

Abstract
Unsolved Mendelian cases often lack obvious pathogenic coding variants, suggesting potential non-coding etiologies. Here, we present a single cell multi-omic framework integrating embryonic mouse chromatin accessibility, histone modification, and gene expression assays to discover cranial motor neuron (cMN) cis-regulatory elements and subsequently nominate candidate non-coding variants in the congenital cranial dysinnervation disorders (CCDDs), a set of Mendelian disorders altering cMN development. We generated single cell epigenomic profiles for ~86,000 cMNs and related cell types, identifying ~250,000 accessible regulatory elements with cognate gene predictions for ~145,000 putative enhancers. Seventy-five percent of elements (44 of 59) validated in an in vivo transgenic reporter assay, demonstrating that single cell accessibility is a strong predictor of enhancer activity. Applying our cMN atlas to 899 whole genome sequences from 270 genetically unsolved CCDD pedigrees, we achieved significant reduction in our variant search space and nominated candidate variants predicted to regulate known CCDD disease genes MAFB, PHOX2A, CHN1, and EBF3 – as well as new candidates in recurrently mutated enhancers through peak- and gene-centric allelic aggregation. This work provides novel non-coding variant discoveries of relevance to CCDDs and a generalizable framework for nominating non-coding variants of potentially high functional impact in other Mendelian disorders.
INTRODUCTION
While the great majority of genetic variants associated with complex disease are common in the population and localize to non-coding sequences, less than 5% of the known Mendelian phenotype entries in OMIM have been attributed to non-coding mutations1-4. However, it remains unsettled the extent to which this disparity in coding:non-coding causal Mendelian variants is explained by the relative effect sizes of coding vs. non-coding variation, difficulty in deciphering the functional impact of non-coding variation, and/or ascertainment due to greater number and size of exome-versus genome-sequenced disease cohorts1,5-8. Nominating pathogenic non-coding variants in Mendelian disease remains a major challenge due to a vastly increased search space (98% of the genome) relative to coding variants. Compounding this challenge is the lack of a generalizable rubric for nominating non-coding pathogenic variants relative to the more readily interpretable molecular and biochemical constraints governing protein coding variant effects.
In recognition of these challenges, large-scale functional genomics projects such as ENCODE and Roadmap Epigenomics have provided valuable and expansive genome-wide functional information across a growing array of potentially disease-relevant tissues and cell types9,10. Such efforts reveal that the non-coding genome is abundant with cis regulatory elements (cREs) - segments of non-coding DNA that regulate gene expression through transcription factor binding and three-dimensional physical interactions with their cognate genes. Biologically active cREs are associated with accessible chromatin, and combinations of accessible cREs vary dramatically among different cell types11. Therefore, understanding the chromatin accessibility landscape of cell types affected in disease is critical to identifying and interpreting disease-causing variation in the non-coding genome.
Disease-relevant developmental processes are disproportionately driven by regulation of gene expression12,13, making congenital genetic disorders attractive candidates for non-coding etiologies. However, sampling developing human cell types remains particularly challenging, as samples are often restricted by cell location, assayable cells, invasiveness of sampling, and/or extremely narrow windows of biologically-relevant regulation of gene expression and development14. Thus, while fetal epigenomic reference sets are emerging for humans, samples are generally assayed at the whole-organ/tissue level and/or at later stages of development, making appropriate sampling and identification of early-born and rare cell types difficult15. By contrast, sample collection and marker-based enrichment in model organisms can achieve substantial representation of disease-relevant cell types at early stages of development16-18.
The congenital cranial dysinnervation disorders (CCDDs) are Mendelian disorders in which movement of extraocular and/or cranial musculature are limited secondary to errors in the development of cranial motor neurons (cMNs) or the growth and guidance of their axons (Figure 1a). Although a known subset of the CCDDs are caused by Mendelian protein-coding variants19-28, a substantial proportion of cases remain unsolved by whole exome sequencing, including pedigrees with Mendelian inheritance patterns and cases with classic phenotypic presentations lacking corresponding mutations in the expected genes (representing potential locus heterogeneity)29. Moreover, most CCDD cases are sporadic or segregate in small dominant families for which non-coding variant prioritization is extremely difficult.
Figure 1. Integrating Mendelian pedigrees with single cell epigenomic data.

a. Schematic depicting subset of human cMNs and their targeted muscles. cMN3 (blue) = oculomotor nucleus which innervates the inferior rectus, medial rectus, superior rectus, inferior oblique, and levator palpebrae superior muscles; cMN4 (purple) = trochlear nucleus which innervates the superior oblique muscle; cMN6 (green) = abducens nucleus which innervates the lateral rectus muscle (bisected); cMN7 (pink) = facial nucleus which innervate muscles of facial expression; cMN12 (black) = hypoglossal nucleus which innervates tongue muscles. Corresponding CCDDs for each cMN are listed under diagram and color coded. CFEOM: congenital fibrosis of the extraocular muscles; CP: congenital ptosis; FNP: fourth nerve palsy; DRS: Duane retraction syndrome; MBS: Moebius syndrome; CFP: congenital facial palsy.
b. Overview of the experimental and computational approach. i) Generating cell type-specific chromatin accessibility profiles. Brightfield and fluorescent images of e10.5 Isl1MN:GFP embryo (left) from which cMNs are microdissected (yellow dotted lines, dissociated, FACS-purified (middle), followed by scATAC and data processing (right; red and blue lines represent adapters, black line represents DNA, orange cylinders represent nucleosomes, grey pentagons represent Tn5). ii) WGS of 270 CCDD pedigrees (left; 899 individuals; sporadic and inherited cases) followed by joint variant calling, QC, and Mendelian variant filtering (right). iii) Integrating genome-wide non-coding variant calls with epigenomic annotations for variant nomination (top). To aid in variant interpretation, we identify cognate genes (2nd row), aggregate candidate variants, generate functional variant effect predictions (3rd row), and validate top predictions in vivo (bottom).
c. UMAP embedding of single cell chromatin accessibility profiles from 86,089 GFP-positive cMNs, sMNs, and their surrounding GFP-negative neuronal tissue colored based on GFP reporter status (left, GFP-positive green, GFP-negative grey), sample (middle, with sample key under UMAP) and cluster (right, with cluster annotations in Supplementary Table 3). Gridlines in middle UMAP apply to left and right UMAPs as well. The inset shows the relative proximity of Cluster 2 cells dissected from the same cell type (cMN7 e10.5) from different technical and biological replicates.
d. Heatmap depicting the proportions of dissected cells within each of the 23 major clusters. Homogeneity/completeness metrics are shown for GFP-positive versus GFP-negative clusters. cMN6 and cMN7 are in close spatial proximity and are commonly co-dissected.
The CCDDs represent an attractive test case for dissecting cell type-specific disorders, as defects in specific cMN populations are highly stereotyped with predictable corresponding human phenotypes30. By contrast, many complex and even some Mendelian diseases are not immediately attributable to an unambiguous, singular cell type of interest, making assaying appropriate cell types a major challenge31-33. Moreover, while sampling and identification of developing cMNs at disease-relevant timepoints is extremely difficult in developing human embryos, cMN birth, migration, axon growth/guidance, and mature anatomy/nerve branches are exquisitely conserved between humans and mice30. Motor neuron reporter mice permit sample collection and marker-based enrichment of cMNs at these key stages of development. Importantly, we have previously demonstrated that such mouse models helped to characterize non-coding pathogenic variants that alter gene expression in HCFP1, a disorder of facial nerve (cMN7) development34. Here, to comprehensively discover the repertoire of cREs underlying proper cMN development, we have generated a chromatin accessibility atlas of developing mouse cMNs and adjacent cell types. We subsequently use this atlas to reduce our candidate variant search space and ultimately interpret and nominate non-coding variants among 270 unsolved CCDD pedigrees (Figure 1b, Supplementary Table 1).
RESULTS
Defining disease-relevant cREs in the developing cMNs
To discover disease-relevant cREs and ultimately reduce our non-coding search space for nominating candidate pathogenic CCDD variants, we generated a single cell atlas of embryonic mouse cMN chromatin accessibility. Using wildtype or transgenic mice expressing GFP under the Isl1MN:GFP or Hb9:GFP motor neuron reporters35,36 (Figure 1ai), we performed fluorescence-assisted microdissection and FACS-based enrichment of GFP-positive primary mouse embryonic oculomotor (cMN3), trochlear (cMN4), abducens (cMN6), facial (cMN7), hypoglossal (cMN12), spinal motor neurons (sMNs), and surrounding GFP-negative non-motor neuron cells (−”neg”), followed by droplet-based single cell ATAC-seq (scATAC). cMN birth and development occur continuously over a period of weeks in early human embryos and days (e9.0-e12.5) in mice34,37. For the known CCDD genes, mRNA expression and/or observed cellular defects typically overlap key developmental timepoints e10.5 and e11.5 in mice – both for cellular identity-related transcription factor38-42 and axon guidance-related22,43,44 variants. Therefore, we captured these two embryonic timepoints for each cMN sample, reasoning that a major proportion of relevant cellular birth and initial axonal wiring would be represented at these ages34,37. At these stages, these cranial nuclei contain only hundreds (cMN3, 4, 6) to thousands (cMN7, 12) of motor neurons per nucleus, per embryo43-45.
We generated scATAC data across 20 unique sample types (cMN3/4, 6, 7, 12, and sMN for GFP-positive and -negative cells, each at e10.5 and e11.5), 9 with biological replicates and 2 with technical replicates for 32 samples in total and sequenced them to high coverage (mean coverage = 48,772 reads per cell). We included GFP-negative cells to reduce uncertainty in peak calling, further increase representation from rare cell types, and capture regional-specific cell types that could harbor elements conferring non-cell-autonomous effects on cMN development. To generate a high-quality set of non-coding elements, we performed stringent quality control (Extended Data Figure 1a-h, Methods). Altogether, we generated high-quality single-cell accessibility profiles for 86,089 (49,708 GFP-positive and 36,381 GFP-negative) cells, in some cases achieving substantial oversampling of cranial motor neurons in the developing mouse embryo (up to 23-fold cellular coverage). Our final dataset revealed prominent signals of expected nucleosome banding, a high fraction of reads in peaks (), transcription start site enrichment, and strong concordance between biological replicates (Figure 1c, Extended Data Figure 1d-h, Supplementary Table 2). In addition to evaluating per-sample and per-cell metrics, we estimated a decrease in global accessibility over developmental time, consistent with observations in other developing cell types (, p-value < 1 × 10−15, linear regression, Supplementary Note 1)46,47.
We performed bulk ATAC on a subset of microdissected and FACS-purified cMN samples to evaluate the concordance between bulk and single cell peak representation. As expected, bulk and single cell cMN ATAC peaks are highly correlated in their matching dissected cell types (Extended Data Figure 2a,b). scATAC peaks were enriched for intronic/distal annotations (relative to exonic/promoter annotations, OR = 1.9, p-value < 2.2 x 10−16, Fisher’s exact test) compared to bulk ATAC intronic/distal annotations, thus better capturing regions that harbor the overwhelming majority of regulatory elements (Extended Data Figure 2c)48. Next, to test the cellular resolution of our scATAC data, we leveraged differences in the strategies used for bulk (cMN3 without cMN4) vs. scATAC dissection (cMN3 and cMN4 combined) and performed cluster analysis on cMN3/4 samples only (ad hoc clusters C1-C20, Extended Data Figure 2a,d,e). We identified significant overlap between ad hoc clusters C18 and C20 scATAC peaks with bulk cMN3 peaks. Moreover, we confirmed accessibility of known cMN3 markers in C18 and C20, and cMN4 markers in C1949,50 (Extended Data Figure 2e). When comparing the scATAC peaks to bulk ATAC peaks in ENCODE9 sampled from major developing brain regions (forebrain, midbrain, hindbrain) at comparable timepoints, we observed diminished overlap for GFP-positive cMN samples relative to GFP-negative samples (Extended Data Figure 3a). Further stratifying scATAC peaks based on cell type specificity scores51 revealed that highly specific scATAC peaks had consistently lower bulk coverage than peaks with low specificity (Extended Data Figure 3b,c), consistent with findings that cell-type specific regulatory elements often act within small populations of cells and may be more difficult to capture and annotate with bulk methods52,53.
To further distinguish between rare, distinct cell types, we adopted an iterative clustering strategy (Methods)51. We first identified 23 major clusters that correspond with “ground truth” dissected cell types based on known anatomy (Figure 1c,d; Supplementary Table 3). Overall, GFP-positive clusters demonstrated much more uniform sample membership than GFP-negative clusters, as reflected by their differences in cluster homogeneity54 ( vs. ) and purity metrics (Figure 1d, Extended Data Figure 4a, Supplementary Table 4, Methods). Upon examining differentially accessible genes and elements through manual curation, review of the literature, and gene ontology analysis, we assigned provisional cell identities to the 23 major clusters, of which 10 clusters are cranial and 5 are spinal motor neurons based on dissection origin, and 9 are cranial and 4 are spinal motor neurons based on putative annotation (Supplementary Table 3). To further resolve the heterogeneity within clusters and to identify functionally and anatomically coherent subpopulations, we performed iterative clustering51 on each major cluster and identified 132 unique subclusters (Extended Data Figure 4bi,ii). Of these, 59 have GFP-positive membership > 90%, representing highly pure motor neuron populations (Extended Data Figure 4c). We observe even more distinct anatomic/temporal membership at the subcluster level, particularly for GFP-negative samples (subcluster homogeneity vs. ). These findings are consistent with highly dynamic and proliferative neurodevelopmental processes during this time period12. Neither major cluster nor subcluster membership was driven by experimental batch (Extended Data Figure 4d).
cMN cRE functional conservation between mouse and human
Common disease risk loci tend to overlap non-coding accessible chromatin in their corresponding cell types - including accessible chromatin that is more readily ascertained in mouse versus human tissues15,51. However, with the exception of a few exemplary elements (e.g., see refs 55-57), the extent of overlap between human/mouse elements underlying Mendelian traits is largely unknown. Therefore, to evaluate the functional conservation of cREs in our cranial motor neuron atlas, we performed in vivo humanized enhancer assays on a curated subset (n = 26) of our candidate scATAC peaks that were absent from the VISTA enhancer database58 and had peak accessibility/specificity in cMNs and general signatures of enhancer function (i.e., evolutionary conservation and non cMN-specific histone modification data59, Supplementary Table 5, Methods). These results validated our approach, as we detected positive enhancer activity (any reporter expression) in 65% (17/26) of candidates. Moreover, 11 of the 17 validated enhancers (65%, 42% overall) recapitulate the anatomic expression patterns (motor neuron expression) predicted from the scATAC accessibility profiles to the resolution of individual nuclei/nerves. By contrast, of 3,229 total non-coding elements assayed in the VISTA enhancer database, only 67 (2.1%) show reproducible evidence of enhancer activity in the cMNs. Thus, high quality single cell accessibility profiles are highly predictive of cell type specific regulatory activity.
Motif enrichment and footprinting reveal putative cMN regulators
To identify transcription factors/motifs responsible for cell type identity, we performed motif enrichment and aggregated footprinting analysis across all 23 major clusters and identified both known lineage-specific motif enrichment as well as new potential cMN transcription factor/motif relationships (Figure 2a,b). For example, we identified significant motif and footprinting enrichment of midbrain transcription factor OTX1 in populations corresponding to developing oculomotor/trochlear motor neurons (cluster cMN3/4.10) and the midbrain-hindbrain boundary (cluster MHB.7)60. We also identified notable footprints for ONECUT2 in multiple motor neuron populations, including cMN3/4, cMN7, and putative pre-enteric neural crest-derived cells (clusters cMN3/4.19, cMN7.11, enteric.17; Figure 2b). Importantly, we detected positive footprint signals for known lineage-specific regulators such as JunD footprints in the spinal and lymphoid lineages61,62 (clusters sMN.15, WBC.18) and GATA1 footprints in the erythroid lineage63 (cluster RBC.20; Figure 2b). Due to the relatively high homogeneity across the motor neuron clusters, we also compared motif enrichment across broader anatomic/functional classes of motor neurons and brain regions (Figure 2c). We identified strong enrichment of regional markers such as DMBX164 in midbrain samples (i.e., cMN3/4, cMN3/4neg). We also found motifs enriched among the ocular motor neurons (i.e., cMN3/4, cMN6) such as PAX5, providing new potential avenues for comparative studies.
Figure 2. Motif enrichment and aggregate footprint analyses distinguish cell type specific TF binding motifs.

a. Heatmap depicting enriched transcription factor binding motifs within differentially accessible peaks by cluster. Each entry is defined by its cluster identity (“clusterID.clusterNumber”). Corresponding cluster IDs and annotations are depicted. Color scale represents hypergeometric test p-values for each cluster and motif. Specific motifs and motif families vary significantly amongst clusters. Cluster annotations are defined in Supplementary Table 3.
b. Aggregated subtraction-normalized footprinting profiles for a subset of cluster-enriched transcription factors (OTX1, ONECUT2, JunD, and GATA1) from (a), centered on their respective binding motifs. Specific clusters display positive evidence for TF motif binding for each motif. Corresponding motif position weight matrices from the CIS-BP database are depicted above each profile. Cluster IDs with corresponding color are below.
c. Motif enrichment comparing broad classes of neuronal subtypes. Midbrain subtype contains motifs from cMN3/4neg cells; hindbrain from cMN6neg, cMN7neg, and cMN12neg cells; somatic MN from cMN3/4, cMN6, and cMN12 GFP-positive cells; branchial MN are from cMN7 GFP-positive cells; midbrain MN are cMN3/4 GFP-positive cells; hindbrain MN are cMN6, cMN7, and cMN12 GFP-positive cells; ocular MN are cMN3/4 and cMN6 GFP-positive cells; lower MN are cMN7, cMN12, and sMN GFP-positive cells. For each graph, the first listed subtype is enriched relative to the second listed subtype.
Assigning cell type specific cREs to their cognate genes
A chief barrier to interpreting non-coding regulatory elements is identifying their cis target genes. While enhancers often regulate adjacent genes, many important regulatory links also occur over much longer distances, including known disease causing events55,57,65-69. Therefore, we generated scRNA data from GFP-positive and -negative cMN3/4, 6, and 7 at e10.5 and e11.5 (Methods) using reporter constructs, microdissection, and collection strategies analogous to those use used to generate the scATAC datasets. We then integrated these scRNA data with the cMN chromatin accessibility data to generate peak-to-gene links at the single cell level for putative cREs within +/− 500kb of a given gene (see Methods70-72). In total, we identified 145,073 known and putative enhancers with peak-to-gene links across the 23 clusters (median = 2 genes per enhancer, range = 1-37; Supplementary Table 6).
