

Abstract
Protein persulfidation is a significant post-translational modification that involves addition of a sulfur atom to the cysteine thiol group and is facilitated by sulfide species. Persulfidation targets reactive cysteine residues within proteins, influencing their structure and/or function across various biological systems. This modification is evolutionarily conserved and plays a crucial role in preventing irreversible cysteine overoxidation, a process that becomes prominent with aging. While, persulfidation decreases with age, its levels in the aged heart and the functional implications of such a reduction in cardiac metabolism remain unknown. Here we interrogated the cardiac persulfydome in wild-type adult mice and age-matched mice lacking the two sulfide generating enzymes, namely cystathionine gamma lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3MST). Our findings revealed that cardiac persulfidated proteins in wild type hearts are less abundant compared to those in other organs, with a primary involvement in mitochondrial metabolic processes. We further focused on one specific target, NDUFB7, which undergoes persulfidation by both CSE and 3MST derived sulfide species. In particular, persulfidation of cysteines C80 and C90 in NDUFB7 protects the protein from overoxidation and maintains the complex I activity in cardiomyocytes. As the heart ages, the levels of CSE and 3MST in cardiomyocytes decline, leading to reduced NDUFB7 persulfidation and increased cardiac NADH/NAD+ ratio. Collectively, our data provide compelling evidence for a direct link between cardiac persulfidation and mitochondrial complex I activity, which is compromised in aging.
Keywords: Persulfidation, Cardiac aging, CSE, 3MST, NDUFB7
Graphical abstract
Highlights
-
•
Cardiac proteins are differentially persulfidated by CSE and 3MST.
-
•
Endogenous cardiac sulfides modify mainly mitochondrial proteins.
-
•
Cardiac NDUFB7 persulfidation maintains complex I activity and preserves NAD + levels.
-
•
Reduced CSE and 3MST expression in aging results in impaired sulfide sensing and impacts on cardiac NAD + bioavailability.
1. Introduction
Protein persulfidation is a highly conserved post-translational protein modification (PTM) of the redox-sensitive amino acid cysteine [1], tightly regulated by intracellular sulfide levels. This PTM declines with aging, and recent studies assert its potential as a defense mechanism against overoxidation and cysteine sulfenylation [1]. Protein persulfides decreasing with age coincide with reduced levels of the three H2S-producing enzymes: cystathionine-γ-lyase (CSE), cystathionine-β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3MST) [1]. CSE and CBS are mainly localized in the cytoplasm whereas 3MST is found both in the cytoplasm and mitochondria, supporting cellular bioenergetics [2,3].
The enzymatic functions of CSE and 3MST have been linked to the physiological function of the cardiovascular system [[2], [4]]. For instance, CSE deficiency in atherogenic mouse models results in deregulated lipid metabolism and accelerated atherosclerosis due to decreased sulfide levels [[5], [6], [7]]. On the other hand, sulfide supplementation and CSE overexpression protect against ischemia-induced heart failure by mitigating oxidative stress and mitochondrial dysfunction [8]. Recent studies on cell-specific deletion of CSE in mice show increased endothelial to mesenchymal transition phenotypes in the myocardium associated with elevated cardiac fibrosis and impaired cardiac function [9]. In addition, CSE overexpression in endothelial cells reverses these effects and preserves cardiac function [9]. Sulfide supplementation also restores endothelial function and pathological vasodilation in patients with vascular dysfunction [10], and, in mice reverses atherogenic phenotypes resulting from CSE deletion [5]. Similarly, 3MST appears crucial for cardiovascular function in aging, as its loss leads to hypertension and cardiac hypertrophy [11]. 3MST is mainly involved in regulating mitochondrial metabolism and mitochondrial protein import [12], and consequently its absence in the heart impacts on mitochondrial respiration and ATP synthesis by influencing the availability of branched chain amino acids [13]. However, whether protein persulfidation from CSE or 3MST is responsible for altering heart mitochondrial function remains unknown. Understanding this link is crucial, given that cardiac bioenergetics heavily rely on mitochondrial oxidative metabolism [14], and aging is linked to both mitochondrial dysfunction and loss of cellular persulfidation. To address this, here we employed global CSE and 3MST mouse lines, aimed to identify CSE and 3MST specific persulfidation targets in the heart and link the loss of age-related cardiac persulfidation to cardiac bioenergetic surveillance mechanisms.
2. Materials and methods
2.1. Materials
Cell culture media were from Gibco (Invitrogen, Darmstadt, Germany), the protease inhibitor cocktail was from Roche (Mannheim, Germany). Copper (II)-TBTA complex and Cyanine5 alkyne were from Lumiprobe (Lumiprobe GmBH, Hannover, Germany) and Daz2 was from Cayman (Cayman Chemical, Michigan, USA). All other chemicals (unless otherwise specified) were from Sigma-Aldrich (Merck, Darmstadt, Germany).
2.2. Animals
Wild type mice in the age of 3 months and 18 months were purchased from Charles Rivers Associates (Boston, USA). CSE [15] and 3MST [16] global knockouts and respective wild type controls were bred in the animal facility of the Biomedical Research Foundation of the Academy of Athens. Mice were housed in conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication no. 85–23). Animals received standard rodent chow diet and all studies were approved by the animal research ethic committee. Animals were randomized using the block randomization method to ensure similar sample sizes per age. No animals were excluded from the analysis.
