

Abstract
Chromodomain helicase DNA-binding protein 1 like (CHD1L) is an oncogene that promotes tumor progression, metastasis, and multidrug resistance. CHD1L expression is indicative of poor outcomes and low survival in cancer patients with various cancer types. Herein, we report a set of CHD1L inhibitors (CHD1Li) discovered from high-throughput screening and evaluated using enzyme inhibition, 3D tumor organoid cytotoxicity and mechanistic assays. The structurally distinct compounds 8-11 emerged as hits with promising bioactivity by targeting CHD1L. CHD1Li were further examined for their stability in human and mouse liver microsomes, which showed compounds 9 and 11 to be the most metabolically stable. Additionally, molecular modeling studies of CHD1Li with the target protein shed light on key pharmacophore features driving CHD1L binding. Taken together, these results expand the chemical space of CHD1Li as a potential targeted therapy for colorectal cancer and other cancers.
Keywords: Cancer, CHD1L, CHD1Li, EMT, Hit to lead, Molecular modeling, Targeted therapy
1. Introduction
Colorectal Cancer (CRC) is the third leading type of cancer and the second most common cause of cancer-related deaths worldwide [1,2]. Age has historically been the main risk factor for CRC, with the average diagnostic age exceeding 50 years [3]. However, the age group at risk for CRC is shifting, as early onset disease is becoming increasingly prevalent, with rates increasing from 11% to 20% in the past 25 years [1,4]. Although recent advances in preventive screening have increased detection and therefore the success of treatments, 22% of CRC patients are not diagnosed until stage IV, and the vast majority of these patients receive chemotherapy as palliative care [1,5–7].
The most effective therapy for early-stage CRC is currently surgical resection [3], while 5-fluorouracil (5-FU) based combination chemotherapy is the first-line standard of care for both metastatic and surgically resectable CRC [5]. Despite the efficacy of surgical intervention at early stages, chemotherapy is the standard of care for advanced disease [8]. Thus, an unmet need exists for targeted antitumor agents that can effectively combat tumor progression and metastasis while synergizing with chemotherapy and targeted therapies.
In 2008, chromodomain helicase DNA-binding protein 1 like (CHD1L), also known as ALC1, was identified as a chromatin remodeling enzyme [9]. It has since gained recognition as an oncogene overexpressed in many cancer types [6,9–11]. Multiple studies consistently link elevated CHD1L expression as a biomarker for poor prognosis, poor survival, and metastasis in various cancers, including CRC [10,12–21]. CHD1L is influenced by key cancer-driving pathways, including Wnt/β-catenin, PI3K/AKT, and Ras/MAPK [22]. Its diverse roles encompass regulating malignant gene expression, cell plasticity and stemness via epithelial-mesenchymal transition (EMT), cell survival, and metastatic potential [23]. Given the critical role of CHD1L in tumor progression, metastasis, and drug resistance, the identification of CHD1L inhibitors (CHD1Li) could lead to effective targeted therapies for CRC and other cancers.
Recently, we reported the high-throughput screening (HTS) discovery and pre-clinical development of the first-in-class CHD1Li for the treatment of cancer, exemplified by the pyrimidine-based lead drug OTI-611 (aka 6.11) [24,25]. The first generation CHD1Li display potent antitumor activity through allosteric inhibition of CHD1L ATPase, which induces programmed cell death. Moreover, inhibition of CHD1L does not cause DNA damage or acute toxicity in mice. In continuation of our efforts to expand the CHD1Li chemical space while improving the structural diversity of pharmacophores, herein, we report the hit-to-lead validation of promising next generation CHD1Li toward the treatment of CRC and other cancers.
2. Materials and methods
2.1. Cell line
The colorectal cancer cell lines SW620, SW480, and HCT116 were purchased from American Type Culture Collection (ATCC) (Manassas, VA). The cell lines were STR profiled and tested for bacteria or mycoplasma contamination prior to use. SW620 cells were transfected with a dual-reporter plasmid and sorted according to their mesenchymal or epithelial phenotype as previously described [26]. The cells were stored in a liquid nitrogen tank until needed, when they were then thawed and passaged twice before use.
2.2. Cell culture
Cells were grown within RPMI 1640 media (Gibco, Ref#: 11875–093) containing 5% fetal bovine serum (Gibco, Ref#: 10437028) in 10 cm tissue culture coated dishes (Fisher Scientific, Ref#: FB012924). Cells were stored in a 37 °C humidified incubator at 5% CO2 for growth. Cells were split by aspirating media from the plate, washing with 5 mL of Phosphate Buffer Solution (PBS) 1X pH 7.4 (Gibco, Ref#: 10010–023), followed by addition of 0.25% Trypsin (Life Technologies, Ref#: 25200072) in media, prior to incubation for 6 min. The trypsin was neutralized with 4 mL of growth media.
2.3. Catalytic enzyme assay
The catalytic enzyme assay was performed as previously described [24,27]. A reaction mixture was prepared at the concentrations of 100 nM of the catalytic domain of CHD1L (cat-CHD1L) and 200 nM/L of c-Myc DNA or mononucleosome (Active Motif, Ref#: 81770) in buffer (50 mM Tris pH 7.5, 50 mM NaCl, 1 mM DTT, 5% glycerol). ATP was added at a concentration of 10 μM to begin the reaction, bringing the total reaction volume to 10 μL. After initiation, the reaction was allowed to proceed for 1 h while incubated at 37 °C. Then, 500 nM/L of Phosphate sensor was added to the reaction to quantify the ATPase activity of the enzyme. An Envision plate reader (Revvity, Waltham, MA) was used to measure the excitation at 430 nm and the emission at 450 nm of the phosphate sensor. The fluorescence generated from this reaction was compared to a standard inorganic phosphate curve to calculate the enzyme kinetics. All statistical analysis was performed using GraphPad Prism.
2.4. Tumor organoid culture
SW620-GFP+, SW480, and HCT116 cell lines were used to grow 3D tumor organoids. After cell lines were split, they were counted by creating a 1:1 ratio mixture of cell suspension and Trypan Blue (Sigma-Aldrich, Ref#: T8154–20 mL). This solution was analyzed and counted using a Bio-Rad TC20 automated cell counter (Bio-Rad, Hercules, CA). 2000 cells in 100 μL of growth media were added to each well in a 96-well clear, round bottom ultra-low attachment (ULA) well plates (Corning, Ref # 7007). Plates were spun at 1000 RPM for 15 min at 25 °C to aggregate the cells. 25 μL of 10% Matrigel (Corning, Ref# 356231) in growth media was added to each well for a final concentration of 2% Matrigel. 100 μL of PBS 1X pH 7.4 was added to the spaces between wells within the plate to reduce evaporation. The plates were incubated at 37 °C for 72 h to grow tumor organoids in each well.
2.5. 3D cytotoxicity assay
The CHD1Li were tested at seven different concentrations to determine their cytotoxic effect on tumor organoids as previously described [24,27]. Compounds were dissolved in DMSO (Sigma-Aldrich, Ref #34869) to make a 10 mM stock solution and stored at − 80°C. After thawing, the stock was diluted with growth media and serially diluted before addition to cells in a 1:6 ratio. After 72 h of growth, cells were treated with CHD1Li in triplicates or quadruplicates at final concentrations ranging from 0.625 μM to 40 μM in 0.4% DMSO. The plate was incubated at 37 °C for 72 h before harvesting. During harvesting, the organoids were transferred to a white, flat bottom 96-well plate (Greiner, Ref#:655083) and treated with 35 μL of 3D Cell Titer Glo (Promega, Ref#: G7572). The plates were then placed on an orbital shaker at 400 RPM for 45 min before they were analyzed using the Envision plate reader. These experiments were conducted on 2 to 3 separate instances on different days to determine result repeatability.
2.6. EMT dual reporter assay
The EMT dual reporter assay was performed as previously described [24,26,27]. SW620-GFP+ cells were plated as 3D organoids and treated with 0–40 μM of CHD1Li as described above. The Opera Phenix HCS System (Revvity) was used to perform high content imaging and analysis of the 3D organoids at settings previously described [26]. This imaging was performed on SW620-GFP+ organoids prior to harvesting for the cytotoxicity assay.
2.7. CSC stemness clonogenic assay
The CSC stemness assay was performed as previously described [28]. SW620-GFP+ Cells were plated at 300 cells/well in 96 well clear, flat bottom, black plates and allowed to attach overnight. The following day, cells were treated with CHD1Li or DMSO control at concentrations ranging from 0.25 to 8 μM in media. The vehicle and treatments were refreshed with 25 μL of treatment or vehicle on days 5 and 8. On day 9, cells were imaged with the Opera Phenix HCS system at settings previously described [28]. Prior to imaging, the colonies were stained with 25 μL of 0.67 μg/mL Hoechst 33342 (Life Technologies, Ref#: H3570) in PBS and incubated for 15 min at 37 °C. The images were analyzed using the Harmony 5.1 software to count colony number, area, and confluence [28].
