimated treatment difference. *ETDs with 95% CI and two-sided p-values were calculated using the same ANCOVA analysis. Missing data were imputed from the observed data in the placebo group using the same ANCOVA model but without treatment as factor.
At week 48, HbA1c had decreased from baseline among patients with type 2 diabetes in the semaglutide group but not in the placebo group (mean −1·39% vs +0·24%); the difference between groups was significant (ETD −1·63 [95% CI −2·20 to −1·06]; p<0·0001; figure 4). Fasting plasma glucose was also significantly reduced from baseline to week 48 (ETD −2·32 mmol/L [95% CI −3·74 to −0·90]; p=0·001), but fasting C-peptide was not (ETR 0·94 [95% CI 0·71–1·25]; p=0·67), with semaglutide compared with placebo.
At week 48, reductions in levels of triglycerides and VLDL cholesterol from baseline were significantly greater with semaglutide than with placebo (ETR 0·83 [95% CI 0·72–0·96]; p=0·013 for triglycerides and 0·83 [95% CI 0·72–0·96]; p=0·012 for VLDL cholesterol), but this was not the case, for example, for total cholesterol (appendix pp 11, 19–20). Blood pressure was reduced from baseline in patients who received semaglutide at 24 weeks but increased again and was not significantly different to placebo at 48 weeks (appendix pp 19–20).
Similar proportions of patients experienced adverse events (42 [89%] patients in the semaglutide group and 19 [79%] in the placebo group, of which six [13%] and two [8%], respectively, were serious; table 2). No serious events were considered related to trial product and there were no deaths. Most adverse events were mild or moderate in severity but there were eight severe events in the semaglutide group versus one in the placebo group; additionally, more adverse events in the semaglutide group than in the placebo group were judged as possibly or probably related to trial product (table 2). No patients withdrew from the trial due to adverse events. A total of five patients had eight adverse events leading to dose reduction (six adverse events in four patients in the semaglutide group, and two adverse events in one patient in the placebo group). Three patients had adverse events leading to premature treatment discontinuation (two with gastrointestinal disorders [nausea] considered probably related to treatment and one with an eye disorder [vitreous detachment] considered unlikely related to treatment), all in the semaglutide group. In total, 64 (90%) patients completed treatment, and three patients (two in the semaglutide group and one in the placebo group) withdrew from the trial.
Table 2.
Adverse events
| Empty Cell | Empty Cell | Semaglutide 2·4 mg group (n=47) | Placebo group (n=24) |
|---|---|---|---|
| On-treatment observation period, weeks | 51·1 (9·9) | 53·5 (5·9) | |
| All patients with adverse events | 42 (89%) | 19 (79%) | |
| Gastrointestinal disorders | 36 (77%) | 8 (33%) | |
| Nausea | 21 (45%) | 4 (17%) | |
| Diarrhoea | 9 (19%) | 2 (8%) | |
| Vomiting | 8 (17%) | 0 (0%) | |
| Abdominal pain | 6 (13%) | 1 (4%) | |
| Decreased appetite | 6 (13%) | 1 (4%) | |
| Eructation | 6 (13%) | 0 (0%) | |
| Abdominal pain upper | 5 (11%) | 2 (8%) | |
| Dyspepsia | 5 (11%) | 0 (0%) | |
| Patients with other adverse events (≥10% in either treatment group) | |||
| Urinary tract infection | 3 (6%) | 4 (17%) | |
| Back pain | 0 | 3 (13%) | |
| Patients with serious adverse events | |||
| On-treatment | 6 (13%) | 2 (8%) | |
| In-trial | 6 (13%) | 2 (8%) | |
| Serious adverse events | |||
| Gastrointestinal disorders | 3 (6%) | 2 (8%) | |
| Eye disorders | 1 (2%) | 0 | |
| Hepatobiliary disorders | 1 (2%) | 0 | |
| Injury, poisoning, and procedural complications | 1 (2%) | 0 | |
| Metabolism and nutrition disorders | 1 (2%) | 0 | |
| Nervous system disorders | 1 (2%) | 0 | |
| Renal and urinary disorders | 1 (2%) | 0 | |
