

Abstract
The Keap1-Nrf2-ARE signaling pathway is an attractive therapeutic target for the prevention and treatment of oxidative stress-associated diseases by activating the cellular expression of cytoprotective enzymes and proteins. Small molecule inhibitors can directly disrupt the Keap1-Nrf2 protein-protein interaction (PPI), resulting in elevated levels of Nrf2 protein and subsequent stimulation of related antioxidant responses. Previously, we found that 1,4-bis(arylsulfonamido)benzene or naphthalene-N,N′-diacetic acid derivatives with an ether type C2-substituent on the benzene or naphthalene core exhibited potent inhibitory activities with IC50’S in the submicromolar or nanomolar range. We here describe a more detailed structure-activity relationship study around the C2 substituents containing various polar linkers shedding new insight on their binding interactions with the Keap1 Kelch domain. The key observation from our findings is that the substituents at the C2-position of the benzene or naphthalene scaffold impact their inhibitory potencies in biochemical assays as well as activities in cell culture. The biochemical FP and TR-FRET assays revealed that the naphthalene derivatives 17b and 18 with an additional carboxylate at the C2 were the most active inhibitors against Keap1-Nrf2 PPI. In the cell-based assay, the two compounds were shown to be potent Nrf2 activators of the transcription of the Nrf2-dependent genes, such as HMOX2, GSTM3, and NQO1.
Keywords: Keap1, Nrf2, Keap1-Nrf2 interaction, Protein-protein interaction Inhibitor, Oxidative stress, Structure-activity relationship, Structural diversity, C2 substituents, TR-FRET
Graphical abstract

Introduction
The human body is equipped with an anti-oxidative defense system to protect cells from high levels of oxidative or electrophilic stress [1]. Nevertheless, sustained oxidative stress causes chronic inflammation, which can lead to the development of a variety of human diseases and conditions including aging, cancer, neurodegenerative and cardiovascular diseases [2–4]. A promising preventive and therapeutic target for these oxidative stress-related chronic diseases and conditions is the Keap1-Nrf2-ARE signaling pathway [3], which plays a critical role in keeping cellular homeostasis by stimulating the expression of cellular cytoprotective proteins and enzymes, such as superoxide dismutase (SOD), NAD(P)H:quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST), and heme oxygenase-1 (HO-1) [5–7]. The signaling pathway is composed of three important cellular components, nuclear factor erythroid 2-related factor 2 (Nrf2), Kelch-like ECH-associated protein 1 (Keap1), and antioxidant response element (ARE) [3].
Nrf2 is the central transcription factor that regulates ARE in the promoter regulatory regions, controlling the transcription and expression of detoxification and antioxidant genes [8]. Keap1 plays a key role in acting as a sensor of chemical and oxidative stresses and regulating Nrf2 activity [9]. Under normal conditions, Keap1 serves as an adaptor protein for the Cullin3 (Cul3)-based ubiquitin E3 ligase complex and simultaneously binds to Nrf2 in the cytoplasm [10]. The close proximity between the E3 ligase complex and Nrf2 promotes the ubiquitination of Nrf2, which subsequently subjects the Nrf2 protein to degradation by the 26S proteasome [11]. Upon exposure to oxidative stress, the cysteine residues in the Keap1 protein are likely modified or oxidized by reactive oxygen or nitrogen species, resulting in conformational changes of the Keap1-Nrf2-Cul3 complex via one of the three possible mechanisms such as Cul3 dissociation [12], hinge and latch [13], or conformation cycling model [14]. These conformational changes lead to the inhibition of the ubiquitin E3 ligase activity, which consequently enables newly synthesized Nrf2 to accumulate, translocate into the nucleus, and activate the expression of the ARE dependent genes [15, 16]. Accordingly, the up-regulation of Nrf2 is regarded as a potential preventive and/or therapeutic strategy for a number of chronic diseases and conditions caused by oxidative stress.
