

ABSTRACT
Neutrophil extracellular traps (NETs) contribute to the pathophysiology of multiple inflammatory and autoimmune diseases. Targeting the NETosis pathway has demonstrated significant therapeutic potency in various disease models. Here, we describe a first-in-class monoclonal antibody (CIT-013) with high affinity for citrullinated histones H2A and H4, which inhibits NETosis and reduces tissue NET burden in vivo with significant anti-inflammatory consequences. We provide a detailed understanding of the epitope selectivity of CIT-013. Detection of CIT-013 epitopes in rheumatoid arthritis (RA) synovium provides evidence that RA is an autoimmune disease with excessive citrullinated NETs that can be targeted by CIT-013. We show that CIT-013 acts upon the final stage of NETosis, binding to its chromatin epitopes when plasma membrane integrity is compromised to prevent NET release. Bivalency of CIT-013 is necessary for NETosis inhibition. In addition, we show that CIT-013 binding to NETs and netting neutrophils enhance their phagocytosis by macrophages in an Fc-dependent manner. This is confirmed using a murine neutrophilic airway inflammation model where a mouse variant of CIT-013 reduced tissue NET burden with significant anti-inflammatory consequences. CIT-013’s therapeutic activity provides new insights for the development of NET antagonists and indicates the importance of a new emerging therapy for NET-driven diseases with unmet therapeutic needs.
KEYWORDS: Citrullinated histones, mechanism of neutrophil extracellular trap inhibition, neutrophil extracellular trap phagocytosis, rheumatoid arthritis, therapeutic monoclonal antibody
Introduction
Neutrophils play an important role in innate immune defense by using several strategies to protect the host against infections. Neutrophils kill pathogens via phagocytosis, release of granule proteins, generation of reactive oxygen species (ROS), and the release of decondensed chromatin termed neutrophil extracellular traps (NETs) by a process called NETosis. Since its discovery by Brinkmann et al,1 the role of NETosis has been associated with the elimination of pathogens.2 However, numerous subsequent studies have shown that aberrant and prolonged NET release is associated with the pathogenesis of a variety of inflammatory disorders, including sepsis, acute respiratory distress syndrome (ARDS), acute lung injury (ALI), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), vasculitis, antiphospholipid syndrome (APS), and asthma (reviewed in Ref. 3–7 The detrimental role of NETs in disease has several elements since NETs are pro-inflammatory, a source of autoantigens and cytotoxic to surrounding tissues and promote thrombosis5. In the past decade, knowledge has accumulated on different aspects of NET biology, including physiological NET inducers,8 cellular events leading to NET release9, and potential NET-targeting therapeutic strategies.10,11 NETosis can be broadly categorized into two main pathways, NADPH oxidase dependent or independent.8,12,13 Both NETosis pathways are highly organized and occur in well-defined phases,14,15 which ultimately lead to cell rupture and expulsion of decondensed chromatin decorated with pro-inflammatory mediators and anti-microbial enzymes such as neutrophil elastase (NE), myeloperoxidase (MPO), and cathepsin G. Pharmacological inhibition of the NETosis pathway and enzymes present in NETs have demonstrated that NET-targeting therapeutics have potential as treatments for diseases in which NET accumulation is an important driver of the pathogenesis.
Histone citrullination by peptidyl arginine deiminase 4 (PAD4) has been closely linked to NETosis through its conversion of arginine residues, mainly at histone N-terminal tails, into citrulline to promote chromatin decondensation.15,16 We previously demonstrated that anti-citrullinated histone antibodies (ACHA) targeting citrullinated histones H2A and H417 inhibit NET release and suppress disease endpoints in various preclinical mouse models.18 Based on this and other preclinical evidence, we have progressed an ACHA termed CIT-013 into clinical development.
In this study, we provide a detailed understanding of the epitope selectivity of CIT-013 and characterize CIT-013’s NETosis-inhibitory mechanism of action, demonstrating that CIT-013 inhibits NET release in the final stages of NETosis immediately after loss of plasma membrane integrity. Furthermore, we show that bivalency is necessary for this inhibitory capacity. We further demonstrate that CIT-013 opsonization of netting neutrophils stimulates phagocytosis by macrophages. Similarly, CIT-013 binding to NETs stimulates phagocytosis by macrophages. In addition, in vivo, we show that CIT-013 reduces tissue NET burden at least in part through enhanced macrophage phagocytosis. Finally, we demonstrate the significant presence of CIT-013’s binding epitopes in RA synovium, providing evidence that RA is an autoimmune disease potentially targeted by the anti-inflammatory effects of CIT-013. Since NETs contribute to the pathophysiology of many autoimmune diseases such as RA, its unique ability to both inhibit NET release and enhance NET clearance indicates the importance of CIT-013 as a promising new therapy.
Materials and methods
Antibody production and purification
A full-length non-good manufacturing practice CIT-013 laboratory-scale batch (L37721) and pilot batch (P3807623) were produced by Lonza using their proprietary GS XceedTM gene expression system in combination with GS-Chinese hamster ovary cells. Both CIT-013 laboratory-scale batch and pilot batch were used in this study.
A CIT-013 laboratory-scale batch was used to generate CIT-013 F(ab’)2 fragments using the PierceTM F(ab’)2 preparation kit (Thermo Fisher Scientific, 44988) according to the manufacturer’s protocol. CIT-013 F(ab’)2 fragments were isolated using PraestoⓇ Jetted A50 ProtA agarose (Purolite) affinity chromatography, and buffer exchange to PBS was performed. Quality of CIT-013 F(ab’)2 fragments was determined using SDS-PAGE and size-exclusion-high-performance liquid chromatography.
For small-scale production and purification of monovalent CIT-013, antibody variable domain-coding sequences of CIT-013 were synthesized and cloned into mammalian expression vectors, encoding a light-chain kappa antibody fragment or a heavy-chain IgG1 antibody fragment. A third expression vector was used for encoding a truncated Fc domain. To promote heterodimerization in the CH3 domains, the CIT-013 heavy-chain IgG1 antibody fragment was modified to encode the T366S:L368A:Y407V mutations creating a hole-like structure, while the truncated Fc domain was modified with incorporated T366W mutation creating a knob-like structure.19 Subsequently, these constructs were used to transiently transfect human embryonic kidney 293F cells using FectoPRO© transfection and booster reagent according to the manufacturer’s protocol. Monovalent CIT-013 was isolated from cell culture supernatants using MabSelect affinity columns in combination with an Akta-FPLC. Additionally, size-exclusion chromatography and buffer exchange to PBS were performed yielding pure monovalent CIT-013.
m-ACHA was produced using a hybridoma cell line as described elsewhere17 and contained comparable target binding, NET inhibitory, and in vivo therapeutic properties to human CIT-013 antibody.
ACPA isolation from RA serum
Serum samples from RA patients were obtained from Sint Maartenskliniek (NL). To determine the effect of anti-citrullinated protein antibodies (ACPAs) on CIT-013-mediated NET inhibition, IgGs were isolated from ACPA-positive (>25 U/ml) or ACPA-negative (<25 U/ml) pooled RA serum using the mAbTrap Kit (Cytiva, 17112801) according to the manufacturer’s protocol. After elution, IgGs were concentrated and the buffer was exchanged to Dulbecco’s phosphate-buffered saline (DPBS, Gibco, 14190–144, pH = 7.2) using Vivaspin columns (Sartorius, VS0601). IgG purity was determined on a NuPAGE 4 to 12%, and Bis-Tris gel (Invitrogen, NP0321BOX) and total IgG concentration was determined using the NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). ACPA content was determined using the Immunoscan CCPlus ELISA (SVAR, RA-96 PLUS RUO) according to the manufacturer’s protocol. Isolated IgGs from ACPA-negative RA sera were used as a dilution medium to obtain desired ACPA concentrations in the final experiment while keeping the IgG concentration similar in each condition.
Peptide ELISA
To determine antibody binding characteristics for citrullinated peptides, we performed a peptide ELISA assay. NUNC maxisorp flat-bottom 96-well plates were coated with 1 µg/ml neutravidin (Thermo Fisher Scientific, 10443985) in 1× PBS (Sigma Aldrich, P5493) overnight at 4°C. Wells were washed three times, blocked with PBS-Tween (Thermo Fisher Scientific, 28352) containing 1% (w/v) bovine serum albumin (BSA, Sigma Aldrich, 10735086001) for 1 h at room temperature (RT) under shaking at 450 rpm, and incubated with 40 ng/ml biotin-conjugated citrullinated peptides (Table 1) in 1× PBS for 1 h at RT under shaking. Wells were washed three times and incubated with indicated concentrations of full-length CIT-013, monovalent CIT-013, CIT-013 F(ab’)2 fragments, m-ACHA, human anti-hen egg lysozyme antibody (cIgG; Crown Bioscience, C0001–5), mouse cIgG (m-cIgG), or ACPA-rich pool of IgG from RA patients in PBS-Tween containing 0.2% (w/v) BSA (referred to as PBS-Tween-BSA) for 1 h at RT under shaking. Subsequently, wells were washed three times and incubated with 50 ng/ml horseradish peroxidase (HRP)-conjugated mouse anti-human IgG (Fc) CH2 domain antibody (Invitrogen, MA516859, clone MK 1 A6), 50 ng/ml HRP-conjugated goat anti-human IgG F(ab’)2 (Abcam, ab87422), or 0.1 µg/ml HRP-conjugated polyclonal goat-anti- mouse immunoglobulin antibody (Dako, P0447) in PBS-Tween-BSA for 1 h at RT under shaking. Subsequently, wells were washed three times with PBS-Tween and TMB (Thermo Fisher Scientific, 74931569A) was added as a substrate for HRP and incubated for 10 min at RT in the dark. The reaction was stopped by adding one-to-one 2 M H2SO4 (Merck, 1007311000), and absorbance was measured at 450 nm (with 620 nm reference) using a Tecan Infinite F50 plate reader. The system was controlled using Magellan F50 software (version 7.2).
Table 1.
Amino acid sequence of non-citrullinated and citrullinated peptides.
| Name | Amino acid position | Amino acid sequence | N-terminal modification | C-terminal modification |
|---|---|---|---|---|
| Histone H2A | 1–12 | SGRGKQGGKARA | Acetyl | Aminohexanoic acid–lysine–biotin |
| Cit-histone H2A | 1–12 | SGXGKQGGKARA | Acetyl | Aminohexanoic acid–lysine–biotin |
| Histone H3 | 1–14 | ARTKQTARKSTGGK | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit-histone H3 | 1–14 | AXTKQTARKSTGGK | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Histone H4 | 1–12 | SGRGKGGKGLGK | Acetyl | Aminohexanoic acid–lysine–biotin |
| Cit-histone H4 | 1–12 | SGXGKGGKGLGK | Acetyl | Aminohexanoic acid–lysine–biotin |
| Cit-tenascin | 2182–2200 | AEMKLXPSNFXNLEGXXKX | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit--enolase | 4–21 | LKIHAXEIFDSXGNPTVE | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit-fibrinogen | 36–48 | GPXVVEXHQSACK | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit-fibrinogen | 60–74 | XPAPPPISGGGYXAX | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit-vimentin | 60–70 | GVYATXSSAVRL | Hydrogen | Aminohexanoic acid–lysine–biotin |
| Cit-filaggrin | 3692–3710 | SHQESTXGRSRGRSGRSGS | Hydrogen | Aminohexanoic acid–lysine–biotin |
X = Citrulline.
Antibody affinity analysis
SPR was performed using the 8-channel Cytiva’s Biacore 8K instrument in a single-cycle kinetic format with 30 nM biotin-conjugated peptides of N-terminal non-citrullinated and citrullinated human histone H2A, histone H3, and histone H4 (Table 1) coupled to a streptavidin biosensor chip. CIT-013 was injected on the sensor chip at RT in a series of concentrations ranging from 1 to 1000 pM, using a running buffer consisting of 10 mM HEPES (pH = 7.4), 150 mM NaCl, 3 mM EDTA, and 0.05% surfactant P20. Resonance was corrected for background using a reference channel without histone peptides. Blanks, consisting of running buffer only, have been subtracted from each condition. Data were analyzed using Biacore Insight software to determine binding affinity and kinetics.
Immunohistochemistry of RA synovial tissue
RA synovial tissue was provided by Tissue Solutions (UK), and hematoxylin & eosin (H&E) staining and immunohistochemistry (IHC) was performed by HistologiX (UK). In brief, samples were snap-frozen and stored at −80°C prior to analysis. Sections were cut at 5–8 µm, air dried at RT for at least 60 min, and fixed in acetone for 10 min. For IHC staining, sections were rinsed in running tap water for 5 min, washed with PBS containing 0.1% (v/v) TweenTM 20, blocked with normal horse serum for 20 min, and incubated with FITC-conjugated cIgG (FITC-cIgG; Ancell Corporation, ANC-295-040) or FITC-CIT-013 for 60 min. After several washes, the sections were blocked in methanol (SLS, CHE2528) containing 0.3% (v/v) hydrogen peroxide (VWR, 23622.298) for 10 min and incubated with polyclonal rabbit anti-FITC antibody (Bio-Rad, 4510–7804) for 30 min. The sections were washed and incubated with ImmPRESS® HRP-conjugated polyclonal horse anti-rabbit IgG antibody (Vector, MP-7401) for 30 min. After several washes, peroxidase was developed with the ImmPACT® DAB substrate (Vector, SK-4501) and counterstained with Mayer’s Haematoxylin (Pioneer, PRC/R/42). H&E staining was performed to study histological features of disease severity. Stained sections were digitally scanned with a 20× objective using the Hamamatsu NanoZoomer 2.0 HT. Histopathology and CIT-013 staining in RA synovia were assessed by a pathologist in a blinded fashion.
