

Abstract
The oxindole scaffold has been the center of several kinase drug discovery programs, some of which have led to approved medicines. A series of two oxindole matched pairs from the literature were identified where TLK2 was a potent off-target kinase. The oxindole has long been considered a promiscuous inhibitor template, but across these 4 specific literature oxindoles TLK2 activity was consistent, while the kinome profile was radically different from narrow to broad spectrum coverage. We synthesized a large series of analogues and through quantitative structure-activity relationship (QSAR) analysis, water mapping of the kinase ATP binding sites, small-molecule x-ray structural analysis and kinome profiling, narrow spectrum, sub-family selective, chemical tool compounds were identified to enable elucidation of TLK2 biology.
Keywords: Kinase Inhibitors, Oxindole, Kinase Chemical Tool, Tousled Like Kinase 2 (TLK2), quantitative structure-activity relationship (QSAR)
1. Introduction
Tousled-like kinase 2 (TLK2) is a ubiquitously expressed, nuclear serine/threonine kinase. Kinase activity of TLK2 and its closest human paralog TLK1 is activated by autophosphorylation, hetero- and homo-dimerization, and oligomerization through conserved coiled-coiled domains.1 TLK kinase activity is highest during S-phase where TLKs phosphorylate ASF1 histone chaperones.2 DNA rapidly associates with histones during replication and ASF1 chaperones regulate the soluble pool of histones H3 and H4 during this process.3 Phosphorylation of ASF1 chaperones by TLKs stabilizes ASF1 protein,4 and promotes interactions between ASF1 and histones as well as histones with downstream chaperones CAF1 and HIRA1. TLK2, but not TLK1, was also shown to regulate G2/M checkpoint recovery after DNA damage through an ASF1A dependent mechanism.5 TLKs have ASF1 independent functions during DNA replication through the regulation of replication forks.6 Depletion of TLKs results in replication fork stalling, the accumulation of single-stranded DNA, and ultimately, DNA damage and cell cycle arrest in G1. These phenotypes were rescued by ectopic expression of WT TLKs but not kinase dead TLKs. Analysis of chromatin accessibility using the Assay for Tranposase-Accessible Chromatin using sequencing (ATAC-seq) following knockdown of TLK1 or TLK2 increased accessibility and transcription of heterochromatin. As a result, loss of TLKs induced alternate lengthening of telomeres thereby activating the cGAS-STING-TBK1 mediated immune response. Consistent with this function, high levels of TLK2 in patients were associated with poor innate and adaptive immune responses in tumors and, potentially, immune evasion.7 Thus, TLKs, and more specifically their kinase activity, are essential for proper chromatin assembly, DNA replication, and maintenance of heterochromatin.
The cellular functions of TLK1 and TLK2 are predominantly overlapping but their clinical implications are more diverse which may suggest other distinct functions. In the present study, we focus on TLK2. TLK2 has been implicated in neurodevelopmental disease and intellectual deficiency.8–10 Neurodevelopmental defects manifested with TLK2 haploinsuffiency and were more severe in a single, homozygous case. TLK2 mutations observed in these studies resulted in loss of TLK2 kinase activity.11 In cellular models of latent gammaherpesvirus infections, knockdown of TLK1 or TLK2 was sufficient to reactivate latent Epstein-Barr virus infection whereas only knockdown of TLK2 resulted in reactivation of latent Kaposi sarcoma-associated herpesvirus.12 Latency allows viral infections to evade the immune system and antiviral therapies resulting in lifelong, incurable infection. Reactivation of virus in combination with antiviral therapy is a promising approach known as “shock and kill” that could potentially cure these latent viral infections.13 TLK2 has also been implicated in breast cancer and glioblastoma where TLK2 is amplified or over-expressed in a high percentage of patients and where higher expression levels are associated with poor patient outcomes.14–16 In cell lines from both cancer types, ectopic expression of TLK2 lead to enhanced aggressiveness and activated SRC signalling whereas knockdown or pharmacological inhibition of TLK2 slowed growth and inhibited invasion. Knockout of TLK2 in mice resulted in late embryonic lethality due to defective trophoblast differentiation and placental failure.17 Conditional knockout of TLK2 to bypass the placental defect led to healthy mice suggesting that TLK2 was otherwise dispensable for development and viability. These findings suggest that inhibition of TLK2 may be tolerable in normal, healthy cells in patients. Thus, TLK2 is a very promising target for drug development in multiple cancer types and as a latency reversing agent in shock and kill approaches for latent viral infections.13–14
While some recent advances have been made around TLK2 chemical tools, with a recent advancement based on indirubin based derivatives, these analogues still inhibit both TLK1 and TLK2 with limited selectivity information.18 There are currently no potent and selective TLK2 inhibitors as chemical probes to investigate TLK2 biology.19 This despite several concerted efforts to map and identify leads within existing ATP-competitive kinase inhibitors and to define inhibitor chemical space.19–28
2. Results
Currently the only small molecule inhibitors available to modulate TLK2 are compounds with very broad kinome coverage including staurosporine, Syk inhibitor R406 and RTK inhibitor sunitinib, all of which inhibit TLK2 as an undesired off-target with low relative potency (Figure 1). However, the sunitinib oxindole based scaffold has demonstrated tractability on TLK2 where small modifications to afford SU14813 still maintain potency on TLK2 even when coverage has been reduced from 187 to 149 kinases. This is further supported by additional results of an enzyme assay-based panel identifying two further oxindoles, GW506033X and SU9516 that had 84% and 57% inhibition of TLK2 at 0.5 μM, respectively.23 The narrower kinome profile of SU9516 provided an attractive starting point for progress towards the development of a selective chemical probe for TLK2. Herein we report the synthesis, modelling, and characterization of oxindole based inhibitors as potent narrow-spectrum TLK2 inhibitors.
Figure 1.

Previously reported kinase inhibitors with off-target of TLK2 inhibition, including two matched pair oxindoles.
Having identified the oxindole scaffold as a promising inhibitor chemotype, we first screened 21 commercially available oxindole based kinase inhibitors (Figure 2)29–35 in a TLK2 enzyme assay (Table S1-S2).23 This enabled us to quickly map the TLK2 vs the close kinome landscape and to prioritize our efforts around the smaller SU9516 and GW506033X oxindoles.
Figure 2.

Screening of literature inhibitors with the oxindole scaffold with activities below 10 μM (see figure S1 for additional screening and structures). Format: Name, primary target, TLK2 IC50
Several interchangeable matched pairs including sunitinib (SU11248), toceranib (SU11654), SU12662 and SU14813 all demonstrated consistent low single digit micromolar activity against TLK2. While all these compounds have promiscuous kinome profiles,20–22 this data increased our confidence that the oxindole may provide a good starting point for TLK2 probe development. The two indirubin derivatives had the same activity against TLK2 (IC50 = 0.44 μM) indicating that oxindoles were bound in the traditional binding mode, with space to grow the solvent exposed region of the molecule.29–35 The only other two oxindoles that showed activity below 1 micromolar were SU9516 and GW506033X, both containing an imidazole head group. The narrow kinome spectrum of SU9516 (10 kinases inhibited to 80% at 1 μM) combined with the potency of GW506033X (IC50 = 0.061 μM) afforded a promising indication that a potent narrow spectrum compound was possible.23 We also noted that the same major kinase off-targets namely, fms-like tyrosine kinase 3 (FLT3) and tropomyosin receptor kinase C (TRKC) were recurring themes and likely a prognostic marker for further promiscuity across the kinome. In addition to screening for TLK2 potency, we screened these two off-targets as a micro-panel to check for selectivity between molecules before kinome-wide screening.36–38
Having decided to focus our attention on SU9516 (1) and related GW506033X, we first sought to explore the structural features required for TLK2 inhibition (Scheme 1). Initially we synthesized a series of direct SU9516 analogs (1-35) using commercially available 5- and/or 6-substituted oxindoles and the Knoevenagel condensation.39–41 The oxindoles along with a corresponding aldehyde were suspended in ethanol and treated with piperidine under reflux to afford condensation products 1-35 in varying yields (15–85%). The products were isolated by direct crystallization from the crude reaction mixture or following chromatographic purification. A second set of analogues was synthesized in a two-step procedure: first using a Friedel Craft acylation by treatment of unsubstituted oxindole with aluminum (III) chloride and the corresponding acid chloride in dichloromethane to afford the 5-position ketone substituted products 36-39 in acceptable to good yields (30–74%).42–44 Second, the ketone products (36-39) were then treated with the corresponding aldehyde, piperidine under reflux to afford condensation products 40-48 in acceptable to good yields (30–74%). To afford a series of sulphonamide analogs, we first treated the unsubstituted oxindole with neat chlorosulphonic acid at −10 °C, affording 49 in good yield (68%).45 The sulfonyl chloride 49 was treated with the corresponding amine to afford the sulphonamide building blocks 50-51 in excellent yields, 78% and 85% respectively. The sulphonamides 50-51 were then treated with the previous Knoevenagel conditions with the respective aldehyde to afford the condensation products 52-55 in good yield (41–75%).
Scheme 1.

Synthetic route to prepare the oxindole analogues - reagents and conditions a) R2CHO, piperidine, EtOH, reflux, 4–24 h; b) R1COCl, AlCl3, CH2Cl2, rt, 48 h; c) ClSO3H, −5 °C, 2 h; d) (R1)2NH, EtN(CH(CH3)2)2 THF, rt, 12 h; e) R1OH, diethyl azadicarboxylate (DEAD), PPh3, rt, −5 °C to rt; f) R1COCl, HATU, EtN(CH(CH3)2)2 , THF, rt, 12 h.
To access a novel 5-position isopropyl ether oxindole, a standard Mitsunobu reaction was performed on the 5-hydroxy oxindole with diethyl azadicarboxylate (DEAD), triphenylphosphine, isopropyl alcohol in THF at 0 °C to afford 5-isopropoxyindolin-2-one (56) in 30% yield.46–48 The ether 56 was then subject to the Knoevenagel conditions with the corresponding aldehyde to afford the desired condensation products 57-59 in good yields (53–62%). Finally, we prepared a series of substituted oxindole amides, furnished with a standard HATU coupling,49 in the presence of DIPEA and THF. The first set (60-64) was afforded in good yields (63–78 %), followed by a series of condensations under standard conditions to render a series of 5-postion substituted amide final products (65-76) in a range of yields (15–71%). The second set of amide substitutions (77-88) were also afforded in good to excellent yields (48–91%) followed by standard Knoevenagel conditions with the appropriate aldehyde to produce the condensation products (89-107) in a range of yields (11–71%).
Having prepared a series of oxindole analogues, these were screened in a TLK2 enzyme assay in a 10-point dose dependent format for TLK2 to determine an IC50 value. The maximum concentration used was 20 μM with an ATP concentration of 10 μM. The corresponding collateral target evaluation was on FLT3 and TRKC which were screened at a 0.5 μM, consistent with previous reports.23 The most advanced compounds were then subjected to a Kinase-Glo assay and additional TLK2 assay. The lead compounds were then screened in a DiscoverX kinome panel assay (>400 kinases) to assess their selectivity across the kinome.20–22
In the first series of analogues we followed up on the initial hit compound (Z)-3-((1H-imidazol-5-yl)methylene)-5-methoxyindolin-2-one (1) which when resynthesized produced a compound with consistent potency on TLK2 (IC50 = 240 nM) (Table 1). The substitution of the imidazole with the pyrrole (2) resulted in a 6-fold drop in potency on TLK2. Introduction of the sterically hindered dimethyl group (3) afforded no activity on TLK2 at the maximum concentration tested but still showed activity on FLT3 and TRKC. The introduction of a pyrazole group (4) lead to a 36-fold reduction of TLK2 activity with respect to 1. The 2-substituted imidazole (5) afforded a compound that was near equipotent with 1 against TLK2. However, none of the simple 5 membered analogues (2-5) improved activity on TLK2. A switch to a 6-member 2-pyridyl (6) and 3-pyridyl (7) head group ring system demonstrated no activity against TLK2 at the maximum concentration tested but also no activity against TRKC and FLT3. Removal of the 5-methoxy group to afford the unsubstituted oxindole (8) lead to a 2-fold drop in potency against TLK2. Interestingly the pyrrole substitution 9 shows the same potency as 8 unlike the corresponding drop observed between 1 and 2.
Table 1.
Matrix array of SU9516 derivatives with 5,6-oxindole substitutions and different heterocyclic head groups.
| ╲ |

|

|

|

|

|

|
|
|
|---|---|---|---|---|---|---|---|---|

|
Name | 1 | 2 | 3 | 4 | 5 | 6 | 7 c |
| TLK2 (IC50)a | 0.24 μM | 1.5 μM | >20 μM | 8.7 μM | 0.38 μM | >20 μM | >20 μM | |
| FLT3 (% Inh.) b | 6.9 | 19 | 1.8 | 57 | 9.6 | 93 | 91 | |
| TRKC (% lnh.) b | 3.7 | 20 | 3.3 | 39 | 6.7 | 85 | 80 | |

|
Name | 8 c | 9 | 10 | 11 | 12 c | 13 | 14 |
| TLK2 (IC50)a | 0.44 μM | 0.47 μM | >20 μM | 5.9 μM | 0.60 μM | >20 μM | >20 μM | |
| FLT3 (% lnh.) b | 19 | 5.9 | 3.0 | 69 | 29 | 100 | 100 | |
| TRKC (% lnh.) b | 7.5 | 3.3 | 6.0 | 45 | 11 | 92 | 99 | |

|
Name | - | 15 | 16 | 17 | 18 | 19 | 20 c |
| TLK2 (IC50)a | 1.7 μM | >20 μM | >20 μM | 2.2 μM | >20 μM | >20 μM | ||
| FLT3 (% Inh.) b | 13 | 24 | 89 | 33 | 100 | 87 | ||
| TRKC (% lnh.l b | 19 | 36 | 75 | 37 | 95 | 77 | ||

|
Name | 21 | 22 | 23 | 24 | 25 c | - | - |
| TLK2 (IC50)a | 1.8 μM | 0.63 μM | >20 μM | >20 μM | 2.1 μM | |||
| FLT3 (% lnh.) b | 13 | 5.3 | 18 | 65 | 20 | |||
| TRKC (% inh.) b | 36 | 11 | 35 | 81 | 45 | |||

|
Name | 26 | 27 | 28 | 29 | 30 c | 31 | 32 c |
| TLK2 (IC50)a | 0.77 μM | 0.15 μM | 0.87 μM | 7.6 μM | 0.62 μM | >20 μM | >20 μM | |
| FLT3 (% lnh.) b | 6.4 | 3.0 | 1.9 | 25 | 11 | 77 | 85 | |
| TRKC (% lnh.) b | 9.0 | 3.1 | 1.1 | 38 | 18 | 67 | 79 | |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2);
E:Z ratio: 7 (50:50); 8 (33:67); 12 (40:60); 20 (28:72); 25 (44:56); 26 (32:68); 30 (47:53); 32 (48:52).39
The corresponding 3,5-dimethyl pyrrole substitution 10 was inactive against TLK2, while the pyrazole 11 showed a 13-fold drop compared with 8. The 2-substituted imidazole analogue 12 was equipotent with 8, while the two pyridyl analogues 13 and 14 demonstrated no activity against TLK2 or TRKC/FLT3. Switching to the 6-methoxy oxindole substitution 15-20 lead to a net reduction of around 10-fold in activity on nearly all analogues with only the pyrrole maintaining TLK2 inhibition at the same level as 2.
Given the decrease in inhibition of TLK2 activity at the 6-position, we decided to explore the 5,6-dimethxoyoxindole core 21-25 to assess potential synergistic effects of a double substitution pattern. The results of the 5 analogues were consist with the mono-substituted 6-methoxy except for the pyrrole 22 that showed a 3-fold jump in potency against TLK2. However, 22 also showed a boost in TRKC and FLT3 inhibition, meaning it was also likely an overall increase in kinome inhibition rather than something solely related to TLK2. We decided to focus our attention on the 5-postion as this seemed to be preferable for TLK2 inhibition. Switching the core to the 5-acetyloxindole 26-32 afforded our first compound 27 with more potency against TLK2 IC50 = 150 nM, than the original starting point (1) (TLK2 IC50 = 240 nM). The imidazole analogue was 3-fold less active against TLK2, but the pyrrole was 60% more potent against TLK2. The 3,5-dimethylpyrolle analogue (28) also demonstrated inhibition of TLK2 for the first time with an IC50 = 870 nM. The pyrazole 29 was significantly weaker nearly 10-fold less potent than the imidazole parent 26. The 2-substituted imidazole (30) is as active as the parent while the pyridyl analogues 31 and 32 showed no activity against TLK2 at the maximum concentration, but there was an increase in TRKC and FLT3 inhibition compared to the other pyridyl analogues synthesized.
There are clear trends observed between pyrrole, 5-position-imidazole, 4-position-imidazole, unsubstituted, 5-position methoxy and acetyl group. The docking of 1, 3, 27 and 28 in the TLK2 protein crystal structure,11 provided an insight into these trends (Figure 3). The differences between 1 and 3 can be simply explained by the steric clash between the 3,5-dimethylpyrrole on 3. However, the reduced solvation by removal of the additional nitrogen is also contributing to the reduced potency of 3 on TLK2 (Figure 3A–B). However, the acetyl substitution of 27 and 28 allowed for a shift in overall binding position that is able to compensate for this steric clash (Figure 3C–D) and enables a closing of the gap of potency from >40-fold between 1 and 3 to equipotent between 26 and 28. We used this knowledge in the design of our next series of ketone-based substituted oxindole analogues.
Figure 3.

Inhibitor docking of A) 1, B) 3, C) 27, D) 28 in TLK2 (PDB: 5O0Y), demonstrating key hinge binding interactions and space in the hydrophobic pocket.
The (Z)-3-((1H-pyrrol-2-yl)methylene)-5-acetylindolin-2-one (27) was the most potent analogue from the initial matrix array (Table 1). The acetyl showed a good trend towards TLK2 inhibition even enabling the 3,5-dimethyl pyrrole substitution 28 to have activity against TLK2 below 1 μM. These two results together with a TLK2 inhibition preference for the pyrrole, unsubstituted 5-position and 4-position imidazole prompted us to focus on the acetyl derivatives (Table 2).
Table 2.
Investigation of derivatives of 27 with pyrrole and imidazole heterocyclic head groups.

| ||||||
|---|---|---|---|---|---|---|
| Name | R1 | X1 | X2 | TLK2 IC50(nM)a | FLT3 (%)b | TRKC (%)b |
| 33 |

|
C-H | C-H | 62 | 5.8 | 9.3 |
| 34 c | N | C-H | 140 | 33 | 46 | |
| 35 | C-H | N | 26 | 30 | 37 | |
| 40 |

|
C-H | C-H | 220 | 3.6 | 2.5 |
| 41 | N | C-H | 230 | 8.9 | 4.3 | |
| 42 | C-H | N | 250 | 14 | 8.7 | |
| 43 |

|
C-H | C-H | 870 | 5.9 | 1.9 |
| 44 | N | C-H | 700 | 17 | 2.8 | |
| 45 |

|
C-H | C-H | 340 | 3.5 | 1.0 |
| 46 | N | C-H | 340 | 15 | 1.9 | |
| 47 |

|
C-H | C-H | 100 | 0.8 | 0.9 |
| 48 | C-H | N | 340 | 9.6 | 5.2 | |
First, we looked at the simple carboxylic acid derivatives 33–35. The direct replacement of the acetyl to form the carboxylic acid 33 demonstrated a 2-fold improvement in TLK2 inhibition, while the imidazole 34 was equipotent. However, the 2-substituted imidazole 35 showed a 6-fold boost in potency against TLK2 (IC50 = 26 nM). Interestingly both imidazole substituted compounds also had good selectivity over the two off-targets FLT3 and TRKC. To pursue the aim of a TLK2 probe quality compound with single digit nanomolar activity against TLK2, we hence investigated larger substituents. The n-butyl substituent analogues 40-42 showed a slight decrease in inhibition compared to 27 with flat SAR across the three analogues. The larger phenyl analogues 43–44 showed no improvement with an overall 5–6-fold decrease compared with 27. Introduction of a 4-fluoro substitution 45-46 mitigated some of the decrease in TLK2 inhibition to 2–3-fold compared with 27 with the same off-target FLT3/TRKC off-target profile. Switching from a phenyl to a furan afforded compounds 47-48 that were near equipotent with 27. Despite decreased ligand efficiency compared with 27, these furan analogues 47-48 presented a potential expansion point for further optimization.
The docking of 44, 46 and 47 supported our initial observations with 27 (Figure 4). The aromatic groups that are able to interact with the catalytic lysine in 44, 46 and 47 appeared to be preferable to a simple alkyl when attempting to drive potency on TLK2 below IC50 = 100 nM (Figure 4). Comparing the unsubstituted 44 to the 4-fluoro substituted 46, we observed a potential halogen bond between the fluorine and a backbone carbonyl which may explain the 3-fold boost in potency against TLK2 (Figure 4A–B).50–52 However, further analysis of the binding and water network of 35 highlighted untapped potential within a network of interactions in the hydrophobic pocket (Figure 5). The carboxylic acid motif of 35 was able to align with the Thr612 which in-turn coordinated a main chain amide, which can be used in the next phase of development. The water network is not disrupted by this series of interactions, but this simulation highlights that the oxindole occupies an optimal hinge binding position.
Figure 4.

Inhibitor docking comparing of A) 44, B) 46 and C) 47 in TLK2 (PDB: 5O0Y) all demonstrating key hinge binding interactions and an expansion in the hydrophobic pocket.
Figure 5.

Left: favorable docking pose of compound 35. Right: visualization of hydration sites from a WaterMap simulation close to docked compound 35.
The relatively flat SAR of the ketone substitution towards TLK2 inhibition led us to expand our search to other 5-postion substitution patterns including two different sulphonamides (52–55) and an isopropyl ether (57-59) (Table 3). Surprisingly, despite the relatively high potency of the acetyl (27), the sulphonamides (60-65) were inactive on TLK2 at the top concentration. The 5-isopropyl ether oxindole showed some potential. The pyrrole analog (57) was 3-fold more potent for TLK2 than the 5-methyl ether version (2). The imidazole (58) was 7-fold less potent on TLK2 than the corresponding 5-methyl ether (1). While the 2-subsitituted imidazole (59) was the only analog to show an improvement over the methyl counterpart with a 2-fold increase in potency against TLK2.
Table 3.
Investigation of sulphonamide and ether derivatives.

| ||||||
|---|---|---|---|---|---|---|
| Name | R1 | X1 | X2 | TLK2 IC50(nM)a | FLT3 (%)b | TRKC (%)b |
| 52 |

|
C-H | C-H | > 20000 | 31 | 1.6 |
| 53 | N | C-H | > 20000 | 55 | 12 | |
| 54 |

|
C-H | C-H | > 20000 | 27 | 3.0 |
| 55 | N | C-H | > 20000 | 62 | 27 | |
| 57 |

|
C-H | C-H | 560 | 8.3 | 2.8 |
| 58 | N | C-H | 1600 | 33 | 11 | |
| 59 | C-H | N | 160 | 25 | 5.9 | |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2)
However, these limited improvements meant we switched to the 5-amide substituted oxindoles 65-76 in hope of driving down potency on TLK2 (Table 4). We first screened a direct n-propyl analog 65 that showed similar potency to 27 with a reduction in off-target potency of FLT3 and TRKC. The addition of a pendent ethyl group to form a pentan-3-yl substitution 66-68 dropped potency by around 10-fold against TLK2 compared to the corresponding n-propyl analog 65. The cyclohexane substitution 69-71 was broadly similar in potency with 27 on TLK2, but with a significant improvement on the off-target profile with a reduction in off-target potency of FLT3 and TRKC compared to 27. The unsubstituted phenyl 72-74 has an asymmetric structure activity relationship (SAR) for TLK2 with the pyrrole 72 5-fold weaker than 27. While the imidazole 73 is 4-fold more potent and both improved off-target selectivity against FLT3 and TRKC. The 2-subsitituted imidazole 74, is 3-fold less potent than 73 and equipotent to 27. The furan substitution analogs 75 and 76 were equipotent with 27 on TLK2 but both demonstrated improved off-target selectivity against FLT3 and TRKC.
Table 4.
Initial investigation of amide derivatives.

| ||||||
|---|---|---|---|---|---|---|
| Name | R1 | X1 | X2 | TLK2IC50(nM)a | FLT3 (%)b | TRKC (%)b |
| 65 |

|
C-H | C-H | 170 | 11 | 4.9 |
| 66 |

|
C-H | C-H | 2400 | 25 | 2.3 |
| 67 | N | C-H | 2300 | 61 | 6.0 | |
| 68 | C-H | N | 2500 | 75 | 15 | |
| 69 |

|
C-H | C-H | 150 | 28 | 11 |
| 70 | N | C-H | 100 | 46 | 16 | |
| 71 | C-H | N | 130 | 58 | 32 | |
| 72 |

|
C-H | C-H | 820 | 21 | 19 |
| 73 | N | C-H | 34 | 44 | 30 | |
| 74 | C-H | N | 110 | 48 | 33 | |
| 75 |

|
C-H | C-H | 120 | 30 | 28 |
| 76 | C-H | N | 130 | 42 | 33 | |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2)
The unsubstituted phenyl 72, is about an order of magnitude less active than close derivatives 73 and 74. This is likely due to the water network coordination as the 5-membered 1,2-diazole, 1,3-diazole and pyrrole ring system are all pointing towards the solvent exposed region of the TLK2 ATP binding site. The interactions in the hydrophobic pocket region were identical for all three compounds, with the designed interaction of Thr612 and the cation-pi interaction between Lys491 and the phenyl ring observed in each of the docked examples (Figure 6A–C). However, we observed through WaterMap that analog 72 was not able to make a stable hydrogen bond network with first solvation shell waters (Figure 7), likely leading to weaker binding.53–56 The lack of solvent coordination coupled with the ability of 74 to hydrophobically stack with Leu468 (Figure 7), which was not possible with 72 and 73, provided a rational explanation for the results we observed.
Figure 6.

Inhibitor docking comparing A) 72, B) 73, C) 74 in TLK2 (PDB: 5O0Y) demonstrating key hinge binding interactions and an expansion in the hydrophobic pocket.
Figure 7.