2.3. Ndufb7 constructs
To synthetize murine Ndufb7 wild type and C80A, C90A or C80A-C90A HA tagged plasmids, we generated multiple overlapping primers based on the murine Ndufb7 (NM_025843.3) sequence (Genomeditech, Shanghai, China). HA coding sequence as well as homology arms were directly introduced into the primers. After amplification, the PCR products of Ndufb7-HA WT, Ndufb7-HA C80A, Ndufb7-HA C90A, and Ndufb7-HA C80, 90A mutant, and HA were separated via agarose electrophoresis. Following amplification and agarose-based separation, the appropriate fragments were subsequently cloned into the BamHI/EcoRI sites of the pcDNA 3.1(+) (Invitrogen) via homologous recombination. For the generation of the lentiviral constructs the appropriate fragments were subsequently cloned into the BamHI/EcoRI sites of the PCDH-CMV-MCS-EF1-T2A-Puro lentiviral vector via homologous recombination (Genomeditech, Shanghai, China). The sequences of all plasmids were further verified by DNA sequencing.
2.4. HEK293 cell culture, treatment and transfection protocols
HEK293 cells were cultured and transfected with Ndufb7, pcDNA, CSE and 3MST plasmids as previously described [17]. Where indicated, cells were treated with 1 mmol/L dithiothreitol (DTT) for 5 min at 37 °C degrees prior to collection.
2.5. HL-1 cell culture and transduction protocol
HL-1 adult mouse cardiac muscle cells were purchased from Sigma-Aldrich (Cat. No. SCC065, Merck, Darmstadt, Germany) and cultured in Claycomb Medium (Cat. No. 51800C, Sigma-Aldrich, Merck, Darmstadt, Germany) supplemented with 10% fetal bovine serum, 0.1 μmol/L norepinephrine and 2 μmol/L l-Glutamine. Cells were seeded in a density of 30.000 cells/cm2 and got transduced with the produced Ndufb7 wild type, C80A, C90A and C80A-C90A lentiviruses for 24 h using 8 μg/mL polybrene (Cat. No. TR-1003, Sigma-Aldrich, Merck, Darmstadt, Germany). The transduced cells were further selected with 2 μg/mL puromycin for 6 days and the transduction efficiency was validated with Immunoblotting.
2.6. Lentiviral preparations
HEK293T cells were cultured in DMEM/F-12 (Cat. No. 21041025, Thermo Fischer Scientific, Harz, Germany) supplemented with 10% fetal bovine serum, 100 U/mL penicillin-streptomycin (Cat. No. 15140122, Thermo Fischer Scientific, Harz, Germany) and 0.01 mol/L HEPES (Cat. No. H0887, Merck, Darmstadt, Germany). Cells were plated in a density of 40.000 cells/cm2 and 24 h later were co-transfected with psPAX2, (Addgene plasmid #12260), pMD2.G (Addgene plasmid #12259) and Ndufb7 wild type, C80A, C90A or C80A-C90A plasmids to produce the respective lentiviruses. For the virus production the media was replaced 24 h after transfection with fresh media and 72 h later the viruses were collected, filtered through a 0.45 μm syringe filter and stored in −80 °C until use. The efficiency of viral infection was tested using the Lenti-X GoStix Plus Takara kit (Cat. No. 631280, Saint-Germain-en-Laye, France) according to the manufacturer instructions.
2.7. Cardiomyocyte isolation
Cardiomyocytes were isolated from 3 to 18 month old wild type mice with a modified Langendorff free method [18]. Gravity isolated cardiomyocytes were snap frozen until further evaluation.
2.8. Detection of cardiac persulfidation
nanoLC-MS/MS: Persulfidomes where detected in pulverized heart tissue as previously described [10].
Antibody array: NDUFB7 persulfidation was adapted from a previous publication [1,17]. NDUFB7 antibody was from Proteintech (Manchester, UK, Cat No. 14912-1-AP)
Immunofluorescent imaging: Pseudocoloring of persulfidation was visualized by a dimedone switch method [1] as previously described [10].
2.9. NADH/NAD+ assays
The assay was performed in pulverized heart tissue, isolated cardiomyocytes, murine HL-1 cardiomyocytes or HEK293 cells with a commercially available NAD/NADH Assay kit from Abcam (Berlin, Germany, Cat.No. ab176723) according to the manufacturers protocol. The values of NADH and NAD+ were normalized to the amount of protein in the samples measured with the Bradford assay from Biorad (Feldkirchen, Germany) and ratios are presented. Where indicated the respected cells were treated with 1 mmol/L Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) for 5 min at 37 °C degrees prior to the assay.
2.10. Oxygen consumption rate
The cellular oxygen consumption rate (OCR) was analyzed using a Seahorse 96 extracellular flux analyzer (Agilent, Waldbronn, Germany). 50.000 HEK293 cells or 8.600 HL-1 murine cardiomyocytes were plated in Seahorse 96-well cell culture plates one day prior to the measurements and equilibrated in Krebs Henseleit buffer (111 mmol/L NaCl, 4.7 mmol/L KCl, 1.25 mmol/L CaCl2, 2 mmol/L MgSO4, 1.2 mmol/L Na2HPO4) supplemented with 1 mmol/L sodium pyruvate for 30 min without CO2 before recordings were made. HEK293 cells were treated with 10 μmol/L oligomycin, 20 μmol/L FCCP, 10 μmol/L rotenone and 10 μmol/L antimycin A. HL-1 murine cardiomyocytes were treated with 10 μmol/L oligomycin, 5 μmol/L FCCP, 10 μmol/L rotenone and 10 μmol/L antimycin A for the mitotest and with one single treatment of rotenone 20 μmol/L to evaluate the Complex I OCR.
