

ABSTRACT
Activins, members of the TGF-beta superfamily, have been isolated and identified in the endocrine system, but have not been substantially investigated in the context of the immune system and endocrine-unrelated cancers. Here, we demonstrated that tumor-bearing mice had elevated systemic activin levels, which correlated directly with tumor burden. Likewise, cancer patients have elevated plasma activin levels compared to healthy controls. We observed that both tumor and immune cells could be sources of activins. Importantly, our in vitro studies suggest that activins promote differentiation of naïve CD4+ cells into Foxp3-expressing induced regulatory T cells (Tregs), particularly when TGF-beta was limited in the culture medium. Database and qRT-PCR analysis of sorted major immune cell subsets in mice revealed that activin receptor 1c (ActRIC) was uniquely expressed on Tregs and that both ActRIC and ActRIIB (activin receptor 2b) were highly upregulated during iTreg differentiation. ActRIC-deficient naïve CD4+ cells were found to be defective in iTreg generation both in vitro and in vivo. Treg suppression assays were also performed, and ActRIC deficiency did not change the function or stability of iTregs. Mice lacking ActRIC or mice treated with monoclonal anti-ActRIC antibody were more resistant to tumor progression than wild-type controls. This phenotype was correlated with reduced expression of Foxp3 in CD4+ cells in the tumor microenvironment. In light of the information presented above, blocking activin-ActRIC signaling is a promising and disease-specific strategy to impede the accumulation of immunosuppressive iTregs in cancer. Therefore, it is a potential candidate for cancer immunotherapy.
KEYWORDS: Activin, cancer immunotherapy, Foxp3, iTregs, TILs, Acvr1c, Alk7
Video Abstract.
© 2023 The Author(s). Published with license by Taylor&Francis Group, LLC
Introduction
Activins, which are members of the transforming growth factor-β) (TGF-β)) superfamily of proteins, are potent regulatory factors that play vital roles in tissue fate determination during embryonic development as well as in regulating homeostatic processes in adults.1–4 Currently, over 45 members of the TGF-β superfamily have been identified, among which inhibins and activins have been found to be closely related but have almost directly opposite biological effects in regulating follicle-stimulating hormone (FSH) synthesis and secretion.5 Inhibins are dimers of an α subunit and a β subunit, whereas activins are homo- or heterodimers of two β subunits. These two subunits are tethered by a disulfide bond.1 Only one isoform of α subunit, but four isoforms of β subunits (βA, βB, βC, and βE) have been identified so far.5–7
Activins signal by binding to a heteromeric tetramer composed of two type I receptors (either ActRIC, gene name Acvr1c, also known as Alk7, or ActRIB/Acvr1b/Alk4) and two type II receptors (either ActRIIA/Acvr2a or ActRIIB/Acvr2b).1,8 Initially, activins bind to type II receptors at the cell surface. This binding, in turn, leads to the recruitment and phosphorylation of type I receptors, which then activate intracellular signaling pathways mainly through Smad proteins or alternatively through MAPK. Primarily, it recruits and phosphorylates receptor-regulated Smad (R-Smad). R-Smads are ligand-specific, with Smad2 and Smad3 mediating activin and TGF-β signaling. After phosphorylation, R-Smads are released and form complexes with the co-Smad, Smad4. The complexes then translocate to the nucleus to regulate the expression of downstream genes.1,9,10 Many groups have reported that Smad2 and Smad3 are important for inducing the expression of the transcription factor (Forkhead box P3) Foxp3, which is a cell lineage-specific marker for regulatory T cells (Tregs).11–13
Tregs play pivotal roles in the maintenance of peripheral immunological tolerance and control of immune responses toward pathogens and tumors.14,15 These Foxp3-expressing CD4+ T cells typically exert their immunosuppressive effects through direct cell-to-cell interactions and via secretion of anti-inflammatory cytokines such as TGF-β, IL-10, and IL-35.11,15,16 Accumulation of Tregs in tumors correlates with poor prognosis and reduced survival in patients with various cancer types.15,17 Conversely, the growth of murine tumors can be inhibited by blocking Tregs, which constitute an extremely high proportion of tumor-infiltrating lymphocytes (TILs) in many murine tumors, such as B16.18 When diphtheria toxin is used to deplete Tregs in tumor-bearing Foxp3-DTR (diphtheria toxin receptor) transgenic mice, these tumors regress.19 Therefore, depletion of Tregs is an attractive strategy to boost antitumor immunity. Foxp3 is a transcription factor that is challenging to inhibit. Hence surface receptors uniquely expressed on Treg cells are of particular interest as they can be targeted with antibodies. It would be even more advantageous if the expression of these surface receptors is specifically induced in the tumor microenvironment.
Activins and their receptors have been reported to play important roles in endocrine tumorigenesis, such as ovarian cancer, breast cancer, and prostate cancer, etc.1,3,20,21 However, their expression patterns and functions are poorly understood in the immune system and endocrine-unrelated cancers.22–25 We previously discovered that ActRIC was a target of the Treg-promoting YAP1 (yes-associated protein1) pathway in mice.26 These findings suggest that blocking the activin-receptor interaction could enhance the efficacy of cancer immunotherapy by inhibiting the immunosuppressive Treg population. However, the challenges surrounding blocking activins directly center on the fact that activins are produced and present in a wide range of tissues. These small proteins, approximately 25 kDa in their mature form, are produced by many types of cells and are widely distributed in the gonads, pancreas, and circulation.1,9 Therefore, depletion of activins would be difficult to achieve and could cause many side effects. Alternatively, blocking one or more of the receptors upregulated in the tumor microenvironment may represent a better strategy.
In this study, our goal was to explore the role of activin in immune homeostasis, its impact on tumor growth, and the identification of the activin receptor induced in the tumor microenvironment. Our data reveal that the interaction between activin and ActRIC promotes the conversion of CD4+CD25low T cells into Foxp3-expressing Tregs, ActRIC emerges as a promising target for cancer immunotherapy.
Methods
Extended materials and methods, including catalog information of all the antibodies and primer sequences, can be found in the supplemental table.
Mice
Wild-type C57BL/6J mice were purchased from the Jackson Laboratory. Rag2 knock-out (KO) and TCRα (T cell receptor alpha chain) KO mice were obtained from Jackson Laboratory and bred in our animal facility. Acvr1ctm1b mice were obtained from Mutant Mouse Resources & Research Centers supported by NIH (MMRRC) and globally knocked out in ActRIC. All animal experiments were performed under protocols approved by the Johns Hopkins University Institutional Animal Care and Use Committee (IACUC). Primarily 6–12 weeks-old female mice were used for all experiments.
Tumor models
Murine B16F10 melanoma cells, MC38 colon adenocarcinoma cells, and EL4 thymoma cells were purchased from the American Type Culture Collection (ATCC) and stored as frozen stocks. Cells were cultured as recommended by ATCC, and cells with passage number between 5–10 were used in the experiments. The right flank region of female animals (approximately 8-weeks old) was shaved one day prior to subcutaneous (s.c.) injection of the indicated cell line cells in 200ul of PBS. 40000 of B16F10, 250,000 MC38, or 200,000 EL4 cells were injected per mouse unless otherwise indicated. Tumor progression was quantified using the following formula
where V is the volume, L is the maximum length, and W is the width perpendicular to the length.27,28 Excised tumors were weighed using a scale and imaged as groups.
Adoptive transfer colitis model
WT or Acvr1c KO naïve CD4+ cells were negatively selected using a naïve CD4+ T cell isolation kit (Miltenyi Biotech) and adoptively transferred to Rag2 KO mice. The mice were then monitored for weight loss for approximately nine weeks as the disease progressed. At the end of the process, the mice were sacrificed, and multiple organs, including the colon, mesenteric lymph nodes (mLN), and spleens (SP) were harvested. Organ-infiltrating lymphocytes were extracted by gradient density centrifugation and analyzed for CD4+ and Foxp3+ populations by flow cytometry.