Snapshot of trajectory waters from WaterMap simulation illustrating favourable solvent interactions of 73. For clarity, waters and hydration sites are shown only at the mouth of the pocket.
To build on the encouraging results of 73 and 74, we decided to explore the amide derivatives with a series of modifications to the phenyl ring system 88-106 and focused on the imidazole and 2-subsitituted imidazole head group (Table 5). First, we probed the 4-position of the phenyl ring, by introducing a nitrile group with an imidazole head group to afford the most potent compound 88 against TLK2 observed so far (IC50 = 12 nM); 13-fold more potent than 27, with good selectivity over FLT3/TRKC. The 3-position nitrile with the imidazole head group 89 afforded 8-fold less potency for TLK2 than 88, but with slightly improved selectivity over FLT3/TRKC, however the 2-subsitituted imidazole 90 only had a 4-fold drop in potency for TLK2. The 2-position nitrile was not well tolerated with 2-subsitituted imidazole 91 having an IC50 over 1 μM on TLK2. The 4-methoxy substitution analogs 92-93 were 7–9-fold less potent on TLK2 than 89. The 3-methoxy with the imidazole head group 94 was consistent with 92–93, but the 2-subsitituted imidazole 95 was 28-fold less potent on TLK2 than 94. In contrast to 91, the 2-methoxy derivatives 96-97 were potent against TLK2 with the imidazole head group 96 being single digit nanomolar (IC50 = 9.1 nM), the most potent compound to date. The 2-subsitituted imidazole derivative 97 was 7-fold less potent on TLK2 but had good selectivity over FLT3/TRKC. The fluorine substitutions around the phenyl ring system 98-102 had relatively flat SAR for TLK2. The 2-position fluoro imidazole 102 was the most potent against TLK2 (IC50 = 110 nM) and all analogs 98-102 had good selectivity against FLT3 and TRKC. The larger trifluoromethyl substitution in the 4-postion was partly tolerated with the imidazole head group 103 showing only a 3-fold drop with respect to 102. However, the 2-subsitituted imidazole 104 showed limited to no activity against TLK2 at the highest concentration tested, potentially due to the inactive geometric E-isomer contribution. The 2-position analogs 105-106 were consistent with 98-102, with equipotency on TLK2 and good selectivity over FLT3. Interestingly, 105-106 both display reduced selectivity over TRKC compared with 103.
Table 5.
Advanced investigation of amide derivatives.

| ||||||
|---|---|---|---|---|---|---|
| Name | R1 | X1 | X2 | TLK2 IC50 (nM)a | FLT3 (%)b | TRKC (%)b |
| 88 c | 4-CN | N | C-H | 12 | 39 | 47 |
| 89 c | 3-CN | N | C-H | 95 | 56 | 52 |
| 90 | C-H | N | 48 | 60 | 46 | |
| 91 | 2-CN | C-H | N | 4000 | 75 | 33 |
| 92 | 4-OMe | N | C-H | 88 | 36 | 45 |
| 93 | C-H | N | 110 | 44 | 50 | |
| 94 | 3-OMe | N | C-H | 100 | 42 | 48 |
| 95 c | C-H | N | 2800 | 84 | 80 | |
| 96 | 2-OMe | N | C-H | 9.1 | 25 | 43 |
| 97 | C-H | N | 67 | 40 | 34 | |
| 98 | 4-F | N | C-H | 190 | 41 | 39 |
| 99 | C-H | N | 140 | 46 | 53 | |
| 100 | 3-F | N | C-H | 200 | 34 | 33 |
| 101 c | C-H | N | 490 | 70 | 60 | |
| 102 | 2-F | C-H | N | 110 | 26 | 24 |
| 103 | 4-CF3 | N | C-H | 330 | 58 | 57 |
| 104 c | C-H | N | 6100 | 88 | 83 | |
| 105 c | 2-CF3 | N | C-H | 400 | 60 | 17 |
| 106 c | C-H | N | 190 | 62 | 20 | |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2);
.E:Z ratio: 88 (50:50); 89 (43:57); 95 (37:63); 101 (35:65); 104 (34:66); 105 (38:62); 106 (34:66).39
The docking of 73, 88 and 96 provided further evidence of the key interaction at the Thr612 residue with the amide bond carbonyl and the catalytic lysine (Lys491) and afford high potency low double and single digit nanomolar compounds against TLK2 (Figure 8). The high affinity was dependent on an aromatic ring system close to the active site but away from the hinge. We looked to exploit these traits to further optimize the analogs towards TLK2 inhibition and potentially increase kinome specificity in the process.
Figure 8.

Inhibitor docking in TLK2 (PDB: 5O0Y) comparing A) 73, B) 88 and C) 96. All demonstrating key hinge binding interactions and a refined expansion in the hydrophobic pocket.
The recently released solved X-ray structures of TLK2 together with our molecular docking simulations, were also helpful in the design of more effective TLK2 inhibitors.10 A 3D-QSAR model was constructed so that the main fields could be overlaid with co-crystallized ligands and verifiable favourable docking poses. We constructed a 3D-QSAR model for TLK2 using the field based QSAR functionality of Schrödinger Maestro (Table 6 and 7). The flexible ligand alignments were performed using a Bemis-Mucko method.52 The 3D-QSAR model allowed recognition of the largest common scaffold and utilized a favorable docking pose of structurally representative derivatives as alignment templates. Torsion angles of larger substituents, including methoxy groups, were manually adjusted when required. The models were constructed with 80% of compounds defined randomly as training sets with the rest of the compounds as a test set (Figure 9). The field values were calculated utilizing a grid spacing approach of 1.0 Å extended 3.0 Å beyond training set limits. Variables with |T-value| < 2 were eliminated from final model building.
Table 6.
Validation of 3D-QSAR models for TLK2
| # Factors | SD | R2 | R^2 CV | R^2 Scramble | Stability | F | P | RMSE | Q^2 | Pearsson-r |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.74 | 0.43 | 0.38 | 0.055 | 0.997 | 56.1 | 1.01E-10 | 0.85 | 0.15 | 0.54 |
| 2 | 0.50 | 0.74 | 0.63 | 0.18 | 0.96 | 105.0 | 2.48E-27 | 0.57 | 0.61 | 0.83 |
| 3 | 0.22 | 0.80 | 0.68 | 0.27 | 0.94 | 98.5 | 1.43E-29 | 0.54 | 0.65 | 0.82 |
| 4 | 0.28 | 0.85 | 0.70 | 0.36 | 0.92 | 104.2 | 3.60E-30 | 0.52 | 0.68 | 0.83 |
| 5 | 0.33 | 0.89 | 0.71 | 0.43 | 0.92 | 116.9 | 7.54E-34 | 0.46 | 0.75 | 0.86 |
| 6 | 0.30 | 0.91 | 0.71 | 0.48 | 0.88 | 122.9 | 3.73E-38 | 0.47 | 0.74 | 0.86 |
Table 7.
Regression Statistics for TLK2 QSAR model
| # Factors | gauss_s | gauss_e | gauss_h | gauss_a | gauss_d |
|---|---|---|---|---|---|
| 1 | 0.53 | 0.062 | 0.13 | 0.19 | 0.09 |
| 2 | 0.27 | 0.070 | 0.32 | 0.26 | 0.09 |
| 3 | 0.28 | 0.069 | 0.31 | 0.24 | 0.10 |
| 4 | 0.29 | 0.067 | 0.28 | 0.25 | 0.12 |
| 5 | 0.30 | 0.068 | 0.25 | 0.25 | 0.14 |
| 6 | 0.31 | 0.068 | 0.23 | 0.27 | 0.13 |
Figure 9.

Scatter plots of field-based 3D-QSAR models showing predicted versus measured binding affinities (nM) of active compounds printed at logarithmic scale with the TLK2 training set (left) and TLK2 all compounds (right).
The R-squared value of the TLK2 model with all four partial least squares regression (PLS) components were 0.75. The model had above average internal predictivities with a Q-square value of 0.75, which was determined by leave-one out (LOO) cross-validation. The stability of the model was 0.92 with four (PLS) components having demonstrated a robust predictivity of structure to activity output.
The shape of the oxindole series had a good alignment in the TLK2 ATP binding pocket with a strong interaction with hinge residues with semi-optimized back pocket QSAR fields (Figure 10A–F). The pyrrole/imidazole head group assisted the hinge interacting with an additional anchor point. The oxindole scaffold was able to form two direct hydrogen bond interactions via the oxindole amide functionality to the main chain amide of Tyr546 and Cys547. The N-H of the pyrrole/imidazole was then able to form an interaction with Glu548. The optimized amide derivatives were also able to interact via the carbonyl of the amide with Thr612 and had a cation-pi interaction with Lys491 forming with the pendent heterocycle. The internal conformational restriction of the oxindole scaffold between the carbonyl and amine of the pendant heterocycle was also assisting in the optimization of TLK2 binding, not only minimizing the entropic loss associated flexibility but also allowed the ligand to adopt a preferred conformation for binding.39,57–58
Figure 10.

QSAR modelling on 73 template: A) Standard docking of 73 in TLK2 (PDB:5O0Y); B) Electrostatics (blue = positive and red = negative); C) hydrogen bonding acceptor fields (magenta = positive and grey = negative); D) hydrogen bonding donor fields (cyan = positive and grey = negative); E) hydrophobic fields (orange = positive and cyan = negative); F) Steric fields (green = positive and yellow = negative).
We further explored the rigidity within the oxindole core with the solving of a series of small molecule crystal structures 1, 2, 15, 19, 72, and 74 (Figure 11, Table S3) and 4, 9, 11, 13 and 16 (Figure S1, Table S3). Structures 1, 2, 16 and 19 were planar due to intramolecular hydrogen bonding between the pyrrole 15 and imidazole 1 and 2 ring and the oxindole carbonyl oxygen atom (Figure 11). The steric hindrance of the pyridine substituent of 19 and lack of hydrogen bond donor resulted in a deviation from planarity for this structure. Whilst the methylene pyrrole/imidazole rings were planar with the oxindole moiety, the benzamide substituents of 72 and 74 were not (Figure 11). The methylene imidazole substituted structures 1 and 15 included a hydrogen bonded dimer between the amine of the imidazole and the oxindole carbonyl instead except 74 which formed flat 1D tapes along the crystallographic -ac plane via oxindole amine, imidazole imine H-bonding. These tapes formed H-bonds to those above and below via hydrogen bonds between the amide functionalities. The methylene pyrazole substituted structures showed no distinct packing motifs with 2 forming interleaved, corrugated tapes via oxindole amine, imidazole imine H-bonding and 16 forming a H-bonded, tetramer structure comprising two crystallographically independent molecules.
Figure 11.

Small molecule crystal structures of 1, 2, 15, 19, 72 and 74.
Table 9.
Optimization of compounds based on QSAR.





Table 11.
Compound results based on QSAR prediction.
| Compound | TLK2 Inhibition Activity (nM) | |||
|---|---|---|---|---|
| Predicted | Found | Log Predicted | log Actual | |
| 117 | 51 | 23 | 7.29 | 7.64 |
| 118 | 56 | 36 | 7.25 | 7.44 |
| 119 | 50 | 77 | 7.30 | 7.11 |
| 120 | 370 | 27 | 6.44 | 7.57 |
| 121 | 68 | 180 | 7.17 | 6.74 |
| 122 | 180 | 47 | 6.75 | 7.33 |
| 123 | 1800 | 630 | 5.76 | 6.20 |
| 124 | 40 | 260 | 7.40 | 6.59 |
| 125 | 8 | 120 | 8.08 | 6.92 |
| 126 | 8 | 15 | 8.12 | 7.82 |
| 127 | 37 | 46 | 7.44 | 7.34 |
| 128 | 180 | 18 | 6.74 | 7.74 |
| 129 | 960 | 2600 | 6.02 | 5.59 |
Table 13.
TLK2 Functional Inhibition Kinase-Glo Results
| Name | TLK2 IC50 (nM) | |
|---|---|---|
| Enzyme | Kinase Glo | |
| 1 | 240 | 280 |
| 97 | 67 | 450 |
| 117 | 15 | 97 |
| 128 | 18 | 250 |
| 129 | 2700 | 7800 |
A series of oxindole derivatives were designed based on the quantitative structure-activity relationships (QSAR) QSAR model utilizing prior SAR knowledge and the small molecule crystal structure information. These were synthesized and tested using the same protocol as described above, with the addition of TLK1 screened at a 0.5 μM concentration, consistent with previous reports.23
We synthesized the series of predicted analogs utilizing similar conditions as before (Scheme 1) with a few alterations in starting materials. The amide intermediates 107-129 were afforded by a routine HATU coupling to the respective 5-aminooxindole in good yields (57–84 %). The amide was then treated with the previous Knoevenagel conditions with the respective aldehyde to afford the condensation products 117-129 in a range of yields (21–71 %) (Scheme 2).
Scheme 2.

Synthetic route to prepare predicted oxindole analogs - reagents and conditions a) R1COCl, EtN(CH(CH3)2)2, THF, rt, 12 h; b) R2CHO, piperidine, EtOH, reflux, 4–24 h.
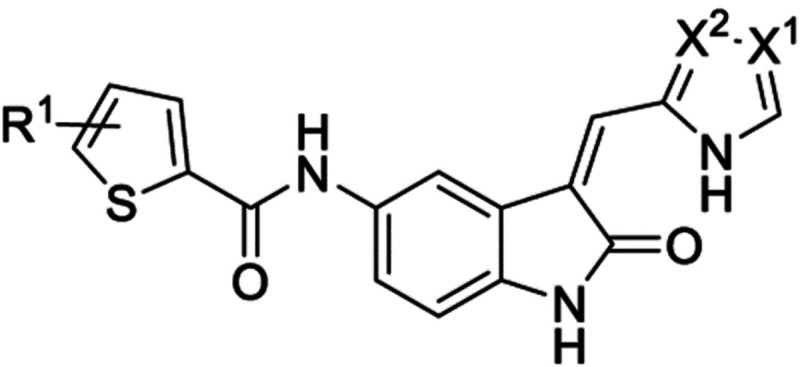
The QSAR modelling suggested that the phenyl ring could be reduced to a 5-member ring and coupled with earlier knowledge of the furan 75 and 76, so we synthesized the thiophenes 117-121 (Table 8). The activities observed were broadly in line with the QSAR model. The unsubstituted thiophene with the imidazole head group 117 was potent on TLK2 (IC50 = 23 nM) but lacked selectivity over FLT3/TRKC, the 2-subtitued imidazole 118 was equipotent but slightly more selective against FLT3/TRKC. The introduction of a 4-methoxy group onto the thiophene 119 reduced potency for TLK2 by 3-fold compared to 117. However, the 4-ethoxy group 120 was equipotent with 117 with equivalent selectivity over FLT3/TRKC. Interestingly 120 was the only compound to show any significant activity on TLK1 in the initial screening (54% inhibition at 1 μM). The final compound in this series, the 4-fluoro 121 was 8-fold less potent against TLK2 compared to 117.
Table 8.
Results of initial set of QSAR predicted compounds.
| |||||||
|---|---|---|---|---|---|---|---|
| Name | R1 | X1 | X2 | TLK2 IC50 (nM)a | FLT3 (%)b | TRKC (%)b | TLK1 (%)b |
| 117 | H | N | C-H | 23 | 12 | 16 | 89 |
| 118 c | H | C-H | N | 36 | 20 | 26 | 80 |
| 119 | 4-OMe | N | C-H | 77 | 20 | 22 | 87 |
| 120 c | 4-OEt | N | C-H | 27 | 9.4 | 8.0 | 54 |
| 121 | 4-F | INI | C-H | ISO | 23 | 12 | 100 |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2);
E:Z ratio: 112 (34:66); 114 (28:72).39
We then optimized these initial compounds to afford a series of more advanced amide derivatives 122-127 (Table 9). The fused thiophene, 122 was equipotent with 117, with no difference in selectivity. This larger 5,5-system was tolerated, so an expansion of the phenyl substitution patterns was synthesized to include 3-trifluoromethyl, 5-fluoro 123 and the fused 2,3-catacol 124, unfortunately these analogs both lost significant activities against TLK2 compared with 117. We also looked at smaller 5-member rings, thiazole 125, oxazole 126 and isothiazole 127 afforded potent compounds against TLK2 with the oxazole approaching towards single digit nanomolar against TLK2.
In a further effort to validate the model and test the limits of the two additional compounds 128 and 129 were synthesised (Table 10). These were based on the cores structure of 73 and tested the predictive power of the model. The addition of the 2-methyl (128) to the imidazole head group afforded an 8-fold increase in potency against TLK2 with no loss of selectivity against FLT3 and TRKC. The model was accurate for the pyrazole 129, the introduction of the extra nitrogen led to an over 15-fold drop in TLK2 inhibition, consistent with previous SAR in this series.
Table 10.
Optimization of compounds based on QSAR.

| |||||||
|---|---|---|---|---|---|---|---|
| Name | R | X1 | X2 | TLK2 IC50 (nM) | FLT3(%) | TRKC (%) | TLK1(%) |
| 128 | Me | N | C-H | 18 | 24 | 35 | 84 |
| 129 | H | C-H | N | 2600 | 87 | 86 | 92 |
8-point dose response (n=1);
percentage inhibition at 0.5 μM (n=2)
In addition to assessing the selectivity profile on the two near off-targets FLT3 and TRKC; we also investigated the wider kinome selectivity profile of ten of the most promising analogs using KinomeSCAN® at 1 μM across over 400 kinases (Table 12, Figure 12).20–22 The original starting point (1) was moderately selective across the kinome hitting 15 kinases over 90%. The 5-acetyl analog 27, was the most promiscuous analog with 62 kinases over 90%, possibly due to the small flexible ketone orientated favourably to towards the common catalytic lysine motif (Figure 3D). Interestingly, 1, 27 and 44 had similar hit kinases above 90%. Common kinases included MST2, CDKL2, ERK8, TRKA, JAK3(JH1domain-catalytic) and TNIK. The amide substitution in the 5-position not only increased selectivity across the kinome but also changed the kinases inhibited. Compounds 72–74 consistently hit MYLK4, BIKE, GRK4, SRPK3 above 90%, another four PRP4, AAK1, PIP5K1A, SRPK2 are revealed as consistent hits if the threshold is decreased to 80% inhibition, in addition to other kinases (see supporting information). The most narrow spectrum inhibitor across the kinome tested in the KinomeSCAN® assay was 97 which hit only 7 kinases above 90%; including BIKE, DRAK2, JAK1(JH2domain-pseudokinase), MAST1, PIP5K1A, GRK4, and TYK2 (JH2domain-pseudokinase). The selectivity of the designed thiophene amide 111 was moderate with 15 kinases inhibited. The addition of a methyl 128 onto the 5-position of the imidazole head group of 73 changed the kinome profile subtly at 90% inhibition, with inhibition reduced on TYK2(JH2domain-pseudokinase), AAK1 but also increased on DRAK2, PIP5K1A, RIOK1, MAST1, LKB1.
Table 12.
Kinome selectivity profiling
| Kinome profiling summary | TLK2 IC50 (nM) | ||
|---|---|---|---|
| Name | >90 | S(10) | |
| 1 | 15 | 0.037 | 240 |
| 27 | 62 | 0.154 | 150 |
| 44 | 15 | 0.037 | 700 |
| 72 | 10 | 0.025 | 820 |
| 73 | 9 | 0.022 | 34 |
| 74 | 11 | 0,027 | 150 |
| 97 | 7 | 0.017 | 67 |
| 111 | 15 | 0.037 | 23 |
| 123 | 11 | 0.027 | IS |
number of kinases inhibited above 90%;
TLK2 enzyme assay result Aligning the results of the prediction with the experimental results, we found a good concordance (Table 11).
Figure 12.

Kinome tree image of KinomeScan data for A) 1, B) 27, C) 44, D) 72, E) 73, F) 74, G) 97, H) 111, I) 128
Finally, we screened the most promising compounds 97, 117, 128, the negative control compound 129, along with the starting point compound 1 in an orthogonal assay measuring the conversion of ATP to ADP.59–61 The results were consistent with both the weak and potent TLK2 binding of the five compounds in the enzyme assay (Figure 13 and Table 13). The potent TLK2 activities of 97, 117 and 128 in addition to their narrow spectrum kinome selectivity makes them attractive starting points for further optimization.
Figure 13.

TLK2 Kinase-Glo curves (n=3) for 1, 97, 117, 128, 129.
3. Discussion
TLK2 represents an attractive target for therapeutic development in a variety of cancer types and in “shock-and-kill” approaches for antiviral therapies. The published TLK2 inhibitors to date only offer compounds with moderate potency and poor kinome selectivity.18,62–63 In the present study, we demonstrate optimization of a chemotype for increased potency and selectivity for the clinically important TLK2 kinase. Since we achieved selectivity over the closest human paralog, TLK1, this work also provides important tool compounds for better characterization of the specific cellular functions of TLK2.64–65
The original target for the oxindole scaffold was receptor tyrosine kinases namely VEGFR and PDGFR.66 However, the scaffold demonstrated varying affinities for a large portion of the kinome.20–23 There have been a number of priority cancer targets including TrkA,67 CDK2,68–70 PDK1,71 and Src,72 among others,73 all inhibited by the oxindole scaffold. While the primary kinase targets were well defined, the oxindole chemotype has historically been considered as a broad-spectrum inhibitor chemotype. However, a LRRK2 program at Novartis by Troxler et al,74 suggested that the replacement of fluorine in the 5-position with a methoxy significantly improved kinome wide selectivity with the oxindole scaffold. This was supported by the narrow spectrum of the initial 5-methoxy substituted hit compound SU9516, when compared to the matched pair oxindoles GW5060033X and Sunitinib.20–23
The two oxindole matched pairs in the literature (Figure 1) demonstrated a tractable affinity to TLK2.20–23 We found several key structural features that could be tuned within the scaffold including: 1) The electronics of the oxindole head group and the internal hydrogen bond; 2) the electronics and sterics of the amide substitution pattern and 3) formation of the key Lys491 interaction with the amide linker.
The use of conformational restriction has been an effective tool in drug discovery.75 This methodology has been utilized to orient ATP competitive inhibitors in the preferred conformation. This can be used to not only increase potency, but also to enhance selectivity over other kinases/targets. There are several examples within kinase inhibitor design including Cyclin G-Associated Kinase (GAK),55,76 Spleen tyrosine kinase (Syk)77 and Cardiac troponin I-interacting kinase (TNNI3K)78–79 where addition or removal of a single nitrogen to alter the inhibitor conformation resulted in an increase by 60–160-fold on target.80 Another method employed in an EGFR inhibitor program involved fixing the pendent substituents with hydrogen bonds between an alcohol and a carbonyl.56 There are a number of other non-kinase examples including the development of a human histamine H4 receptor (hH4R) inhibitor agonist,81 and design of a selective CB2 cannabinoid receptor agonist.82
The adding or removing of a nitrogen has been proven to have a significant effect on activities across different systems, not only by altered conformation, but also in reduced de-solvation energy potential and as a hydrogen bond donor/acceptor.76 The oxindole scaffold has multiple potential contact points within the TLK2 ATP binding site. However, we found that that the most crucial nitrogen was on the pendant head group contained within the pyrrole/imidazole and formed not only an interaction at the hinge with Glu548 but also, an internal hydrogen bond with the carbonyl of the oxindole. This internal hydrogen bond pre-organizes the orientation of the molecule as an optimal ATP memetic (Figure 14). The solvent exposed shell is a key modulator of activity, with differing profiles depending on the nitrogen location on the ‘head group’ 5-member ring. We can rationally explain these differences by modelling the first water network solvent shell.55,84–85 Comparing Figure 14D to Figure 14E–F using WaterMap, both imidazoles are able to coordinate using the second nitrogen, while the pyrrole is not leading to a 10–20 fold drop in potency on TLK2. In addition to the conformation and head group nitrogen and oxindole hinge interactions our modelling highlights that to achieve a high affinity compound, a favourable fit into the hydrophobic back pocket is required. The key hydrogen bond interaction to the catalytic lysine (Lys491) affords a considerable boost in potency. The challenge then is to maintain selectivity across the kinome, this is achieved in part by a series of cationic-pi interactions also donated by Lys491, but also favourable fine-tuned π−π stacking interactions. This is demonstrated with a highlighted section of the 5-position substitution and the possibility to form a cationic- π interaction with Lys491. The small molecule crystal structures (Figure 4 & Table S10) support the theoretical observations with the TLK2 ATP binding site.
Figure 14.