| Respiratory, thoracic, and mediastinal disorders | 1 (2%) | 0 | |
| Musculoskeletal and connective tissues disorders | 0 | 1 (4%) | |
| Psychiatric disorders | 0 | 1 (4%) | |
| Fatal events | 0 | 0 | |
| Severity* | |||
| Mild | 37 (79%) | 17 (71%) | |
| Moderate | 26 (55%) | 10 (42%) | |
| Severe | 8 (17%) | 1 (4%) | |
| Relationship to trial product* | |||
| Probable | 22 (47%) | 4 (17%) | |
| Possible | 23 (49%) | 9 (38%) | |
| Unlikely | 31 (66%) | 18 (75%) | |
| Leading to withdrawal of trial product | 3 (6%) | 0 | |
| Leading to withdrawal from trial | 0 | 0 | |
Data are n (%) unless otherwise stated. Data are from all exposed patients on treatment (safety analysis set). %=proportion of patients with at least one adverse event. Numbers are patients with at least one adverse event.
Patients could have more than one adverse event.
As expected, the most common adverse events associated with semaglutide were gastrointestinal (table 2). The most frequently reported gastrointestinal adverse events were mild-to-moderate transient nausea, diarrhoea, and vomiting, which mainly occurred during treatment initiation or dose escalation (appendix p 12). The median durations of nausea, diarrhoea, and vomiting were 8 days (IQR 3–341), 7 days (3–34), and 3 days (1–41), respectively, in patients treated with semaglutide. Among the 53 patients with type 2 diabetes, on-treatment hypoglycaemic adverse events (as per the American Diabetes Association classification) were reported for 12 (34%) patients in the semaglutide group and five (28%) in the placebo group, of which only one event with semaglutide (and none with placebo) was classed as a severe symptomatic episode. No hypoglycaemic episodes were reported for patients without type 2 diabetes.
Hepatic function remained stable after semaglutide treatment and did not result in decompensating events. Ten hepatic events were identified in six patients, nine of which were in five patients who received semaglutide (appendix p 21). All events were non-serious and mild or moderate in severity. The MELD score fluctuated during the trial in both treatment groups but was similar at week 48 (appendix p 13). The change in MELD scores between baseline and week 48 was not clinically meaningful in either group (mean 0·2 [SE 1·3] in the semaglutide group and 0·1 [3·1] in the placebo group); there were no MELD scores over 15 points at any stage during treatment. All patients with measurements were classed as Child–Pugh A at baseline and week 48. Renal function, as measured by estimated glomerular filtration rate, remained stable in patients who received semaglutide and decreased slightly with placebo (ratios to baseline 1·00 and 0·97, respectively; p=0·53).
In the semaglutide group, albumin increased transiently at week 24 but returned to baseline at week 48 and remained stable in the placebo group. Bilirubin increased with semaglutide but not placebo, while international normalised ratio increased slightly in both groups and thrombocytes remained unchanged (appendix p 22). None of the values at week 48 were significantly different from baseline.
Discussion
In this phase 2 study of patients with NASH-related compensated cirrhosis, semaglutide 2·4 mg once weekly did not significantly improve fibrosis or achievement of NASH resolution compared with placebo. However, in patients with cirrhosis, semaglutide did lead to improvements in cardiometabolic risk parameters (weight loss, glycaemic control, and lipids), did not lead to new safety concerns, and was well tolerated based on the established profile of the GLP-1RA class. Despite the lack of histological changes with semaglutide, improvements were seen in non-invasive markers of disease activity. We also noted a clinically significant reduction in liver fat by MRI-PDFF.