ELISA
Both human and mouse blood samples were collected in ethylenediaminetetraacetic acid (EDTA)-coated tubes. Blood was centrifuged at 500 g for 10 min to separate plasma from cells. Plasma was aliquoted and stored at −80°C before being used for ELISA. The Mouse/Human/Rat Quantikine ELISA Kit (R&D Systems) was used according to the manufacturer’s protocols to derive absolute concentrations of activins using a standard curve (performed along with all samples). The samples were diluted to remain within a detectable range where appropriate.
Human blood
Consented human blood samples were obtained from the Johns Hopkins University School of Medicine under an institution-approved IRB protocol. Human blood leukopaks were obtained from Johns Hopkins School of Medicine, Division of Gastroenterology and Hepatology. Whole blood in EDTA-coated tubes was spun down to obtain cell-free plasma.
Cell sorting
Homogenized spleen, lymph node, and/or tumor-infiltrating cells were stained in FACS buffer with the defining antibodies, as indicated. Cells were sorted using a BD FACSAriaTM Fusion flow cytometer with the assistance of the Sidney Kimmel Comprehensive Cancer Center Flow Cytometry Core staff into complete RPMI medium (Gibco), supplemented with 5% FBS, before being spun down for use in tissue culture or stored in TRIzol (ThermoFisher Scientific).
Cell culture
T cells were activated with plate-bound anti-CD3 and soluble anti-CD28 antibodies in the presence of IL-2 (200 U/mL). RPMI medium, supplemented with 5–10% FBS, sodium pyruvate, nonessential amino acids, antibiotics, and β-mercaptoethanol was used. 105−106 cells were plated in a tissue culture-treated 24-well plate containing 1 ml media. For Treg induction, up to 10 ng/ml of recombinant TGF-β was used. Murine or human activins were obtained from R&D Systems and added to the culture at concentrations up to 50ug/ml.
Flow cytometry
Cell Trace Violet (Invitrogen) was used to track cell proliferation, according to the manufacturer’s protocol. Cells were stained with a viability dye for 20 min at room temperature in PBS, fluorophore-conjugated antibodies against surface markers for 15 min at 4°C in FACS buffer, and fluorophore-conjugated antibodies against intracellular markers for 45 min at 4°C in permeabilization wash buffer. Both BD Cytofix and ThermoFisher Transcription Factor fixation kits were used, depending on the antibodies used.
qRT-PCR
Cells were lysed using TRIzol reagent (Invitrogen). After phase separation with chloroform, RNA was purified using a Direct-zol RNA Purification Kit (Zymo Research), according to the manufacturer’s protocol. Equal amounts of total RNA (as measured by NanoDrop Spectrophotometer) were converted to cDNA using the SuperScript III Reverse Transcription Kit (Invitrogen), as described by the manufacturer. Gene expression was quantified using the SYBR Green RT-qPCR Master Mix (ThermoFisher Scientific) with gene-specific primers. Primer sequences were either obtained from the Harvard Primer Bank or designed using the NCBI Primer Blast Software. All primers were validated with dose-dependent amplification using qRT-PCR, with melt curves, and with DNA gel electrophoresis.
Monoclonal antibodies production
Monoclonal antibodies targeting ActRIC were generated using the hybridoma technology. In brief, C57BL6 mice were immunized with the extracellular domain (ECD) of activin receptor 1c. The antibody-producing B lymphocytes were isolated from these mice and fused with the immortal P3X63Ag8.653 mouse myeloma cell line to form hybrid cells, called hybridomas. The hybridomas of each clone were expanded in vitro. The nude mice were primed with i.p. injection of 0.5 ml pristane per mouse 1 week prior to inoculation with hybridomas. The cultured hybridomas were then collected, washed, and resuspended in PBS at 2.5106 cells/ml. 2 ml of the cell suspension per mouse were i.p. injected into the primed nude mice using a 22-G needle. Wait for 1–2 weeks for ascites to form. The ascites was then collected and purified for monoclonal antibodies using a HiTrapTM protein HP column (GE Healthcare Bio-Sciences AB, Sweden).
Statistical analysis
All statistical analyses were performed using the GraphPad Prism 9 software. Unpaired two-tailed Student’s t-test was used to compare means between two groups, and one-way or two-way analysis of variance (ANOVA) was used to determine the statistical significance of data that had more than two groups. Values are presented as the mean ± SEM, where appropriate. * represents p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Results
Activins are elevated in peripheral blood and tumor microenvironment
To test whether activin levels were elevated systemically in the presence of tumors, peripheral blood samples were collected from healthy WT mice, pregnant mice, and mice bearing various tumors, including B16F10, EL4, and MC38, and spontaneous tumors that developed in aged mice. ELISA was performed to measure the plasma levels of activins. Compared to healthy mice, most of the tumor-bearing mice, in addition to the pregnant mice, had upregulated activin levels in their peripheral blood (Figure 1a). However, the B16F10 tumor model did not induce significantly elevated plasma activin levels, possibly due to localized production and/or a high rate of activin consumption by aggressive tumor cells in the tumor environment. Next, peripheral blood samples were collected on different days post MC38 and EL4 tumor cell inoculation and plasma levels of activin A were measured. Activin A levels significantly increased at later stages of cancer progression, indicating that the plasma levels of activin A directly correlate with tumor burden (Figures 1b,c).
Figure 1.
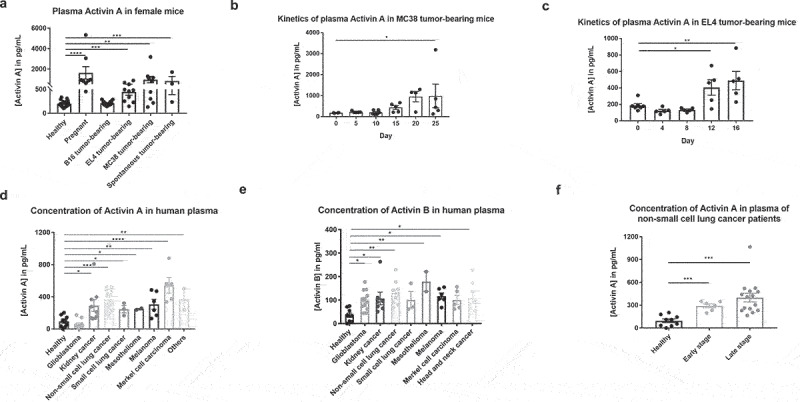
Activins are elevated in peripheral blood and tumor microenvironment in tumor bearing mice and cancer patients. (a) peripheral blood samples were collected from healthy mice, pregnant mice, and mice carrying various tumors. Plasma levels of activins were measured by ELISA kits. (b-c) peripheral blood samples were collected from mice at indicated days after MC38 (b), EL4 (c) tumor implantation, and measured for plasma levels of activins using ELISA. (d-f), human plasma levels of activins, were measured by ELISA from samples obtained from healthy donors and cancer patients. Activin a (d), activin B (e) and activin a levels of patients with different stages of non-small cell lung cancer (f) were measured respectively.
Similar phenomena were observed in human samples. Plasma obtained from healthy donors and cancer patients was tested for activin A and B levels using ELISA. Patients with melanoma, kidney, lung, melanoma, Merkel cell carcinomas, and head and neck cancer had upregulated activin A and B levels in the peripheral blood (Figures 1d,e), except for activin A in patients with glioblastoma. Consistent with the results in mice, early-stage non-small cell lung cancer patients exhibited less upregulation of activin A compared to late-stage patients (Figure 1f). This result suggests that activin levels are upregulated in proportion to cancer progression and could potentially be used as a biomarker of disease burden.