2.8. Microsomal assay
The microsomal stability of hit CHD1Li 8-11 was evaluated in CD-1 mouse microsomes (Gibco, Ref#: MSMCPL) and human microsomes (Gibco, Ref#: HMMCPL) as previously described [29]. At the final concentrations, the 100 μL reaction solution consisted of 0.5 mg/mL microsomes, phosphate buffer pH 7.4 (44 mM KH2PO4, 56 mM K2HPO4), 1.94 mg/mL UDPGA (Sigma Aldrich, Ref# U6751), 25 μg alamethicin (Sigma-Aldrich, A4665), 1 mM MgCl2, 1 mM NADPH (Sigma-Aldrich, Ref# 481973–50MG), 1% DMSO, and 10 μM of either CHD1Li or testosterone (Sigma, T1500–1 G) for control tubes. All the reaction components except NADPH were pre-incubated for 5 min at 37 °C on a heat block. The reactions were activated by adding NADPH and incubated at 37 °C for the remainder of the reaction. At time points 0, 5, 15, 30, 45, and 60 min, 100 μL of the reaction solution was removed and mixed with acetonitrile at a 1:1 ratio to halt the reaction. Samples from each time point were centrifuged at 3000 RPM for 5 min and the supernatant was sampled for mass spectrometry analysis.
2.8.1. Mass spectrometry analysis
A Prominence HPLC (Shimadzu, Kyoto, Japan) coupled to a QTrap 4500 mass spectrometer (AB Sciex, Farmingham, MA) was used for this analysis. The mass spectrometer was operated in the ESI+ mode and all settings were optimized by manually tuning to infused standard solutions. Global mass spectrometer parameter settings were selected to give the highest average sensitivity for all compounds of interest. Global mass spectrometer settings are as follows: curtain gas, 40 psi; collision gas, high; ion spray voltage, 4500 V; source temperature, 700 °C; ion source gas 1 and 2, 50 psi; entrance potential, 10 V; and collision cell exit potential, 14 V. The collision energy (CE) and declustering potential (DP) were optimized separately for each compound (see Table S4 for LC-MS/MS transitions). Data was collected using the multiple reaction monitoring (MRM) mode. Quantitation was performed using an external calibration curve for each compound. Analytical separation was achieved with a Phenomenex kinetex C18 column [2.1 × 100 mm, 2.6 μm]. The column was held at 40 °C and eluted at 0.6 mL/min with a gradient of 0.1% formic acid (A) and 0.1% formic acid in 9:1 acetonitrile:water (B) with a total runtime of 10 min. Chromatographic separation was achieved with a linear gradient (time, % of solvent B): 0–0.5 min, 5% B; 0.5–4.5 min, 5–55% B; 4.5–6.5 min, 55–94% B; 6–7 min, 94–5% B; and then isocratic for 3 min at 5% B to re-equilibrate the column. The first 3 min of eluent was diverted to waste to reduce contamination of the front end of the mass spectrometer. Calibration curves were prepared from the stock standard solutions. Serial dilutions of stock standards were prepared in DMSO. Calibration standards were prepared in 20% DMSO in water. Calibration curves typically contained 25 fmols – 3 pmols on column for testosterone, and 25 fmols – 2 pmols on column for CHD1Li 8-11. The corresponding mass transitions are shown in Table S4.
2.9. Molecular modeling
All calculations were performed using the Schrodinger molecular ¨ modeling suite (version 2022–2) and OPLS4 force field.
2.9.1. Ligand and protein preparation
The 3D low energy conformation of hit compounds 8-11 was created with the LigPrep module. Ligands ionization and tautomeric states were generated using Epik at pH 7.0 ± 2.0. The 3D model of fl-CHD1L (UniProt# Q86WJ1) in the active state was downloaded from AlphaFold database [30] and processed using the protein preparation workflow. A reliability check of the minimized protein structure was conducted prior to its use for further calculations.
2.9.2. Binding site elucidation
The minimized fl-CHD1L structure was characterized for potential druggable sites using the SiteMap module. The minimum site points per site were set at 15 while using a more restrictive requirement for hydrophobicity to exclude sites occupying free space. The returned sites were ranked based on the site score, drugability score, size, and volume.
2.9.3. Molecular docking and binding free energy (ΔGbind) estimation
The minimized structures of hit CHD1Li 8-11 and fl-CHD1L were used as inputs for molecular docking calculations using the induced-fit docking (IFD) protocol at the best site identified from SiteMap calculations. The receptor grid box was defined as the centroid of amino acid residues bordering the selected binding site with an inner and outer box size of 10 and 30 Å, respectively. The ligand ring conformations were sampled at 2.5 kcalmol−1, followed by a short minimization of the protein structure to RMSD 0.18 Å and Prime refinement of residues within 5 Å of binding site. Ligands were then redocked into receptor structures within 30 kcalmol−1 using extra-precision Glide docking.
Subsequently, the ΔGbind for the resulting CHD1L complexes were estimated using Prime molecular mechanics/generalized Born surface area (MM/GBSA) module. The variable-dielectric generalized Born model (VSGB), which uses water as an implicit solvent, was selected as the solvation model. All atoms of binding site residues were minimized during the calculation to account for ligand-induced conformational changes. The best pose for each hit compound was determined using the docking score, glide emodel, glide energy, IFD score and ΔGbind.
2.9.4. Molecular dynamics (MD) simulation
MD simulation was conducted as previously described [25]. Briefly, the requisite model systems for CHD1Li 9 and 11 were created using the system builder. Each system was solvated with SPC solvent models in an orthorhombic box with 10 Å periodic boundary conditions on all sides. System neutralization was achieved with NaCl salt at ~50 mM. The Desmond MD simulation was performed over a total of 500 ns simulation period using the NPT ensemble at constant temperature (300 K) and pressure (1.01 bar). Simulation trajectories were recorded at 500 ps intervals and using relevant Python scripts.
2.10. Statistical analysis
Statistical analysis and graphs were executed in GraphPad Prism 10 for MacOS (version 10.0.2). A non-linear regression model was used to create a logarithmic curve to model the inhibitor vs response variable slope and calculate the half maximal inhibitory concentration (IC50) of each compound within a 95% confidence interval. Normalization to control and EC2-fold calculations were performed in Microsoft Excel for MacOS (version 16.76).
3. Results
3.1. Hit to lead validation workflow
30 compound hits out of the 52 hits discovered from our previous HTS showed repeat activity in a secondary screen; thus, hits were reexamined for their potential as CHD1Li using a tailored hit-to-lead validation schema to prioritize additional hits as leads for further optimization (Fig. 1). The previously discovered pyrimidine-based lead, CHD1Li 6.0, was used as reference and positive control. The compounds were first tested against the cat-CHD1L recombinant enzyme ATPase assay to confirm inhibitory activity and eliminate false positives from the HTS. Once confirmed as hits, they are tested for their ability to induce cytotoxicity or inhibit or reverse EMT, using CRC tumor organoids models.
Fig. 1.
