

Abstract
Protein acetylation is a central event in orchestrating diverse cellular processes. However, current strategies to investigate protein acetylation in cells are often nonspecific or lack temporal and magnitude control. Here, we developed an acetylation tagging system, AceTAG, to induce acetylation of targeted proteins. The AceTAG system utilizes bifunctional molecules to direct the lysine acetyltransferase p300/CBP to proteins fused with the small protein tag FKBP12F36V, resulting in their induced acetylation. Using AceTAG, we induced targeted acetylation of a diverse array of proteins in cells, specifically histone H3.3, the NF-κB subunit p65/RelA, and the tumor suppressor p53. We demonstrate that targeted acetylation with the AceTAG system is rapid, selective, reversible and can be controlled in a dose-dependent fashion. AceTAG represents a useful strategy to modulate protein acetylation and should enable the exploration of targeted acetylation in basic biological and therapeutic contexts.
Graphical Abstract
INTRODUCTION
Lysine acetylation is one of the most frequent post-translational modifications (PTMs), occurring on >10000 sites on human proteins, and plays critical roles in human biology.1 This covalent modification is reversible, and its dynamic equilibrium is mediated by a combined ~40 lysine deacetylases (KDACs) and acetyltransferases (KATs).2,3 Acetylation not only imparts direct functional consequences on proteins but can also interplay with other PTMs, such as phosphorylation, ubiquitination, methylation, and SUMOylation, and its dysregulation has been implicated in diverse human diseases, including various cancers,4,5 neurodegenerative disorders,6,7 autoimmune conditions,8,9 and metabolic diseases.5,10,11 Despite its frequency and diverse roles, a substantial hurdle for accurate investigations of acetylation is the dearth of tools to selectively modulate protein acetylation in cells. Although conventional methods, such as genetic or chemical manipulation of KDACs or KATs, have yielded much insight into the myriad roles of acetylation,12,13 they induce global alterations to their substrates, potentially complicating the interpretation of the biological effects relating to a single protein.14,15 Alternatively, methods for site-specific modification of substrates, to either mimic or block acetylation,16,17 could have undesirable effects on protein structure, obfuscate analysis of alternate PTMs on the same residues, and do not permit acute or graded investigations of acetylation in cells. To overcome these obstacles, we envisioned a potentially generalizable, chemical-based strategy to dynamically and selectively induce acetylation on a protein of interest (POI) directly in cells.
Encouraged by the transformative utility of chemical inducers of dimerization18,19 and ubiquitin-inducing small molecules,20 as well as recent examples of phosphorylation modifying bifunctional molecules,21,22 we hypothesized that a similar heterobifunctional system could be employed to modulate the acetylation of any POI on-demand by proximity-induced acetylation. Acetylation can impart inhibitory effects on a protein as well as activating or gain-of-function outcomes; thus, a potential hurdle for the development of such heterobifunctional small molecules is the identification of functionally “silent” ligands that selectively bind to target proteins without overt effects on protein function to prevent counterproductive or complicated outputs. Considering this, and inspired by various technologies that enable the selective targeting of genetically tagged proteins with small molecules,23–25 we conceptualized a strategy to direct protein acetylation to targeted POIs, even in the absence of available small molecule ligands.
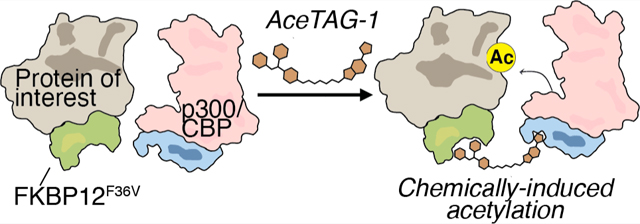
Here, we report the development of an “acetylation tagging” system, or AceTAG, which consists of a heterobifunctional molecule formed by covalently linking a KAT-binding ligand to an FKBP12F36V-binding ligand, resulting in chemically induced proximity between cellular acetylation machinery and a targeted protein. Using AceTAG, we demonstrate that the KAT p300/CBP can be recruited directly to histone H3.3, p65/RELA, and p53, resulting in their targeted acetylation at functionally relevant sites in cells. We demonstrate AceTAG-mediated acetylation is dose-controlled, reversible, rapid, and selective for tagged protein targets.
RESULTS AND DISCUSSION
Bifunctional AceTAG Molecules Mediate Binding between p300/CBP and FKBP12F36V.
Among KATs, the E1A-associated protein (p300) and its paralogue CREB-binding protein (CBP) are two of the most prominent members, playing key roles as transcriptional co-activators essential for myriad cellular processes, including growth and development, stress response, oncogenesis, and DNA damage.26 p300/CBP collectively regulate >2/3 of the known acetylation sites in humans,1 indicative of a broad substrate tolerability, and therefore represent high-priority candidates for the design of generalizable chemically induced acetylation strategies. Recently, inhibitors targeting the conserved p300/CBP bromodomain (BRD)27 have been co-opted into bifunctional molecules to target chromatin machinery. This includes their conjugation to DNA-targeting polyamides28 to coordinate histone acetylation in vitro and alter gene expression in cellulo as well as their use to recruit p300/CBP to gene loci for targeted gene transcription via catalytically inactive Cas9,29 the latter presumably through proximity-induced histone acetylation. However, it remained an open question as to whether a potentially generalizable strategy might be developed to recruit p300/CBP directly to protein targets, resulting in their targeted acetylation in living cells.