Neutrophil isolation
Peripheral blood from healthy volunteers was collected in VACUETTE® lithium-heparin tubes. All blood donors, including the donors from which monocytes were isolated, gave informed consent in accordance with the Declaration of Helsinki. Neutrophils were isolated by Ficoll-Paque Plus (Sigma Aldrich, 17-1440-03) density gradient centrifugation (40 min at 400×g) followed by dextran sedimentation in 0.9% NaCl solution (B. Braun, 25900) containing 6% dextran T500 (Pharmacosmos, 551005009007). Erythrocytes were lysed with an ammonium-chloride-potassium lysis buffer (155 mM NH4Cl (Sigma Aldrich, A9434), 10 mM KHCO3 (Sigma Aldrich 237,205), and 0.1 mM Na2EDTA (Sigma Aldrich, E5134, pH = 7.2) for 3 min at RT. Finally, neutrophils were washed and resuspended in RPMI 1640 containing GlutaMAX (Thermo Fisher Scientific, 61870044) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS, Serena, S-FBS-SA-015), 50 U/ml penicillin (pen), and 50 µg/ml streptomycin (strep) (Thermo Fisher Scientific, 15070063). To determine the cell concentration, neutrophils were diluted in 0.4% trypan blue solution (Thermo Fisher Scientific, 15250061) and counted with the Cellometer Auto T4 Bright Field Cell Counter. The system was controlled using the Cellometer Auto Counter software (version 3.3.9.5). Neutrophil purity was confirmed by flow cytometry according to the described protocol using 125 ng/ml APC/Cy7-conjugated mouse anti-human CD45 antibody (Biolegend, 368516, clone: 2D1), 330 ng/ml PerCP/Cy5.5-conjugated mouse anti-human CD16 antibody (Biolegend, 302028, clone: 3G8), 830 ng/ml APC-conjugated mouse anti-human CD66b antibody (Invitrogen, 17-0666-42, clone: G10F5), and 1000x diluted fixable viability dye eFluor 506 (Thermo Fisher Scientific, 65-0866-14). When deviating purity and characteristics of isolated neutrophils were encountered, donors were excluded from the experiment.
Neutrophil phagocytosis assay
Neutrophils were resuspended in RPMI 1640 (without phenol red) (Thermo Fisher Scientific, 11835030) supplemented with 2% (v/v) FBS, 50 U/ml pen, 50 µg/ml strep, 10 mM HEPES (Thermo Fisher Scientific, 15630080), and 1 mM CaCl2 (referred to as neutrophil assay buffer hereafter) in the absence or presence of indicated concentrations of cIgG or CIT-013. As a positive control for blocking phagocytosis, neutrophils were pre-incubated with 50 µM Cytochalasin D (Cyto D, Thermo Fisher Scientific, PHZ1063) for 15 min at 37°C and 5% CO2. Neutrophils (1×105 per well) were transferred to a clear flat-bottom black 96-well plate, and pHrodoTM Green E. coli BioparticlesTM Conjugates (Invitrogen, P35366) or pHrodoTM Green S. aureus BioparticlesTM Conjugates (Invitrogen, P35367) were added with a final concentration of 300 µg/ml. Neutrophils and bacterial bioparticle conjugates were centrifuged at RT for 3 min at 350×g and incubated for 90 min at 37°C. Phagocytosis was detected by measuring fluorescence (Ex/Em: 509 nm/533 nm) using CLARIOstar Plus using Multi-user Reader Control and MARS data analysis software (version 3.32).
Degranulation assay
Neutrophils were resuspended in neutrophil assay buffer in the absence or presence of 169 nM cIgG or CIT-013. Cyto D (10 µM) was used as a positive control to block degranulation. Neutrophils (3×105 per well) were transferred to a 24-well plate and stimulated with 1 µg/ml lipopolysaccharide (LPS, Sigma Aldrich, L2630, serotype O111:B4) for 180 min at 37°C and 5% CO2. Subsequently, membrane expression of degranulation markers was determined by flow cytometry according to the described protocol using 0.5 μg/ml PE-conjugated mouse anti-human CD11b antibody (Biolegend, 301306, clone: ICRF44), 1 μg/ml PerCP/Cyanine5.5-conjugated mouse anti-human CD66b antibody (Biolegend, 305108, clone: G10F5), and 0.2 µg/ml DAPI (Biolegend, 422801), used as a cell viability marker.
To measure LL-37 secretion, neutrophils were resuspended in neutrophil assay buffer. Neutrophils (2×104 per well) were transferred to a poly-L-lysine (Sigma Aldrich, P4832) pre-coated clear flat-bottom black 96-well plate and stimulated with 12.5 µM calcium ionophore (A23187, Thermo Fisher Scientific, A1493) in the absence or presence of 169 nM cIgG or CIT-013. After 15 min, supernatant was transferred to a V-bottom 96-well plate and centrifuged for 3 min at 400×g to pellet cell debris. LL-37 secretion was measured in the supernatant with a LL-37 ELISA detection kit (Hycult Biotech, HK321–02) according to the manufacturer’s protocol.
ROS assay
Before the performance of the ROS assay, heat-killed (HK) S. aureus (Invivogen, tlrl-hksa) and HK E. coli (Invivogen, tlrl- hkeb2) were Ig-opsonized in 1× DPBS containing 10% (v/v) heat-inactivated (30 min at 56°C) human plasma for 30 min at 37°C and by gentle agitation. Subsequently, bacteria were washed twice with 1× DPBS and resuspended in KRPG buffer (145 mM NaCl (VWR, 1.06404.1000), 5.7 mM Na3PO4 (Sigma Aldrich, 342483), 4.86 mM KCl (VWR, 1.04936.0500), 0.54 mM CaCl2 (Sigma Aldrich, E506), 1.2 mM MgSO4 (Sigma Aldrich, M7506), and 5.5 mM Glucose (Sigma Aldrich, G8270), pH = 7.35). Neutrophils were resuspended in pre-heated (37°C) KRPG buffer in the absence or presence of 169 nM cIgG or CIT-013. As a positive control for blocking NADPH oxidase-dependent ROS production, neutrophils were pre-incubated with 10 µM diphenyleneiodonium chloride (DPI, Sigma Aldrich, D2926) for 30 min at 37°C and 5% CO2. Neutrophils (1×105 per well) were transferred to a clear flat-bottom black 96-well plate and stimulated with 10 nM phorbol 12-myristate 13-acetate (PMA, Sigma Aldrich, P1585) or a multiplicity of 1000 heat-killed opsonized bacteria. ROS was measured with the Amplex RedTM Red Hydrogen Peroxide/Peroxidase Assay kit (Thermo Fisher Scientific, A22188) according to the manufacturer’s protocol. ROS production was detected over time by measuring fluorescence (Ex/Em: 530 nm/590 nm) with CLARIOstar Plus using Multi-user Reader Control and MARS data analysis software.
Flow cytometry
For the quality control of neutrophil and monocyte isolation or the degranulation assay, cells were resuspended in 1× DPBS containing 1% (w/v) BSA and 0.1% (w/v) NaN3 (Sigma Aldrich, S2002) (referred to as FACS buffer hereafter) and pre-incubated with 50x diluted Human TruStain FcX (Biolegend, 422302) for 15 min at RT. Subsequently, fluorescence-conjugated antibodies or viability dyes were resuspended in FACS buffer and cells were stained for 45 min at RT. Next, cells were centrifugated at RT for 5 min at 350×g followed by two washing steps with FACS buffer before acquisition with BD FACSCantoTM II. The system was controlled using BD FACSDiva software (version 8.0.1), and analysis of flow cytometry data was performed using FlowJo software (version 10.8.0). Activated neutrophils (i.e., from the degranulation assay or used as positive control in the binding assay) or non-stimulated neutrophils were fixed in 1× DPBS containing 2% (v/v) paraformaldehyde (PFA, Thermo Fisher Scientific, 28906) and measured the next day.
CIT-013 binding to leukocytes, erythrocytes, and thrombocytes
Two milliliter blood was incubated with ammonium-chloride-potassium lysis buffer for 10 min at RT to lyse erythrocytes. Cells were centrifuged for 5 min at 350×g and washed twice with neutrophil assay buffer. Leukocytes were incubated in neutrophil assay buffer containing 20 µg/ml HiLyteTM Fluor 488-conjugated cIgG (HL488-cIgG) or HL488-CIT-013 in combination with 1 µg/ml PerCP/Cyanin5.5-conjugated mouse anti-human CD3 antibody (Biolegend, 317335, clone: OKT3), 2 µg/ml APC-conjugated mouse anti-human CD14 antibody (Biolegend, 367118, clone: 63D3), 1 µg/ml Pacific BlueTM-conjugated mouse anti-human CD20 antibody (Biolegend, 302328, clone: 2H7), or 0.5 µg/ml PE-conjugated mouse anti-human CD66b antibody (Biolegend, 392904, clone: 6/40c) for 15 min at 37°C and 5% CO2. Subsequently, leukocytes were centrifuged for 3 min at 400×g and washed once with FACS buffer before flow cytometric acquisition. As a positive control for CIT-013 binding, leukocytes were stimulated with 12.5 µM A23187 for 1 h and 45 min after which netting neutrophils were stained with HL488-cIgG or HL488-CIT-013 in combination with anti-CD66b antibody. To determine the binding of CIT-013 to erythrocytes, whole blood was incubated in neutrophil assay buffer containing 20 µg/ml HL488-cIgG or HL488-CIT-013 in combination with 2 µg/ml APC-conjugated mouse anti-human CD235ab antibody (Biolegend, 306607, clone: HIR2). For thrombocyte isolation, whole blood was centrifuged for 15 min at 140×g and thrombocyte-rich plasma was isolated, washed with neutrophil assay buffer, and centrifuged for 10 min at 750×g. Thrombocytes were incubated in neutrophil assay buffer with 20 µg/ml HL488-cIgG or HL488-CIT-013 in combination with 21 ng/ml PE-conjugated mouse anti-human CD41 antibody (Biolegend, 303706, clone: HIP8).
Preparation of immune complexes
ICs were formed by incubating 5 µg/ml human serum albumin (HSA, VWR 31020) with 282.5 µg/ml polyclonal rabbit anti-HSA antibody (Sigma Aldrich, A0433) (molecular ratio of 1:25) in neutrophil assay buffer for at least 90 min at 37°C. ICs were homogenized by vortexing, and NET formation was induced by adding 50 µl of ICs in a final volume of 200 µl of buffer per well.
Quantitative immunofluorescence live imaging NET assay
The quantitative immunofluorescence live imaging NET assay was performed as described elsewhere with minor differences.20 In short, clear flat-bottom black 96-well plates were coated with 0.01–0.001% poly-L-lysine in distilled water (Thermo Fisher Scientific, 15230204) for at least 60 min at RT and washed three times with 1× DPBS before seeding 2 × 104 neutrophils per well in neutrophil assay buffer. Neutrophils were stimulated with indicated concentrations of A23187, PMA, or ICs in the absence or presence of indicated concentrations of cIgG, full-length CIT-013, monovalent CIT-013, or CIT-013 F(ab’)2 fragments. When no concentrations are indicated, 12.5 µM A23187, 100 nM PMA, and 169 nM antibodies were used. To study whether rheumatoid factor (RF) or ACPA interferes with CIT-013-mediated NET inhibition, indicated concentrations of RF (Lee Biosolutions, 508–27) or ACPA-rich pool of IgG from RA patients were added to the wells simultaneously with A23187. SytoxTM Green (Thermo Fisher Scientific, S7020) was added to the wells with a final concentration of 20 nM to visualize the NETs. NET release was recorded with the IncuCyte® S3 platform for at least 180 min at 37°C and 5% CO2 using a 20× objective. During multiple timepoints, phase contrast as well as green fluorescence (Ex/Em: 504 nm/523 nm) images were taken at four spots per well.
Analysis of immunofluorescence live imaging NET assay data
Analysis of NET release and permeable neutrophils was performed using IncuCyte® software (version 2021A). In short, SytoxTM Green positive pixels were identified in the green channel with adaptive segmentation and a threshold GCU of 2–5. The edge split tool was turned on with an edge sensitivity between −20 and 0 for accurate quantification of closely spaced objects. “Hole fill” was set on 100 µm2, and “adjust size” was set between −3 and 1 pixel. To determine NETs, filters were applied to exclude objects with an area of > 270–346 µm2, an eccentricity of > 0.3, a mean intensity of < 16.5–20.5, and an integrated intensity of > 7000–10,000. The processing definition of the analysis was optimized for every single experiment with filter settings in the above-described range. Raw data of the percentage of SytoxTM Green confluency per image, referred to as the NET area (%), and the percentage of NETs were exported, and graphs were created as described elsewhere.
Citrullinated nucleosome ELISA
To determine the presence of citrullinated nucleosomes in NETs, we performed a NET assay as described elsewhere in this study but in the absence of SytoxTM Green. Neutrophils were stimulated with NETosis inducing stimuli in neutrophil assay buffer for 240 min at 37°C and 5% CO2. NETs were digested by adding 15 U/ml micrococcal nuclease solution (Thermo Fisher Scientific, EN0181) for 15 min at 37°C and 5% CO2, and EDTA (VWR, E177) was added (pH = 8.0, final concentration of 9.8 mM) to stop nuclease activity. To pellet cell debris, supernatants were transferred to a U-bottom 96-well plate and centrifuged at 20×g for 5 min at RT. NET harvest, present in the supernatant, was used to quantify citrullinated nucleosomes by ELISA.
NUNC maxisorp flat-bottom 96-well plates were coated with 0.35 µg/ml polyclonal rabbit anti-histone H3 (citrulline R2+R8+R17) antibody (Abcam, ab5103) in 1× PBS overnight at 4°C. Wells were washed three times with PBS containing 0.05% (v/v) TweenTM 20 (referred to as PBS-Tween hereafter) followed by blocking with PBS-Tween containing 5% (w/v) ELK (referred to as PBS-Tween-ELK hereafter) for 2 h at RT. PBS-Tween-ELK was removed, and supernatant containing NET harvest (non-diluted) was incubated for 1 h at RT under shaking (450 rpm). Subsequently, wells were washed three times with PBS-Tween and incubated with 400 ng/ml m-ACHA in PBS-Tween-ELK for 1 h at RT under shaking. After three washes with PBS-Tween, wells were incubated with 0.1 µg/ml HRP-conjugated polyclonal goat-anti-mouse immunoglobulin antibody in PBS-Tween-ELK for 1 h at RT under shaking. The detection of HRP was performed as described elsewhere in this study. The citrullinated nucleosome ELISA was also used to determine m-ACHA epitope in bronchoalveolar lavage fluid (BALF), which was diluted 50× in PBS-Tween-ELK.