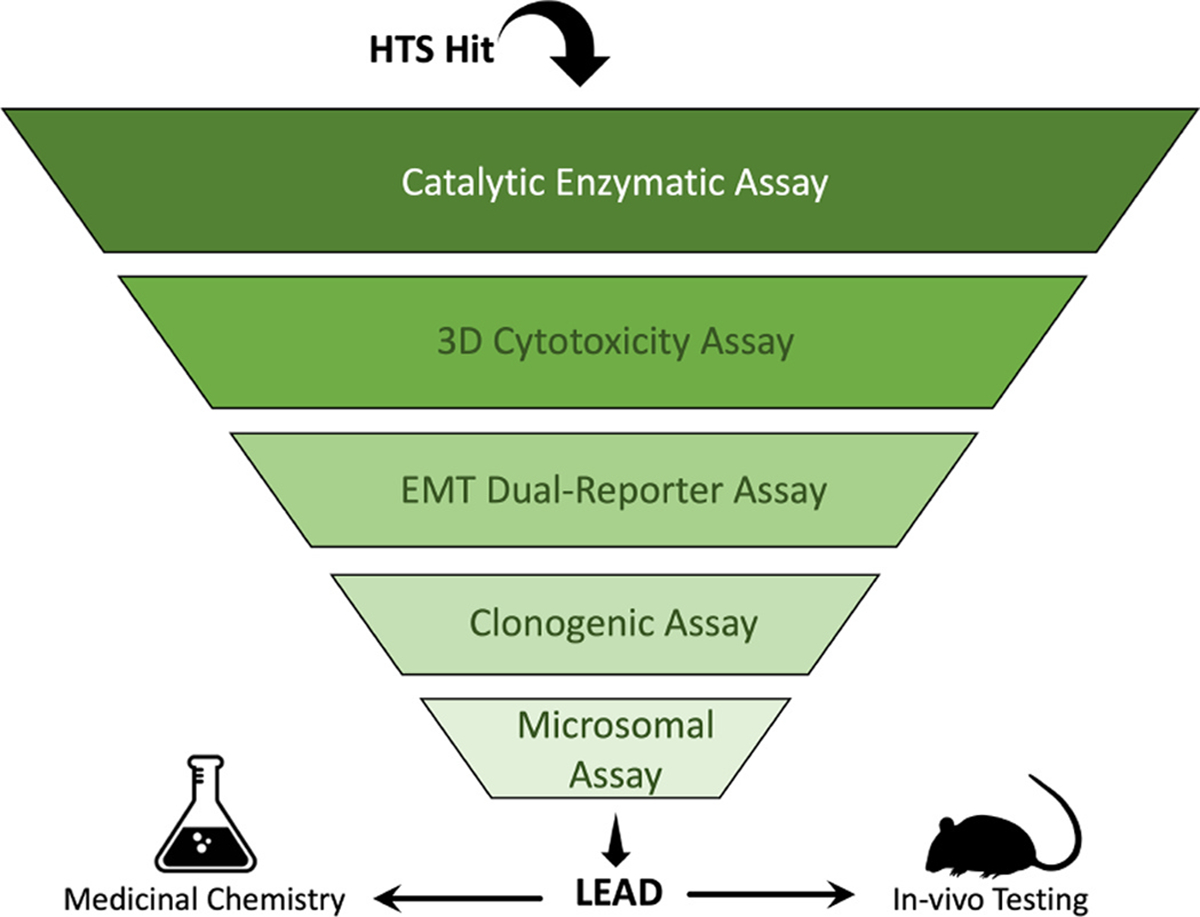
Testing scheme for CHD1Li drug discovery. CHD1Li are selected from either HTS hits or analogs developed from a previously identified hit. They are then subjected to different in vitro assays in the “testing funnel” to filter out the less promising hits or analogs. Highly promising compounds are advanced for further testing and the most promising (leads) are taken through in vivo testing or used as structural templates for lead optimization using medicinal chemistry. Results from all tests are analyzed for existing SAR to design new analogs and improve efficacy.
Promising hits were next examined for their ability to inhibit CSC stemness, using the clonogenic colony formation assay [28]. The mesenchymal phenotype of SW620 cells (SW620-GFP+) was selected for all cell-based experiments in this validation scheme performed after the 3D cytotoxicity assay, since these cells display enhanced tumorigenic properties, including multidrug resistance and CSC stemness [26]. Therefore, this phenotype has the highest potential for colony formation, metastasis, and represents an aggressive CRC tumor cell model [26, 31]. Finally, top hits from these experiments were evaluated for their metabolic stability in mouse liver microsomes [32,33]. The results from the hit-to-lead validation experiments are summarized in Table 1.
Table 1.
Hit to lead validation results.
| Compound Number | Enzyme Inhibition IC50 (μM) | 3D Cytotoxicity IC50 (μM) | EMT Reversal | GFP IC50 (μM) | RFP EC(2-fold) | CSC Stemness IC50 (μM) | Microsomal Stability t1/2 (min) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| SW620 | SW480 | HCT116 | Human | Mouse | ||||||
| 6.0 | 3.33 | 3.04 | 3.23 | 5.94 | Full | 2.69 | 22.9 | 1.09 | - | 67.16 |
| 8 | 2.76 | 19.3 | 14.3 | 10.6 | Partial | > 40 | 13.3 | 13.10 | 9.95 | 7.92 |
| 9 | 4.61 | 28.3 | 15.7 | 19.9 | Partial | > 40 | 8.3 | 8.34 | 103.47 | 101.99 |
| 10 | 2.29 | 29.7 | 34.1 | 27.3 | Partial | > 40 | 35.4 | 12.80 | 6.06 | 6.13 |
| 11 | 4.57 | 3.31 | 1.73 | 5.86 | Full | 8.5 | 9.5 | 0.34 | 96.59 | 108.49 |
| 12 | 7.97 | > 40 | - | - | Partial | > 40 | 38.5 | - | - | - |
| 13 | 1.33 | > 40 | - | - | Partial | > 40 | > 40 | - | - | - |
| 14 | 5.00 | > 40 | - | - | Partial | > 40 | 23.6 | - | - | - |
| 15 | 6.15 | > 40 | - | - | Partial | >40 | 10.0 | - | - | - |
| 16 | 5.53 | > 40 | - | - | Partial | >40 | 29.3 | - | - | - |
| 17 | 4.30 | > 40 | - | - | Partial | > 40 | > 40 | - | - | - |
| 18 | 4.35 | > 40 | - | - | Partial | >40 | > 40 | - | - | - |
| 19 | 4.03 | > 40 | - | - | Partial | >40 | 30.2 | - | - | - |
| 20 | 8.33 | > 40 | - | - | Partial | 28.2 | >40 | - | - | - |
| 21 | 3.30 | > 40 | - | - | Partial | >40 | > 40 | - | - | - |
| 22 | 2.90 | > 40 | - | - | Partial | >40 | > 40 | - | - | - |
| 23 | 2.40 | > 40 | - | - | Partial | >40 | > 40 | - | - | - |
| 24 | 8.67 | > 40 | - | - | No | > 40 | >40 | - | - | - |
| 25 | 13.13 | > 40 | - | - | No | > 40 | >40 | - | - | - |
| 26 | 6.50 | > 40 | - | - | No | > 40 | >40 | - | - | - |
| 27 | 3.80 | > 40 | - | - | No | >40 | .40 | - | - | - |
| 28 | 4.90 | > 40 | - | - | No | >40 | >40 | - | - | - |
| 29 | 7.20 | > 40 | - | - | No | >40 | >40 | - | - | - |
| 30 | 7.17 | — | - | - | - | - | - | - | - | - |
| 31 | 8.15 | - | - | - | - | - | - | - | - | - |
| 32 | > 20 | - | - | - | - | - | - | - | - | - |
| 33 | > 20 | - | - | - | - | - | - | - | - | - |
| 34 | > 20 | - | - | - | - | - | - | - | - | - |
| 35 | > 20 | - | - | - | - | - | - | - | - | - |
| 36 | > 20 | - | - | - | - | - | - | - | - | - |
CHDILi 6.0 is a lead pharmacophore and reference compound (green), a dash indicates the experiment was not performed for that compound.
3.2. CHD1L enzyme inhibition
Confirmed hits from primary HTS were re-evaluated for their dose dependent inhibition of cat-CHD1L ATPase. Compounds 32-36 were deprioritized due to their limited potency with inhibition concentration 50% (IC50) values > 20 μM. All the other compounds displayed relatively potent CHD1L inhibition with IC50 < 15 μM (Table 1). Hit compounds 8-11 displayed similar inhibitory activity compared to CHD1Li 6.0 and were prioritized for further validation (Table 1 and Fig. 2A).
Fig. 2.