Recently, in an approach called degradation TAG (dTAG), it has been shown that synthetic ligands that recognize the engineered FKBP12 variant FKBP12F36V can be conjugated to E3 ligands,30 resulting in selective ubiquitination and proteasome-mediated degradation of FKBP12F36V-tagged proteins.25 We hypothesized a similar strategy could be adopted for targeted protein acetylation (Figure 1a). To this end, we appended the previously reported FKBP12F36V-binding ligand25,31 to the 5-isoxazoly-benzimidizole p300/CBP BRDinhibitor27 to generate a series of compounds, AceTAG 1–3, with different linker compositions (Figure 1b and Figure S1a). We first assessed the ability of AceTAG 1–3 to mediate complex formation between soluble recombinant FKBP12F36V and the BRD domain of p300 (BRD-p300) in vitro using an AlphaScreen assay31 (Figure S1b). We observed characteristic bell-shaped autoinhibitory curves for all AceTAG analogues, the result of AceTAG molecules saturating both FKBP12F36V and BRD-p300, effectively outcompeting ternary complex formation.32 We noted that AceTAG-1 has increased potency relative to other analogues, with maximum complex formation occurring at ~1 μM (Figure 1c). In addition, the luminescence signal is considerably diminished upon co-treatment of either terminal binding molecules (FKBP-c and p300-c) in a concentration-dependent fashion (Figure 1d,e and Figure S1c), confirming that observed ternary complex formation is dependent on the simultaneous binding of both BRD-p300 and FKBP12F36V. To verify that AceTAG molecules can engage their protein binding partners in cells, we constructed “fully functionalized” photoaffinity probes33 of both the FKBP12F36V (FKBP-p) and p300/CBP (p300-p) ligands and confirmed respective binding to recombinantly expressed p300 and FKBP12F36V can be efficiently competed when cells are co-treated with increasing concentrations of AceTAG-1 (Figure 1f,g). Together, these biochemical data indicate that AceTAG 1–3 effectively engage FKBP12F36V and p300/CBP and are capable of inducing complex formation.
Figure 1.

AceTAG molecules bind p300/CBP and FKBP12F36V and can mediate ternary complex formation. (a) Schematic overview of targeted acetylation tagging (AceTAG) strategy. Hetereobifunctional AceTAG molecules induce acetylation of FKBP12F36V-POI fusions through recruitment of KAT p300/CBP. (b) Structures of AceTAG molecules composed of an FKBP12F36V binding ligand and a p300/CBP-binding ligand connected by a linker. (c) AceTAG molecules induce ternary complex between FKBP12F36V and the bromodomain of p300 (BRD-p300) as determined by AlphaScreen (Figure S1b). (d, e) AceTAG-1 (300 nM) mediated ternary complex formation between FKBP12F36V and BRD-p300 can be blocked upon co-incubation of increasing concentrations of p300-binding (d) or FKBP12F36V-binding ligands (e, Figure S1c). Data in (c–e) represent as mean ± s.d. of n = 3 replicates. (f, g) Confirmation of AceTAG-1 target engagement in cells. FKBP12F36V-H3.3 (f) and full length p300 (g) were recombinantly expressed with HA epitope tags by transient transfection in HEK293T cells, which were then co-treated with a photoaffinity probe derived from the FKBP12F36V ligand (f, FKBP-p) or p300/CBP ligand (g, p300-p) and increasing concentrations of AceTAG-1, photo-cross-linked, and lysed, and proteomes were conjugated to an azide–rhodamine tag by CuAAC chemistry and analyzed by in-gel fluorescence staining. The results in (f, g) are representative of three independent biological replicates (n = 3). Full images of gels are shown in Figure S6.
AceTAG Molecules Induce Targeted Protein Acetylation in Cells.
We next evaluated whether AceTAG molecules could induce acetylation of FKBP12F36V-tagged proteins in cells. We chose first to target histone H3.3 given that p300/CBP is a predominant histone acetyl transferase. Although the specific sites on H3 targeted by p300/CBP are not firmly established, recent proteomic studies have indicated that pharmacological inhibition and genetic ablation of p300/CBP result in decreased levels of H3K18ac, H3K27ac, and H3K36ac.15 In addition, in vitro studies suggest H3K14, H3K18, H3K23, H3K64, and H3K122 are also major acetylation targets.34–36 We first stably transfected HeLa cells with H3.3-FKBP12F36V-HA and confirmed that the H3.3FKBP12F36V chimera localizes primarily to chromatin along with endogenous H3.3 (Figure S2a). We next evaluated AceTAG compounds AceTAG 1–3 for their relative ability to induce acetylation at H3.3 K18 using acetyl histone H3 K18 selective antibodies. We observed AceTAG-mediated K18 acetylation for all analogues, with AceTAG-1 inducing modestly higher levels of acetylation relative to the other analogues (Figure 2a and Figure S2b).
Figure 2.