Addressing features of the metabolic syndrome is essential in patients with NASH-related cirrhosis. Not only are cardiovascular morbidity and mortality common in this population, but type 2 diabetes and obesity increase the risk of fibrosis progression, hepatic decompensation, and hepatocellular carcinoma.6–8 In this trial, semaglutide reduced bodyweight and HbA1c was decreased in patients with type 2 diabetes. This is reflective of findings seen in trials of semaglutide in people with type 2 diabetes and those with overweight or obesity.16–18 Semaglutide has also been shown to positively affect cardiovascular risk in patients with type 2 diabetes.15 Thus, semaglutide treatment may provide an opportunity to address multiple factors associated with adverse outcomes in advanced NASH.
The GLP-1RAs liraglutide and semaglutide have previously been investigated in patients with NASH, but mainly in those without cirrhosis. In the randomised, phase 2 LEAN trial in 52 patients with NASH and overweight, liraglutide 1·8 mg once daily led to NASH resolution in 39% of patients versus 9% with placebo (p=0·019) after 48 weeks.28 In a larger, placebo-controlled, randomised, 72-week, phase 2 trial in 320 patients with NASH F1–3, semaglutide 0·4 mg once daily led to a significantly greater proportion of patients achieving NASH resolution with no worsening of fibrosis versus placebo (59% vs 17%; p<0·001), with a non-significant difference in the proportion of patients with improvement in fibrosis stage (43% vs 33%; p=0·48).20
In the phase 2 trial of semaglutide in patients with NASH and F1–3, a maximum dose of 0·4 mg once daily was investigated,20 which contrasts with the 2·4 mg once weekly schedule used in the current trial. Semaglutide 2·4 mg once weekly has been shown to be effective and well tolerated for weight reduction in patients with overweight or obesity,18 and therefore was considered appropriate for this trial given the high disease burden of participants, including BMI of 27 kg/m2 or more. A once-weekly dosing schedule was also anticipated to be likely to improve the burden of drug administration compared with once-daily dosing. It was estimated that maximum plasma concentrations of semaglutide after once-weekly 2·4 mg doses would be similar to those achieved with 0·4 mg once daily (unpublished data).
Overall, the safety profile of semaglutide seen in this trial was consistent with previous trials in patients with type 2 diabetes,16,17 overweight or obesity,18 and NASH,20 with mild-to-moderate, transient gastrointestinal effects accounting for most on-treatment adverse events. We did not observe effects on hepatic or renal function with semaglutide treatment, and there were no decompensating events. Although the study was not powered to assess decompensating events, these data might be considered reassuring in the vulnerable patient population studied.
Strengths of this trial include its robust design and high completion rate. There were various limitations. The change in the primary endpoint may be considered a limitation, especially since the sample size calculation was done on the basis of MRE; thus, the study was likely underpowered with a relatively small size (61 patients with evaluable biopsy), which might have limited its power to detect a difference between treatments for the revised primary endpoint. The treatment duration of 48 weeks used in the current study is in line with other phase 2b and 3 trials in patients with NASH and compensated cirrhosis.29,30 However, this is a shorter duration than the 72-week treatment period used in the phase 2 trial of semaglutide in patients with NASH and F1–3.20 It may be that a longer duration of treatment, as seen with entecavir for patients with cirrhosis due to hepatitis B,31 could have provided more scope to establish if semaglutide had significant effects on NASH and its components, as well as fibrosis regression, in patients with NASH and compensated cirrhosis. Trials investigating the treatment of NASH over a longer follow-up period are currently ongoing (eg, NCT03439254) and could indicate whether a longer treatment duration has a significant effect on NASH and its components in patients with compensated cirrhosis.