Representative oxindoles with three key head groups showing first shell solvent front interactions using WaterMap A) 72, B) 73, C) 74, D) 72, E), 73 and F) 74 in TLK2 (PDB: 5O0Y). Waters are presented with two different representations balls A-C (green - low energy and red – higher energy) and full formulae D-F.
Conclusions
Oxindole based inhibitors are often considered to be promiscuous across the kinome, this mainly derived from several well- known broad-spectrum inhibitors including sunitinib and GW506033X.86 Here we demonstrate fine-tuning and optimisation to derive a narrow spectrum oxindole based TLK2 inhibitor, utilizing cutting edge modelling to prospectively design inhibitors. The result was compound 126 and 128 that were 10 to 15-fold more potent than the starting point 1 with maintenance of kinome selectivity and selectivity over the sub-family member TLK1. The QSAR and water network modelling provided new insights into the development of selective chemical probes for TLK2 and beyond. The comprehensive data sets also afford a more complete level of knowledge of the oxindole previously unavailable to the medicinal chemist’s toolbox.
Experimental
Molecular Modelling
Ligand preparation: Structures of small molecules were parametrized and minimized using the LigPrep module of Schrodinger 2019–4 suite employing OPLS3e force field.
Protein preparation: The TLK2 kinase a crystal structure (PDB:5O0Y) co-crystallized with substrate analog (phosphothiophosphoric acid-adenylate ester) was downloaded from RCSB-databanks. The structure was H-bond optimized and minimized using standard protein preparation procedure of Schrödinger suite.
TLK2 Molecular docking: The ligand docking was performed using SP settings of Schrodinger docking protocol with softened vdw potential (scaling 0.6). The initial docking of resulted quite poor convergence, thus oxindole-indole scaffold can bind in several conformations. To ensure correct docking mode of the oxindole (2-indolone) scaffold to kinase folds was confirmed using LPDB module of the Schrödinger package and oxindole as a query. After visual inspection of PDB:2X2K, 2X2M, 4QMO and 1PF8. To further improve convergence of docking poses a hydrogen bond constraint to mainchain NH of hinge residue was required, as experimentally observed in the case of other oxindole containing scaffolds (NH of C507 in the case of TLK2). The grid box was centered using coordinate center of the core structure of corresponding x-ray ligand as template. Graphical illustrations were generated using, Maestro software of Schrödinger. Flexible ligand alignments were performed using a Bemis-Mucko method, enabling recognition of the largest common scaffolds using a favorable docking pose of structurally representative derivative as alignment template (73). Torsion angles of larger substituents, such as methoxy groups, were manually adjusted to fit template structures when needed.
TLK2 3D-QSAR model: The model for the TLK2 kinase was constructed using the field based QSAR functionality of Schrödinger Maestro 2019–4. The models were constructed by defining 80% of compounds randomly as training sets and rest of the compounds were considered as test set. Grid spacing of 1.0 Å extended 3.0 beyond training set limits was used to the calculate field values. Subsequently, predicted versus measured affinities were plotted to identify outliers. The statistical metrics of models: R-square values with four PLS components were 0.79. The model shows acceptable internal predictivity (Q-square value 0.70), determined by leave-one-out (LOO) cross-validation. Stability value above 0.9 with four partial least squares regression (PLS) components let us to suggest that the quality and predictivity of the model inside the studied applicability domain was robust.
Crystallography
Each compound had a suitable crystal was selected and mounted on a MITIGEN holder (MiTeGen, Ithaca, NY, USA) in perfluoroether oil on a Rigaku FRE+ diffractometer equipped with a Mo target, VHF Varimax confocal mirrors, an AFC12 goniometer and HyPix 6000 detector (1, 2, 15, 19, 72 and 74) or a Rigaku 007HF diffractometer equipped with a Cu target, HF Varimax confocal mirrors, an AFC11 goniometer and HyPix 6000 detector (15, 74). The crystal was kept at a steady T = 100(2) K during data collection. CrysAlisPro87 was used to record images, process all data and apply empirical absorption corrections. Unit cell parameters were refined against all data. The structures were solved by the ShelXT88 structure solution program using the using dual methods solution method and using Olex289 as the graphical interface. The models were refined with version 2018/3 of ShelXL,88 using full matrix least squares minimization on F2 minimization. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model, except for those bonded to N-atoms which were located in the difference map and refined with a riding model. Structure 72 crystallised as an ethanol solvate. The data for 72 were processed as a 2-component non-merohedral twin (rotation (UB1, UB2) = 179.9982° around [0.00 0.71 0.71] (reciprocal) or [−0.39 0.85 0.35] (direct) lattice vectors). CCDC deposit numbers: 2309069–2309077, see supporting information.
Chemistry
General chemistry
All reactions were performed using flame-dried round-bottomed flasks or reaction vessels. Where appropriate, reactions were carried out under an inert atmosphere of nitrogen with dry solvents, unless otherwise stated. Yields refer to chromatographically and spectroscopically pure isolated yields. Reagents were purchased at the highest commercial quality and used without further purification. Reactions were monitored by thin-layer chromatography carried out on 0.25-mm. Merck silica gel plates (60F-254) using ultraviolet light as a visualizing agent. NMR spectra were recorded on a Varian Inova 400 (Varian, Palo Alto, CA, USA) or Inova 500 spectrometer (Varian, Palo Alto, CA, USA) and calibrated using residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26, 13C NMR = 77.16). The following abbreviations or combinations thereof were used to explain the multiplicities observed: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Liquid chromatography (LC) and high resolution mass spectra (HRMS) were recorded on a ThermoFisher hybrid LTQ FT (ICR 7T) (ThermoFisher, Waltham, MA USA). Compound spectra of 1-129 and mass spectrometry method are included in the Supplementary Materials. All compounds were >98% pure by 1H/13C NMR and LCMS.
3-[(3,5-Dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-2H-indol-2-one (SU5416/Semaxanib) (10) was consistent with commercial product and previous report.90 2-oxoindoline-5-sulfonyl chloride (49) was prepared consistent with previous report.45 5-isopropoxyindolin-2-one (56) was prepared consistent with previous report.74
General Procedures
Procedure A. Knoevenagel condensation
The oxindole (1.0 equiv), aldehyde (1.1 equiv), piperidine (2.4 equiv) were suspended in ethanol at reflux overnight. The solvent was removed and the resulting solid re-suspended DCM/EtOAc 1/1 and flushed through a plug of silica (DCM/EtOAc 1/1). The product was the purified by silica gel flash column chromatography (hexane:EtOAc) to afford the title compound.
Procedure B. Friedel-Crafts acylation
To a suspension of AlCl3 (6.0 equiv) in CH2Cl2 was added the appropriate acyl chloride (3.0 equiv) and the slurry was stirred at room temperature for 30 min, followed by the addition of oxindole (1.0 equiv) in CH2Cl2. The resulting mixture was stirred at room temperature for 2 days. Upon completion, the solution was cooled to 0 °C before quenching the reaction with ice/water, followed extraction of the organics with CH2Cl2. The combined organics were washed with saturated solution of NaHCO3, dried over Na2SO3 and the solvent was removed by rotary evaporation before purification by silica gel flash column chromatography (cyclohexane:EtOAc 9:1 to 1:1) to afford the title compound.
Procedure C. Friedel-Crafts acylation
The appropriate acyl chloride (3.0 equiv) was added to grinded AlCl3 (5.0 equiv) and the slurry was heated to 150 °C, then oxindole (1.0 equiv) was added portionwise. The resulting mixture was heated at 185 °C for 5 min. Upon completion, the mixture was cooled to 0 °C and was quenched with ice/water. The acylated product precipitated and was collected by filtration. Purification by silica gel flash column chromatography (cyclohexane:EtOAc 9:1 to 1:1) afforded the title compound.
General Procedure D. Sulphonamide coupling
To a solution of 2-oxoindoline-5-sulfonyl chloride (1.0 equiv) in THF the corresponding amine (1.1 equiv) and DIPEA (3.0 equiv) at room temperature, and the resulting mixture was stirred for 18 h at room temperature. Upon completion, the mixture was diluted with EtOAc and saturated solution of NaHCO3 and layers were separated. The organics were further extracted from the aqueous with EtOAc and the combined organics were washed with brine, dried over Na2SO4, filtered and the solvent was removed by rotary evaporation. Purification by silica gel flash column chromatography (hexane:EtOAc or CH2Cl2:MeOH mixtures) afforded the desired compound.
General Procedure E. Mitsunobu reaction
A dry three-neck flask was charged with 5-hydroxyindolin-2-one (1.0 equiv), PPh3 (1.4 equiv) and the appropriate alcohol (1.4 equiv) in anhydrous THF (0.25M). The solution was cooled to 0 °C and diethyl azadicarboxylate (1.4 equiv) in PhMe was added dropwise. The resulting mixture was warmed to room temperature and stirred for 18 h. Upon completion, the solvent reduced in vacuo, before purification by silica gel flash column chromatography (hexane:EtOAc 8:2) to afford the title compound.
General Procedure F. Amide HATU coupling
To a solution of amine (1.0 equiv) in THF, HATU (1.3 equiv), the corresponding carboxylic acid (1.1 equiv) and DIPEA (3.0 equiv) at room temperature, and the resulting mixture was stirred for 18 h at room temperature. Upon completion, the mixture was diluted with EtOAc and saturated solution of NaHCO3 and layers were separated. The organics were further extracted from the aqueous with EtOAc and the combined organics were washed with brine, dried over Na2SO4, filtered and the solvent reduced in vacuo. Purification by silica gel flash column chromatography (hexane:EtOAc or CH2Cl2:MeOH mixtures) afforded the desired compound.
(3Z)-3-[(1H-imidazol-5-yl)methylidene]-5-methoxy-2,3-dihydro-1H-indol-2-one (1):
Compound 1 was prepared by procedure A to afford a purple solid (177 mg, 0.734 mmol, 80%). MP 210–212 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.81 (s, 1H), 10.82 (s, 1H), 8.02 (s, 1H), 7.88 (s, 1H), 7.61 (s, 1H), 7.36 − 7.35 (m, 1H), 6.80 − 6.76 (m, 2H), 3.76 (s, 3H). HRMS m/z [M+H]+ calcd for C13H12N3O2: 242.0930, found 242.0918, LC tR = 2.94 min, > 98% Purity. Consistent with previous report.85
(3Z)-5-methoxy-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (2):
Compound 2 was prepared by procedure A to afford a dark orange solid (168 mg, 0.699 mmol, 76%). MP 220–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 10.84 (s, 1H), 7.56 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.29 (q, J = 2.3 Hz, 1H), 6.75 (dt, J = 3.6, 1.7 Hz, 1H), 6.58 (dd, J = 8.4, 2.3 Hz, 1H), 6.45 (d, J = 2.3 Hz, 1H), 6.34 − 6.29 (m, 1H), 3.76 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.8, 159.3, 140.3, 129.6, 124.7, 124.2, 119.6, 119.1, 117.9, 117.1, 111.1, 107.0, 96.1, 55.3. HRMS m/z [M+H]+ calcd for C14H13N2O2: 241.0977, found 241.0961, LC tR = 4.38 min, > 98% Purity.
(3Z)-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylidene]-5-methoxy-2,3-dihydro-1H-indol-2-one (3):
Compound 3 was prepared by procedure A to afford a red solid (200 mg, 0.745 mmol, 81%). MP 231–233 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.45 (s, 1H), 10.57 (s, 1H), 7.58 (s, 1H), 7.40 (d, J = 2.4 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.67 (dd, J = 8.4, 2.4 Hz, 1H), 5.99 (d, J = 2.5 Hz, 1H), 3.77 (s, 3H), 2.31 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.5, 154.8, 135.6, 132.0, 131.6, 126.9, 126.6, 123.7, 113.3, 112.5, 111.9, 109.6, 104.1, 55.6, 13.5, 11.4. HRMS m/z [M+H]+ calcd for C16H17N2O2: 269.1290, found 269.1282, LC tR = 4.79 min, > 98% Purity.
(3Z)-5-methoxy-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (4):
Compound 4 was prepared by procedure A to afford a red solid (160 mg, 0.663 mmol, 72%). MP 192–194 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 7.87 (s, 2H), 7.73 (s, 1H), 7.40 (d, J = 2.2 Hz, 1H), 6.86 − 6.77 (m, 3H), 3.77 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.0, 154.8, 136.6, 134.5, 130.3, 126.0, 123.2, 114.9, 113.6, 111.0, 109.8, 106.3, 55.9. HRMS m/z [M+H]+ calcd for C13H12N3O2: 242.0930, found 242.0920, LC tR = 3.85 min, > 98% Purity.
(3Z)-3-(1H-imidazol-2-ylmethylidene)-5-methoxy-2,3-dihydro-1H-indol-2-one (5):
Compound 5 was prepared by procedure A to afford a red solid (164 mg, 0.680 mmol, 74%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H), 7.86 (s, 1H), 7.55 (t, J = 1.5 Hz, 2H), 7.34 (d, J = 1.2 Hz, 1H), 6.82 (d, J = 1.4 Hz, 2H), 3.77 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 168.9, 155.2, 143.7, 133.7, 132.6, 125.0, 124.7, 124.1, 120.6, 115.1, 110.5, 106.0, 55.6. HRMS m/z [M+H]+ calcd for C13H12N3O2: 242.0930, found 242.0917, LC tR = 2.80 min, > 98% Purity.
(3Z)-5-methoxy-3-(pyridin-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (6):
Compound 6 was prepared by procedure A to afford a dark red solid (176 mg, 0.698 mmol, 76%). MP 176–178 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.96 − 8.86 (m, 1H), 8.82 (d, J = 2.7 Hz, 1H), 7.96 (td, J = 7.6, 1.8 Hz, 1H), 7.88 (dt, J = 7.8, 1.1 Hz, 1H), 7.56 (s, 1H), 7.47 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 6.88 (dd, J = 8.4, 2.7 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 3.77 (s, 3H). 13C NMR (100 MHz, DMSO- d6) δ 169.3, 154.2, 153.2, 149.6, 137.3, 137.3, 133.7, 129.9, 128.6, 124.2, 122.3, 116.3, 114.0, 109.8, 55.3. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0966, LC tR = 3.88 min, > 98% Purity.
(3Z)-5-methoxy-3-(pyridin-3-ylmethylidene)-2,3-dihydro-1H-indol-2-one (7):
Compound 7 was prepared by procedure A to afford a red solid (104 mg, 0.412 mmol, 45%, ratio of E:Z - 50:50). MP 206–208 °C; (Z)-5-Methoxy-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (500 MHz, DMSO-d6) δ 10.5 (d, J = 6.0 Hz, 1H), 9.19 (dd, J = 1.8, 1.1 Hz, 1H), 8.91 (dddd, J = 8.1, 2.3, 1.6, 0.6 Hz, 1H), 8.58 (dd, J = 4.8, 1.7 Hz, 1H), 7.84 (s, 1H), 7.49 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 7.41 (d, J = 2.5 Hz, 1H), 6.82 (dd, J = 8.4, 2.5 Hz, 1H), 6.74 (dd, J = 8.4, 0.5 Hz, 1H), 3.77 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 167.1, 154.7, 152.4, 150.3, 137.9, 134.9, 133.0, 129.9, 129.3, 125.3, 123.2, 115.4, 110.1, 106.3, 55.6. (E)-5-Methoxy-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (500 MHz, DMSO-d6) δ 10.5 (d, J = 6.0 Hz, 2H), 8.862 − 8.857 (m, 1H), 8.65 (ddd, J = 4.9, 1.7, 0.6 Hz, 1H), 8.12 (dddd, J = 7.9, 2.4, 1.7, 0.8 Hz, 1H), 7.62 (s, 1H), 7.56 (ddd, J = 7.9, 4.7, 0.5 Hz, 1H), 6.89 – 6.89 (m, 1H), 6.86 (dd, J = 8.4, 2.5 Hz, 1H), 6.80 (dd, J = 8.4, 0.5 Hz, 1H), 3.60 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 168.1, 154.0, 150.2, 149.7, 136.9, 136.3, 132.3, 130.5, 129.9, 123.6, 121.4, 115.6, 110.7, 108.9, 55.3. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0966, LC tR = 3.10 min, > 98% Purity.
(3Z)-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (8):
Compound 8 was prepared by procedure A to afford a bright yellow solid (156 mg, 0.739 mmol, 49%, ratio of E:Z - 33:67). MP 249–251 °C; (Z)-3-((1H-Pyrazol-5-yl)methylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.39 (bs, 1H), 9.31 (d, J = 7.6 Hz, 1H), 8.00 (s, 1H), 7.90 (s, 1H), 7.47 (s, 1H), 7.16 (td, J = 7.6, 1.4 Hz, 1H), 6.96 (td, J = 7.6, 1.1 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 169.9, 141.5, 138.1, 137.0, 128.2, 126.9, 126.4, 126.1, 122.6, 121.28, 120.6, 108.7. (E)-3-((1H-Pyrazol-5-yl)methylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 7.99 (s, 1H), 7.83 (s, 1H), 7.66 (d, J = 6.9 Hz, 1H), 7.21−7.15 (m, 1H), 7.01 (td, J = 7.6, 1.0 Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 168.6, 139.8, 139.0, 138.1, 127.9, 124.3, 123.9, 121.3, 120.6, 120.2, 119.2, 109.5. HRMS m/z [M+H]+ calcd for C12H10N3O: 212.0824, found 212.0814, LC tR = 2.82 min, > 98% Purity.
(3Z)-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (9):
Compound 9 was prepared by procedure A to afford a dark orange solid (159 mg, 0.756 mmol, 67%). MP 218–220 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 10.89 (s, 1H), 7.74 (s, 1H), 7.63 (dt, J = 7.4, 0.9 Hz, 1H), 7.35 (td, J = 2.6, 1.4 Hz, 1H), 7.15 (td, J = 7.6, 1.2 Hz, 1H), 6.99 (td, J = 7.6, 1.1 Hz, 1H), 6.88 (dt, J = 7.8, 0.9 Hz, 1H), 6.83 (dt, J = 3.6, 1.7 Hz, 1H), 6.35 (dt, J = 3.7, 2.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.2, 139.0, 129.6, 126.9, 126.3, 125.6, 125.2, 121.2, 120.3, 118.5, 116.8, 111.4, 109.5. HRMS m/z [M+H]+ calcd for C13H11N2O: 211.0871, found 211.0865, LC tR = 4.47 min, > 98% Purity.
(3Z)-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (11):
Compound 11 was prepared by procedure A to afford an orange solid (90 mg, 0.426 mmol, 38%). MP 190–192 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.56 (s, 1H), 10.52 (s, 1H), 7.94 (s, 1H), 7.72 (d, J = 7.5 Hz, 1H), 7.48 (s, 1H), 7.27 − 7.24 (m, 1H), 7.00 (td, J = 7.6, 1.1 Hz, 1H), 6.87 (d, J = 7.7 Hz, 1H), 6.85 − 6.80 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.5, 146.3, 142.5, 140.4, 129.7, 126.2, 125.8, 125.0, 121.9, 121.0, 110.6, 109.4. HRMS m/z [M+H]+ calcd for C12H10N3O: 212.0824, found 212.0812, LC tR = 3.85 min, > 98% Purity.
(3Z)-3-(1H-imidazol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (12):
Compound 12 was prepared by procedure A to afford a bright yellow solid (195 mg, 0.923 mmol, 82% ratio of E:Z - 40:60 - not able to clearly distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. 3-((1H-Imidazol-2-yl)methylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.52 (s, 1H), 9.40 (dd, J = 7.7, 1.3 Hz, 1H), 7.45 (s, 2H), 7.39 (s, 1H), 7.24 − 7.22 (m, 1H), 7.00 (td, J = 7.6, 1.1 Hz, 1H), 6.85 (d, J = 7.7 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.6, 143.4, 142.4, 129.6, 128.9, 127.1, 124.3, 121.8, 121.0, 120.9, 120.2, 109.1. Minor diastereoisomer. 3-((1H-Imidazol-2-yl)methylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 14.10 (s, 1H), 7.83 (d, J = 7.4 Hz, 1H), 7.80 (s, 1H), 7.55 (bs, 1H), 7.34 (s, 1H), 7.26 − 7.23 (m, 1H), 7.03 (td, J = 7.6, 1.0 Hz, 1H), 6.92 (d, J = 7.8 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 168.9, 143.7, 140.1, 132.5, 127.1, 124.4, 124.0, 123.7, 122.0, 121.0, 120.7, 110.0. HRMS m/z [M+H]+ calcd for C12H10N3O: 212.0824, found 212.0813, LC tR = 2.60 min, > 98% Purity.
(3Z)-3-(pyridin-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (13):
Compound 13 was prepared by procedure A to afford a dark orange solid (160 mg, 0.720 mmol, 64%). MP 191–193 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 9.03 − 8.97 (m, 1H), 8.88 (ddd, J = 4.8, 1.8, 0.8 Hz, 1H), 7.94 (td, J = 7.7, 1.8 Hz, 1H), 7.86 (dt, J = 7.9, 1.1 Hz, 1H), 7.57 (s, 1H), 7.46 (ddd, J = 7.5, 4.7, 1.2 Hz, 1H), 7.28 (td, J = 7.7, 1.3 Hz, 1H), 6.98 (td, J = 7.7, 1.1 Hz, 1H), 6.87 (dt, J = 7.7, 0.9 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 153.2, 149.7, 143.6, 137.2, 133.7, 130.8, 129.3, 128.4, 127.9, 124.1, 121.5, 121.2, 109.6. HRMS m/z [M+H]+ calcd for C14H11N2O: 223.0871, found 223.0860, LC tR = 3.85 min, > 98% Purity.
(3Z)-3-(pyridin-3-ylmethylidene)-2,3-dihydro-1H-indol-2-one (14):
Compound 14 was prepared by procedure A to afford a light yellow solid (45 mg, 0.203 mmol, 18%). MP 155–157 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 8.87 (dt, J = 1.9, 0.9 Hz, 1H), 8.65 (dd, J = 4.9, 1.6 Hz, 1H), 8.12 (dddd, J = 7.9, 2.4, 1.7, 0.8 Hz, 1H), 7.62 (s, 1H), 7.55 (ddd, J = 7.9, 4.8, 0.9 Hz, 1H), 7.37 (dd, J = 7.8, 1.1 Hz, 1H), 7.27 − 7.23 (m, 1H), 6.91 − 6.85 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 168.1, 150.1, 149.8, 143.2, 136.4, 132.1, 130.6, 130.6, 129.4, 123.7, 122.3, 121.3, 120.6, 110.3. HRMS m/z [M+H]+ calcd for C14H11N2O: 223.0871, found 223.0861, LC tR = 2.60 min, > 98% Purity.
(3Z)-6-methoxy-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (15):
Compound 15 was prepared by procedure A to afford an orange solid (137 mg, 0.570 mmol, 62%). MP 225–226 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 10.84 (s, 1H), 7.56 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.29 (td, J = 2.6, 1.4 Hz, 1H), 6.75 (dt, J = 3.6, 1.7 Hz, 1H), 6.58 (dd, J = 8.4, 2.3 Hz, 1H), 6.45 (d, J = 2.3 Hz, 1H), 6.32 (dt, J = 3.7, 2.4 Hz, 1H), 3.76 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.8, 159.3, 140.3, 129.6, 124.7, 124.2, 119.6, 119.1, 117.9, 117.1, 111.1, 107.0, 96.1, 55.3. HRMS m/z [M+H]+ calcd for C14H13N2O2: 241.0977, found 241.0962, LC tR = 4.41 min, > 98% Purity.
(3Z)-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylidene]-6-methoxy-2,3-dihydro-1H-indol-2-one (16):
Compound 16 was prepared by procedure A to afford an orange solid (200 mg, 0.745 mmol, 81%). MP 231–233 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 10.72 (s, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.40 (s, 1H), 6.56 (dd, J = 8.4, 2.3 Hz, 1H), 6.45 (d, J = 2.3 Hz, 1H), 5.96 (d, J = 2.5 Hz, 1H), 3.75 (s, 3H), 2.30 (s, 3H), 2.27 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.9, 158.5, 139.4, 134.4, 130.1, 126.5, 121.6, 119.1, 118.6, 113.1, 112.0, 106.7, 95.8, 55.2, 13.5, 11.3. HRMS m/z [M+H]+ calcd for C16H17N2O2: 269.1290, found 269.1272, LC tR = 4.67 min, > 98% Purity.
(3Z)-6-methoxy-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (17):