AceTAG molecules induce rapid, targeted acetylation of H3.3-FKBP12F36V in cells. (a) Concentration-dependent acetylation of H3.3FKBP12F36V lysine residues by AceTAG-1. H3.3-FKBP12F36V HeLa cells were treated with increasing concentrations of AceTAG-1 for 2 h, and acetylation of K18, K27, and K23 was monitored by immunoblot. Shown in the right panel is quantitation of immunoblot signal of K18Ac relative to α-tubulin as the mean ± s.e.m. of n = 3 biologically independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t tests comparing DMSO- to AceTAG-1-treated samples. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. (b) Immunoblot analysis of controls for AceTAG-mediated acetylation of H3.3. Cells were treated with the histone deacetylase inhibitor SAHA (5 μM) or co-treated with AceTAG-1 (625 nM) and DMSO, an FKBP12F36V binding ligand (50 μM, Figure S1c), or the p300/CBP KAT domain inhibitor A-485 (1 μM) for 2 h which block AceTAG-1-induced acetylation. The results in (a, b) are representative of two independent biological replicates (n = 2). (c) Immunoblot analysis of AceTAG-1 (625 nM) treated H3.3-FKBP12F36V HeLa cells over the indicated time course. (d) Washout experiments showing decreasing acetylation upon removal of AceTAG-1 from cells. Immunoblot of H3.3-FKBP12F36V HeLa cells pretreated with AceTAG-1 (625 nM) or vehicle for the indicated time, washed with DPBS, and resuspended in fresh media (without AceTAG-1) for the indicated time. Shown in the right panel is quantitation of immunoblot signal of K18Ac relative to α-tubulin as the mean ± s.e.m. of n = 3 biologically independent experiments. The results in (c, d) are representative of three independent biological replicates (n = 3). Full images of blots are shown in Figures S7 and S8.
We observed that AceTAG-mediated acetylation of H3.3FKBP12F36V is dose-dependent, with highest levels of acetylation achieved between 625 nM and 3 μM, near the concentration needed for maximum ternary complex formation in vitro (Figure 1c). In addition, acetylation levels demonstrate a decreasing trend at the highest AceTAG-1 concentrations, consistent with acetylation being a consequence of ternary complex induction in cells (Figure 2a). We also confirmed increased H3.3-FKBP12F36V acetylation upon incubation of cells with the broad histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA), indicating the fusion protein is a substrate of endogenous HDACs (Figure 2b). We next examined levels at other known H3.3 sites using available antibodies, including K9, K14, K23, K27, and K79 (Figure 2a and Figure S2c), detecting induced acetylation only at K18, K23, and K27 (Figure 2a). Notably, we did not observe changes in acetylation of untagged, endogenous H3.3 after AceTAG-1 treatment in either WT HeLa or H3.3-FKBP12F36V HeLa cells, suggesting that AceTAG-1 does not modulate p300/CBP KAT activity (Figure S2d,e). In addition, acetylation can be blocked by co-incubation with either A-485, a p300/CBP KAT inhibitor,37 with excess FKBP12F36V ligand (FKBP-c, Figure 2b) or with excess p300/CBP ligand (p300-c, Figure S2f). Collectively, these data indicate that AceTAG-induced acetylation in cells is dependent on p300/CBP enzymatic activity as well as binding to both p300/CBP and the FKBP12F36V-tagged protein.
We next set out to assess the kinetics of AceTAG-mediated acetylation in cells by monitoring acetylation levels of H3.3 K18 over multiple time points. Surprisingly, we observed that induced acetylation occurs almost immediately, after only ~5 min exposure to AceTAG-1 (Figure 2c) as well as after continuous incubation (up to 24 h). In addition, when AceTAG-1 is removed from cells, H3.3 K18 acetylation steadily dissipates, reaching near basal levels within 2 h after compound removal (Figure 2d). This re-equilibration is likely the result of endogenous KDAC activity, together indicating that the targeted acetylation is reversible and dependent on the heterobifunctional compound.
AceTAG Strategy Extendable to Multiple Proteins.
Satisfied with the successful compound-induced acetylation of H3.3, we next used our AceTAG system to assess chemically mediated acetylation of other targeted proteins. To avoid potential obfuscation by endogenous target proteins, we generated FKBP12F36V-RelA and FKBP12F36V-p53 constructs, two proteins wherein p300/CBP-mediated acetylation is known to impart consequences38,39 on their transcriptional activity. Subsequently, FKBP12F36V-RelA was stably transfected in a HeLa RelA−/− cell line while FKBP12F36V-p53 was stably transfected in H1299 non-small cell carcinoma (NSCLC) cells, which have a homozygous partial deletion of TP53 and lack p53 protein expression. Treatment of FKBP12F36V-RelA expressing cells with AceTAG-1 resulted in dose-dependent acetylation at K310, a site previously proposed to be a predominant target of p300/CBP (Figure 3a,b),40–42 with minimal effects on neighboring K314/315 (Figure S3a). In FKBP12F36V-p53 H1299 cells, we monitored acetylation of the C-terminal domain of p53, specifically at K305, K373, and K382, sites previously suggested to be substrates of p300/CBP,43–46 where strong AceTAG-1-dependent acetylation was also observed (Figure 3c,d). Consistent with our previous observations, we observe apparent acetylation autoinhibition for both targets at higher AceTAG-1 concentrations (Figure 3a, c) and blockade of induced acetylation upon co-treatment of cells with p300/CBP KAT inhibitor A-485 or competing FKBP ligand (Figure 3b,d). In addition, a significant level of p53 acetylation is detectable after ~10 min of AceTAG compound treatment (Figure S3b), similar to the kinetics observed for H3.3. Finally, we note that AceTAG-1-induced acetylation does not appear dependent upon the positioning of the FKBP12F36V tag, as similar acetylation effects were observed with C-terminally tagged p53 (Figure S3c).