3D high-magnification live imaging immunofluorescence confocal microscopy of netting neutrophils
Neutrophils were washed twice with 1× DPBS followed by cell membrane labeling according to the protocol provided by the PKH26 Red Fluorescent Cell Linker Kit for General Cell Membrane Labeling (Sigma Aldrich, MIDI26-1KT). Subsequently, neutrophils were washed twice with neutrophil assay buffer and resuspended in neutrophil assay buffer containing 0.5 µM SPY-650 DNA dye (Tebu-Bio, SC501). After incubation for 45 min at 37°C, neutrophils were washed twice in neutrophil assay buffer and seeded at 2×104 neutrophils per well in poly-L-lysine pre-coated clear flat-bottom black 96-well plates. Neutrophils were stimulated as described above in the absence or presence of 25 µg/ml HL488-cIgG or HL488-CIT-013. Binding of CIT-013 to netting neutrophils was recorded over time at 37°C and 5% CO2 with different immunofluorescence confocal microscopes using 20×, 40x, and 100× objectives, including Opera Phenix HCS system, controlled using Harmony® High Content Imaging and Analysis Software (version 4.9) or Nikon Ti2 system, with NL5, a Hamamatsu Fusion camera, an Oxxius L6Cc laser, and a Tokay Hit incubator stage controlled by NL5 from MicroManager software (version 2.0.1 20210829). Every 3 min, a set of three z-stack image sequences was created containing green (Ex/Em: 504 nm/523 nm), orange (Ex/Em: 550 nm/567 nm), and red (Ex/Em: 652 nm/674 nm). Images were processed using ImageJ/Fiji software (version 2.3.0/1.53f).
Analysis of the chromatin area
The chromatin area of netting neutrophils in images captured with confocal microscopy has been analyzed by MATLAB (version: R2010a) using a previously published code.14 This code follows the change in chromatin of single cells during the time course of NETosis. For all three conditions (No Ab, cIgG, and CIT-013), data from two donors have been analyzed in a blinded fashion. For each video, netting neutrophils have been excluded from the analysis if they did not undergo the full process of NETosis, could not be identified as single cells, were too close to the edge of the imaging window, or lead to a code error (i.e., high migration, cell detachment, and spontaneous gray value change). Single values of chromatin area have been excluded if the code failed to detect the correct area (i.e., wrong cell or part of the nucleus detected). For each cell, the relative change in chromatin area has been determined by normalizing the chromatin area to the chromatin area of the first analyzable frame.
Primary monocyte isolation and differentiation
Peripheral blood from healthy volunteers was collected in buffy coats and 1:1 diluted with 1× DPBS. RosetteSep Human Monocyte Enrichment cocktail (Stem Cell Technologies, 15068) was added to the diluted blood (ratio 1:50) followed by 20 min of incubation at RT. Monocytes were isolated by Ficoll-Paque Plus density gradient centrifugation (25 min at 800×g) and washed three times with 1× DPBS containing 2 mM EDTA and 0.1% (v/v) BSA. Monocytes were diluted in 0.4% trypan blue solution and counted with the Cellometer Auto T4 Bright Field Cell Counter before cryopreserving them for later use. Monocyte purity was confirmed by flow cytometry according to the described protocol elsewhere in this study using 1 µg/ml FITC-conjugated mouse anti-human CD14 antibody (BD Biosciences, 345784, clone: MφP9). When deviating purity and characteristics of isolated monocytes were encountered, donors were excluded from the experiment.
Frozen vials of monocytes were thawed, and cells were resuspended in 10 ml of chilled serum-free IMDM (Thermo Fisher Scientific, 12440046) containing pen-strep (referred to as sf-IMDM). After 10 min of centrifugation at 200×g, monocytes were resuspended in preheated sf-IMDM and seeded in a clear flat-bottom black 96-well plate (2×104 cells per well after purity correction) and incubated for 1 h at 37°C and 5% CO2. Subsequently, IMDM containing 17% (v/v) FBS and 100 ng/ml recombinant human M-CSF (Immunotools 11,343,117) was added to the wells (ratio 1:1), and monocytes were incubated for 3 days at 37°C and 5% CO2. On day 4, medium was discarded and fresh IMDM containing 8.5% (v/v) FBS, pen-strep, and M-CSF was added. Differentiated macrophages were used for experiments on day 7. To confirm monocyte differentiation toward M2-like macrophages, cells were stimulated with 100 ng/mL LPS (serotype O111:B4) overnight at 37°C and 5% CO2 and IL-6 and IL-10 present in the supernatants were determined with the respective Human Uncoated ELISA Kits (Invitrogen, IL-6: 88-7066-88 and IL-10: 88-7104-88) according to the manufacturer’s protocol.
NET harvest for macrophage phagocytosis
Neutrophils were resuspended in neutrophil assay buffer, plated in 6-well culture plates (4×106 per well), and stimulated with 12.5 µM A23187 for 3 h at 37°C and 5% CO2. To remove A23187, NETs were carefully washed twice with 1 ml of DPBS followed by NET digestion with 0.8 U/ml micrococcal S7 nuclease in RPMI supplemented with pen-strep for 5 min at 37°C. Digestion was terminated with 10 mM EDTA, and NET containing supernatant was collected per donor. The concentration of NETs was determined with a Quant-IT PicoGreen dsDNA assay kit (Thermo Fisher Scientific, P11496) according to the manufacturer’s protocol. The presence of citrullinated nucleosomes in the NETs was determined using an ELISA as described above. Finally, the NET digestion was checked by DNA electrophoresis. Briefly, 0.5 µg of DNA was loaded with 10× orange G (Sigma Aldrich, O7252) in glycerol (Sigma Aldrich, G5516) on a 0.8% agarose gel in TAE buffer (Thermo Fisher Scientific, 10041223) and was run for 2 h at 100 V. The DNA was stained with SYBR® Safe DNA Gel Stain (Thermo Fisher Scientific, S33102), and the gel was imaged using the Chemidoc ultraviolet transilluminator. The GeneRuler 1 kb Plus DNA ladder (Thermo Fisher Scientific, SM1333) was used to determine the size of the NET fragments.
Engulfment of NETs and netting neutrophils by macrophages
To study NET phagocytosis by macrophages, M2-like macrophages were stained with 1× DPBS containing 0.5 μg/mL CellMask Orange membrane stain (Thermo Fisher Scientific, C10045) and 5 μM Hoechst 33342 (Thermo Fisher Scientific, 62249) for 15 min at 37°C and 5% CO2, washed three times with 1× DPBS, and incubated with 1× DPBS containing 10 µM chloroquine (Sigma Aldrich, C6628) (inhibitor of lysozyme activity resulting in accumulation of internalized NETs, which improves visualization and quantification) for 15 min at 37°C and 5% CO2. Before NET exposure to macrophages, SytoxTM Green was added to the NETs with a final concentration of 20 nM in the absence or presence of 169 nM cIgG, CIT-013, or CIT-013 F(ab’)2 fragments. To study whether RF interferes with CIT-013-mediated NET phagocytosis, indicated concentrations of RF were added to the wells simultaneously with CIT-013. Subsequently, 2500 ng/ml NETs (in RPMI 1640) were exposed to macrophages in IMDM containing 8.5% (v/v) FBS and pen-strep for 1 h at 37°C and 5% CO2. To block Fc gamma receptor (FcR), macrophages were preincubated with a mixture of the following antibodies: 10 µg/ml mouse anti-human CD64 (FcR1) blocking antibody (Biolegend, 305002, clone: 10.1), 10 µg/ml mouse anti-human CD32a (FcR2a), blocking antibody (Stem Cell Technologies, 60012, clone: IV.3), and 10 µg/ml mouse anti-human CD16 (FcR3) blocking antibody (Thermo Fisher Scientific, 16-0166-85, clone: 3G8). In addition, preincubation of macrophages with 10 µg/ml Cyto D was used as a positive control to block phagocytosis. Both FcR blocking antibodies and Cyto D were present during NET phagocytosis. After NET phagocytosis, macrophages were washed three times with 1× DPBS and fixed with 1× DPBS containing 2% (v/v) PFA before imaging with the Operetta CLS high content immunofluorescence microscopes. Images were taken at seven spots per well consisting of a set of three fluorescence images containing green (Ex/Em: 504 nm/523 nm), orange (Ex/Em: 550 nm/567 nm), and blue (Ex/Em: 360 nm/461 nm).
To determine the engulfment of netting neutrophils, we performed the assay according to the protocol described above with minor differences. Neutrophils were stimulated for the NETosis pathway with 12.5 µM A23187 for 5 min at 37°C and 5% CO2, followed by two washes with neutrophil assay buffer to remove A23187. Activated neutrophils were added at a 2:1 ratio to the M2-like macrophages (stained with CellMask Orange and Hoechst) in the presence of cIgG or CIT-013 and 20 nM SytoxTM Green, and cells were co-incubated for 1.5 h at 37°C and 5% CO2.
Analysis of NET and netting neutrophil phagocytosis by macrophages was performed in a blinded fashion using Harmony 4.1 software. In short, green fluorescence intensity was measured in the macrophage (based on plasma membrane stain) minus the nucleus area (based on DNA stain). Macrophages containing green fluorescence signals above background noise were counted positive. For quantification, phagocytic index was calculated by multiplying the percentage of phagocytic macrophages (number of positive macrophages divided by the total number of macrophages per field of view) with the number of detected positive areas per macrophage.
Macrophage fixation was omitted when 3D high-magnification live imaging immunofluorescence confocal microscopy was performed. 3D microscopy of the phagocytosis of netting neutrophils was performed with Hoechst, SytoxTM Green, and PKH26 Red Fluorescent Cell dye to label neutrophil membrane and SPY650-FastActTM (F-actin stain; 1000× diluted).
Neutrophilic airway inflammation mouse model
The animal experiments were approved by the Aptuit Committee on Animal Research and Ethics and were performed by Aptuit, an EvoTec company, in accordance with the recommendation of the Association for Assessment and Accreditation of Laboratory Animal Care. Mice were randomly assigned to control or treatment groups.
Eight-week-old female Balb/c mice were purchased from Charles River Laboratories. Mice were sensitized with 100 µg of Dermatophagoides pteronyssinus (house dust mite, HDM, Stallergenes Greer, 371590, batch XPB82D3A2.5) and 25 µg of complete Freund’s adjuvant (CFA, Sigma Aldrich, F5881) via subcutaneous injection (50 µl). Fourteen days after sensitization, mice received 1 mg/kg dexamethasone (Dex) (Sigma Aldrich, D4902) or vehicle orally (p.o.) or 20 mg/kg m-ACHA (TDD22.101-03,019-10) or 20 mg/kg mouse anti-hen egg lysozyme IgG1 antibody (m-cIgG; Crown Bioscience, C0005) or vehicle intravenously (i.v.) one hour before allergen challenge. Mice were challenged intranasally (i.n.) with 100 µg of HDM or received vehicle and were sacrificed 24 hours later.
After sacrifice, the murine lungs were lavaged three times with 400 µl of DPBS (Ca2+-, and Mg2+-free) at a slow and steady rate. The BALF was centrifuged for 5 min at 400×g and 4°C. The acellular supernatant was collected and stored at −80°C until further use. The cell pellet was resuspended in 1× DPBS and cells counted by an automated cell counter (DASIT Sysmex XT-2000Iv).
Lung histopathology
Lung tissue was collected after bronchoalveolar lavage. Lungs were inflated and fixed with 10% phosphate buffered formalin for 24 hours at RT. The tissue was dehydrated using increasing ethanol solutions (Honeywell, 02855 and 02870) followed by 100% toluene (Honeywell, 32249) and finally embedded in paraffin (Leica, 39602012) at 56–58°C. Two longitudinal 5-µm sections (10 µm apart) were collected and stained with hematoxylin and eosin (PRC, PRC/R/42). Histopathology was assessed by a pathologist who was blinded for the treatment groups. Semi-quantitative scores were based on the entire section according to the following criteria: grade 0, no significant pathology; 1, minimal severity and focal lesions; 2, minimal severity and multi-focal lesions; 3, moderate severity; and 4, marked severity. The average of both sections was taken as a final score for each animal. The numbers of macrophages and eosinophils were counted based on morphology. Macrophages with abundant cytosolic vacuoles were counted as phagocytic macrophages. For each animal, the cells were counted in 10 blood vessels (<300 µm diameter) for the perivascular zone, in 10 bronchioles (<600 µm diameter) for the peribronchiolar zone, and 10 parenchymal fields with transect 1500 µm without large blood vessels (>100 µm diameter) or bronchioles for the alveolar zone.
Immunofluorescence staining of murine lung tissue
Longitudinal 4-µm sections were taken from the paraffin-embedded lung tissue and deparaffinized. Epitopes were retrieved by a 32-min incubation in cell conditioning 1 solution (Ventana Medical Systems) at 93°C using the Ventana Discovery Ultra automated staining platform. To detect NETs in inflamed lung tissue, slides were incubated with 20 µg/ml polyclonal rabbit anti-histone H3 (citrulline R2+R8+R17) antibody and 2 µg/ml polyclonal goat anti-mouse MPO antibody (Bio-Techne, AF3667) for 1 h at 37°C. After washing, slides were stained with 4 µg/ml Alexa Fluor 488-conjugated polyclonal donkey anti-rabbit IgG antibody (Abcam, ab150073) and Alexa Fluor 555-conjugated polyclonal donkey anti-goat IgG (Abcam, ab150134) for 32 min at 37°C. To detect m-ACHA epitope in inflamed lung tissue, slides were incubated with 65.4 µg/ml FITC-conjugated m-ACHA, followed by 10 µg/ml rabbit anti-FITC antibody (BioRad, 4510–7804) and 10 µg/ml Alexa FluorTM Plus 647-conjugated donkey anti-rabbit IgG (H+L) antibody (Invitrogen, A32795). Stained slides were manually washed with EZ Prep solution (Ventana Medical Systems, 950–102) and deionized water before incubation with 300 nM DAPI (Thermo Fisher Scientific, D3571) for 30 min at RT. Slides were mounted using ProLong Gold Antifade Mountant (Thermo Fisher Scientific, P36934) and scanned at 40× using a Zeiss Axio Scan Z1 slide scanner. NETs (structures containing citrullinated histone H3 and MPO) were scored by a pathologist blinded to the treatment groups. Autofluorescence signals were present in images (green channel), but actual NETs were considered extracellular based on the morphology or minimal distance of 3 nuclear radii from the nearest nucleus. For each animal, five blood vessels of diameter <300 µm were assessed for the perivascular region, five bronchioles with a diameter of <600 µm for the peribronchiolar area, and 10 parenchymal fields (transect 500 µm) without large (>200 µm) blood vessels and bronchioles for the alveolar area. The scores were 0 for negative, 1 for mild, 2 for moderate, and 3 for severe incidence of NETs in the various areas.