Compound 17 was prepared by procedure A to afford an orange solid (189 mg, 0.783 mmol, 85%). MP 209–211 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.06 (s, 1H), 7.80 (s, 1H), 7.64 (dd, J = 19.1, 8.0 Hz, 3H), 6.85 (s, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.47 (s, 1H), 3.78 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 160.8, 141.8, 140.2, 138.0, 125.0, 121.3, 118.6, 116.5, 110.7, 107.6, 96.5, 55.4. HRMS m/z [M+H]+ calcd for C13H12N3O2: 242.0930, found 242.0917, LC tR = 3.54 min, > 98% Purity.
(3Z)-3-(1H-imidazol-2-ylmethylidene)-6-methoxy-2,3-dihydro-1H-indol-2-one (18):
Compound 18 was prepared by procedure A to afford an orange solid (126 mg, 0.522 mmol, 57%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.94 (s, 1H), 11.33 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.61 (s, 1H), 7.50 (s, 1H), 7.29 (s, 1H), 6.60 (dd, J = 8.4, 2.3 Hz, 1H), 6.48 (d, J = 2.3 Hz, 1H), 3.78 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 169.6, 160.6, 143.9, 141.7, 132.0, 123.8, 121.9, 121.5, 120.1, 116.7, 107.5, 96.4, 55.4. HRMS m/z [M+H]+ calcd for C13H12N3O2: 242.0930, found 242.0916, LC tR = 2.82 min, > 98% Purity.
(3Z)-6-methoxy-3-(pyridin-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (19):
Compound 19 was prepared by procedure A to afford a mustard solid (151 mg, 0.599 mmol, 65%). MP 182–184 °C; δ 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 8.98 (d, J = 8.6 Hz, 1H), 8.89 − 8.78 (m, 1H), 7.92 (td, J = 7.7, 1.9 Hz, 1H), 7.81 (dt, J = 8.0, 1.1 Hz, 1H), 7.42 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 7.39 (s, 1H), 6.55 (dd, J = 8.7, 2.5 Hz, 1H), 6.42 (d, J = 2.4 Hz, 1H), 3.79 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.0, 161.7, 153.6, 149.5, 145.5, 137.1, 130.4, 129.6, 128.9, 128.0, 123.7, 114. 6, 106.5, 96.0, 55.3. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0970, LC tR = 3.85 min, > 98% Purity.
(Z)-6-methoxy-3-(pyridin-3-ylmethylene)indolin-2-one (20)
Compound 20 was prepared by procedure A to afford a mustard solid (93 mg, 0.369 mmol, 40%, ratio of E:Z - 28:72). MP 177–179 °C; (Z)-6-Methoxy-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.73 (bs, 1H), 8.84 (d, J = 2.3 Hz, 1H), 8.62 (dd, J = 4.9, 1.6 Hz, 1H), 8.08 (dt, J = 7.9, 2.1 Hz, 1H), 7.53 (dd, J = 7.9, 4.8 Hz, 1H), 7.43 (s, 1H), 7.32 (d, J = 8.5 Hz, 1H), 6.45 − 6.42 (m, 2H), 3.75 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 168.9, 161.5, 149.77, 149.75, 145.1, 136.2, 130.9, 129.0, 128.6, 123.7, 123.1, 113.4, 106.8, 96.7, 55.4. (E)-6-Methoxy-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 9.13 (d, J = 2.2 Hz, 1H), 8.82 (dt, J = 8.1, 2.0 Hz, 1H), 8.54 (dd, J = 4.8, 1.7 Hz, 1H), 7.62 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.46 (dd, J = 8.1, 4.8 Hz, 1H), 6.57 (dd, J = 8.4, 2.4 Hz, 1H), 6.40 (d, J = 2.3 Hz, 1H), 3.78 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 167.7, 161.0, 152.1, 149.7, 142.7, 137.5, 130.2, 129.9, 128.6, 123.5, 121.5, 117.1, 106.9, 96.0, 55.4. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0965, LC tR = 2.70 min, > 98% Purity.
(Z)-3-(1H-imidazol-5-ylmethylidene)-5,6-dimethoxy-2,3-dihydro-1H-indol-2-one (21):
Compound 21 was prepared by procedure A to afford a bright red solid (160 mg, 0.590 mmol, 76%, ratio of E:Z - 50:50). MP 222–224 °C; (Z)-3-((1H-Imidazol-5-yl)methylene)-5,6-dimethoxyindolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 13.71 (bs, 1H), 7.94 (s, 1H), 7.68 (s, 1H), 7.56 (s, 1H), 7.38 (s, 1H), 6.53 (s, 1H), 3.77 (s, 3H), 3.77 (s, 3H). (E)-3-((1H-Imidazol-5-yl)methylene)-5,6-dimethoxyindolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.14 (s, 1H), 9.20 (s, 1H), 7.97 (s, 1H), 7.81 (d, J = 1.1 Hz, 1H), 7.30 (s, 1H), 6.47 (s, 1H), 3.78 (s, 3H), 3.76 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 170.6, 150.0, 143.1, 137.9, 136.7, 125.1, 124.3, 122.2, 114.1, 113.1, 104.8, 94.6, 56.5, 55.6. HRMS m/z [M+H]+ calcd for C14H14N3O3: 272.1035, found 272.1023, LC tR = 2.76 min, > 98% Purity.
(3Z)-5,6-dimethoxy-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (22):
Compound 22 was prepared by procedure A to afford a bright red solid (93 mg, 0.369 mmol, 40%). MP 167–169 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.27 (s, 1H), 10.65 (s, 1H), 7.61 (s, 1H), 7.35 (s, 1H), 7.29 (q, J = 2.2 Hz, 1H), 6.73 (dt, J = 3.6, 1.7 Hz, 1H), 6.52 (s, 1H), 6.32 (dt, J = 4.0, 2.4 Hz, 1H), 3.77 (d, J = 2.2 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.6, 149.1, 144.4, 133.2, 129.7, 124.6, 124.4, 118.9, 117.9, 116.3, 111.1, 104.3, 95.4, 56.3, 55.8. HRMS m/z [M+H]+ calcd for C15H15N2O3: 271.1083, found 271.1072, LC tR = 4.29 min, > 98% Purity.
(3Z)-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylidene]-5,6-dimethoxy-2,3-dihydro-1H-indol-2-one (23):
Compound 23 was prepared by procedure A to afford a dark red solid (167 mg, 0.560 mmol, 72%). MP 282–285 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.28 (s, 1H), 10.53 (s, 1H), 7.45 (s, 1H), 7.42 (s, 1H), 6.51 (s, 1H), 5.96 (d, J = 2.5 Hz, 1H), 3.78 (s, 3H), 3.75 (s, 3H), 2.30 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 13C NMR (101 MHz, DMSO-d6) δ 169.7, 148.4, 144.3, 134.3, 132.3, 130.1, 126.6, 121.9, 117.2, 114.0, 112.0, 104.3, 95.3, 56.6, 55.8, 13.5, 11.4. HRMS m/z [M+H]+ calcd for C22H13N4O: 349.1089, found 349.1088, LC tR = 4.61 min, > 98% Purity.
(3Z)-5,6-dimethoxy-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (24):
Compound 24 was prepared by procedure A to afford a dark red solid (160 mg, 0.590 mmol, 76%). MP 230–232 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.49 (s, 1H), 10.26 (s, 1H), 8.85 (s, 1H), 7.89 (d, J = 2.3 Hz, 1H), 7.29 (s, 1H), 6.75 (d, J = 2.3 Hz, 1H), 6.51 (s, 1H), 3.80 (s, 3H), 3.77 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 170.2, 150.9, 146.6, 143.2, 137.8, 129.7, 125.7, 122.6, 113.2, 112.6, 109.9, 94.9, 56.5, 55.6. HRMS m/z [M+H]+ calcd for C14H14N3O3: 272.1035, found 272.1028, LC tR = 3.21 min, > 98% Purity.
(Z)-3-(1H-imidazol-2-ylmethylidene)-5,6-dimethoxy-2,3-dihydro-1H-indol-2-one (25):
Compound 25 was prepared by procedure A to afford a dark red solid (95 mg, 0.350 mmol, 45%, ratio of E:Z - 44:56). MP >300 °C; (E)-3-((1H-Imidazol-2-yl)methylene)-5,6-dimethoxyindolin-2-one. 1H NMR (500 MHz, DMSO-d6) δ 10.28 (bs, 1H), 9.26 (s, 1H), 7.41 (s, 2H), 7.21 (s, 1H), 6.50 (s, 1H), 3.80 (s, 3H), 3.77 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 170.2, 151.1, 143.5, 143.2, 137.8, 132.1, 125.6, 117.5, 113.28, 113.26, 94.8, 56.5, 55.6. (Z)-3-((1H-Imidazol-2-yl)methylene)-5,6-dimethoxyindolin-2-one. 1H NMR (500 MHz, DMSO-d6) δ 14.01 (bs, 1H), 7.69 (s, 1H), 7.56 (s, 1H), 7.50 (bs, 1H), 7.30 (bs, 1H), 6.54 (s, 1H), 3.78 (s, 3H), 3.77 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 169.4, 150.6, 144.7, 144.0, 134.5, 124.5, 122.2, 120.0, 115.1, 105.5, 95.5, 56.3, 55.8. HRMS m/z [M+H]+ calcd for C14H14N3O3: 272.1035, found 272.1022, LC tR = 2.74 min, > 98% Purity.
(Z)-5-acetyl-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (26):
Compound 26 was prepared by procedure A to afford a bright orange solid (154 mg, 0.608 mmol, 71%, E:Z ratio 32:68). MP 262–265 °C; (Z)-3-((1H-Pyrazol-5-yl)methylene)-5-acetylindolin-2-one. 1H NMR (500 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.12 (d, J = 1.8 Hz, 1H), 8.02 (s, 1H), 7.94 (d, J = 1.1 Hz, 1H), 7.82 (dd, J = 8.1, 1.9 Hz, 1H), 7.56 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 2.57 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 196.8, 170.5, 145.4, 139.7, 137.0, 130.4, 128.9, 127.5, 122.7, 119.7, 119.4, 108.6, 26.5. (E)-3-((1H-Pyrazol-5-yl)methylene)-5-acetylindolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.33 (d, J = 1.7 Hz, 1H), 8.06 (s, 1H), 8.01 (s, 1H), 7.85 (dd, J = 8.2, 1.7 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 2.58 (s, 3H). HRMS m/z [M+H]+ calcd for C14H12N3O2: 254.0930, found 254.0918, LC tR = 2.74 min, > 98% Purity.
(3Z)-5-acetyl-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (27):
Compound 27 was prepared by procedure A to afford a dark orange solid (117 mg, 0.467 mmol, 54%). MP 218–220 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 9.08 − 8.92 (m, 1H), 8.85 (ddd, J = 4.8, 1.9, 0.8 Hz, 1H), 7.92 (td, J = 7.7, 1.8 Hz, 1H), 7.84 (dt, J = 7.9, 1.1 Hz, 1H), 7.43 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 7.25 (td, J = 7.6, 1.3 Hz, 1H), 6.95 (td, J = 7.7, 1.1 Hz, 1H), 6.84 (dt, J = 7.7, 0.8 Hz, 1H), 3.31 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 153.2, 149.7, 143.6, 137.3, 133.7, 130.8, 129.3, 128.4, 127.9, 124.1, 121.5, 121.2, 109.6, 39.3. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0963, LC tR = 3.99 min, > 98% Purity.
(3Z)-5-acetyl-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylidene]-2,3-dihydro-1H-indol-2-one (28):
Compound 28 was prepared by procedure A to afford a dark orange solid (149 mg, 0.532 mmol, 62%). MP >300 °C; δ 1H NMR (400 MHz, DMSO-d6) δ 13.33 (s, 1H), 11.14 (s, 1H), 8.35 (d, J = 1.6 Hz, 1H), 7.82 − 7.68 (m, 2H), 6.96 (d, J = 8.2 Hz, 1H), 6.05 (d, J = 2.5 Hz, 1H), 2.59 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 197.0, 169.8, 141.8, 136.8, 133.1, 130.5, 126.9, 126.6, 125.9, 124.7, 118.5, 113.0, 111.3, 108.9, 26.7, 13.6, 11.5. HRMS m/z [M+H]+ calcd for C17H17N2O2: 281.1290, found 281.1278, LC tR = 4.62 min, > 98% Purity.
(3Z)-5-acetyl-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (29):
Compound 29 was prepared by procedure A to afford a mustard solid (33 mg, 0.130 mmol, 15%). MP 240–242 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.76 (s, 1H), 10.94 (s, 1H), 9.75 (s, 1H), 7.96 (s, 1H), 7.92 (dd, J = 8.2, 1.8 Hz, 1H), 7.56 (s, 1H), 6.97 (d, J = 8.1 Hz, 1H), 6.86 (s, 1H), 2.58 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 196.6, 170.0, 146.5, 146.2, 130.7, 130.1, 127.2, 127.0, 124.0, 121.8, 120.4, 111.0, 109.1, 26.5. HRMS m/z [M+H]+ calcd for C14H12N3O2: 254.0930, found 254.0920, LC tR = 3.30 min, > 98% Purity.
(Z)-5-acetyl-3-(1H-imidazol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (30):
Compound 30 was prepared by procedure A to afford a bright yellow solid (134 mg, 0.529 mmol, 62%, E:Z ratio 47:53 - not able to distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. 3-((1H-Imidazol-2-yl)methylene)-5-acetylindolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 10.94 (bs, 1H), 10.14 (d, J = 1.8 Hz, 1H), 7.91 (dd, J = 8.2, 1.9 Hz, 1H), 7.56 (bs, 1H), 7.48 (bs, 1H), 7.46 (s, 1H), 6.96 (d, J = 8.2 Hz, 1H), 2.58 (s, 3H). Minor diastereoisomer. 3-((1H-Imidazol-2-yl)methylene)-5-acetylindolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 13.97 (bs, 1H), 10.95 (bs, 1H), 8.54 (d, J = 1.6 Hz, 1H), 8.06 (s, 1H), 7.88 (dd, J = 8.2, 1.7 Hz, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 1.3 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 2.60 (s, 3H). HRMS m/z [M+H]+ calcd for C14H12N3O2: 254.0930, found 254.0917, LC tR = 2.86 min, > 98% Purity.
(3Z)-5-acetyl-3-(pyridin-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (31):
Compound 31 was prepared by procedure A to afford a mustard solid (190 mg, 0.719 mmol, 63%). MP 250–252 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.00 (s, 1H), 9.77 (d, J = 1.8 Hz, 1H), 8.88 (dt, J = 4.8, 1.2 Hz, 1H), 7.96 (td, J = 7.6, 1.8 Hz, 1H), 7.93 − 7.80 (m, 2H), 7.62 (s, 1H), 7.49 (ddd, J = 7.5, 4.7, 1.3 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 2.55 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 196.4, 169.7, 152.9, 149.7, 147.6, 137.5, 134.9, 131.5, 130.6, 128.9, 128.9, 128.4, 124.6, 121.3, 109.5, 26.4. HRMS m/z [M+H]+ calcd for C16H13N2O2: 265.0977, found 265.0966, LC tR = 3.72 min, > 98% Purity.
(3Z)-5-acetyl-3-(pyridin-3-ylmethylidene)-2,3-dihydro-1H-indol-2-one (32):
Compound 32 was prepared by procedure A to afford a yellow solid (88 mg, 0.333 mmol, 39%, E:Z ratio 48:52 - not able to distinguish E/Z isomers). MP 209–211 °C; Major diastereoisomer. 5-Acetyl-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 11.10 (bs, 1H), 9.24 (d, J = 2.2 Hz, 1H), 8.93 − 8.91 (m, 2H), 8.61 (dd, J = 4.8, 1.7 Hz, 1H), 8.08 (s, 1H), 7.94 − 7.91 (m, 1H), 7.51 (dd, J = 8.1, 4.8 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 2.58 (s, 3H). Minor diastereoisomer. 5-Acetyl-3-(pyridin-3-ylmethylene)indolin-2-one. 1H NMR (600 MHz, DMSO-d6) δ 11.08 (bs, 1H), 8.69 (dd, J = 4.8, 1.6 Hz, 1H), 8.38 (d, J = 1.6 Hz, 1H), 8.23 − 8.15 (m, 1H), 7.99 (d, J = 1.7 Hz, 1H), 7.94 − 7.91 (m, 1H), 7.73 (s, 1H), 7.59 (dd, J = 7.7, 5.0 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 2.42 (s, 3H). HRMS m/z [M+H]+ calcd for C16H13N2O2: 265.0977, found 265.0967, LC tR = 2.46 min, > 98% Purity.
(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indole-5-carboxylic acid (33):
Compound 33 was prepared by procedure A to afford a dark orange solid (90 mg, 0.354 mmol, 42%, E/Z ratio 30:70). MP >220 °C (decomp.); (Z)-3-((1H-pyrrol-2-yl)methylene)-2-oxoindoline-5-carboxylic acid. 1H NMR (600 MHz, DMSO-d6) δ 13.28 (bs, 1H), 11.07 (bs, 1H), 8.18 (d, J = 1.5 Hz, 1H), 7.77 (dd, J = 8.0, 1.4 Hz, 1H), 7.77 (s, 1H), 7.34 − 7.33 (m, 1H), 6.88 − 6.85 (m, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.34 (dt, J = 3.7, 2.3 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 170.3, 169.7, 140.1, 129.7 (s, 2C), 128.6, 126.1, 125.4, 124.1, 120.3, 119.5, 117.1, 111.3, 108.3. (E)-3-((1H-pyrrol-2-yl)methylene)-2-oxoindoline-5-carboxylic acid. 1H NMR (600 MHz, DMSO-d6) δ 11.91 (bs, 1H), 10.58 (bs, 1H), 8.59 (d, J = 1.5 Hz, 1H), 7.81 (dd, J = 8.0, 1.4 Hz, 1H), 7.52 (s, 1H), 7.21 − 7.20 (m, 1H), 7.15 − 7.05 (m, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.42 − 6.41 (m, 1H). 13C NMR (125 MHz, DMSO-d6) δ 170.3, 169.7 142.6, 132.2, 129.8 (s, 2C), 127.6, 124.8, 124.0, 123.1, 120.9, 113.5, 111.5, 108.2. HRMS m/z [M+H]+ calcd for C14H11N2O3: 255.0770, found 255.0762, LC tR = 3.79 min, > 98% Purity.
(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indole-5-carboxylic acid (34):
Compound 34 was prepared by procedure A to afford a yellow solid (104 mg, 0.408 mmol, 48%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.90 (s, 1H), 8.25 (s, 1H), 8.07 (s, 1H), 7.96 (s, 1H), 7.83 (dd, J = 8.1, 1.7 Hz, 1H), 7.54 (s, 1H), 6.89 (d, J = 8.1 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 170.3, 167.9, 145.3, 138.3, 130.6, 129.8, 128.2, 126.6, 124.4, 123.9, 122.4, 120.6, 108.6. HRMS m/z [M+H]+ calcd for C13H10N3O3: 256.0722, found 256.0716, LC tR = 2.46 min, > 98% Purity.
(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indole-5-carboxylic acid (35):
Compound 35 was prepared by procedure A to afford a yellow solid (113 mg, 0.443 mmol, 52%). MP >260 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 14.34 (s, 1H), 8.15 (d, J = 1.4 Hz, 1H), 7.83 (dd, J = 8.0, 1.4 Hz, 1H), 7.57 (s, 1H), 7.53 (s, 1H), 7.36 − 7.25 (m, 1H), 6.81 (d, J = 7.9 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 174.4, 169.3, 143.9, 137.0, 134.7, 132.2, 130.5, 128.2, 125.0, 123.0, 122.8, 120.5, 108.8. HRMS m/z [M+H]+ calcd for C13H10N3O3: 256.0722, found 256.0714, LC tR = 2.46 min, > 98% Purity.
5-butyrylindolin-2-one (36):
Compound 36 was prepared by procedure B to afford a colourless solid (901 mg, 4.431 mmol, 59%). MP 130–132 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 7.89 − 7.82 (m, 1H), 7.82 − 7.76 (m, 1H), 6.89 (d, J = 8.1 Hz, 1H), 3.54 (s, 2H), 2.90 (t, J = 7.2 Hz, 2H), 1.61 (h, J = 7.3 Hz, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 198.5, 176.8, 148.2, 130.4, 129.0, 126.1, 124.1, 108.7, 39.4, 35.5, 17.6, 13.7. HRMS m/z [M+H]+ calcd for C12H14NO2: 204.1025, found 204.1017, LC tR = 3.18 min, > 98% Purity.
5-benzoylindolin-2-one (37):
Compound 37 was prepared by procedure C to afford a colourless solid (1.319 g, 5.555 mmol, 74%). MP 186–188 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 7.73 − 7.59 (m, 5H), 7.57 − 7.51 (m, 2H), 6.95 (d, J = 8.6 Hz, 1H), 3.57 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 194.7, 176.7, 148.3, 138.0, 131.9, 131.3, 130.0, 129.2 (s, 2C), 128.4 (s, 2C), 126.2, 126.0, 108.7, 35.6. HRMS m/z [M+H]+ calcd for C15H12NO2: 238.0868, found 238.0858, LC tR = 3.36 min, > 98% Purity.
5-(4-fluorobenzoyl)indolin-2-one (38):
Compound 38 was prepared by procedure B to afford a colourless/light green solid (571 mg, 2.237 mmol, 30%). MP 155–157 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.76 − 7.55 (m, 3H), 7.51 (td, J = 7.4, 1.7 Hz, 1H), 7.40 − 7.31 (m, 2H), 6.95 (d, J = 8.6 Hz, 1H), 3.57 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 191.4, 176. 8, 158.8 (d, J = 247.6 Hz), 149.3, 132.8 (d, J = 8.2 Hz), 131.2, 130.0 (d, J = 14.9 Hz), 129.9, 127.20 (d, J = 16.1 Hz), 126.5, 125.5, 124.7 (d, J = 3.4 Hz), 116.1 (d, J = 21.4 Hz), 109.0, 35.5. HRMS m/z [M+H]+ calcd for C15H11FNO2: 256.0774, found 256.0763, LC tR = 3.40 min, > 98% Purity.
5-(furan-2-carbonyl)indolin-2-one (39):
Compound 39 was prepared by procedure B to afford a beige solid (836 mg, 3.579 mmol, 49%). Consistent with previous report.86
(3Z)-5-butanoyl-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (40):
Compound 40 was prepared by procedure A to afford a mustard solid (43 mg, 0.153 mmol, 31%). MP 153–155 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.22 (s, 1H), 11.23 (s, 1H), 8.30 (d, J = 1.6 Hz, 1H), 7.99 (s, 1H), 7.82 (dd, J = 8.2, 1.7 Hz, 1H), 7.40 (q, J = 1.8, 1.3 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.92 (dt, J = 3.6, 1.7 Hz, 1H), 6.39 (dt, J = 3.7, 2.3 Hz, 1H), 2.99 (t, J = 7.2 Hz, 2H), 1.66 (h, J = 7.3 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 198.8, 169.6, 142.6, 130.6, 129.6, 127.8, 127.5, 126.5, 125.3, 121.3, 118.6, 115.6, 111.8, 109.2, 17.6 (s, 2C), 13.8. HRMS m/z [M+H]+ calcd for C17H17N2O2: 281.1290, found 281.1286, LC tR = 4.85 min, > 98% Purity.
(3Z)-5-butanoyl-3-(1H-imidazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (41):
Compound 41 was prepared by procedure A to afford a bright yellow solid (58 mg, 0.207 mmol, 42%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.52 (s, 1H), 10.36 (d, J = 1.9 Hz, 1H), 8.53 (s, 1H), 7.75 − 7.61 (m, 3H), 7.51 (s, 1H), 6.85 (d, J = 8.1 Hz, 1H), 2.96 (t, J = 7.3 Hz, 2H), 1.74 − 1.68 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 174.3, 170.5, 165.8, 156.3, 148.0, 145.3, 130.8, 124.3, 122.2, 121.8, 114.0, 111.9, 108.1, 48.6, 25.6, 14.0. HRMS m/z [M+H]+ calcd for C16H16N3O2: 282.1243, found 282.1233, LC tR = 3.36 min, > 98% Purity.
(3Z)-5-butanoyl-3-(1H-imidazol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (42):
Compound 42 was prepared by procedure A to afford a bright yellow solid (77 mg, 0.274 mmol, 56%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.98 (s, 1H), 11.48 (s, 1H), 8.56 (d, J = 1.6 Hz, 1H), 8.08 (s, 1H), 7.89 (dd, J = 8.2, 1.7 Hz, 1H), 7.59 (d, J = 2.1 Hz, 1H), 7.39 (d, J = 1.1 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 3.03 (t, J = 7.2 Hz, 2H), 1.65 (q, J = 7.3 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 199.0, 169.4, 143.6, 143.5, 132.9, 131.1, 129.2, 125.9, 124.1, 122.6, 121.2, 120.8, 109.9, 40.1, 17.6, 13.8. HRMS m/z [M+H]+ calcd for C16H16N3O2: 282.1243, found 282.1236, LC tR = 3.29 min, > 98% Purity.
(3Z)-5-benzoyl-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (43):
Compound 43 was prepared by procedure A to afford a red solid (29 mg, 0.092 mmol, 22%). MP 232–234 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.21 (s, 1H), 11.29 (s, 1H), 8.10 (d, J = 1.6 Hz, 1H), 7.97 (s, 1H), 7.78 − 7.70 (m, 2H), 7.69 − 7.64 (m, 1H), 7.61 − 7.53 (m, 3H), 7.41 (q, J = 1.8, 1.2 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.93 (dt, J = 3.6, 1.7 Hz, 1H), 6.38 (dt, J = 3.7, 2.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 195.1, 169.6, 142.6, 138.0, 132.1, 130.4, 130.0, 129.6, 129.4 (s, 2C), 128.5 (s, 2C), 128.1, 126.6, 125.4, 121.6, 119.9, 115.2, 111.8, 109.1. HRMS m/z [M+H]+ calcd for C20H15N2O2: 315.1134, found 315.1120, LC tR = 4.90 min, > 98% Purity.
(3Z)-5-benzoyl-3-(1H-imidazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (44):
Compound 44 was prepared by procedure A to afford a dark orange solid (24 mg, 0.076 mmol, 24%). MP 255–257 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 9.96 (s, 1H), 7.95 (d, J = 1.0 Hz, 1H), 7.78 − 7.71 (m, 4H), 7.69 − 7.55 (m, 5H), 6.99 (d, J = 8.1 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 194.9, 170.3, 145.6, 138.3, 137.8, 136.9, 131.7, 131.3, 129.9, 129.5, 129.3 (s, 2C), 128.6, 128.2 (s, 2C), 126.6, 122.34, 120.6, 108.9. HRMS m/z [M+H]+ calcd for C19H14N3O2: 316.1086, found 316.1076, LC tR = 3.51 min, > 98% Purity.
(3Z)-5-[(4-fluorophenyl)carbonyl]-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (45):
Compound 45 was prepared by procedure A to afford a dark orange solid (37 mg, 0.111 mmol, 28%). MP 242–244 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.19 (s, 1H), 11.35 (s, 1H), 8.14 (d, J = 1.6 Hz, 1H), 7.98 (s, 1H), 7.65 (ddd, J = 13.9, 5.5, 2.7 Hz, 1H), 7.62 − 7.51 (m, 2H), 7.46 − 7.30 (m, 3H), 7.02 (d, J = 8.1 Hz, 1H), 6.94 (dt, J = 3.7, 1.7 Hz, 1H), 6.39 (dt, J = 4.1, 2.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 191.7, 169.6, 159.0 (d, J = 248.1 Hz), 143.5, 132.9 (d, J = 8.2 Hz), 130.4, 130.2 (d, J = 3.0 Hz), 130.0, 129.6, 128.3, 127.3 (d, J = 15.7 Hz), 126.8, 125.7, 124.7 (d, J = 3.4 Hz), 121.8, 119.3, 116.2 (d, J = 21.4 Hz), 114.9, 111.8, 109.3. HRMS m/z [M+H]+ calcd for C20H14N2O2F: 333.1039, found 333.1035, LC tR = 4.88 min, > 98% Purity.
(3Z)-5-[(4-fluorophenyl)carbonyl]-3-(1H-imidazol-5-ylmethylidene)-2,3-dihydro-1H-indol-2-one (46):
Compound 46 was prepared by procedure A to afford a bright yellow solid (51 mg, 0.153 mmol, 39%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.04 (d, J = 1.6 Hz, 1H), 7.98 (d, J = 39.4 Hz, 1H), 7.75 (s, 1H), 7.70 (dd, J = 8.2, 1.9 Hz, 1H), 7.66 (tdd, J = 7.5, 5.3, 1.8 Hz, 1H), 7.53 (dd, J = 7.2, 1.9 Hz, 1H), 7.50 (d, J = 5.3 Hz, 1H), 7.42 (s, 1H), 7.39 (d, J = 3.9 Hz, 1H), 7.37 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 192.1, 173.7, 170.8, 158.9 (d, J = 246.5 Hz), 145.3, 137.1, 132.1 (d, J = 8.5 Hz), 129.9, 129.8 (d, J = 3.6 Hz, 2C), 129.6, 129.6, 128.2 (d, J = 16.6 Hz, 2C), 128.1, 124.5 (d, J = 3.5 Hz), 123.6, 115.9 (d, J = 21.5 Hz), 108.6. HRMS m/z [M+H]+ calcd for C19H13N3O2F: 334.0992, found 334.0979, LC tR = 3.57 min, > 98% Purity.
(Z)-3-((1H-pyrrol-2-yl)methylene)-5-(furan-2-carbonyl)indolin-2-one (47):