Because the accuracy of peak-to-gene links inferred from separate assays of ATAC and RNA data (“diagonal integration”)73 depends heavily on cell pairings, we performed multiple analyses to ensure that both our ATAC-RNA pairings and gene expression estimates were well calibrated. We compared our imputed single cell gene expression estimates to independently collected in-house bulk RNAseq experiments from cMN3, 4, 6, and 7 at e10.5 and e11.5 annotated with ground truth dissection labels (Methods). We identified strong positive concordance between imputed gene expression and measured bulk RNAseq signal in the appropriate cell types (Figure 3a,b). We also found that our ATAC-RNA pairings and peak-to-gene links were sensitive to the cellular composition of our scRNA integration data. If the identical master peakset was compared to scRNA data from e10.5 to e11.5 mouse brain (“MOCA neuro”) or e9.5 to e13.5 mouse heart (“MOCA cardiac”)74 in place of our cMN-enriched scRNA data, we found fewer significant peak-to-gene links and fewer concordant cognate genes (Figure 3c-f; Methods).
Figure 3. Effects of RNA input data on peak-to-gene accuracy.
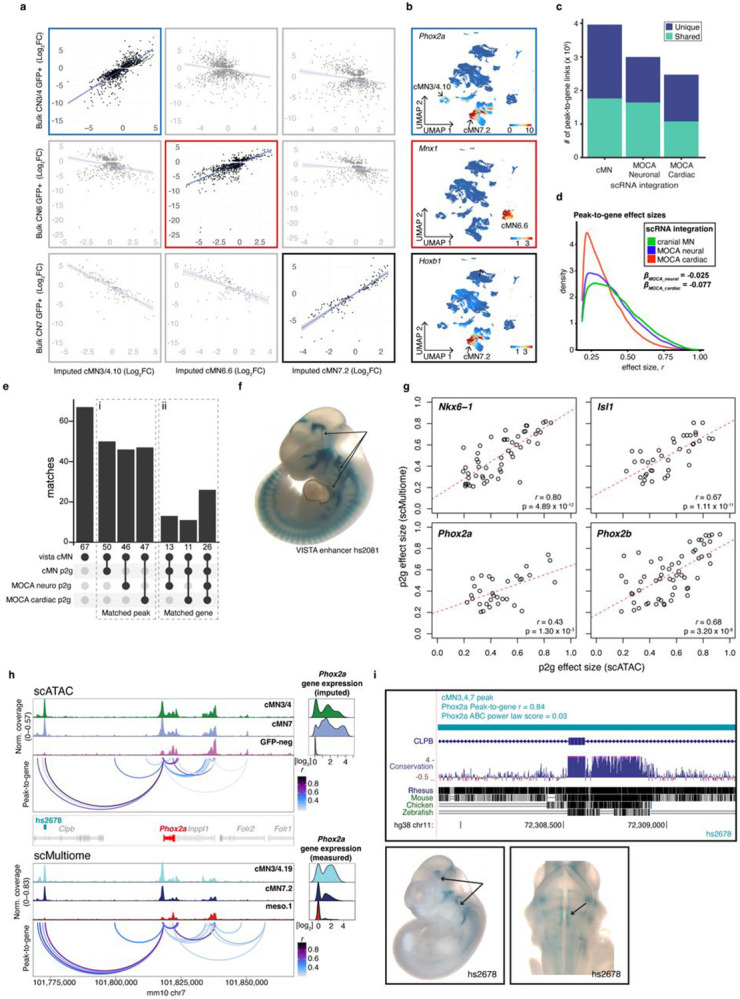
a. Scatterplots depicting imputed gene expression values projected onto scATAC clusters cMN3/4.10, cMN6.6, and cMN7.2 (x axis) versus measured gene expression values from independently collected bulk RNA-seq samples (y axis). Imputed gene expression shows a significant positive relationship when compared with corresponding bulk samples (cMN3/4, cMN6, and cMN7, respectively).
b. Feature plots depicting imputed gene expression for three classic cMN marker genes (Phox2a (top, boxed in blue), Mnx1 (middle, boxed in red), and Hoxb1 (bottom, boxed in black))37. Expression is restricted to corresponding clusters cMN3/4.10 (Phox2a), cMN6.6 (Mnx1), and cMN7.2 (Phox2a, Hoxb1) as expected.
c. Stacked barplot depicting total number of unique and shared peak-to-gene links using three distinct scRNA integration datasets against the common scATAC cMN peakset. cMN: scRNA-seq data from age- and dissection-matched, oversampled cranial motor neurons (this work). MOCA Neuro: age-matched, uniformly sampled embryonic neural tissue from the MOCA database. MOCA Cardiac: non-age-matched, uniformly sampled embryonic cardiac tissue from the MOCA dataset74.
d. Distribution of peak-to-gene effect sizes using different scRNA integration datasets (shared links only). Estimated effect sizes are significantly stronger using cMN scRNA integration when compared to MOCA neuro and MOCA cardiac integration.
e. Barplot depicting peak-to-gene elements from the three scRNA integrations overlapping 67 experimentally validated cMN enhancers (“vista cMN”, left). i. “Matched peak” indicates overlapping peaks irrespective of predicted cognate gene (middle). ii. “Matched gene” indicates both overlapping peaks and identical cognate gene within the VISTA cMN enhancers (right, note that the vista cMN enhancers to not have defined target genes). Toggling between scRNA integrations can alter or eliminate target gene predictions. i and ii represent intersect and distinct peaks, respectively.
f. In vivo enhancer assay for cMN VISTA enhancer hs2081 (lateral view). This enhancer overlaps a predicted peak-to-gene link using both cMN and MOCA cardiac scRNA input. However, enhancer activity is positive in cranial nerves 3, 7, and 12 (arrows) and negative in embryonic heart (dotted lines).
g. Comparing scATAC versus scMultiome peak-to-gene effect sizes for four motor neuron transcription factors (Nkx6-1, Isl1, Phox2a, and Phox2b)37. Each circle represents a peak. All four genes show a positive linear relationship across both assays.
h. scATAC (top) and scMultiome (bottom) accessibility profiles with peak-to-gene connections for a 100kb window centered around Phox2a. scATAC profiles are parsed by sample while scMultiome profiles are parsed by predicted cluster label. Peak-to-gene predictions are highly concordant across both assays. Novel cMN enhancer hs2678 is accessible in cMN3/4 and cMN7 and is predicted to enhance Phox2a by both scATAC (r = 0.84) and scMultiome (r = 0.69) peak-to-gene estimates.
i. (Top) hs2678 orthologous region in the human genome. hs2678 is 70.3 kb distal to human PHOX2A and is embedded in coding and intronic sequence of CLPB. (Bottom) In vivo enhancer assay using human hs2678 sequence is positive in cMN3 and cMN7 (arrows), recapitulating known Phox2a gene expression patterns41. Reporter expression views are shown as lateral (left) and dorsal through the 4th ventricle (right).
Next, we performed a joint ATAC-RNA coassay (“scMultiome”) on a subset of e11.5 GFP-positive cells represented in our main scATAC dataset (cMN3/4, cMN7, cMN12, sMN), thereby allowing us to benchmark our inferred ATAC-RNA pairings against direct experimental measurements (“vertical integration”; Extended Data Figure 5a-d). We found that scMultiome peak-to-gene links were highly concordant with our original scATAC peak-to-gene links (Figure 3g-i). We then examined the single cell accessibility profiles of four highly characterized cMN enhancers with known connection to the Isl1 gene – a cMN master regulator embedded in a gene desert (Figure 4a-c)58,75. Strikingly, both by diagonal and vertical integration, we found that for these four enhancers (mm933, CREST1/hs1419, CREST3/hs215, and hs1321), chromatin accessibility alone was a significant predictor of in vivo Isl1 expression patterns in the anatomically appropriate cMN (Figure 4d,e; Extended Data Figure 5d; Wald test p-value = 0.011; Methods).
Figure 4. Exceptional gene regulation of cranial motor neuron master regulator Isl1.
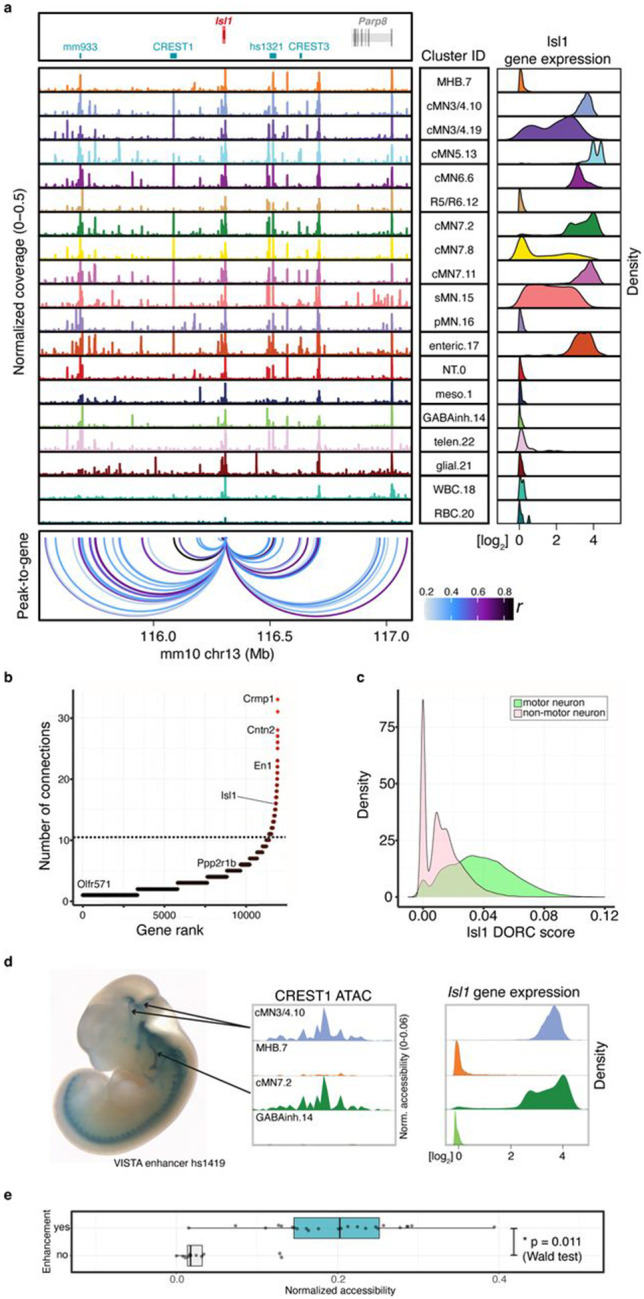
a. Pseudobulked chromatin accessibility profiles for all annotated clusters over a 1.5 Mb window centered about Isl1. Imputed gene expression profiles for each cluster are shown to the right. Isl1 is located within a gene desert with the nearest up- and downstream flanking genes 1.2 and 0.7 Mb away, respectively. Peak-to-gene predictions match known Isl1 enhancers (CREST1 in motor neurons and CREST3 in sensory neurons75; mm933 in multiple cranial motor nerves, dorsal root ganglion, and nose; hs1321 in multiple cranial motor nerves and forebrain) and identify additional putative enhancers surrounding Isl1.
b. The number of normalized regulatory connections for each rank ordered gene. Isl1 ranks in the top 1% of all genes with at least one regulatory connection. The inflection point of the plotted function is demarcated with a dotted line.
c. Per-cell Domain of Regulatory Chromatin (DORC) scores for Isl1 gene. DORC scores are significantly higher for cells from motor neuron clusters relative to non-motor neuron clusters (p-value < 1 x 10−15, ANOVA).
d. (Left) Lateral whole mount In vivo reporter assay testing CREST1 (VISTA enhancer hs1419) enhancer activity. CREST1 drives expression in cranial nerves 3, 4, and 7 (black lines; there is also expression in trigeminal motor nerve). (Right) Single cell ATAC profiles and imputed gene expression for a subset of corresponding clusters. CREST1 accessibility and Isl1 gene expression are positively correlated with in vivo expression patterns.
e. Boxplot depicting normalized accessibility levels for enhancers CREST1, CREST3, mm933, and hs1321 within nine scATAC clusters corresponding to distinct anatomic regions. Manually scored enhancer activity (“enhancement”) is significantly correlated with normalized accessibility (p-value = 0.011, Wald test). Center line: median; box limits: upper and lower quartiles; whiskers – 1.5 x interquartile range.
Lastly, we integrated histone modification signatures into our enhancer predictions by performing H3K27Ac scCUT&Tag on e11.5 GFP-positive cMN3/4, cMN6, and cMN7 and e10.5 cMN7 (7 replicates total) and generated Activity-by-Contact (ABC) enhancer predictions for each cell type (Methods76,77). Of 6,072 total ABC enhancers, 4,925 (81%) directly overlapped our peak-to-gene links, including multiple in vivo ground truth enhancers (Extended Data Figure 6a, Figure 3i, Figure 4a, Supplementary Table 7). Because availability of cell type specific experimental data can be a limiting factor in accurate enhancer prediction, we assessed the relative contribution of cell type-specific chromatin accessibility versus histone modification data to ABC prediction accuracy. Specifically, among 67 annotated cMN enhancers in the VISTA enhancer database (visualized at e11.5 by presence of beta-galactosidase in the nucleus and/or nerve), 49 had some evidence of expression in cranial nerve (CN)7. Among these, we identified seven that had both visible CN7 expression and ABC cMN7 enhancer predictions at e11.5. For all seven enhancers (100%), ABC cognate gene predictions were concordant with peak-to-gene predictions. We then reran our ABC predictions, replacing either our cMN7 ATAC data with mouse embryonic limb e11.5 ATAC data (ENCODE ENCSR377YDY; “Limb ATAC”) or our cMN7 histone modification data with mouse limb histone modification data (ENCODE ENCSR897WBY; “Limb H3K27Ac”) and compared predictions. Substituting limb ATAC for cMN7 ATAC data resulted in only 14% (1/7) concordance, while substituting limb H3K27Ac for cMN7 H3K27Ac data resulted in 57% (4/7) concordance (Extended Data Figure 6b). Thus, for this curated set of data, we find that cell type-specific ATAC signal is a better predictor of reproducible cognate gene predictions than cell type-specific histone modification signal or non-cell-type-specific ATAC signal.
Embryonic mouse chromatin accessibility atlas
In summary, we generated a chromatin accessibility atlas of the developing cMNs and surrounding cell types (reference tracks in the UCSC Genome Browser will be provided here). We combined GFP-positive (n = 49,708) and -negative (n = 36,381) cells to improve joint peak calling performance and to capture potential regional heterogeneity of non-motor neuron cell types as well as motor neuron progenitors78. Cluster analysis revealed 9 putative cMN, 4 putative sMN, and multiple non-MN/non-neuronal clusters (of 23 total). Although sMNs are not directly implicated in CCDDs, they may provide value for comparative studies with cMNs79,80. We also performed iterative clustering to identify 132 subclusters, of which 58 are highly pure groups of motor neurons. Although we are currently unable to annotate subclusters, more detailed spatial and developmental profiling of the cMN subnuclei may help to identify functionally-relevant groups of cells and/or cell states. Finally, a high quality and cell type-specific catalog of cMN elements and their cognate genes can be used to interpret and prioritize CCDD variants, as we describe below.
Human phenotypes and genome sequencing
We enrolled and phenotyped 899 individuals (356 affected, 543 family members) across 270 pedigrees with CCDDs. 202 probands were sporadic (simplex) cases enrolled as trios, while 42 and 19 pedigrees displayed clear dominant or recessive inheritance patterns, respectively (Supplementary Table 8). Of note, the dominant pedigrees included 3 with CFP that we have reported to harbor pathogenic SNVs in a non-coding peak, “cRE2”, within the HCFP1 locus on chromosome 334. The CCDDs included congenital fibrosis of the extraocular muscles (CFEOM), congenital ptosis (CP), Marcus Gunn jaw winking (MGJW), fourth nerve palsy (FNP), Duane retraction syndrome (DRS), congenital facial palsy (CFP), and Moebius syndrome (MBS) (Supplementary Table 8). Importantly, these CCDD phenotypes can be connected to maldevelopment of their disease-relevant cMNs: CFEOM to cMN3/4, CP to the superior branch of cMN3, FNP to cMN4, DRS to cMN6, CFP to cMN7, and MBS to cMNs 6 and 7 (Figure 1a, Supplementary Table 1). Affected individuals could have isolated or syndromic CCDDs.
We performed whole genome sequencing (WGS) and variant calling of the 899 individuals (Methods). First, to generate a comprehensive and unbiased set of genetically plausible candidates, we performed joint single nucleotide variant (SNV) and insertion/deletion (indel) genotyping, quality control, and variant frequency estimation from > 15,000 WGS reference samples in the Genome Aggregation Database (gnomAD)81,82. We identified 54,804,014 SNV/indels across the cohort. Of these, 1,150,021 (2.1%) were annotated as exonic, 18,761,202 (34.2%) intronic, 34,512,518 (63.0%) intergenic, and 364,300 (0.7%) within promoters. We next performed initial SNV/indel variant filtering based on established and custom criteria, including genotype quality, allele frequency, and conservation (Methods)83,84. We incorporated family structures to include or exclude genetically plausible candidates that are consistent with known modes of Mendelian inheritance. Applying this approach to the 54,804,014 SNVs/indels across our cohort, we identified 26,000 plausible candidates (mean = 101 variants per pedigree). We also performed short read structural variant (SV) discovery using an ensemble SV algorithm (GATK-SV) that was comparable to SVs generated in gnomAD and the 1000 Genomes Project81,85 and identified 221,857 total SVs (including transposable elements and other complex events). These WGS from deeply phenotyped CCDD pedigrees present a rich catalog of otherwise unannotated candidate Mendelian disease variants, as reflected in our report of noncoding SNVs and duplications as a cause of isolated facial weakness34.