Figure 3.

AceTAG targeted acetylation can be applied to multiple POIs. (a) AceTAG-1 induces acetylation of FKBP12F36V-p65/RelA fusion in HeLa cells. FKBP12F36V-RelA-HA RelA−/− HeLa cells were treated with increasing concentrations of AceTAG-1 (2 h) and lysed, and FKBP12F36V-RelA was enriched and blotted with noted antibodies. Shown in the right panel is quantitation of immunoblot signal for p65 K310Ac relative to FKBP12F36V-RelA-HA as the mean ± s.e.m. of n = 2 biologically independent experiments. (b) Immunoblot analysis of controls for AceTAG-mediated acetylation of RelA. Cells were treated with TSA (2 μM) and nicotinamide (10 mM) as a positive control for p65 acetylation42 or co-treated with AceTAG-1 (625 nM) and DMSO, an FKBP12F36V binding ligand (Figure S1c), or the p300/CBP KAT domain inhibitor A485 for 2 h which block AceTAG-1-induced acetylation. (c) AceTAG-1 induces acetylation of FKBP12F36V-p53 fusion in H1299 cells. FKBP12F36V-p53HA H1299 was treated with increasing concentrations of AceTAG-1 (2 h) and lysed, and FKBP12F36V-p53 was enriched and blotted with noted antibodies. Shown in the right panel is quantitation of immunoblot signal for p53 K382Ac relative to FKBP12F36V-p53-HA as the mean ± s.e.m. of n = 3 biologically independent experiments. (d) Immunoblot analysis of controls for AceTAG-mediated acetylation of p53. The results in (b, d) are representative of two independent biological replicates (n = 2). Full images of blots are shown in Figures S9 and S10. Statistical significance (black asterisks) was calculated with unpaired two-tailed Student’s t tests comparing DMSO- to AceTAG-1-treated samples. Statistical significance (red asterisks) of the autoinhibition effect was calculated with unpaired two-tailed Student’s t tests comparing 625 nM AceTAG-1- and 15 or 50 μM AceTAG-1-treated samples. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Selectivity of AceTAG-Induced Acetylation.
To more robustly characterize the site selectivity of induced acetylation of targeted proteins using AceTAG, we monitored AceTAG-1-induced acetylation in cells by quantitative mass spectrometry (MS). In these experiments, H3.3-FKBP12F36V-HA was enriched from stably transfected HeLa cells treated with (1) DMSO, (2) AceTAG-1, or (3) SAHA and trypsinized for protein identification and quantitation by using tandem mass tags (TMT) (Figure S4a). In line with immunoblotting experiments (Figure 2a and Figure S2c), we measured substantial increases in acetylation at K18, K23, and K27 but detected no acetylation at other identified lysines on H3.3 (Figure 4a and Figure S4b). Together, these data suggest that when p300/CBP is recruited to H3.3 in cells by using our AceTAG system, the preferred acetylation sites are K18, K23, and K27. We next sought to evaluate the selectivity of AceTAG-induced acetylation across the human proteome. Treatment of HeLa cells with AceTAG-1 resulted in no observable acetylation changes in well-established p300/CBP substrates, including c-Myc47 and STAT348 (Figure S5a) or broader acetylation perturbation via immunoblot assays (Figure 4b). To assess targeted acetylation selectivity more globally and quantitatively, we performed MS-based acetylproteomic analysis of AceTAG-treated H3.3-FKBP12F36V HeLa cells. Briefly, acetylated peptides from H3.3-FKBP12F36V HeLa cells treated either with (1) DMSO, (2) AceTAG-1, or (3) SAHA were digested, enriched, identified, and quantified by using TMT (Figure S5b). Relative to DMSO, we observed no substantial changes in acetylation of any detected protein in AceTAG-treated cells while SAHA treatment resulted in increases across ~30 proteins (Figure 4c,d, Figure S5c–e, and Table S1). We note that no substantial changes in endogenous histone lysine acetylation was observed upon AceTAG treatment, including H3.3, likely due to the substantially lower H3.3-FKBP12F36V acetylation and expression levels (~25-fold) relative to endogenous histones (Figure S5f). Collectively, these data suggest that AceTAG-1 does not broadly affect p300/CBP KAT activity on other substrates, and AceTAG-1 mediated acetylation is endowed with exquisite selectivity for FKBP12F36V-tagged proteins.
Figure 4.