Compound 47 was prepared by procedure A to afford a yellow solid (19 mg, 0.062 mmol, 14%). MP >220 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 13.82 (s, 1H), 8.53 (s, 1H), 8.13 (s, 1H), 8.08 (d, J = 1.6 Hz, 1H), 7.83 (s, 1H), 7.78 (dd, J = 8.2, 1.7 Hz, 1H), 7.37 (d, J = 3.5 Hz, 1H), 7.33 (s, 1H), 6.95 (d, J = 8.1 Hz, 1H), 6.91 − 6.80 (m, 1H), 6.78 (dd, J = 3.6, 1.7 Hz, 1H), 6.34 (d, J = 3.0 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 180.6, 174.2, 165.6, 151.9, 147.6, 133.6, 130.0, 129.2, 126.6, 125.4, 119.8, 118.9, 116.2, 115.5, 114.1, 112.4, 111.3, 109.9. HRMS m/z [M+H]+ calcd for C18H13N2O3: 305.0926, found 305.0920, LC tR = 4.44 min, > 98% Purity.
(3Z)-5-[(furan-2-yl)carbonyl]-3-(1H-imidazol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (48):
Compound 48 was prepared by procedure A to afford a yellow solid (23 mg, 0.075 mmol, 17%). MP >270 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 10.73 (s, 2H), 10.55 (s, 1H), 8.53 (s, 1H), 8.09 (d, J = 1.6 Hz, 1H), 7.81 (dd, J = 8.1, 2.0 Hz, 1H), 7.73 (d, J = 3.6 Hz, 1H), 7.53 (s, 1H), 7.36 (s, 1H), 6.96 (d, J = 8.1 Hz, 1H), 6.85 (dd, J = 3.5, 1.7 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 180.4, 173.9, 165.5, 151.6, 147.5, 131.8, 129.7, 129.2, 127.6, 122.5, 120.1, 117.3, 114.9, 114.0, 112.4, 111.2, 108.6. HRMS m/z [M+H]+ calcd for C17H12N3O3: 306.0879, found 306.0867, LC tR = 2.96 min, > 98% Purity.
N,N-dimethyl-2-oxoindoline-5-sulfonamide (50):
Compound 50 was prepared by procedure D to afford a beige solid (447 mg, 1.985 mmol, 55%). MP 198–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 7.67 − 7.46 (m, 2H), 7.04 − 6.96 (m, 1H), 3.60 (s, 2H), 2.57 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 176.4, 148.0, 128.4, 126.9, 126.9, 123.7, 109.1, 37.6 (s, 2C), 35.7. HRMS m/z [M+H]+ calcd for C10H13N2O3S: 241.0649, found 241.0633.
5-((4-methylpiperazin-1-yl)sulfonyl)indolin-2-one (51):
Compound 51 was prepared by procedure D to afford a beige/brown solid (496 mg, 1.679 mmol, 39%). MP 170–172 °C; HRMS m/z [M+H]+ calcd for C13H18N3O3S: 296.1069, found 296.1054. Consistent with previous report.88
(3Z)-N,N-dimethyl-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indole-5-sulfonamide (52):
Compound 52 was prepared by procedure A to afford an orange solid (54 mg, 0.170 mmol, 41%). MP 258–260 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 11.33 (s, 1H), 8.11 (s, 1H), 8.05 (d, J = 1.7 Hz, 1H), 7.53 (dd, J = 8.2, 1.8 Hz, 1H), 7.44 (q, J = 2.3 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 6.94 (dt, J = 3.7, 1.7 Hz, 1H), 6.41 (dt, J = 4.1, 2.3 Hz, 1H), 2.61 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 142.1, 129.6, 128.9, 127.1, 127.1, 126.6, 125.9, 122.0, 117.9, 114.7, 112.0, 109.5, 37.7 (2C, s). HRMS m/z [M+H]+ calcd for C15H16N3O3S: 318.0912, found 318.0897, LC tR = 4.10 min, > 98% Purity.
(3Z)-3-(1H-imidazol-5-ylmethylidene)-N,N-dimethyl-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide (53):
Compound 53 was prepared by procedure A to afford an orange solid (52 mg, 0.217 mmol, 52%). MP 275–277 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H), 10.87 (s, 1H), 9.92 (d, J = 1.9 Hz, 1H), 8.07 (t, J = 0.9 Hz, 1H), 8.02 (d, J = 1.1 Hz, 1H), 7.62 (d, J = 0.7 Hz, 1H), 7.58 (dd, J = 8.2, 2.0 Hz, 1H), 7.04 − 6.99 (m, 1H), 2.65 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 170.0, 145.1, 138.3, 136.9, 129.4, 128.4, 127.14, 127.09, 125.9, 123.0, 120.1, 108.9, 37.8. HRMS m/z [M+H]+ calcd for C14H15N4O3S: 319.0865, found 319.0850, LC tR = 2.54 min, > 98% Purity.
(3Z)-5-[(4-methylpiperazin-1-yl)sulfonyl]-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-2-one (54):
Compound 54 was prepared by procedure A to afford a bright yellow solid (82 mg, 0.220 mmol, 65%). MP >260 °C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 11.35 (s, 1H), 8.09 (s, 1H), 8.02 (d, J = 1.7 Hz, 1H), 7.51 (dd, J = 8.2, 1.8 Hz, 1H), 7.44 (q, J = 2.2 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 6.95 (dt, J = 3.6, 1.7 Hz, 1H), 6.41 (dt, J = 4.0, 2.3 Hz, 1H), 2.89 (t, J = 4.7 Hz, 4H), 2.36 (t, J = 4.9 Hz, 4H), 2.13 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 142.2, 129.6, 129.0, 127.2, 127.2, 126.6, 125.9, 122.1, 117.8, 114.6, 112.0, 109.5, 53.5, 45.8 (s, 2C), 45.3 (s, 2C). HRMS m/z [M+H]+ calcd for C18H21N4O3S: 373.1334, found 373.1326, LC tR = 3.10 min, > 98% Purity.
(3Z)-3-(1H-imidazol-5-ylmethylidene)-5-[(4-methylpiperazin-1-yl)sulfonyl]-2,3-dihydro-1H-indol-2-one (55):
Compound 55 was prepared by procedure A to afford a bright yellow solid (67 mg, 0.228 mmol, 67%). MP >260 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 10.85 (s, 1H), 9.96 (d, J = 1.9 Hz, 1H), 7.97 (s, 1H), 7.94 (s, 1H), 7.59 (s, 1H), 7.52 (dd, J = 8.1, 2.0 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 2.95 (s, 4H), 2.36 (d, J = 5.2 Hz, 4H), 2.12 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 170.2, 144.7, 140.9, 137.0, 129.9, 127.8, 127.7, 127.0, 125.6, 123.4, 118.5, 108.6, 53.7 (s, 2C), 45.9 (s, 2C), 45.3. HRMS m/z [M+H]+ calcd for C17H20N5O3S: 374.1287, found 374.1276, LC tR = 2.29 min, > 98% Purity.
(Z)-3-((1H-pyrrol-2-yl)methylene)-5-isopropoxyindolin-2-one (57):
Compound 57 was prepared by procedure A to afford a red solid (48 mg, 0.179 mmol, 68%). MP 149–151 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.39 (s, 1H), 10.68 (s, 1H), 7.76 (s, 1H), 7.34 (td, J = 2.6, 1.3 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 6.79 (dt, J = 3.6, 1.7 Hz, 1H), 6.75 (d, J = 8.3 Hz, 1H), 6.70 (dd, J = 8.4, 2.3 Hz, 1H), 6.35 (dt, J = 3.6, 2.3 Hz, 1H), 4.54 (p, J = 6.0 Hz, 1H), 1.26 (s, 3H), 1.25 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 152.8, 132.9, 129.5, 126.6, 126.2, 125.5, 120.1, 117.4, 115.4, 111.4, 110.0, 107.0, 70.1, 22.0 (s, 2C). HRMS m/z [M+H]+ calcd for C16H17N2O2: 269.1290, found 269.1283, LC tR = 4.97 min, > 98% Purity.
(3Z)-3-(1H-imidazol-5-ylmethylidene)-5-(propan-2-yloxy)-2,3-dihydro-1H-indol-2-one (58):
Compound 58 was prepared by procedure A to afford an orange solid (37 mg, 0.137 mmol, 53%). MP 211–213 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.79 (s, 1H), 10.81 (s, 1H), 8.01 (s, 1H), 7.87 (s, 1H), 7.59 (s, 1H), 7.36 − 7.34 (m, 1H), 6.76 (d, J = 3.0 Hz, 2H), 4.57 − 4.51 (m, 1H), 1.27 (s, 3H), 1.25 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 13C NMR (100 MHz, DMSO-d6) δ 169.1, 153.0, 139.5, 138.1, 133.6, 128.1, 125.4, 123.1, 120.6, 116.5, 110.3, 107.7, 70.1, 22.0. HRMS m/z [M+H]+ calcd for C15H16N3O2: 270.1243, found 270.1230, LC tR = 3.42 min, > 98% Purity.
(3Z)-3-(1H-imidazol-2-ylmethylidene)-5-(propan-2-yloxy)-2,3-dihydro-1H-indol-2-one (59):
Compound 59 was prepared by procedure A to afford a bright orange solid (44 mg, 0.163 mmol, 62%). MP 256–258 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 9.17 (d, J = 2.6 Hz, 1H), 7.85 (s, 1H), 7.55 − 7.34 (m, 3H), 6.79 (d, J = 1.4 Hz, 2H), 4.59 (p, J = 6.0 Hz, 1H), 1.30 − 1.25 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 169.0, 153.2, 143.7, 133.7, 132.5, 125.0, 124.7, 124.2, 120.6, 117.4, 110.6, 108.3, 69.9, 22.0 (s, 2C). HRMS m/z [M+H]+ calcd for C15H16N3O2: 270.1243, found 270.1230, LC tR = 3.29 min, > 98% Purity.
N-(2-oxoindolin-5-yl)butyramide (60):
Compound 60 was prepared by procedure F to afford a colourless solid (552 mg, 2.531 mmol, 75%). MP 165–167 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.26 (s, 1H), 9.70 (s, 1H), 7.55 − 7.47 (m, 1H), 7.32 (dd, J = 8.3, 2.0 Hz, 1H), 6.71 (d, J = 8.3 Hz, 1H), 3.44 (s, 2H), 2.23 (t, J = 7.3 Hz, 2H), 1.59 (h, J = 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 170.6, 139.1, 133.5, 126.0, 118.4, 116.5, 108.8, 38.3, 36.1, 18.7, 13.6. HRMS m/z [M+H]+ calcd for C12H15N2O2: 219.1134, found 219.1125, LC tR = 2.51 min, > 98% Purity.
2-ethyl-N-(2-oxoindolin-5-yl)butanamide (61):
Compound 61 was prepared by procedure F to afford a colourless solid (590 mg, 2.395 mmol, 71%). MP 228–230 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 9.68 (s, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 3.45 (s, 2H), 2.16 (tt, J = 9.4, 5.1 Hz, 1H), 1.59 − 1.48 (m, 2H), 1.47 − 1.36 (m, 2H), 0.84 (t, J = 7.4 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 13C NMR (100 MHz, DMSO-d6) δ 176.3, 173.4, 139.2, 133.3, 126.0, 118.69, 116.8, 108.8, 49.8, 36.0, 25.3 (s, 2C), 11.9 (s, 2C). HRMS m/z [M+H]+ calcd for C14H19N2O2: 247.1447, found 247.1436, LC tR = 3.06 min, > 98% Purity.
N-(2-oxoindolin-5-yl)cyclohexanecarboxamide (62):
Compound 62 was prepared by procedure F to afford a colourless solid (780 mg, 2.632 mmol, 78%). MP 223–225 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.25 (s, 1H), 9.62 (s, 1H), 7.57 − 7.45 (m, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H), 6.70 (d, J = 8.3 Hz, 1H), 3.44 (s, 2H), 2.27 (tt, J = 11.7, 3.3 Hz, 1H), 2.03 − 1.67 (m, 4H), 1.68 − 1.60 (m, 1H), 1.39 (qd, J = 13.4, 12.9, 3.7 Hz, 2H), 1.29 − 1.13 (m, 3H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 173.8, 139.0, 133.6, 125.9, 118.4, 116.5, 108.8, 44.8, 36.0, 29.2 (s, 2C), 25.42, 25.26 (s, 2C). HRMS m/z [M+H]+ calcd for C15H19N2O2: 259.1447, found 259.1435, LC tR = 3.25 min, > 98% Purity.
N-(2-oxoindolin-5-yl)benzamide (63):
Compound 63 was prepared by procedure F to afford a colourless solid (647 mg, 2.565 mmol, 76%). MP 115–117 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.37 (s, 1H), 10.14 (s, 1H), 7.99 − 7.90 (m, 2H), 7.67 (d, J = 2.0 Hz, 1H), 7.59 − 7.55 (m, 1H), 7.55 − 7.44 (m, 3H), 6.79 (d, J = 8.3 Hz, 1H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) 176.4, 165.2, 139.8, 135.1, 133.2, 131.4, 128.4 (s, 2C), 127.6 (s, 2C), 125.9, 119.9, 117.8, 108.8, 36.1. HRMS m/z [M+H]+ calcd for C15H13N2O2: 253.0977, found 253.0967, LC tR = 2.93 min, > 98% Purity.
N-(2-oxoindolin-5-yl)furan-2-carboxamide (64):
Compound 55 was prepared by procedure F to afford a beige solid (605 mg, 2.498 mmol, 74%). 1H NMR (600 MHz, DMSO-d6) δ 10.35 (s, 1H), 10.07 (s, 1H), 7.90 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 2.2 Hz, 1H), 7.49 (dd, J = 8.4, 2.1 Hz, 1H), 7.29 (d, J = 3.4 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 6.68 (dd, J = 3.5, 1.7 Hz, 1H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 156.0, 147.7, 145.5, 139.9, 132.4, 126.0, 120.0, 117.8, 114.3, 112.1, 108.8, 36.1. HRMS m/z [M+H]+ calcd for C13H11N2O3: 243.0770, found 243.0759, LC tR = 2.51 min, > 98% Purity.
N-[(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]butanamide (65):
Compound 65 was prepared by procedure A to afford a bright yellow solid (85 mg, 0.288 mmol, 63%). MP 260–262 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 10.80 (s, 1H), 9.74 (s, 1H), 7.95 (d, J = 2.0 Hz, 1H), 7.59 (s, 1H), 7.35 (q, J = 2.2 Hz, 1H), 7.17 (dd, J = 8.3, 2.0 Hz, 1H), 6.92 (dt, J = 3.5, 1.6 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.35 (dt, J = 3.7, 2.3 Hz, 1H), 2.27 (t, J = 7.3 Hz, 2H), 1.62 (h, J = 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.7, 169.3, 134.8, 133.5, 129.5, 126.1, 125.7, 125.1, 120.7, 118.8, 117.0, 111.4, 110.5, 109.4, 38.2, 18.7, 13.7. HRMS m/z [M+H]+ calcd for C17H18N3O2: 296.1399, found 296.1392, LC tR = 4.14 min, > 98% Purity.
2-ethyl-N-[(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]butanamide (66):
Compound 66 was prepared by procedure A to afford a yellow solid (86 mg, 0.266 mmol, 66%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 10.79 (s, 1H), 9.72 (s, 1H), 8.02 − 7.92 (m, 1H), 7.60 (s, 1H), 7.33 (td, J = 2.6, 1.4 Hz, 1H), 7.16 (dd, J = 8.3, 2.0 Hz, 1H), 6.90 (dt, J = 3.6, 1.7 Hz, 1H), 6.83 − 6.73 (m, 1H), 6.33 (dt, J = 3.7, 2.3 Hz, 1H), 2.18 (tt, J = 9.4, 5.1 Hz, 1H), 1.55 (ddq, J = 13.2, 9.0, 7.4 Hz, 2H), 1.48 − 1.35 (m, 2H), 0.86 (t, J = 7.4 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 173.5, 169.3, 134.9, 133.3, 129.5, 126.2, 125.7, 125.1, 120.7, 119.1, 117.0, 111.4, 110.8, 109.4, 49.8, 25.3 (2C, s), 11.9 (2C, s). HRMS m/z [M+H]+ calcd for C19H22N3O2: 324.1712, found 324.1702, LC tR = 4.56 min, > 98% Purity.
2-ethyl-N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]butanamide (67):
Compound 67 was prepared by procedure A to afford a yellow solid (70 mg, 0.216 mmol, 53%). MP 285–287 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.74 (s, 1H), 9.84 (s, 1H), 8.02 (s, 1H), 7.98 (s, 1H), 7.85 (d, J = 22.8 Hz, 1H), 7.71 (s, 1H), 7.24 (dd, J = 8.2, 2.0 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 2.21 (tt, J = 9.5, 5.0 Hz, 1H), 1.56 (dddd, J = 13.5, 7.3, 5.8, 1.9 Hz, 2H), 1.44 (ddd, J = 13.0, 7.3, 5.2 Hz, 2H), 0.87 (t, J = 7.4 Hz, 6H). 13C NMR (150 MHz, DMSO-d6) δ 174.4, 173.6, 140.3, 135.8, 133.5, 132.5, 124.4, 121.2, 120.4, 120.0, 111.5, 109.6, 108.2, 49.7, 25.3 (s, 2C), 11.9 (s, 2C). HRMS m/z [M+H]+ calcd for C18H21N4O2: 325.1665, found 325.1660, LC tR = 3.29 min, > 98% Purity.
2-ethyl-N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]butanamide (68):
Compound 68 was prepared by procedure A to afford a yellow solid (74 mg, 0.228 mmol, 56%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 14.06 (s, 1H), 11.08 (s, 1H), 9.80 (s, 1H), 7.99 (s, 1H), 7.56 (d, J = 4.2 Hz, 2H), 7.46 − 7.27 (m, 2H), 6.86 (d, J = 8.2 Hz, 1H), 2.20 (tt, J = 9.8, 5.0 Hz, 1H), 1.56 (dq, J = 15.5, 7.6 Hz, 2H), 1.45 (dq, J = 13.2, 6.7, 6.0 Hz, 2H), 0.87 (t, J = 7.5 Hz, 6H). 13C NMR (150 MHz, DMSO-d6) δ 173.7, 169.0, 143.4, 135.9, 133.8, 132.6, 124.0, 123.8, 123.7, 121.1, 120.9, 112.0, 110.0, 49.8, 25.3, 11.9. HRMS m/z [M+H]+ calcd for C18H21N4O2: 325.1665, found 325.1656, LC tR = 3.24 min, > 98% Purity.
N-[(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]cyclohexanecarboxamide (69):
Compound 69 was prepared by procedure A to afford a yellow solid (71 mg, 0.274 mmol, 71%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.37 − 13.27 (m, 1H), 10.78 (s, 1H), 9.66 (s, 1H), 7.96 (d, J = 1.9 Hz, 1H), 7.57 (s, 1H), 7.33 (td, J = 2.6, 1.3 Hz, 1H), 7.14 (dd, J = 8.3, 2.0 Hz, 1H), 6.89 (dt, J = 3.7, 1.7 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 6.33 (dt, J = 3.7, 2.3 Hz, 1H), 2.33 − 2.25 (m, 1H), 1.76 (td, J = 13.9, 3.4 Hz, 4H), 1.64 (d, J = 11.3 Hz, 1H), 1.41 (qd, J = 13.6, 13.1, 3.5 Hz, 2H), 1.31 − 1.14 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.9, 169.3, 134.8, 133.6, 129.5, 126.1, 125.7, 125.0, 120.6, 118.8, 117.0, 111.4, 110.6, 109.4, 44.7, 29.2 (2C, s), 25.4, 25.3 (2C, s). HRMS m/z [M+H]+ calcd for C20H22N3O2: 336.1712, found 336.1703, LC tR = 4.71 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]cyclohexanecarboxamide (70):
Compound 70 was prepared by procedure A to afford a yellow solid (89 mg, 0.265 mmol, 68%). >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.70 (s, 1H), 10.89 (s, 1H), 9.71 (s, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.70 (s, 2H), 7.21 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 2.31 (tt, J = 11.7, 3.4 Hz, 1H), 2.08 − 1.67 (m, 4H), 1.65 (dd, J = 10.5, 4.0 Hz, 1H), 1.42 (qd, J = 13.7, 13.1, 3.5 Hz, 2H), 1.35 − 1.06 (m, 3H). 13C NMR (100 MHz, DMSO-d6) δ 173.9, 168.9, 146.2, 139.5, 138.6, 135.5, 133.8, 128.0, 124.3, 123.7, 119.8, 111.2, 109.7, 44.7, 29.2 (2C, s), 25.4, 25.3 (2C, s). HRMS m/z [M+H]+ calcd for C19H21N4O2: 337.1665, found 337.1657, LC tR = 3.47 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]cyclohexanecarboxamide (71):
Compound 71 was prepared by procedure A to afford a yellow solid (81 mg, 0.241 mmol, 62%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 14.05 (s, 1H), 11.07 (s, 1H), 9.74 (s, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 2.1 Hz, 1H), 7.51 (s, 1H), 7.38 (dd, J = 8.4, 2.0 Hz, 1H), 7.34 (t, J = 1.2 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 2.31 (tt, J = 11.7, 3.5 Hz, 1H), 1.78 (ddt, J = 23.2, 12.4, 3.3 Hz, 4H), 1.68 − 1.63 (m, 1H), 1.42 (qd, J = 12.5, 3.2 Hz, 2H), 1.27 (qt, J = 12.4, 3.1 Hz, 2H), 1.20 (tt, J = 12.5, 3.2 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 174.0, 168.9, 143.4, 135.7, 134.1, 132.6, 123.8, 123.8, 123.8, 120.9, 120.8, 111.6, 110.0, 44.8, 29.2 (2C, s), 25.4, 25.3 (2C, s). HRMS m/z [M+H]+ calcd for C19H21N4O2: 337.1665, found 337.1652, LC tR = 2.88 min, > 98% Purity.
N-[(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]benzamide (72):
Compound 55 was prepared by procedure A to afford a mustard solid (60 mg, 0.182 mmol, 46%). MP 254–256 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 10.87 (s, 1H), 10.17 (s, 1H), 8.08 (d, J = 1.9 Hz, 1H), 8.03 − 7.94 (m, 2H), 7.65 (s, 1H), 7.62 − 7.50 (m, 3H), 7.43 − 7.33 (m, 2H), 6.92 (dt, J = 3.6, 1.7 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.36 (dt, J = 3.7, 2.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.3, 165.1, 135.4, 135.0, 133.1, 131.4, 129.5, 128.4 (s, 2C), 127.5 (s, 2C), 126.2, 125.8, 125.1, 120.7, 120.3, 116.9, 112.0, 111.5, 109.4. HRMS m/z [M+H]+ calcd for C20H16N3O2: 330.1242, found 330.1234, LC tR = 4.45 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (73):
Compound 73 was prepared by procedure A to afford an orange solid (78 mg, 0.236 mmol, 60%, E:Z ratio 43:57 - not able to distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.25 (bs, 1H), 10.19 (bs, 1H), 8.11 (d, J = 2.1 Hz, 1H), 8.01 − 7.95 (m, 3H), 7.84 (s, 1H), 7.74 (s, 1H), 7.55 − 7.49 (m, 3H), 7.45 (dd, J = 8.3, 2.0 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H). Minor diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.32 (bs, 1H), 9.31 (bs, 1H), 8.00 − 7.98 (m, 1H), 7.81 (s, 1H), 7.59 − 7.55 (m, 2H), 7.53 − 7.50 (m, 3H), 7.49 (s, 1H), 7.40 (dd, J = 8.2, 2.2 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H). HRMS m/z [M+H]+ calcd for C19H15N4O2: 331.1195, found 331.1181, LC tR = 3.20 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (74):
Compound 74 was prepared by procedure A to afford an orange solid (89 mg, 0.269 mmol, 68%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.22 (s, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.98 (dd, J = 7.0, 1.9 Hz, 2H), 7.62 − 7.51 (m, 6H), 7.35 (d, J = 1.4 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.0, 165.3, 143.4, 136.4, 134.9, 133.6, 132.6, 131.5, 128.4 (s, 2C), 127.6 (s, 2C), 124.0, 123.8, 123.7, 122.3, 120.9, 113.3, 109.9. HRMS m/z [M+H]+ calcd for C19H15N4O2: 331.1195, found 331.1186, LC tR = 3.23 min, > 98% Purity.
N-[(3Z)-2-oxo-3-(1H-pyrrol-2-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]furan-2-carboxamide (75):
Compound 75 was prepared by procedure A to afford an orange solid (74 mg, 0.232 mmol, 56%). MP 226–228 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.38 (s, 1H), 10.94 (s, 1H), 10.13 (s, 1H), 8.01 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 1.7 Hz, 1H), 7.64 (s, 1H), 7.39 (dd, J = 8.3, 2.0 Hz, 1H), 7.37 (t, J = 1.9 Hz, 1H), 7.33 (d, J = 3.5 Hz, 1H), 6.91 (dd, J = 3.6, 1.4 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.71 (dd, J = 3.5, 1.8 Hz, 1H), 6.36 (dd, J = 3.7, 2.4 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 174.0, 169.4, 156.1, 147.7, 145.5, 135.8, 132.3, 129.5, 126.2, 125.8, 125.2, 120.6, 120.3, 117.0, 114.2, 112.1, 111.5, 109.5. HRMS m/z [M+H]+ calcd for C18H14N3O3: 320.1035, found 320.1018, LC tR = 3.90 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]furan-2-carboxamide (76):
Compound 76 was prepared by procedure A to afford an orange solid (72 mg, 0.225 mmol, 54%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 14.06 (s, 1H), 11.14 (s, 1H), 10.14 (s, 1H), 8.06 (d, J = 2.0 Hz, 1H), 7.94 (dd, J = 1.7, 0.8 Hz, 1H), 7.59 (s, 1H), 7.58 − 7.52 (m, 2H), 7.35 (t, J = 1.1 Hz, 1H), 7.31 (dd, J = 3.5, 0.8 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.71 (dd, J = 3.5, 1.7 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 13C NMR (150 MHz, DMSO) δ 169.0, 156.2, 147.6, 145.6, 143.4, 136.5, 132.9, 132.6, 124.1, 123.9, 123.6, 122.3, 121.0, 114.4, 113.3, 112.1, 110.0. HRMS m/z [M+H]+ calcd for C17H13N4O3: 321.0988, found 321.0979, LC tR = 2.35 min, > 98% Purity.
4-cyano-N-(2-oxoindolin-5-yl)benzamide (77):
Compound 77 was prepared by procedure F to afford a colourless solid (786 mg, 2.835 mmol, 84%). MP >240 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 10.35 (s, 1H), 8.08 (d, J = 8.1 Hz, 2H), 8.01 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 2.1 Hz, 1H), 7.52 (dd, J = 8.3, 2.1 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 163.7, 140.1, 139.1, 132.7, 132.4 (s, 2C), 128.4 (s, 2C), 126.0, 120.0, 118.4, 117.8, 113.7, 108.9, 36.1. HRMS m/z [M+H]+ calcd for C16H12N3O2: 278.0929, found 278.0920, LC tR = 2.90 min, > 98% Purity.
3-cyano-N-(2-oxoindolin-5-yl)benzamide (78):
Compound 78 was prepared by procedure F to afford a colourless solid (814 mg, 2.936 mmol, 87%). MP 214–216 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 10.30 (s, 1H), 8.37 (t, J = 1.7 Hz, 1H), 8.23 (dt, J = 7.9, 1.4 Hz, 1H), 8.04 (dt, J = 7.7, 1.4 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.66 (d, J = 2.1 Hz, 1H), 7.51 (dd, J = 8.4, 2.1 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 163.2, 140.1, 136.0, 134.8, 132.7, 132.4, 131.2, 129.8, 126.1, 120.0, 118.4, 117.7, 111.5, 108.9, 36.1. HRMS m/z [M+H]+ calcd for C16H12N3O2: 278.0929, found 278.0919, LC tR = 2.92 min, > 98% Purity.
2-cyano-N-(2-oxoindolin-5-yl)benzamide (79):
Compound 79 was prepared by procedure F to afford a colourless solid (664 mg, 2.395 mmol, 71%). MP 218–220 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.53 (s, 1H), 10.18 (s, 1H), 8.23 (s, 1H), 7.91 (ddd, J = 33.5, 5.4, 3.1 Hz, 1H), 7.85 (q, J = 7.8 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.26 − 7.19 (m, 2H), 6.93 (d, J = 8.2 Hz, 1H), 3.55 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.5, 167.3, 143.7, 134.6, 133.6, 132.5, 131.6, 127.8, 127.2, 126.4, 124.7, 124.0, 123.3, 122.8, 109.1, 35.9. HRMS m/z [M+H]+ calcd for C16H12N3O2: 278.0929, found 278.0921, LC tR = 3.08 min, > 98% Purity.
4-methoxy-N-(2-oxoindolin-5-yl)benzamide (80):