MPO and double-stranded DNA quantification in BALF
Double-stranded DNA (dsDNA) was measured in 10 times diluted BALF using the Quant-iT PicoGreen dsDNA assay kit according to the manufacturer’s protocol. Fluorescence was measured on the CLARIOstar (Ex/Em: 483/15 nm and 530/30 nm) or on a SpectraMax iD5 (Ex/Em: 480 nm and 520/4 nm). MPO was measured in BALF using the myeloperoxidase mouse ELISA kit (Invitrogen, EMMPO) according to the manufacturer’s protocol.
Statistics
Both generation of graphics and statistical analyses were performed using GraphPad Prism 9 software (version 9.3.1). The quantitative results are reported as mean and, where indicated, ± standard error of the mean (SEM), and semi-quantitative results as median. Differences were considered significant at P <.05. Data below the lower limit of quantification (LLOQ) were set at LLOQ. If more than 30% of the data points were below LLOQ, the condition was not included in statistical tests. The normal distribution of each data set was assessed using the Shapiro-Wilk test except for the data from the mouse model, which was assessed using the D’Agostino & Pearson test. The comparison of two unpaired groups without Gaussian distribution was performed using the two-tailed Mann-Whitney test, while two unpaired groups with Gaussian distribution were compared with a two-tailed unpaired t test. The comparison of two paired groups without Gaussian distribution was performed using the two-tailed Wilcoxon matched-pairs signed rank test. The comparison of two paired groups with Gaussian distribution was performed using a two-tailed paired t test. The comparison of three or more paired groups with Gaussian distribution was performed using repeated measures (RM) one-way or ordinary two-way analysis of variance (ANOVA), while three or more paired groups without Gaussian distribution were compared using the Friedman test. The comparison of three or more unpaired groups with Gaussian distribution was performed using ordinary one-way ANOVA, while the comparison of three or more unpaired groups without Gaussian distribution was performed using the Kruskal–Wallis test. With the comparison of three or more groups, Dunnett’s, Tukey’s, Dunn’s, or Holm-Šidák’s multiple comparison test or uncorrected Fisher’s LSD test was used as indicated. Non-linear regression was used to calculate R2, EC50, and IC50. When no individual data points are shown, the number of sampled units, n, is indicated in the figure legends. The sample size was determined based on the robustness of the signal and the variation of the data points. No outliers were excluded.
Results
CIT-013 specifically binds its epitopes with high affinity
We previously reported the development of the mouse hybridoma-derived ACHA (m-ACHA) into a humanized and fully optimized clinical development candidate ACHA (now called CIT-013).18 CIT-013 and m-ACHA show comparable characteristics when tested for binding to the N-terminal peptide of citrullinated histone H2A (Figure S1a and Table 1). Surface plasmon resonance (SPR) analysis of antibody affinity using biotinylated histone peptides (Table 1) linked to the surface of a streptavidin-coated biosensor chip was performed to calculate binding affinities of CIT-013. CIT-013 binds to the non-citrullinated peptide of N-terminal histone H2A with an equilibrium dissociation constant (KD) of 508 pM, while a higher binding affinity was observed for the peptide of citrullinated N-terminal histone H2A and H4 (KD of 9.37 pM and 106 pM, respectively) (Table 2). Binding of CIT-013 to native histone H2A could not be confirmed by ELISA (Figure S1b). KD values could not be determined for non-citrullinated N-terminal peptides of histone H3 and H4, nor for the citrullinated N-terminal peptide of histone H3.
Table 2.
CIT-013’s affinity for non-citrullinated and citrullinated N-terminal histone peptides.
| Ligand | Kon (M s−1) | Koff (s−1) | KD (pM) |
|---|---|---|---|
| Histone H2A | 1.5 × 106 | 7.61 × 10–4 | 508 |
| Histone H3 | - | - | NB |
| Histone H4 | - | - | NB |
| Cit-histone H2A | 6.07 × 106 | 5.69 × 10−5 | 9.37 |
| Cit-histone H3 | - | - | NB |
| Cit-histone H4 | 5.44 × 105 | 5.78 × 10−5 | 106 |
NB = no binding detected.
Recently, it has been described that ACPAs with anti-inflammatory properties show polyreactivity for multiple citrullination proteins present in RA patients.21–23 We tested peptides from citrullinated proteins common in RA to determine CIT-013’s epitope specificity. While CIT-013 shows strong binding to the N-terminal citrullinated peptides of histones H2A and H4, no binding was observed for the tested peptides of citrullinated tenascin, citrullinated -enolase, citrullinated fibrinogen or , citrullinated vimentin, and citrullinated filaggrin (Figure S1c and Table 1). These data demonstrate that CIT-013 selectively binds the N-terminus of citrullinated histone H2A and H4 with low picomolar affinity.
CIT-013’s epitope is present in RA synovial tissue
NETs and NET-associated markers have been shown to be elevated in serum from RA patients and correlate with disease activity.24,25 Significantly higher DNA levels observed in RA synovial fluid (SF) also show a strong correlation with SF neutrophil counts,26 while NE-DNA complexes and NE activity are significantly elevated in RA SF.27 Furthermore, SF neutrophils from RA patients have an increased propensity to release NETs with RA autoantibodies and increased pro-inflammatory cytokines conducive for the induction of NETosis in the absence of microbial stimuli.28 We have previously demonstrated that a precursor molecule of CIT-013 shows therapeutic benefit in RA mouse models.18 To further investigate CIT-013 as a potential RA therapeutic, we assessed the presence of CIT-013’s epitopes in human RA synovium. FITC-conjugated CIT-013 (FITC-CIT-013) was used to visualize CIT-013’s citrullinated epitopes in synovial tissue from RA patients with FITC-conjugated cIgG (FITC-cIgG) as a control. Six synovia were categorized into minimal active (Figure 1a), moderate active (Figure 1b), and moderate to marked active (Figure 1c) synovitis depending upon their histological features (Table 3). CIT-013 staining correlated positively with the observed histological severity (Figure 1, boxes on the right and Table 3). Consistent with previous reports indicating neutrophilia and elevated NETosis in RA synovial fluid, histological staining showed intense CIT-013 staining particularly in tissues with clear signs of moderate or moderate to marked active synovitis with the presence of synovial hyperplasia and lymphoid-rich follicular zones. CIT-013 staining in these tissues was particularly found around synovial lining and sublining with infiltrating cells around lymphoid foci. In synovial tissue with minimal active synovitis, CIT-013’s target was minimally present with minor staining in acute inflammatory infiltrates around the microvasculature. These data highlight the significance of citrullination-associated NET formation in RA synovitis and support the proposal that excessive citrullinated NETs can be targeted as a therapeutic approach via the epitope selectivity of CIT-013.
Figure 1.
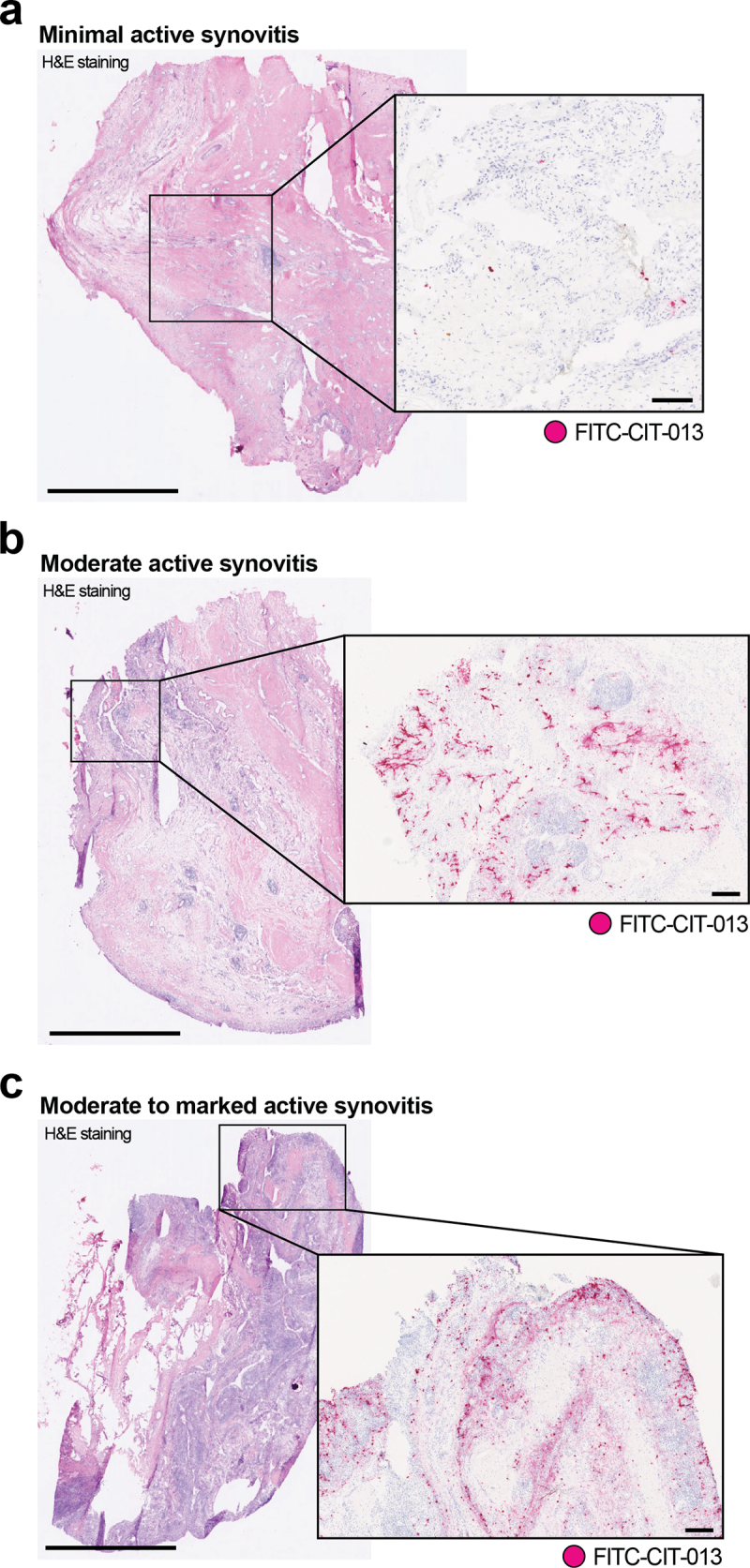
Elevated CIT-013 epitope in RA synovial tissue with moderate to marked active synovitis.
Three representative images of H&E-stained synovial biopsies (left) from distinct RA patients with minimal active synovitis (a), moderate active synovitis (b), and moderate to marked active synovitis (c). For the detection of CIT-013’s epitope in RA synovial tissue (magenta staining in boxes on the right), samples were subsequently incubated with FITC-conjugated CIT-013, followed by a rabbit anti-FITC antibody and finally with an HRP-conjugated horse anti-rabbit antibody. Histological severity grade and incidence of CIT-013 staining were scored by a pathologist (Table 3). The scale bar for H&E staining is 2 mm, and the scale bar for CIT-013 staining is 200 µm.
Table 3.
Histological severity grade of RA synovia and incidence of CIT-013 staining.
| Biopsy | Histological severity grade | Key histological features | Incidence of CIT-013 staining | Histological localization of CIT-013 staining |
|---|---|---|---|---|
| 1 | No active synovitis | Quiescent synovium with a few scattered inflammatory cells within connective tissue | Occasional positive punctate cells | Infiltrating acute cells |
| 2* | Minimal synovitis | Scattered lymphoid foci and scattered mixed inflammatory infiltrate, cartilage and bone admixed with the synovial matrix | Minimal diffuse staining, occasional positive punctate cells | Infiltrating acute cells |
| 3 | Minimal active synovitis | Focal areas of synovial hyperplasia with diffuse inflammatory infiltrates in the sub-lining | Minimal diffuse staining, occasional positive punctate cells | Infiltrating acute cells |
| 4 | Minimal reactive synovitis | Low-grade acute synovitis with a low-grade fibrogranular reaction around detritic foci, and low-grade acute inflammatory infiltrate around microvasculature | Occasional positive punctate cells | Infiltrating acute cells |
| 5* | Moderate active synovitis | Lymphoid foci with microvascular sinusoid formation, hyperplastic synovial lining with zones of lymphoid, mononuclear and macrophage infiltration in sub-lining, peri-vascular acute inflammatory infiltrates, and diffuse stromal cell reaction | Moderate to marked multi-focal staining | Synovial lining, sub-lining, peri-vascular, peri-lymphoid, and demarcation of follicular zones |
| 6* | Moderate to marked active immune synovitis | Marked lymphoid-rich (follicular) synovitis, multiple lymphoid foci with abundant microvascular sinusoid formation, markedly hyperplastic synovial lining with dense zones of lymphoid, mononuclear and macrophage infiltration in sub-lining, peri-vascular acute inflammatory infiltrates, and diffuse stromal cell reaction | Marked zonal staining | Synovial lining, sub-lining, peri-vascular, peri-lymphoid, and demarcation of follicular zones |
*These biopsies are demonstrated in Figure 1.
CIT-013 does not bind blood cells and does not inhibit neutrophil functions other than NETosis
To further evaluate CIT-013 binding, we assessed epitopes and off-target binding on healthy human whole blood by flow cytometry. Failure of HL488-CIT-013 to bind leukocyte, thrombocyte, or erythrocyte populations (Figure 2a and S2a) demonstrates that these cells do not contain binding sites for CIT-013 on their cellular membrane. Neutrophils stimulated to undergo NETosis (confirmed by immunofluorescence microscopy) were used as positive control and showed significant detectable binding of HL488-CIT-013 (Figure 2a and S2b). The possibility that CIT-013 might inhibit innate immune neutrophil functions other than NETosis29 was also investigated. CIT-013 does not interfere with neutrophil phagocytosis of bacterial particles (Figure 2b and S3a), ROS production (Figure 2c and S3b), and degranulation (Figure 2d, S3c, and S3d). Together, these data indicate that CIT-013 does not bind any healthy blood cells and does not interfere with the neutrophil’s anti-microbial activity, but does interact with netting neutrophils.
Figure 2.