It should also be noted that, although patient baseline characteristics were generally well balanced, a greater proportion of patients in the semaglutide group had Ishak score 6, whereas more placebo recipients had a score of 4 or 5, and hepatic collagen proportion was also higher in the semaglutide group. Furthermore, MRE score was higher in the semaglutide group and baseline levels of liver enzymes and pro-C3 were also somewhat higher with semaglutide versus placebo. It may be, therefore, that patients in the placebo group had a more heterogeneous, lower-grade fibrosis than the semaglutide group, and this may have affected the ability to show a treatment difference. The high rate of fibrosis improvement in the placebo group was similar to that seen in the trial of semaglutide in NASH F1–3,20 and may be related to sampling variability as well as the inherent inconsistency between conventional pathology assessments of treatment response in patients with NASH-related cirrhosis.
Restrictions due to the COVID-19 pandemic affected the conduct of the trial to some extent but both treatment groups were similarly impacted, primarily involving changing site visits to telephone or remote audio contacts. A limitation was the use of a single pathologist to assess histology slides, which were permitted to be up to 12 months or 360 days old, with no re-read of baseline assessments at the end of trial. This approach was originally intended to evaluate secondary histology endpoints only. When the primary endpoint was changed during the trial, it was not feasible to alter the pathology procedures. It should be emphasised that the change in the primary endpoint was done to align with evolving regulatory standards,25,26 and this happened before any unblinding and trial analysis. To support histology analysis, the primary endpoint and the secondary endpoint of NASH resolution were also assessed by machine learning software (PathAI), which assessed lower proportions of patients as having met these endpoints than with pathologist evaluation. This phenomenon has been observed previously with PathAI and is not fully understood, but may be driven by PathAI determining that the “no worsening of NASH” part of the endpoint was not fulfilled. Ultimately, machine learning software may facilitate greater consistency in the interpretation of histological outcomes in NASH.32
The mainstay of NASH treatment is weight loss.10,11 Patients who received semaglutide in the current trial had a mean weight loss of almost 9% from baseline without any evidence of a negative effect on safety. Weight-loss results in the current trial are similar to those observed in previous 72-week phase 2 trials of semaglutide in NASH F1–3 in which semaglutide treatment led to a mean weight change of −5% with semaglutide 0·1 mg once daily to −13% in the semaglutide 0·4 mg once daily group.20 It is, however, important that future studies understand the type of weight loss (adipose vs muscle; central vs peripheral), as detailed changes in body composition were not assessed in the current study. Sarcopenia (muscle wasting) may be masked if coexistent with morbid obesity and is a predictor of poor outcomes in cirrhosis, such as hepatic decompensation, poor quality of life, and premature mortality.12,33 Although the current study did not assess measures of physical function, the previously discussed phase 2b trial in NASH F1–3 highlighted that semaglutide led to improvements in the physical component of the short form-36 quality-of-life questionnaire in parallel with weight loss.
Larger trials are needed to investigate whether semaglutide improves liver-related morbidity and mortality in patients with NASH and compensated cirrhosis. The phase 2b ATLAS trial indicated that a combination of cilofexor and firsocostat led to improvements in NASH activity and a reduction in fibrosis score in patients with bridging fibrosis or compensated cirrhosis (F3–4).34 A combination of these treatments with semaglutide is currently under investigation (NCT04971785). In a similar population, the STELLAR trials of selonsertib failed to show an antifibrotic effect in patients with NASH and F3–4.30 Similarly, the FALCON clinical trial programme in patients with NASH and F3–4 did not show a statistical advantage for pegbelfermin over placebo in terms of histology, but did indicate improvements in non-invasive measures of fibrosis, steatosis, or inflammation,29 while simtuzumab also failed to show an antifibrotic effect in patients with bridging fibrosis and compensated cirrhosis.35
In conclusion, in this phase 2 study of patients with NASH and compensated cirrhosis, semaglutide 2·4 mg once weekly did not significantly improve fibrosis or achievement of NASH resolution, but was well tolerated, did not raise any new safety concerns, and led to improved cardiometabolic parameters and non-invasive markers of liver fat and liver injury associated with fibrosis progression.
Supplementary Material
Research in context.