Compound 80 was prepared by procedure F to afford a colourless solid (738 mg, 2.614 mmol, 77%). MP 125–127 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.33 (s, 1H), 9.96 (s, 1H), 7.97 − 7.91 (m, 2H), 7.68 − 7.64 (m, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.08 − 7.01 (m, 2H), 6.79 (d, J = 8.3 Hz, 1H), 3.83 (s, 3H), 3.49 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 13C NMR (151 MHz, DMSO) δ 176.4, 164.6, 161.8, 139.6, 133.3, 129.5 (s, 2C), 127.1, 125.9, 120.0, 117.9, 113.6 (s, 2C), 108.8, 55.4, 36.1. HRMS m/z [M+H]+ calcd for C16H15N2O3: 283.1083, found 283.1072, LC tR = 3.01 min, > 98% Purity.
3-methoxy-N-(2-oxoindolin-5-yl)benzamide (81):
Compound 81 was prepared by procedure F to afford a colourless solid (820 mg, 2.905 mmol, 86%). MP 125–127 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 10.08 (s, 1H), 7.65 (s, 1H), 7.52 (dq, J = 7.8, 1.8, 1.2 Hz, 2H), 7.48 − 7.46 (m, 1H), 7.43 (t, J = 7.9 Hz, 1H), 7.14 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 3.83 (s, 3H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 164.9, 159.2, 139.8, 136.5, 133.1, 129.5, 125.9, 120.0, 119.8, 117.9, 117.1, 112.8, 108.8, 55.3, 36.1. HRMS m/z [M+H]+ calcd for C16H15N2O3: 283.1083, found 283.1073, LC tR = 3.08 min, > 98% Purity.
2-methoxy-N-(2-oxoindolin-5-yl)benzamide (82):
Compound 82 was prepared by procedure F to afford a colourless oil (676 mg, 2.395 mmol, 71%). 1H NMR (600 MHz, DMSO-d6) δ 10.33 (s, 1H), 9.98 (s, 1H), 7.65 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 7.5, 1.8 Hz, 1H), 7.49 (tdd, J = 8.8, 6.9, 2.0 Hz, 2H), 7.17 (d, J = 8.4 Hz, 1H), 7.06 (td, J = 7.5, 0.9 Hz, 1H), 6.77 (d, J = 8.3 Hz, 1H), 3.89 (s, 3H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 164.1, 156.5, 139.6, 133.2, 132.0, 129.7, 126.1, 125.0, 120.5, 119.2, 117.0, 112.0, 108.9, 55.9, 36.1. HRMS m/z [M+H]+ calcd for C16H15N2O3: 283.1083, found 283.1072, LC tR = 3.21 min, > 98% Purity.
4-fluoro-N-(2-oxoindolin-5-yl)benzamide (83):
Compound 83 was prepared by procedure F to afford a colourless solid (812 mg, 3.005 mmol, 89%). MP 150–152 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.38 (s, 1H), 10.17 (s, 1H), 8.04 − 7.99 (m, 2H), 7.65 (d, J = 2.0 Hz, 1H), 7.50 (dd, J = 8.4, 2.1 Hz, 1H), 7.35 (t, J = 8.8 Hz, 2H), 6.80 (d, J = 8.3 Hz, 1H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 164.1, 164.0 (d, J = 248.8 Hz), 139.9, 133.1, 131.5 (d, J = 3.0 Hz), 130.3 (d, J = 9.0 Hz, 2C), 126.0, 120.1, 117.9, 115.3 (d, J = 21.8 Hz, 2C), 108.8, 36.1. HRMS m/z [M+H]+ calcd for C15H12FN2O2: 271.0883, found 271.0872, LC tR = 3.09 min, > 98% Purity.
3-fluoro-N-(2-oxoindolin-5-yl)benzamide (84):
Compound 84 was prepared by procedure F to afford a colourless solid (830 mg, 3.071 mmol, 91%). MP 89–91 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.40 (s, 1H), 10.23 (s, 1H), 7.80 (dd, J = 7.8, 1.3 Hz, 1H), 7.75 (dt, J = 9.8, 2.1 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.57 (td, J = 8.0, 5.8 Hz, 1H), 7.52 (dd, J = 8.4, 2.1 Hz, 1H), 7.43 (td, J = 8.5, 2.6 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 163.7 (d, J = 2.3 Hz), 161.9 (d, J = 244.2 Hz), 140.0, 137.4 (d, J = 6.8 Hz), 132.9, 130.6 (d, J = 8.1 Hz), 126.0, 123.8 (d, J = 2.7 Hz), 120.1, 118.3 (d, J = 21.2 Hz), 117.9, 114.4 (d, J = 22.9 Hz), 108.8, 36.1. HRMS m/z [M+H]+ calcd for C15H12FN2O2: 271.0883, found 271.0873, LC tR = 3.12 min, > 98% Purity.
2-fluoro-N-(2-oxoindolin-5-yl)benzamide (85):
Compound 85 was prepared by procedure F to afford a colourless solid (438 mg, 1.621 mmol, 48%). MP 85–87 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 10.26 (s, 1H), 7.74 − 7.59 (m, 2H), 7.56 (d, J = 9.0 Hz, 1H), 7.48 (d, J = 8.2 Hz, 1H), 7.33 (t, J = 9.8 Hz, 2H), 6.79 (t, J = 6.5 Hz, 1H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 162.4, 158.9 (d, J = 248.2 Hz), 139.9, 133.0, 132.3 (d, J = 8.2 Hz), 129.9 (d, J = 2.8 Hz), 126.1, 125.21 (d, J = 15.3 Hz), 124.6 (d, J = 3.1 Hz), 119.2, 117.0, 116.2 (d, J = 21.8 Hz), 108.9, 36.1. HRMS m/z [M+H]+ calcd for C15H12FN2O2: 271.0883, found 271.0866, LC tR = 3.14 min, > 98% Purity.
N-(2-oxoindolin-5-yl)-4-(trifluoromethyl)benzamide (86):
Compound 86 was prepared by procedure F to afford a colourless solid (897 mg, 2.801 mmol, 83%). MP 140–142 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.34 (s, 1H), 7.84 (dd, J = 8.0, 1.3 Hz, 1H), 7.78 (t, J = 7.5 Hz, 1H), 7.75 − 7.63 (m, 2H), 7.60 (d, J = 2.0 Hz, 1H), 7.43 (dd, J = 8.4, 2.1 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 176.4, 165.2, 139.9, 136.4 (q, J = 2.2 Hz), 133.0, 131.2 (d, J = 272.0 Hz), 128.5, 126.3 (m, 1C), 126.2, 125.8 (d, J = 31.3 Hz), 125.2, 122.5, 119.1, 116.9, 108.9, 36.1. HRMS m/z [M+H]+ calcd for C16H12F3N2O2: 321.0851, found 321.0841, LC tR = 3.69 min, > 98% Purity.
N-(2-oxoindolin-5-yl)-2-(trifluoromethyl)benzamide (87):
Compound 87 was prepared by procedure F to afford a colourless solid (778 mg, 2.429 mmol, 72%). MP >240 °C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 10.34 (s, 1H), 7.84 (dd, J = 7.9, 1.2 Hz, 1H), 7.81 − 7.75 (m, 1H), 7.73 − 7.64 (m, 2H), 7.60 (d, J = 2.0 Hz, 1H), 7.44 (dd, J = 8.4, 2.1 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 176.4, 165.2, 139.9, 136.5 (q, J = 2.4 Hz), 133.0, 132.6, 129.9, 128.5, 126.3 (q, J = 4.5 Hz), 126.2, 125.8 (d, J = 31.3 Hz), 123.8 (d, J = 273.9 Hz), 119.1, 116.9, 108.9, 36.1. HRMS m/z [M+H]+ calcd for C16H12F3N2O2: 321.0851, found 321.0839, LC tR = 3.23 min, > 98% Purity.
4-cyano-N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (88):
Compound 88 was prepared by procedure A to afford a red/orange solid (68 mg, 0.191 mmol, 53%, E:Z ratio 50:50 - not able to distinguish E/Z isomers). MP >300 °C; Diastereoisomer 1. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-4-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.50 (s, 1H), 9.48 (d, J = 2.2 Hz, 1H), 8.14 − 8.12 (m, 2H), 8.05 − 8.02 (m, 2H), 7.60 (s, 1H), 7.58 (s, 1H), 7.35 (bs, 1H), 6.94 (d, J = 8.3 Hz, 1H). Diastereoisomer 2. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-4-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.55 (s, 1H), 10.46 (s, 1H), 8.14 − 8.12 (m, 2H), 8.12 − 8.10 (m, 1H), 8.05 − 8.02 (m, 2H), 7.56 (s, 2H), 7.52 (bs, 1H), 7.41 (s, 1H), 7.39 (bs, 1H), 6.86 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C20H14N5O2: 356.1148, found 356.1138, LC tR = 3.13 min, > 98% Purity.
3-cyano-N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (89):
Compound 89 was prepared by procedure A to afford a red/orange solid (73 mg, 0.205 mmol, 73%, E:Z ratio 43:57 - not able to distinguish E/Z isomers). MP >250 °C (decomp.); Major diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-3-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (bs, 1H), 10.55 (s, 1H), 9.48 (d, J = 2.2 Hz, 1H), 8.43 (s, 1H), 8.29 − 8.26 (m, 1H), 8.07 (tt, J = 7.9, 1.4 Hz, 2H), 7.78 − 7.74 (m, 1H), 7.58 (m, 1H), 7.58 − 7.56 (m, 1H), 7.41 (s, 1H), 6.86 (d, J = 8.3 Hz, 1H). Minor diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2oxoindolin-5-yl)-3-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 14.06 (bs, 1H), 11.18 (bs, 1H), 10.45 (s, 1H), 8.43 − 8.42 (m, 1H), 8.29 − 8.26 (m, 1H), 8.10 (d, J = 2.1 Hz, 1H), 8.06 − 7.98 (m, 1H), 7.78 − 7.74 (m, 1H), 7.60 (s, 1H), 7.57 − 7.56 (m, 1H), 7.41 (s, 1H), 7.36 (s, 1H), 6.95 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C20H14N5O2: 356.1148, found 356.1142, LC tR = 3.13 min, > 98% Purity.
3-cyano-N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (90):
Compound 90 was prepared by procedure A to afford a yellow/mustard solid (69 mg, 0.194 mmol, 54%). MP >250 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 13.70 (s, 1H), 11.02 (s, 1H), 10.40 (s, 1H), 8.42 (t, J = 1.7 Hz, 1H), 8.27 (dt, J = 8.0, 1.5 Hz, 1H), 8.13 (d, J = 2.0 Hz, 1H), 8.07 (dt, J = 7.7, 1.4 Hz, 1H), 8.03 (s, 1H), 7.79 (d, J = 3.7 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.73 (s, 1H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.1, 163.3, 139.7, 138.9, 136.4, 135.9, 134.9, 133.0, 132.4, 131.2, 129.9, 128.1, 124.4, 123.0, 121.2, 120.0, 118.4, 112.6, 111.5, 109.8. HRMS m/z [M+H]+ calcd for C20H14N5O2: 356.1148, found 356.1140, LC tR = 3.18 min, > 98% Purity.
2-cyano-N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (91):
Compound 91 was prepared by procedure A to afford an orange solid (76 mg, 0.214 mmol, 59%, E:Z ratio 25:75 - not able to distinguish E/Z isomers ). MP >300 °C; (Z)-N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-2-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 13.28 (bs, 1H), 11.07 (bs, 1H), 8.18 (s, 1H), 7.78 − 7.77 (m, 1H), 7.77 (s, 1H), 7.33 (s, 1H), 6.87 (s, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.35 − 6.34 (m, 1H). (E)-N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-2-cyanobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 11.91 (bs, 1H), 10.59 (bs, 1H), 8.59 (s, 1H), 7.81 (dd, J = 8.0, 1.4 Hz, 1H), 7.52 (s, 1H), 7.20 (s, 1H), 7.10 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.41 (s, 1H). HRMS m/z [M+H]+ calcd for C20H14N5O2: 356.1148, found 356.1135, LC tR = 2.45 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-4-methoxybenzamide (92):
Compound 92 was prepared by procedure A to afford a yellow solid (70 mg, 0.194 mmol, 55%). MP 275–277 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.69 (s, 1H), 10.91 (s, 1H), 10.04 (s, 1H), 8.10 (s, 1H), 8.00 (s, 1H), 7.99 − 7.95 (m, 2H), 7.74 (s, 2H), 7.43 (dd, J = 8.3, 2.0 Hz, 1H), 7.09 − 7.05 (m, 2H), 6.87 (d, J = 8.3 Hz, 1H), 3.84 (s, 3H). 1H NMR (400 MHz, DMSO-d6) δ 13C NMR (150 MHz, DMSO-d6) δ 176.3, 168.9, 164.6, 161.8, 136.0, 133.5, 129.4 (s, 2C), 127.0, 124.4, 121.3, 120.4, 119.9, 117.8, 113.6 (s, 2C), 112.7, 109.6, 108.7, 55.4. HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1291, LC tR = 2.74 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-4-methoxybenzamide (93):
Compound 93 was prepared by procedure A to afford a bright orange solid (91 mg, 0.253 mmol, 71%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 10.06 (s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 8.02 − 7.94 (m, 2H), 7.66 − 7.49 (m, 3H), 7.36 (s, 1H), 7.12 − 7.04 (m, 2H), 6.95 − 6.87 (m, 1H), 3.84 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.0, 164.7, 161.8, 143.4, 136.2, 133.8, 132.6, 129.5 (2C, s), 126.9, 123.9, 123.8, 123.8, 122.3, 120.9, 113.6 (2C, s), 113.2, 109.9, 55.4. HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1292, LC tR = 3.25 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-3-methoxybenzamide (94):
Compound 94 was prepared by procedure A to afford a yellow solid (98 mg, 0.272 mmol, 70%). MP 138–140 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.71 (s, 1H), 10.99 (s, 1H), 10.18 (s, 1H), 8.12 (d, J = 2.0 Hz, 1H), 8.03 (s, 1H), 7.78 (s, 1H), 7.72 (s, 1H), 7.58 − 7.55 (m, 1H), 7.52 (dd, J = 2.7, 1.5 Hz, 1H), 7.48 − 7.44 (m, 1H), 7.44 − 7.41 (m, 1H), 7.16 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.1, 164.9, 159.2, 139.7, 138.8, 136.3, 136.2, 133.3, 129.6, 128.1, 124.3, 122.9, 121.4, 120.1, 119.8, 117.2, 112.8, 109.7, 55.3. HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1288, LC tR = 2.78 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-3-methoxybenzamide (95):
Compound 95 was prepared by procedure A to afford a red solid (55 mg, 0.153 mmol, 43%, E:Z ratio 37:63 - not able to distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-3- methoxybenzamide. 1H NMR (600 MHz, DMSO-d6) δ 12.90 (bs, 1H), 11.15 (bs, 1H), 10.53 (s, 1H), 10.21 (s, 1H), 9.44 (d, J = 2.2 Hz, 1H), 7.58 − 7.40 (m, 5H), 7.40 (s, 1H), 7.15 (tdd, J = 8.2, 2.7, 0.9 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 3.85 (s, 3H). Minor diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-3-methoxybenzamide. 1H NMR (600 MHz, DMSO-d6) δ 14.07 (s, 1H), 10.19 (s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 7.60 (s, 1H), 7.58 − 7.40 (m, 5H), 7.35 (d, J = 1.2 Hz, 1H), 7.17 − 7.14 (m, 1H), 6.93 (d, J = 8.3 Hz, 1H), 3.84 (s, 3H). HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1291, LC tR = 3.28 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-5-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-2-methoxybenzamide (96):
Compound 96 was prepared by procedure A to afford a mustard/yellow solid (59 mg, 0.164 mmol, 46%). MP >250 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 13.70 (s, 1H), 10.94 (d, J = 74.7 Hz, 1H), 10.44 (s, 1H), 8.21 − 7.99 (m, 6H), 7.76 (s, 2H), 7.52 − 7.39 (m, 1H), 6.90 (d, J = 8.3 Hz, 1H), 3.34 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 206.5, 169.0, 163.8, 139.7, 139.0, 136.5, 132.9, 132.5 (s, 2C) 128.4 (s, 2C) 124.5, 123.0, 121.3, 120.1, 118.4, 113.8, 112.6, 109.7, 30.7. HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1290, LC tR = 2.96 min, > 98% Purity.
N-[(3Z)-3-(1H-imidazol-2-ylmethylidene)-2-oxo-2,3-dihydro-1H-indol-5-yl]-2-methoxybenzamide (97):
Compound 97 was prepared by procedure A to afford a bright red solid (49 mg, 0.136 mmol, 38%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 14.08 (s, 1H), 11.13 (s, 1H), 10.08 (s, 1H), 8.07 (d, J = 2.0 Hz, 1H), 7.74 (dd, J = 7.6, 1.8 Hz, 1H), 7.68 (dd, J = 8.4, 2.0 Hz, 1H), 7.65 (s, 1H), 7.57 (d, J = 2.2 Hz, 1H), 7.53 − 7.51 (m, 1H), 7.36 (t, J = 1.2 Hz, 1H), 7.20 (dd, J = 8.5, 0.9 Hz, 1H), 7.09 (td, J = 7.5, 1.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 169.0, 163.9, 156.6, 143.5, 136.2, 133.7, 132.6, 132.3, 130.0, 124.2, 124.2, 124.0, 123.7, 121.3, 120.9, 120.6, 112.3, 112.1, 110.0, 56.0. HRMS m/z [M+H]+ calcd for C20H17N4O3: 361.1301, found 361.1290, LC tR = 2.84 min, > 98% Purity.
4-fluoro-N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (98):
Compound 98 was prepared by procedure A to afford a yellow/orange solid (57 mg, 0.164 mmol, 44%). MP >250 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 13.72 (s, 1H), 11.00 (s, 1H), 10.22 (s, 1H), 8.12 (s, 1H), 8.07 − 8.04 (m, 2H), 8.03 (s, 1H), 7.75 (d, J = 32.5 Hz, 2H), 7.48 − 7.39 (m, 1H), 7.39 − 7.36 (m, 2H), 6.89 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.1, 164.1, 164.0 (d, J = 250.0 Hz), 139.7, 138.8, 136.3, 133.3, 131.4 (d, J = 2.8 Hz), 130.3 (d, J = 8.9 Hz, 2C), 128.1, 124.4, 122.9, 121.4, 120.2, 115.4 (d, J = 21.8 Hz, 2C), 112.8, 109.7. HRMS m/z [M+H]+ calcd for C19H14FN4O2: 349.1101, found 349.1092, LC tR = 2.80 min, > 98% Purity.
4-fluoro-N-[(3Z)-3-[(1H-imidazol-2-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (99):
Compound 99 was prepared by procedure A to afford an orange solid (87 mg, 0.250 mmol, 68%). MP >300 °C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.23 (s, 1H), 8.14 − 8.01 (m, 3H), 7.59 (s, 1H), 7.56 (dd, J = 8.4, 2.0 Hz, 1H), 7.45 − 7.27 (m, 3H), 6.93 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.0, 164.2, 164.0 (d, J = 249.0 Hz), 143.4, 136.4, 133.5, 132.6, 131.3 (d, J = 3.0 Hz), 130.3, 130.2, 124.0, 123.8 (2C, d, J = 19.9 Hz), 122.3, 121.0, 115.3 (2C, d, J = 21.8 Hz), 113.3, 109.9. HRMS m/z [M+H]+ calcd for C19H14FN4O2: 349.1101, found 349.1091, LC tR = 2.74 min, > 98% Purity.
3-fluoro-N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (100):
Compound 100 was prepared by procedure A to afford an orange solid (67 mg, 0.192 mmol, 52%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.71 (s, 1H), 11.00 (s, 1H), 10.28 (s, 1H), 8.12 (s, 1H), 8.03 (s, 1H), 7.84 (dt, J = 7.8, 1.2 Hz, 1H), 7.83 − 7.75 (m, 2H), 7.73 (s, 1H), 7.60 (td, J = 8.0, 5.8 Hz, 1H), 7.50 − 7.38 (m, 2H), 6.90 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.1, 163.8 (d, J = 2.7 Hz), 162.0 (d, J = 244.4 Hz), 139.7, 138.9, 137.2 (d, J = 6.8 Hz), 136.4, 133.1, 130.6 (d, J = 8.1 Hz), 128.1, 124.4, 123.8 (d, J = 2.6 Hz), 123.0, 121.4, 120.1, 118.4 (d, J = 21.1 Hz), 114.4 (d, J = 22.8 Hz), 112.8, 109.8. HRMS m/z [M+H]+ calcd for C19H14FN4O2: 349.1101, found 349.1090, LC tR = 2.80 min, > 98% Purity.
3-fluoro-N-[(3Z)-3-[(1H-imidazol-2-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (101):
Compound 101 was prepared by procedure A to afford a yellow solid (75 mg, 0.215 mmol, 58%, E:Z ratio 35:65), MP >210 °C (decomp.); (Z)-N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-3-fluorobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.54 (s, 1H), 10.32 (s, 1H), 9.46 (d, J = 2.2 Hz, 1H), 7.86 − 7.83 (m, 1H), 7.81 − 7.77 (m, 1H), 7.59 − 7.56 (m, 1H), 7.53 (dd, J = 8.3, 2.2 Hz, 1H), 7.47 − 7.42 (m, 2H), 7.41 (s, 1H), 6.85 (d, J = 8.3 Hz, 1H). (E)-N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-3-fluorobenzamide. 1H NMR (600 MHz, DMSO-d6) δ 14.06 (s, 1H), 10.29 (s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 7.86 − 7.83 (m, 1H), 7.81 − 7.77 (m, 1H), 7.60 (s, 1H), 7.59 − 7.56 (m, 1H), 7.47 − 7.42 (m, 2H), 7.35 (s, 1H), 6.94 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C19H14FN4O2: 349.1101, found 349.1088, LC tR = 3.33 min, > 98% Purity.
2-fluoro-N-[(3Z)-3-[(1H-imidazol-2-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (102):
Compound 102 was prepared by procedure A to afford an orange solid (48 mg, 0.138 mmol, 37%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 10.33 (s, 1H), 8.09 (d, J = 2.0 Hz, 1H), 7.69 (td, J = 7.4, 1.8 Hz, 1H), 7.62 − 7.57 (m, 2H), 7.54 (dd, J = 8.4, 2.0 Hz, 1H), 7.45 − 7.22 (m, 3H), 6.92 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.0, 162.5, 158.9 (d, J = 248.8 Hz), 143.4, 136.4, 133.4, 132.5 (d, J = 8.4 Hz), 129.9 (d, J = 3.0 Hz), 125.0, 124.9, 124.6 (d, J = 3.4 Hz), 124.1, 123.9, 123.6, 121.5, 121.1–120.9 (m), 116.2 (d, J = 22.0 Hz), 112.4, 110.1. HRMS m/z [M+H]+ calcd for C19H14FN4O2: 349.1101, found 349.1084, LC tR = 2.83 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-4-(trifluoromethyl)benzamide (103):
Compound 100 was prepared by procedure A to afford a yellow solid (58 mg, 0.146 mmol, 47%). MP >300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.71 (s, 1H), 11.01 (s, 1H), 10.44 (s, 1H), 8.17 (d, J = 8.0 Hz, 2H), 8.14 (d, J = 2.0 Hz, 1H), 8.03 − 8.02 (m, 1H), 7.93 (d, J = 8.2 Hz, 2H), 7.80 (s, 1H), 7.73 (s, 1H), 7.43 (dd, J = 8.3, 2.0 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.1, 164.0, 139.7, 138.9, 138.7, 136.4, 133.0, 131.3 (q, J = 32.8, 31.9 Hz), 128.5 (s, 2C), 128.1, 125.5 (dd, J = 4.9, 2.2 Hz, 2C), 124.9, 124.4, 123.0 (d, J = 6.1 Hz), 121.4, 120.1, 112.8, 109.8. HRMS m/z [M+H]+ calcd for C20H14F3N4O2: 399.1069, found 399.1058, LC tR = 3.25 min, > 98% Purity.
(Z)-N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-4-(trifluoromethyl)benzamide (104):
Compound 104 was prepared by procedure A to afford a yellow solid (51 mg, 0.128 mmol, 41%, E:Z ratio 34:66 - not able to distinguish E/Z isomers). MP >210 °C (decomp.); Major diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-4- (trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 12.87 (bs, 1H), 10.55 (bs, 1H), 10.48 (s, 1H), 9.48 (d, J = 2.2 Hz, 1H), 8.18 − 8.17 (m, 2H), 7.92 − 7.91 (m, 2H), 7.61 − 7.56 (m, 2H), 7.41 (s, 1H), 6.86 (d, J = 8.3 Hz, 1H). Minor diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-4- (trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 14.06 (bs, 1H), 11.16 (bs, 1H), 10.45 (s, 1H), 8.18 − 8.17 (m, 2H), 7.97 − 7.92 (m, 2H), 7.61 (s, 1H), 7.46 (bs, 2H), 7.35 (s, 1H), 6.95 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C20H14F3N4O2: 399.1069, found 399.1058, LC tR = 3.20 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-2-(trifluoromethyl)benzamide (105):
Compound 105 was prepared by procedure A to afford a yellow solid (46 mg, 0.116 mmol, 37%, E:Z ratio 38:62 - not able to distinguish E/Z isomers). MP 225–227 °C; Major Diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-2-(trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.47 (bs, 1H), 10.42 (bs, 1H), 9.29 (d, J = 2.1 Hz, 1H), 7.88 (s, 2H), 7.86 − 7.68 (m, 4H), 7.60 (dd, J = 8.3, 2.2 Hz, 1H), 7.50 (s, 1H), 6.82 (d, J = 8.3 Hz, 1H). Minor Diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-2- (trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 10.53 (bs, 1H), 8.09 (d, J = 2.0 Hz, 1H), 8.00 (s, 1H), 7.95 (s, 1H), 7.86 − 7.68 (m, 5H), 7.34 (dd, J = 8.3, 2.0 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C20H14F3N4O2: 399.1069, found 399.1058, LC tR = 2.89 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-2-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-2-(trifluoromethyl)benzamide (106):
Compound 106 was prepared by procedure A to afford a yellow solid (56 mg, 0.141 mmol, 45%, E:Z ratio 34:66 - not able to distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-2-(trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 12.93 (bs, 1H), 10.54 (bs, 1H), 10.50 (bs, 1H), 9.45 (d, J = 2.1 Hz, 1H), 7.87 − 7.83 (m, 1H), 7.81 − 7.77 (m, 1H), 7.73 − 7.68 (m, 3H), 7.50 (s, 1H), 7.41 (s, 1H), 7.36 (s, 1H), 6.85 (d, J = 8.3 Hz, 1H). Minor diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)-2- (trifluoromethyl)benzamide. 1H NMR (600 MHz, DMSO-d6) δ 14.07 (bs, 1H), 11.17 (bs, 1H), 10.51 (bs, 1H), 8.04 (d, J = 2.0 Hz, 1H), 7.81 − 7.77 (m, 1H), 7.70 − 7.68 (m, 3H), 7.58 − 7.57 (m, 2H), 7.52 (d, J = 2.0 Hz, 1H), 7.37 − 7.34 (m, 1H), 6.93 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C20H14F3N4O2: 399.1069, found 399.1057, LC tR = 2.86 min, > 98% Purity.
N-(2-oxoindolin-5-yl)thiophene-2-carboxamide (107):
Compound 107 was prepared by procedure F to afford a red solid (514 mg, 1.991 mmol, 59%). MP 105–107 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 10.10 (s, 1H), 7.97 (dd, J = 3.7, 1.1 Hz, 1H), 7.82 (dd, J = 5.0, 1.1 Hz, 1H), 7.63 − 7.57 (m, 1H), 7.46 (dd, J = 8.4, 2.1 Hz, 1H), 7.21 (dd, J = 5.0, 3.7 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 159.6, 140.3, 139.9, 132.6, 131.5, 128.7, 128.0, 126.1, 120.1, 117.9, 108.9, 36.1. HRMS m/z [M+H]+ calcd for C13H11N2O2S: 259.0541, found 259.0532, LC tR = 2.84 min, > 98% Purity.
3-methoxy-N-(2-oxoindolin-5-yl)thiophene-2-carboxamide (108):
Compound 108 was prepared by procedure F to afford a beige solid (662 mg, 2.296 mmol, 68%). MP 75–77 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.37 (s, 1H), 9.14 (s, 1H), 7.80 (d, J = 5.5 Hz, 1H), 7.59 − 7.55 (m, 1H), 7.43 (dd, J = 8.3, 2.2 Hz, 1H), 7.18 (d, J = 5.6 Hz, 1H), 6.77 (d, J = 8.3 Hz, 1H), 4.06 (s, 3H), 3.48 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 159.1, 156.7, 139.8, 132.2, 130.4, 126.2, 119.6, 117.4, 117.1, 115.6, 108.9, 59.5, 36.0. HRMS m/z [M+H]+ calcd for C14H13N2O3S: 289.0647, found 289.0637, LC tR = 3.05 min, > 98% Purity.