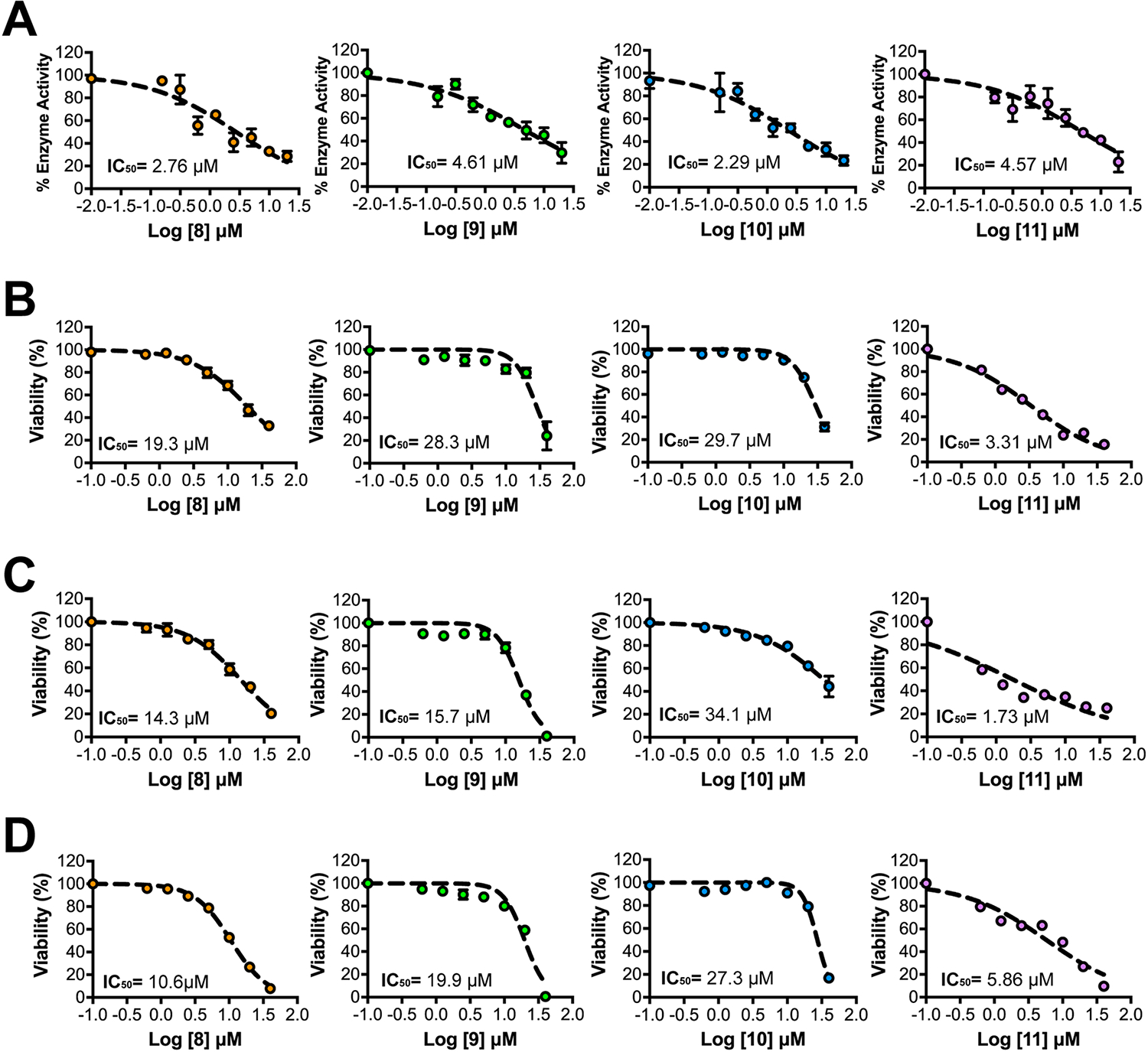
Top HTS hits exhibit dose-dependent inhibition of CHD1L and tumor organoid viability. (A) Graphical representation of dose-response inhibition of the cat-CHD1L enzyme by CHD1Li (N = 2). Each data point is averaged from 2 experiments performed in quadruplicate and error bars are the calculated standard error of the mean (SEM). Graphical representation of dose-response cytotoxicity induced by top CHD1Li (N = 3) in (B) SW620-GFP+, (C) SW480, and (D) HCT116 CRC tumor organoids. Each data point is averaged from 3 experiments performed in triplicate and error bars are the calculated standard error of the mean (SEM).
3.3. 3D cytotoxicity
As a measure of antitumor activity, we assessed prioritized hits for their ability to induce cytotoxicity using SW620 tumor organoids cultured uniformly in 96-well plates [26,27,34]. Despite the promising IC50 values for inhibition of CHD1L ATPase, many compounds in the screen showed poor cytotoxicity even at a high dose of 40 μM, which is likely due to unfavorable physicochemical properties (Table 1). For example, compounds 18 and 20 were poorly soluble in DMSO and aqueous solutions likely prohibiting cellular activity. However, 8-11 displayed relatively good cytotoxicity with IC50 < 30 μM further validating their selection as CHD1Li (Fig. 2B). Notably, 11 exhibited the best cell based activity with an IC50 of 3.3 μM, which is comparable to CHD1Li 6.0 but up to 9-fold greater than 8-10 (Fig. 2B). The cellular activity of CHD1Li 11 is likely facilitated due to its better physicochemical properties i.e., molecular weight, lipophilicity, and aqueous solubility (Table S1). The cytotoxicity of CHD1Li were subsequently tested in two additional CRC lines, SW480 and HCT116, to evaluate their ability to induce cell death across differing phenotypes and oncogenic mutations (Fig. 2C, D). SW480 and SW620 cells were originally isolated from the same patient’s primary tumor site and metastatic site, respectively [35]. Together, the three cell lines used represent distinct CRC phenotypes with different genetic aberrations and tumorigenic and metastatic potential. Although the potency of each CHD1Li differed slightly between CRC cell types, the ranking of each compound in terms of cytotoxicity remained unchanged, with compound 11 displaying the most potent activity, followed by 8, 9, and finally 10 (Fig. 2B–D).
3.4. EMT reversal
We have previously demonstrated that CHD1Li exhibit dose-dependent inhibition or reversal of EMT in CRC tumor organoids [24, 27]. Using this approach, EMT inhibition or reversal was evaluated in SW620-GFP+ organoids using a fluorescent dual-reporter plasmid, measuring downregulation of mesenchymal biomarker vimentin with concomitant upregulation of the epithelial biomarker E-cadherin (Fig. 3) [26]. We observed partial effects or inhibition of EMT with some CHD1Li such as 8 and 10 while 9 and 11 effectively reversed EMT (Fig. 3A). CHD1Li 11 displayed the most potent reversion of EMT, which led to tumor organoid killing at higher doses (Fig. 3). This suggests that 11 may be effective at reducing the metastatic potential of aggressive mesenchymal tumor cells.
Fig. 3.
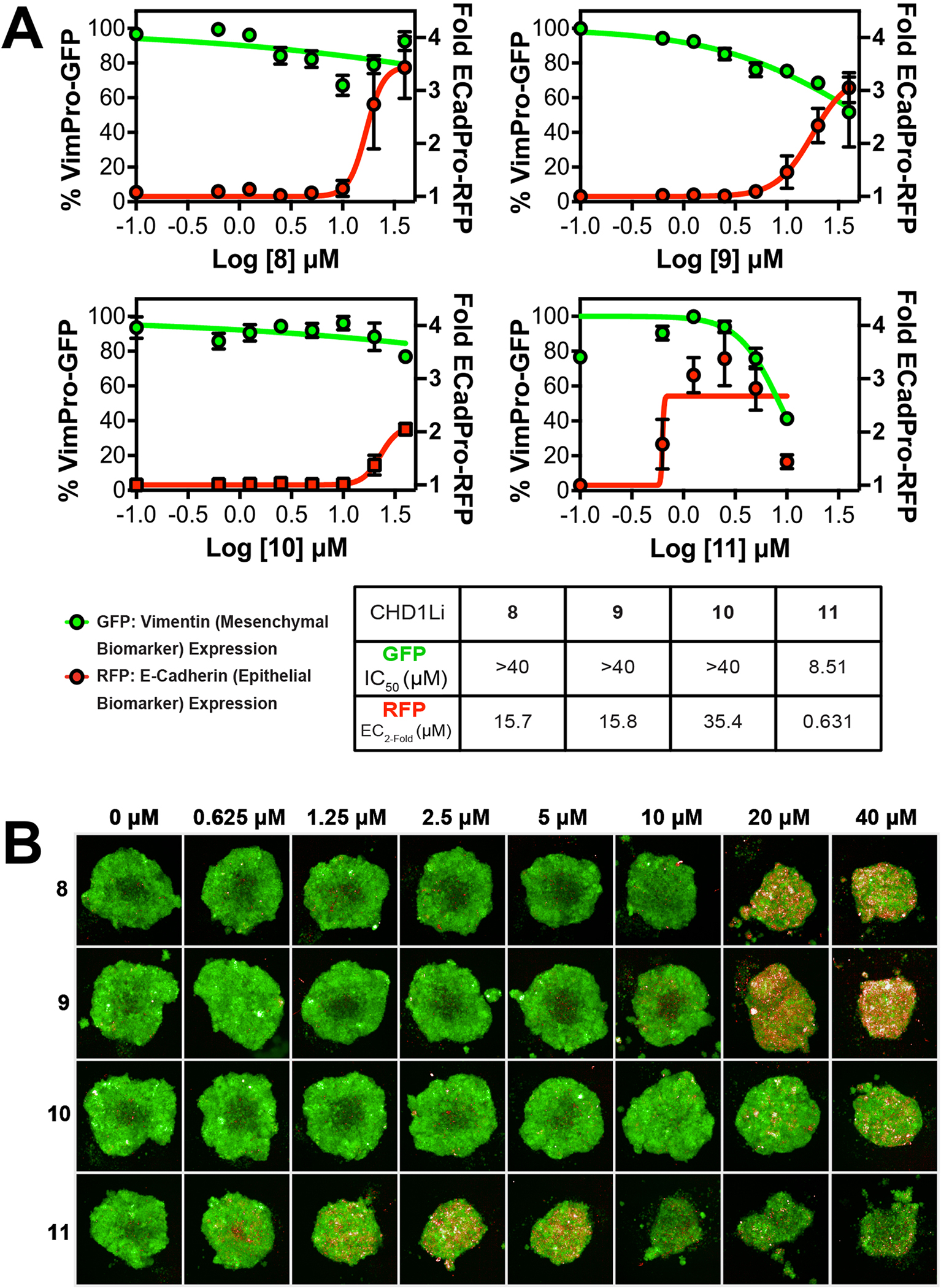
CHD1Li Modulates EMT. (A) Graphical representation of EMT reversal by each of the top HTS hits (N = 3) Each data point is the average of 3 experiments performed in triplicate and error bars are the calculated standard error of the mean (SEM). (B) Representative fluorescent images (10x air objective) of 3D SW620-GFP+ tumor organoids treated with CHD1Li for 72 h at concentrations of 0–40 μM. All images obtained using Opera Phenix using the RFP and GFP channel.