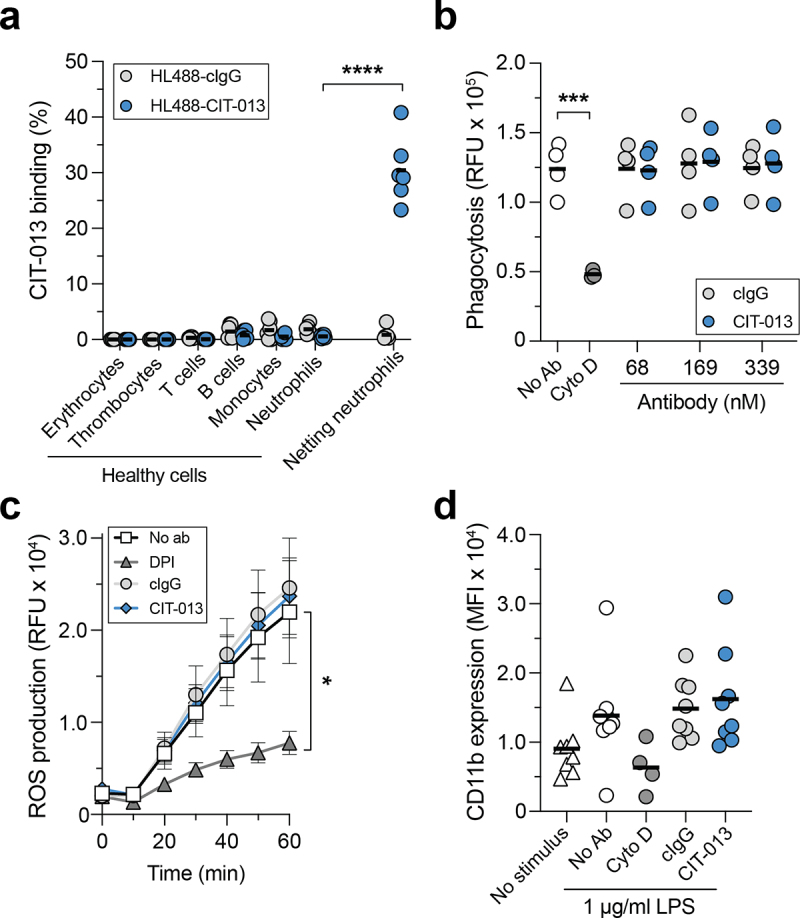
CIT-013 does not bind blood cells and does not affect neutrophil anti-microbial activity other than NETosis.
(a) Binding of HiLyteTM Fluor 488-conjugated isotype control Ab (HL488-cIgG) or HL488-CIT-013 to erythrocytes, thrombocytes, T cells, B cells, monocytes, and neutrophils was measured using flow cytometry. Neutrophils stimulated with A23187 for 120 min (netting neutrophils) were used as positive control. (b) Neutrophil phagocytosis of fluorescent pHrodoTM Green S. aureus BioparticlesTM Conjugates in the absence (No Ab) or presence of cIgG, CIT-013, or Cyto D (actin polymerization inhibitor as a positive control inhibitor of phagocytosis). (c) ROS production by neutrophils stimulated with Ig-opsonized heat-killed S. aureus in the absence or presence of cIgG, CIT-013, or DPI (NADPH oxidase inhibitor as a positive control inhibitor for ROS production) (n = 6). Statistics were performed on the area under the curve. (d) CD11b membrane degranulation marker on neutrophils stimulated with LPS in the absence or presence of cIgG, CIT-013, or Cyto D (positive control inhibitor of degranulation) at t = 180 min. *P<.05, ***P<.001, and ****P<.0001, two-tailed paired t test (a), two-tailed unpaired t test (b), and RM one-way ANOVA using Dunnett’s multiple comparisons test (c). RFU = relative fluorescence units; MFI = mean fluorescence intensity.
CIT-013 specifically inhibits citrullination-dependent NETosis in a dose-dependent manner
A23187 and PMA are often used to induce NETosis in vitro. Although both stimuli are non-physiological, they are useful in an in vitro setting because they activate distinct NETosis pathways and their robust NETosis induction leads to low donor-to-donor variation. A23187 induces calcium influx, leading to PAD4 activation and release of NETs rich in citrullinated histones, while PMA activates the NADPH oxidase complex leading to ROS production and release of NETs with low levels of citrullinated histones.8 We used the cell membrane impermeable DNA dye SytoxTM green in combination with quantitative live imaging microscopy to study the ability of CIT-013 to inhibit NETosis in response to A23187 or PMA. Using this technique, a NETosis event is defined as a SytoxTM Green area that is significantly larger than the plasma membrane of the neutrophil (Figure 3a; red and yellow arrows and S4a), indicating that chromatin has been expelled into the extracellular environment.20 Based on this method, neutrophils that die without expelling their DNA into the extracellular space (permeable neutrophils; Figure 3a; blue arrow and S4a) were excluded from analysis. Speed and magnitude of NETosis response were dependent on the concentration of each stimulus (Figure S4b and S4c), with NET release in response to A23187 occurring faster and PMA-induced NETosis resulting in a higher proportion of neutrophils releasing NETs (Figure 3a, b, and S4d). To investigate possible differences in histone citrullination between NETs derived from PMA- and A23187-stimulated neutrophils, we subjected harvested NETs to an ELISA in which citrullinated nucleosomes were captured with an anti-citrullinated histone H3 antibody and detected with m-ACHA. This approach detected significant N-terminal histone citrullination in NETs following A23187 stimulation and low levels of citrullination in NETs induced by PMA stimulation (Figure 3c). As CIT-013 binds citrullinated histone H2A and H4 with high affinity (Table 2), we hypothesized that CIT-013 would selectively target A23187-induced NETosis. Indeed, NET release in response to A23187 was completely inhibited with CIT-013 (Figure 3d, Supplemental movie 1) with an IC50 of 4.6 nM (Figure 3e), whereas PMA-induced NET release was not inhibited (Figure 3f). Previously, we have shown that a precursor ACHA monoclonal antibody is able to inhibit NETosis in response to physiological stimuli such as activated platelets, gout synovial fluid, and RA synovial fluid.18 Here, we show that immune complexes (ICs) can induce citrullination-dependent NETosis (Figure S4e and S4f) which can be inhibited by CIT-013 (Figure 3g). The therapeutic relevance of inhibiting IC-induced NETosis is highlighted by acute and chronic inflammatory diseases, such as RA, SLE, ARDS, and ALI, where ICs are formed that might be able to trigger NETosis.30–32
Figure 3.
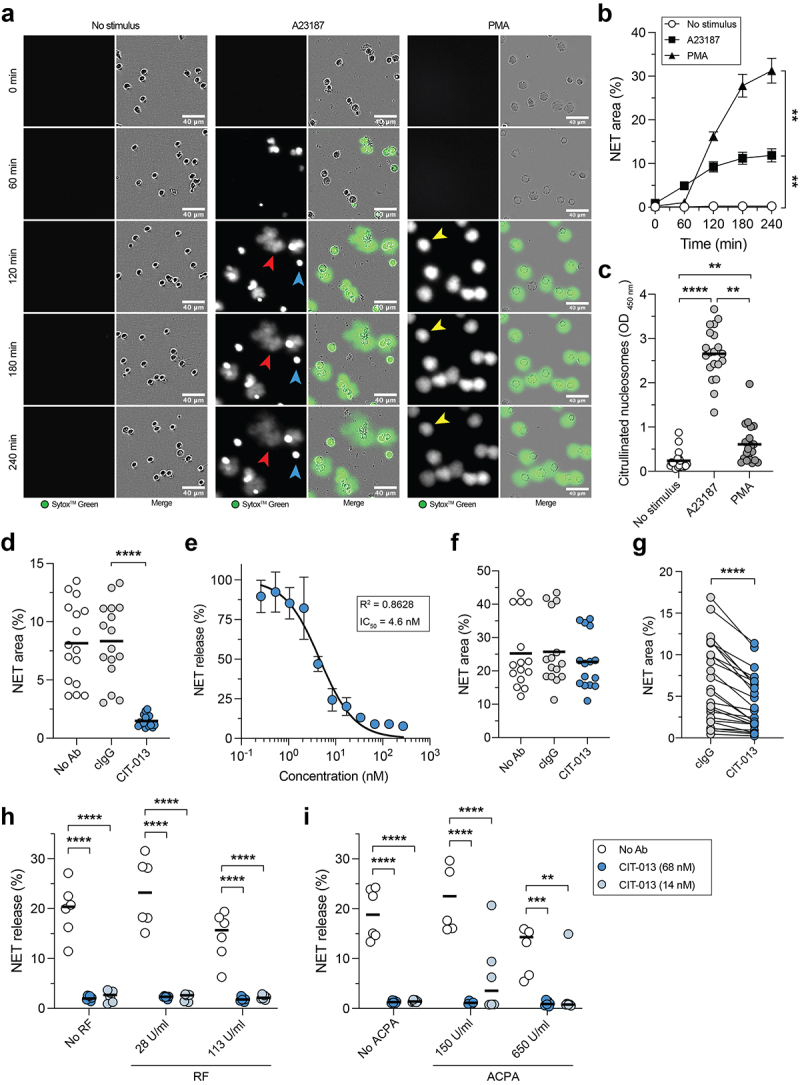
CIT-013 specifically inhibits the release of citrullination rich NETs.
A23187- and PMA-induced NET release was measured by a quantitative immunofluorescence live imaging NET assay using the impermeable DNA dye SytoxTM Green. (a) Representative images of NET release in response to A23187 (red arrows) and PMA (yellow arrows) at different time points. Permeable neutrophils with intracellular chromatin are indicated with blue arrows. (b) Quantification of NET release over time (n = 5). Statistics were performed on t = 240 min. (c) Citrullinated nucleosome detection in NET harvest at t = 240 min. (d) A23187-induced NET release at t = 240 min in the absence (No Ab) or presence of cIgG or CIT-013. (e) Dose-dependent inhibition of A23187-induced NET release with CIT-013 (n = 3). Data were normalized to cIgG (set as 100% NET release). (f) PMA-induced NET release at t = 240 min in the absence or presence of cIgG or CIT-013. (g) Immune complex-induced NET release at t = 240 min in the presence of cIgG or CIT-013. A23187-induced NET release in the absence or presence of CIT-013 in combination with RF (h) or ACPA-rich pool of IgG from RA patients (i) at t = 60 and 180 min, respectively. Concentrations in (i) represent the level of ACPA present in the assay, not the level of IgGs. **P<.01, ***P<.001, and ****P<.0001, Repeated measures one-way ANOVA with Dunnett’s (b) or Tukey’s (d) multiple comparisons test, Friedman test with Dunn’s multiple comparisons test (c), two-tailed Wilcoxon matched-pairs signed rank test (g), and ordinary two-way ANOVA with Dunnett’s multiple comparisons test (h and i).
High levels of RF and ACPA are present in RA patients.33–36 Since RF binds Fc portions of IgG and ACPAs bind various citrullinated proteins, including histones (Figure S1d), both RF and ACPA could influence CIT-013’s NET inhibitory efficacy. Therefore, we determined NET inhibition in the presence of disease-relevant levels of RF and ACPA. CIT-013-mediated NET inhibition was not affected by RF (Figure 3h) nor by ACPA (Figure 3i).
Together, these data demonstrate that CIT-013 is a specific inhibitor of citrullination-dependent NETosis, even in the presence of disease-relevant immunoglobulins like RF or ACPA. Importantly, IC-induced NETosis can be inhibited by CIT-013. ICs are important disease-relevant stimuli of NETosis, and therefore, inhibition of IC-induced NETosis further underscores the therapeutic potential of CIT-013.
CIT-013’s bivalency is necessary for its NET-inhibitory capacity
To understand the features of CIT-013 necessary to inhibit NET formation, we investigated whether antibody Fc domain and antibody bivalency are requisites. To study this, we generated CIT-013 F(ab’)2 fragments of ~110 kDa and a monovalent CIT-013 antibody of ~100 kDa composed of a full-length constant heavy (CH) chain and a truncated CH chain lacking the variable heavy (VH) and CH1 domains (Figure 4a). Heterodimerization of a full-length CH chain with a truncated CH chain was achieved by a hole and knob mutation in the CH3 Fc domains.19 While NET release in response to A23187 was inhibited by full-length CIT-013 as well as CIT-013 F(ab’)2 fragments in a dose-dependent manner, monovalent CIT-013 completely lacked NETosis inhibitory capacity (Figure 4b). Although the binding efficacy of monovalent CIT-013 to peptides of citrullinated histone H2A was decreased relative to full-length CIT-013 (EC50 of 89.8 nM and 0.3 nM respectively), significant binding by monovalent CIT-013 was detected at concentrations greater that 5 nM (Figure S5a). Binding efficacy of full-length CIT-013 compared to CIT-013 F(ab’)2 fragments was similar (Figure S5b). Together, these findings show that bivalency of CIT-013 is necessary for its NETosis inhibiting capacity.
Figure 4.
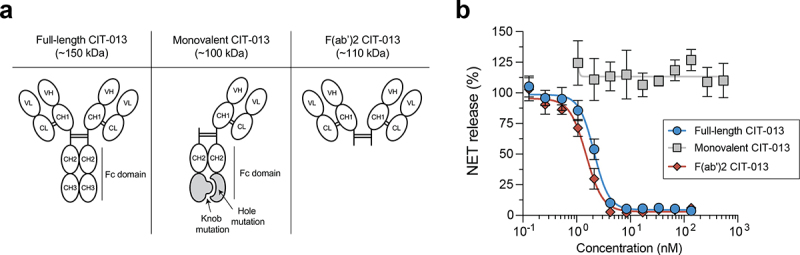
Bivalency is necessary for the NET-inhibitory capacity of CIT-013.
(a) Schematic design of a full-length CIT-013 antibody, a monovalent CIT-013 antibody, and a CIT-013 F(ab’)2 fragment. Monovalent CIT-013 was produced by co-expression of a light chain, a heavy chain containing a hole mutation (T366S:L368A:Y407V), and a truncated heavy chain containing a knob mutation (T366W). (b) Neutrophils were stimulated with A23187 in the presence of full-length CIT-013, monovalent CIT-013, or CIT-013 F(ab’)2 fragments (n = 3). Data were normalized to cIgG (set as 100% NET release). CL = constant light; VL = variable light; CH = constant heavy; VH = variable heavy; Fc = constant fragment.