Activins are homo- or heterodimers composed of two β subunits. To date, βA, βB, βC, and βE subunits have been identified and are encoded by the Inhba, Inhbb, Inhbc, and Inhbe genes, respectively. To determine whether activins are produced by tumor or immune cells, mRNA was extracted from B16F10, MC38, and EL4 tumor cell lines, as well as healthy C57BL/6J spleens. The transcription of the Inhba, Inhbb, Inhbc, and Inhbe genes was tested in these samples. The results suggested that splenocytes and MC38 express mRNA encoding subunits of activin A, splenocytes, and EL4 cells were high in Inhbb and Inhbc, but only splenocytes were high in Inhbe (Figure 2a).
Figure 2.
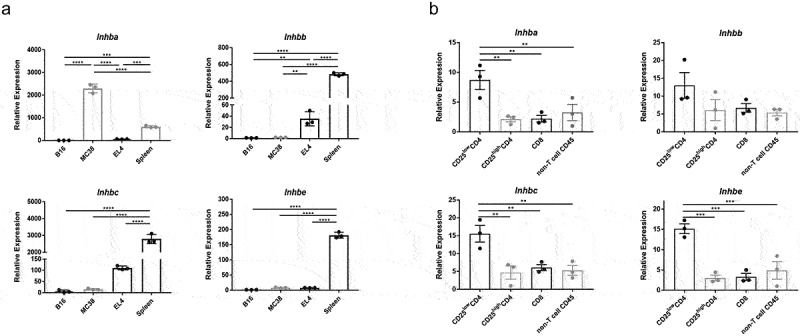
RNA expression of activins is detected in tumor cells and immune cells. (a) differential gene expression of the inhibin beta protein subunits – monomers of the dimeric activins – was compared using qRT-PCR. RNA was isolated from B16F10, MC38, and EL4 cell lines as well as healthy C57BL/6J mouse spleens. (b) MC38 tumor-infiltrating lymphocytes were sorted into major immune cell subsets as indicated and tested for the expression of the different activin subunit genes.
To further elucidate which immune cell subset produces activins in the tumor microenvironment, MC38 tumor-infiltrating lymphocytes were sorted accordingly, as shown in Figure 2b. Non-Treg conventional CD4+(CD25LowCD4+ or Foxp3-CD4+) cells showed upregulated expression of all four inhibin β subunits (Figure 2b). This observation, although not statistically significant, also seemed to hold true for cells sorted from healthy spleens (Figure S1B). Collectively, this suggests that the tumor microenvironment promotes the expression of activins in non-Treg CD4+ cells. A survey of the Immgen Skyline project RNAseq database, however, indicated that many immune cell types, including neutrophils, CD4+ conventional T cells, and myeloid cells, express inhibin β subunits that make up activins. (Figure S1A). Thus, environmental cues are likely to govern activin secretion by various immune cells.
Activins promote the development of iTregs in vitro and in vivo
Based on these observations, it was suggested that the levels of activins in the peripheral blood increased with tumor progression. Thus, we suspect that activins play an important role in enhancing tumor growth. Activins belong to the TGF-β superfamily of proteins, the main components of which are known to be essential for the development and function of Foxp3+ Tregs. We hypothesized that activins play a role in increasing Tregs by inducing Foxp3 expression. To test this, naïve CD4+ T cells isolated from WT C57BL/6J mice were cultured in vitro under iTreg differentiation conditions in the presence or absence of activin A or B at a constant concentration along with titration of TGF-β. The addition of activins did not significantly affect iTreg development when there was sufficient TGF-β in the culture medium. However, when TGF-β was limited, the addition of either activin A or activin B significantly increased the percentage of CD25highFoxp3+ cells in CD4+ cells (Figure 3a). Meanwhile, Ki67 staining revealed that these Foxp3+ cells exhibited higher proliferation rates (Figure 3b).
Figure 3.
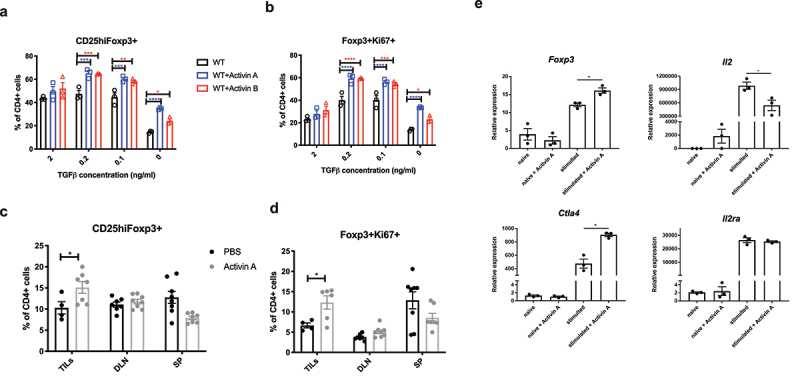
Activins promote the development of induced Tregs (iTregs) in vitro and in vivo. (a-b) naive CD4+ T cells were isolated from WT C57BL/6J mice, and then cultured in vitro with plate-bound anti-CD3, soluble anti-CD28, recombinant IL-2 and the indicated gradient concentrations of TGF-b for 3 days. The percentage of CD25highFoxp3+ (a) and Foxp3+Ki67+ (b) in CD4+ cells were analyzed by flow cytometry. (c-d) WT C57BL/6J mice were inoculated with B16F10 tumors and treated with 50ng per mouse recombinant activin A or PBS every other day for 21 days. TILs and specified organs (draining lymph nodes shown as DLN, spleens shown as SP) were harvested from these mice and analyzed for the frequency of CD25highFoxp3+ (c) and Foxp3+Ki67+ (d) in CD4+ cells by flow cytometry. (e) naive CD4+ T cells were isolated from WT C57BL/6J mice, and then stimulated with plate-bound anti-CD3, soluble anti-CD28, recombinant IL-2 in the absence or presence of recombinant activin A. 36 hours later, RNA was analyzed for the expression of indicated genes using qRT-PCR.
After demonstrating the direct effect of activins in inducing iTreg differentiation in vitro, we aimed to test their in vivo efficacy. WT C57BL/6J mice bearing B16F10 tumors were treated with recombinant activin A or PBS. Tumor growth was not significantly different between the two groups (data not shown). Nevertheless, upon conducting flow cytometric analysis on cells harvested from the specified organs, we observed a significantly higher frequency of Tregs among the tumor-infiltrating lymphocytes (TILs) in the activin A group (Figure 3c). Furthermore, we also noted increased Ki67 expression in these CD4+Foxp3+ TILs (Figure 3d).