2.11. Immunoblotting and DNA content
Immunoblotting was performed as previously described [17]. CSE antibody was from Proteintech (Cat. No. 12217-1-AP, Manchester, UK), 3MST antibody was from Sigma-Aldrich (Cat. No. HPA001240, Merck, Darmstadt, Germany), GAPDH antibody was from Santa Cruz (Cat. No. sc-47724, Santa Cruz Biotechnology Inc, Heidelberg, Germany), Vinculin monoclonal antibody was from Thermo Fisher Scientific (Cat. No. MA5-11690, Invitrogen, Harz, Germany) and the HA antibody was from Abcam (Berlin, Germany, Cat. No. ab1424). DNA was isolated with the PureLink Genomic DNA mini kit (Cat. Mo K1820-00, Invitrogen, Thermo Fischer Scientific, Harz, Germany).
2.12. SIRT3 activity
Activity was measured in HEK293 cells with a commercially available fluorometric kit from Abcam (Berlin, Germany, Cat. No. ab156067), NAD addition was omitted.
2.13. Statistical analysis
All data are presented as the mean ± SD. Normal distribution was examined using the d’Agostino–Pearson omnibus normality test. When 2 groups were compared, the Student's t-test was used when samples were normally distributed and Mann Whitney test for non-parametric analysis. When more than 2 groups were compared, one-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test was used. For studies comparing more than two variables two-way ANOVA followed by Bonferroni's multiple comparison test was used. For non parametric analysis Kruskal-Wallis with Dunn's multiple comparisons test was used. Details are presented on each figure legend. All analyses were performed using GraphPad PRISM data analysis software (version 10.2; GraphPad Software).
3. Results
3.1. CSE and 3MST derived sulfides modify the persulfidation of the mitochondrial protein NDUFB7
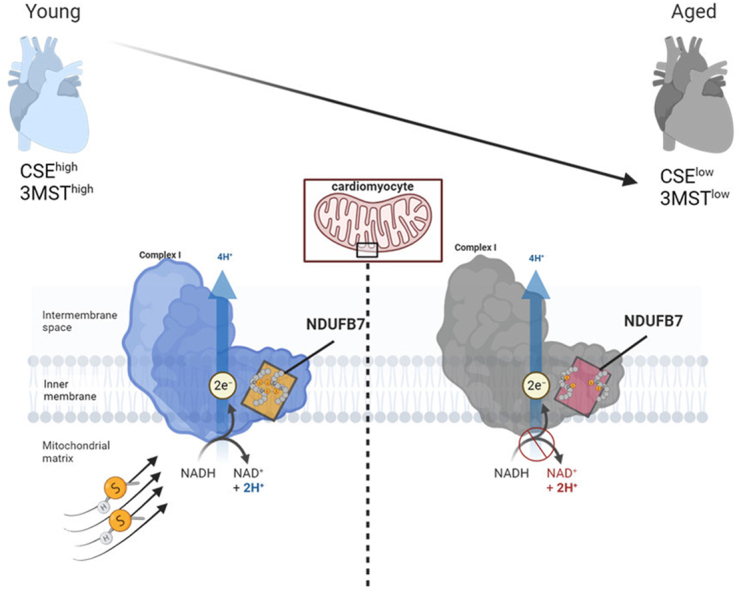
Persulfidation was identified by nanoLC tandem MS methods in pulverized heart tissue isolated from wild type adult mice, or age-matched mice lacking CSE and 3MST. In total 369 cysteine residues in 157 proteins were identified to be persulfidated in wild type hearts (Table 1). In CSE knock out hearts 56 cysteines in 32 proteins were found less persulfidated (p < 0.1) with only 4 of them being significantly reduced (p < 0.05) compared to wild type hearts. GO enrichment analysis showed that the CSE dependent persulfidated proteins were involved mainly in iron and metal cluster binding (GO:0051539, 0051536, 0051540) as well as mitochondrial electron transfer activity and NADH dehydrogenase activity (GO:0009055, 0008137, 0050136, 0003955, 0003954) (Fig. 1A). 3MST knock out had a greater impact on cardiac persulfidation, as 115 cysteines in 58 proteins exhibited reduced persulfidation (p < 0.1), with 24 being significantly reduced (p < 0.05) (Table 1). Those belonged mainly to mitochondrial electron transfer activity processes (GO: 0009055) and receptor, protein or heparin binding processes (GO:0043394, 0051087, 0043167, 0043169) (Fig. 1B). Among the CSE and 3MST knockout hearts, 25 common proteins with a tendency of significance were identified and only 4 proteins were found to exhibit significantly reduced persulfidation, with the most interesting target being the NADH:Ubiquinone Oxidoreductase Subunit B7 (NDUFB7) (Table 1).
Fig. 1.

Reduced persulfidation in the hearts of CSE and 3MST knock out mice affects NDUFB7 persulfidation and NADH/NAD+ ratio. (A-B) Volcano plots showing persulfidated cysteines in proteins, in hearts from wild type (WT) compared to (A) CSE or (B) 3MST knock out (KO) mice and respective GO enrichment analysis of the modified proteins. n = 4/group. Unpaired Student's t-test. Red/Green: persulfidated cysteines in the respective proteins with a p < 0.1 enriched in the WT samples, blue: persulfidated proteins with a p < 0.1 enriched in the CSE (A) or the 3MST (B) samples. (C) Persulfidated NDUFB7 S-SH detected with a dimedone switch method and plotted as the relative fluorescence intensity of Daz2:Cy5/NBF in whole hearts from mice as in panels A and B. n = 6/group. one-way ANOVA, Tukey's multiple comparisons test. (D) NADH/NAD+ ratio in whole heart homogenates from mice as in panels A and B. n = 4/group. Kruskal-Wallis, Dunn's multiple comparisons test. (E) Representative Western blot analysis for HA tagged NDUFB7, GAPDH and persulfidated NDUFB7 S-SH detected with a dimedone switch method and plotted as the relative fluorescence intensity of Daz2:Cy5/NBF in HEK293 cells transfected with wild type (WT) NDUFB7, or NDUFB7 mutated at Cys80 to alanine (C80A), Cys90 to alanine (C90A) or double mutated and co-transfected with pcDNA or a CSE and a 3MST expressing plasmid. Dithiothreitol (DTT) was used as a negative control for the removal of endogenous persulfidation. n = 6/group. two-way ANOVA, Tukey's multiple comparisons test. (F) Representative Western blot analysis for CSE, 3MST, HA tagged NDUFB7, Vinculin and persulfidated NDUFB7 S-SH detected as the relative fluorescence intensity of Daz2:Cy5/NBF in murine HL-1 cardiomyocytes infected with lentiviruses expressing a wild type (WT) NDUFB7, or NDUFB7 mutated at Cys80 to alanine (C80A), Cys90 to alanine (C90A) or double mutated. DTT was used as a negative control. n = 6/group. one-way ANOVA, Tukey's multiple comparisons test.