CIT-013 blocks NET release into the extracellular environment
To understand the characteristics and kinetics of NETosis inhibition by CIT-013, we performed live imaging confocal microscopy following A23187 NETosis stimulation in the presence of HL488-CIT-013. Healthy neutrophils are characterized by their multi-lobulated nucleus (Figure 5a), but major chromatin changes occur when the NETosis pathway is activated. NETosis is a highly organized process and can be separated into three distinct phases: (1) chromatin re-organization within the nucleus (Figure 5b, blue arrow in phase 1), (2) breaching of the nuclear membrane followed by chromatin decondensation within the cytoplasm (Figure 5b, red arrow in phase 2), and (3) plasma membrane rupture (Figure 5b, yellow arrow in phase 3a), leading to chromatin release and diffusion into the extracellular space.14 HL488-CIT-013 does not bind healthy (non-stimulated) human neutrophils (Figure 5a) nor netting neutrophils displaying chromatin re-organization within the nucleus (Figure 5b, phase 1) or chromatin decondensation within the cytoplasm (Figure 5b, phase 2). In these neutrophils, the chromatin area was also not affected by CIT-013 (Figure 5c, d). HL488-CIT-013’s first binding event was observed specifically after initial loss of plasma membrane integrity and exposure of decondensed chromatin to the extracellular environment (Figure 5b, phase 3b). After initial HL488-CIT-013 binding, HL488-CIT-013 diffuses over time beyond the rupture point into the netting neutrophil (Figure 5b, green arrow in phase 3c, Figure S6, and Supplemental movie 2). Consequently, CIT-013 binding inhibits NET distribution into the extracellular environment, keeping most of the chromatin inside the neutrophil (Figure 5e), which is not observed in the presence of HL488-cIgG (Figure 5e, f). Finally, three-dimensional confocal imaging confirmed that HL488-CIT-013 binding colocalizes with the site of initial plasma membrane integrity loss and chromatin escape (Figure 5g). These data demonstrate that CIT-013 binds neutrophils uniquely during the terminal stage of the NETosis pathway when the plasma membrane ruptures to prevent NET expulsion into the extracellular space.
Figure 5.
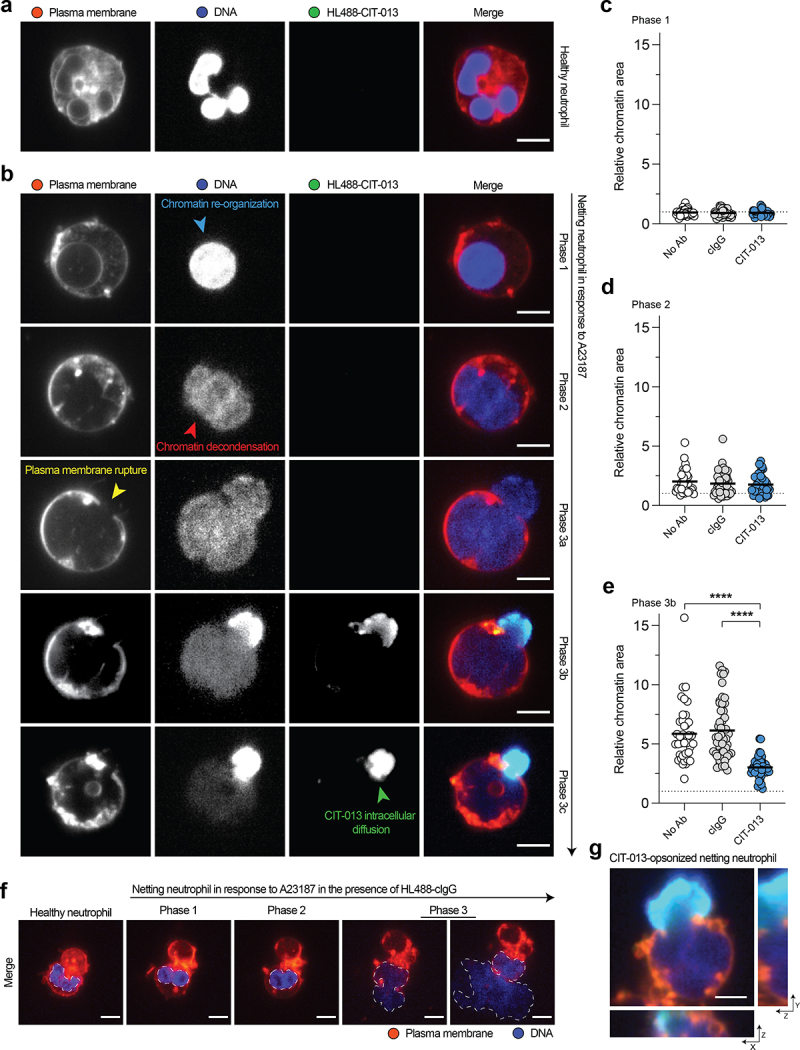
CIT-013 blocks chromatin release from netting neutrophils.
(a) Representative image of a non-stimulated neutrophil characterized by its multi-lobed nucleus in the presence of HiLyteTM Fluor 488-CIT-013 (HL488-CIT-013). (b) Representative images of an A23187-induced netting neutrophil in the presence of HL488-CIT-013. This neutrophil showed a re-organized rounded nucleus (blue arrow) in phase 1, nuclear membrane rupture and chromatin decondensation (red arrow) in phase 2, and plasma membrane rupture and chromatin release into the extracellular environment (yellow arrow) in phase 3a of the NETosis pathway. HL488-CIT-013 binding to netting neutrophils occurs in phase 3b followed by intracellular diffusion of HL488-CIT-013 beyond the rupture point (green arrow) in phase 3c. Quantification of the relative chromatin area of netting neutrophils in phase 1 (c), phase 2 (d), and phase 3b (e) of the NETosis pathway in the absence (No Ab) or presence of HL488-cIgG or HL488-CIT-013. The dotted line represents the relative chromatin area of non-stimulated neutrophils. ****P<.0001, Kruskal–Wallis test with Dunn’s multiple comparisons test. (f) Representative image of a netting neutrophil in the presence of HL488-cIgG. (g) Orthogonal representative projection of a netting neutrophil in phase 3b of the NETosis pathway confirming colocalization of CIT-013 with the site of plasma membrane rupture and chromatin escape. The scale bar in (a), (b), and (g) is 5 µm and in (f) is 10 µm.
CIT-013 binding to NETs and netting neutrophils enhances their phagocytosis by macrophages
Considering CIT-013 possessing an IgG1 Fc region, we investigated whether CIT-013 binding to epitopes present on NETs or netting neutrophils enhanced their phagocytosis. To examine this, NETosis was activated with A23187 in neutrophils in the presence of cIgG or CIT-013 (resulting in CIT-013-opsonized netting neutrophils; Figure 5g) and co- cultured with human macrophages. Using three-dimensional live imaging confocal microscopy, we observed that CIT-013 significantly enhanced internalization of opsonized netting neutrophils by macrophages (Figure 6a, b and S7a). These data demonstrate that CIT-013-opsonized netting neutrophils, and thus neutrophils whose NETosis cascade has been inhibited by CIT-013, are phagocytosed by macrophages. To investigate whether CIT-013 enhances phagocytosis of already existing NETs by macrophages as well, citrullinated NETs from A23187-stimulated neutrophil cultures were harvested and cleaved with S7 nuclease to generate NET fragments with a size ranging from 1,500 to > 20,000 base pairs (Figure S7b). NET digestion is a physiological process that occurs in blood and tissue and significantly facilitates the process of NET engulfment by macrophages37. To enhance detection of internalized material, chloroquine was added to impair lysosomal degradation of internalized NET fragments. NET fragments in the presence or absence of cIgG were phagocytosed by macrophages to a minimal level, while binding of CIT-013 to NET fragments significantly increased NET fragment phagocytosis by macrophages (Figure 6c and S7c). Visualizing CIT-013-opsonized NET fragment phagocytosis over time (Figure 6d, white arrows and Supplemental movie 3) identified accumulation of engulfed NETs in intracellular vesicles (Figure 6d, yellow arrows). FcR involvement was demonstrated by pre-treatment of macrophages with FcR blocking antibodies, which resulted in a significant decrease in phagocytosis of CIT-013-opsonized NET fragments (Figure 6e). In addition, NET phagocytosis was back to baseline levels when using CIT-013 F(ab’)2 fragments (Figure 6e).
Figure 6.
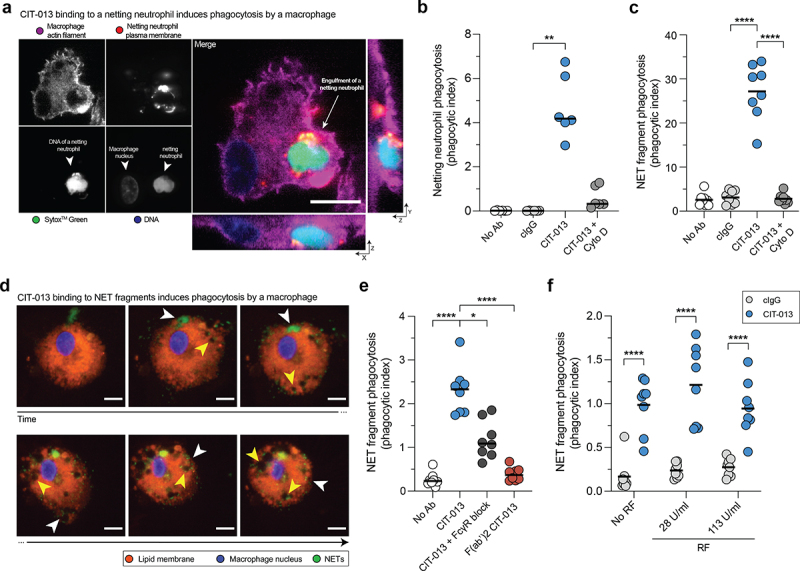
Phagocytosis of CIT-013-opsonized NETs and netting neutrophils by macrophages.
Neutrophils were stimulated with A23187 in the absence (No Ab) or presence of cIgG (resulting in NETs) or CIT-013 (resulting in CIT-013-opsonized netting neutrophils) and co-cultured with macrophages. Pre-incubated macrophages with Cyto D were used as control for phagocytosis inhibition. (a) Orthogonal representative projection of a CIT-013-opsonized netting neutrophil that was internalized by a macrophage at t = 60 min. (b) Quantification of phagocytosis of netting neutrophils by macrophages at t = 60 min. (c) Quantification of phagocytosis of NET fragments in the presence of cIgG or CIT-013 in combination with Cyto D at t = 60 min. (d) Representative images of CIT-013-opsonized NET fragments that were internalized by a macrophage (white arrows) and accumulated in intracellular vesicles (yellow arrows) over time. (e) Quantification of phagocytosis of NET fragments in the presence of CIT-013 F(ab’)2 fragments or CIT-013 in combination with a mix of blocking antibodies against FcRI, FcRIIA, and FcRIII (FcR block) at t = 60 min. (f) Quantification of phagocytosis of NET fragments in the presence of CIT-013 in combination with RF at t = 60 min. *P<.05, **P<.01, and ****P<.0001, Friedman test with Dunn’s multiple comparisons test (b), RM one-way ANOVA with Dunnett’s multiple comparisons test (c and e), and ordinary two-way ANOVA with uncorrected Fisher’s LSD test (f). The scale bar in (a) and (d) is 10 µm.
Furthermore, to study the influence of RF on CIT-013-mediated phagocytosis efficacy, we determined NET fragment phagocytosis in the presence of disease-relevant levels of RF. CIT-013-mediated NET fragment phagocytosis was not affected by RF (Figure 6f).
These data demonstrate that CIT-013 binding to NETs, even in the presence of disease-relevant immunoglobulins like RF, stimulates Fc-mediated NET phagocytosis by macrophages.
m-ACHA enhances phagocytosis by macrophages and diminishes inflammatory infiltrate in vivo
To translate the mechanistic observations described here into an in vivo setting, we investigated the effects of m-ACHA, a murine variant of CIT-013, in a mouse model of neutrophilic airway inflammation.38 This model was chosen because of two essential reasons that are highlighted in the following discussion section. The m-ACHA molecule contains similar target binding (Figure S1a), NET inhibitory capacity (Figure S8), and in vivo therapeutic properties compared to CIT-013.17,18 Mice were sensitized with HDM and CFA on day 0. Fourteen days after sensitization, mice received m-ACHA, mouse cIgG (m-cIgG), the corticosteroid Dex, or vehicle one hour before HDM challenge. Twenty-four hours after the challenge, mice were sacrificed for collection of BALF and preparation of lungs for immunohistopathology. m-ACHA’s epitope was confirmed to be present in BALF (Figure 7a) and inflamed airway tissue (Figure 7b). Consistent with the previously described anti-inflammatory activity of m-ACHA,18 a significant reduction in alveolitis (Figure S9a) and neutrophils in the airways (Figure 7c and S9b) and BALF (Figure 7d) was associated with m-ACHA administration. Interestingly, the effect on airway neutrophilia of m-ACHA was more pronounced compared to the administration of a high dose of Dex. This is consistent with the previously reported corticosteroid resistance of this model.38 Administration of m-ACHA and Dex resulted in a near equivalent reduction of airway mononuclear cells (figure S9c and S9d).
Figure 7.