Integrating epigenomic filters with human WGS variants
To further refine the 26,000 CCDD candidate SNVs/indels, we eliminated from further analysis 37 pedigrees definitively solved by coding variants and reported separately, and then applied cell type-specific filters from our scATAC peakset to each CCDD phenotype (Methods). We identified 5,353 unique segregating SNVs/indels (3,163 de novo/dominant, 1,173 homozygous recessive, and 1,017 compound heterozygous) that overlapped cMN-relevant peaks of accessible chromatin (23.6 and 13.6 candidates per monoallelic and biallelic pedigree, respectively). Applying an analogous cell type-aware framework for SVs, we identified 115 candidates (72 deletions, 27 duplications, 1 inversion, 13 mobile element insertions, and 2 complex rearrangements encompassing multiple classes of SVs). There was substantial overlap between candidate variants and CCDD-relevant cMN peaks when compared to size-matched randomized peaks (median de novo Z-score = 10.9, median dominant inherited Z-score = 30.1, p-value < 2.0 x 10−4, permutation test; Supplementary Table 9). Using these 5,468 cell type-aware non-coding CCDD candidate SNVs/indels/SVs and ATAC-based cMN enhancers, we next identified strong candidate variants using gene-centric and peak-centric approaches.
We adopted a gene-centric aggregation approach by first identifying non-coding candidate variants connected to a restricted set of 16 known CCDD disease genes19,21-26,28,42,86-93. We identified non-coding variants connected to four: MAFB, PHOX2A, CHN1, and EBF3 (Table 1). We also identified compound heterozygous variants connected to ISL1 in a proband with CFP; ISL1 is not a known disease gene but is a master cMN regulator (Extended Data Figure 7a,b). Extending this approach to the entire genome, we identified 559 genes with multiple connected peaks containing dominant candidate variants (“multi-hit genes”, range of connected variants per gene = 2-6, Supplementary Table 10).
Table 1. Non-coding candidate variants and putative target genes.
1Coding loss-of-function intolerance - https://doi.org/10.1038/s41586-020-2308-7; 2Coding dosage sensitivity - https://doi.org/10.1016/j.cell.2022.06.036; 3Non-coding mutational constraint (1 kb windows) - https://doi.org/10.1101/2022.03.20.485034; †Multi-hit gene; ††Multi-hit peak; †††non-coding deletion; *mean value across deleted interval; (Y) denotes established CCDD gene for stated phenotype; AD: autosomal dominant/de novo, ar(h): autosomal recessive homozygous, ar(ch): autosomal recessive compound heterozygous, I: intronic, P: promoter, D: distal, LoF: loss-of-function, GoF: gain-of-function.
| CCDD | Pedigree | Inheritance | Non-coding variant(hg38) |
Peak Type | Nearest gene | Target gene | Distance to target (kb) |
Reporter ID | Peak to gene r | Peak to gene FDR |
gnomAD allele frequency |
Predicted mechanism |
SAD Z-score | Target gene loeuf1 |
Target gene pHaplo2 |
Target gene pTriplo2 |
Non-coding Z-score3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CFEOM | S25 | AD | chr10:129794079 TTGAG>T | D | EBF3 | EBF3† (Y-DRS) | 170 | hs2776 | 0.24 | 2.90E-07 | 8.37E-05 | LoF | −11.77 | 0.15 | 1.00 | 1.00 | 3.10 |
| MGJW | S176 | AD | chr10:129884231 C>A | I | EBF3 | EBF3† (Y-DRS) | - | hs2775 | 0.29 | 3.89E-10 | 4.88E-05 | GoF | 0.11 | 0.15 | 1.00 | 1.00 | 3.74 |
| Ptosis | S95 | AD | chr10:129944464 G>C | I | EBF3 | EBF3† (Y-DRS) | - | hs2774 | 0.21 | 7.76E-06 | - | GoF | 0.98 | 0.15 | 1.00 | 1.00 | 5.14 |
| DRS | S12 | ar(h) | chr11:72394626 C>G | I | CLPB | PHOX2A (Y-CFEOM) | 156 | - | 0.26 | 1.09E-08 | 1.41E-03 | GoF | 0.18 | 0.80 | 0.76 | 0.98 | 2.32 |
| Ptosis | S32 | AD | chr2:175005662 C>T†† | P | CHN1 | CHN1 (Y-DRS) | - | - | 0.48 | 1.31E-28 | 1.39E-04 | LoF | −0.38 | 0.57 | 0.41 | 0.72 | 2.59 |
| CFEOM/DRS | S251 | AD | chr2:175006051 GCTT>G†† | P | CHN1 | CHN1 (Y-DRS) | - | - | 0.48 | 1.31E-28 | - | GoF | 2.29 | 0.57 | 0.41 | 0.72 | 2.08 |
| DRS | S230 | AD | chr20:40866929-40945626††† | D | TOP1 | MAFB (Y-DRS) | 256 | hs2769 hs2770 | 0.23* | 1.19E-05* | - | - | - | 0.40 | 0.94 | 1.00 | 2.19* |
| CFP | S205 | ar(ch) | chr5:51172762 T>A | D | ISL1 | ISL1 | 221 | hs1321 | 0.74 | 1.36E-86 | 2.26E-03 | LoF | −0.41 | 0.23 | 0.95 | 0.85 | −2.28 |
| CFP | S205 | ar(ch) | chr5:51172961 T>G | D | ISL1 | ISL1 | 221 | hs1321 | 0.74 | 1.36E-86 | 2.33E-03 | LoF | −0.12 | 0.23 | 0.95 | 0.85 | −2.28 |
| DRS | S190, S238 | ar(h) | chr22:27493955-27497536††,††† | D | MN1 | MN1 | 307 | hs2757 | - | - | 1.38E-04 | - | - | 0.48 | 0.99 | 0.92 | 0.29* |
| DRS | S191 | ar(ch) | chr17:1455690 G>A†† | I | CRK | CRK | - | - | - | - | - | GoF | 0.44 | 0.34 | 0.97 | 1.00 | 0.30 |
| DRS | S191 | ar(ch) | chr17:1456361 G>A†† | P | CRK | CRK | - | - | - | - | 1.51E-03 | LoF | −1.24 | 0.34 | 0.97 | 1.00 | - |
| DRS | S211 | ar(ch) | chr17:1455565 C>T†† | I | CRK | CRK | - | - | - | - | 1.19E-04 | GoF | 0.49 | 0.34 | 0.97 | 1.00 | 0.30 |
| DRS | S211 | ar(ch) | chr17:1456436G C>G†† | P | CRK | CRK | - | - | - | - | 3.77E-04 | LoF | −12.28 | 0.34 | 0.97 | 1.00 | - |
| DRS | S211 | ar(ch) | chr17:1456438 G>A†† | P | CRK | CRK | - | - | - | - | 3.77E-04 | LoF | −2.06 | 0.34 | 0.97 | 1.00 | - |
| DRS | WL | AD | chr17:48003752 A>C†† | D | CDK5RAP3 | CDK5RAP3 | 22 | hs2777 | 0.57 | 8.04E-43 | - | GoF | 4.31 | 0.97 | 0.24 | 0.54 | 1.94 |
| MBS | S174 | ar(ch) | chr17:48003557 C>G†† | D | CDK5RAP3 | CDK5RAP3 | 22 | hs2777 | 0.57 | 8.04E-43 | 4.04E-03 | LoF | −0.15 | 0.97 | 0.24 | 0.54 | 1.94 |
| MBS | S174 | ar(ch) | chr17:48003826 C>T†† | D | CDK5RAP3 | CDK5RAP3 | 22 | hs2777 | 0.57 | 8.04E-43 | 9.42E-04 | GoF | 1.69 | 0.97 | 0.24 | 0.54 | 1.94 |
| CFP | S156 | AD | chr3:128459417G>C†† | D | DNAJB8 | GATA2 | 7 | - | 0.28 | 6.08E-10 | - | LoF | −4.88 | 0.34 | 0.98 | 0.87 | - |
| CFP | S180 | AD | chr3:128459454A>G†† | D | DNAJB8 | GATA2 | 7 | - | 0.28 | 6.08E-10 | 3.95E-05 | GoF | 2.88 | 0.34 | 0.98 | 0.87 | - |
| CFP | S194 | AD | chr3:128459455G>A†† | D | DNAJB8 | GATA2 | 7 | - | 0.28 | 6.08E-10 | - | GoF | 11.40 | 0.34 | 0.98 | 0.87 | - |
EBF3, which encodes the EBF transcription factor 3, is an example of both a CCDD gene and a multi-hit gene. Monoallelic EBF3 loss-of-function (LoF) coding mutations cause Hypotonia, Ataxia, and Delayed Development Syndrome (HADDS)94, and two individuals are reported with HADDS and DRS, one with a coding missense variant and one with a splice site variant92,95. We identified a series of coding and noncoding EBF3 variants (Supplementary Table 11). Two probands with DRS have large de novo multi-gene deletions (Figure 5a), and one proband with fourth nerve palsy has a de novo stop-gain coding variant (Figure 5b). These three individuals also have phenotypes consistent with HADDS. We also identified three inherited non-coding variants with peak-to-gene connections to EBF3 (Figure 5b). Pedigrees S25 (distal indel), S176 (intronic SNV), and S95 (intronic SNV) segregate non-coding candidate variants with isolated CFEOM, MGJW, and ptosis, respectively. The multiple ocular CCDD phenotypes we observed potentially reflect pleiotropic consequences of EBF3 variants, a phenomenon previously observed for coding mutations in other CCDD genes96. Moreover, the differences in syndromic versus isolated phenotypes may reflect more cell type-specific effects of non-coding variants. Indeed, multiple Mendelian disorders with non-coding etiologies are restricted to isolated cell types or organ systems57,65,97-100. Notably, EBF3 is broadly expressed across cMNs (Figure 5c) and is one of the most constrained genes in the human genome as measured by depletion of coding LoF variants in gnomAD and SV dosage sensitivity (loeuf = 0.1500 and pHaplo = 0.9996, respectively; Figure 5d)82,101,102. We observed exceptional conservation of non-coding elements within EBF3 introns, comparable to or exceeding exonic conservation. This includes the ultraconserved element UCE318 (Figure 5b,e) located in intron 6 with a peak-to-gene link to EBF3 (r = 0.69, FDR = 6.2 x 10−69). We also detected a peak-to-gene link from VISTA enhancer hs737 to EBF3 (r = 0.60, FDR = 4.8 x 10−49), an element located > 1.2 Mb upstream of the gene that was previously reported to be linked to EBF3 and to harbor de novo variants associated with autism with hypotonia and/or motor delay103. We did not observe any candidate variants in UCE318, consistent with extreme depletion of both disease-causing and polymorphic variation within ultraconserved elements104, nor in hs737, consistent with its non-CCDD phenotype.
Figure 5. An integrated coding/non-coding candidate allelic series for EBF3.

a. Window depicting the terminal arm of chr10q (top). Large de novo deletions in two trios (middle, bottom) with simplex syndromic DRS (S233, S131) overlap multiple coding genes including EBF3 (boxed), an exceptionally conserved gene at the coding and non-coding level.
b. Nominated coding and non-coding SNVs and indels connected to EBF3. For each variant, the subject’s WGS ID code, CCDD phenotype (and if isolated or syndromic), the variant coordinate in NG_030038.1 (and if coding or noncoding and if familial or de novo) is indicated. Variants 5 and 8 are reported previously in DECIPHER and elsewhere92,95. Peak-to-gene links containing variants connected to EBF3 are depicted by curved lines. EBF3 contains highly conserved non-coding intronic elements, including ultra-conserved element UCE 318 in intron 6, whose sequence drives strong expression in the embryonic hindbrain (VISTA enhancer hs232, see (e) below).
c. Imputed gene expression profiles for Ebf3. Ebf3 is broadly expressed among the cMNs.
d. EBF3 is exceptionally intolerant to loss-of-function, gene dosage, and missense variation. Density plots depict genome-wide distribution of loss-of-function constraint (“loeuf”, “pLI”)82,125, probability of haploinsufficiency (“pHaplo”)101, and missense constraint (“z-score”)126. Respective scores exceeding thresholds of 0.35, 0.9, 0.84, and 2.0 are colored red. EBF3 (dotted lines) ranks as the 563rd, 861st, 3rd, and 508th most constrained gene in the genome, respectively. Distributions are rescaled for consistent sign and ease of visualization.
e. Lateral view of in vivo reporter assay testing UCE 318 (VISTA enhancer hs232), a putative EBF3 enhancer (peak-to-gene r = 0.42, FDR = 6.72 x 10−22). Strong reporter expression is observed in the embryonic hindbrain (arrow).
Second, we took a peak-centric approach by examining all 5,468 (5,353 SNV/indels, 115 SVs) cell type aware non-coding variants, irrespective of cognate gene. When aggregating variants within appropriate cMN peak with corresponding CCDD phenotype, we identified 28 peaks harboring variants in more than one pedigree (“multi-hit peaks”). Fourteen multi-hit peaks contained variants obeying a dominant mode of inheritance (28 unique dominant/de novo variants with one variant present in two unrelated families, and including the 3 pathogenic chromosome 3 “cRE2” SNVs that cause CFP34), and 14 multi-hit peaks contained variants obeying a recessive mode of inheritance (35 unique recessive variants; Supplementary Table 12). Moreover, 10 of these multi-hit peaks were also linked to multi-hit genes. Because enhancers confer cell type-specific function, we reasoned that true functional non-coding SNV/indels are less likely than coding variants to cause syndromic, multi-system birth defects. Interestingly, when stratifying pedigrees by isolated/syndromic status, we found a significant overrepresentation of isolated CCDD phenotypes for our dominant multi-hit peaks (OR = 5.9, p-value = 2.3 x 10−3, Fisher’s exact test), but not for our recessive multi-hit peaks (OR 0.8, p-value = 0.64).
Among the multi-hit peaks, we identified 3.6 kb homozygous non-coding deletions centered over peak hs2757 in two probands with DRS; in each case, the consanguineous parents were heterozygous for the deletion. The probands had extended runs of homozygosity with a shared 16 kb haplotype surrounding the deletion, consistent with a founder mutation (Figure 6a-c). hs2757 is broadly accessible in multiple cMN populations, including cMN6, and is located 307 kb upstream of its nearest gene, MN1; MN1 imputed gene expression estimates revealed widespread expression across all sampled cell types, including cMN6 (Figure 6d)82,101. Monoallelic LoF coding mutations in MN1 cause CEBALID syndrome, a disorder affecting multiple organ systems. A subset of individuals with coding variants in MN1 are reported to have CEBALID syndrome with DRS89. MN1 is exceptionally constrained against LoF variation and dosage changes (loeuf = 0.087; pHaplo = 0.9901, Figure 6e)82,101 We performed in vivo enhancer testing on hs2757 which revealed reporter expression in a subset of tissues with known Mn1 expression105, including expression in the hindbrain overlapping the anatomic territory of cMN6 (Figure 6f). Surprisingly, in this case we did not observe a peak-to-gene link between hs2757 and Mn1 and did observe links with genes C130026L21Rik (whose sequence maps to a different chromosome in human) and Pitpnb (Supplementary Table 12). Multiple scenarios may explain this result, such as active Mn1 enhancement occurring prior to the mouse e10.5-e11.5 window investigated here. Alternatively, our regression-based peak-to-gene estimates may be less sensitive at detecting enhancers for ubiquitously expressed genes, a phenomenon previously observed for other enhancer prediction methods76.
Figure 6. MN1 enhancer deletions across multiple CCDD pedigrees.

a. IGV screenshot depicting 3.6 kb non-coding deletions in two probands with DRS from separate consanguineous pedigrees (S190, S238).
b. ddPCR copy number estimates of deletions. For each pedigree, the affected proband is homozygous recessive for the deletion with one heterozygous allele inherited from each parent. Error bars denote 95% confidence intervals.
c. Genomic context of the non-coding deletions. The deletions (red bar below chr 22 ideogram) fall within extended runs of homozygosity (grey bars above ideogram, 19.5 Mb, 18.8 Mb, respectively, of which 16 kb surrounding the deletion is shared between the probands) and eliminates putative enhancer hs2757 (green bar below ideogram) located 307 kb from nearest gene MN1.
d. hs2757 chromatin accessibility (left) and Mn1 imputed gene expression (right) profiles in the cMNs and surrounding cell types. Mn1 is widely expressed across multiple midbrain/hindbrain cell types, and hs2757 is accessible across multiple cell types, including cMN6.
e. Density plots depicting genome-wide distribution of loss-of-function constraint (“loeuf”, “pLI”)82,125, and probability of haploinsufficiency (“pHaplo”)101 metrics. Respective scores exceeding thresholds of 0.35, 0.9, 0.84, and 2.0 are colored red. MN1 (dotted lines) ranks as the 131rd, 605th, and 402nd most constrained gene in the genome, respectively. Distributions are rescaled for consistent sign and ease of visualization.
f. In vivo reporter assay testing hs2757 enhancer activity (humanized sequence). Lateral (left) and dorsal (right) whole mount lacZ staining reveals hs2757 consistently drives expression in midbrain and hindbrain tissue, including the anatomic territory of cMN6.
Mechanistic insights of non-coding disease variants
Mendelian disease variant interpretation often relies on variant level predictions of pathogenicity106,107. However, such prediction algorithms are typically agnostic to cell type- or disease-specific information. More recent approaches have incorporated cell type-specific epigenomic data to annotate non-coding variants in common diseases53,108,109. To leverage our cell type-specific accessibility profiles for variant level functional interpretation, we trained a convolutional neural network110 to generate cell type-specific predictions of chromatin accessibility for each cranial motor neuron population. When evaluating held-out test data, we consistently observed high concordance between our accessibility predictions and true scATAC coverage for each cell type (median Pearson’s r = 0.84; range = 0.81 to 0.95; Figure 7a; Extended Data Figure 8a-c). Thus, to predict the effects of participant variants on element accessibility, we used our trained model to generate cell-type specific SNP Accessibility Difference (SAD)110 scores.
Figure 7. scATAC-trained convolutional neural network accurately predicts cell type specific accessibility status and human mutation effects in a transiently developing cell type.