Given that NDUFB7 was found modified at Cys80 and Cys90, the two main cysteines of the Cx9C-containing subunit that form intra-molecular disulfide bonds which are important for the NADH dehydrogenase function of the protein [19], we focused on this in more detail. Indeed, by performing an antibody array persulfidation assay as previously described [1,10], we were able to confirm reduced NDUFB7 persulfidation in CSE and 3MST knock out heart samples (Fig. 1C), accompanied by increased NADH/NAD+ ratio (Fig. 1D). To examine whether Cys80 and Cys90 are the key modified cysteines on NDUFB7, we generated murine Ndufb7 cysteine 80 to alanine and cysteine 90 to alanine single mutants, as well as a cysteine 80 and 90 to alanine double mutant. We chose to overexpress those mutants either in HEK293 cells, which do not basally express CSE or 3MST (Fig. 1E), resulting in low levels of endogenous persulfidation, or raise lentiviruses to overexpress them in CSE and 3MST expressing murine cardiomyocytes (Fig. 1F). All mutants expressed equal amounts of NDUFB7 protein when overexpressed in HEK293 cells (Fig. 1E) or HL-1 murine cardiomyocytes (Fig. 1F). Our data show that persulfidation signals were increased in wild type or single mutated NDUFB7 co-transfected with CSE and 3MST HEK293 cells, whilst the double mutation completely abolished the DAZ2:Cy5 signal, an indication that these 2 cysteine residues sense both CSE- and 3MST-derived sulfides (Fig. 1E). However, in murine HL-1 cardiomyocytes, mutation of C80A, C90A or double C80A-C90A mutation resulted in reduced persulfidation signals, with a trend for a stronger effect in the double mutant (Fig. 1F). Taken together, these data indicate a CSE and 3MST-dependent regulatory role of persulfidation in mitochondrial proteins in the heart.
3.2. Persulfidation controls NAD+ levels through NDUFB7 activity
Next, we aimed to link NDUFB7 persulfidation, with mitochondrial complex I activity and the capacity of the cell to generate NAD+. When oxygen consumption rates were monitored in HEK293 cells overexpressing CSE and 3MST, to induce persulfidation, double mutation of the NDUFB7 resulted in reduced complex I activity and overall lower basal respiratory capacity (Fig. 2A). Interestingly, in the murine HL-1 cardiomyocytes although single and double mutants exhibited similar effects to the complex I dependent oxygen consumption rate, differential responses were observed considering the basal, maximal and ATP dependent mitochondrial respiration (Fig. 2B). In particular, the C80A and C80A-C90A double mutation of Ndufb7 exhibited similar effects on basal, ATP dependent and maximal oxygen consumption rate, the C90A mutation had no statistically significant impact. Collectively these data propose that while Complex I dependent mitochondrial respiration is equally regulated by the persulfidation of the C80 and C90, complex I independent mitochondrial respiration is more sensitive to the modification of the C80. These results were paralleled with the NDUFB7 dependent NADH to NAD+ conversion, as increased persulfidation in wild type NDUFB7 resulted in reduced NADH/NAD+ ratio and as such increased function of the protein, an increase that was abolished in the presence of the reducing agent TCEP. Single mutants showed reduced function similar to the double mutation of the two reactive cysteines in HEK293 cells (Fig. 2C). Notably, in contrast to the superior contribution of the C80A mutation to the HL-1 murine cardiomyocyte mitochondrial respiration, both C80A and C90A mutations resulted in significant elevation of the NADH/NAD+ ratio (Fig. 2D), with no evident cumulative effect on the double mutant. Taken together the data indicate that single de-persulfidation of NDUFB7 results in impaired NADH/NAD+ ratio and compromised Complex I activity, although particularly the C80 cysteine residue is more important for mitochondrial basal respiration. We next treated cells expressing the wild type NDUFB7 and either transfected with pcDNA or with CSE and 3MST, with excessive amounts of H2O2, which is responsible for the irreversible oxidative modification of cysteine thiols. Interestingly, while H2O2 increased the NADH/NAD+ ratio in pcDNA transfected cells, expression of CSE and 3MST maintained the capacity of mitochondria to convert NADH to NAD+, indicating that endogenous sulfides act to protect against cysteine overoxidation of NDUFB7 and preserve complex I activity (Fig. 2E). Last, given the importance of NAD+ in the regulation of sirtuin activity, which impacts on cardiac acetylation and heart failure phenotypes [20], we evaluated SIRT3 activity omitting the addition of NAD+. Our data reveal that in HEK293 cells expressing CSE and 3MST maintained high SIRT3 activity levels only in NDUFB7 non-mutated conditions, whilst non-persulfidatable mutants exhibited reduced SIRT3 activity (Fig. 2F). Taken together our data demonstrate that CSE and 3MST derived persulfides collectively modify key proteins of complex I, including NDUFB7, preserving its activity by shielding its subunits against cysteine hyperoxidation.