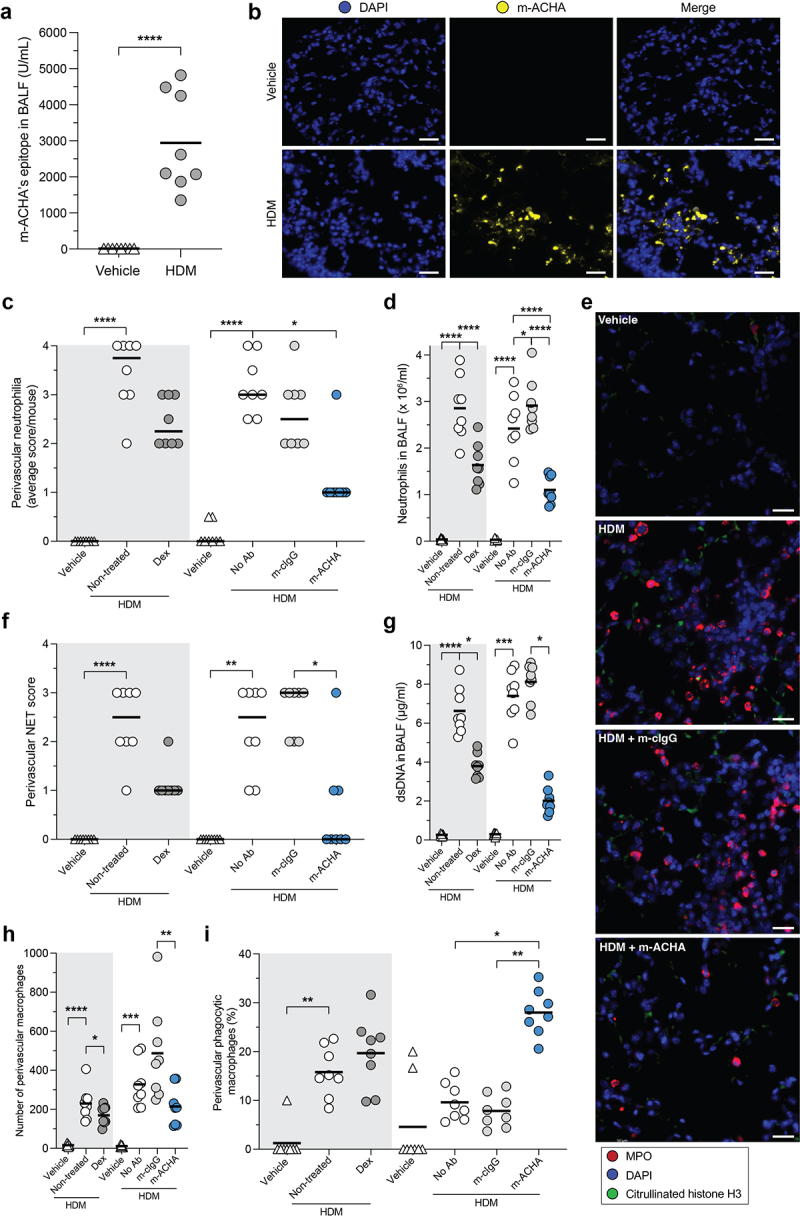
Reduced inflammatory infiltrate and enhanced phagocytosis induced by m-ACHA in a neutrophilic airway inflammation mouse model.
CFA/HDM-sensitized Balb/c mice were challenged (i.n.) with HDM or vehicle. One hour before challenge, mice received Dex or vehicle p.o. (gray area) or received m-ACHA, mouse isotype control antibody (m-cIgG), or vehicle i.v. (white area). BALF and lung tissue were collected 24 hours after challenge. (a) m-ACHA epitope levels in BALF from vehicle and HDM challenged mice which did not receive any antibody treatment. (b) Representative fluorescence IHC images from lung tissue of vehicle and HDM challenged mice stained with DAPI (DNA in blue) and m-ACHA (yellow). The scale bar is 20 µm. (c) Semi-quantitative scores of perivascular neutrophilia in H&E-stained lung sections (2 sections per mouse). (d) Neutrophil numbers in BALF. (e) Anatomically matched and randomly selected fluorescence IHC images from lung tissue stained with anti-citrullinated histone H3 antibody (green), anti-MPO antibody (red), and DAPI. The scale bar is 20 µm. (f) Semi-quantitative scoring of NETs (admixing of citH3 and MPO) in the perivascular zone of immunofluorescent-stained lung sections. Autofluorescence signals were present in images (green channel), but NETs were considered extracellular based on the morphology or minimal distance of 3 nuclear radii from the nearest nucleus. (g) dsDNA levels in BALF. The total number of macrophages (h) and the percentage of phagocytic macrophages (i) in the perivascular zone. Graphs are depicted as median in (c and f) and as mean in (a, d, and g−i) of eight mice per group. Comparisons were made per administration route (p.o. or i.v.). *P <.05, **P <.01, ***P <.001, ****P <.0001, unpaired two-tailed t test (a), Kruskal-Wallis test with Dunn’s multiple comparison test (c and f−h), and ordinary one-way ANOVA with Holm-Šidák’s multiple comparisons test (d and i).
To quantify airway tissue NETs following allergen challenge with the various treatments, semi-quantitative analysis was carried out on NET structures containing both citrullinated histone H3 and MPO (Figure 7e and S9e). Administration of m-ACHA profoundly reduced HDM challenge-associated airway NET burden (Figure 7f, S9f, and S9g) and significantly reduced NET-associated components in BALF, including dsDNA (Figure 7g) and MPO (Figure S9h). To investigate whether enhanced NET phagocytosis may have contributed to the lower NET levels detected in m-ACHA-treated mice, airway macrophages were counted based on morphology and those with abundant cytosolic vacuoles classified as phagocytic macrophages. While m-ACHA treatment does not increase the total number of airway macrophages (Figure 7h, S9i, and S9j), a significant increase in airway phagocytic macrophages was observed in m-ACHA-treated mice compared to m-cIgG-treated mice (Figure 7i, S9k, and S9l). Although mice following administration of HDM challenge plus Dex showed reduced airway NET levels, this treatment group did not show a significant increase in macrophage phagocytosis. Based on these results, we propose that NET tissue burden in lung tissue of this acute neutrophilic airway inflammation model is at least in part reduced by m-ACHA through its unique ability to enhance phagocytosis of NETs and netting neutrophils.
Discussion
Our data show that the therapeutic monoclonal antibody CIT-013 binds with picomolar affinity to its citrullinated N-terminal histone targets, H2A and H4, and thereby can specifically inhibit citrullination-dependent NET release in a dose-dependent manner. CIT-013 does not affect innate immune neutrophil functions distinct from NETosis. We also demonstrate that loss of plasma membrane integrity during the final phase of the NETosis pathway allows CIT-013 to bind netting neutrophils and thereby prevent NET expulsion in a manner that requires CIT-013’s bivalency. Furthermore, we show that CIT-013 has a unique mode of action comprising NETosis inhibition and enhanced phagocytosis of CIT-013 opsonized NETs and netting neutrophils by human macrophages in an Fc-dependent manner. Finally, the detection of CIT-013 epitopes in RA synovium provides evidence and confirms that RA is an autoimmune disease with excessive citrullinated-NETs that can be targeted by CIT-013. The in vivo anti-inflammatory effect of m-ACHA, following in part due to binding NETs and enhancing phagocytic clearance, further demonstrates the therapeutic relevance of the mechanisms of action of CIT-013 and indicates the importance of this first-in-class therapeutic antibody as a potential new therapy in RA.
Although NETs play a role in the defense against pathogens, de-regulated NETosis can enhance autoantigen exposure and activate inflammatory immune responses.5,39 The association of NETs with a range of human pathologies has generated growing interest in developing therapeutic strategies that target NET biology.10,11 NETs with high levels of histone citrullination have been linked to the development and severity of different autoimmune and inflammatory disorders, including RA,40 APS,41 SLE,42 sepsis,43 asthma,44 and Covid-19.45 Increasing awareness of the etiological importance of NETosis increases the need to develop and test novel therapeutic approaches that target NETosis, decrease the half-life of tissue NETs, and mitigate the pro-inflammatory consequences of NETs in disease.
CIT-013 has a unique mechanism of action with both Fab regions necessary to inhibit NET formation. It is likely that in particular CIT-013’s strong binding to two epitopes (citrullinated histone H2A as well as H4) within a single nucleosome or on two adjacent nucleosomes contributes to its enhanced NET-inhibitory efficacy by (1) crosslinking different nucleosomes to inhibit further chromatin expansion and the outward motive force necessary for NET expulsion and/or (2) forming a large intracellular chromatin-immune complex unable to be expelled. Retention of the decondensing chromatin in netting neutrophils will have numerous anti-inflammatory consequences including reduced tissue deposition of NETs and their cytotoxic components such as histones, enzymes (e.g., MPO and NE) and anti-microbial proteins. Furthermore, inhibition of NETosis will reduce autoantigen exposure. Consistent with this, blocking NETosis by PAD4 inhibition has been demonstrated to have anti-inflammatory benefit in various murine disease models, including wound healing,46 thrombosis,47 atherosclerosis,48 sepsis-induced endotoxic shock,49 colitis,50 ischemia reperfusion-related tissue injury,51 fibrosis,52 inflammatory arthritis,53 and SLE.54
CIT-013’s high affinity for the citrullinated N-termini of histone H2A and H4, present in citrullination-rich NETs and netting neutrophils, in combination with CIT-013’s functional Fc region, enables the antibody to induce enhanced phagocytosis by macrophages. In fact, it might be plausible that enhanced phagocytosis by macrophages also occurs when CIT-013 binds non-citrullinated NETs since CIT-013 also shows moderate affinity toward native histone H2A. We hypothesize that this action will further reduce tissue NET burden with important anti-inflammatory consequences. The importance of efficiently clearing NETs is illustrated in SLE where patients with impaired NET degradation capacity show a correlation with high anti-NET autoantibody titers, lupus nephritis,55 and symptom flares.56
Recently, additional studies confirm our previous work17 showing that ACPAs in RA patients contain anti-inflammatory properties in mouse models of RA.21–23 This proves that antibodies against citrullinated epitopes, like CIT-013, have potential for the development of new therapeutics. Indeed, ACPA derived from RA patients protect mice from collagen antibody-induced arthritis, showing strong interaction with citrullinated α-enolase and Fc;R2B on macrophages and resulting in increased IL-10 secretion and reduced osteoclastogenesis.22 These ACPAs do not bind citrullinated histone H2A and have a NET-independent mechanism of action. Besides, these ACPAs are polyreactive with low affinity for their epitopes, while CIT-013 is an optimized and well-developed clinical development candidate which shows high specificity and affinity with a powerful dual mechanism of action, even in the presence of RA-derived immunoglobulins, like RF or ACPA.
An important milestone in evaluating a first-in-class therapeutic approach is translating in vitro observations to the in vivo setting. With this in mind, we have demonstrated in vivo anti-inflammatory activity of m-ACHA (a murine variant of CIT-013), and this effect is associated with reduced tissue NETs and increased macrophage phagocytic activity. This implies that reduction in tissue NETs results solely from enhanced NET phagocytosis, but the in vitro data presented here, as well as in vivo data from our previous studies,18 suggest that these effects result from both modes of action of CIT-013. It is also possible that reduced tissue NET burden following m-ACHA treatment and allergen challenge is a consequence of reduced recruitment of neutrophils into the airways; however, we believe that the weight of evidence supports the inverse interpretation, i.e., that reduced tissue NET burden decreases pro-inflammatory signals that recruit neutrophils and monocytes to tissue. In fact, we previously showed, using an acute peritonitis mouse model, that the therapeutic action of CIT-013 is primarily based on the reduction of NET burden, while reduced peritoneal neutrophil infiltration was not observed.18
Finally, our data suggest that the novel mode of action of CIT-013 is the basis for observed differentiation from corticosteroid treatment in the airway neutrophilia model examined. As far as we know, only CIT-013 is able to both reduce tissue NETs and enhance macrophage phagocytosis, which results in a significant anti-inflammatory effect.18
Small molecules that interfere with NETosis have been shown to result in therapeutic effects in various in vivo studies. One focus has been the inhibition of histone citrullination by small-molecule inhibitors of PAD4 such as BMS-P5,57 Cl-amidine,49 BB-Cl-amidine,58 and GSK484,59 all of which inhibit NET release resulting in reduced NET-dependent inflammation in different disease models. Besides PAD4 inhibitors, other reagents have been shown to directly interfere with NETosis, including thrombomodulin,60 metformin,61 and gasdermin D inhibitors.62 These studies, which engage the NETosis pathway at distinct points, present a consistent outcome of important anti-inflammatory benefit. These effects are encouraging, especially since these small-molecule approaches target NETosis only, with no reported impact on the phagocytic clearance of NETs. We believe that the monoclonal antibody profile of CIT-013 described here offers a more comprehensive therapeutic approach, targeting both NET production and the clearance of pro-inflammatory NETs and netting neutrophils. This is supported by our in vivo studies, demonstrating that NET-targeting therapeutics have potential as effective treatments in diseases where NET biology is a key driver of the pathology.18 Furthermore, this dual profile will be appropriate across various stages of disease progression where patient diagnosis will be associated with varying levels of pre-established tissue NET burden. Small-molecule strategies targeting NETosis have not yet progressed into clinical development, perhaps also indicating that a monoclonal antibody therapeutic that only binds netting neutrophils and NETs may have a superior safety profile over small molecules targeting, for example, PAD enzymes or gasdermin D which are not NETosis specific and can penetrate all tissues and cells.
A limitation of this study is that we had to use a mouse model of neutrophilic airway inflammation instead of a mouse model of RA to study CIT-013’s mechanism of action. There are two essential reasons for this. First, we lack insight into the time frame of NET formation and macrophages infiltration in inflamed tissue in a chronic collagen-induced arthritis mouse model or any other mouse model of RA, which makes it impossible to estimate at which timepoint during disease, CIT-013-induced NET phagocytosis would be detectable. Second, macrophages are abundantly present in the lung, which makes this model ideal to study CIT-013-induced phagocytosis.
The data presented here together with data from our previous study18 suggest that in a clinical setting, CIT-013 will both inhibit the formation of NETs and accelerate clearance of pre-existing NETs and netting neutrophils. The observation that synovial tissues from RA patients showed strong CIT-013 staining indicates that RA is an appropriate target disease for CIT-013 clinical investigation. We believe that reducing tissue NET burden, endothelial damage, and NET-associated cardiovascular pathology will improve patient quality of life. While this study, as well as our previous studies,17,18 draws attention to the therapeutic potential of CIT-013 in RA, it is worth noting that a broad range of immune-mediated inflammatory diseases have symptoms and severities that are associated with the pathology of de-regulated NETosis. In addition to autoimmune diseases such as RA and lupus, we propose CIT-013 as a novel therapeutic with potential in severe neutrophilic asthma, chronic obstructive pulmonary disease (COPD), ARDS, cardiovascular disease, and dermatological conditions with significant unmet need such as hidradenitis suppurativa (HS). Citryll has recently completed Phase 1 healthy volunteer clinical studies for its lead NET-targeting antibody CIT-013 and is transitioning into the next stage of its clinical development with the initiation of Phase 2 proof-of-concept trials in RA and HS.
Supplementary Material
Acknowledgments
This work is dedicated to Dr Jos Raats who unfortunately has passed away unexpectedly in March 2022. Without him, the ACHA program as it is today would not have seen the light.
We thank Dr J. van Rosmalen for designing, cloning, production, and purification of monovalent CIT-013. We also thank Jeroen Cole, Anna Feher, and Peter Drent from Confocal.nl and Pim French and Maurice de Wit from Erasmus Medical Centre for their support with confocal microscopy. We thank Tim van Breemen and Josephine Stein for their contribution to mechanism of action-related experiments.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Funding Statement
Part of this work was supported by a loan under the Innovation Credit Scheme (IK18012) obtained from the Netherlands Enterprise Agency. Luise Erpenbeck was supported by the DFG (ER 723/2-1).