3-ethoxy-N-(2-oxoindolin-5-yl)thiophene-2-carboxamide (109):
Compound 109 was prepared by procedure F to afford a beige solid (796 mg, 2.633 mmol, 78%). MP 98–100 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.33 (s, 1H), 9.20 (s, 1H), 7.78 (d, J = 5.5 Hz, 1H), 7.56 − 7.51 (m, 1H), 7.39 (dd, J = 8.4, 2.1 Hz, 1H), 7.16 (d, J = 5.5 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.49 (s, 2H), 1.45 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 159.0, 155.8, 139.8, 132.3, 130.4, 126.4, 119.0, 117.5, 116.9, 115.9, 109.1, 68.0, 36.1, 14.8. HRMS m/z [M+H]+ calcd for C15H15N2O3S: 303.0803, found 303.0795, LC tR = 3.56 min, > 98% Purity.
3-fluoro-N-(2-oxoindolin-5-yl)thiophene-2-carboxamide (110):
Compound 110 was prepared by procedure F to afford a beige solid (532 mg, 1.926 mmol, 57%). MP 101–103 °C; HRMS m/z [M+H]+ calcd for C13H10FN2O2S: 277.0447, found 277.0438, LC tR = 3.35 min, > 98% Purity.
N-(2-oxoindolin-5-yl)thieno[3,2-b]thiophene-2-carboxamide (111):
Compound 111 was prepared by procedure F to afford a beige solid (366 mg, 1.164 mmol, 69%). MP 125–127 °C; HRMS m/z [M+H]+ calcd for C15H11N2O2S2: 315.0262, found 315.0251, LC tR = 3.45 min, > 98% Purity.
2-fluoro-N-(2-oxoindolin-5-yl)-5-(trifluoromethyl)benzamide (112):
Compound 112 was prepared by procedure F to afford a beige solid (651 mg, 1.925 mmol, 57%). MP 96–97 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.46 (s, 1H), 10.38 (s, 1H), 8.03 (dd, J = 6.2, 2.5 Hz, 1H), 7.97 (dt, J = 7.7, 3.2 Hz, 1H), 7.65 − 7.54 (m, 2H), 7.46 (dd, J = 8.3, 2.1 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 162.0–160.0 (m, 1C), 140.1, 132.6, 129.6 (dd, J = 9.6, 3.9 Hz), 127.3 (p, J = 3.9 Hz), 126.2, 126.1 (d, J = 17.0 Hz), 125.4 (qd, J = 32.8, 3.2 Hz), 124.5, 122.7, 119.4, 117.7 (d, J = 23.5 Hz), 117.2, 109.0, 36.1. HRMS m/z [M+H]+ calcd for C16H11F4N2O2: 339.0757, found 339.0740, LC tR = 3.80 min, > 98% Purity.
N-(2-oxoindolin-5-yl)benzo[d][1,3]dioxole-4-carboxamide (113):
Compound 113 was prepared by procedure F to afford a beige solid (750 mg, 2.531 mmol, 75%). MP 105–107 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.70 (s, 1H), 7.60 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 8.4, 2.0 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 7.10 (d, J = 7.7 Hz, 1H), 6.96 (t, J = 7.9 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 6.15 (s, 2H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.3, 161.8, 147.8, 145.2, 139.9, 132.7, 126.1, 121.7, 121.1, 119.4, 117.7, 117.2, 110.9, 108.90, 101.7, 36.1. HRMS m/z [M+H]+ calcd for C16H13N2O4: 297.0875, found 297.0865, LC tR = 3.18 min, > 98% Purity.
N-(2-oxoindolin-5-yl)thiazole-2-carboxamide (114):
Compound 114 was prepared by procedure F to afford a beige solid (735 mg, 2.835 mmol, 84%). MP 108–110 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.71 (s, 1H), 10.39 (s, 1H), 8.38 (s, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.4, 2.1 Hz, 1H), 7.53 (s, 1H), 6.79 (d, J = 8.4 Hz, 1H), 3.50 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.4, 155.1, 152.9, 142.5, 140.4, 131.8, 128.3, 126.1, 120.3, 117.8, 108.9, 36.0. HRMS m/z [M+H]+ calcd for C12H10N3O2S: 260.0494, found 260.0484, LC tR = 2.72 min, > 98% Purity.
N-(2-oxoindolin-5-yl)oxazole-2-carboxamide (115):
Compound 115 was prepared by procedure F to afford a beige solid (345 mg, 1.418 mmol, 70%). MP 87–89 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.65 (s, 1H), 10.39 (s, 1H), 8.13 − 8.07 (m, 2H), 7.72 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.4, 2.1 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 3.49 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 176.8, 164.6, 157.9, 144.4, 140.7, 132.4, 126.7, 126.5, 120.7, 118.3, 109.3, 36.5. HRMS m/z [M+H]+ calcd for C12H10N3O3: 244.0722, found 244.0709, LC tR = 2.25 min, > 98% Purity.
N-(2-oxoindolin-5-yl)isothiazole-5-carboxamide (116):
Compound 116 was prepared by procedure A to afford a beige solid (328 mg, 1.265 mmol, 75%). MP 178–180 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 10.38 (s, 1H), 8.71 (d, J = 1.8 Hz, 1H), 8.09 (d, J = 1.8 Hz, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 8.4, 2.1 Hz, 1H), 6.82 (d, J = 8.3 Hz, 1H), 3.51 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 176.33, 163.32, 159.18, 157.40, 140.49, 131.77, 126.22, 123.87, 120.32, 117.97, 108.97, 36.03. HRMS m/z [M+H]+ calcd for C12H10N3O2S: 260.0494, found 260.0485, LC tR = 2.57 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]thiophene-2-carboxamide (117):
Compound 117 was prepared by procedure A to afford an orange solid (89 mg, 0.265 mmol, 68%). MP>300 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.71 (s, 1H), 11.00 (s, 1H), 10.22 (s, 1H), 8.06 (d, J = 2.0 Hz, 1H), 8.03 (s, 1H), 8.01 − 8.00 (m, 1H), 7.84 (dd, J = 5.0, 1.1 Hz, 1H), 7.80 (s, 1H), 7.71 (s, 1H), 7.39 (dd, J = 8.3, 2.0 Hz, 1H), 7.23 (dd, J = 5.0, 3.7 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO -d6) δ 169.1, 159.7, 140.2, 139.7, 138.8, 136.3, 132.8, 131.6, 128.8, 128.1 (s, 2C), 124.5, 123.0, 121.4, 120.1, 112.9, 109.8. HRMS m/z [M+H]+ calcd for C17H13N4O2S: 337.0759, found 327.0721, LC tR = 2.78 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-2-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]thiophene-2-carboxamide (118):
Compound 118 was prepared by procedure A to afford an orange solid (92 mg, 0.274 mmol, 71%, E:Z ratio 34:66 - not able to distinguish E/Z isomers). MP >300 °C; Major diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)thiophene-2- carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (bs, 1H), 10.55 (bs, 1H), 10.26 (bs, 1H), 9.42 (d, J = 2.2 Hz, 1H), 8.05 (d, J = 3.9 Hz, 1H), 7.83 (dd, J = 5.0, 1.1 Hz, 1H), 7.49 (dd, J = 8.3, 2.2 Hz, 1H), 7.50 − 7.44 (bs, 2H), 7.41 (s, 1H), 7.22 (dd, J = 5.0, 3.7 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H). Minor diastereoisomer. N-(3-((1H-imidazol-2-yl)methylene)-2-oxoindolin-5-yl)thiophene-2- carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 14.07 (bs, 1H), 11.16 (bs, 1H), 10.22 (bs, 1H), 8.06 (d, J = 2.0 Hz, 1H), 8.01 (dd, J = 3.8, 1.2 Hz, 1H), 7.85 (dd, J = 5.0, 1.1 Hz, 1H), 7.62 (s, 1H), 7.57 (d, J = 2.1 Hz, 1H), 7.52 (dd, J = 8.4, 2.1 Hz, 1H), 7.36 (bt, J = 1.2 Hz, 1H), 7.23 (dd, J = 5.0, 3.7 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H). HRMS m/z [M+H]+ calcd for C17H13N4O2S: 337.0759, found 337.0753, LC tR = 3.07 min, > 98% Purity.
(Z)-N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-3-methoxythiophene-2-carboxamide (119):
Compound 119 was prepared by procedure A to afford an orange solid (45 mg, 0.123 mmol, 35%). MP 268–270 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.72 (s, 1H), 10.99 (s, 1H), 9.20 (s, 1H), 8.03 (s, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.84 − 7.80 (m, 2H), 7.69 (s, 1H), 7.50 − 7.47 (m, 1H), 7.20 (d, J = 5.6 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 4.09 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.1, 159.2, 156.8, 139.7, 138.7, 136.2, 132.5, 130.5, 128.1, 124.6, 123.1, 120.7, 120.0, 117.1, 115.5, 112.3, 109.7, 59.6. HRMS m/z [M+H]+ calcd for C18H15N4O3S: 367.0865, found 367.0854, LC tR = 2.78 min, > 98% Purity.
3-ethoxy-N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]thiophene-2-carboxamide (120):
Compound 120 was prepared by procedure A to afford an orange solid (52 mg, 0.137 mmol, 41%, E:Z ratio 28:72 - not able to distinguish E/Z isomers). MP Decomp. >250 °C; Major diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-3-ethoxythiophene- 2-carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 12.74 (bs, 1H), 10.39 (bs, 1H), 9.32 (s, 1H), 9.23 (bs, 1H), 7.96 − 7.95 (m, 2H), 7.87 (dd, J = 8.3, 2.2 Hz, 1H), 7.81 (d, J = 5.5 Hz, 1H), 7.51 (s, 1H), 7.21 (d, J = 5.5 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 4.41 − 4.37 (m, 2H), 1.59 (t, J = 6.9 Hz, 3H). Minor diastereoisomer. N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)-3-ethoxythiophene- 2-carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 13.73 (bs, 1H), 10.99 (bs, 1H), 9.25 (s, 1H), 8.02 (d, J = 14.3 Hz, 2H), 7.83 (s, 1H), 7.81 − 7.79 (m, 1H), 7.71 (s, 1H), 7.37 (dd, J = 8.4, 2.1 Hz, 1H), 7.19 (d, J = 5.5 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 4.39 − 4.35 (m, 2H), 1.47 (t, J = 7.0 Hz, 3H). HRMS m/z [M+H]+ calcd for C19H17N4O3S: 381.1021, found 381.1007, LC tR = 3.03 min, > 98% Purity.
3-fluoro-N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]thiophene-2-carboxamide (121):
Compound 121 was prepared by procedure A to afford an orange solid (37 mg, 0.104 mmol, 29%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.69 (s, 1H), 11.00 (s, 1H), 9.83 − 9.72 (m, 1H), 8.03 (s, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 5.5, 4.0 Hz, 1H), 7.81 (s, 1H), 7.70 (s, 1H), 7.38 (dd, J = 8.3, 2.0 Hz, 1H), 7.16 (d, J = 5.5 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.10, 157.67, 156.86, 154.22, 139.70, 138.82, 136.49, 132.43, 129.77 (d, J = 9.9 Hz), 128.08, 124.46, 123.10, 121.51, 119.97, 118.30 (d, J = 26.4 Hz), 113.03, 109.72. HRMS m/z [M+H]+ calcd for C17H12FN4O2S: 355.0665, found 355.0652, LC tR = 2.68 min, > 98% Purity.
(Z)-N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)thieno[3,2-b]thiophene-2-carboxamide (122):
Compound 122 was prepared by procedure A to afford a light orange solid (45 mg, 0.110 mmol, 36%). MP >250 °C (decomp.); 1H NMR (600 MHz, DMSO-d6) δ 13.68 (s, 1H), 10.95 (s, 1H), 10.34 (s, 1H), 8.34 (s, 1H), 8.09 (d, J = 2.0 Hz, 1H), 8.01 (s, 1H), 7.89 (d, J = 5.2 Hz, 1H), 7.78 (s, 1H), 7.54 (d, J = 5.2 Hz, 1H), 7.40 (dd, J = 8.2, 2.0 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.0, 160.1, 142.0, 141.6, 139.4, 138.5, 136.3, 132.7, 131.8, 124.5, 121.4 (s, 2C), 121.2, 120.6, 120.4 (s, 2C), 120.3, 112.7, 109.7. HRMS m/z [M+H]+ calcd for C19H13N4O2S2: 393.0480, found 393.0468, LC tR = 3.08 min, > 98% Purity.
2-fluoro-N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-5-(trifluoromethyl)benzamide (123):
Compound 123 was prepared by procedure A to afford an orange solid (21 mg, 0.062 mmol, 21%). MP 202–204 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.73 (s, 1H), 10.99 (s, 1H), 10.75 (s, 1H), 8.22 (s, 1H), 8.03 (s, 1H), 7.85 (d, J = 2.3 Hz, 1H), 7.79 − 7.75 (m, 2H), 7.35 (dd, J = 9.4, 5.8 Hz, 2H), 6.91 (d, J = 8.4 Hz, 1H). 13C NMR (150 MHz, DMSO) δ 169.1, 164.6, 154.3, 139.7, 139.0, 136.1, 133.3, 128.9, 128.1, 126.7, 125.3, 124.6, 123.5, 123.0, 121.7, 121.4, 120.1, 119.8, 111.4, 110.1. HRMS m/z [M+H]+ calcd for C20H13F4N4O2: 417.0975, found 417.0962, LC tR = 3.22 min, > 98% Purity.
(Z)-N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)benzo[d][1,3]dioxole-4-carboxamide (124):
Compound 124 was prepared by procedure A to afford a brown/red solid (47 mg, 0.126 mmol, 37%). MP 189–191 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.71 (s, 1H), 10.96 (s, 1H), 9.72 (s, 1H), 8.30 (s, 1H), 8.06 − 8.03 (m, 1H), 8.00 (d, J = 4.2 Hz, 1H), 7.85 − 7.66 (m, 2H), 7.45 (dt, J = 8.3, 3.1 Hz, 1H), 7.27 (dd, J = 8.1, 1.1 Hz, 1H), 7.12 (dd, J = 7.7, 1.1 Hz, 1H), 6.98 (t, J = 7.9 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 6.18 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 169.0, 161.9, 147.8, 145.3, 139.5, 136.3, 134.6, 132.9, 128.5, 124.6, 121.7, 121.1, 120.6, 120.3, 117.5, 112.0, 111.0, 109.7, 101.7, 43.8. HRMS m/z [M+H]+ calcd for C20H15N4O4: 375.1093, found 375.1081, LC tR = 2.85 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-1,3-thiazole-2-carboxamide (125):
Compound 125 was prepared by procedure A to afford a mustard green solid (60 mg, 0.178 mmol, 46%). MP 271–273 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.70 (s, 1H), 11.01 (s, 1H), 10.70 (s, 1H), 8.17 (s, 1H), 8.13 − 8.10 (m, 2H), 8.03 (s, 1H), 7.74 (d, J = 24.9 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.1, 164.1, 157.6, 157.6, 144.0, 139.7, 138.9, 136.6, 132.2, 126.4, 124.5, 123.0, 121.5, 120.1, 112.7, 109.8. HRMS m/z [M+H]+ calcd for C16H12N5O2S: 338.0712, found 338.0700, LC tR = 2.52 min, > 98% Purity.
N-[(3Z)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-1,3-oxazole-2-carboxamide (126):
Compound 126 was prepared by procedure A to afford a dark orange solid (57 mg, 0.177 mmol, 43%). MP >300 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.68 (s, 1H), 10.99 (s, 1H), 10.73 (s, 1H), 8.38 (dd, J = 6.0, 0.8 Hz, 1H), 8.15 − 7.96 (m, 2H), 7.73 (d, J = 14.6 Hz, 2H), 7.52 (dd, J = 4.0, 0.8 Hz, 1H), 7.47 (d, J = 7.7 Hz, 1H), 6.83 (dd, J = 29.0, 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.1, 155.1, 153.0, 142.6, 139.7, 138.9, 136.7, 132.0, 128.4, 128.0, 124.4, 123.1, 121.5, 119.9, 112.7, 109.8. HRMS m/z [M+H]+ calcd for C16H12N5O3: 322.0940, found 322.0930, LC tR = 2.21 min, > 98% Purity.
N-[(3Z/E)-3-[(1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]-1,2-thiazole-5-carboxamide (127):
Compound 127 was prepared by procedure A to afford an orange solid (48 mg, 0.142 mmol, 37%, E:Z ratio 30:70). MP >300 °C; (Z)-N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)isothiazole-5-carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 12.74 (s, 1H), 10.60 (s, 1H), 10.46 (s, 1H), 9.41 (d, J = 2.1 Hz, 1H), 8.72 (dd, J = 3.5, 1.7 Hz, 1H), 8.16 (d, J = 1.8 Hz, 1H), 8.03 (s, 1H), 7.95 (s, 1H), 7.54 (s, 1H), 7.50 − 7.44 (m, 1H), 6.86 (d, J = 8.2 Hz, 1H). (E)-N-(3-((1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)isothiazole-5-carboxamide. 1H NMR (600 MHz, DMSO-d6) δ 13.71 (s, 1H), 11.05 (s, 1H), 10.59 (s, 1H), 8.12 (s, 0H), 8.07 (s, 1H), 8.04 (s, 1H), 7.82 (s, 1H), 7.73 (s, 1H), 7.42 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H).HRMS m/z [M+H]+ calcd for C16H12N5O2S: 338.0712, found 338.0699, LC tR = 2.41 min, > 98% Purity.
N-[(3Z)-3-[(4-methyl-1H-imidazol-5-yl)methylidene]-2-oxo-2,3-dihydro-1H-indol-5-yl]benzamide (128):
Compound 128 was prepared by procedure A to afford a light orange solid (59 mg, 0.171 mmol, 43%). MP >270 °C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 13.91 (s, 1H), 11.02 (s, 1H), 10.23 (s, 1H), 8.15 (d, J = 2.0 Hz, 1H), 8.07 − 8.02 (m, 2H), 7.99 (s, 1H), 7.71 (d, J = 0.7 Hz, 1H), 7.67 − 7.62 (m, 1H), 7.62 − 7.56 (m, 2H), 7.51 (dd, J = 8.4, 2.0 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 2.50 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.5, 165.2, 147.6, 138.2, 135.9, 134.8, 133.1, 131.5, 128.4 (s, 2C), 127.5 (s, 2C), 124.8, 124.4, 122.3, 121.4, 118.1, 113.4, 109.5, 13.1. HRMS m/z [M+H]+ calcd for C20H17N4O2: 345.1352, found 345.1342, LC tR = 2.76 min, > 98% Purity.
N-[(3Z)-2-oxo-3-(1H-pyrazol-5-ylmethylidene)-2,3-dihydro-1H-indol-5-yl]benzamide (129):
Compound 129 was prepared by procedure A to afford a dark red/purple solid (87 mg, 0.263 mmol, 66%). MP 218–220 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.54 (s, 1H), 10.53 (s, 1H), 10.21 (s, 1H), 7.99 − 7.97 (m, 2H), 7.91 (d, J = 2.2 Hz, 1H), 7.72 − 7.44 (m, 6H), 6.88 (s, 1H), 6.86 (d, J = 8.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 169.6, 165.3, 165.2, 139.2, 136.8, 135.0, 134.9, 132.5, 131.5, 131.4, 128.42, 128.36, 127.59, 127.55, 125.3, 123.9, 121.7, 109.8, 109.1. HRMS m/z [M+H]+ calcd for C19H15N4O2: 331.1195, found 331.1185, LC tR = 3.56 min, > 98% Purity.
Acknowledgement
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. The US National Institutes of Health (NIH) is acknowledged for support (1U24DK11604–01). We thank Biocenter Finland/DDCB for financial support and the CSC-IT Center for Science Ltd. (Finland) for allocation of computational resources. We also thank Dr. Brandie Ehrmann and Ms. Diane E. Wallace for LC-MS/HRMS support provided by in the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill. The core is supported by the National Science Foundation under Grant No. (CHE-1726291). In addition, we thank the University of North Carolina’s Department of Chemistry NMR Core Laboratory for the use of their NMR spectrometers, along with Dr. Marc ter Horst and Dr. Andrew Camp for their assistance with NMR spectroscopy. The core is supported by the National Science Foundation under Grant No. (CHE-1828183).
Footnotes
The authors declare no competing financial interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.xxxxxx
References
- 1.Klimovskaia I. M.; Young C.; Strømme C. B.; Menard P.; Jasencakova Z.; Mejlvang J.; Ask K.; Ploug M.; Nielsen M. L.; Jensen O. L.; Groth A. Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat Commun. 2014, 5, 3394. doi: 10.1038/ncomms4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silljé H. H.; Takahashi K.; Tanaka K.; Van Houwe G.; Nigg E. A. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999, 18, 5691–5702. doi: 10.1093/emboj/18.20.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammond C. M.; Strømme C. B.; Huang H.; Patel D. J.; Groth A. Histone Chaperones as Cardinal Players in Development. Nat Rev Mol Cell Biol. 2017, 18, 141–158. doi: 10.3389/fcell.2022.767773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pilyugin M.; Demmers J.; Verrijzer C. P.; Karch F.; Moshkin Y. M. Phosphorylation-mediated control of histone chaperone ASF1 levels by Tousled-like kinases PLoS One. 2009, 4, e8328. doi: 10.1371/journal.pone.0008328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruinsma W.; van den Berg J.; Aprelia M.; Medema R. H. Tousled-like kinase 2 regulates recovery from a DNA damageinduced G2 arrest EMBO Rep. 2016, 17, 659–670. doi: 10.15252/embr.201540767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S.; Segura-Bayona S.; Villamor-Payà M.; Saredi G.; Todd M. A. M.; Attolini C. S.; Chang T. Y.; Stracker T. H.; Groth A. Tousled-like kinases stabilize replication forks and show synthetic lethality with checkpoint and PARP inhibitors. Sci Adv. 2018, 4, eaat4985. doi: 10.1126/sciadv.aat4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segura-Bayona S.; Villamor-Payà M.; Attolini C. S.; Koenig L. M.; Sanchiz-Calvo M.; Boulton S. J.; Stracker T. H. Tousled-Like Kinases Suppress Innate Immune Signaling Triggered by Alternative Lengthening of Telomeres. Cell Rep. 2020, 32, 107983. doi: 10.1016/j.celrep.2020.107983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lelieveld S. H.; Reijnders M. R.; Pfundt R.; Yntema H. G.; Kamsteeg E. J.; de Vries P.; de Vries B. B.; Willemsen M. H.; Kleefstra T.; Löhner K.; Vreeburg M.; Stevens S. J.; van der Burgt I.; Bongers E. M.; Stegmann A. P.; Rump P.; Rinne T.; Nelen M. R.; Veltman J. A.; Vissers L. E.; Brunner H. G.; Gilissen C. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016, 19, 1194–1196. doi: 10.1038/nn.4352. [DOI] [PubMed] [Google Scholar]
- 9.Töpf A.; Oktay Y.; Balaraju S.; Yilmaz E.; Sonmezler E.; Yis U.; Laurie S.; Thompson R.; Roos A.; MacArthur D. G.; Yaramis A.; Güngör S.; Lochmüller H.; Hiz S.; Horvath R. Severe neurodevelopmental disease caused by a homozygous TLK2 variant. Eur J Hum Genet. 2020, 28, 383–387. doi: 10.1038/s41431-019-0519-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reijnders M. R. F.; Miller K. A.; Alvi M.; Goos J. A. C.; Lees M. M.; de Burca A.; Henderson A.; Kraus A.; Mikat B.; de Vries B. B. A.; Isidor B.; Kerr B.; Marcelis C.; Schluth-Bolard C.; Deshpande C.; Ruivenkamp C. A. L.; Wieczorek D.; Deciphering Developmental Disorders Study, Baralle D.; Blair E. M.; Engels H.; Lüdecke H. J.; Eason J.; Santen G. W. E.; Clayton-Smith J.; Chandler K.; Tatton-Brown K.; Payne K.; Helbig K.; Radtke K.; Nugent K. M.; Cremer K.; Strom T. M.; Bird L. M.; Sinnema M.; Bitner-Glindzicz M.; van Dooren M. F.; Alders M.; Koopmans M.; Brick L.; Kozenko M.; Harline M. L.; Klaassens M.; Steinraths M.; Cooper N. S.; Edery P.; Yap P.; Terhal P. A.; van der Spek P. J.; Lakeman P.; Taylor R. L.; Littlejohn R. O.; Pfundt R.; Mercimek-Andrews S.; Stegmann A. P. A.; Kant S. G.; McLean S.; Joss S.; Swagemakers S. M. A.; Douzgou S.; Wall S. A.; Küry S.; Calpena E.; Koelling N.; McGowan S. J.; Twigg S. R. F.; Mathijssen I. M. J.; Nellaker C.; Brunner H. G.; Wilkie A. O. M. De Novo and Inherited Loss-of-Function Variants in TLK2: Clinical and Genotype-Phenotype Evaluation of a Distinct Neurodevelopmental Disorder. Am J Hum Genet. 2018, 102, 1195–1203. doi: 10.1016/j.ajhg.2018.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mortuza G. B.; Hermida D.; Pedersen A. K.; Segura-Bayona S.; López-Méndez B.; Redondo P.; Rüther P.; Pozdnyakova I.; Garrote A. M.; Muñoz I. G.; Villamor-Payà M.; Jauset C.; Olsen J. V.; Stracker T. H.; Montoya G. Molecular basis of Tousled-Like Kinase 2 activation. Nat Commun. 2018. 9, 2535. doi: 10.1038/s41467-018-04941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dillon P. J.; Gregory S. M.; Tamburro K.; Sanders M. K.; Johnson G. L.; Raab-Traub N.; Dittmer D. P.; Damania B. Tousled-like kinases modulate reactivation of gammaherpesviruses from latency. Cell Host Microbe. 2013, 13, 204–214. doi: 10.1016/j.chom.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim Y.; Anderson J. L.; Lewin S. R. Getting the “kill” into “shock and kill”: strategies to eliminate latent HIV. Cell Host Microbe. 2018, 23, 14–26. doi: 10.1016/j.chom.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin M.; Yao Z.; Zhao N.; Zhang C. TLK2 enhances aggressive phenotypes of glioblastoma cells through the activation of SRC signaling pathway. Cancer Biol Ther. 2019, 20, 101–108. doi: 10.1080/15384047.2018.1507257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J. A.; Anurag M.; Veeraraghavan J.; Schiff R.; Li K.; Wang X. S. Amplification of TLK2 Induces Genomic Instability via Impairing the G2-M Checkpoint. Mol Cancer Res. 2016, 14, 920–927. doi: 10.1158/1541-7786.MCR-16-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J. A.; Tan Y.; Wang X.; Cao X.; Veeraraghavan J.; Liang Y.; Edwards D. P.; Huang S.; Pan X.; Li K.; Schiff R.; Wang X. S. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat Commun. 2016, 7, 12991. doi: 10.1038/ncomms12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segura-Bayona S.; Knobel P. A.; González-Burón H.; Youssef S. A.; Peña-Blanco A.; Coyaud É.; López-Rovira T.; Rein K.; Palenzuela L.; Colombelli J.; Forrow S.; Raught B.; Groth A.; de Bruin A.; Stracker T. H. Differential requirements for Tousled-like kinases 1 and 2 in mammalian development. Cell Death Differ. 2017, 24, 1872–1885. doi: 10.1038/cdd.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S. B.; Chang T. Y.; Lee N. Z.; Yu Z. Y.; Liu C. Y.; Lee H. Y. Design, synthesis and biological evaluation of bisindole derivatives as anticancer agents against Tousled-like kinases. Eur J Med Chem. 2022, 227, 113904. doi: 10.1016/j.ejmech.2021.113904. [DOI] [PubMed] [Google Scholar]