Fig. 2.

CSE and 3MST-triggered persulfidation preserves mitochondrial complex I activity and NADH/NAD+ levels. (A-B) Oxygen consumption rate (OCR) in (A) HEK293 cells co-transfected with CSE and 3MST as well as wild type (WT) NDUFB7, or NDUFB7 mutated at Cys80 to alanine (C80A), Cys90 to alanine (C90A) or double mutated (B) murine HL-1 cardiomyocytes infected with lentiviruses expressing a WT or mutated NDUFB7. Complex I oxygen consumption rate was evaluated in response to 10 μmol/L Rotenone. n = 6–14/group. one-way ANOVA, Tukey's multiple comparisons test. (C) NADH/NAD+ ratio in HEK293 cells as in panel A. (D) NADH/NAD+ ratio in HL-1 cardiomyocytes as in panel B. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was used as a disulfide bond breaker. n = 6/group. two-way ANOVA, Tukey's multiple comparisons test (C). one-way ANOVA, Tukey's multiple comparisons test (D). (E) NADH/NAD+ ratio in HEK293 cells transfected with NDUFB7 and either pcDNA or CSE and 3MST plasmids and treated with solvent (Sol.) or 500 μmol/L of H2O2 for up to 72 h n = 6/group. one-way ANOVA, Tukey's multiple comparisons test. (F) SIRT3 activity in HEK293 cells transfected with CSE and 3MST as well as a WT or mutated NDUFB7. n = 6/group. one-way ANOVA, Tukey's posthoc analysis.
3.3. Loss of persulfidation in cardiomyocytes isolated from middle aged murine hearts alters NAD+ homeostasis
In line with the reduced protein expression of CSE and 3MST in primary isolated murine cardiomyocytes from 18 compared to 3 month old wild type mice (Fig. 3A), global persulfidation was reduced in 18 month age hearts (Fig. 3B), an age that is linked to cardiac dysfunctional phenotypes [[21], [22], [23], [24]]. Although, the global NDFUB7 levels did not change in the cardiomyocytes from middle aged murine hearts (Fig. 3C), persulfidation of NDUFB7 was significantly reduced in cardiomyocytes isolated from 18 month old mice, an effect that could not be reversed after treatment of the isolated cells for 10 min with the rapid releasing H2S donor, NaHS (Fig. 3D). These results were consistent with the increased NADH/NAD+ levels observed in the middle aged murine cardiomyocytes. Interestingly, NaHS treatment was able to increase the NAD+ levels only in the young cardiomyocytes but not in the middle aged, strengthening the evidence that the irreversible oxidative modifications occurring in aging cannot be rescued by sulfide supplementation (Fig. 3E).
Fig. 3.

Reduced NDUFB7 persulfidation during aging impairs NADH/NAD+ ratio. (A) Representative immunoblotting analysis for CSE and 3MST and relative densitometric analysis normalized to the DNA content in primary murine isolated cardiomyocytes from 3 or 18 month old wild type mice. n = 4/group, Unpaired Student's t-test. (B) Representative confocal images and quantification showing the results of the dimedone switch method for the detection of persulfidation plotted as the intensity of the Daz2:Cy5 persulfidation signal (blue to red, upper panel) and DAPI (grey), NBF–Cl (red) and a-actinin (blue) in the lower panel in samples as in A. n = 4–6/group, Unpaired Student's t-test. (C) Representative immunobloting and respective quantification for NDUFB7 and VINCULIN for samples as in panel A. n = 4–5/group. (D-E) Persulfidated NDUFB7 S-SH detected with a dimedone switch method and plotted as the relative fluorescence intensity of Daz2:Cy5/NBF (D) and NADH/NAD ratio (E) in samples as in panel A, treated for 10 min with solvent (Sol.) or 100 μmol/L NaHS. n = 6/group. two-way ANOVA, Bonferroni's posthoc analysis.
4. Conclusion
Protein persulfidation is a significant reversible and regulatory PTM that in the heart relies heavily on the enzymatic activities of CSE and 3MST. In this study, we aimed to compare the cardiac persulfidome derived from CSE or 3MST knock out animals and understand how such a modification impacts on cardiac signaling. Strikingly, endogenous persulfidation of the murine heart was found to be less abundant compared to what has been described for other organs such as the liver [25] or the endothelium [10]. Only 157 proteins were found to be modified in the wild type myocardium, with 4 of them commonly targeted significantly from CSE and 3MST derived sulfides. 3MST played a more dominant role to cardiac persulfidation, with a total of 24 significant proteins and 20 of them solely modified from 3MST-derived sulfides. Of note 3MST/H2S also can serve as an inorganic electron donor at the respiratory chain, complementing and balancing the bioenergetic role of Krebs cycle-derived electron donors [26]. Interestingly, CSE and 3MST affected distinct proteins in different cellular compartments i.e. electron transport flavoproteins for CSE and primarily tricarboxylic acid cycle related proteins as malate dehydrogenase 1 (MDH1), fructose-bisphosphate aldolase (ALDOA), pyruvate kinase (PKM) and succinate dehydrogenase A (SDHA) for 3MST, indicating of a complementary role of the two proteins in the regulation of cardiac persulfidation and mitochondrial metabolism. When focusing on common CSE and 3MST targets, we, surprisingly, observed that the deletion of both enzymes reduced persulfidation of NADH:Ubiquinone Oxidoreductase Subunits/NDUF subunits, which are key components of mitochondrial complex I activity.