Evidence before this study
PubMed was searched on July 19, 2022, for articles published since Jan 1, 2017, without language restrictions, using the search terms “non-alcoholic steatohepatitis” or “NASH” and “cirrhosis” in the title. The retrieved articles were manually reviewed for relevance and their reference lists examined for additional sources of relevant information. Patients with non-alcoholic steatohepatitis (NASH)-related cirrhosis are at high risk of potentially life-threatening liver-related morbidities, as well as cardiovascular events. Current first-line treatment in patients with compensated cirrhosis and overweight or obesity involves lifestyle interventions to achieve weight loss and treat cardiometabolic comorbidities, but there is no approved NASH-specific pharmacotherapeutic treatment. Selonsertib, simtuzumab, and pegbelfermin are among the agents that have been investigated in NASH-related cirrhosis, but the primary efficacy endpoints of these trials were not met. Compared with placebo, the glucagon-like peptide-1 receptor agonist semaglutide improved NASH resolution and metabolic parameters in patients with non-cirrhotic NASH (fibrosis stage 1–3) and could be of benefit to patients with NASH-related cirrhosis.
Added value of this study
This placebo-controlled, randomised phase 2 trial is the first study to assess the efficacy and safety of semaglutide 2·4 mg once weekly in patients with NASH-related compensated cirrhosis (fibrosis stage 4). There was no difference between semaglutide and placebo for the primary endpoint (fibrosis improvement without worsening of NASH) or the supportive secondary endpoint of NASH resolution. However, compared with placebo, semaglutide led to reductions in liver enzymes, liver steatosis (but not stiffness), and levels of the exploratory hepatic collagen biomarker pro-collagen 3 peptide.
Patients treated with semaglutide lost more weight, had lower concentrations of triglycerides and VLDL cholesterol, and those with type 2 diabetes also had reductions in HbA1c levels, compared with placebo. No new safety concerns were raised with semaglutide in this population, with no decompensating events or deaths; as expected, the main adverse events were mild to moderate and transient gastrointestinal events.
Implications of all the available evidence
Semaglutide 2·4 mg once weekly did not improve fibrosis without worsening of NASH. However, addressing features of the metabolic syndrome is essential in individuals with NASH-related cirrhosis, as is weight loss in those who have overweight or obesity, and there was evidence of improvement in cardiometabolic parameters and non-invasive markers of liver injury with semaglutide treatment. The relatively small size of the current trial may have limited its ability to demonstrate an effect on fibrosis and NASH resolution.
Acknowledgments
This study was sponsored by Novo Nordisk A/S. PNN was supported by the National Institute of Health Research (NIHR) Birmingham Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR or the Department of Health. RL receives funding support from NCATS (5UL1TR001442), NIDDK (U01DK061734, U01DK130190, R01DK106419, R01DK121378, R01DK124318, P30DK120515), NHLBI (P01HL147835), and NIAAA (U01AA029019). Medical writing support was provided by Stephen Purver of Apollo, OPEN Health Communications, and was funded by Novo Nordisk A/S in accordance with Good Publication Practice guidelines. The authors thank the patients participating in this trial, the investigators, all trial site staff, and all Novo Nordisk A/S employees involved in the trial.