We then measured the mRNA levels of Foxp3, CTLA-4, IL-2, and IL-2 receptor A(CD25) in CD4+ cells stimulated with or without recombinant activin A. Upon stimulation of naïve CD4+ cells with anti-CD3 and anti-CD28 in the presence of IL-2, recombinant activin A enhanced Foxp3 and Ctla4 mRNA expression. Conversely, activin A suppressed IL-2 transcription (Figure 3e), while IL-2 receptor A expression remained unchanged. These observations suggest that activin A reprograms activated CD4+ T cells toward a more suppressive phenotype, which is in concordance with previous findings that activin A enhances SMAD2/3 phosphorylation in cell line cells and primary murine CD4+ cells.26,29
ActRIC and ActRIIB mediate activin signaling in CD4+ cells
Activins are systemically present in numerous tissues and mediate a wide range of biological actions on the growth and differentiation of many cell types. The inhibition of activins is difficult to achieve and can provoke a wide variety of side effects. The regulation of activin receptors is a potentially better solution to focus specifically on activin signaling in CD4+ cells. To determine which receptors may receive an activin signal in CD4+ cells, the Immgen Skyline RNAseq database was again surveyed. The results suggested that Acvr1c was uniquely expressed by the CD4+ and Treg subsets of immune cells (Figure S2A). To confirm this, Foxp3-YFP reporter animals were used to sort immune cell subsets from the spleens for qRT-PCR analysis of activin receptor isoforms. Acvr1c was uniquely upregulated in the Treg (CD4+Foxp3+) subset (Figure 4a). A similar trend was observed in B16F10 melanoma tumor-draining lymph nodes (Figure S2B). Additionally, the Acvr1c mRNA level only increased significantly with the increase of Foxp3 when naïve CD4+ cells were skewed to iTregs under the conditions of plate-bound anti-CD3, soluble anti-CD28, and IL-2 plus TGF-β Figure 4b). When TGF-β was not included in the culture medium, Acvr1c only increased slightly on day 4 as the CD4+ T cells were nonspecifically stimulated.
Figure 4.
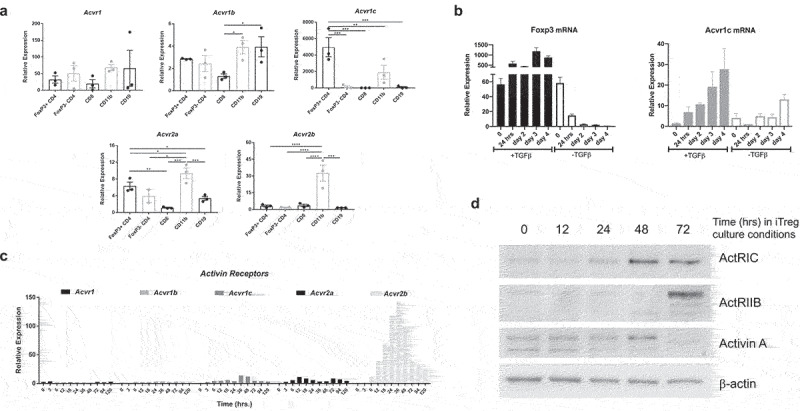
ActRIC and ActRIIB mediate activin signaling in CD4+ cells. (a) healthy spleen cells from Foxp3-YFP reporter mice were sorted into major immune cell subsets. RNA from these samples was tested for the expression of the different activin receptors. (b) naive CD4+ T cells were isolated from WT C57BL/6J mice, and then stimulated with plate-bound anti-CD3, soluble anti-CD28, recombinant IL-2 in the absence or presence of recombinant TGF-b. Cells were collected at indicated time points and analyzed for the mRNA expression of Foxp3 and Acvr1c using qRT-PCR. (c) WT naïve CD4 cells were differentiated under iTregs skewing conditions (plate-bound anti-CD3, anti-CD28, IL2, and TGF- β), and cells were collected at indicated times. RNA from these samples was tested for the expression of the different activin receptor transcripts using qRT-PCR. (d) WT naïve CD4+ cells were differentiated under iTregs skewing conditions before being collected at indicated timepoints for immunoblot analysis.
However, activin ligands interact with a dual receptor system at the cell surface involving both type I and type II receptors. In addition to ActRIC as a type I receptor, there must be another type II receptor involved.9,30 The expression kinetics of the five well-known activin receptors were investigated during iTreg differentiation. The transcription of Acvr2b was also upregulated (Figure 4c). The results of western blot analysis also validated that the expression of ActRIC and ActRIIB increased when iTregs were differentiated (Figure 4d). It has been well studied that activins initially bind to type II receptors and then recruit type I receptors to form a heterodimeric complex, which phosphorylate smad2 and smad3 proteins. Between these two types of receptors, activated type I receptors interact with Smads and are also called activin receptor-like kinases (Alk). Given that ActRIIB has a wider spectrum of ligands and is expressed by different immune cell subsets, while ActRIC is more specific for activins8,31 and appears to be unique to CD4+ T cells. Therefore, we hypothesized that ActRIC is a better target for blocking the effects of activins in the process of naïve CD4+ cells being induced to become Tregs.
Activin signaling through ActRIC regulates iTregs differentiation, rather than stability or function
To assess the role of ActRIC during iTreg development, we used a global knockout mouse strain, Acvr1ctm1b in which a critical exon of Acvr1c was deleted. During homeostasis, Acvr1c global knock-out animals were overall healthy (data not shown) and exhibited normal immune cell development and homeostasis (Figure S3A). The in vitro iTreg differentiation study, in the absence or presence of recombinant activin A at a constant concentration along with titration of TGF-β was repeated with an additional group of naïve CD4+ cells isolated from Acvr1c KO mice. The KO cells exhibited a defect in their ability to rescue Foxp3 expression upon the addition of activin A when TGF-β was not in excess, as quantified by flow cytometry (Figure 5a).
Figure 5.
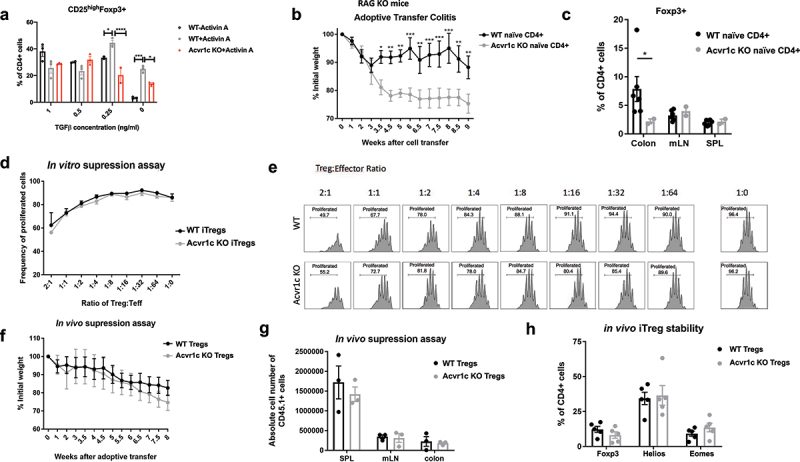
Activin signaling through ActRIC regulates iTregs differentiation, rather than stability or function. (a) naïve CD4+ T cells were isolated from WT or Acvr1c KO mice and cultured under iTreg conditions in the absence or presence of activins. (b) naïve CD4+ T cells were isolated from WT or Acvr1c KO mice and adoptively transferred to Rag2 KO mice. The animals were monitored for weight loss over the course of disease progression. (c) the induction of Foxp3 in different organs was analyzed by flow cytometry. (d, e) naïve CD4+ cells were isolated from WT or Acvr1c KO mice bearing Foxp3-GFP reporter and cultured under iTreg condition. The induced Tregs were sorted using the Foxp3-GFP reporter, and then cocultured with the cell Trace Violet-labeled naïve CD4+ cells at indicated ratios. Proliferation of the T conventional cells was observed three days later. The results are summarized in (d), and individual plots are shown in (e). (f, g) Treg cells (CD45.2) from WT or Acvr1c KO animals were adoptively transferred with naïve CD45.1+ CD4+ cells into Rag KO animals. The recipients’ weight was monitored for 8 weeks (F), after which the indicated organs were harvested for flow cytometry analysis to determine the number of CD45.1+ cells (G). (h) iTregs generated under in vitro skewing conditions and sorted by the Foxp3-GFP reporter were adoptively transferred into TCRα KO animals. One week later, the expression frequencies of Foxp3, helios, and Eomes were quantified using flow cytometry.