The NADH:ubiquinone oxidoreductase (complex I) catalyses the first step of the oxidative phosphorylation and consists of 45 proteins in humans [27]. Embedded in the mitochondrial inner membrane, it couples the oxidation of NADH to the reduction of ubiquinone and the translocation of protons across the inner membrane. Given its size and role in energy conversion it is not surprising that complex I deficiencies are the most frequently encountered class of mitochondrial disorders [28]. In the mitochondrial inter-membrane space (IMS) two families of short proteins reside that include repeated double cysteine motifs (Cx9C or Cx3C). The cysteines are oxidized in the IMS, forming disulfide bridges that stabilize a hairpin confirmation in which α-helices between the cysteines align in an anti-parallel manner [19]. Among those proteins the NDUF family surveil mitochondrial NAD+ availability. Indeed, we were able to identify that persulfidation targets NDUF proteins and in particular NDUFB7 in cysteines 80 and 90 which are located in the Cx9C motif and are involved in disulfide bonds formation [19]. Detailed subsequent studies in which the reactive cysteines were mutated showed that persulfidation of the two specific cysteines is responsible to preserve NDUFB7 function, protect against overoxidation, maintain complex I activity and high levels of mitochondrial nicotinamide adenine dinucleotide-NAD+, with cysteine C80 exhibiting a greater contribution to the basal mitochondrial respiration in cardiomyocytes. Whether physiologically both cysteines are simultaneously persulfidated, or single presulfidation suffice to preserve complex I activity remains to be further investigated. NAD+ is a pyrimidine dinucleotide which preserves energy homeostasis and redox balance through numerous mitochondrial metabolic processes, while it serves as rate-limiting substrate for enzymes in non-redox reactions involved in DNA repair, epigenetic regulation and post-translation modifications [29]. The high energy demands of the heart makes it particularly susceptible to dysregulated NAD+ metabolism and NAD+ depletion is linked to disease pathogenesis [30]. During aging, which represents a form of chronic stress, NAD+ pools decline in various tissues, including the heart, which is correlated with a decline in mitochondrial function [31]. Of note, in stressed cardiomyocytes reduced levels of NAD+ and increased NAD+ catabolizing enzymes contribute to cell death in heart failure [32]. Thus, NAD+ replenishment through NAD+ precursors supplementation or inhibition of NAD+ catabolic enzymes have shown to reverse vascular dysfunction induced by aging and improve cardiac function and, thus, are investigated as anti-aging strategies [20,33,34]. Simultaneously to reduced NAD+ levels observed in aging, the NADH/NAD+ ratio is significantly increased [35]. Notably, increase in NADH levels initiate reductive stress responses [36], and have also been linked to pathological myocardial remodeling [37]. Further experiments would be requiered to confirm the existence of reductive stress in cardiomyocytes with impaired complex I activity due to lack of NDUFB7 persulfidation. There are different NAD+ catabolizing enzymes which belong to three different classes: the sirtuin family of deacetylases (SIRTs), the poly(ADP-ribose) polymerases (PARPs) and the (cyclic ADP-ribose) synthases (cADPRs), like CD38, which are mostly expressed in the heart and whose function is cell type- and context-dependent [33]. SIRTs are of particular interest given their impact on global acetylation of proteins, which dictates cellular phenotypes and transcriptional programs. This is not the first report linking hydrogen sulfide bioavailability to NAD+ levels, as in vessels H2S dependent NAD+ and SIRT1 regulation has been proposed to be a novel anti-aging strategy [38]. In our studies, we made the first link of persulfidation control of SIRTs activity through its impact on NAD+ availability in the murine heart, a finding that is very promising for future studies on how loss of persulfidation in aging affects cardiomyocyte transcription and function. Given that changes in NAD+ metabolism during aging are incompletely understood, our studies have shed some light on novel mechanisms affecting endogenous NAD+ pools that impact on cardiac cell homeostasis.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (CRC1366, Project B1 to S.-I.B., Project ID: 456687919; the Emmy Noether Programme BI 2163/1-1 to S.-I.B.; CRC1531, Project A2 to S.-I.B. and Project B3 to J.H., Project ID: 456687919; the Johanna Quandt Young Academy at Goethe to S.-I.B.; the Cardio-Pulmonary Institute, EXC 2026, Project ID: 390649896); the National Natural Science Foundation of China to J.H., Project ID: 82070501; the Hellenic Foundation for Research and Innovation under the “First Call for Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project number: HFRI-FM17-886) to A.P. and by an Onassis Foundation grant under the Onassis Foundation-BRFAA call to A.P. M.-K.D. was supported with a scholarship from Onassis Foundation.
CRediT authorship contribution statement
Maria-Kyriaki Drekolia: Formal analysis, Investigation, Methodology. Christina Karantanou: Formal analysis, Investigation, Methodology, Writing – original draft. Ilka Wittig: Data curation, Methodology. Yuanyuan Li: Methodology. Dominik C. Fuhrmann: Methodology. Bernhard Brüne: Methodology. Antonia Katsouda: Methodology. Jiong Hu: Funding acquisition, Investigation, Methodology, Writing – review & editing. Andreas Papapetropoulos: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. Sofia-Iris Bibli: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Declaration of competing interest
There are no conflicts of interest to declare.
Acknowledgements
The authors are indebted to Britta Heckmann for expert technical assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.103014.
Contributor Information
Andreas Papapetropoulos, Email: apapapet@pharm.uoa.gr.