Disclosure statement
Citryll employees have financial interest.
Author contribution
P.V., M.L., S.K., A.K., S.D., D.M, and R.G.S.C. designed the experimental strategies. M.L., S.K., D.M., M.F., I.R., A.K., S.D., T.B., K.W., R.P., P.Z., L.E., and E.N. performed experiments and analyzed the data. M.L. and D.M. performed statistical analysis. P.V., M.L., S.K., D.M., M.F., E.N. L.E., H.E., E.M., and R.G.S.C. provided scientific input. M.L., S.K., D.M., H.E., E.M., and R.G.S.C. drafted and/or designed the manuscript. R.G.S.C is the senior investigator of this study. All authors critically reviewed the paper.
Data availability statement
All data are available under a material transfer agreement with Citryll B.V.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2023.2281763
Abbreviations
- A23187
Calcium ionophore
- ACHA
Anti-citrullinated histone antibodies
- ACPA
Anti-citrullinated protein antibodies
- ALI
Acute lung injury
- APS
Antiphospholipid syndrome
- ARDS
Acute respiratory distress syndrome
- BALF
Bronchoalveolar lavage fluid
- COPD
Chronic obstructive pulmonary disease
- CFA
Complete freund’s adjuvant
- cIgG
Isotype control IgG
- Cyto D
Cytochalasin D
- Dex
Dexamethasone
- DPI
Diphenyleneiodonium chloride
- FBS
Fetal bovine serum
- FcR
Fc gamma receptor
- HDM
House dust mite
- HK
Heat-killed
- HL488
HiLyteTM Fluor 488
- HS
Hidradenitis suppurativa
- HSA
Human serum albumin
- ICs
Immune complexes
- IHC
Immunohistochemistry
- KD
Equilibrium dissociation constant
- m-ACHA
Mouse anti-citrullinated histone antibody (murine variant of CIT-013)
- MPO
Myeloperoxidase
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NE
Neutrophil elastase
- NETs
Neutrophil extracellular traps
- PAD4
Peptidyl arginine deiminase 4
- PMA
Phorbol 12-myristate 13-acetate
- RA
Rheumatoid arthritis
- RF
Rheumatoid factor
- ROS
Reactive oxygen species
- RT
Room temperature
- SF
Synovial fluid
- SLE
Systemic lupus erythematosus
- SPR
Surface Plasmon Resonance
References
- 1.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A.. Neutrophil Extracellular Traps Kill Bacteria. Sci. 2004;303(5663):1532–22. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 2.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y.. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–62. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tambralli A, Gockman K, Knight JS. NETs in APS: Current knowledge and future perspectives. Curr Rheumatol Rep. 2020;22(10):67. doi: 10.1007/s11926-020-00936-1. [DOI] [PubMed] [Google Scholar]
- 4.Li RHL, Tablin F. A comparative review of neutrophil extracellular traps in sepsis. Front Vet Sci. 2018;5:291. doi: 10.3389/fvets.2018.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–47. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 6.Bosmann M, Ward PA. Protein-based therapies for acute lung injury: Targeting neutrophil extracellular traps. Expert Opin Ther Targets. 2014;18(6):703–14. doi: 10.1517/14728222.2014.902938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morán G, Uberti B, Quiroga J. Role of cellular metabolism in the formation of neutrophil extracellular traps in airway diseases. Front Immunol. 2022;13:1–12. doi: 10.3389/fimmu.2022.850416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, Brinkmann V, von Bernuth H, Zychlinsky A. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6:e24437. doi: 10.7554/eLife.24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muñoz LE, Boeltz S, Bilyy R, Schauer C, Mahajan A, Widulin N, Grüneboom A, Herrmann I, Boada E, Rauh M, et al. Neutrophil Extracellular Traps Initiate Gallstone Formation. Immunity. 2019;51(3):443–50.e4. doi: 10.1016/j.immuni.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194–211. doi: 10.1007/s12016-020-08804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnado A, Crofford LJ, Oates JC. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J Leukoc Biol. 2016;99(2):265–78. doi: 10.1189/jlb.5BT0615-234R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. 2012;92(4):841–49. doi: 10.1189/jlb.1211601. [DOI] [PubMed] [Google Scholar]
- 13.Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:1–9. doi: 10.3389/fimmu.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neubert E, Meyer D, Rocca F, Günay G, Kwaczala-Tessmann A, Grandke J, Senger-Sander S, Geisler C, Egner A, Schön MP, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9(1):3767. doi: 10.1038/s41467-018-06263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, Goldman RD, Wagner DD, Waterman CM. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326–37. doi: 10.1073/pnas.1909546117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–13. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chirivi RGS, Jenniskens GJ, Raats JMH. Anti-Citrullinated Protein Antibodies as Novel Therapeutic Drugs in Rheumatoid Arthritis. J Clin Cell Immunol. 2013;1(S6):1–13. doi: 10.4172/2155-9899.S6-006. [DOI] [Google Scholar]
- 18.Chirivi RGS, Van Rosmalen JWG, Van der Linden M, Euler M, Schmets G, Bogatkevich G, Kambas K, Hahn J, Braster Q, Soehnlein O, et al. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol Immunol. 2021;18(6):1528–44. doi: 10.1038/s41423-020-0381-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16(7):677–81. doi: 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Linden M, Westerlaken GHA, Van Der Vlist M, Van Montfrans J, Meyaard L. Differential signalling and kinetics of neutrophil extracellular trap release revealed by quantitative live imaging. Sci Rep. 2017;7(1):6529. doi: 10.1038/s41598-017-06901-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raposo B, Afonso M, Israelsson L, Wähämaa H, Stålesen R, Wermeling F, Haj Hensvold A, Grönwall C, Rethi B, Klareskog L, et al. Divergent and dominant anti- inflammatory effects of patient-derived anticitrullinated protein antibodies (ACPA) in arthritis development. Ann Rheum Dis. 2023;82(5):724–26. doi: 10.1136/ard-2022-223417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Y, Ge C, Moreno-Giró À, Xu B, Beusch CM, Sandor K, Su J, Cheng L, Lönnblom E, Lundqvist C, et al. A subset of antibodies targeting citrullinated proteins confers protection from rheumatoid arthritis. Nat Commun. 2023;14(1):1–19. doi: 10.1038/s41467-023-36257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez AM, Brewer RC, Moon J-S, Acharya S, Kongpachith S, Wang Q, Jahanbani S, Wong HH, Lanz TV, Love ZZ, et al. Anti-citrullinated protein antibodies with diverse specificities ameliorate collagen antibody-induced arthritis in a time-dependent manner. Arthritis Rheumatol. 2023;online ahead of print. doi: 10.1002/art.42679. [DOI] [PubMed] [Google Scholar]
- 24.Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014;16(3):R122. doi: 10.1186/ar4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bach M, Moon J, Moore R, Pan T, Nelson JL, Lood C. A neutrophil activation biomarker panel in prognosis and monitoring of patients with rheumatoid arthritis. Arthritis Rheumatol. 2020;72(1):47–56. doi: 10.1002/art.41062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, Grant MM, Milward M, Lundberg K, Buckley CD, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. 2015;67(12):3135–45. doi: 10.1002/art.39313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, Hoffmann V, Fox DA, Buckner JH, Kaplan MJ. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. 2020;5(13):e139388. doi: 10.1172/jci.insight.139388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5(178):178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Linden M, Meyaard L. Fine-tuning neutrophil activation: Strategies and consequences. Immunol Lett. 2016;178:3–9. doi: 10.1016/j.imlet.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 30.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraaij T, Tengström FC, Kamerling SWA, Pusey CD, Scherer HU, Toes REM, Rabelink TJ, van Kooten C, Teng YKO. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev. 2016;15(6):577–84. doi: 10.1016/j.autrev.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 32.Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, Laskay T. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J Immunol. 2014;193(4):1954–65. doi: 10.4049/jimmunol.1400478. [DOI] [PubMed] [Google Scholar]
- 33.Limon M. The assessment of rheumatoid factor levels and hemogram parameters as predictors for rheumatologic disease. Int J Of Rheum Dis. 2023;26(6):1041–47. doi: 10.1111/1756-185X.14683. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen SF, Bojesen SE, Schnohr P, Nordestgaard BG. Elevated rheumatoid factor and long term risk of rheumatoid arthritis: a prospective cohort study. BMJ. 2012;345(sep06 2):e5244–e5244. doi: 10.1136/bmj.e5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boeters DM, Mangnus L, Ajeganova S, Lindqvist E, Svensson B, Toes REM, Trouw LA, Huizinga TWJ, Berenbaum F, Morel J, et al. The prevalence of ACPA is lower in rheumatoid arthritis patients with an older age of onset but the composition of the ACPA response appears identical. Arthritis Res Ther. 2017;19(1):1–10. doi: 10.1186/s13075-017-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willemze A, Böhringer S, Knevel R, Levarht EWN, Stoeken-Rijsbergen G, Houwing-Duistermaat JJ, Van Der Helm-van Mil AHM, Huizinga TWJ, Toes REM, Trouw LA. The ACPA recognition profile and subgrouping of ACPA-positive RA patients. Ann Rheum Dis. 2012;71(2):268–74. doi: 10.1136/annrheumdis-2011-200421. [DOI] [PubMed] [Google Scholar]
- 37.Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191(5):2647–56. doi: 10.4049/jimmunol.1300436. [DOI] [PubMed] [Google Scholar]
- 38.Russell V, Brana M, Pompilio D, Ambrosi G, Andreetta F, Venturini E, Chalidou A, Manni ME. Steroid-resistant inflammation in a mouse model of severe asthma is not inhibited by the combination of theophylline and budesonide. Eur Respir J. 2018;52:A1048. doi: 10.1183/13993003.congress-2018.PA1048. [DOI] [Google Scholar]
- 39.Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189(6):2689–95. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Neil LJ, Barrera-Vargas A, Sandoval-Heglund D, Merayo-Chalico J, Aguirre-Aguilar E, Aponte AM, Ruiz-Perdomo Y, Gucek M, El-Gabalaawy H, Fox DA, et al. Neutrophil-mediated carbamylation promotes articular damage in rheumatoid arthritis. Sci Adv. 2020;6(44):eabd2688. doi: 10.1126/sciadv.abd2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Zuo Y, Zhang S, Makris UE, Karp DR, Li Z. Additional risk factors associated with thrombosis and pregnancy morbidity in a unique cohort of antiphospholipid antibody-positive patients. Chin Med J (Engl). 2022;135(6):658–64. doi: 10.1097/CM9.0000000000001964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casey KA, Smith MA, Sinibaldi D, Seto NL, Playford MP, Wang X, Carlucci PM, Wang L, Illei G, Yu B, et al. Modulation of cardiometabolic disease markers by type I interferon inhibition in systemic lupus erythematosus. Arthritis Rheumatol. 2021;73(3):459–71. doi: 10.1002/art.41518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian Y, Russo RM, Li Y, Karmakar M, Liu B, Puskarich MA, Jones AE, Stringer KA, Standiford TJ, Alam HB. Serum citrullinated histone H3 concentrations differentiate patients with septic verses non-septic shock and correlate with disease severity. Infection. 2021;49(1):83–93. doi: 10.1007/s15010-020-01528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuczia P, Zuk J, Iwaniec T, Soja J, Dropinski J, Malesa-Wlodzik M, Zareba L, Bazan JG, Undas A, Bazan-Socha S. Citrullinated histone H3, a marker of extracellular trap formation, is increased in blood of stable asthma patients. Clin Transl Allergy. 2020;10(1):31. doi: 10.1186/s13601-020-00337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuo Y, Yalavarthi S, Navaz SA, Hoy CK, Harbaugh A, Gockman K, Zuo M, Madison JA, Shi H, Kanthi Y, et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight. 2021;6:e150111. doi: 10.1172/jci.insight.150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–19. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110(21):8674–79. doi: 10.1073/pnas.1301059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Carmona-Rivera C, Moore E, Seto NL, Knight JS, Pryor M, Yang ZH, Hemmers S, Remaley AT, Mowen KA, et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front Immunol. 2018;9:1–13. doi: 10.3389/fimmu.2018.01680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biron BM, Chung CS, O’Brien XM, Chen Y, Reichner JS, Ayala A. Cl-Amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9(1):22–32. doi: 10.1159/000448808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, Davis T, Matesic LE, Thompson PR, Hofseth LJ. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am J Physiol - Gastrointest Liver Physiol. 2011;300(6):G929–G38. doi: 10.1152/ajpgi.00435.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018;93(2):365–74. doi: 10.1016/j.kint.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. 2017;214(2):439–58. doi: 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K, Yamamoto K. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep. 2015;5(1):1–7. doi: 10.1038/srep13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. 2013;123(7):2981–93. doi: 10.1172/JCI67390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–18. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, Bengtsson AA, Blom AM. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188(7):3522–31. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 57.Li M, Lin C, Deng H, Strnad J, Bernabei L, Vogl DT, Burke JJ, Nefedova Y. A novel peptidylarginine deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol Cancer Ther. 2020;19(7):1530–38. doi: 10.1158/1535-7163.MCT-19-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ai P, Pan H, Chen K, Zheng J, Gao Z, Jin G. Viral mimetic poly(I: C) induces neutrophil extracellular traps via PAD4 to promote inflammation and thrombosis. Biochem Biophys Res Commun. 2021;565:64–71. doi: 10.1016/j.bbrc.2021.05.091. [DOI] [PubMed] [Google Scholar]
- 59.Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, Chong BH. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. 2019;10(1):1322. doi: 10.1038/s41467-019-09160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shrestha B, Ito T, Kakuuchi M, Totoki T, Nagasato T, Yamamoto M, Maruyama I. Recombinant thrombomodulin suppresses histone-induced neutrophil extracellular trap formation. Front Immunol. 2019;10:2535. doi: 10.3389/fimmu.2019.02535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menegazzo L, Scattolini V, Cappellari R, Bonora BM, Albiero M, Bortolozzi M, Romanato F, Ceolotto G, Vigili de Kreutzeberg S, Avogaro A, et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018;55(6):593–601. doi: 10.1007/s00592-018-1129-8. [DOI] [PubMed] [Google Scholar]
- 62.Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, Martins TV, Cólon DF, Borges VF, Brauer VS, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. 2021;138(25):2702–13. doi: 10.1182/blood.2021011525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available under a material transfer agreement with Citryll B.V.