To evaluate the role of ActRIC in vivo, a classic adoptive transfer model that induces colitis in the absence of functional Tregs was used. When naïve CD4+ T cells are introduced into Rag2 KO mice, they cause inflammation in the colon, which is associated with weight loss. The development of even a small number of iTregs can lessen disease severity in recipient animals. In the experiment, when Rag2 KO recipients received Acvr1c KO naïve CD4+ T cells instead of WT cells, the mice that received Acvr1c KO cells developed much more severe colitis. This outcome was attributed to the failure of iTreg generation emphasizing the role of Acvr1c in regulating immune responses and inflammation (Figures 5b,c).
The next question that we asked was whether activin A signaling through ActRIC exclusively promotes iTreg differentiation or also has an impact on Treg function. To answer this question, both in vitro and in vivo suppression assays were conducted. Acvr1c KO mice were crossed with the Foxp3-GFP reporter mice. Naïve CD4+ cells were isolated from these mice and WT Foxp3-GFP mice and differentiated into Tregs in vitro for three days. Foxp3+ Tregs were then sorted using the Foxp3-GFP reporter and co-cultured with CD45.1+ genetically marked and Cell Trace Violet-labeled CD4+ T conventional cells (CD4+CD25low) under stimulation conditions at the indicated ratios (Figure 5d,e). Acvr1c KO Tregs had no apparent defect in their ability to suppress the proliferation of T effector cells. Similar results were observed in the assay with Tregs directly sorted from Foxp3-GFP animals (Figure S3B). In vivo, co-injection of Tregs and naïve T cells partially protects Rag2 KO mice from colitis because Tregs can delay or prevent the activation and expansion of conventional T cells. When Tregs from WT or KO animals were co-transferred with CD45.1+ naïve CD4+ cells, Acvr1c KO Tregs did not have any defect in their ability to suppress the short-term proliferation of CD45.1+ T conventional cells (Figure S3C and S3D). When this experiment was extended to two months, the group receiving Acvr1c KO Tregs exhibited similar weight loss as the control group receiving WT Tregs (Figure 5f). Additionally, the absolute number of CD45.1+ responder T cells was unaffected in the different organs (Figure 5g). These findings concur with the conclusions from the in vitro experiments that the activity of activin on Tregs via ActRIC enhances iTreg development, but is not necessary for functional suppression. They also imply that activin-ActRIC blockade may have high tumor specificity with minimal colonic inflammatory toxicity, as is seen with both anti-CTLA-4 and anti-PD-1 blockades.
Finally, the stability of Acvr1c KO Tregs was comparable to that of WT Tregs. The number and frequency of Foxp3-expressing KO Treg cells transferred into the long-term colitis model was approximately equal to that of the WT group across different organs (Figure S3E). When Acvr1c KO-induced Tregs (sorted using the Foxp3-GFP reporter after in vitro iTreg differentiation) were adoptively transferred into TCRα KO mice for one week, their stability was also comparable to that of the WT controls (Figure 5h). Additionally, when these sorted iTregs were maintained in tissue culture without any cytokines for four days, both groups maintained high Foxp3 expression (data not shown). Thus, while the deletion of ActRIC hinders the induction of Tregs, this receptor seems to play a minimal role once Foxp3 is expressed.
Acvr1c KO animals exhibit resistance to tumor progression in a CD4+ T cell dependent manner
Since Foxp3+ Tregs are important in mediating the immunosuppressive tumor microenvironment, we investigated whether blockade of the activin-ActRIC signaling pathway would impact tumor growth. The easiest way to test this was to evaluate tumor growth in Acvr1c KO vs. WT mice. In both the MC38 colon cancer model (Figure 6a,b) and the B16F10 melanoma tumor model (Figure S4A,B), tumor growth was significantly more inhibited in the Acvr1c KO group compared to the WT group. When TILs, DLNs, and spleens were harvested for flow cytometric analysis, we found a reduced frequency of Tregs in the Acvr1c KO group, which was significant only for TILs (Figure 6d). Meanwhile, these Foxp3+ TILs were less proliferative (Figure 6e). In addition to Foxp3, CD4+ cells in TILs of the Acvr1c KO group also exhibited lower GITR expression (Figure 6f). These results suggested that there were fewer inhibitory helper T cells in the Acvr1c KO group. Staining using our own antibody revealed substantial ActRIC expression on the surface of CD4+CD25 high cell, again exclusively significant for TILs in WT group (Figure 6b). When cytokine production was investigated by intracellular staining, both CD4+ and CD8+ cells in TILs showed higher levels of TNF-α and IFN-γ Figure 6h–k). In summary, these results revealed that ActRIC deficiency made the tumor microenvironment less immunosuppressive, leading to a higher accumulation of functional cytotoxic lymphocytes. Therefore, depletion of CD8 cells abrogated the tumor inhibition achieved by anti-PD-1+ anti-ActRIC treatment, as illustrated by the red group in Figure S4C-E.
Figure 6.
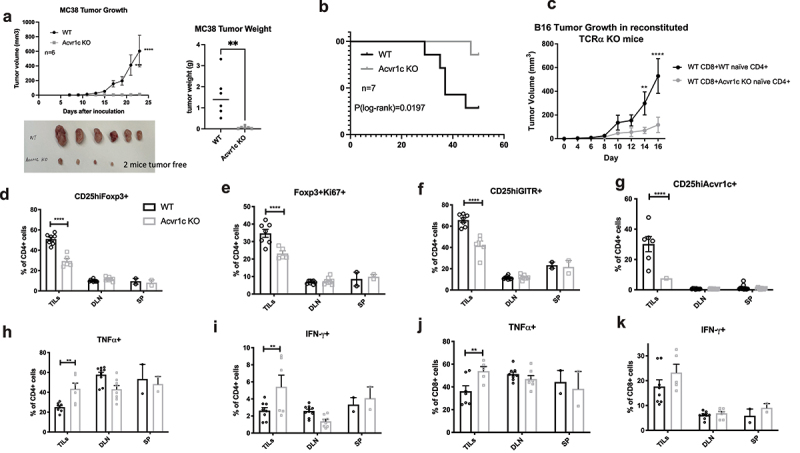
Tumor growth was inhibited in Acvr1c KO mice. (a) results of one representative experiment of MC38 tumor growth in WT vs. Acvr1c KO mice. Global Acvr1c KO and WT female mice were challenged subcutaneously with 250,000 MC38 tumor cells. Disease progression was monitored over time by measuring the tumor size using caliper. The tumors were excised 23 days post-injection and weighed. (b) survival analysis of WT vs. Acvr1c KO mice with MC38 tumors. (c) TCRα KO mice were reconstituted with WT CD8+ cells and WT or Acvr1c KO naïve CD4+ cells 1 week prior to MC38 tumor challenge. Tumor size was monitored over time. (d-k) the tumor infiltrating lymphocytes (TILs) extracted from the tumors, together with the cells processed from the draining lymph nodes and spleens, were analyzed for the frequency of indicated markers by flow cytometry. The percentage of CD25highFoxp3+ (D), Foxp3+Ki67+(E), CD25highGITR+(F) and CD25highAcvr1c+ (G) cells were analyzed in WT v.S. Acvr1c KO CD4+ cells. The production of TNFα and IFN-γ by CD4+ cells (H, I) or CD8+ cells (J, K) were also measured by intracellular cytokine staining.