Sofia-Iris Bibli, Email: Iris.Bibli@medma.uni-heidelberg.de.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Zivanovic J., Kouroussis E., Kohl J.B., Adhikari B., Bursac B., Schott-Roux S., Petrovic D., Miljkovic J.L., Thomas-Lopez D., Jung Y., Miler M., Mitchell S., Milosevic V., Gomes J.E., Benhar M., Gonzalez-Zorn B., Ivanovic-Burmazovic I., Torregrossa R., Mitchell J.R., Whiteman M., Schwarz G., Snyder S.H., Paul B.D., Carroll K.S., Filipovic M.R. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metabol. 2020;31:207. doi: 10.1016/j.cmet.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cirino G., Szabo C., Papapetropoulos A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol. Rev. 2023;103:31–276. doi: 10.1152/physrev.00028.2021. [DOI] [PubMed] [Google Scholar]
- 3.Bibli S.-I., Fleming I. Oxidative post-translational modifications: a focus on cysteine S-culfhydration and the regulation of endothelial fitness, Antioxid. Redox Signal. 2021;35:1494–1514. doi: 10.1089/ars.2021.0162. [DOI] [PubMed] [Google Scholar]
- 4.Katsouda A., Markou M., Zampas P., Varela A., Davos C.H., Vellecco V., Cirino G., Bucci M., Papapetropoulos A. CTH/MPST double ablation results in enhanced vasorelaxation and reduced blood pressure via upregulation of the eNOS/sGC pathway. Front. Pharmacol. 2023;14 doi: 10.3389/fphar.2023.1090654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibli S.-I., Hu J., Sigala F., Wittig I., Heidler J., Zukunft S., Tsilimigras D.I., Randriamboavonjy V., Wittig J., Kojonazarov B., Schürmann C., Siragusa M., Siuda D., Luck B., Abdel Malik R., Filis K.A., Zografos G., Chen C., Wang D.W., Pfeilschifter J., Brandes R.P., Szabo C., Papapetropoulos A., Fleming I. Cystathionine γ lyase sulfhydrates the RNA binding protein human antigen R to preserve endothelial cell function and delay atherogenesis. Circulation. 2019;139:101–114. doi: 10.1161/CIRCULATIONAHA.118.034757. [DOI] [PubMed] [Google Scholar]
- 6.Mani S., Li H., Untereiner A., Wu L., Yang G., Austin R.C., Dickhout J.G., Lhoták Š., Meng Q.H., Wang R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127:2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208. [DOI] [PubMed] [Google Scholar]
- 7.Yuan S., Yurdagul A., Peretik J.M., Alfaidi M., Al Yafeai Z., Pardue S., Kevil C.G., Orr A.W. Cystathionine γ-lyase modulates flow-dependent vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2018;38:2126–2136. doi: 10.1161/ATVBAHA.118.311402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calvert J.W., Elston M., Nicholson C.K., Gundewar S., Jha S., Elrod J.W., Ramachandran A., Lefer D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z., Xia H., Sharp T.E., LaPenna K.B., Katsouda A., Elrod J.W., Pfeilschifter J., Beck K.-F., Xu S., Xian M., Goodchild T.T., Papapetropoulos A., Lefer D.J. Hydrogen sulfide modulates endothelial-mesenchymal transition in heart failure. Circ. Res. 2023;132:154–166. doi: 10.1161/CIRCRESAHA.122.321326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bibli S.-I., Hu J., Looso M., Weigert A., Ratiu C., Wittig J., Drekolia M.K., Tombor L., Randriamboavonjy V., Leisegang M.S., Goymann P., Delgado Lagos F., Fisslthaler B., Zukunft S., Kyselova A., Justo A.F.O., Heidler J., Tsilimigras D., Brandes R.P., Dimmeler S., Papapetropoulos A., Knapp S., Offermanns S., Wittig I., Nishimura S.L., Sigala F., Fleming I. Mapping the endothelial cell S-sulfhydrome highlights the crucial role of integrin sulfhydration in vascular function. Circulation. 2021;143:935–948. doi: 10.1161/CIRCULATIONAHA.120.051877. [DOI] [PubMed] [Google Scholar]
- 11.Peleli M., Bibli S.-I., Li Z., Chatzianastasiou A., Varela A., Katsouda A., Zukunft S., Bucci M., Vellecco V., Davos C.H., Nagahara N., Cirino G., Fleming I., Lefer D.J., Papapetropoulos A. Cardiovascular phenotype of mice lacking 3-mercaptopyruvate sulfurtransferase. Biochem. Pharmacol. 2020;176 doi: 10.1016/j.bcp.2020.113833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsouda A., Valakos D., Dionellis V.S., Bibli S.-I., Akoumianakis I., Karaliota S., Zuhra K., Fleming I., Nagahara N., Havaki S., Gorgoulis V.G., Thanos D., Antoniades C., Szabo C., Papapetropoulos A. MPST sulfurtransferase maintains mitochondrial protein import and cellular bioenergetics to attenuate obesity. J. Exp. Med. 2022;219 doi: 10.1084/jem.20211894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z., Xia H., Sharp T.E., LaPenna K.B., Elrod J.W., Casin K.M., Liu K., Calvert J.W., Chau V.Q., Salloum F.N., Xu S., Xian M., Nagahara N., Goodchild T.T., Lefer D.J. Mitochondrial H2S regulates BCAA catabolism in heart failure. Circ. Res. 2022;131:222–235. doi: 10.1161/CIRCRESAHA.121.319817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picca A., Mankowski R.T., Burman J.L., Donisi L., Kim J.-S., Marzetti E., Leeuwenburgh C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018;15:543–554. doi: 10.1038/s41569-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A.K., Mu W., Zhang S., Snyder S.H., Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagahara N., Nagano M., Ito T., Shimamura K., Akimoto T., Suzuki H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria. Sci. Rep. 2013;3:1986. doi: 10.1038/srep01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittig J., Drekolia M.-K., Kyselova A., Delgado Lagos F., Bochenek M.L., Hu J., Schäfer K., Fleming I., Bibli S.