A variety of electrophilic Nrf2 inducers are in different stages of clinical development with some receiving market approval [3, 17]. These include dimethyl fumarate (DMF), bardoxolone methyl (CDDO-Me), and omaveloxolone [18–20]. The dimethyl fumarate (Tecfidera®), approved by the US FDA in 2013, shows efficacy for the treatment of relapsing multiple sclerosis (MS). The alkylation agent is believed to react with cysteine thiols of Keap1, activating the Nrf2-ARE pathway [17, 21]. However, nonspecific S-alkylation with cysteine thiols in other proteins and enzymes could cause severe adverse effects [22]. The triterpenoid CDDO-methyl ester and omaveloxolone are potent Nrf2 activators, which result from the reaction between its highly active α,β-unsaturated α-cyano ketone and Cys151 of Keap1 [23]. Bardoxolone methyl first entered phase II clinical trial for the treatment of type 2 diabetes mellitus and advanced kidney disease. Unfortunately, the phase III clinical study of CDDO-Me was terminated due to cardiovascular toxicity issues [19, 24]. FDA also recently rejected bardoxolone’s treatment of Alport syndrome, a rare genetic disorder with progressive kidney function deterioration [25]. However, omaveloxolone, a structural analog of bardoxolone, was recently approved by FDA for the treatment of Friedreich’s ataxia, a rare genetic neurodegenerative disorder with impaired Nrf2 signaling [20].
Direct disruption of the Keap1-Nrf2 protein-protein interaction (PPI) is an alternative approach for the discovery of Nrf2 inducers. Small molecule inhibitors non-covalently bind to the Keap1 Kelch domain where ETGE and DLG motifs of the Nrf2 Neh2 domain interacts, preventing its ubiquitation and its subsequent degradation, thus increasing the level of the Nrf2 protein in cells [13, 26]. Different from covalent cysteine modifiers, these non-electrophilic compounds most likely would have the advantages of better target specificity, less off-target effects, and lower toxicity [27]. In 2013, our group first reported LH601A (1) as a direct inhibitor of Keap1-Nrf2 PPI (Figure 1) [28]. To date, there have been several series of Keap1-Nrf2 PPI small molecule inhibitors reported, including our tetrahydroisoquinoline (THIQ, 1) [28], 3-phenylpropanoic acid (2) [29], 1,2-xylylenediamine (3) [30], 9H-fluoren-9-one (4) [31], 1,4-diaminonaphthalene (5a-e) [32–36], and 1,4-diamino-2-benzyloxybenzene (6a-b) [35, 37], as shown in Figure 1. In particular, optimization studies on the 1,4-diaminonaphthalene scaffold have been actively conducted by several research groups over the years, leading to the discovery of highly potent Keap1-Nrf2 PPI inhibitors [38]. Very recently, we reported a series of 1,4-bis(arylsulfonamido)benzene or naphthalene derivatives with C2 substituents (e.g., 6a-b and 5d-e) that exhibited excellent Keap1-Nrf2 inhibitory activities in the submicromolar or nanomolar range [35, 36].
Fig. 1.
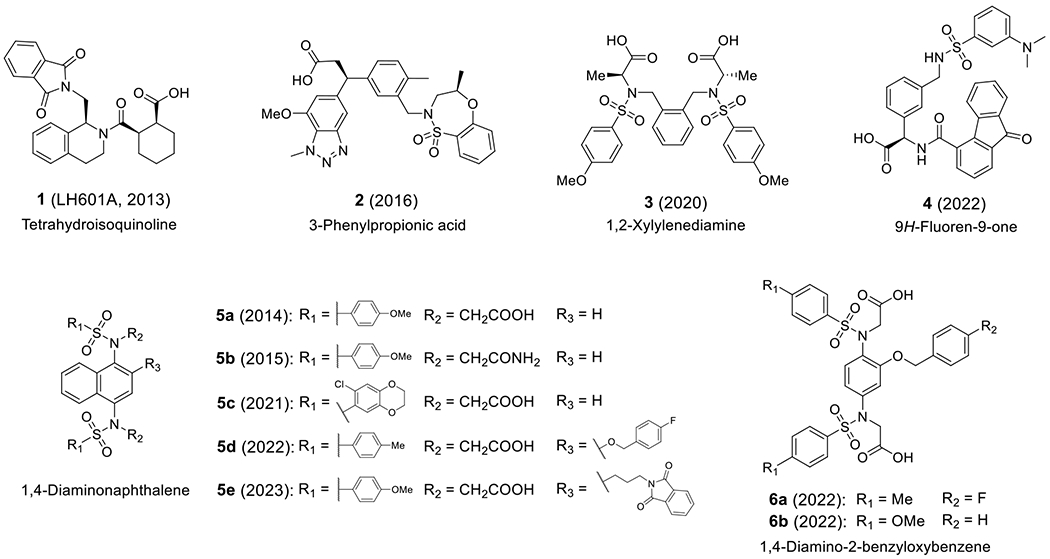
Representative direct inhibitors of Keap1-Nrf2 protein-protein interaction.