However, since the Acvr1c KO mice used in our studies had a global ActRIC knockout, attributing the slowed tumor growth solely to the disturbance of iTregs that derived from CD4+ CD25low cells posed a challenge. To gain further insight, we reconstituted TCRα KO animals with WT CD8+ and either WT or Acvr1c KO naïve CD4+ cells. These recipient animals were challenged with B16F10 tumor cells, and all mice shared C57BL/6 background, with minimal autoimmunity observed during the experiment’s relatively short duration. Notably, the group receiving Acvr1c KO naïve CD4+ cells exhibited significantly delayed tumor growth compared to the WT controls (Figure 6c). The separation of the two tumor growth curves mirrored to that observed in Figure S4A, strongly suggesting that the blockade of iTreg development due to ActRIC deficiency was the primary factor contributing to the inhibition of tumor growth.
Monoclonal antibodies targeting ActRIC were generated and showed therapeutic effects in treating mouse tumors
We generated anti-mouse ActRIC monoclonal antibodies using hybridoma technology, immunizing mice with the extracellular domain (ECD) of ActRIC. Prior to administering the anti-ActRIC mAb to mice with tumors, we conducted in vitro experiments to validate the binding affinity and blocking efficiency of two candidate clones (both in isotype of mouse IgG1). Among these clones, 5D11 clone exhibited superior binding affinity when interacting with 293T cells transfected with plasmids encoding Acvr1c (Figure 7a) and demonstrated greater blocking efficiency in inhibiting the effect of activin A on inducing Foxp3 expression in CD4+ cells skewed toward iTregs (Figure 7b). Based on these observations, we selected the 5D11 clone for use in a subsequent in vivo tumor study.
Figure 7.
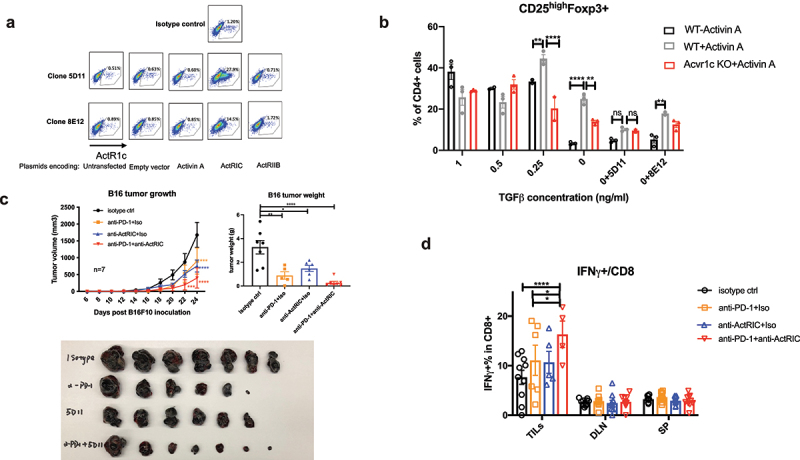
Monoclonal antibodies targeting ActRIC were generated and showed therapeutic effects in treating mouse tumors. (a) 293 T cells were transfected with plasmids encoding for empty vector, activin A, activin receptor 1c (ActRIC) or activin receptor 2b(ActRIIB). After rest for a day, the transfected 293T cells were collected and stained by using the two clones of monoclonal antibody targeting ActRIC as the primary antibody and a fluorescence conjugated anti-mouse IgG1 antibody as the secondary antibody. (b) naïve CD4+ T cells were isolated from WT or Acvr1c KO mice and cultured under iTreg conditions in the absence or presence of activins as described previously. The two clones of monoclonal antibodies were tested for their blocking efficiency. (C) WT C57/BL6J mice were inoculated with B16F10 tumor cells subcutaneously at 40,000 cells/mouse. 6 days later, the i.P. treatment with indicated antibodies started when the tumors became palpable. Tumor growth kinetics are graphed. 24 days post the inoculation, the tumors were excised, weighed and imaged. (d) the frequency of IFN-γ producing CD8 cells in TILs, DLN, and spleens harvested from the treated mice with B16 tumors was analyzed by flow cytometry.
As depicted in Figure 7c, monotherapy with anti-ActRIC significantly delayed B16F10 tumor growth compared to the isotype control. The effect was even more pronounced when anti-ActRIC was used in combination with an anti-PD-1 antibody. One possible mechanism for this additive effect is that activin-ActRIC signaling operates through a distinct pathway from the PD-1 ligand receptor. Based on our understanding, anti-ActRIC antibodies target CD4+ regulatory T cells and anti-PD-1 antibodies directly unleash suppressed cytotoxic CD8+ T cells32. Although both antibody treatments ultimately enhance the functionality of CD8+ T cells in eradicating cancer cells, as demonstrated in Figure 7d, it’s important to emphasize that these antibodies exert their effects on distinct immune cell populations, additively contributing to their common objective.
We used the Human Protein Atlas Immune Cell RNA sequencing database to investigate the expression of activin and its receptors in human immune cells. The expression profiles of relevant genes from the dataset generated by Schmiedel et al. are presented in Figure S5A.33 Similar to our findings in mice, we observed the expression of inhibin β subunits across various immune cell populations, while ACVR1C and ACVR2B were notably expressed in Treg cells. Furthermore, an in vitro study on human naïve CD4+ cells isolated from healthy donor PBMCs, Figure S5B, revealed that although the effect was not as pronounced as in mice, activins promoted FOXP3 induction. Notably, the anti-ActRIC antibody abrogated the effects of activin in human CD4+ cells (>95% homology between human and murine ActRIC ECD). While further investigation is warranted, these data suggest that human activins may interact with ACVR1C in a manner similar to their interactions in mice, Figure 8.
Figure 8.
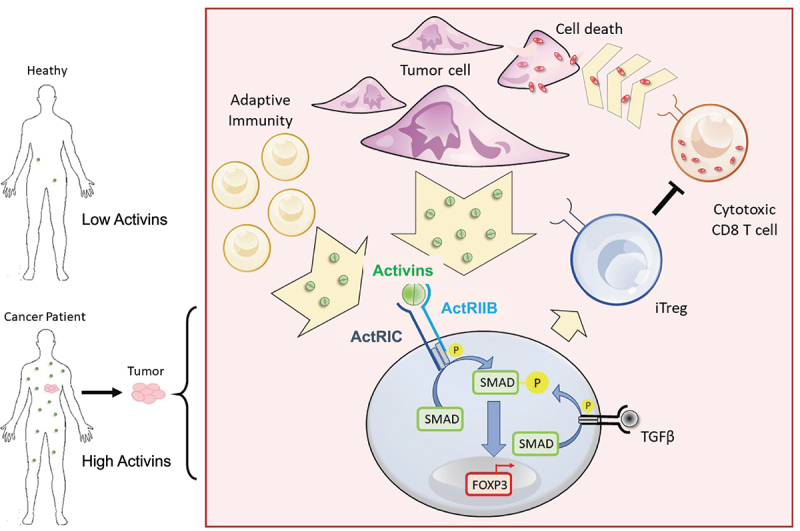
Graphical abstract describes the ability of activins to promote FOXP3 induction through ActRIC in cancer. Created with BioRender.com.
Discussion
While cancer immunotherapy has shown promise, there is room for improvement, as only a subset of patients benefits from these treatments. Our research reveals that systemic activin levels are elevated in various solid tumor cancer patients, extending beyond reproductive or endocrine-related cancers, where activins were previously implicated. Within the tumor microenvironment, it appears that activated CD4+ T cells are responsible for producing this signaling molecule, as evidenced by the reduction in tumor burden when blocking the activin pathway in the B16 tumor model, even in cases where systemic activin levels are not notably elevated.