a. Neural net predicted chromatin accessibility profiles (red) compared to actual scATAC sequencing coverage (black) for a region of mouse chromosome 6 in three cell types (cMN7 e10.5, cMN7 e11.5, and cMN12 e11.5). The grey box highlights a transient 678 bp peak (cRE2) that is accessible in cMN7 e10.5, but not cMN7 e11.5 or cMN12 e11.5. SNVs within the human orthologous peak cRE2 cause congenital facial weakness, a disorder of cMN7.
b. Neural net-trained in silico saturation mutagenesis predictions for specific nucleotide changes in human cRE2 for cMN7 e10.5, cMN7 e11.5, and cMN12 e11.5. Predicted loss-of-function nucleotide changes are colored in blue and gain-of-function in red. Predictions for four known loss-of-function pathogenic variants (chr3:128178260 G>C, chr3:128178261 G>A, chr3:128178262 T>C, chr3:128178262 T>G) are boxed. All four pathogenic variants are predicted loss-of-function for cMN7 e10.5, but not cMN7 e11.5 or cMN12 e11.5.
c. Pseudobulk accessibility profiles of cRE2 (red box) CN7 e10.5 for wildtype and two CRISPR-mutagenized mouse lines ( and ) show a qualitative reduction in cRE2 scATAC sequencing coverage, consistent with in silico saturation mutagenesis predictions. Each pseudobulk profile represents normalized sequencing coverage across two biological replicates.
d. Locus-specific footprinting evidence overlapping cRE2. A 792 bp window showing sequencing coverage for cMN7 e10.5 after correcting for Tn5 insertion bias. The NR2F1 transcription factor binding site is mutated in individuals with HCFP1-CFP and overlaps a local minimum in scATAC coverage. TOBIAS footprinting scores for cRE2 wildtype, , and are depicted in solid, dashed, and dotted lines, respectively. Wildtype footprinting scores are higher than mutant scores.
e. Stacked barplot depicting wildtype versus mutant scATAC read counts over a 7.7 kb window for cMN7 e10.5 in heterozygote embryos. cRE2 mutant alleles are consistently depleted across two biological replicates (; p-value = 2.4 x 10−14, binomial test).
Our peak-centric approach successfully re-identified the HCFP1 cRE2 SNVs that we reported to be pathogenic for CFP34, and scATAC data revealed that cRE2 was accessible in cMN7 at mouse e10.5 but not e11.5 (Figure 7a). Examining cRE2 SNV SAD scores, we found that all four Cluster A LoF variants were predicted to close the chromatin (SAD Z-scores of −4.88, −3.60, −6.29, and −3.93). Moreover, these predicted variant effects were specific to cMN7 at e10.5 (but not e11.5, Figure 7b), further underscoring the importance of accurately parsing both cell type and developmental cell state. We then experimentally corroborated the predicted variant effect on chromatin accessibility by performing scATAC on two CRISPR-mutagenized mouse lines harboring HCFP1 cRE2 Cluster A SNVs (previously reported and new mouse models)34. Consistent with our machine learning predictions, we observed subtle yet consistent reductions in cis chromatin accessibility for both mutant lines when compared to wildtype (4/4 replicates total; mean normalized mutant / wildtype coverage = 0.59; Figure 7c). We also found positive evidence for site-specific footprinting overlapping the cRE2 NR2F1 binding site in wildtype, but not in the two mutant lines (Figure 7b,d), consistent with results from targeted antibody-based assays34. Finally, to circumvent batch and normalization effects across separate experiments, we performed scATAC on embryos from wildtype-by-mutant crosses from and directly measured the resultant heterozygous mutant allele fraction in cis (“binomial ATAC”; Figure 7e). This approach generates an internally calibrated estimate of effect size and is sufficiently powered to detect true differences at relatively low sequencing coverage (i.e., chromatin accessibility profiles of rare or transiently developing cell types). We found a significant depletion of Fam5 mutant alleles across multiple replicates, again consistent with a LoF mode of pathogenicity (wildtype / mutant counts = 4.2; p-value = 2.4 x 10−14; binomial test). These multiple lines of evidence, both at the epigenome-wide level and at a well-characterized individual locus provide support that our machine learning model is well calibrated and not overfitted.
We next examined the predictions of the neural net at epigenome-wide level, and among our 5,353 cell type-aware candidate SNVs/indels, identified 114 additional variants with normalized absolute SAD Z-scores > 2; that is, variants predicted to significantly increase or decrease accessibility in cis within their disease-relevant cellular context, including 7 variants linked to multi-hit genes (Supplementary Table 13). When incorporating these SAD scores, we identified several cell type-aware candidate variants and peaks with convergent lines of evidence. First, several of the non-coding variants connected to known CCDD genes had significant SAD scores (Table 1). The EBF3 non-coding variants chr10:129794079TTGAG>T, chr10:129884231C>A, and chr10:129944464G>C had SAD scores of −11.77, +0.11, and +0.98, respectively. The variant connected to CHN1 segregated in a parent and child with a mixed CFEOM-DRS phenotype was predicted to increase accessibility (SAD Z-score = +2.29). This is notable because CHN1 coding variants result in atypical DRS through a gain-of-function mechanism23,43,111. Second, combining multiple layers of evidence can be used to elevate candidate variants connected to potentially novel CCDD disease genes. For example, compound heterozygous variants in two DRS probands in the multi-hit CRK promoter region had significant negative scores consistent with LoF (SAD Z-scores = −13.69, −2.06; Supplementary Table 12). Such highly annotated non-coding variants are attractive candidates for downstream functional validation, as they provide distinct, refutable predictions for gene targets, cell types, and effect on accessibility.
Nominated cell type-specific variants alter expression in vivo
Although we show that single cell chromatin accessibility is a strong predictor of cMN enhancer activity, even highly conserved and presumably functional enhancers can be surprisingly robust to mutagenesis8,112-114. Therefore, to evaluate the functional consequences of our nominated CCDD variants, we selected 33 elements harboring cell type-aware candidate SNVs for in vivo humanized enhancer assays. For testing, we prioritized these variants based on multiple annotations from our framework, including conservation, significant SAD scores, multi-hit peaks/genes, and cognate gene predictions (Supplementary Table 14). We first screened the wildtype human enhancer sequences and detected positive enhancer activity in 82% (27/33) of candidates. Combining these with the 26 previously tested, we found enhancer activity in 44/59 total (75%). Importantly, we note that these elements were not selected randomly and therefore not intended to reflect generalizable patterns across the genome.
Next, we tested 4 of the 27 positive elements by introducing the nominated CCDD SNVs into the wildtype sequence. Remarkably, one mutant enhancer harboring multiple candidate variants for DRS and MBS (“hs2777-mut”) showed visible gain of expression compared to wildtype (“hs2777”), including in midbrain, hindbrain, and neural tube (Extended Data Figure 9a,b). Wildtype hs2777 is accessible across multiple cell types and has peak-to-gene links to seven genes (Cdk5rap3, Nfe2l1, Sp2, Tbx21, Npepps, Socs7, and Snx11), and ABC enhancer prediction for Cdk5rap3, specifically to cMN7 at e10.5. hs2777-mut contains four SNVs (1 DRS, 2 MBS, 1 off-target, mutating 0.21% of original wildtype base pairs; Extended Data Figure 9c,d). To better decompose the individual effects of these variants, we performed in silico saturation mutagenesis across the entire hs2777 sequence (Extended Data Figure 9e). We observed notable gain-of-function effects for two of the three on-target SNVs (DRS “Variant C”, and MBS “Variant D”; chr17:48003826C>T and chr17:48003752A>C) within the affected cell types, with corresponding SAD Z-scores ranging from +1.12 to +4.34.
DISCUSSION
We have developed a publicly available atlas of developing cranial motor neuron chromatin accessibility and have combined it with cell type-specific histone modification and in vivo transgenesis information to generate a reference set of enhancers with cognate gene predictions in a set of rare, transiently developing cell types. Such a resource can be used to discover highly specific cREs and target genes underlying the molecular regulatory logic of cMN development. Furthermore, we can leverage known properties of the cMNs to inform comparative studies across diverse cell types. For example, the ocular cMNs are known to be selectively resistant to degeneration (compared to sMNs) in diseases such as ALS. Therefore, understanding the differentially accessible cREs that underlie differences between cMNs/sMNs could render important clues to the mechanisms of selective resistance/vulnerability and ultimately open new therapeutic avenues80. Finally, a deeply sampled, highly specific chromatin accessibility atlas may help to learn generalizable features that predict enhancer activity in additional cell types. Importantly, cranial nerve expression is a core readout for tested cREs in the VISTA enhancer database, thereby providing invaluable ground truth data at an overlapping developmental timepoint (e11.5)58.
We used this reference to nominate and prioritize non-coding variants in the CCDDs, a set of Mendelian disorders altering cMN development and demonstrate that principled prioritization approaches can select appropriate candidates for downstream functional validation (e.g., transgenic reporter assays, non-coding in vivo disease models, etc.), which are otherwise often costly and labor-intensive with high rates of failure. To aid in interpretation, we connected non-coding variants to their cognate genes using imputed gene expression values from separate assays (diagonal integration). This approach allowed us to leverage existing information of cognate coding genes, including known disease associations and coding constraint82. Moreover, such integrated cell type-aware datasets provide important context to cell type-agnostic estimates of non-coding constraint (discussed in ref. 115). When applying this framework to our CCDD cohort, we achieved a search space reduction of 4 orders of magnitude, making non-coding candidate sets human-readable and tractable for functional and mechanistic studies (23.6 candidates per monoallelic pedigree; 13.6 per biallelic pedigree). Furthermore, we incorporated multiple lines of evidence such as allelic aggregation, cognate gene identification, mutational constraint, and functional prediction. This approach successfully re-identified the pathogenic variants in our cohort at the GATA2 cRE2 locus34 and led us to nominate novel candidate disease variants (Table 1). We also identified compelling individual candidate variants and peaks without multiple hits. Such candidates will be easier to resolve with larger cohort sizes and larger families. Indeed, our ability to reduce candidate variant numbers was limited by the large proportion of unsolved small dominant pedigrees in our cohort, which are notoriously difficult to analyze. Moreover, while de novo and recessive mutations are clearly an important source of causal pathogenic variation in sporadic cases, such cases are also more likely to involve non-genetic etiologies.
Although a given peak can harbor hundreds of predicted transcription factor binding motifs, we demonstrate in principle that locus-specific footprinting can implicitly reduce a ~1 kb peak to a ~10 bp individual transcription factor binding site of interest. Given sufficient sequencing coverage116 and data quality, such approaches could immediately be applied to other rare diseases and cell types. Alternatively for common diseases, causal non-coding variants are more abundant, but also confounded by linkage disequilibrium. In this case, locus-specific footprinting (in concert with careful demarcation of element boundaries, chromatin accessibility QTL analysis117, and statistical fine-mapping118) may further resolve causal common variants and identify affected transcription factor binding sites across the genome – all inferred from a single assay. Proof of feasibility of such approaches in rare diseases could also influence data collection strategies for common diseases119.
Through our analysis, we also encountered potential limitations affecting non-coding variant interpretation. We in part leveraged sequence conservation and constraint to prioritize pathogenic variants. However, while the known genes and cREs underlying cMN development are highly conserved, a conservation-based strategy may not identify pathogenic variants in human-specific and/or rapidly evolving sequences114,120,121. Strikingly, we also found that even relatively subtle differences in cellular composition and ATAC/RNA collection strategies can distort cognate gene estimates. These findings should inform appropriate sampling strategies in the future, such as single cell multiomic assays. Unbiased genetic strategies such as partitioned LD score regression can be extremely useful towards defining disease-relevant cell types, though such approaches are effectively restricted to common diseases122. Moreover, we find that even when sampling the appropriate cell type, subtle differences in cell state can profoundly influence variant interpretation. We provide a concrete example at the well-characterized non-coding GATA2 locus34, where pathogenic variant effects are no longer detectable in the same cell type within a mere 24 hours of development (i.e., embryonic day 10.5 versus 11.5). Moreover, we sampled cMNs at e10.5 and e11.5 based on developmental patterns of previously described protein-coding mutations, but we do not exclude the possibility that novel disease mutations may also be relevant at different timepoints. Therefore, while our genetic framework can generalize to other disorders, we suspect that appropriate prospective or retrospective epigenomic cell sampling will benefit from highly detailed biological knowledge of each specific disease process.
Finally, the interpretation of non-coding variants can benefit from our knowledge of coding variants as they share challenges in common – namely, practical limitations in allelic expansion and functional validation. Here, we present generalizable approaches that aggregate plausible alleles based on physical (“peak-centric”) and biological (“gene-centric”) proximity to facilitate allelic expansion in a principled manner. These challenges may be further alleviated by expanding rare disease data sharing platforms123 to more comprehensively incorporate non-coding variation. Finally, development of functional perturbation assays that balance both scalability113 and specificity124 will disproportionately benefit validation of non-coding variants, which are naturally more abundant and cell type-specific than coding variants. The outputs of such assays would also iteratively provide training material for further refined functional prediction algorithms.
Rapid advances in next generation sequencing technologies have led to a renaissance in Mendelian gene discovery. As access to WGS and functional genomics data becomes less limiting, alternative analytical and experimental frameworks will be needed to finally resolve Mendelian cases and disorders that are otherwise recalcitrant to traditional exome-based approaches.
METHODS
Mouse husbandry, dissection, dissociation, FACS
We performed husbandry, dissection, dissociation, and fluorescence-activated cell sorting (FACS) as described previously128. Briefly, we crossed C57BL/6 (JAX # 000664) female mice with either 129S1/C57BL/6J IslMN:GFP (JAX # 01795235) or Hb9:GFP (JAX # 005029128) male mice and separated them following one night of breeding. Pregnant females were sacrificed at 10.5 or 11.5 days post-conception and whole embryos were grossly dissected in chilled 1x PBS (ThermoFisher) then immediately placed in 1x B27 supplement (Gibco 17504044) in Hibernate E (Fisher NC0285514). Next, GFP-positive cranial motor neurons, GFP-positive spinal motor neurons, and GFP-negative surrounding cells were microdissected in pre-chilled HBSS (ThermoFisher) and placed in 1x B-27 supplement, 1x Glutamax (ThermoFisher 35050061), and 100 U/mL Penicillin-Streptomycin (PenStrep, ThermoFisher 15140122) in Hibernate E (medium 2). Microdissected tissues were dissociated using papain and ovomucoid solutions prepared from Papain Dissociation System (Worthington Biochemical LK003150). Tissues were resuspended in papain solution. Samples were then incubated at 37°C for 30 minutes and agitated every 10 minutes to ensure complete dissociation. Following incubation, samples were spun down at 300 rcf for 5 minutes, the supernatant was removed, and dissociated tissues were resuspended in 500 uL of ovomucoid solution (plus or minus 100 μL depending on quantity of tissue). Tissues were again spun down at 300 rcf for 5 minutes and resuspended in 500 μL of medium 2 (plus or minus 100 μL depending on quantity of tissue) and transferred to a 5mL polystyrene round bottom tube on ice. Live GFP-positive singlets were separated from GFP-negative cells (GFP-negative limb buds from embryos used as negative control to set gates) using an ARIA-561 FACS machine at the Immunology Research Core at Harvard Medical School (for ATAC-seq samples), and an BD FACS Aria II at the Jimmy Fund Core at the Dana-Farber Cancer Institute (for bulk and single cell RNA-seq samples). GFP-positive cells were collected either into 200 uL of media containing 1x Glutamax, 100 U/mL PenStrep, and 2% 2-Mercaptoethanol (Gibco 21985023) in Neurobasal-A Medium (ThermoFisher 10888022) for ATAC-seq, or into 96 well fully-skirted Eppendorf plates containing a starting volume of 5 ul/well of Hibernate E for single cell RNAseq, or directly into 1.5 ml tubes containing Qiagen RNeasy Lysis buffer/Buffer RLT (Qiagen 79216) for the bulk RNAseq. Embryos were not selected based on sex. Embryos were excluded if they did not match expected developmental stage as estimated from morphological features.
Single cell ATAC-seq: Nuclei Isolation, tagmentation, and sequencing
We performed fluorescence-assisted microdissection to collect samples cMN3/4, cMN7, and sMN from Isl1MN:GFP mice and likewise to collect samples of cMN6, cMN12, and sMN from Hb9:GFP mice, each at both e10.5 and e11.5. We performed FACS-purification as described above to collect GFP-positive motor neurons, as well as GFP-negative cells surrounding the motor neurons to better distinguish between motor neuron versus non motor neuron regulatory elements (for a total of 20 sample types, 9 with biological replicates and 2 with technical replicates for 32 samples in all). Nuclei were isolated in accordance with Low Cell Input Nuclei Isolation guidelines provided by ‘Demonstrated Protocol – Nuclei Isolation for Single Cell ATAC Sequencing Rev A’ from 10x Genomics. Cell suspensions were spun down at 300 rcf for 5 min at 4°C in a fixed angle centrifuge, the supernatant was removed, and the pellet was resuspended in 50 uL of 0.04% BSA in PBS. The cell solution was then transferred to 0.2 mL tube and centrifuged at 300 rcf for 5 minutes at 4 °C in a swinging bucket centrifuge. Without contacting the bottom of the tube, 45 uL of supernatant was removed, and the cell pellet was resuspended in 45 uL of chilled Lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 Substitute, 0.01% Digitonin, 1% BSA, in nuclease-free water). Nuclei suspensions were incubated on ice for 3 minutes and 50 uL of wash buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA, 0.1% Tween-20, in nuclease free water) was added to the suspensions without mixing. Nuclei suspensions were then spun down in a swinging bucket centrifuge at 500 rcf for 5 minutes at 4 °C, 95 uL of supernatant was removed, and 45 uL of nuclei buffer was added. Samples were again spun down in a swinging bucket centrifuge at 500 rcf for 5 minutes at 4 °C, all supernatant was removed without contacting the bottom of the tube, and nuclei were resuspended in 7 uL of nuclei buffer. 2 uL of this final nuclei suspension was added to 3 uL of nuclease-free water, and 5 uL of trypan blue, and cell viability was inspected using the Countess II FL Automated Cell Counter (Thermo Fisher Scientific AMQAF1000). We performed scATAC transposition, droplet formation, and library construction as described in protocol CG000168 using v1 reagents (10x Genomics). scATAC libraries were sequenced on the Illumina NextSeq 500 system using standard Illumina chemistry. Paired inserts were minimum 2 x 34 bp in length excluding indices, and libraries were distributed to achieve an estimated coverage of ≥ 25,000 read pairs per cell in accordance with 10x Genomics guidelines (actual mean coverage was 48,772 reads per cell). Samples failing quality control were excluded (e.g., failed TapeStation output).
scATAC preprocessing, peak calling, dimensionality reduction, and cluster analysis
We performed a modified workflow based on Cusanovich et al.129. Briefly, we generated fastq files from bcl using cellranger mkfastq. We initially included all single cell ATAC barcodes perfectly matching an allowlist provided by 10x Genomics. We also included fixed barcodes if they had a maximum Hamming distance of 1 and if they were present in the top 2% of barcode counts. As a final check, we manually inspected the distribution of fixed barcodes in reduced dimension space to ensure a roughly even distribution across all cells. We aligned individual samples to the mm10 reference genome using Bowtie2129, generated sample level .bam files, filtered reads with MAPQ < 10, and performed PCR deduplication. We established heuristic coverage per cell thresholds for each sample separately. To generate cell counts, we performed hard filtering based on log10[nfrags/barcode] for each sample separately.