In this study, we describe a more detailed structure-activity relationship (SAR) analysis of C2-substituted 1,4-bis(arylsulfonamido)benzene or naphthalene-N,N’-diacetic acid analogs, by replacing the ether moiety of the substituents with various polar linkers. It provides a better understanding of binding interactions in the Keap1-Nrf2 binding interface and improves the structural diversity of the C2-substituented benzene or naphthalene series. The key finding is that the linker-modified 1,4-bis(arylsulfonamido)benzene or naphthalene-N,N’-diacetic acids displayed an improvement in activity using either a fluorescence polarization (FP) or a time-resolved fluorescence energy transfer (TR-FRET) assay. Among them, compounds 17b and 18 possessing an acetate group at C2 are the most potent inhibitors against Keap1-Nrf2 PPI and were shown to activate such Nrf2-regulated genes as NQO1, GSTM3, and HMOX2.
Results and Discussion
Design strategy.
Our previous SAR studies demonstrated that 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid series with the ether type C2-substituents have potent inhibitory activities in the submicromolar range, whereas the lack of this substituent led to a significant decrease in potency [32, 35]. However, several optimization strategies for the C2-substituents—including modification of the oxygen-containing linker length, addition of substituents to the para-position of the benzyl ring, and replacement of the benzyl ring with a naphthalene or biphenyl ring—did not result in any significant effect on the inhibitory potency against Keap1-Nrf2 protein-protein interaction (PPI) [35]. Furthermore, although the benzyloxy group as a C2-substituent connected to the benzene core gave rise to the increased activity in both FP and TR-FRET assays, the fragments did not exhibit any noticeable binding interaction in the central P3 subpocket of Keap1 Kelch domain [35]. Therefore, we turned our attention to optimize the O-linked C2-substituents in a two-step process as illustrated in Figure 2 based on the chemical structure of compound 6b, one of the most potent 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid derivatives with an IC50 value of 786 nM in the FP assay (Figure 2) [35]. First, C2-substituents with different polar functional groups were introduced to 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid, in an effort to enhance the binding affinity to Keap1 protein in the central P3 pocket via polar interaction (e.g. hydrogen bond) with uncharged polar or charged amino acids located in the polar P1 and P2 subpockets (Figure 4). Specifically, the ether linker was replaced with a sulfonic acid, an ester, or an ether group containing acetic acid, acetamide, or polar heterocycles (morpholine, phthalimide, and tetrazole). Furthermore, considering the previous SAR studies, the benzene cores with the optimized C2-substituents were changed to naphthalene rings to potentially further enhance the inhibitory activity for achieving better π-cation interactions with arginine residues in the central pocket.
Fig. 2.

Optimization of ether-linked benzene analog 6b over two steps, C2-substituent and core modifications
Fig. 4.

Docking results of selected inhibitors with the Keap1 Kelch domain (PDB code: 4XMB); (A) 5a, (B) 8g, (C) 18. The partial charge of Keap1 protein was represented in blue for polar surface areas and red for lipophilic surface areas. Hydrogen bonds were indicated as red dashed lines, while π-π stacking or π-cation interactions were shown as yellow lines.