Chemically induced acetylation selectivity assessment using quantitative proteomics. (a) Fold changes in H3.3 acetylated lysine abundances of H3.3-FKBP12F36V HeLa cells treated with AceTAG-1. Cells were treated with DMSO, AceTAG-1 (625 nM), or SAHA (5 μM) for 2 or 24 h and lysed; H3.3 was enriched, trypsinized, and labeled with isobaric TMT reagents and then subjected to quantitative MS (outlined in Figure S3a and Table S1). Data represent a median of n = 6 biological replicates for AceTAG-1-treated cells and a median of n = 2 biological replicates for SAHA-treated cells. Note that peptides may not have been detected in every replicate but are required to be detected and quantified in at least two biological replicates to be included in this analysis. No acetylation was detected for K36, K56, K79, and K122. Statistical significance was calculated with unpaired two-tailed Student’s t tests comparing DMSO- to AceTAG-1- or SAHA-treated samples. *p < 0.05; ***p < 0.001; ****p < 0.0001. See Figure S4b for representative spectra. (b) AceTAG-1 does not induce broad changes in acetylation as indicated by immunoblot analysis of H3.3-FKBP12F36V HeLa cells treated with increasing concentrations of AceTAG-1 by using a pan acetyl-lysine antibody. The results are representative of three independent biological replicates (n = 3). Full images of blots are shown in Figure S11. (c, d) Volcano plots showing that AceTAG-1 does not induce broad changes in acetylation in the proteome as determined by quantitative MS. H3.3-FKBP12F36V HeLa cells treated with DMSO, AceTAG-1 (600 nM, c), or SAHA (2 μM, d) for 2 or 16 h, lysed, trypsinized, and acetylated peptides enriched and labeled with TMT reagents, and then subjected to quantitative MS (Table S1). The vertical dashed lines correspond to 2-fold change in enrichment relative to DMSO, and the horizontal line corresponds to a P value of 0.05 for statistical significance. Red circles correspond to protein targets with >2-fold change (P < 0.05) relative to DMSO. Each point represents an individual acetylated peptide plotted as a mean of n = 3 biological replicates for DMSO and AceTAG-1 treatments and n = 2 biological replicates for SAHA treatments combined in one TMT 10-plex experiment. An independent replicated TMT experiment is shown in Figure S5. Examples of histone-derived sites with increased acetylation upon SAHA treatment are noted in (d).
CONCLUSION
Here, we describe a method for the selective acetylation of targeted proteins in live cells using heterobifunctional small molecules. We demonstrate that AceTAG molecules can bind to and recruit the lysine acetyltransferase p300/CBP to multiple protein targets genetically fused with FKBP12F36V, effectively inducing acetylation in live cells. We show AceTAG-mediated acetylation is rapid, occurring within minutes of molecule addition, selective for tagged proteins, reversible, and dependent upon KAT enzymatic activity. In addition, AceTAG molecules possess submicromolar efficacy in cells and, as expected, possess characteristics associated with heterobifunctional small molecules, including autoinhibition at increased concentrations both in vitro and in cellulo as well as reduced ternary complex formation with competing FKBP12F36V and p300/CBP-binding ligands.
Despite the consequential roles of protein acetylation, methods to study the effects of acetylation on specific protein targets in cells are limited. Common approaches include genetic or pharmacological ablation of acetylation machinery which affects global substrates, potentially complicating any downstream analyses, or substrate mutagenesis, which lacks dynamic control and can perturb substrate structure or potentially interfere with competing PTMs. Thus, the ability to selectively induce acetylation of targeted proteins overcomes many of these challenges. Toward this end, chemical-based approaches have yielded ligand-directed acetyl-donating reagents for stoichiometric, nonenzymatic transfer of acyl groups to lysine residues on specific proteins like androgen receptor,49 histone H2B,50,51 dihydrofolate reductase,52,53 and phosphoglycerate mutase 1.54 Though capable of targeting endogenous POIs, such reagents are “single use”, allowing for the transfer of one acetyl group per molecule and, further, are limited to proteins with available ligands that bind within proximity to targeted recipient lysine residues. In addition, recent approaches have established that recruitment of p300/CBP to specific DNA sequences with bifunctional molecules can induce transcriptional modulation.28,29,55 However, these approaches rely on DNA-targeting polyamides or sequence targeting using guide RNA’s via engineered Cas9. Our AceTAG system enables the direct targeting of tagged proteins for acetylation, even in the absence of available targeting ligands, in principle enabling the study p300/CBP-mediated acetylation on a wide range of targets.
There are some important points when considering implementation of AceTAG to study protein acetylation. First, we recognize that our current studies have focused on three protein targets. Although current evidence deems p300/CBP as having the broadest substrate scope of all KATs,1 it is unclear how generalizable this approach might be to induce acetylation of other substrates or even neo-substrates. Thus, future attention should be given to exploring a broader subset of targets from diverse functional classes. In addition, even though we conducted a brief exploration of linker length and composition, we do observe linker-dependent ternary complex formation and targeted acetylation (Figure 1c–e and Figure S2b). Like other bifunctional molecules,56–58 we suspect that each protein target will likely have unique linker preferences, necessitating thorough linker optimization studies. In addition, we recognize that the functional effects of acetylation on protein targets are often site-specific, and it is not yet clear if site-selective acetylation can be achieved by using heterobifunctional molecule systems. In our studies, we note consistent acetylation of observable sites on p53 using both C- and N-terminal FKBP12F36V fusion constructs, suggesting that selectivity is largely driven by the substrate recognition of the recruited acetylation machinery. In this regard, future studies will focus on exploration of AceTAG selectivity on additional targets as well as its potential tunability for siteselective acetylation. Furthermore, AceTAG should be fully compatible with CRISPR-knockin technologies, which would benefit various investigations of acetylation in cells. We also note that although a strong “hook effect” is observed during our in vitro experiments (Figure 1c), we observe a somewhat blunted effect in cells while monitoring induced acetylation of H3.3, p65, and p53 (Figures 2a and 3a,c). Given that these targets are endogenous substrates of p300/CBP, we hypothesize there might be naturally occurring recognition that imparts positive cooperativity, effectively delaying autoinhibition, as observed in other heterobifunctional systems.32,59 In addition, as we show in our washout studies (Figure 2d), there is likely competing HDAC activity of chemically induced acetylation, which could complicate equilibrium interpretation. Lastly, the utility of our AceTAG system is limited by the availability and reliability of methods to monitor acetylation. Currently, the majority of proteins with evidence of acetylation lack site-specific antibodies while acetylation detection by MS-based proteomics typically require pan anti-acetyl lysine antibodies for enrichment and detection, which themselves have limited coverage.60 However, as we demonstrate in the case of H3.3, the use of the FKBP12F36V-HA fusion constructs enables the direct enrichment and monitoring of target proteins by quantitative MS, partially alleviating the dependence on antibodies to explore new target areas.