We performed LSI-based clustering to generate sample-level clades as described previously130. In order to enrich peak representation from rare neuronal populations, we manually assigned between 3-7 clades to each sample and then performed peak calling on each clade using MACS2130. We first performed cell QC based on heuristic filters (low FRiP and accessible peaks-per-cell outliers), then peak QC (filtering peaks in a low proportion of remaining cells per clade). All post-QC cells and peaks were then combined to generate a master peak-by-cell callset. Samples failing any stage of QC were excluded (e.g., inadequate read coverage).
We performed LSI-based dimensionality reduction (log-scaled TF-IDF transformation followed by singular value decomposition) on our binarized peak-by-cell matrix as based on previously described methods130. We used umap() (https://github.com/lmcinnes/umap) to further reduce the dimensionality of our data to 3-dimensional UMAP coordinates. We then performed cluster analysis using Seurat’s SNN-graph approach. Once the major clusters were defined, we repeated our dimensionality reduction and cluster analysis on each major cluster to generate subclusters.
Cluster homogeneity, completeness, and purity
In order to formalize the agreement between our dissection/FACS labels (“class”) and our cluster/subcluster labels (“cluster”), we calculated homogeneity h, completeness c, and Vmeasure , using the sabre package131:
Where is the set of dissection/FACS class labels; is the set of clusters or subclusters; is the number of single cells belonging to class and cluster or subcluster ; is the total number of single cells; and is the ratio of weights attributed to and ( is the weighted harmonic mean of and ). As becomes very large or very small, approaches and , respectively. Here we set to 1.
We also generated a per-cluster purity metric, p to quantify the maximum cellular representation of each cluster/subcluster:
Homogeneity, completeness, and Vmeasure calculations across varying conditions of and are summarized in Supplementary Table 4.
Motif Enrichment and aggregated footprinting analysis
We used the mouse motifs from the cisBP database from the chromVARmotifs database to compute cluster and sample specific motif footprinting and enrichments (mouse_pwms_v2). For each motif, we identified all sites in peaks where a motif was present. Clusters 3, 4, 5, and 9 were excluded from footprint analysis. We next identified differentially accessible peaks for each group of interest using ArchR’s getMarkerFeatures() function, normalizing for differences across groups with transcriptional start site (TSS) Enrichment and log10(nFrags). We selected peaks for each group that met an FDR threshold of below 0.01 and a LogF2C of >=1. Aggregated footprint plots were generated for select motifs using plotFootprints(), by first normalizing the Tn5 bias by subtracting it from the footprinting signal. For site-specific footprints, we used TOBIAS to generate Tn5-bias corrected bigwigs and footprint scores across the genome for each cell type131. For bias estimation and correction we excluded ENCODE denylist regions from mm10-blacklist.v2.bed (https://github.com/Boyle-Lab/).
In vivo lacZ enhancer validation
We selected 25 putative wildtype enhancers for downstream experimental validation based on the following criteria. First, we selected elements with significant cell type specificity scores51. Next, we excluded any elements that did not lift over to the human genome (hg19). We then identified elements with evidence of H3K27Ac marks in the ENCODE portal131 and no existing experimental data in the VISTA enhancer browser132 (freeze September 2019). Finally, we performed manual curation in order to select for elements with high conservation, against elements in repetitive regions, and ensured representation of elements from cMNs 3, 4, 6, 7, 12, and sMNs.
We performed in vivo enhancer testing using the enSERT transgenesis method described by Osterwalder et al.133. Briefly, the orthologous human sequence each candidate enhancer was cloned into a pCR4-Shh::lacZ-H11 vector (Addgene plasmid # 139098) containing the mouse Shh minimal promoter, lacZ reporter gene, and H11 safe harbor locus homology arms. The cloned construct, Cas9 protein, and H11-sgRNAs were delivered via mouse embryonic pronuclear injection (mouse FVB/NJ JAX #001800) and transferred to female hosts. Embryos were collected at e11.5, stained with X-gal, and evaluated for reporter activity.
For candidate variant testing, we generated enhancer clones bearing the human reference or variant allele as described above. In the case of compound heterozygous variants, we cloned both variants into the same construct in cis. In the case of full enhancer deletion candidates, we cloned only the wildtype enhancer.
Bulk ATAC-seq
We performed bulk ATAC-seq as described previously127 for FACS-purified cells from six anatomic/temporal regions: IslMN:GFP-positive cMN3 at e10.5 and e11.5, cMN7 at e10.5, sMN e10.5 and e11.5, and IslMN:GFP-negative hindbrain at e11.5. We processed the bulk ATAC sequencing data by running the .fastq files through the Encode ATAC-seq pipeline (https://github.com/ENCODE-DCC/atac-seq-pipeline) using default parameters. To analyze peaks for each bulk sample, we used Irreproducible Discovery Rate (IDR) optimal peaks, generated between pseudoreplicates or biological replicates when appropriate. After generating peaksets for each bulk sample, we created a bulk master peakset by concatenating all the individual peaksets and merging with bedtools merge. We further generated bulk peaksets specific to each sample using bedtools subtract, allowing for ≤ 50% overlap between peaks.
Single Cell RNA-seq
Husbandry and collection strategy was identical to the scATAC strategy described above, except that we combined GFP-positive and -negative cells from the same dissections. We performed single cell RNA-seq for FACS-purified eGFP-positive motor neurons from 6 anatomic/temporal regions: cMN3+4 and cMN7 from Isl1MN:GFP mice and cMN6 from Hb9:GFP mice, all at both e10.5 and e11.5 (for total of 10 samples). In most samples we spiked in 10% surrounding eGFP-negative hindbrain cells as an internal control for comparison to non-motor neurons. Samples were submitted to the Klarman Cell Observatory/Regev Lab at the Broad Institute of MIT and Harvard for processing on a 10X Genomics Chromium platform. The 10X Genomics Chromium Single Cell 3’ Reagent Kit (using v2 single index chemistry, CG00052) was used for mRNA capture and library preparation. Samples were multiplexed for a read-depth goal of 50,000 reads/cell (actual mean coverage was 94,829 reads/cell). Sequencing was performed on a HiSeq 4000 by Broad Genomic Services using standard Illumina chemistry. The data was then aligned in the Engle lab using Cell Ranger v2.1.1 against the ENSEMBL Mus musculus genomic reference build GRCm38.87 (modified to include eGFP and tdTomato sequences). Quality control was performed in Seurat to remove doublets and low-read cells. Analysis was done in Seurat where samples were integrated with Canonical Correlation Analysis (CCA)134. Motor neurons were identified from eGFP, Isl1 and expression of other motor neuron markers (eGFP was regressed out to avoid affecting clusters),
Bulk RNA-seq
We performed bulk RNA-seq for FACS-purified eGFP+ cells from 7 anatomic/temporal regions: cMN3, cMN4, cMN6, cMN7 at each corresponding brainstem level, at both e10.5 and e11.5 (except for cMN6 that was only collected at e11.5 due to cell number limitations at e10.5; with two biological replicates from all times/regions and 1 additional technical replicate of cMN6, for a total of 15 samples). Samples from multiple litters were merged to reach a threshold for appropriate cell number and sent to Rutgers RUCDR for library preparation and sequencing. For the e11.5 samples, 200 ng/sample of RNA was isolated with Oligo-dT beads, enriching for mRNA. Depletion of beta globin mRNA and ribosomal RNA was performed. For the e10.5 samples and the e11.5 cMN6 samples, due to the lower total RNA from fewer starting cells in these nuclei at these ages, whole-transcriptome Nugen Amplification was performed. Samples were sequenced with a 100 bp paired-end strategy to sequence full-length transcripts on an Illumina HiSeq2500 for an approximate read-depth of 60 million paired-end reads/sample. This generated R1 and R2 reads for each of 2 lanes of data/sample that were subsequently concatenated. STAR (Spliced Transcripts Alignment to a Reference)134, a splice-aware tool, was used to align reads to ENSEMBL Mus musculus genomic reference build GRCm38.87, and RSEM (RNA-Seq by Expectation Maximization)135 was used to generate the count files. We then used DESeq2136 to make comparisons.
Generating peak-to-gene links
For our original RNA inputs for peak-to-gene links, we performed scRNA-seq on cMN3+4, cMN6, and cMN7 dissections (GFP-positive and -negative) at e10.5 and e11.5. Our husbandry and collection strategy was identical to the scATAC strategy described above, except that we combined GFP-positive and -negative cells from the same dissections. We performed scRNA seq as described in protocol CG000168 using v2 single index chemistry and sequenced on the Illumina HiSeq 4000. To benchmark our scRNAseq results, we also performed bulk RNAseq on cMN3, cMN6, and cMN7.
We integrated multiple scRNA-seq datasets from GFP-positive and -negative cells from cMN3/4, 6, and 7 dissections at e10.5 and e11.5 into a single Seurat object using Seurat’s integration framework76,135. We excluded cells with more than 5% of reads aligning to the mitochondrial genome. After examining the distribution of the number of unique features and number of unique reads per cell for each sample, we manually filtered cells with low feature counts. Finally, we normalized each sample using the NormalizeData() function, identified the top 10,000 variable features per sample, and scaled each sample using the ScaleData() function.
Next, we excluded scATAC clusters (clusters 3, 4, 5, and 9) with high proportions of GFP-positive sMN and cMN12 dissected cells, as those samples are not represented in our scRNA dataset. We then performed unconstrained scATAC-RNA integration on all remaining cells using addGeneIntegrationMatrix() in ArchR135.
We then evaluated the projected gene expression values from our scATAC-RNA integration for three high-confidence scATAC clusters (cMN3/4.10, cMN6.6, and cMN7.2). We selected these clusters due to unambiguous sample membership based on microdissection origin (purity), FACS labels (corresponding to cMN7, cMN6, and cMN3/4, respectively), and known marker locus accessibility/expression. We compared imputed gene expression from these clusters to corresponding bulk RNAseq samples that were independently dissected and FACS purified. Specifically, we performed differential expression analysis on bulk RNAseq data (DEseq v1.34.0136) and on imputed gene expression on scATACseq data (using getMarkerFeatures() function in ArchR). We fit a linear model of the log2[fold-change] expression for all combinations of bulk samples and single cell clusters, and confirmed a significant positive correlation between projected gene expression for marker genes in each cluster against its corresponding bulk counterpart.
We calculated peak-to-gene correlations using ArchR’s addPeak2GeneLinks() function, with reducedDims = “IterativeLSI_ArchR". We included all high confidence links (FDR < 0.0001) with a minimum correlation coefficient of ≥ 0.1, within +/− 500 kb of a given gene, which we reasoned would include the vast majority of putative enhancers76,137, including those active in only a subset of cells.
We then benchmarked this cMN peak-to-gene set against two alternative scATAC-RNA integrations using subsetted scRNAseq data from the Mouse Organogenesis Cell Atlas (MOCA)137. First we created a neuronal dataset set by integrating our oversampled cMN scATAC profiles with more uniformly sampled sci-RNA neuronal clusters from MOCA (annotated as “Cholinergic Neurons”, “Excitatory Neurons”, “Inhibitory Neurons”, “Neural Progenitor Cells”, “Postmitotic Premature Neurons”, “Primitive Erythroid Lineage”, and “Stromal Cells”). We removed any cells that were not collected at e10.5 and e11.5 to age-match our scATAC set. We also performed an scATAC-RNA integration using a more distantly related cell type with minimal sampling overlap, (sci-RNA MOCA Cluster 34 annotated as “Cardiac Muscle Lineage”) and included non-age-matched cells for this integration. We then generated peak-to-gene links as described above and quantified the total number of links across different RNA integrations.
To quantify and compare the distribution of peak-to-gene links across different genes, we tabulated significant peak-to-gene links (r > 0.1 and FDR < 10−4) +/− 50 kb of each gene’s TSS. In the case of peaks connected to multiple genes, we selected the link with the lowest FDR value. Next, we generated modified Domain of Regulatory Chromatin (DORC) scores first described by Ma et al.138 by normalizing all reads in our peak-by-cell matrix by unique fragment count. We then summed these normalized values for all peak-to-gene connections within +/− 500 kb of each gene TSS for every cell.
Single cell Multiome (scMultiome)
We performed timed matings, microdissections, dissociation, and FACS to collect GFP-positive cMN3/4, cMN7, cMN12, and sMN cells at e11.5 as described above. Instead of generating separate reactions for each cell type, we pooled these cells prior to dissociation, selected GFP-positive cells via FACS, and performed Low Cell Input Nuclei Isolation (10x Genomics CG000365) and Single Cell Multiome ATAC + Gene Expression assay (10x Genomics CG000338) on a total of two pooled replicates. We performed sequencing on a NextSeq 500 for Multiome ATAC and Gene Expression libraries separately, using a custom sequencing recipe for ATAC provided by Illumina. We performed QC, dimensionality reduction, and generated peak-to-gene links as described above using functionality in Signac and ArchR70,139. In order to facilitate direct comparison across modalities, we calculated scMultiome fragment depth against our high confidence scATAC peakset. We calculated multimodal weights for each cell using a weighted nearest neighbour approach140 and performed ab initio graph-based clustering on our scMultiome cell set. In order to annotate these clusters, we generated cell-cell anchors by defining scMultiome clusters as the query set and our well-annotated scATAC clusters as the reference set. Because each multiome cluster was typically dominated by a single predicted scATAC cluster, we annotated each multiome cluster based on its maximum predicted scATAC membership.
Single cell CUT&Tag
We collected cranial motor neurons (GFP-positive cMN3+cMN4 e11.5, cMN6 e11.5, cMN7 e10.5, and cMN7 e11.5) as described above and performed a modified scCUT&Tag protocol74,125. Briefly, we collected GFP-positive cells directly into fresh antibody buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 1x protease inhibitor (Sigma 11873580001), 2 mM EDTA, 0.05% digitonin, 0.01 % NP-40, 1× protease inhibitors and 2% filtered BSA). We centrifuged samples at 450 rcf for 5 minutes, washed in 200 uL antibody buffer, centrifuged at 600 rcf for 3 minutes, resuspended in 1:50 H3K27Ac primary antibody (monoclonal Rabbit anti-mouse, Abcam ab177178), and incubated overnight at 4°C with gentle rotation. Nuclei were centrifuged at 600 rcf for 3 minutes, washed in 200 uL Dig-Wash-BSA buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM spermidine, 1x protease inhibitor, 0.05% digitonin, 0.01 % NP-40, 1x protease inhibitor and 2% filtered BSA), centrifuged at 600 rcf for 3 minutes, resuspended in 1:50 IgG secondary antibody (guinea pig anti-rabbit Novus Biologicals, NBP1-72763), and incubated 1 hour at room temperature with gentle rotation. Nuclei were then centrifuged at 600 rcf for 3 minutes, washed 3x in Dig300-Wash-BSA (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 1x protease inhibitor, 0.05% digitonin, 0.01% NP-40, 1x protease inhibitors and 2% filtered BSA), resuspended in 1:20 pAG-Tn5 (EpiCypher 15-1017), and incubated 1 hour at room temperature with gentle rotation. Nuclei were centrifuged at 450 rcf for 3 minutes, washed 3x in Dig300-Wash-BSA, resuspended in 200 uL tagmentation buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 1x protease inhibitor, 0.05% digitonin, 0.01 % NP-40, 1x protease inhibitor, 2% filtered BSA, and 10 mM MgCl2), incubated 1 hour at 37°C with agitation every 15 minutes. Tagmentation was halted with Stop buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine, 1x protease inhibitor, 0.05% digitonin, 0.01% NP-40, 1x protease inhibitors, 2% filtered BSA, and 25 mM EDTA), centrifuged at 450 rcf for 3 minutes, washed in diluted nuclei buffer (1x ATAC Nuclei Buffer (10x Genomics, PN-2000207) and 2% filtered BSA), centrifuged at 450 rcf for 3 minutes, and resuspended in diluted nuclei buffer. Intact nuclei were stained with DAPI and were visualized and counted under fluorescent microscopy. 70 μL of ATAC master mix (8 μL tagmented nuclei, 7 μL ATAC Buffer B (10x Genomics, PN-2000193), 56.5 μL Barcoding Reagent B (10x Genomics, PN-2000194), 1.5 μL Reducing Agent B (10x Genomics, PN-2000087), 2 μL Barcoding Enzyme (10x Genomics, PN-2000139) was loaded for GEM generation according to the 10x Genomics scATAC v1.1 protocol. Nuclei were diluted if necessary (up to a maximum of 25,000 total nuclei per reaction). Subsequent GEM generation and cleanup steps were performed according to the 10x Genomics scATAC v1.1 protocol. Library prep was also performed using the standard protocol, except that total PCR cycles were increased to 16. All centrifugation steps were performed using a swing-bucket rotor.
Activity-by-contact (ABC) enhancer predictions
We generated enhancer predictions for four cell types, GFP-positive cMN3+4 e11.5, cMN6 e11.5, cMN7 e10.5, and cMN7 at e11.5, adapting the Activity-By-Contact (ABC) model v0.2 described previously139,140. We defined potential enhancer regions by merging scATAC peaksets for each sample. We provided sample-specific H3K27Ac read counts from scCUT&Tag experiments described above. We also provided imputed RNA expression tables for each cell type from the scATAC-scRNA integration described above. We estimated contact frequencies based on the ABC power law function. We evaluated our enhancer predictions against 67 VISTA enhancers classified as positive for “cranial nerve”, of which 12 had ABC enhancer predictions. Importantly, our ABC predictions also correctly identify the peak and cognate gene for the CREST1 enhancer (VISTA enhancer hs1419), for which both the enhancer locus and cognate gene are known140.
Participant whole genome sequencing, reprocessing, SNV/indel calling and quality control.
Research participants were enrolled into the long-term genetic study of CCDDs at Boston Children’s Hospital (BCH; clinicaltrials.gov identifier NCT03059420). The Institutional Review Board at BCH approved the study. Informed consent was obtained from each participant or legal guardian. Individual-level data was de-identified and studies were performed in compliance with US 45.CFR.46 and the Declaration of Helsinki. WGS was performed at Baylor Human Genome Sequencing Center through the Gabriella Miller Kids First Pediatric Research Program (dbGaP Study Accession: phs001247). Joint variant calling for all samples was performed at the Broad Institute. We uploaded raw 30X coverage PCR-free WGS data to the Broad Institute’s secure Google Cloud server and reprocessed these data through the Broad Institute’s production pipeline. We realigned raw read data to the GRCh38 human reference sequence using BWA-MEM and reprocessed using the Broad’s Picard Toolkit. We then performed variant calling on the resultant BAM files using the Genome Analysis Toolkit (GATK 4.0 HaplotypeCaller). In the final step of variant calling, we jointly genotyped each site in the genome alongside a collection of over 20,000 reference genomes assembled by the Broad Institute. Joint variant calling provides two crucial advantages over individual or batched genotyping141. First, it dramatically improves variant calling accuracy due to i) clearer distinction between homozygous sites versus missing data; ii) greater sensitivity to detect rare variants, and iii) greater specificity against spurious variants. Second, joint calling by its design generates a well-calibrated estimate of allele frequency within our cohort against the large gnomAD database. Assuming that the allele frequency of a bona fide Mendelian disease-causing variant is lower than its disease prevalence, this information allows us to exclude variants with implausibly high allele frequencies141,142. Finally, we performed variant filtering using GATK’s Variant Quality Score Recalibrator and applied custom hard filters as required.