3.5. Inhibition of CHD1L-mediated CSC stemness
Complementary to CHD1Li effects on EMT, we utilized the clonogenic assay to evaluate the CHD1Li’s ability to inhibit CSC stemness [28]. The IC50 of stemness for all compounds ranged between nM to low μM (Table 1) with the most potent CHD1Li being compound 9 and 11 (Fig. 4A,B). However, 11 superior and 3-fold more potent compared CHD1Li 6.0, and this is also consistent with its ability to reverse EMT (Fig. 3). Mesenchymal cells expressing vimentin possess enhanced CSC stemness, which facilitates cell survival and increased invasion and metastatic potential [36]. As such, the compounds that only induced partial EMT reversal had reduced potency against stemness compared to CHD1Li 11.
Fig. 4.
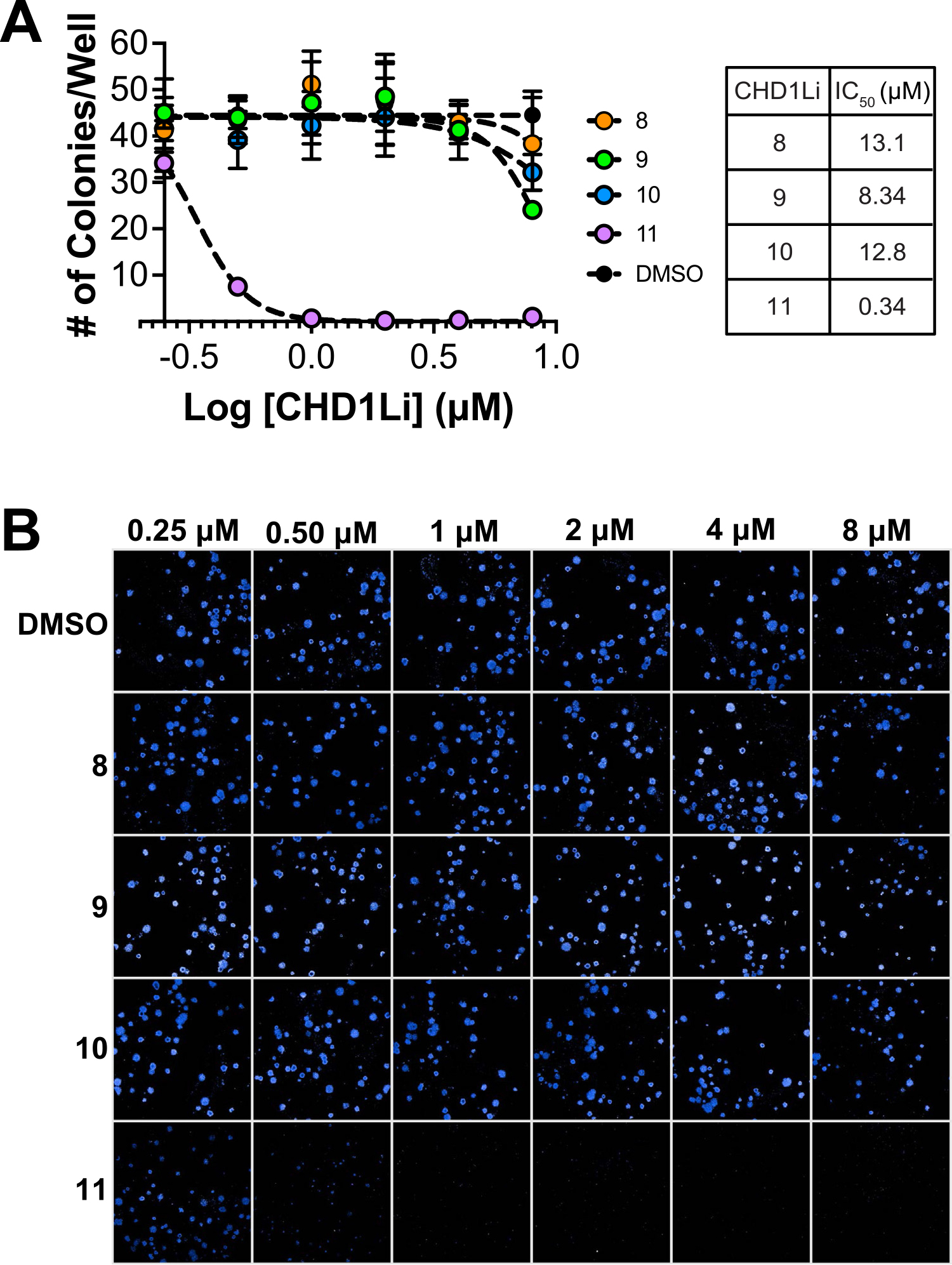
CHD1Li Inhibit CSC Stemness in CRC cells. (A) Graphical representation of dose-response inhibition of CSC stemness by the CHD1Li (N = 2). Error bars are the calculated standard error of the mean (SEM) of 2 experiments performed in triplicate. (B) Representative images (25 stitched fields of view on 10x air objective) of 2D SW620-GFP+ colonies treated with CHD1Li for 10 days at 0.25–8 μM and stained with Hoechst dye for imaging. All images were obtained using the Opera Phenix.
3.6. Microsomal stability
A knowledge of metabolic stability is an important step during early-stage drug development for lead prioritization [37]. We have previously observed a correlation between in vitro microsomal stability and in vivo pharmacokinetics and efficacy for CHD1Li [25]. In vitro evaluation of hit CHD1Li 8-11 stability towards biotransformation showed that CHD1Li 9 and 11 were the most stable when exposed to human and mouse microsomes with half-lives of ~100 min. Compounds 9 and 11 also exhibited up to 16-fold superior stability compared to CHD1Li 8 and10 (Table 1). The poor stability in CHD1Li 8 and 10 are attributed to the S-methyl unit as shown by the mass transition analysis (Table S4). Sterically unhindered S-methyl units are considered as metabolic soft spots due to their vulnerability to cytochrome P450-mediated oxidative metabolism [38]. Hence, optimization studies for these CHD1Li should focus on addressing the thiomethyl functionalities. Similarly, the ability of CHD1Li 11 to exist as either the pyrimidinol or pyrimidinone tautomer (Table S1) requires attention to avoid idiosyncratic toxicity outcomes [39,40].
3.7. In silico CHD1L binding studies
3.7.1. Binding site elucidation
We have previously characterized the potential binding sites on CHD1L using a reported crystal structure and prioritized an allosteric site on the C-ATPase domain as the most plausible binding site for CHD1Li 6.0 and analogs using molecular dynamics simulations [25]. However, in this study, we utilized an AI generated homology model retrieved from the AlphaFold database to improve the accuracy of our predicted CHD1Li complexes [30].
Structural analysis of the apo AlphaFold homology model before and after protein preparation provided a full length (fl)-CHD1L structure in an active conformation with improved protein packing and without missing loops, steric clashes, or unusual B-factors [41–43]. More importantly, SiteMap calculations returned five potential binding sites (Table S2), including the ATP binding site, a large allosteric site on the macro domain, two shallow sites on the N-ATPase domain, and the previously identified CHD1Li allosteric binding site on the C-ATPase domain. Again, the C-ATPase allosteric site had the highest drugability score (1.039) and site score (1.016) overall, confirming it as the most plausible binding site (Fig. 5A).
Fig. 5.