Finally, we believe that AceTAG-based methods can be applied to investigate functional consequences of acetylation in cells. Toward this end, we have shown that targeted acetylation occurs at functional residues on proteins such as H3.3, p65/RelA, and p53, providing evidence that AceTAG is an attractive tool to study the various roles of acetylation in cells. Looking forward, we envision similar strategies can be extended to target endogenous proteins for acetylation, bypassing the requirement of genetic manipulation altogether. Given the manifold roles acetylation can have on protein function, we suspect that the identification of functionally “silent” ligands or binders61 will prove necessary for the generation of such acetylation-targeting chimeric small molecules. In this regard, powerful ligand discovery technologies, including chemoproteomic-based methods62,63 and DNA-encoded libraries (DELs),64 should prove fruitful. In conclusion, we envision AceTAG will not only serve as useful tool to study basic biology but also could enable the exploration of chemically induced acetylation as a therapeutic strategy.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the Scripps NMR, MS, and Genetic Perturbation Screening and Genomics core facilities. We thank Dr. Michael Erb (Scripps Research) and Timothy Bishop (Scripps Research) for their useful discussions. J.M.W. was supported by NIH/NIAID T32 AI007244. The authors gratefully acknowledge Scripps Research for funding.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c07850.
Supplementary figures and tables; experimental details (PDF)
Compiled acetylproteomics data for AceTAG-1 and SAHA treated WT HeLa and H3.3-FKBP12 (F36V) cells (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c07850
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE65 partner repository with the data set identifier PXD027617.
Contributor Information
Wesley W. Wang, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Li-Yun Chen, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Jacob M. Wozniak, Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, United States
Appaso M. Jadhav, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Hayden Anderson, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States.
Taylor E. Malone, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States
Christopher G. Parker, Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458, United States; Department of Chemistry, The Scripps Research Institute, La Jolla, California 92037, United States
REFERENCES
- (1).Narita T; Weinert BT; Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol 2019, 20 (3), 156–174. [DOI] [PubMed] [Google Scholar]
- (2).Sheikh BN; Akhtar A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet 2019, 20 (1), 7–23. [DOI] [PubMed] [Google Scholar]
- (3).Seto E; Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor Perspect. Biol 2014, 6 (4), No. a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Singh BN; Zhang GH; Hwa YL; Li JP; Dowdy SC; Jiang SW Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther 2010, 10 (6), 935–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ropero S; Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol 2007, 1 (1), 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Morris M; Knudsen GM; Maeda S; Trinidad JC; Ioanoviciu A; Burlingame AL; Mucke L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci 2015, 18 (8), 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Saha RN; Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006, 13 (4), 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mazzone R; Zwergel C; Artico M; Taurone S; Ralli M; Greco A; Mai A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenet 2019, 11 (1), 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Forster N; Gallinat S; Jablonska J; Weiss S; Elsasser HP; Lutz W. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J. Immunol 2007, 178 (11), 6941–6948. [DOI] [PubMed] [Google Scholar]
- (10).Iyer A; Fairlie DP; Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol. Cell Biol 2012, 90 (1), 39–46. [DOI] [PubMed] [Google Scholar]
- (11).Guan KL; Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci 2011, 36 (2), 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ali I; Conrad RJ; Verdin E; Ott M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev 2018, 118 (3), 1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schiedel M; Conway SJ Small molecules as tools to study the chemical epigenetics of lysine acetylation. Curr. Opin. Chem. Biol 2018, 45, 166–178. [DOI] [PubMed] [Google Scholar]
- (14).Choudhary C; Kumar C; Gnad F; Nielsen ML; Rehman M; Walther TC; Olsen JV; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325 (5942), 834–840. [DOI] [PubMed] [Google Scholar]
- (15).Weinert BT; Narita T; Satpathy S; Srinivasan B; Hansen BK; Scholz C; Hamilton WB; Zucconi BE; Wang WW; Liu WR; Brickman JM; Kesicki EA; Lai A; Bromberg KD; Cole PA; Choudhary C. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018, 174 (1), 231–244 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tracy TE; Sohn PD; Minami SS; Wang C; Min SW; Li Y; Zhou Y; Le D; Lo I; Ponnusamy R; Cong X; Schilling B; Ellerby LM; Huganir RL; Gan L. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron 2016, 90 (2), 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Su H; Yang F; Wang Q; Shen Q; Huang J; Peng C; Zhang Y; Wan W; Wong CCL; Sun Q; Wang F; Zhou T; Liu W. VPS34 Acetylation Controls Its Lipid Kinase Activity and the Initiation of Canonical and Non-canonical Autophagy. Mol. Cell 2017, 67 (6), 907–921. [DOI] [PubMed] [Google Scholar]