We performed rigorous QC at multiple stages of variant calling, performed filtering based on standard sequencing quality metrics (e.g., uniformity of coverage, transition/transversion ratio, indel length profiles, etc.), and compared them to our internal database of reference genomes. We used heterozygosity of common variants on chrX and coverage of sites on chrY to confirm reported gender and to identify sex chromosome aneuploidy. We also extracted variant calls from 12,000 well-covered variant sites and used these variants for principal component analysis together with a large reference panel to infer the geographical ancestry of samples, to infer pairwise relatedness of the samples, to identify unexpected duplicates, and to determine cryptic relatedness and unexpected patterns of relatedness within reported families. The data/analyses presented in the current publication have been deposited in and are available from the dbGaP database under dbGaP accession phs001247.v1.p1. Adult participants and guardians of children provided written informed consent for participation. No participant compensation was provided.
Structural Variants
We generated an SV callset using the ensemble GATK-SV pipeline as described previously (https://github.com/broadinstitute/gatk-sv)142-146. Briefly, we performed joint genotyping and harmonized SV calls from multiple detection tools (Manta, Wham, MELT, GATK-gCNV, and cn.MOPS143-147), as well as manual read inspection using IGV148, and estimated SV allele frequencies against gnomAD SV v2.1. We first excluded any SVs with cohort AF ≥ 0.005, irrespective of coding or non-coding status. When evaluating for de novo and inherited SV candidates, we restricted our callset to 45 and 49 curated pedigrees, respectively. One SV (deletion chr22:27493955-27497536) was identified through manual curation. These SVs were subsequently used for downstream analysis incorporating pedigree non-coding element information.
We also performed a separate bespoke analysis for genome-wide transposon insertions (L1, Alu, and SVA) profiling on the GMKF WGS dataset using xTea149. Raw transposon insertions with different features and confidence levels were annotated and processed to generate both rare and de novo insertion lists for further variant interpretation. Beyond basic feature annotations (transposon family, breakpoint, and gene annotations), all insertions were annotated with 1) population allele frequencies (AFs) derived from the 1000 genomes project, gnomAD SV, euL1db, and other polymorphic insertion collections from the literature81,150-152; 2) overlapping repeats annotated by RepeatMasker and homopolymers; 3) other gene annotations such as pLI score, OMIM disease-causing genes, and potential CCDD-related genes. For putative pathogenic rare insertions, we first applied population AF threshold of 0.01 to remove common polymorphic insertions. We then filtered nested insertions–where a putative insertion landed in an existing insertion from the same transposon family–as they are error-prone in short read sequencing platforms. Finally, we filtered for all high confidence annotations (“two_side_tprt_both” and “two_side_tprt”) in affected samples for downstream genetic analysis. For de novo insertions, raw calls of transposon insertions were examined and only those present in the affected proband but fully absent in both parents (i.e., without a single supporting read) were retained. Trio families with any member bearing abnormal high number of transposon calls were filtered, as these outlier samples carried excessive noisy signals (clipped and discordant reads) and consequently false positive calls could affect de novo insertion calling. We then removed insertions that have been reported in populational datasets and known polymorphic insertion collections in the literature. We also filtered out error-prone nested insertions. Finally, high-confidence insertions (feature = “two_side_tprt_both”) in affected participants were reported as the de novo insertions for further genetic interpretation (Supplementary Table 15).
Applying cell-type aware filters for human non-coding mutations
Our original WGS callset contained 49,824,956 variant calls for 899 individuals across 270 distinct families with CCDDs. We loaded these unfiltered variant calls in .vcf format into Hail (https://github.com/hail-is/hail) as a MatrixTable. Multi-allelic variants were split so that all variants are represented in a bi-allelic format. In splitting multi-allelic variants, spanning deletions were not kept. This resulted in 54,804,014 bi-allelic variants. These variants were annotated with TOPMed allele frequencies, gnomAD genomes allele frequencies and allele counts, GERP scores and ClinVar variant pathogenicity labels. Using native and custom Hail functions, we generated scripts to filter the MatrixTable’s variant calls based on custom specifications for variant annotations, variant locus, and call quality filters.
We set the following hard filters for all searches:
gnomAD AF152 ( < 1 x 10−3 for dominant/de novo; < 1 x 10−2 for recessive)
TopMED AF153 ( < 1 x 10−3 for dominant/de novo; < 1 x 10−2 for recessive)
GERP154 > 2
Only return variants that pass all quality filters in the VCF
Genotype Quality: > 20
Allele Balance: > 0.15 (heterozygous calls)
To generate a list of cell type specific genomic regions of interest for each disease group, we used data from single cell ATAC-seq experiments performed on mouse cranial motor neurons at e10.5 and e11.5. From here we implicitly assume that: i) we have correctly mapped each disease-relevant cell type (at the appropriate timepoint) to its appropriate cognate phenotype; ii) biologically active cREs are accessible; and iii) patterns of chromatin accessibility are correlated across species148. Peaks called on each cMN sample were lifted over from mm10 to hg38, and the converted intervals were concatenated into a single file and overlapping peaks were combined using bedtools merge. For disease types with > 1 cMN of interest, the master list of intervals for each cranial nerve were again merged using bedtools merge to create a list of intervals defining regions accessible in one or both cMNs. This final master list of intervals was used to narrow the total genomic search space for each disease group, with only variants contained in the regions specific to the cMN(s) of interest being retained.
Modes of Inheritance
In order to leverage pedigree information, we first stratified our 270 pedigrees into 7 major disease categories that shared cell type specific aetiology (CFEOM, FNP, DRS, CFP, Moebius, Ptosis, Ptosis/MGJWS). We further stratified these pedigree groups into subgroups based on 4 inheritance/phenotype patterns (familial/syndromic; familial/isolated; trio/syndromic; trio/isolated). We incorporated inheritance by only retaining variants that matched appropriate mode(s) of inheritance in at least one family in a given subgroup. For example, for trios we searched variants obeying de novo, dominant (if either parent was affected), compound heterozygous, and/or homozygous recessive modes of inheritance. For de novo variants, we used Hail’s likelihood-based caller (https://github.com/ksamocha/de_novo_scripts). For familial cases, we manually inspected each pedigree structure and specified custom variant searches based on plausible modes of inheritance, including de novo, dominant, compound heterozygous, homozygous recessive, and dominant with incomplete penetrance. In the case of compound heterozygous variant configurations affecting non-coding elements, we defined each scATAC peak as our unit of heredity. Within this framework, one variant in a peak had to be inherited from an unaffected father, and a different variant in the same peak had to be inherited from an unaffected mother. Finally, we performed cohort-level filtering by eliminating any rare candidate variants that were also present in any unaffected individuals in the cohort (for dominant / de novo searches) or that were present in a homozygous state in any unaffected individual (for recessive searches). We removed one outlier pedigree which had an excessive number of candidate variant calls.
For SV genetic interpretation, we performed inheritance based searches for dominant/de novo modes of inheritance in the appropriate pedigrees, using the same custom search parameters as described for the SNV/indel framework. We identified all de novo and inherited variants overlapping disease-relevant peaks for each eligible pedigree using the findOverlapPairs() function from the GenomicRanges package.
For TE genetic interpretation, we imported the list of TEs called with xTEA149 into Hail as a MatrixTable. We performed inheritance-based searches for dominant/de novo modes of inheritance, again using the same custom search parameters as described for the SNV/indel framework. We converted the TE MatrixTable from hg19 coordinates to hg38, and filtered out calls with invalid/unknown contigs, and only included highest confidence calls (Feature info = “two_side_tprt_both”). We applied estimated gnomAD AF thresholds of 0.01 and 0 for dominant inherited and de novo alleles, respectively. We used the same cell type-specific peak interval/disease group combination described above but added +/− 15bp padding to each peak to account for uncertainty in the insertion point.
To identify multi-hit peaks, we aggregated candidate variant results within each cell type/disease pairing by peak and selected for any peaks with SNVs/indels and/or SVs present in ≥ 2 families. For multi-hit tabulation, we excluded any SVs > 100 kb or with clear coding etiology. Variants within multi-hit peaks were required to obey the same broad mode of inheritance (i.e., dominant or recessive). In addition, dominant and recessive multi-hit variants could not be present in any unaffected individual across the cohort in the heterozygous and homozygous configuration, respectively. Candidate variants in any previously solved pedigrees were excluded from final tabulation19,21,22,27,34,87,88,90,92,155-161.
Permutation testing
To assess the statistical significance of the results that lie within the regions drawn from scATAC sequencing of developing cranial motor neurons, we performed permutation tests to determine whether the regions corresponding to specific cranial motor neurons were enriched for variants. We analyzed dominant inherited and de novo variants separately.
First, we performed a search to find variants using the same thresholds for frequency, conservation, quality, and inheritance, but without limiting the search space to only genomic intervals defined in the scATAC peaks. We then split these results by disease group based on the phenotype of the family to create the genome-wide distribution of candidate variants for each disease group. After examining the distribution of the number of genome-wide de novo variants per individual after filtering for thresholds, we removed four individuals from the results due to existing significantly outside of the distribution (with the threshold drawn at >75 de novos per individual).
We then conducted permutation tests on each disease group, using regioneR.162 We used the original set of genomic locations from the cranial motor neuron(s) scATAC data to randomly generate a new list of peaks. The new list of randomly generated peaks was restricted to the same peak sizes and number of peaks as the original list, and could not overlap. We used the hg38 masked genome from BSGenomes in order to restrict the locations where the randomized peaks could be located. We then counted the number of variants within these new regions. This process was repeated for 5000 iterations for each disease group for both de novo and dominant inherited variants.
ddPCR copy number validation
We performed ddPCR droplet generation and droplet reading using the QX200 droplet digital PCR system with Biorad ddPCR Supermix for Probes (Bio-Rad #186-3010). We performed copy number genotyping for non-coding element hs2757 in pedigrees S190 and S138 using ddPCR Copy Number Assay (Bio-Rad dHsaCNS845311073) and TaqMan Copy Number Reference Assay, human, TERT (Life Tech 4403315) as an internal control. We used the following thermocycler protocol: 1 x [95°C for 10 min]; 40 x [94°C for 30s, 60°C for 1 min]; 1 x [98°C for 10 min], 1 x [4°C hold]. Genotyping was performed in duplicate for all samples.
Convolutional neural network training and prediction
We generated accessibility predictions using Basenji110,162 after training the network with mouse motor neuron scATAC-seq data. We generated separate predictions for each biological replicate (32 replicates total). To preprocess scATAC-seq data before training the neural network, we first generated bigwigs from the scATAC-seq bam files using mm10 as the reference FASTA. We clipped bigwig coverage at 150 to trim outliers. We generated training, validation, and test sequences with a split of 80% training sequences, 10% validation, and 10% test. We identified regions that should not be included in training sequences with a bed file containing regions that were hard masked in the mm10 fasta file combined with the Encode denylist. The mm10 FASTA file was filtered to only include chromosomes 1-19, X, and Y.
We trained the network retaining the model architecture from the original Basenji manuscript, with seven dilated layers. For this work, the dense output layer contained 32 units (one for each sample). Training was stopped when the correlation coefficient for validation predictions vs. validation experimental data failed to improve after 12 iterations (patience = 12), and the weights from the best iteration were saved as the final model. The complete architecture and list of hyperparameters can be found at https://github.com/arthurlee617/noncoding-mendel under params.json.
Using this trained network, we generated SNP activity difference (SAD) scores for each human candidate variant by calculating the total difference in predicted reference vs. alternate coverage over a 131,072 bp window centered about each variant site (hg38). Here we made the implicit assumption that a network trained on mouse accessibility data was portable across species within the same cell type110,163. We also included four solved CFP pathogenic variants as truth data. For ease of interpretation, we converted all SNV predictions from raw counts differences to Z-scores, which fit a normal distribution. To calculate Z-scores for individual candidate indels, we used the SNV derived scores for our null distribution.
Non-coding CRISPR mice and binomial ATAC
We performed scATAC-seq for GFP-positive cMN7 e10.5 from two CRISPR-mutagenized mouse lines ( and ) corresponding to human non-coding pathogenic variants described previously. is reported previously, corresponding to the pathogenic SNV (chr6:88224892A>G) mouse line163. (chr6:88224893C>T) was mutated on a C57Bl6 background via CRISPR-Cas9 homology directed repair at the Boston Children’s Hospital Gene Manipulation & Genome Editing Core and subsequently crossed onto the mixed IslMN:GFP line described above. For each mutant line, we generated two biological replicates (4 replicates total) on embryos from [homozygous mutant x homozygous mutant] timed matings and compared to our wildtype cMN7 e10.5 replicates. For ad hoc comparison across these samples, we performed iterative LSI dimensionality reduction and batch correction using Harmony164 and normalised coverage by log10(nfrags). We note that also harbours an off-target C>T variant 54bp downstream from the target site (i.e., in addition to the on-target variant). This off-target nucleotide is not mutated in any affected samples. However, we do not explicitly exclude the possibility that this off-target variant contributes to the difference in accessibility relative to wildtype. For binomial ATAC, we performed [wildtype x homozygous mutant] timed matings for GFP-positive cMN7 from the e10.5 line, again across two biological replicates.
To test the cis effects of the mutant allele on accessibility, we tabulated reference versus mutant allele counts and performed a two-sided exact binomial test:
where the number of trials, corresponds to sequencing coverage, the number of successes, corresponds to reference allele count, and the expected probability of success, corresponds to the expected sampling probability of the reference allele under the null hypothesis : .
Extended Data
Extended Data Figure 1. Per-cell and -sample quality metrics for scATAC data.

a. Representative FACS gating strategy for WT GFP-positive and GFP-negative cMN7 at e10.5. Left: Forward scatter area (FSC-A) and side scatter area (SSC-A), corresponding to cell size and granularity/complexity, are used to enrich for intact cells and exclude debris. Middle: forward scatter width (FSC-W) and FSC-A are used to exclude doublets. Right: Green fluorescent protein area (GFP-A) and 633 nm-excitation (APC-A) are used to enrich for GFP-positive and GFP-negative cells. GFP-negative gates are calibrated by dissociated limb buds prior to collection as a negative control. All samples are fresh, live cells without fixative or nuclear staining.
b. Representative TapeStation trace showing tagmented DNA fragment sizes prior to library preparation.
c. Representative histogram of per-cell scATAC reads in a single sample. Read cutoff is shown by a dotted line and determined heuristically for each sample.
d. Insert size distributions (top) and transcriptional start site (TSS) enrichment (bottom) for all samples and replicates. Insert sizes consistently show a characteristic nucleosome banding pattern (~147 bp wavelength). Samples IDs are shown in Supplementary Table 2.
e. Correlation matrix depicting all possible pairwise sample correlations (Spearman’s rho) for scATAC coverage in all rank-ordered peaks. Scatterplots for selected sample pairs from the four highlighted boxes within the matrix are shown on the right. Correlations decrease with increasing biological distance (top to bottom).
f. Representative clade diagram depicting the relative accessibility (red is positive, blue is negative) of 5kb genomic windows (rows) across individual cells within a given sample (columns). Distinct clades (colored bars) were determined heuristically for each sample for downstream peak calling. The number of clades per sample were selected to maximize representation of common and rare cell types.
g. Ridgeplot depicting density of per-cell fraction of reads in peaks (FRiP) for each dissected sample and replicate at e10.5 (red) and e11.5 (blue). Samples IDs are shown in Supplementary Table 2. Mean FRiP values are consistently higher for e11.5 samples (p-value = 4 x 10−5, binomial test).
h. Distribution of FRiP values for GFP-positive motor neurons (green) versus GFP-negative surrounding brain tissue (pink). GFP-negative cells display significantly greater dispersion compared to GFP-positive cells, particularly at e10.5. (p-value = 1.1x10−286, Brown-Forsythe Test). See Supplementary Note 1 for additional information.
Extended Data Figure 2. Comparing and contrasting bulk versus single cell ATAC profiles.