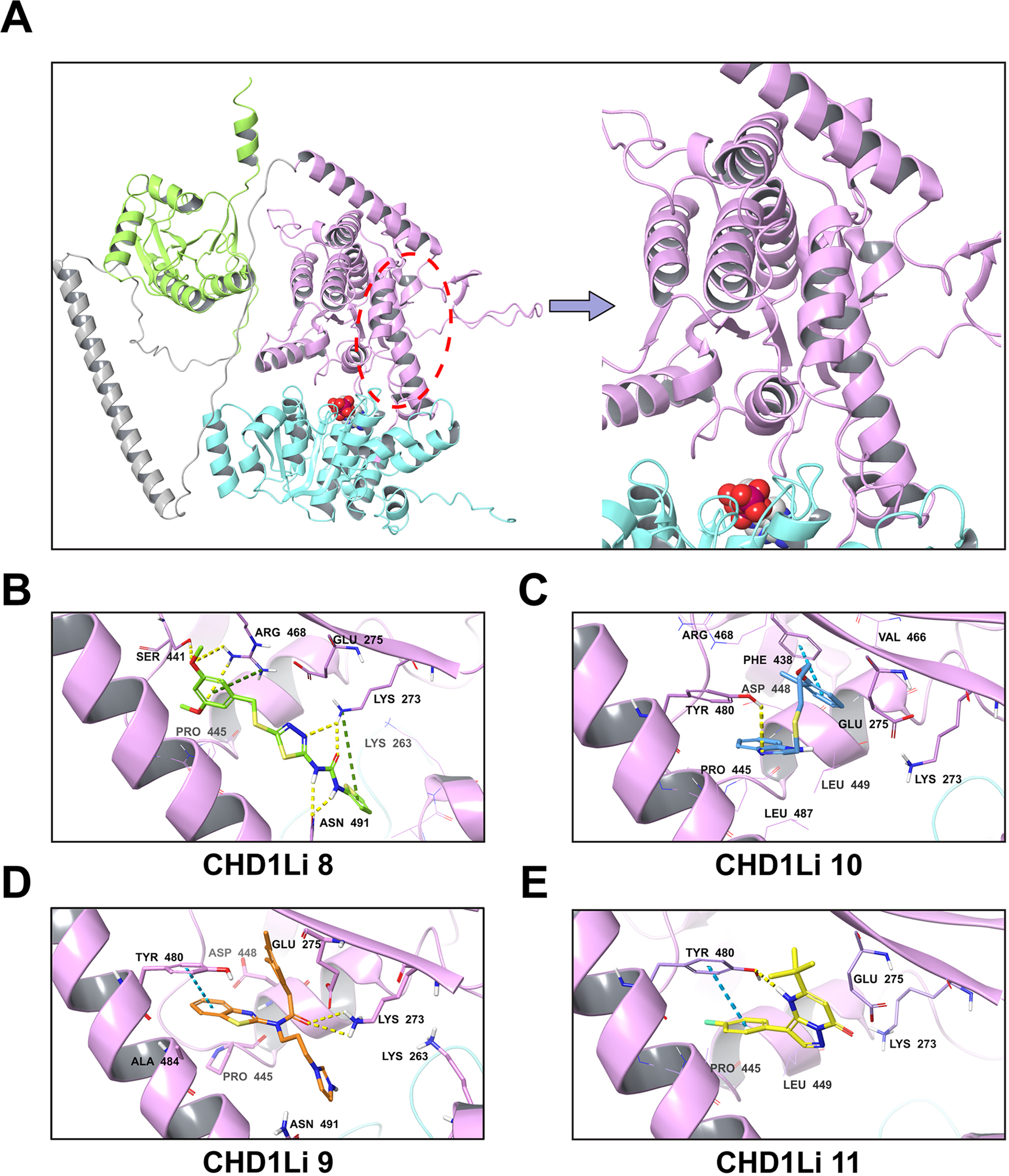
Predicted fl-CHD1L allosteric binding site and binding poses of hit CHD1Li. (A) fl-CHD1L structure showing the most plausible CHD1Li binding site. The domain architecture is depicted in cartoon representation as N-ATPase (cyan), C-ATPase (purple), macro domain (green) and linker region (gray). The proposed C-ATPase allosteric binding site is marked with a red ellipse while the ATP binding site is depicted by the bound ADP shown in CPK representation. (B-E). Each panel shows the 3D representation of the predicted binding pose for the CHD1Li as indicated. The non-bonded interactions are depicted as hydrogen bond (yellow-dashed), pi-cation (green-dashed), and pi-pi stacking (blue-dashed).
3.7.2. CHD1Li target interaction
To gain molecular level insight on the possible target interaction profile of hit CHD1Li 8-11 and identify the corresponding pharmacophore features furnishing enzyme inhibition, the compounds were docked at the C-ATPase allosteric site using the IFD protocol. The higher docking and IFD scores for CHD1Li 8 and 10 (Table S3) suggests that they bind more tightly to the target compared to CHD1Li 9 and 11. The MM/GBSA binding free energy, which is often a more accurate descriptor of native poses [44], is also higher for CHD1Li 8 and 10 as compared to 9 and 11. These results agree with the enzymatic data, which showed CHD1Li 8 and 10 as more potent CHD1Li than 9 and 11 (Fig. 2). Nevertheless, the weaker cell-based potencies of CHD1Li 8-10 as compared to 11 despite their potent CHD1L inhibition can be attributed to structural features limiting cell membrane permeability [45,46]. For instance, with a calculated pKa of 7.12 (Table S1) and a protonated imidazole nitrogen, CHD1Li 9 will exist at an ~50:50% ratio of ionized and neutral forms at physiological pH likely impacting its cell activity and potency due to limited diffusion into cells of the ionized form.
Analysis of the docked complexes showed the CHD1Li structural features favoring target binding (Fig. 5B–E). CHD1Li 8 formed strong H-bond interactions (within 3 Å) with Asn 491 and Lys 273 via its urea and thiadiazole units, and other H-bond interactions with Ser 441 and Arg 468 via the 3,5-dimethoxy unit (Fig. 5B). The pi-cation interactions of electron-rich phenyl and thiophene rings with Lys 273 and Arg 468, and the hydrophobic contacts with Lys 263, Glu 275, and Pro 445 presumably stabilized the ligand in the allosteric binding site. Although CHD1Li 10 (Fig. 5C) only formed pi-pi stacking and H-bond interactions with Phe 438 and Tyr 480, respectively, the hydrophobic propyl linker allowed a robust network of hydrophobic contacts to favor high CHD1L affinity comparable to CHD1Li 8. On the other hand, the similar enzyme inhibition of CHD1Li 9 and 11 is justified by their identical pi-pi stacking interaction with Tyr 480 (Fig. 5D,E). Moreover, CHD1Li 9 featured an H-bond with Lys 273 while CHD1Li 10 interacted similarly with Tyr 480. These results suggest that a combination of H-bond acceptors and hydrophobic groups favor CHD1L binding. Thus, it is inferable that CHD1Li 9 and 11 can be optimized by incorporating pharmacophore features that favor these desired interactions. This is supported by CHD1Li 8, the most potent hit, having the highest number of H-bond acceptors and overall interactions with CHD1L (Fig. 5B), and the most stable target complexation (Table S2).
3.7.3. Molecular dynamics simulation
Having established the possible protein-ligand interactions, we conducted a 500 ns long MD simulation for CHD1Li 9 and 11 (Fig. 6). The goal was to analyze the time-dependent evolution of these interactions and determine ligand stability in terms of root-mean square deviation (RMSD) from the reference structure, i.e., the IFD pose. MD simulation is crucial to comprehensively account for the structural flexibility and ligand-induced conformational changes in a drug-target model system beyond IFD [47].
Fig. 6.
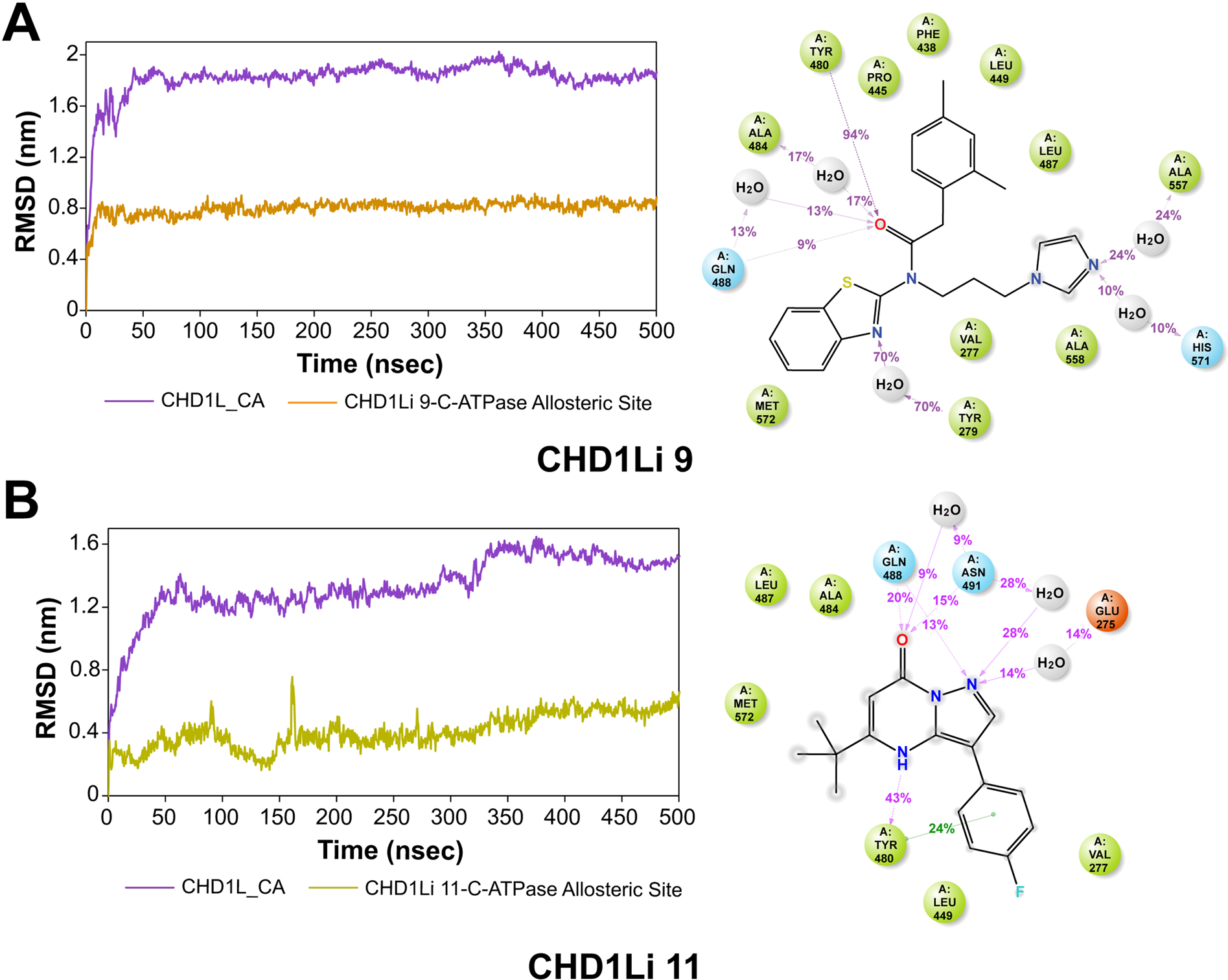
CHD1Li molecular dynamics with CHD1L. The MD trajectory analysis involved the assessment of CHD1Li 9 (A) and 11 (B). The left panels depict the RMSD evolution of both the protein and the complex, while the right panel illustrates a 2D representation of protein-CHD1Li contacts throughout the 500 ns simulation period. In the 2D representation, hydrogen bonds are denoted in purple, hydrophobic residues are represented by green spheres, and polar residues are indicated by blue spheres.