- (18).Stanton BZ; Chory EJ; Crabtree GR Chemically induced proximity in biology and medicine. Science 2018, 359 (6380), No. eaao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gerry CJ; Schreiber SL Unifying principles of bifunctional, proximity-inducing small molecules. Nat. Chem. Biol 2020, 16 (4), 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Burslem GM; Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181 (1), 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yamazoe S; Tom J; Fu Y; Wu W; Zeng L; Sun C; Liu Q; Lin J; Lin K; Fairbrother WJ; Staben ST Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study. J. Med. Chem 2020, 63 (6), 2807–2813. [DOI] [PubMed] [Google Scholar]
- (22).Siriwardena SU; Munkanatta Godage DNP; Shoba VM; Lai S; Shi M; Wu P; Chaudhary SK; Schreiber SL; Choudhary A. Phosphorylation-Inducing Chimeric Small Molecules. J. Am. Chem. Soc 2020, 142 (33), 14052–14057. [DOI] [PubMed] [Google Scholar]
- (23).Crabtree GR; Schreiber SL Three-part inventions: Intracellular signaling and induced proximity. Trends Biochem. Sci 1996, 21 (11), 418–422. [DOI] [PubMed] [Google Scholar]
- (24).Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M; Simpson D; Mendez J; Zimmerman K; Otto P; Vidugiris G; Zhu J; Darzins A; Klaubert DH; Bulleit RF; Wood KV HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 2008, 3 (6), 373–382. [DOI] [PubMed] [Google Scholar]
- (25).Nabet B; Roberts JM; Buckley DL; Paulk J; Dastjerdi S; Yang A; Leggett AL; Erb MA; Lawlor MA; Souza A; Scott TG; Vittori S; Perry JA; Qi J; Winter GE; Wong KK; Gray NS; Bradner JE The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 2018, 14 (5), 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dancy BM; Cole PA Protein lysine acetylation by p300/CBP. Chem. Rev 2015, 115 (6), 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hay DA; Fedorov O; Martin S; Singleton DC; Tallant C; Wells C; Picaud S; Philpott M; Monteiro OP; Rogers CM; Conway SJ; Rooney TP; Tumber A; Yapp C; Filippakopoulos P; Bunnage ME; Muller S; Knapp S; Schofield CJ; Brennan PE Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc 2014, 136 (26), 9308–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Taniguchi J; Feng Y; Pandian GN; Hashiya F; Hidaka T; Hashiya K; Park S; Bando T; Ito S; Sugiyama H. Biomimetic Artificial Epigenetic Code for Targeted Acetylation of Histones. J. Am. Chem. Soc 2018, 140 (23), 7108–7115. [DOI] [PubMed] [Google Scholar]
- (29).Chiarella AM; Butler KV; Gryder BE; Lu DB; Wang TA; Yu XF; Pomella S; Khan J; Jin J; Hathaway NA Dose-dependent activation of gene expression is achieved using CRISPR and small molecules that recruit endogenous chromatin machinery. Nat. Biotechnol 2020, 38 (1), 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yang W; Rozamus LW; Narula S; Rollins CT; Yuan R; Andrade LJ; Ram MK; Phillips TB; van Schravendijk MR; Dalgarno D; Clackson T; Holt DA Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. J. Med. Chem 2000, 43 (6), 1135–1142. [DOI] [PubMed] [Google Scholar]
- (31).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Douglass EF; Miller CJ; Sparer G; Shapiro H; Spiegel DA A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc 2013, 135 (16), 6092–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Conway LP; Jadhav AM; Homan RA; Li W; Rubiano JS; Hawkins R; Lawrence RM; Parker CG Evaluation of fully-functionalized diazirine tags for chemical proteomic applications. Chem Sci. 2021, 12 (22), 7839–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Henry RA; Kuo YM; Andrews AJ Differences in Specificity and Selectivity Between CBP and p300 Acetylation of Histone H3 and H3/H4. Biochemistry 2013, 52 (34), 5746–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Di Cerbo V; Mohn F; Ryan DP; Montellier E; Kacem S; Tropberger P; Kallis E; Holzner M; Hoerner L; Feldmann A; Richter FM; Bannister AJ; Mittler G; Michaelis J; Khochbin S; Feil R; Schuebeler D; Owen-Hughes T; Daujat S; Schneider R. Acetylation of histone H3 at lysine 64 regulates nucleosome dynamics and facilitates transcription. eLife 2014, 3, No. e01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tropberger P; Pott S; Keller C; Kamieniarz-Gdula K; Caron M; Richter F; Li GH; Mittler G; Liu ET; Buhler M; Margueron R; Schneider R. Regulation of Transcription through Acetylation of H3K122 on the Lateral Surface of the Histone Octamer. Cell 2013, 152 (4), 859–872. [DOI] [PubMed] [Google Scholar]
- (37).Lasko LM; Jakob CG; Edalji RP; Qiu W; Montgomery D; Digiammarino EL; Hansen TM; Risi RM; Frey R; Manaves V; Shaw B; Algire M; Hessler P; Lam LT; Uziel T; Faivre E; Ferguson D; Buchanan FG; Martin RL; Torrent M; Chiang GG; Karukurichi K; Langston JW; Weinert BT; Choudhary C; de Vries P; Kluge AF; Patane MA; Van Drie JH; Wang C; McElligott D; Kesicki E; Marmorstein R; Sun C; Cole PA; Rosenberg SH; Michaelides MR; Lai A; Bromberg KD Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550 (7674), 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chen L; Fischle W; Verdin E; Greene WC Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293 (5535), 1653–1657. [DOI] [PubMed] [Google Scholar]