Activins serve diverse biological functions. The results from in vitro studies suggest that both activin A and B have the potential to enhance the function of TGF-β in the conversion of CD4+ CD25low T cells into iTregs. Since there was still a baseline level of TGF-β in the culture medium containing fetal bovine serum (FBS)13, it remains unclear whether activin alone was able to induce a moderate degree of conversion to iTregs. Nevertheless, compelling evidence supports the additive effect of activins on the conversion of non-Tregs into iTregs induced by low concentrations of TGF-β. This discovery addresses a crucial need in the field-identifying cancer biomarkers that predict the immune environment, therapeutic response, and adverse events. The availability of validated plasma ELISA kits for activins opens the possibility for the straightforward identification of patients with elevated activins through pre-treatment blood testing. This identification may offer the potential for these patients to benefit from immunotherapy targeting these signaling molecules or their receptors. Thus, blocking activin signaling, which normally promotes the induction of Treg cells in the tumor microenvironment, could prove effective for a broad spectrum of solid tumor patients. Several studies have associated elevated activin family signaling molecules with poor prognosis in the oncology, particularly in cancers of the female reproductive system.1,34 Additionally, activin has been linked to skin carcinogenesis by inducing tumor-promoting Langerhans cells and Tregs, attracting macrophages, and reprogramming them into tumor-associated macrophages.35 However, the full potential of the activin pathway as an immunotherapeutic target remains underexplored. While current therapies focus on reactivating immune cells or inhibiting immune suppression signals, this approach offers the prospect of inhibiting the generation of iTregs that suppress anti-tumor immunity.
As activins have a variety of endocrine functions, their signaling in the tumor microenvironment is likely regulated by receptor levels. The activin Type I receptor, ActRIC, is of particular interest because it is uniquely expressed by CD4+ T cells. Although ActRIIB is highly upregulated by induced Tregs, type II activin receptor subunits require multimerization with type I activin receptor subunits, such as ActRIC, to phosphorylate Smads. Hypothesizing that blocking ActRIC would be most specific for the modulation of activin-mediated Foxp3 induction in CD4+ T cells, mice with genetic deletion of ActRIC were used to examine this potential immunosuppressive mechanism in vivo. Acvr1c KO animals showed no discernible developmental or immune defects and remained healthy. Notably, when challenged with B16 or MC38 subcutaneous tumors, these mice displayed delayed disease progression and tumor growth. Furthermore, when tumor-bearing mice received monoclonal antibodies that blocked ActRIC, especially in combination with anti-PD-1 therapy, a significant decrease in tumor burden was observed. These observations strongly indicate that activins’ impact on T cells is relevant in the tumor microenvironment, promoting immunosuppression. While further research is needed to understand the roles of activins and ActRIC in myeloid and NK cell compartments, our findings suggest a novel and promising target for cancer immunotherapy by reversing the conversion of non-Tregs to iTregs. Additionally, the use of ActRIC-blocking antibodies holds promise for reducing side effects due to the target’s unique expression profile on iTregs. However, there is a risk of autoimmunity, and ActRIC’s presence in adult tissues like the ovaries, breast, pancreatic islet beta-cells, and adipose tissue2,5,7,21,36,37 necessitates caution about effects on the reproductive system and pancreas. Despite these concerns, this approach may offer potential advantages in combating obesity.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the generous time and expertise of the flow cytometry staff, Ada Tam, Richard L. Blosser, and Jessica Gucwa. Chirag Patel helped with methodology development and experimental design. We thank Dr. Suzanne L. Topalian, Tracee McMiller, and members of the Topalian Lab for processing and archiving the human plasma samples.
Funding Statement
This work was supported by the Bloomberg~Kimmel Institute for Cancer Immunotherapy (BKI) fund at the Johns Hopkins School of Medicine.
Abbreviations
| TGF-β | transforming growth factor-β |
| ELISA | enzyme-linked immunosorbent assay |
| Tregs | regulatory T cells |
| iTregs | induced regulatory T cells |
| FSH | follicle-stimulating hormone |
| ActRIC | activin receptor 1c |
| ActRIIB | activin receptor 2b |
| Acvr1c | gene name for murine ActRIC |
| ACVR1C | gene name for murine ActRIC |
| ECD | extracellular Domain |
| Alk7 | activin receptor like kinase 7 |
| TILs | tumor-infiltrating lymphocytes |
| Foxp3 | Forkhead box P3 |
| DTR | diphtheria toxin receptor |
| TCRα | T cell receptor alpha chain |
| DLN | draining lymph nodes |
| mLN | mesentenric lymph nodes |
| SP(L) | spleen (lymphocytes) |
Disclosure statement
Y.Z., A.L., J.F., and D.P. are inventors of a licensed patent related to this work and are eligible to receive royalties. The terms of this arrangement have been reviewed and approved by Johns Hopkins University School of Medicine in accordance with its policy on objectivity in research.
Data availability statement
The authors confirm that the data supporting the findings of this study are available in the article and its supplementary materials. Raw data generated at the Bloomberg~Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University School of Medicine, are available from the corresponding author Y.Z. and D.P. on request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2023.2297503.
References
- 1.Risbridger GP, Schmitt JF, Robertson DM.. Activins and inhibins in endocrine and other tumors. Endocr Rev. 2001;22(6):836–12. doi: 10.1210/edrv.22.6.0450 [DOI] [PubMed] [Google Scholar]
- 2.Jörnvall H, Reissmann E, Andersson O, Mehrkash M, Ibáñez CF.. ALK7, a receptor for nodal, is dispensable for embryogenesis and left-right patterning in the mouse. Mol Cell Biol. 2004;24(21):9383–9389. doi: 10.1128/mcb.24.21.9383-9389.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonomi L, Brown M, Ungerleider N, Muse M, Matzuk MM, Schneyer A. Activin B regulates islet composition and islet mass but not whole body glucose homeostasis or insulin sensitivity. Am J Physiol Endocrinol Metab. 2012;303(5):E587–596. doi: 10.1152/ajpendo.00177.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu AQ, Popova EY, Barnstable CJ. Activin signals through SMAD2/3 to increase photoreceptor precursor yield during embryonic stem cell differentiation. Stem Cell Rep. 2017;9(3):838–852. doi: 10.1016/j.stemcr.2017.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makanji Y, Zhu J, Mishra R, Holmquist C, Wong WPS, Schwartz NB, Mayo KE, Woodruff TK. Inhibin at 90: from discovery to clinical application, a historical review. Endocr Rev. 2014;35(5):747–794. doi: 10.1210/er.2014-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuchida K, Nakatani M, Yamakawa N, Hashimoto O, Hasegawa Y, Sugino H. Activin isoforms signal through type I receptor serine/threonine kinase ALK7. Mol Cell Endocrinol. 2004;220(1–2):59–65. doi: 10.1016/j.mce.2004.03.009 [DOI] [PubMed] [Google Scholar]