-I. Endothelial-dependent S-sulfhydration of tissue factor pathway inhibitor regulates blood coagulation. Redox Biol. 2023;62 doi: 10.1016/j.redox.2023.102694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ackers-Johnson M., Li P.Y., Holmes A.P., O'Brien S.-M., Pavlovic D., Foo R.S. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 2016;119:909–920. doi: 10.1161/circresaha.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szklarczyk R., Wanschers B.F.J., Nabuurs S.B., Nouws J., Nijtmans L.G., Huynen M.A. NDUFB7 and NDUFA8 are located at the intermembrane surface of complex I. FEBS Lett. 2011;585:737–743. doi: 10.1016/j.febslet.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 20.Martin A.S., Abraham D.M., Hershberger K.A., Bhatt D.P., Mao L., Cui H., Liu J., Liu X., Muehlbauer M.J., Grimsrud P.A., Locasale J.W., Payne R.M., Hirschey M.D. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich's ataxia cardiomyopathy model. JCI Insight. 2017;2 doi: 10.1172/jci.insight.93885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai D.-F., Rabinovitch P.S. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009;19:213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai D.-F., Santana L.F., Vermulst M., Tomazela D.M., Emond M.J., MacCoss M.J., Gollahon K., Martin G.M., Loeb L.A., Ladiges W.C., Rabinovitch P.S. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esfahani N.S., Wu Q., Kumar N., Ganesan L.P., Lafuse W.P., Rajaram M.V.S. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell. 2021;20 doi: 10.1111/acel.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schefer V., Talan M.I. Oxygen consumption in adult and AGED C57BL/6J mice during acute treadmill exercise of different intensity. Exp. Gerontol. 1996;31:387–392. doi: 10.1016/0531-5565(95)02032-2. [DOI] [PubMed] [Google Scholar]
- 25.Mustafa A.K., Gadalla M.M., Sen N., Kim S., Mu W., Gazi S.K., Barrow R.K., Yang G., Wang R., Snyder S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Módis K., Coletta C., Erdélyi K., Papapetropoulos A., Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. Faseb. J. 2013;27:601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- 27.Gowthami N., Sunitha B., Kumar M., Keshava Prasad T.S., Gayathri N., Padmanabhan B., Srinivas Bharath M.M. Mapping the protein phosphorylation sites in human mitochondrial complex I (NADH: ubiquinone oxidoreductase): a bioinformatics study with implications for brain aging and neurodegeneration. J. Chem. Neuroanat. 2019;95:13–28. doi: 10.1016/j.jchemneu.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Vercellino I., Sazanov L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022;23:141–161. doi: 10.1038/s41580-021-00415-0. [DOI] [PubMed] [Google Scholar]
- 29.Abdellatif M., Sedej S., Kroemer G. NAD+ metabolism in cardiac health, aging, and disease. Circulation. 2021;144:1795–1817. doi: 10.1161/CIRCULATIONAHA.121.056589. [DOI] [PubMed] [Google Scholar]
- 30.Mericskay M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 2016;109:207–215. doi: 10.1016/j.acvd.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Braidy N., Guillemin G.J., Mansour H., Chan-Ling T., Poljak A., Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Pillai J.B., Isbatan A., Imai S., Gupta M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 33.Abdellatif M., Trummer-Herbst V., Koser F., Durand S., Adão R., Vasques-Nóvoa F., Freundt J.K., Voglhuber J., Pricolo M.-R., Kasa M., Türk C., Aprahamian F., Herrero-Galán E., Hofer S.J., Pendl T., Rech L., Kargl J., Anto-Michel N., Ljubojevic-Holzer S., Schipke J., Brandenberger C., Auer M., Schreiber R., Koyani C.N., Heinemann A., Zirlik A., Schmidt A., von Lewinski D., Scherr D., Rainer P.P., von Maltzahn J., Mühlfeld C., Krüger M., Frank S., Madeo F., Eisenberg T., Prokesch A., Leite-Moreira A.F., Lourenço A.P., Alegre-Cebollada J., Kiechl S., Linke W.A., Kroemer G., Sedej S. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abd7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Picciotto N.E., Gano L.B., Johnson L.C., Martens C.R., Sindler A.L., Mills K.F., Imai S., Seals D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15:522–530. doi: 10.1111/acel.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan Y., Liang B., Liu X.-L., Liu W.-J., Huang B.-H., Yang S.-B., Gao Y.-Z., Meng J.-S., Li M.-J., Ye T., Wang C.-Z., Hu X.-K., Xing D.-M. Targeting NAD+: is it a common strategy to delay heart aging? Cell Death Dis. 2022;8:230. doi: 10.1038/s41420-022-01031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shanmugam G., Wang D., Gounder S.S., Fernandes J., Litovsky S.H., Whitehead K., Radhakrishnan R.K., Franklin S., Hoidal J.R., Kensler T.W., Dell'Italia L., Darley-Usmar V., Abel E.D., Jones D.P., Ping P., Rajasekaran N.S. Reductive stress causes pathological cardiac remodeling and diastolic dysfunction. Antioxidants Redox Signal. 2020;32:1293–1312. doi: 10.1089/ars.2019.7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das A., Huang G.X., Bonkowski M.S., Longchamp A., Li C., Schultz M.B., Kim L.-J., Osborne B., Joshi S., Lu Y., Treviño-Villarreal J.H., Kang M.-J., Hung T.-T., Lee B., Williams E.O., Igarashi M., Mitchell J.R., Wu L.E., Turner N., Arany Z., Guarente L., Sinclair D.A. Impairment of an endothelial NAD+-H2S signaling network is a reversible cause of vascular aging. Cell. 2018;173:74–89.e20. doi: 10.1016/j.cell.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.