Declaration of interests
RL, MFA, MJA, EL, VR, AJS, JMS, and PNN were investigators in the trial and received grants from the study sponsor paid to their institutions to conduct the study. RL is co-founder of LipoNexus Inc, and a consultant to Aardvark Therapeutics, Altimmune, Anylam/Regeneron, Amgen, Arrowhead Pharmaceuticals, AstraZeneca, Bristol Myers Squibb, CohBar, Eli Lilly, Galmed, Gilead, Glympse bio, Hightide, Inipharma, Intercept, Inventiva, Ionis, Janssen, Madrigal, Metacrine, NGM Biopharmaceuticals, Novartis, Novo Nordisk, Merck, Pfizer, Sagimet, Theratechnologies, 89bio, Terns Pharmaceuticals, and Viking Therapeutics. In addition, his institutions have received research grants from Arrowhead Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Galectin Therapeutics, Galmed Pharmaceuticals, Gilead, Hanmi, Intercept, Inventiva, Ionis, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, Novo Nordisk, Pfizer, Sonic Incytes, and Terns Pharmaceuticals. MFA has received research/grant support from Allergan, Boeringher Ingelheim, BMS, Celgene, Durect, Enanta, Enyo, Galmed, Genentech, Gilead, Hanmi, Intercept, Inventiva, Madrigal, Novo Nordisk, Poxel, TARGET Pharma, and Viking; has acted as a consultant for Hanmi, NGM, BMS, 89bio, Madrigal, Intercept, Merck, Inventiva, Novo Nordisk, Sonic Incytes, and Theratechnologies; has served as a speaker for Clinical Care Options, Medscape, Fishawack LLC, and Terra Firma; has received royalties from UptoDate; and holds stock in Pfizer. MJA has received consultation fees, speaker fees, and research grants from Novo Nordisk and speaker fees and consultation fees from Norgine. JS has acted as a consultant for Apollo Endoscopy, Bristol Myers Squibb, Boehringer Ingelheim, Echosens, Genfit, Gilead Sciences, Intercept Pharmaceuticals, Madrigal, Merck, Nordic Bioscience, Novartis, Pfizer, Roche, Sanofi, and Siemens Healthcare GmbH; received research funding from Boehringer Ingelheim, Gilead Sciences, and Siemens Healthcare GmbH; and received speaker honorarium from Falk Foundation and Madrigal. MJ, MSK, and NK are employees of Novo Nordisk A/S; MSK and MJ are shareholders in Novo Nordisk A/S. EL has received research/grant support from 89bio, AbbVie, Akero Therapeutics, Allergan, Alnylam Pharmaceuticals, Amgen, Ascelia Pharma, Assemblybio, AstraZeneca, Axcella Health, Biocryst Pharmaceuticals, Bird Rock Bio, Boehringer Ingelheim, Bristol Myers Squibb, Conatus Pharmaceuticals, Cymabay Therapeutics, CytoDyn, DSM, Durect Corporation, Eli Lilly and Company, Enanta Pharmaceuticals, Enyo Pharma, Exalenz Bioscience, Galectin Therapeutics, Galmed Pharmaceuticals, Genfit, Genentech, Gilead Sciences, GlaxoSmithKline, Hanmi Pharmaceuticals, Hightide Biopharma, Intercept Pharmaceuticals, Inventiva, Janssen Pharmaceuticals, Laboratory for Advanced Medicine, Loxo Oncology, Madrigal Pharmaceuticals, Merck & Co, Metacrine, NGM Biopharmaceuticals, Northsea Therapeutics, Novartis, Novo Nordisk, Pfizer, Poxel, Roche, Sagimet Biosciences, Synlogic Therapeutics, Terns Pharmaceuticals, Viking Therapeutics, and Zydus Pharmaceuticals; has acted as a consultant for Akero, Boehringer Ingelheim, BMS, Intercept, Novo Nordisk, Metacrine, Sagimet, and Terns; and has received speaker honorarium from Gilead Sciences, AbbVie, and Intercept. AJS is President of Sanyal Biotechnology and has stock options in Exhalenz, Genfit, Hemoshear, Durect, Indalo, Northsea, Tiziana, and Rivus. He has served as a consultant to Genfit, Gilead, Malinckrodt, Pfizer, Salix, Boehringer Ingelheim, Novartis, Bristol Myers Squibb, Merck, Hemoshear, Lilly, Novo Nordisk, Terns, Albireo, Janssen, Poxel, 89bio, Siemens, AstraZeneca, NGM Bio, Amgen, Regeneron, Genentech, Alnylam, Roche, Madrigal, Inventiva, Covance, Prosciento, Histoindex, and PathAI. His institution has received grant support from Gilead, Malinckrodt, Boehringer Ingelheim, Novartis, Bristol Myers Squibb, Merck, Lilly, Novo Nordisk, Fractyl, Madrigal, and Inventiva. He has received royalties from Elsevier and UptoDate. PNN has received grants from Novo Nordisk and Boehringer Ingelheim; and has acted as a consultant on behalf of the University of Birmingham for Novo Nordisk, Boehringer Ingelheim, Gilead, Intercept Pharmaceuticals, Pfizer, and Poxel Pharmaceuticals. VR has acted as a consultant for Boehringer Ingelheim, Novo Nordisk, Poxel, Enyo, Madrigal, Terns, Intercept, NGM Bio, and Pfizer; and received research grants from Gilead Sciences and Intercept Pharmaceuticals.