- 7.Goebel EJ, Ongaro L, Kappes EC, Vestal K, Belcheva E, Castonguay R, Kumar R. The orphan ligand, activin C, signals through activin receptor-like kinase 7. Elife. 2022;11. doi: 10.7554/eLife.78197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. 2016;8(8):a022053. doi: 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuchida K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, Inokuchi K. Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal. 2009;7(1):15. doi: 10.1186/1478-811x-7-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui X, Shang, S., Lv, X., Zhao, J., Qi, Y., Liu, Z.. Perspectives of small molecule inhibitors of activin receptor‑like kinase in anti‑tumor treatment and stem cell differentiation (review). Mol Med Rep. 2019;19:5053–5062. doi: 10.3892/mmr.2019.10209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iizuka-Koga M, Nakatsukasa, H., Ito, M., Akanuma, T., Lu, Q., Yoshimura, A.. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J Autoimmun. 2017;83:113–121. doi: 10.1016/j.jaut.2017.07.002 [DOI] [PubMed] [Google Scholar]
- 12.Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, Di Rella F, de Candia P, De Rosa V. Molecular mechanisms controlling Foxp3 expression in health and autoimmunity: from epigenetic to post-translational regulation. Front Immunol. 2019;10:3136. doi: 10.3389/fimmu.2019.03136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber S, Stahl FR, Schrader J, Luth S, Presser K, Carambia A, Flavell RA, Werner S, Blessing M, Herkel J, et al. Activin a promotes the TGF-β-induced conversion of CD4+CD25− T cells into Foxp3+ induced regulatory T cells. J Immunol. 2009;182(8):4633–4640. doi: 10.4049/jimmunol.0803143 [DOI] [PubMed] [Google Scholar]
- 14.Halvorsen EC, Mahmoud SM, Bennewith KL. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev. 2014;33:1025–1041. doi: 10.1007/s10555-014-9529-x [DOI] [PubMed] [Google Scholar]
- 15.Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19(1):116. doi: 10.1186/s12943-020-01234-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei X, Zhang J, Gu Q, Huang M, Zhang W, Guo J, Zhou X. Reciprocal expression of IL-35 and IL-10 defines two distinct effector Treg subsets that are required for maintenance of immune tolerance. Cell Rep. 2017;21(7):1853–1869. doi: 10.1016/j.celrep.2017.10.090 [DOI] [PubMed] [Google Scholar]
- 17.Overacre-Delgoffe AE, Chikina, M., Dadey, R. E., Yano, H., Brunazzi, E. A., Shayan, G.. Interferon-γ drives Treg fragility to promote anti-tumor immunity. Cell. 2017;169(6):1130–1141.e1111. doi: 10.1016/j.cell.2017.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carmenate T, Ortíz Y, Enamorado M, García-Martínez K, Avellanet J, Moreno E, Graça L, León K. Blocking IL-2 signal in vivo with an IL-2 Antagonist Reduces tumor growth through the control of regulatory T cells. J Immunol. 2018;200(10):3475–3484. doi: 10.4049/jimmunol.1700433 [DOI] [PubMed] [Google Scholar]
- 19.Fisher SA, Aston WJ, Chee J, Khong A, Cleaver AL, Solin JN, Ma S, Lesterhuis WJ, Dick I, Holt RA, et al. Transient Treg depletion enhances therapeutic anti-cancer vaccination. Immunity Inflam & Disease. 2017;5(1):16–28. doi: 10.1002/iid3.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morianos I, Papadopoulou G, Semitekolou M, Xanthou G. Activin-A in the regulation of immunity in health and disease. J Autoimmun. 2019;104:102314. doi: 10.1016/j.jaut.2019.102314 [DOI] [PubMed] [Google Scholar]
- 21.Bertolino P, Holmberg R, Reissmann E, Andersson O, Berggren P-O, Ibáñez CF. Activin B receptor ALK7 is a negative regulator of pancreatic β-cell function. Proc Natl Acad Sci U S A. 2008;105(20):7246–7251. doi: 10.1073/pnas.0801285105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michael IP, Saghafinia S, Tichet M, Zangger N, Marinoni I, Perren A, Hanahan D. ALK7 signaling manifests a homeostatic tissue barrier that is abrogated during Tumorigenesis and metastasis. Dev Cell. 2019;49(3):409–424.e406. doi: 10.1016/j.devcel.2019.04.015 [DOI] [PubMed] [Google Scholar]
- 23.Wu B, Zhang, S., Guo, Z., Bi, Y., Zhou, M., Li, P.Wan, Y. Y.. The TGF-β superfamily cytokine activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation. Immunity. 2021;54(2):308–323.e306. doi: 10.1016/j.immuni.2020.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kita A, Kasamatsu A, Nakashima D, Endo-Sakamoto Y, Ishida S, Shimizu T, Kimura Y, Miyamoto I, Yoshimura S, Shiiba M, et al. Activin B regulates adhesion, invasiveness, and migratory activities in oral cancer: a potential biomarker for metastasis. J Cancer. 2017;8(11):2033–2041. doi: 10.7150/jca.18714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones CP, Gregory LG, Causton B, Campbell GA, Lloyd CM. Activin a and TGF-β promote TH9 cell–mediated pulmonary allergic pathology. J Allergy Clin Immunol. 2012;129(4):1000–1010.e1003. doi: 10.1016/j.jaci.2011.12.965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni X, Tao J, Barbi J, Chen Q, Park BV, Li Z, Zhang N, Lebid A, Ramaswamy A, Wei P, et al. YAP is essential for Treg-mediated suppression of antitumor immunity. Cancer Discov. 2018;8(8):1026–1043. doi: 10.1158/2159-8290.Cd-17-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feldman JP, Goldwasser R, Mark S, Schwartz J, Orion I. A mathematical model for tumor volume evaluation using two-dimensions. J Appl Quant Meth. 2009;4:455–462. [Google Scholar]
- 28.Sápi J, Kovács L, Drexler DA, Kocsis P, Gajári D, Sápi Z. Tumor Volume Estimation and Quasi-Continuous Administration for Most Effective Bevacizumab Therapy. PLoS ONE. 2015;10(11):e0142190. doi: 10.1371/journal.pone.0142190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L, Zhang W, Liang H-F, Zhou Q-D, Ding Z-Y, Yang H-Q, Liu W-B, Wu Y-H, Man Q, Zhang B-X, et al. Activin a induces growth arrest through a SMAD- dependent pathway in hepatic progenitor cells. Cell Commun Signal. 2014;12(1):18. doi: 10.1186/1478-811x-12-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-β signal transduction. J Cell Sci. 2001;114(24):4359–4369. doi: 10.1242/jcs.114.24.4359 [DOI] [PubMed] [Google Scholar]
- 31.Khalil AM, Dotimas H, Kahn J, Lamerdin JE, Hayes DB, Gupta P, Franti M. Differential binding activity of TGF- family proteins to select TGF- receptors. J Pharmacol Exp Ther. 2016;358(3):423–430. doi: 10.1124/jpet.116.232322 [DOI] [PubMed] [Google Scholar]
- 32.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175(6):1701–1715 e1716. doi: 10.1016/j.cell.2018.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walentowicz P, Krintus M, Sadlecki P, Grabiec M, Mankowska-Cyl A, Sokup A, Walentowicz-Sadlecka M. Serum inhibin a and inhibin B levels in epithelial ovarian cancer patients. PLoS ONE. 2014;9(3):e90575. doi: 10.1371/journal.pone.0090575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antsiferova M, Huber M, Meyer M, Piwko-Czuchra A, Ramadan T, MacLeod AS, Havran WL, Dummer R, Hohl D, Werner S, et al. Activin enhances skin tumourigenesis and malignant progression by inducing a pro-tumourigenic immune cell response. Nat Commun. 2011;2(1):576. doi: 10.1038/ncomms1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bondestam J, Huotari M-A, Morén A, Ustinov J, Kaivo-Oja N, Kallio J, Horelli-Kuitunen N, Aaltonen J, Fujii M, Moustakas A, et al. cDNA cloning, expression studies and chromosome mapping of human type I serine/threonine kinase receptor ALK7 (ACVR1C). Cytogenet Cell Genet. 2001;95(3–4):157–162. doi: 10.1159/000059339 [DOI] [PubMed] [Google Scholar]
- 37.Zhao M, Okunishi K, Bu Y, Kikuchi O, Wang H, Kitamura T, Izumi T. Targeting activin receptor–like kinase 7 ameliorates adiposity and associated metabolic disorders. JCI Insight. 2023;8(4). doi: 10.1172/jci.insight.161229 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available in the article and its supplementary materials. Raw data generated at the Bloomberg~Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University School of Medicine, are available from the corresponding author Y.Z. and D.P. on request.