- (39).Reed SM; Quelle DE p53 Acetylation: Regulation and Consequences. Cancers 2015, 7 (1), 30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chen LF; Williams SA; Mu Y; Nakano H; Duerr JM; Buckbinder L; Greene WC NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol 2005, 25 (18), 7966–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li H; Wittwer T; Weber A; Schneider H; Moreno R; Maine GN; Kracht M; Schmitz ML; Burstein E. Regulation of NF-kappa B activity by competition between RelA acetylation and ubiquitination. Oncogene 2012, 31 (5), 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Buerki C; Rothgiesser KM; Valovka T; Owen HR; Rehrauer H; Fey M; Lane WS; Hottiger MO Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008, 36 (5), 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gu W; Roeder RG Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90 (4), 595–606. [DOI] [PubMed] [Google Scholar]
- (44).Liu L; Scolnick DM; Trievel RC; Zhang HB; Marmorstein R; Halazonetis TD; Berger SL p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol 1999, 19 (2), 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sakaguchi K; Herrera JE; Saito S; Miki T; Bustin M; Vassilev A; Anderson CW; Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12 (18), 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang YH; Tsay YG; Tan BCM; Lo WY; Lee SC Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem 2003, 278 (28), 25568–25576. [DOI] [PubMed] [Google Scholar]
- (47).Faiola F; Liu X; Lo S; Pan S; Zhang K; Lymar E; Farina A; Martinez E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol. Cell. Biol 2005, 25 (23), 10220–10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhuang S. Regulation of STAT signaling by acetylation. Cell. Signalling 2013, 25 (9), 1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhang Y; Mantravadi PK; Jobbagy S; Bao W; Koh JT Antagonizing the Androgen Receptor with a Biomimetic Acyltransferase. ACS Chem. Biol 2016, 11 (10), 2797–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fujiwara Y; Yamanashi Y; Fujimura A; Sato Y; Kujirai T; Kurumizaka H; Kimura H; Yamatsugu K; Kawashima SA; Kanai M. Live-cell epigenome manipulation by synthetic histone acetylation catalyst system. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (4), No. e2019554118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Amamoto Y; Aoi Y; Nagashima N; Suto H; Yoshidome D; Arimura Y; Osakabe A; Kato D; Kurumizaka H; Kawashima SA; Yamatsugu K; Kanai M. Synthetic Posttranslational Modifications: Chemical Catalyst-Driven Regioselective Histone Acylation of Native Chromatin. J. Am. Chem. Soc 2017, 139 (22), 7568–7576. [DOI] [PubMed] [Google Scholar]
- (52).Hamajima W; Fujimura A; Fujiwara Y; Yamatsugu K; Kawashima SA; Kanai M. Site-Selective Synthetic Acylation of a Target Protein in Living Cells Promoted by a Chemical Catalyst/Donor System. ACS Chem. Biol 2019, 14 (6), 1102–1109. [DOI] [PubMed] [Google Scholar]
- (53).Mizumoto S; Xi SQ; Fujiwara Y; Kawashima SA; Yamatsugu K; Kanai M. Hydroxamic Acid-Piperidine Conjugate is an Activated Catalyst for Lysine Acetylation under Physiological Conditions. Chem. - Asian J 2020, 15 (6), 833–839. [DOI] [PubMed] [Google Scholar]
- (54).Zhang X; Jiang L; Huang K; Fang C; Li J; Yang J; Li H; Ruan X; Wang P; Mo M; Wu P; Xu Y; Peng C; Uesugi M; Ye D; Yu FX; Zhou L. Site-Selective Phosphoglycerate Mutase 1 Acetylation by a Small Molecule. ACS Chem. Biol 2020, 15 (3), 632–639. [DOI] [PubMed] [Google Scholar]
- (55).Chen TJ; Gao D; Zhang RS; Zeng GH; Yan H; Lim EJ; Liang FS Chemically Controlled Epigenome Editing through an Inducible dCas9 System. J. Am. Chem. Soc 2017, 139 (33), 11337–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hughes SJ; Ciulli A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017, 61 (5), 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bemis TA; La Clair JJ; Burkart MD Unraveling the Role of Linker Design in Proteolysis Targeting Chimeras. J. Med. Chem 2021, 64 (12), 8042–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).McEnaney PJ; Parker CG; Zhang AX; Spiegel DA Antibody-Recruiting Molecules: An Emerging Paradigm for Engaging Immune Function in Treating Human Disease. ACS Chem. Biol 2012, 7 (7), 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Casement R; Bond A; Craigon C; Ciulli A. Mechanistic and Structural Features of PROTAC Ternary Complexes. Methods Mol. Biol 2021, 2365, 79–113. [DOI] [PubMed] [Google Scholar]
- (60).Svinkina T; Gu HB; Silva JC; Mertins P; Qiao J; Fereshetian S; Jaffe JD; Kuhn E; Udeshi ND; Carr SA Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Antiacetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol. Cell Proteomics 2015, 14 (9), 2429–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Schreiber SL A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines. Isr. J. Chem 2019, 59 (1–2), 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Parker CG; Galmozzi A; Wang YJ; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534 (7608), 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Goodnow RA; Dumelin CE; Keefe AD DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discovery 2017, 16 (2), 131–147. [DOI] [PubMed] [Google Scholar]
- (65).Perez-Riverol Y; Csordas A; Bai JW; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.