Funding
Novo Nordisk A/S.
Footnotes
See Online for appendix
See the appendix (p 3) for further information.
Data sharing
Data will be shared with bona fide researchers who submit a research proposal approved by the independent review board. Individual participant data will be shared in data sets in a de-identified and anonymised format. Data will be made available after research completion and approval of the product and product use in the European Union and the USA. Information about data access request proposals can be found at novonordisk-trials.com.
References
- 1.Taylor RS, Taylor RJ, Bayliss S, et al. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterology 2020; 158: 1611–25. [DOI] [PubMed] [Google Scholar]
- 2.Sanyal AJ, Van Natta ML, Clark J, et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N Engl J Med 2021; 385: 1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantovani A, Csermely A, Petracca G, et al. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: an updated systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2021; 6: 903–13. [DOI] [PubMed] [Google Scholar]
- 4.Ng CH, Lim WH, Lim GEH, et al. Mortality outcomes by fibrosis stage in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2022; published online May 2. 10.1016/j.cgh.2022.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018; 67: 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang JD, Ahmed F, Mara KC, et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 2020; 71: 907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021; 184: 2537–64. [DOI] [PubMed] [Google Scholar]
- 8.Alexopoulos AS, Crowley MJ, Wang Y, et al. Glycemic control predicts severity of hepatocyte ballooning and hepatic fibrosis in nonalcoholic fatty liver disease. Hepatology 2021; 74: 1220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loomba R, Ratziu V, Harrison SA, NASH Clinical Trial Design International Working Group. Expert panel review to compare FDA and EMA guidance on drug development and endpoints in nonalcoholic steatohepatitis. Gastroenterology 2022; 162: 680–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016; 64: 1388–402. [DOI] [PubMed] [Google Scholar]
- 11.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018; 67: 328–57. [DOI] [PubMed] [Google Scholar]
- 12.European Association for the Study of the Liver. EASL Clinical Practice Guidelines on nutrition in chronic liver disease. J Hepatol 2019; 70: 172–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higuchi M, Tamaki N, Kurosaki M, et al. Longitudinal association of magnetic resonance elastography-associated liver stiffness with complications and mortality. Aliment Pharmacol Ther 2022; 55: 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moctezuma-Velazquez C, Márquez-Guillén E, Torre A. Obesity in the liver transplant setting. Nutrients 2019; 11: 2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375: 1834–44. [DOI] [PubMed] [Google Scholar]
- 16.Aroda VR, Ahmann A, Cariou B, et al. Comparative efficacy, safety, and cardiovascular outcomes with once-weekly subcutaneous semaglutide in the treatment of type 2 diabetes: insights from the SUSTAIN 1–7 trials. Diabetes Metab 2019; 45: 409–18. [DOI] [PubMed] [Google Scholar]
- 17.Thethi TK, Pratley R, Meier JJ. Efficacy, safety and cardiovascular outcomes of once-daily oral semaglutide in patients with type 2 diabetes: the PIONEER programme. Diabetes Obes Metab 2020; 22: 1263–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilding JPH, Batterham RL, Calanna S, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med 2021; 384: 989–1002. [DOI] [PubMed] [Google Scholar]