- 19.Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; Buser-Doepner C.; Campbell R. M.; Carter A. J.; Cohen P.; Copeland R. A.; Cravatt B.; Dahlin J. L.; Dhanak D.; Edwards A. M.; Frederiksen M.; Frye S. V.; Gray N.; Grimshaw C. E.; Hepworth D.; Howe T.; Huber K. V.; Jin J.; Knapp S.; Kotz J. D.; Kruger R. G.; Lowe D.; Mader M. M.; Marsden B.; Mueller-Fahrnow A.; Müller S.; O’Hagan R. C.; Overington J. P.; Owen D. R.; Rosenberg S. H.; Roth B.; Ross R.; Schapira M.; Schreiber S. L.; Shoichet B.; Sundström M.; Superti-Furga G.; Taunton J.; Toledo-Sherman L.; Walpole C.; Walters M. A.; Willson T. M.; Workman P.; Young R. N.; Zuercher W. J. The promise and peril of chemical probes. Nat Chem Biol. 2015, 11, 536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J., A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005, 23, 329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 21.Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008, 26, 127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 22.Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011, 29, 1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 23.Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011, 29, 1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elkins J. M.; Fedele V,; Szklarz M.; Abdul Azeez K. R.; Salah E.; Mikolajczyk J.; Romanov S.; Sepetov N.; Huang X. P.; Roth B. L.; Al Haj Zen A.; Fourches D.; Muratov E.; Tropsha A.; Morris J.; Teicher B. A.; Kunkel M.; Polley E.; Lackey K. E.; Atkinson F. L.; Overington J. P.; Bamborough P.; Müller S.; Price D. J.; Willson T. M.; Drewry D. H.; Knapp S.; Zuercher W. J. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol. 2016, 34, 95–103. doi: 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- 25.Drewry D. H.; Wells C. I.; Andrews D. M.; Angell R.; Al-Ali H.; Axtman A. D.; Capuzzi S. J.; Elkins J. M.; Ettmayer P.; Frederiksen M.; Gileadi O.; Gray N.; Hooper A.; Knapp S.; Laufer S.; Luecking U.; Michaelides M.; Müller S.; Muratov E.; Denny R. A.; Saikatendu K. S.; Treiber D. K.; Zuercher W. J.; Willson T. M. Progress towards a public chemogenomic set for protein kinases and a call for contributions. PLoS One. 2017, 12, e0181585. doi: 10.1371/journal.pone.0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wells C. I.; Al-Ali H.; Andrews D. M.; Asquith C. R. M.; Axtman A. D.; Dikic I.; Ebner D.; Ettmayer P.; Fischer C.; Frederiksen M.; Futrell R. E.; Gray N. S.; Hatch S. B.; Knapp S.; Lücking U.; Michaelides M.; Mills C. E.; Müller S.; Owen D.; Picado A.; Saikatendu K. S.; Schröder M.; Stolz A.; Tellechea M.; Turunen B. J.; Vilar S.; Wang J.; Zuercher W. J.; Willson T. M.; Drewry D. H. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int J Mol Sci. 2021, 22, 566. doi: 10.3390/ijms22020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klaeger S.; Heinzlmeir S.; Wilhelm M.; Polzer H.; Vick B.; Koenig P. A.; Reinecke M.; Ruprecht B.; Petzoldt S.; Meng C.; Zecha J.; Reiter K.; Qiao H.; Helm D.; Koch H.; Schoof M.; Canevari G.; Casale E.; Depaolini S. R.; Feuchtinger A.; Wu Z.; Schmidt T.; Rueckert L.; Becker W.; Huenges J.; Garz A. K.; Gohlke B. O.; Zolg D. P.; Kayser G.; Vooder T.; Preissner R.; Hahne H.; Tonisson N.; Kramer K.; Gotze K.; Bassermann F.; Schlegl J.; Ehrlich H. C.; Aiche S.; Walch A.; Greif P. A.; Schneider S.; Felder E. R.; Ruland J.; Medard G.; Jeremias I.; Spiekermann K.; Kuster B., The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knapp S.; Arruda P.; Blagg J.; Burley S.; Drewry D. H.; Edwards A.; Fabbro D.; Gillespie P.; Gray N. S.; Kuster B.; Lackey K. E.; Mazzafera P.; Tomkinson N. C.; Willson T. M.; Workman P.; Zuercher W. J. A public-private partnership to unlock the untargeted kinome. Nat Chem Biol. 2013, 9, 3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 29.Sun L.; Liang C.; Shirazian S.; Zhou Y.; Miller T.; Cui J.; Fukuda J. Y.; Chu J. Y.; Nematalla A.; Wang X.; Chen H.; Sistla A.; Luu T. C.; Tang F.; Wei J.; Tang C. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003, 46, 1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- 30.Pryer N. K.; Lee L. B.; Zadovaskaya R.; Yu X.; Sukbuntherng J.; Cherrington J. M.; London C. A. Proof of target for SU11654: inhibition of KIT phosphorylation in canine mast cell tumors. Clin Cancer Res. 2003, 9, 5729–5734. [PubMed] [Google Scholar]
- 31.Patyna S.; Laird A. D.; Mendel D. B.; O’farrell A. M.; Liang C.; Guan H.; Vojkovsky T.; Vasile S.; Wang X.; Chen J.; Grazzini M.; Yang C. Y.; Haznedar J. O.; Sukbuntherng J.; Zhong W. Z.; Cherrington J. M.; Hu-Lowe D. SU14813: a novel multiple receptor tyrosine kinase inhibitor with potent antiangiogenic and antitumor activity. Mol Cancer Ther. 2006, 5, 1774–1782. doi: 10.1158/1535-7163.MCT-05-0333. [DOI] [PubMed] [Google Scholar]
- 32.Hoessel R.; Leclerc S.; Endicott J. A.; Nobel M. E.; Lawrie A.; Tunnah P.; Leost M.; Damiens E.; Marie D.; Marko D.; Niederberger E.; Tang W.; Eisenbrand G.; Meijer L. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999, 1, 60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 33.Leclerc S.; Garnier M.; Hoessel R.; Marko D.; Bibb J. A.; Snyder G. L.; Greengard P.; Biernat J.; Wu Y. Z.; Mandelkow E. M.; Eisenbrand G.; Meijer L. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem. 2001, 276, 251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 34.Bertrand J. A.; Thieffine S.; Vulpetti A.; Cristiani C.; Valsasina B.; Knapp S.; Kalisz H. M.; Flocco M. Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J Mol Biol. 2003, 333, 393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 35.Mapelli M.; Massimiliano L.; Crovace C.; Seeliger M. A.; Tsai L. H.; Meijer L.; Musacchio A. Mechanism of CDK5/p25 binding by CDK inhibitors. J Med Chem. 2005, 48, 671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- 36.Asquith C. R. M.; Treiber D. K.; Zuercher W. J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G associated kinase (GAK). Bioorg. Med. Chem. Lett. 2019, 29, 1727–1731. doi: 10.1016/j.bmcl.2019.05.025. [DOI] [PubMed] [Google Scholar]
- 37.Bembenek S. D.; Hirst G.: Mirzadegan T. Determination of a Focused Mini Kinase Panel for Early Identification of Selective Kinase Inhibitors. J Chem Inf Model. 2018, 58, 1434–1440. doi: 10.1021/acs.jcim.8b00222. [DOI] [PubMed] [Google Scholar]
- 38.Brandt P.; Jensen A. J.; Nilsson J. Small kinase assay panels can provide a measure of selectivity. Bioorg. Med. Chem. Lett. 2009, 19, 5861–5863. doi: 10.1016/j.bmcl.2009.08.083. [DOI] [PubMed] [Google Scholar]
- 39.Sun L.; Tran N.; Tang F.; App H.; Hirth P.; McMahon G.; Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998, 41, 2588–4603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- 40.Jones G. The Knoevenagel Condensation. Organic Reactions. 2004, 204–273. 10.1002/0471264180.or015.02 [DOI] [Google Scholar]
- 41.Knoevenagel E. Condensation von Malonsäure mit aromatischen Aldehyden durch Ammoniak und Amine [Condensation of malonic acid with aromatic aldehydes via ammonia and amines]. Berichte der deutschen chemischen Gesellschaft. 1898, 31, 2596–2619. doi: 10.1002/cber.18980310308 [DOI] [Google Scholar]
- 42.Rueping R.; Nachtsheim B. J. A Review of New Developments in the Friedel-Crafts Alkylation - From Green Chemistry to Asymmetric Catalysis. Beilstein J. Org. Chem. 2010, 6, No. 6. 10.3762/bjoc.6.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedel C.; Crafts J. M. Sur une Méthode Générale Nouvelle de Synthèse d’Hydrocarbures, d’Acétones, etc., Compt. Rend. 1877, 84, 1392–1395. [Google Scholar]
- 44.Friedel C.; Crafts J. M., Sur une Méthode Générale Nouvelle de Synthèse d’Hydrocarbures, d’Acétones, etc., Compt. Rend. 1877, 84, 1450–1454. [Google Scholar]
- 45.Xiao J.; Westbroek W.; Motabar O.; Lea W. A.; Hu X.; Velayati A.; Zheng W.; Southall N.; Gustafson A. M.; Goldin E.; Sidransky E.; Liu K.; Simeonov A.; Tamargo R. J.; Ribes A.; Matalonga L.; Ferrer M.; Marugan J. J. Discovery of a novel noniminosugar acid α glucosidase chaperone series. J Med Chem. 2012, 55, 7546–7559. doi: 10.1021/jm3005543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitsunobu O.; Yamada Y. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. doi: 10.1246/bcsj.40.2380 [DOI] [Google Scholar]
- 47.Mitsunobu O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis. 1981, (1), 1–28. doi: 10.1055/s-1981-29317 [DOI] [Google Scholar]
- 48.Lepore S. D.; He Y. Use of Sonication for the Coupling of Sterically Hindered Substrates in the Phenolic Mitsunobu Reaction. J. Org. Chem. 2003, 68, 8261–8263. doi: 10.1021/jo0345751. [DOI] [PubMed] [Google Scholar]
- 49.Carpino L. A.; Imazumi H.; El-Faham A.; Ferrer F. J; Zhang C.; Lee Y.; Foxman B. M.; Henklein P.; Hanay C.; Mügge C.; Wenschuh H.; Klose J.; Beyermann M.; Bienert M. The Uronium/Guanidinium Peptide Coupling Reagents: Finally the True Uronium Salts. Angew. Chem. Int. Ed. Engl. 2002, 41, 441–445. doi: . [DOI] [PubMed] [Google Scholar]
- 50.Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601 doi: 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Auffinger P.; Hays F. A.; Westhof E.; Ho P. S.; Halogen bonds in biological molecules. Proc Natl Acad Sci USA. 2004, 101, 16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stumpfe D.; Bajorath J. Exploring Activity Cliffs in Medicinal Chemistry. J Med Chem. 2012, 55, 2932–2942. doi: 10.1021/jm201706b. [DOI] [PubMed] [Google Scholar]
- 52.Bemis G. W.; Murcko M. A. The Properties of Known Drugs. 1. Molecular Frameworks J Med Chem. 1996, 39, 2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 53.Kohlmann A.; Zhu X.; Dalgarno D. Application of MM-GB/SA and WaterMap to SRC Kinase Inhibitor Potency Prediction. ACS Med Chem Lett. 2012, 3, 94–99. doi: 10.1021/ml200222u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higgs C.; Beuming T.; Sherman W. Hydration Site Thermodynamics Explain SARs for Triazolylpurines Analogues Binding to the A2A Receptor. ACS Med Chem Lett. 2010, 1, 160–164. doi: 10.1021/ml100008s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asquith C. R. M.; Tizzard G. J.; Bennett J. M.; Wells C. I.; Elkins J. M.; Willson T. M.; Poso A.; Laitinen T. Targeting the Water Network in Cyclin G-Associated Kinase (GAK) with 4-Anilino-quin(az)oline Inhibitors. ChemMedChem. 2020, 15, 1200–1215. doi: 10.1002/cmdc.202000150 [DOI] [PubMed] [Google Scholar]
- 56.Asquith C. R. M.; Maffuid K. A.; Laitinen T.; Torrice C. D.; Tizzard G. J.; Crona D. J.; Zuercher W. J. Targeting an EGFR Water Network with 4-Anilinoquin(az)oline Inhibitors for Chordoma. ChemMedChem. 2019, 14, 1693–1700. doi: 10.1002/cmdc.201900428. [DOI] [PubMed] [Google Scholar]
- 57.Fang Z.; Song Y.; Zhan P.; Zhang Q.; Liu X. Conformational Restriction: An Effective Tactic in ‘Follow-On’-Based Drug Discovery Future Med. Chem. 2014, 8, 885–901. doi: 10.4155/fmc.14.50. [DOI] [PubMed] [Google Scholar]
- 58.Asquith C. R. M.; Laitinen T.; Bennett J. M.; Wells C. I.; Elkins J. M.; Zuercher W. J.; Tizzard G. J.; Poso A. Design and analysis of the 4-anilino-quin(az)oline kinase inhibition profiles of GAK/SLK/STK10 using quantitative structure activity relationships. ChemMedChem. 2020, 15, 26–49. doi: 10.1002/cmdc.201900521. [DOI] [PubMed] [Google Scholar]
- 59.Kleman-Leyer K. M.; Klink T. A.; Kopp A. L.; Westermeyer T. A.; Koeff M. D.; Larson B. R.; Worzella T. J.; Pinchard C. A.; van de Kar S. A.; Zaman G. J.; Hornberg J. J.; Lowery R. G. Characterization and optimization of a red-shifted fluorescence polarization ADP detection assay. Assay Drug Dev Technol. 2009, 7, 56–67. doi: 10.1089/adt.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zegzouti H.; Zdanovskaia M.; Hsiao K.; Goueli S. A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. Assay Drug Dev Technol. 2009, 7, 560–572. doi: 10.1089/adt.2009.0222. [DOI] [PubMed] [Google Scholar]
- 61.Li H.; Totoritis R. D.; Lor L. A.; Schwartz B.; Caprioli P.; Jurewicz A. J.; Zhang G. Evaluation of an antibody-free ADP detection assay: ADP-Glo. Assay Drug Dev Technol. 2009, 7, 598–605. doi: 10.1089/adt.2009.0221. [DOI] [PubMed] [Google Scholar]
- 62.Johnson D.; Hussain J.; Bhoir S.; Chandrasekaran V.; Sahrawat P.; Hans T.; Khalil M. I.; De Benedetti A.; Thiruvenkatam V.; Kirubakaran S. Synthesis, kinetics and cellular studies of new phenothiazine analogs as potent human-TLK inhibitors. Org Biomol Chem. 2023, 21, 1980–1991. doi: 10.1039/d2ob02191a. [DOI] [PubMed] [Google Scholar]
- 63.Ronald S.; Awate S.; Rath A.; Carroll J.; Galiano F.; Dwyer D.; Kleiner-Hancock H.; Mathis J. M.; Vigod S.; De Benedetti A. Phenothiazine Inhibitors of TLKs Affect Double-Strand Break Repair and DNA Damage Response Recovery and Potentiate Tumor Killing with Radiomimetic Therapy. Genes Cancer. 2013, 4, 39–53. doi: 10.1177/1947601913479020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uitdehaag J. C.; Verkaar F.; Alwan H.; de Man J.; Buijsman R. C.; Zaman G. J. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol. 2012, 166, (3), 858–876. doi: 10.1111/j.1476-5381.2012.01859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rokosz L. L.; Beasley J. R.; Carroll C. D.; Lin T.; Zhao J.; Appell K. C.; Webb M. L. Kinase inhibitors as drugs for chronic inflammatory and immunological diseases: progress and challenges Expert Opin. Ther. Targets 2008, 12, 883–903. doi: 10.1517/14728222.12.7.883. [DOI] [PubMed] [Google Scholar]
- 66.Sun L.; Tran N.; Liang C.; Tang F.; Rice A.; Schreck R.; Waltz K.; Shawver L. K.; McMahon G.; Tang C. Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. J Med Chem. 1999, 42, 5120–5130. doi: 10.1021/jm9904295. [DOI] [PubMed] [Google Scholar]
- 67.Wood E. R.; Kuyper L.; Petrov K. G.; Hunter R. N 3rd.; Harris P. A.; Lackey K. Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg Med Chem Lett. 2004, 14, 953–957. doi: 10.1016/j.bmcl.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 68.Bramson H. N.; Corona J.; Davis S. T.; Dickerson S. H.; Edelstein M.; Frye S. V.; Gampe R. T Jr.; Harris P. A.; Hassell A.; Holmes W. D.; Hunter R. N.; Lackey K. E.; Lovejoy B.; Luzzio M. J.; Montana V.; Rocque W. J.; Rusnak D.; Shewchuk L.; Veal J. M.; Walker D. H.; Kuyper L. F. Oxindole-based inhibitors of cyclin-dependent kinase 2 (CDK2): design, synthesis, enzymatic activities, and X-ray crystallographic analysis. J Med Chem. 2001, 44, 4339–4358. doi: 10.1021/jm010117d. [DOI] [PubMed] [Google Scholar]
- 69.Luk K. C.; Simcox M. E.; Schutt A.; Rowan K.; Thompson T.; Chen Y.; Kammlott U.; DePinto W.; Dunten P.; Dermatakis. A. A new series of potent oxindole inhibitors of CDK2. Bioorg Med Chem Lett. 2004, 14, 913–917. doi: 10.1016/j.bmcl.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 70.Dermatakis A.; Luk K. C.; DePinto W. Synthesis of potent oxindole CDK2 inhibitors. Bioorg Med Chem. 2003, 11, 1873–1881. doi: 10.1016/s0968-0896(03)00036-1. [DOI] [PubMed] [Google Scholar]
- 71.Islam I.; Brown G.; Bryant J.; Hrvatin P.; Kochanny M. J.; Phillips G. B.; Yuan S.; Adler M.; Whitlow M.; Lentz D.; Polokoff M. A.; Wu J.; Shen J.; Walters J.; Ho E.; Subramanyam B.; Zhu D.; Feldman R. I.; Arnaiz D. O. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 2: optimization of BX-517. Bioorg Med Chem Lett. 2007, 17, 3819–3825. doi: 10.1016/j.bmcl.2007.05.060. [DOI] [PubMed] [Google Scholar]
- 72.Guan H.; Laird A. D.; Blake R. A.; Tang C.; Liang C. Design and synthesis of aminopropyl tetrahydroindole-based indolin-2-ones as selective and potent inhibitors of Src and Yes tyrosine kinase. Bioorg Med Chem Lett. 2004, 14, 187–190. doi: 10.1016/j.bmcl.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 73.Kaur M.; Singh M.; Chadha N.; Silakari O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur J Med Chem. 2016, 123, 858–894. doi: 10.1016/j.ejmech.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 74.Troxler T.; Greenidge P.; Zimmermann K.; Desrayaud S.; Drückes P.; Schweizer T.; Stauffer D.; Rovelli G.; Shimshek D. R. Discovery of novel indolinone-based, potent, selective and brain penetrant inhibitors of LRRK2. Bioorg Med Chem Lett. 2013, 23, 4085–4090. doi: 10.1016/j.bmcl.2013.05.054. [DOI] [PubMed] [Google Scholar]
- 75.Fang Z.; Song Y.; Zhan P.; Zhang Q.; Liu X. Conformational restriction: an effective tactic in ‘follow-on’-based drug discovery. Future Med Chem. 2014, 6, 885–901. doi: 10.4155/fmc.14.50. [DOI] [PubMed] [Google Scholar]
- 76.Asquith C. R. M.; Laitinen T.; Bennett J. M.; Godoi P. H.; East M. P.; Tizzard G. J.; Graves L. M.; Johnson G. L.; Dornsife R. E.; Wells C. I.; Elkins J. M.; Willson T. M.; Zuercher W. J. Identification and Optimization of 4-Anilinoquinolines as Inhibitors of Cyclin G Associated Kinase. ChemMedChem 2018, 13, 48–66. doi: 10.1002/cmdc.201700663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ellis J. M.; Altman M. D.; Bass A.; Butcher J. W.; Byford A. J.; Donofrio A.; Galloway S.; Haidle A. M.; Jewell J.; Kelly N.; Leccese E. K.; Lee S.; Maddess M.; Miller J. R.; Moy L. Y.; Osimboni E.; Otte R. D.; Reddy M. V.; Spencer K.; Sun B.; Vincent S. H.; Ward G. J.; Woo G. H.; Yang C.; Houshyar H.; Northrup A. B. Overcoming mutagenicity and ion channel activity: optimization of selective spleen tyrosine kinase inhibitors. J Med Chem. 2015, 58, 1929–1939. doi: 10.1021/jm5018169. [DOI] [PubMed] [Google Scholar]
- 78.Lawhorn B. G.; Philp J.; Zhao Y.; Louer C.; Hammond M.; Cheung M.; Fries H.; Graves A. P.; Shewchuk L.; Wang L.; Cottom J. E.; Qi H.; Zhao H.; Totoritis R.; Zhang G.; Schwartz B.; Li H.; Sweitzer S.; Holt D. A.; Gatto G. J. Jr,; Kallander, L. S. Identification of Purines and 7-Deazapurines as Potent and Selective Type I Inhibitors of Troponin I-Interacting Kinase (TNNI3K). J Med Chem. 2015, 58, 7431–7448. doi: 10.1021/acs.jmedchem.5b00931. [DOI] [PubMed] [Google Scholar]
- 79.Asquith C. R. M.; Laitinen T.; Wells C. I.; Tizzard G. J.; Zuercher W. J. New Insights into 4-Anilinoquinazolines as Inhibitors of Cardiac Troponin I-Interacting Kinase (TNNi3K). Molecules. 2020, 25, 1697. doi: 10.3390/molecules25071697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azuaje J.; Jespers W.; Yaziji V.; Mallo A.; Majellaro M.; Caamaño O.; Loza M. I.; Cadavid M. I.; Brea J.; Åqvist J.; Sotelo E.; Gutiérrez-de-Terán H. Effect of Nitrogen Atom Substitution in A3 Adenosine Receptor Binding: N-(4,6-Diarylpyridin-2-yl)acetamides as Potent and Selective Antagonists. J Med Chem. 2017, 60, 7502–7511. doi: 10.1021/acs.jmedchem.7b00860. [DOI] [PubMed] [Google Scholar]
- 81.Geyer R.; Nordemann U.; Strasser A.; Wittmann H. J.; Buschauer A. Conformational Restriction and Enantioseparation Increase Potency and Selectivity of Cyanoguanidine-Type Histamine H4 Receptor Agonists. J Med Chem. 2016, 59, 3452–3470. doi: 10.1021/acs.jmedchem.6b00120. [DOI] [PubMed] [Google Scholar]
- 82.El Bakali J.; Muccioli G. G.; Body-Malapel M.; Djouina M.; Klupsch F.; Ghinet A.; Barczyk A.; Renault N.; Chavatte P.; Desreumaux P.; Lambert D. M.; Millet R. Conformational Restriction Leading to a Selective CB2 Cannabinoid Receptor Agonist Orally Active Against Colitis. ACS Med Chem Lett. 2014, 6, 198–203. doi: 10.1021/ml500439x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pennington L. D.; Moustakas D. T. The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem. 2017, 60, 3552–3579. doi: 10.1021/acs.jmedchem.6b01807. [DOI] [PubMed] [Google Scholar]
- 84.Robinson D. D.; Sherman W.; Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. ChemMedChem. 2010, 5, 618–627. doi: 10.1002/cmdc.200900501. [DOI] [PubMed] [Google Scholar]
- 85.Robinson D.; Bertrand T.; Carry J. C.; Halley F.; Karlsson A.; Mathieu M.; Minoux H.; Perrin M. A.; Robert B.; Schio L.; Sherman W. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J Chem Inf Model. 2016, 56, 886–894. doi: 10.1021/acs.jcim.5b00641. [DOI] [PubMed] [Google Scholar]
- 86.Hu Y.; Kunimoto R.; Bajorath J. Mapping of inhibitors and activity data to the human kinome and exploring promiscuity from a ligand and target perspective. Chem Biol Drug Des. 2017, 89, 834–845. doi: 10.1111/cbdd.12919. [DOI] [PubMed] [Google Scholar]
- 87.CrysAlisPro Software System Rigaku Oxford Diffraction, (2018)
- 88.Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A, 2015, 71, 3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. doi: 10.1107/S0021889808042726 [DOI] [Google Scholar]
- 90.Fong T. A.; Shawver L. K.; Sun L.; Tang C.; App H.; Powell T. J.; Kim Y. H.; Schreck R.; Wang X.; Risau W.; Ullrich A.; Hirth K. P.; McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999, 59, 99–106. [PubMed] [Google Scholar]
- 91.Lane M. E.; Yu B.; Rice A.; Lipson K. E.; Liang C.; Sun L.; Tang C.; McMahon G.; Pestell R. G.; Wadler S. A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res. 2001, 61, 6170–6177. [PubMed] [Google Scholar]
- 92.Dumas M. E.; Chen G. Y.; Kendrick N. D.; Xu G.; Larsen S. D.; Jana S.; Waterson A. G.; Bauer J. A.; Hancock W.; Sulikowski G. A.; Ohi R. Dual inhibition of Kif15 by oxindole and quinazolinedione chemical probes. Bioorg Med Chem Lett. 2019, 29, 148–154. doi: 10.1016/j.bmcl.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim M. H.; Tsuhako A. L.; Co E. W.; Aftab D. T.; Bentzien F.; Chen J.; Cheng W.; Engst S.; Goon L.; Klein R. R.; Le D. T.; Mac M.; Parks J. J.; Qian F.; Rodriquez M.; Stout T. J.; Till J. H.; Won K. A.; Wu X.; Yakes F. M.; Yu P.; Zhang W.; Zhao Y.; Lamb P.; Nuss J. M.; Xu W. The design, synthesis, and biological evaluation of potent receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2012, 22, 4979–4985. doi: 10.1016/j.bmcl.2012.06.029. [DOI] [PubMed] [Google Scholar]