The convergence of CHD1Li 9 at an average RMSD of 0.8 nm within the first 10 ns and the preservation over the entire 500 ns simulation period indicates a highly stable complexation without the ligand diffusing away from the allosteric binding pocket (Fig. 6A). Both CHD1Li 9 and 11 had sustained binding site residence, which is inferable from the H-bond interactions with the amide oxygen atom of Tyr 480, which were sustained over a relatively longer period of time (Fig. 6 A,B right panels). While hydrophobic interactions are important for CHD1Li binding, increased H-bond acceptors appear to be beneficial at improving tight CHD1L binding and residence time.
4. Discussion
CHD1L overexpression and its oncogenic function are well characterized [23]. Notably, CHD1L functions at the interface of tumor progression and tumor cell survival, particularly by promoting malignant gene expression, cell cycle progression, inhibition of programed cell death, and promoting the increased invasion and metastatic potential, all of which contribute to multidrug resistance of chemotherapy and targeted therapy in the clinic [10,23]. Many of these tumorigenic properties have been linked to CHD1L’s role in EMT, where CRC cells gain CSC stemness. Hence, CHD1L inhibition can reduce tumor viability, reverse EMT, and decrease CSC stemness CRC and potentially other cancers. Consequently, the drug discovery process described herein produced two new CHD1Li 9 and 11 as leads for further development.
CHD1Li 11 is particularly noteworthy as it potently inhibits the target resulting in, EMT reversal, inhibition of stemness, and tumor cell death in metastatic CRC cells and tumor organoids. Furthermore, 11 was the most stable to metabolic degradation in human and mouse microsomes. In addition to antitumor properties, a small molecule drug capable of eliciting inhibitory effects on tumor progression is desirable for treating aggressive cancers, as it can prevent drug resistance, tissue invasion and metastasis. The antimetastatic potential of CHD1Li is substantiated in a report by Mu et al., which demonstrated that CHD1L knockdown significantly inhibited lung metastasis in an orthotopic model of breast cancer [12].
5. Conclusion
Building on our previous CHD1Li lead drug development, the structural diversity of CHD1Li characterized herein is expected to facilitate a new series of CHD1Li antitumor agents for CRC and other forms of cancer. Furthermore, CHD1Li display single agent antitumor activity while also eliciting the inhibition pathways driving tumor progression and multidrug resistance. Thus, we anticipate that CHD1Li will be an effective therapeutic strategy alone or when combined with standard of care chemotherapy or targeted therapies, increasing their potential impact for treating a variety of cancers.
Supplementary Material
Acknowledgments
The authors thank the CU AMC Drug Discovery and Development Share Resource (D3SR). The D3SR is supported by the University of Colorado Anschutz Center for Drug Discovery, which was established in 2020 with generous grant funding from The ALSAM Foundation and through CU AMC institutional support. The D3SR is supported in part by the University of Colorado Cancer Center, an NIH NCI designated center (P30-CA046934).
Funding
This research was supported by a grant from the NIH NCI awarded to D.V.L. (R01-CA251361).
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: D. V.L. is the founder of Onconaut Therapeutics Incorporated, which is developing CHD1Li as a lead drug therapies for the treatment of cancer. D.V.L, H.E., P.A., Q.Z. are inventors on patents pertaining to this research.
CRediT authorship contribution statement
S.C. and P.A. executed the majority of experiments, conducted data analysis, wrote and edited the paper. Q.Z. conducted biological experiments and data analysis and contributed to writing the paper. H.E. contributed to data analysis, figure design and preparation, and paper editing. D.C.A assisted with molecular modeling experiments. B.M. conducted all microsomal assay LCMS analysis. J.T.K. expressed and purified recombinant cat-CHD1L enzyme. T.K. assisted with experiments and data analysis. U.B.K contributed to microsomal experimental design and data analysis. D.V.L conceived and supervised the research, co-designed experiments, conducted data analysis, and wrote and edited the paper.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.biopha.2023.116037.
References
- [1].Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A, Colorectal cancer statistics, 2023, CA: A Cancer J. Clin. 73 (3) (2023) 233–254. [DOI] [PubMed] [Google Scholar]
- [2].Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I, Flanders WD, Brawley OW, Gapstur SM, Jemal A Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States, CA: A Cancer J. Clin. 68 (1) (2018) 31–54. [DOI] [PubMed] [Google Scholar]
- [3].Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ, Colorectal carcinoma: a general overview and future perspectives in colorectal cancer, Int. J. Mol. Sci. 18 (1) (2017) 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Society AC, Colorectal Cancer Facts & Figures 2020–2022. Society AC, Ed. American Cancer Society: Atlanta, Georgia, 2020. [Google Scholar]
- [5].Wolpin BM, Mayer RJ, Systemic treatment of colorectal cancer, Gastroenterology 134 (5) (2008) 1296–1310.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ji X, Li J, Zhu L, Cai J, Zhang J, Qu Y, Zhang H, Liu B, Zhao R, Zhu Z, CHD1L promotes tumor progression and predicts survival in colorectal carcinoma, J. Surg. Res. 185 (0031) (2013) 84–91. [DOI] [PubMed] [Google Scholar]
- [7].Martin R, Paty P, Fong Y, Grace A, Cohen A, DeMatteo R, Jarnagin W, Blumgart L, Simultaneous liver and colorectal resections are safe for synchronous colorectal liver metastasis, J. Am. Coll. Surg. 197 (2) (2003) 233–241. [DOI] [PubMed] [Google Scholar]
- [8].Holch J, Stintzing S, Heinemann V, Treatment of metastatic colorectal cancer: standard of care and future perspectives, Visc. Med 32 (3) (2016) 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ma NF, Hu L, Fung JM, Xie D, Zheng BJ, Chen L, Tang DJ, Fu L, Wu Z, Chen M, Fang Y, Guan XY, Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma, Hepatology 47 (2) (2008) 503–510. [DOI] [PubMed] [Google Scholar]
- [10].Soltan MA, Eldeen MA, Eid RA, Alyamani NM, Alqahtani LS, Albogami S, Jafri I, Park MN, Alsharif G, Fayad E, Mohamed G, Osman R, Kim B, Zaki MSA, A pan-cancer analysis reveals CHD1L as a prognostic and immunological biomarker in several human cancers, Front. Mol. Biosci. 10 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cheng W, Su Y, Xu F, CHD1L: a novel oncogene, Mol. Cancer 12 (1) (2013) 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mu QJ, Li HL, Yao Y, Liu SC, Yin CG, Ma XZ, Chromodomain helicase/ATPase DNA-binding protein 1-like gene (CHD1L) expression and implications for invasion and metastasis of breast cancer, PLOS One 10 (11) (2015), e0143030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wu J, Zong Y, Fei X, Chen X, Huang O, He J, Chen W, Li Y, Shen K, Zhu L, Presence of CHD1L over-expression is associated with aggressive tumor biology and is a novel prognostic biomarker for patient survival in human breast cancer, PLOS One 9 (8) (2014), e98673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Su Z, Zhao J, Xian G, Geng W, Rong Z, Wu Y, Qin C, CHD1L is a novel independent prognostic factor for gastric cancer, Clin. Transl. Oncol. 16 (8) (2014) 702–707. [DOI] [PubMed] [Google Scholar]