- 19.Targher G, Mantovani A, Byrne CD. Mechanisms and possible hepatoprotective effects of glucagon-like peptide-1 receptor agonists and other incretin receptor agonists in non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol 2023; 8: 179–91. [DOI] [PubMed] [Google Scholar]
- 20.Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med 2021; 384: 1113–24. [DOI] [PubMed] [Google Scholar]
- 21.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005; 41: 1313–21. [DOI] [PubMed] [Google Scholar]
- 22.De Franchis R, Baveno VI Faculty. Expanding consensus in portal hypertension: report of the Baveno VI consensus workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol 2015; 63: 743–52. [DOI] [PubMed] [Google Scholar]
- 23.Dulai PS, Sirlin CB, Loomba R. MRI and MRE for non-invasive quantitative assessment of hepatic steatosis and fibrosis in NAFLD and NASH: clinical trials to clinical practice. J Hepatol 2016; 65: 1006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Child CG, Turcotte JG. Surgery and portal hypertension. Major Probl Clin Surg 1964; 1: 1–85. [PubMed] [Google Scholar]
- 25.European Medicines Agency. Reflection paper on regulatory requirements for the development of medicinal products for chronic non-infectious liver diseases (PBC, PSC, NASH). EMA/CHMP/299976/2018. Nov 15, 2018. https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-regulatory-requirements-development-medicinal-products-chronic-non-infectious-liver_en.pdf (accessed March 3, 2023).
- 26.Food and Drug Administration, CDER. Guidance for Industry. Noncirrhotic nonalcoholic steatohepatitis with liver fibrosis: developing drugs for treatment. FDA-2018-D-3632. Dec 2018. https://www.fda.gov/media/119044/download (accessed March 3, 2023). [Google Scholar]
- 27.Loomba R, Sanyal AJ, Kowdley KV, et al. Factors associated with histologic response in adult patients with nonalcoholic steatohepatitis. Gastroenterology 2019; 156: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016; 387: 679–90. [DOI] [PubMed] [Google Scholar]
- 29.Abdelmalek MF, Sanyal AJ, Nakajima A, et al. Efficacy and safety of pegbelfermin in patients with nonalcoholic steatohepatitis and compensated cirrhosis: results from the phase 2b, randomized, double-blind, placebo-controlled FALCON 2 study. The Liver Meeting, Nov 12–15, 2021. Abstract LP8. [Google Scholar]
- 30.Harrison SA, Wong VW, Okanoue T, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J Hepatol 2020; 73: 26–39. [DOI] [PubMed] [Google Scholar]
- 31.Marcellin P, Asselah T. Long-term therapy for chronic hepatitis B: hepatitis B virus DNA suppression leading to cirrhosis reversal. J Gastroenterol Hepatol 2013; 28: 912–23. [DOI] [PubMed] [Google Scholar]
- 32.Taylor-Weiner A, Pokkalla H, Han L, et al. A machine learning approach enables quantitative measurement of liver histology and disease monitoring in NASH. Hepatology 2021; 74: 133–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Sherif O, Armstrong M. Peculiarities of cirrhosis due to nonalcoholic steatohepatitis (NASH). Semin Liver Dis 2020; 40: 1–10. [DOI] [PubMed] [Google Scholar]
- 34.Loomba R, Noureddin M, Kowdley KV, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology 2021; 73: 625–43. [DOI] [PubMed] [Google Scholar]
- 35.Harrison SA, Abdelmalek MF, Caldwell S, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 2018; 155: 1140–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be shared with bona fide researchers who submit a research proposal approved by the independent review board. Individual participant data will be shared in data sets in a de-identified and anonymised format. Data will be made available after research completion and approval of the product and product use in the European Union and the USA. Information about data access request proposals can be found at novonordisk-trials.com.