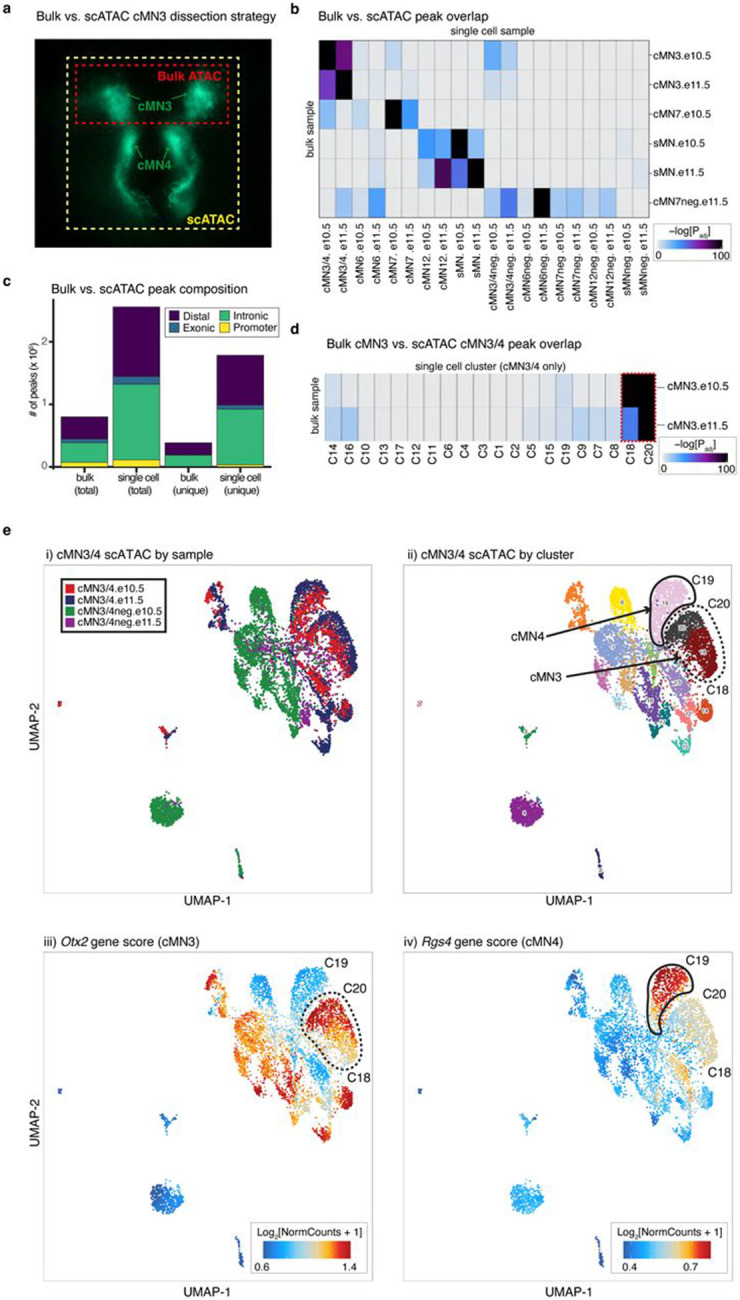
a. Fluorescence microscopy image illustrating cMN3 and cMN4 microdissection strategies. For scATAC experiments, cMN3 and cMN4 were microdissected en bloc (yellow box). For bulk ATAC microdissections, only cMN3 was excised (red box). All other cMN microdissection strategies were identical across bulk and scATAC.
b. Heatmap depicting enrichment of sample-specific bulk ATAC versus scATAC peaks. Color scale represents hypergeometric test p-values using the peakAnnoEnrichment() function in ArchR. Samples marked with “neg” are GFP-negative cells surrounding the motor neurons of interest. All other samples are GFP-positive motor neurons.
c. Stacked barplot depicting relative proportions of different classes of accessible chromatin (“distal”, “exonic”, “intronic”, and “promoter”). scATAC peaks are enriched for total number of peaks, total number of unique peaks, and cell type-specific peak annotations (distal and intronic).
d. Heatmap depicting enrichment of overlapping peaks for bulk cMN3 dissections versus ad hoc clusters (C1-C20) generated from scATAC cMN3/4 dissections only. Color scale represents hypergeometric test p-values. Ad hoc clusters C18 and C20 with the highest peak enrichment for bulk cMN3 are outlined by dashed red lines.
e. In silico microdissection of scATAC cMN3/4 clusters corroborates physical microdissections. Left to right, UMAP embeddings of scATAC cMN3/4 dissections colored by i) dissected sample; ii) ad hoc clusters; and gene scores for iii) cMN3 marker gene Otx2126; and iv) cMN4 marker gene Rgs4127. Putative cMN3 (C18 and C20) and cMN4 (C19) clusters inferred from dissection origin, marker genes, and GFP status are denoted by dashed and solid red lines, respectively.
Extended Data Figure 3. Cranial motor neuron scATAC peaks are underrepresented in regional bulk datasets.
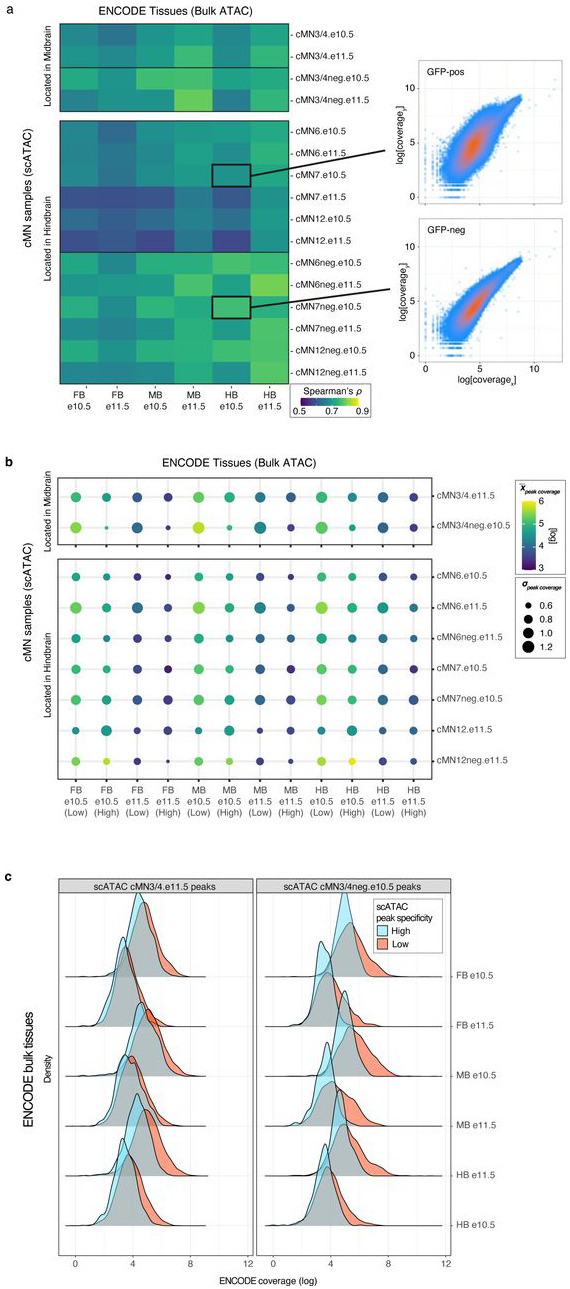
a. (Left) Heatmap depicting correlation coefficients (Spearman’s ) between scATAC peaks from cMN microdissections versus bulk ATAC peaks from ENCODE e10.5 and e11.5 mouse developing forebrain (FB), midbrain (MB), and hindbrain (HB) dissections. Anatomically concordant bulk brain regions are more highly correlated with scATAC non-motor neuron samples (‘−neg’) than scATAC cranial motor neuron samples. (Right) Scatterplots depicting rank-ordered per-peak sequencing coverage for bulk vs. scATAC samples.
b. Bubble chart depicting ENCODE bulk ATAC coverage in scATAC cMN peaks from a subset of samples, stratified by cell type specificity scores (‘High’ vs. ‘Low’). Colors reflect mean peak coverage (with lighter color reflecting higher coverage), while area reflects standard deviation. Bulk tissues tend to have higher coverage in low specificity peaks when compared to highly cell type specific peaks.
c. Density plots depicting distribution of ENCODE bulk peak coverage within cMN3/4 scATAC peaks from (b), stratified by specificity scores. High specificity scATAC peaks (blue) have consistently lower bulk coverage compared to low specificity peaks (red).
Extended Data Figure 4. scATAC cluster purity across major clusters and subclusters.
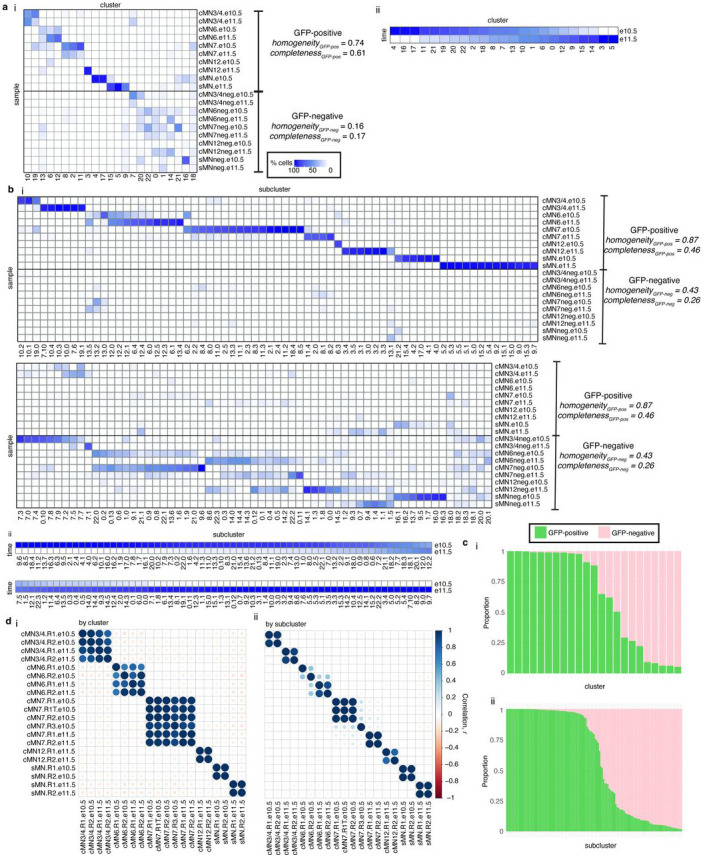
a. Heatmaps depicting purity of the 23 major scATAC clusters, stratified by i) sample and ii) embryonic age. cMN7 cells migrate past cMN6, are in close spatial proximity at these developmental ages, and are commonly co-dissected. Samples are GFP-positive unless otherwise marked (‘neg’). Clusters with higher membership from GFP-positive samples have higher purity than clusters with higher membership from GFP-negative samples. Most clusters feature cells from both e10.5 and e11.5 dissections, consistent with ongoing cell birth and proliferation. Homogeneity/completeness metrics calculated for GFP-positive versus GFP-negative samples are shown.
b. Heatmaps depicting purity of the 132 scATAC subclusters, stratified by i) sample and ii) embryonic age. As observed with the major clusters in (a), subclusters with high GFP-positive membership have greater purity than high GFP-negative subclusters. In contrast to the major clusters, a greater proportion of subclusters have skewed temporal membership (e10.5 vs. e11.5), potentially reflecting transient cell states.
c. Stacked barplots depicting proportion of GFP-positive and -negative cells within each i) cluster and ii) subcluster. Most clusters and subclusters are skewed towards pure (i.e., > 90%) GFP-positive or −negative membership. Here Cluster/subcluster IDs are not shown for ease of visualization. Detailed cluster annotations are available in Supplementary Table 3.
d. Correlation matrix depicting pairwise correlations between all biological replicates among i) major clusters and ii) subclusters. Cluster/subcluster membership is highly correlated across biological replicates from different batches, particularly for subclusters.
Extended Data Figure 5. Single cell multiome reproducibility and quality control.
a. Chromatin fragment length distribution (left), transcription start site (TSS) enrichment (middle), and joint UMAP embedding (right) comparing scMultiome biological replicates (red and blue). Replicates are highly concordant.
b. Histogram (left) and UMAP embedding (right) depicting distribution of scMultiome prediction ID scores of annotations transferred from the scATAC reference set to the scMultiome query set using the TransferData() function in Seurat128. The distribution is heavily skewed towards higher scores.
c. scMultiome annotations based on prediction IDs. Most predicted annotations correspond to Isl1MN:GFP-positive cell types, consistent with scMultiome dissection strategy.
d. Direct comparison of peak-to-gene links from scATAC versus scMultiome for motor neuron master regulator Isl1. scATAC peak-to-gene links are generated from imputed gene expression values (“GeneIntegrationMatrix”) whereas scMultiome links are generated from direct gene expression measurements (“GeneExpressionMatrix”). Ground truth enhancer CREST1 is highly accessible in Isl1-positive clusters with strong peak-to-gene links across both modalities.
Extended Data Figure 6. Toggling input data for Activity-by-Contact enhancer prediction.

a. Whole mount in vivo enhancer reporter expression for the seven VISTA Enhancers that are annotated for cranial nerve (CN) expression, inspected for and have CN7 expression, and have positive Activity-by-Contact (ABC) enhancer predictions for CN7 at e11.5. Peak-to-gene predictions match ABC predictions in all cases (7/7). Replacing CN7 e11.5 H3K27Ac or ATAC data with these data from a distantly related cell type (mouse embryonic limb e11.5) results in either a matching or a non-matching cognate gene prediction. Substituting cMN7 e11.5 histone modification data with “Limb H3K27Ac” histone modification data alters predictions for 3 out of 7 enhancers. Substituting cMN7 scATAC data with “Limb ATAC” data alters predictions for 6 out of 7 enhancers. Neither substituted input correctly identifies the CREST1 enhancer (VISTA enhancer hs1419). Positive evidence of CN7 enhancement is depicted by arrows.
b. Stacked barplot summarizing consequences of toggled input data.
Extended Data Figure 7. Compound heterozygous non-coding candidate variants in an ISL1 enhancer.

a. An affected trio with isolated congenital facial palsy, a CCDD affecting cMN7 (left), in which the affected offspring harbors compound heterozygous non-coding candidate SNVs (depicted by blue and red bars) affecting highly conserved nucleotides in enhancer hs2757 (right). The enhancer is predicted to regulate Isl1 (peak-to-gene r = 0.744, ABC power law = 0.024). Variant coordinates are in NG_023040.1.
b. In vivo reporter assay testing hs2757 enhancer activity. Enhancement is present in cranial nerve 7 (arrows), an Isl1 positive cell type. Reporter expression views are shown as lateral (left) and dorsal through the 4th ventricle (right).
Extended Data Figure 8. Quality metrics for Basenji convolutional neural network accessibility predictions.

a. Precision-recall (PRC, left) and receiver-operating characteristic (ROC, right) curves measuring favorable performance (as measured by positive predictive value, sensitivity, true positive rate, and false positive rate) of Basenji accessibility predictions for cMN7 e10.5. AU denotes area under curve. Dotted lines represent the baseline classification rate.
b. Scatterplot depicting Basenji accessibility predictions vs. true scATAC sequencing coverage for cMN7 e10.5. Each point represents a 128 bp test bin whose sequence was excluded from training. Measured and predicted coverage are positively correlated (Pearson’s R = 0.833).
c. Boxplot summarizing area under PRC (AUPRC) and ROC (AUROC), and Pearson’s R for all samples and replicates. Quality metrics are consistent across samples. Data points depicted in (a) and (b) are highlighted in red. Centre line – median; box limits – upper and lower quartiles; whiskers – 1.5 x interquartile range.
Extended Data Figure 9. Cell type-aware candidate variants alter reporter expression in vivo.
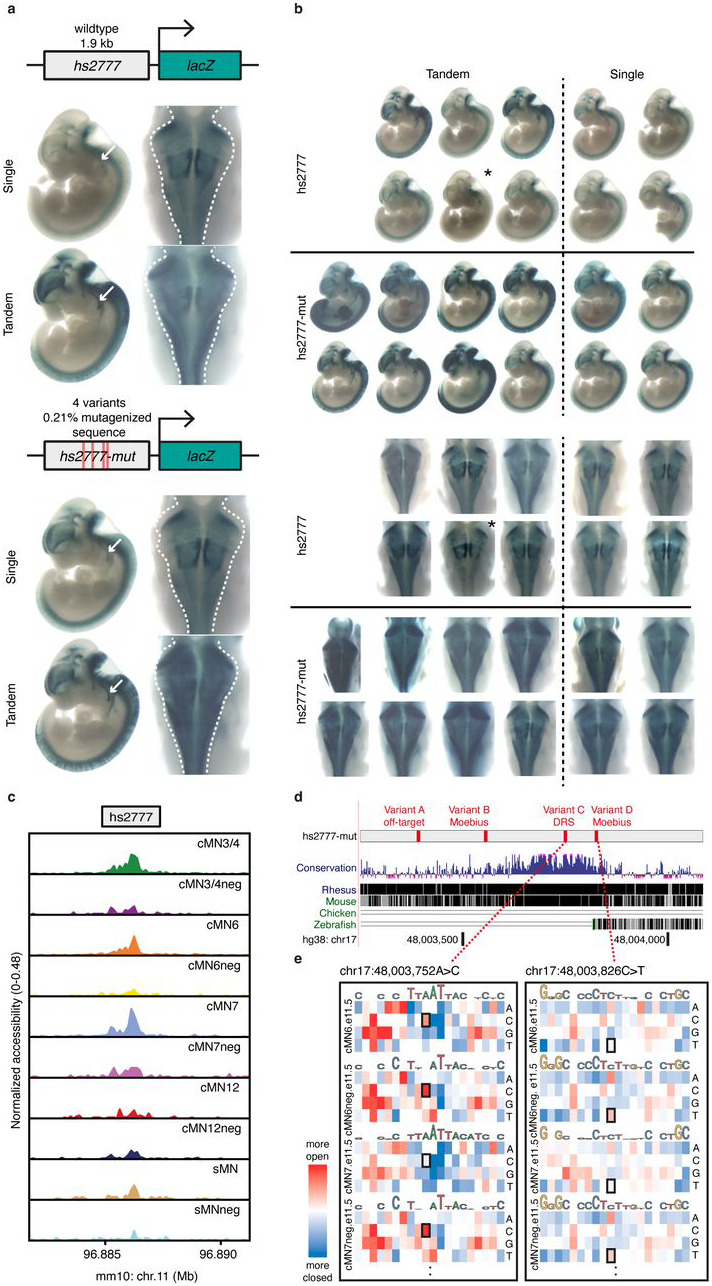
a. Representative whole mount in vivo enhancer reporter expression for (top) hs2777 wildtype and (bottom) hs2777-mut enhancer constructs. For each reporter insertion, dosage is labelled (“single”, “tandem”). Reporter expression views are shown as lateral (left) and dorsal through the 4th ventricle (right). Cranial nerve 7 (white arrows) and surrounding hindbrain tissue (dashed lines) show visible gain of reporter expression.
b. Additional replicates as in (a), matched by injection batch (top and bottom). hs2777-mut constructs reproducibly show increased reporter expression across midbrain, hindbrain, and neural tube. Random insertions are denoted by an asterisk.
c. hs2777 chromatin accessibility profiles in the cranial motor neurons and surrounding cell types. The wildtype element is accessible across multiple cMNs and surrounding cells.
d. UCSC screenshot depicting location of hs2777-mut variants: “Variant A” (chr17:48003393G>A, off-target), “Variant B” (chr17:48003557C>G, Moebius), “Variant C” (chr17:48003752A>C, DRS), and “Variant D” (chr17:48003826C>T, Moebius). hs2777-mut overlaps conserved non-coding sequence, particularly for Variants C and D.
e. Neural net-trained in silico saturation mutagenesis predictions for all possible nucleotide changes in hs2777 for selected samples cMN6 e11.5, cMN6neg e11.5, cMN7 e11.5, and cMN7neg e11.5. Predicted loss-of-function nucleotide changes are colored in blue and gain-of-function in red. Specific nucleotide changes corresponding to in vivo Variants C and D are boxed. Samples marked with “neg” are GFP-negative cells surrounding the motor neurons of interest. All other samples are GFP-positive motor neurons. Variants C and D are predicted to increase accessibility in relevant samples consistent with their corresponding phenotypes; DRS alters cMN6 but not cMN7 development (Variant C), while MBS alters both (Variant D).