- [15].He LR, Ma NF, Chen JW, Li BK, Guan XY, Liu MZ, Xie D, Overexpression of CHD1L is positively associated with metastasis of lung adenocarcinoma and predicts patients poor survival, Oncotarget 6 (31) (2015) 31181–31190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Su FR, Ding JH, Bo L, Liu XG, Chromodomain helicase/ATPase DNA binding protein 1-like protein expression predicts poor prognosis in nasopharyngeal carcinoma, Exp. Ther. Med. 8 (6) (2014) 1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Chen L, Chan TH, Liu M, Kong KL, Qiu JL, Li Y, Yuan YF, Guan XY, SPOCK1 is regulated by CHD1L and blocks apoptosis and promotes HCC cell invasiveness and metastasis in mice, Gastroenterology 144 (1) (2013) 179–191, e4. [DOI] [PubMed] [Google Scholar]
- [18].Hyeon J, Ahn S, Park CK, CHD1L Is a marker for poor prognosis of hepatocellular carcinoma after surgical resection, Korean J. Pathol. 47 (1) (2013) 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen L, Chan TH, Yuan YF, Hu L, Huang J, Ma S, Wang J, Dong SS, Tang KH, Xie D, Li Y, Guan XY, CHD1L promotes hepatocellular carcinoma progression and metastasis in mice and is associated with these processes in human patients, J. Clin. Investig. 120 (4) (2010) 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].He WP, Zhou J, Cai MY, Xiao XS, Liao YJ, Kung HF, Guan XY, Xie D, Yang GF, CHD1L protein is overexpressed in human ovarian carcinomas and is a novel predictive biomarker for patients survival, BMC Cancer 12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang W, Wu J, Fei X, Chen W, Li Y, Shen K, Zhu L, CHD1L promotes cell cycle progression and cell motility by up-regulating MDM2 in breast cancer, Am. J. Transl. Res. 11 (3) (2019) 1581–1592. [PMC free article] [PubMed] [Google Scholar]
- [22].Bianchini G, De Angelis C Licata, L. Gianni, L. Treatment, landscape of triple-negative breast cancer – expanded options, evolving needs, Nat. Rev. Clin. Oncol. 19 (2) (2022) 91–113. [DOI] [PubMed] [Google Scholar]
- [23].Xiong X, Lai X, Li A, Liu Z, Ma N, Diversity roles of CHD1L in normal cell function and tumorigenesis, Biomark. Res. 16 (1) (2021), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abbott JM, Zhou Q, Esquer H, Pike L, Broneske TP, Rinaldetti S, Abraham AD, Ramirez DA, Lunghofer PJ, Pitts TM, Regan DP, Tan AC, Gustafson DL, Messersmith WA, LaBarbera DV, First-in-class inhibitors of oncogenic CHD1L with preclinical activity against colorectal cancer, Mol. Cancer Ther. 19 (8) (2020) 1598–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Prigaro BJ, Esquer H, Zhou Q, Pike LA, Awolade P, Lai X-H, Abraham AD, Abbott JM, Matter B, Kompella UB, Messersmith WA, Gustafson DL, LaBarbera DV, Design, synthesis, and biological evaluation of the first inhibitors of oncogenic CHD1L, J. Med. Chem. 65 (5) (2022) 3943–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Esquer H, Zhou Q, Nemkov T, Abraham AD, Rinaldetti S, Chen YC, Zhang X, Orman MV, D’Alessandro A, Ferrer M, Messersmith WA, LaBarbera DV, Isolating and targeting the real-time plasticity and malignant properties of epithelial-mesenchymal transition in cancer, Oncogene 40 (16) (2021) 2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Prigaro BJ, Esquer H, Zhou Q, Pike LA, Awolade P, Lai XH, Abraham AD, Abbott JM, Matter B, Kompella UB, Messersmith WA, Gustafson DL, LaBarbera DV, Design, synthesis, and biological evaluation of the first inhibitors of oncogenic CHD1L, J. Med. Chem. 65 (5) (2022) 3943–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Esquer H, Zhou Q, Abraham AD, LaBarbera DV, Advanced high-content-screening applications of clonogenicity in cancer, SLAS Discov. 25 (7) (2020) 734–743. [DOI] [PubMed] [Google Scholar]
- [29].Abraham AD, Esquer H, Zhou Q, Tomlinson N, Hamill BD, Abbott JM, Li L, Pike LA, Rinaldetti S, Ramirez DA, Lunghofer PJ, Gomez JD, Schaack J, Nemkov T, D’Alessandro A, Hansen KC, Gustafson DL, Messersmith WA, LaBarbera DV, Drug design targeting T-cell factor-driven epithelial–mesenchymal transition as a therapeutic strategy for colorectal cancer, J. Med. Chem. 62 (22) (2019) 10182–10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold, Nature 596 (7873) (2021) 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chaffer CL, San Juan BP, Lim E, Weinberg RA, EMT, cell plasticity and metastasis, Cancer Metastas. Rev. 35 (4) (2016) 645–654. [DOI] [PubMed] [Google Scholar]
- [32].Masimirembwa CM, Bredberg U, Andersson TB, Metabolic stability for drug discovery and development: pharmacokinetic and biochemical challenges, Clin. Pharmacokinet. 42 (6) (2003) 515–528. [DOI] [PubMed] [Google Scholar]
- [33].Abraham AD, Esquer H, Zhou Q, Tomlinson N, Hamill BD, Abbott JM, Li L, Pike LA, Rinaldetti S, Ramirez DA, Lunghofer PJ, Gomez JD, Schaack J, Nemkov T, D’Alessandro A, Hansen KC, Gustafson DL, Messersmith WA, LaBarbera DV, Drug design targeting T-cell factor-driven epithelial-mesenchymal transition as a therapeutic strategy for colorectal cancer, J. Med. Chem. 62 (22) (2019) 10182–10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou Q, Abraham AD, Li L, Babalmorad A, Bagby S, Arcaroli JJ, Hansen RJ, Valeriote FA, Gustafson DL, Schaack J, Messersmith WA, LaBarbera DV, Topoisomerase IIα mediates TCF-dependent epithelial-mesenchymal transition in colon cancer. Oncogene 35 (38) (2016) 4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hewitt RE, McMarlin A, Kleiner D, Wersto R, Martin P, Tsokos M, Stamp GW, Stetler-Stevenson WG, Validation of a model of colon cancer progression, J. Pathol. 192 (4) (2000) 446–454. [DOI] [PubMed] [Google Scholar]
- [36].Usman S, Waseem NH, Nguyen TKN, Mohsin S, Jamal A, Teh MT, Waseem A, Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis, Cancers 13 (19) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Z, Tang W, Drug metabolism in drug discovery and development, Acta Pharm. Sin. B 8 (5) (2018) 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Trunzer M, Faller B, Zimmerlin A, Metabolic soft spot identification and compound optimization in early discovery phases using metasite and LC-MS/MS validation, J. Med. Chem. 52 (2) (2009) 329–335. [DOI] [PubMed] [Google Scholar]
- [39].Stepan AF, Mascitti V, Beaumont K, Kalgutkar AS, Metabolism-guided drug design, MedChemComm 4 (4) (2013) 631–652. [Google Scholar]
- [40].Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD, Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States, Chem. Res. Toxicol. 24 (9) (2011) 1345–1410. [DOI] [PubMed] [Google Scholar]
- [41].Caldararu O, Kumar R, Oksanen E, Logan DT, Ryde U, Are crystallographic B-factors suitable for calculating protein conformational entropy? Phys. Chem. Chem. Phys. 21 (33) (2019) 18149–18160. [DOI] [PubMed] [Google Scholar]
- [42].Shehu A, Kavraki LE, Modeling structures and motions of loops in protein molecules, Entropy 14 (2) (2012) 252–290. [Google Scholar]
- [43].Rossi KA, Weigelt CA, Nayeem A, Krystek SR Jr, Loopholes and missing links in protein modeling, Protein Sci. 16 (9) (2007) 1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fischer A, Smieško M, Sellner M, Lill MA, Decision making in structure-based drug discovery: visual inspection of docking results, J. Med. Chem. 64 (5) (2021) 2489–2500. [DOI] [PubMed] [Google Scholar]
- [45].Yang NJ, Hinner MJ, Getting across the cell membrane: an overview for small molecules, peptides, and proteins, in: Gautier A, Hinner MJ (Eds.), In Site-Specific Protein Labeling: Methods and Protocols, Springer New York, New York, NY, 2015, pp. 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem. 45 (12) (2002) 2615–2623. [DOI] [PubMed] [Google Scholar]
- [47].De Vivo M, Masetti M, Bottegoni G, Cavalli A, Role of molecular dynamics and related methods in drug discovery, J. Med. Chem. 59 (9) (2016) 4035–4061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.