ACKNOWLEDGEMENTS
We are indebted to all study participants and their families. We thank Ryosuke Fujiki, Tulsi Patel, Ben Weisburd, Julie Jurgens, Orit Rozenblatt-Rozen, Aviv Regev, Andrew Hill, and Jay Shendure for important technical discussions. We thank Max Tischfield, Sarah Izen, Alicia Nugent, Alon Gelber, and Matthew Bauer for technical assistance with bulk and scRNA-seq experiments. Next generation sequencing for single cell experiments was performed at the Molecular Genetics Core at Boston Children’s Hospital. WGS of the CCDD cohort was performed at Baylor College of Medicine through the Gabriella Miller Kids First Pediatric Research Program (dbGaP Study Accession: phs001247). New mouse lines were generated by the Gene Manipulation & Genome Editing Core at Boston Children’s Hospital. FACS experiments were performed at the Blavatnik Institute Department of Immunology Flow Cytometry Core Facility at Harvard Medical School, the Boston Children's Hospital Hem/Onc-HSCI Flow Cytometry Research Facility, and the Dana-Farber Flow Cytometry Hematologic Neoplasia and Jimmy Fund Cores at Dana-Farber Cancer Institute.
The work was supported by the Gabriella Miller Kids First Pediatric Research Program NHBLI X01HL132377 (E.C.E.), NEI R01EY027421 (D.G.M., M.E.T., E.C.E.), NICHD R01HD114353 (L.A.P), NHGRI R01HG003988 (L.A.P.), NIMH R01MH115957 (M.E.T., H.B.), DP2-AG072437 (E.A.L.), NINDS K08-NS099502 (M.F.R.), NHLBI T32-HL007627 (M.F.R), NIGMS T32-GM007748 (M.F.R.), Project ALS A13-0416 (E.C.E.), Boston Children’s Hospital - Broad Institute Collaborative Grant (E.C.E.), Boston Children’s Hospital Manton Center Rare Disease Fellowships (A.S.L, B.Z.) and Manton Center Pilot Project Award (B.Z.), Suh Kyungbae Foundation (E.A.L.), the Abramson Fund for Undergraduate Research (C.L.), and the Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (NIH U54HD090255). The research of M.K. and L.A.P. was conducted at the E.O. Lawrence Berkeley National Laboratory and performed under U.S. Department of Energy Contract DE-AC02-05CH11231, University of California. E.C.E. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
COMPETING INTEREST STATEMENT
D.G.M. is a paid advisor to GlaxoSmithKline, Insitro, and Overtone Therapeutics, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Google, Merck, Microsoft, Pfizer, and Sanofi-Genzyme. M.E.T. has received research support and/or reagents from Microsoft, Illumina Inc, Pacific Biosciences, and Ionis Pharmaceuticals. Otherwise, the authors declare that they have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the interpretation of this article.
Data availability
All data generated in this work are available through the Gene Expression Omnibus accession number GSExxxxxx.
Code availability
Custom code to perform analyses from this work is available at https://github.com/arthurlee617/noncoding-mendel.
REFERENCES
- 1.Smedley D. et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. The American Journal of Human Genetics vol. 99 595–606 Preprint at 10.1016/j.ajhg.2016.07.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amberger J. S. & Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes. Curr. Protoc. Bioinformatics 58, 1.2.1–1.2.12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visscher P. M. et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 101, 5–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hekselman I. & Yeger-Lotem E. Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nature Reviews Genetics Preprint at 10.1038/s41576-019-0200-9 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Short P. J. et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 555, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilissen C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Gordon C. T. & Lyonnet S. Enhancer mutations and phenotype modularity. Nature genetics vol. 46 3–4 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Osterwalder M. et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasperini M., Tome J. M. & Shendure J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson M. E. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meehan T. F. et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 49, 1231–1238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziffra R. S. et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Domcke S. et al. A human cell atlas of fetal chromatin accessibility. Science 370, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montoro D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaFave L. M. et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 38, 212–228.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pijuan-Sala B. et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada K. et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat. Genet. 35, 318–321 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Yamada K. et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Invest. Ophthalmol. Vis. Sci. 45, 2218–2223 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Nakano M. et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat. Genet. 29, 315–320 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Tischfield M. A. et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyake N. et al. Human CHN1 Mutations Hyperactivate 2-Chimaerin and Cause Duane’s Retraction Syndrome. Science vol. 321 839–843 Preprint at 10.1126/science.1156121 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohlhase J. et al. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 11, 2979–2987 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Al-Baradie R. et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71, 1195–1199 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tischfield M. A. et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 37, 1035–1037 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Jen J. C. Mutations in a Human ROBO Gene Disrupt Hindbrain Axon Pathway Crossing and Morphogenesis. Science vol. 304 1509–1513 Preprint at 10.1126/science.1096437 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webb B. D. et al. HOXB1 founder mutation in humans recapitulates the phenotype of Hoxb1−/− mice. Am. J. Hum. Genet. 91, 171–179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida K. et al. Congenital fibrosis of the extraocular muscles (CFEOM) syndrome associated with progressive cerebellar ataxia. Am. J. Med. Genet. A 143A, 1494–1501 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Whitman M. C. & Engle E. C. Ocular congenital cranial dysinnervation disorders (CCDDs): insights into axon growth and guidance. Hum. Mol. Genet. 26, R37–R44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tychsen L. The Cause of Infantile Strabismus Lies Upstairs in the Cerebral Cortex, Not Downstairs in the Brainstem. Archives of Ophthalmology vol. 130 1060 Preprint at 10.1001/archophthalmol.2012.1481 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Maass P. G. et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 47, 647–653 (2015). [DOI] [PubMed] [Google Scholar]
- 33.De Strooper B., De Strooper B. & Karran E. The Cellular Phase of Alzheimer’s Disease. Cell vol. 164 603–615 Preprint at 10.1016/j.cell.2015.12.056 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Tenney A. P. et al. Non-coding variants alter Gata2 expression in rhombomere 4 motor neurons and cause dominant hereditary congenital facial paresis. Nat. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewcock J. W., Genoud N., Lettieri K. & Pfaff S. L. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Wichterle H., Lieberam I., Porter J. A. & Jessell T. M. Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Cordes S. P. Molecular genetics of cranial nerve development in mouse. Nat. Rev. Neurosci. 2, 611–623 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Studer M., Lumsden A., Ariza-McNaughton L., Bradley A. & Krumlauf R. Altered segmental identity and abnormal migration of motor neurons in mice lacking Hoxb-1. Nature 384, 630–634 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Chisaka O., Musci T. S. & Capecchi M. R. Developmental defects of the ear, cranial nerves and hindbrain resulting from targeted disruption of the mouse homeobox gene Hox-1.6. Nature 355, 516–520 (1992). [DOI] [PubMed] [Google Scholar]
- 40.Koshiba-Takeuchi K. et al. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat. Genet. 38, 175–183 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Pattyn A., Morin X., Cremer H., Goridis C. & Brunet J. F. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development 124, 4065–4075 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Park J. G. et al. Loss of MAFB Function in Humans and Mice Causes Duane Syndrome, Aberrant Extraocular Muscle Innervation, and Inner-Ear Defects. Am. J. Hum. Genet. 98, 1220–1227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nugent A. A. et al. Mutant α2-chimaerin signals via bidirectional ephrin pathways in Duane retraction syndrome. J. Clin. Invest. 127, 1664–1682 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng L. et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 82, 334–349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michalak S. M. et al. Ocular Motor Nerve Development in the Presence and Absence of Extraocular Muscle. Invest. Ophthalmol. Vis. Sci. 58, 2388–2396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Argelaguet R. et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lara-Astiaso D. et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thurman R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bally-Cuif L., Cholley B. & Wassef M. Involvement of Wnt-1 in the formation of the mes/metencephalic boundary. Mech. Dev. 53, 23–34 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Grillet N., Dubreuil V., Dufour H. D. & Brunet J.-F. Dynamic expression of RGS4 in the developing nervous system and regulation by the neural type-specific transcription factor Phox2b. J. Neurosci. 23, 10613–10621 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cusanovich D. A. et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nott A. et al. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science vol. 366 1134–1139 Preprint at 10.1126/science.aay0793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corces M. R. et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nature Genetics vol. 52 1158–1168 Preprint at 10.1038/s41588-020-00721-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nowosad J. & Stepinski T. F. Spatial association between regionalizations using the information-theoretical V-measure. International Journal of Geographical Information Science vol. 32 2386–2401 Preprint at 10.1080/13658816.2018.1511794 (2018). [DOI] [Google Scholar]
- 55.Spielmann M. et al. Homeotic Arm-to-Leg Transformation Associated with Genomic Rearrangements at the PITX1 Locus. The American Journal of Human Genetics vol. 91 629–635 Preprint at 10.1016/j.ajhg.2012.08.014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klopocki E. et al. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am. J. Hum. Genet. 88, 70–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lettice L. A. et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 12, 1725–1735 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Visel A., Minovitsky S., Dubchak I. & Pennacchio L. A. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis C. A. et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simeone A., Acampora D., Gulisano M., Stornaiuolo A. & Boncinelli E. Nested expression domains of four homeobox genes in developing rostral brain. Nature 358, 687–690 (1992). [DOI] [PubMed] [Google Scholar]
- 61.Davidson C. L., Cameron L. E. & Burshtyn D. N. The AP-1 transcription factor JunD activates the leukocyte immunoglobulin-like receptor 1 distal promoter. Int. Immunol. 26, 21–33 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Sequential expression of JUN B, JUN D and FOS B proteins in rat spinal neurons: Cascade of transcriptional operations during nociception. Neurosci. Lett. 129, 221–224 (1991). [DOI] [PubMed] [Google Scholar]
- 63.Evans T., Reitman M. & Felsenfeld G. An erythrocyte-specific DNA-binding factor recognizes a regulatory sequence common to all chicken globin genes. Proc. Natl. Acad. Sci. U. S. A. 85, 5976–5980 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gogoi R. N. et al. The paired-type homeobox gene Dmbx1 marks the midbrain and pretectum. Mech. Dev. 114, 213–217 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Benko S. et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 41, 359–364 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Lupiáñez D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox J. J., Willatt L., Homfray T. & Woods C. G. A SOX9 duplication and familial 46,XX developmental testicular disorder. N. Engl. J. Med. 364, 91–93 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Gonen N. et al. Sex reversal following deletion of a single distal enhancer of Sox9. Science 360, 1469–1473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kurth I. et al. Duplications of noncoding elements 5’ of SOX9 are associated with brachydactyly-anonychia. Nature genetics vol. 41 862–863 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Granja J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corces M. R. et al. The chromatin accessibility landscape of primary human cancers. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Granja J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Argelaguet R., Cuomo A. S. E., Stegle O. & Marioni J. C. Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 39, 1202–1215 (2021). [DOI] [PubMed] [Google Scholar]
- 74.Cao J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uemura O. et al. Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev. Biol. 278, 587–606 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Fulco C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bartosovic M., Kabbe M. & Castelo-Branco G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee S. et al. A regulatory network to segregate the identity of neuronal subtypes. Dev. Cell 14, 877–889 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.An D. et al. Stem cell-derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee H. et al. Multi-omic analysis of selectively vulnerable motor neuron subtypes implicates altered lipid metabolism in ALS. Nat. Neurosci. 24, 1673–1685 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Collins R. L. et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karczewski K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whiffin N. et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 19, 1151–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harrison S. M., Biesecker L. G. & Rehm H. L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 103, e93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Byrska-Bishop M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shinwari J. M. A. et al. Recessive mutations in COL25A1 are a cause of congenital cranial dysinnervation disorder. Am. J. Hum. Genet. 96, 147–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Snijders Blok L. et al. De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum. Genet. 137, 375–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frints S. G. M. et al. Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita. Hum. Mutat. 40, 2270–2285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mak C. C. Y. et al. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain 143, 55–68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jurgens J. A. et al. Novel variants in TUBA1A cause congenital fibrosis of the extraocular muscles with or without malformations of cortical brain development. Eur. J. Hum. Genet. 29, 816–826 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cederquist G. Y. et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum. Mol. Genet. 21, 5484–5499 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Novo Mutations in EBF3 Cause a Neurodevelopmental Syndrome. Am. J. Hum. Genet. 100, 138–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Whitman M. C. et al. Decreased ACKR3 (CXCR7) function causes oculomotor synkinesis in mice and humans. Hum. Mol. Genet. 28, 3113–3125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deisseroth C. A. et al. An Integrated Phenotypic and Genotypic Approach Reveals a High-Risk Subtype Association for EBF3 Missense Variants Affecting the Zinc Finger Domain. Ann. Neurol. 92, 138–153 (2022). [DOI] [PubMed] [Google Scholar]
- 95.Firth H. V. et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khan A. O. & Al-Mesfer S. Recessive COL25A1 mutations cause isolated congenital ptosis or exotropic Duane syndrome with synergistic divergence. J. AAPOS 19, 463–465 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Weedon M. N. et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nature Genetics vol. 46 61–64 Preprint at 10.1038/ng.2826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Klopocki E. et al. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. Journal of Medical Genetics vol. 45 370–375 Preprint at 10.1136/jmg.2007.055699 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Ferrara A. M. et al. A Novel Mechanism of Inherited TBG Deficiency: Mutation in a Liver-Specific Enhancer. The Journal of Clinical Endocrinology & Metabolism vol. 100 E173–E181 Preprint at 10.1210/jc.2014-3490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Disruption of Autoregulatory Feedback by a Mutation in a Remote, Ultraconserved PAX6 Enhancer Causes Aniridia. Am. J. Hum. Genet. 93, 1126–1134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Collins R. L. et al. A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gray P. A. et al. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306, 2255–2257 (2004). [DOI] [PubMed] [Google Scholar]
- 103.Padhi E. M. et al. Coding and noncoding variants in EBF3 are involved in HADDS and simplex autism. Hum. Genomics 15, 44 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bejerano G. et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Liu W. et al. The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development 135, 3959–3968 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kircher M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ionita-Laza I., McCallum K., Xu B. & Buxbaum J. D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 48, 214–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiou J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nasser J. et al. Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kelley D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miyake N. et al. Expansion of the CHN1 strabismus phenotype. Invest. Ophthalmol. Vis. Sci. 52, 6321–6328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Snetkova V. et al. Ultraconserved enhancer function does not require perfect sequence conservation. Nature Genetics vol. 53 521–528 Preprint at 10.1038/s41588-021-00812-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kircher M. et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 10, 3583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shin T. et al. Rare variation in noncoding regions with evolutionary signatures contributes to autism spectrum disorder risk. medRxiv (2023) doi: 10.1101/2023.09.19.23295780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen S. et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv 2022.03.20.485034 (2022) doi: 10.1101/2022.03.20.485034. [DOI] [Google Scholar]
- 116.Buenrostro J. D., Wu B., Chang H. Y. & Greenleaf W. J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liang D. et al. Cell-type-specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. Nat. Neurosci. 24, 941–953 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weissbrod O. et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 52, 1355–1363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.International Common Disease Alliance. From Maps to Mechanisms to Medicine: Using human genetics to propel the understanding and treatment of common diseases. [White paper] (2020). [Google Scholar]
- 120.Vollger M. R. et al. Segmental duplications and their variation in a complete human genome. Science (2022) doi: 10.1126/science.abj6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prabhakar S. et al. Human-Specific Gain of Function in a Developmental Enhancer. Science (2008) doi: 10.1126/science.1159974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Finucane H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Philippakis A. A. et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum. Mutat. 36, 915–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kvon E. Z. et al. Comprehensive In Vivo Interrogation Reveals Phenotypic Impact of Human Enhancer Variants. Cell 180, 1262–1271.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lek M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Samocha K. E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stuart T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fujiki R., Lee J. Y., Jurgens J. A., Whitman M. C. & Engle E. C. Isolation and Culture of Oculomotor, Trochlear, and Spinal Motor Neurons from Prenatal Islmn:GFP Transgenic Mice. J. Vis. Exp. (2019) doi: 10.3791/60440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bentsen M. et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 11, 4267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Osterwalder M. et al. Characterization of Mammalian In Vivo Enhancers Using Mouse Transgenesis and CRISPR Genome Editing. Methods Mol. Biol. 2403, 147–186 (2022). [DOI] [PubMed] [Google Scholar]
- 133.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y. & Greenleaf W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li B. & Dewey C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gasperini M. et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 1516 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Ma S. et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stuart T., Srivastava A., Madad S., Lareau C. A. & Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hao Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.DePristo M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Minikel E. V. et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 8, 322ra9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Kronenberg Z. N. et al. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol. 11, e1004572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gardner E. J. et al. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McKenna A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klambauer G. et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Robinson J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chu C. et al. Comprehensive identification of transposable element insertions using multiple sequencing technologies. Nat. Commun. 12, 3836 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sudmant P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mir A. A., Philippe C. & Cristofari G. euL1db: the European database of L1HS retrotransposon insertions in humans. Nucleic Acids Res. 43, D43–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Evrony G. D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taliun D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Davydov E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Natera-de Benito D. et al. Recessive variants in COL25A1 gene as novel cause of arthrogryposis multiplex congenita with ocular congenital cranial dysinnervation disorder. Hum. Mutat. 43, 487–498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McMillin M. J. et al. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 92, 150–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Guan J. et al. SIX2 haploinsufficiency causes conductive hearing loss with ptosis in humans. J. Hum. Genet. 61, 917–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kruszka P. et al. Phenotype delineation of ZNF462 related syndrome. Am. J. Med. Genet. A 179, 2075–2082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Patak J. et al. MAGEL2-related disorders: A study and case series. Clin. Genet. 96, 493–505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Verloes, A. et al. Baraitser-Winter cerebrofrontofacial syndrome: delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 23, 292–301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dobyns W. B. et al. MACF1 Mutations Encoding Highly Conserved Zinc-Binding Residues of the GAR Domain Cause Defects in Neuronal Migration and Axon Guidance. Am. J. Hum. Genet. 103, 1009–1021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gel B. et al. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics btv562 Preprint at 10.1093/bioinformatics/btv562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kelley D. R. Cross-species regulatory sequence activity prediction. PLoS Comput. Biol. 16, e1008050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Korsunsky I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated in this work are available through the Gene Expression Omnibus accession number GSExxxxxx.
Custom code to perform analyses from this work is available at https://github.com/arthurlee617/noncoding-mendel.