Chemistry.
The ester 7a and sulfonate 7b were prepared according to the synthetic procedure shown in Scheme 1. The benzyl protected analogs 20a-b were synthesized in three steps, including a nucleophilic aromatic substitution of 3-fluoro-4-nitroaniline with benzyl alcohol, reduction of the nitro group, and sulfonamide formation. The TFA-mediated removal of the benzyl protecting group followed by esterification of phenolic alcohol with benzoyl chloride or benzenesulfonyl chloride gave 21a-b, which were alkylated with t-butyl bromoacetate, and then deprotected under acidic conditions to yield the target products 7a-b. As shown in Scheme 1, other 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid derivatives 8a-h containing various polar C2-substituents were prepared from the key intermediates 24a-b generated through the alkylation of 20a-b with t-butyl 2-bromoacetate and benzyl deprotection of 23a-b. Addition of sulfonyl chloride or substituted alkyl bromide reagents followed by the removal of t-butyl protecting groups afforded the final products 8a-h.
Scheme 1. Synthesis of benzene core series 7a-b and 8a-h with various O-linked fragmentsa.

aReagents and conditions: (a) benzyl alcohol, NaH, THF, 0 °C to rt, 6 h, 54%; (b) Fe, AcOH, H2O, 80 °C, 30 min; (c) R1-substituted benzenesulfonyl chloride, pyridine, DCM, rt, overnight, 24-47% (over 2 steps); (d) 10% Pd/C, H2, MeOH, rt, 4 h-overnight, 19-72%; (e) benzoyl chloride or benzenesulfonyl chloride, TEA, DCM, rt, 2 h, 36-94%; (f) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 65-88%; (g) TFA, DCM, rt, 3 h-overnight, 33%-quant.; (h) sulfonyl chloride reagents, TEA, DCM, rt, 3 h-overnight, 25-74% (for 25a-c and 25e); (i) substituted alkyl bromide, K2CO3, DMF, rt, 6 h-overnight, 28-87% (for 25d and 25f-h).
Additionally, the oxygen-linked benzene analogs 9a-d were prepared from the diethyl acetate analogue 27, as outlined in Scheme 2. Treatment of 20a with ethyl bromoacetate and the removal of benzyl protecting group gave key intermediate 27. The phenolic hydroxyl group was subjected to nucleophilic substitution with several aliphatic halides, followed by saponification to produce the target compounds 9a-d. 1-Methylpiperazine analog 10 was also prepared from the key intermediate 27. Alkylation of 27 with t-butyl 2-bromoacetate and deprotection of t-butyl ester in 29 with TFA followed by an amide coupling reaction with 1-methylpiperazine using EDCI and HOBt gave intermediate 30. Base-catalyzed hydrolysis of 30 in the presence of NaOH yielded the final target compound 10.
Scheme 2. Preparation of O-linked benzene core series 9a-d and 10 from key compound 27a.

aReagents and conditions: (a) ethyl bromoacetate, K2CO3, DMF, rt, overnight, 31%; (b) TFA, rt, overnight, 38%; (c) aliphatic halides, K2CO3, DMF, rt, 3 h-overnight, 31-60%; (d) NaOH, MeOH, H2O, 60 °C, 5 h-overnight, 35-65%; (e) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 87%; (f) TFA, DCM, rt, 1 h, 98%; (g) N-methyl piperazine, EDCI, HOBt, TEA, DCM, rt, overnight, 74%.
A general procedure to synthesize reverse ester analogues 11a-b is summarized in Scheme 3. Under the ester coupling conditions of EDCI and DMAP, 5-amino-2-nitrobenzoic acid was treated with phenol or benzyl alcohol to form esters 31a-b, respectively. Nitro reduction and subsequent sulfonamide formation provided disulfonamides 32a-b, which underwent alkylation with t-butyl 2-bromoacetate followed by TFA deprotection of the t-butyl ester to afford the target compounds 11a-b.
Scheme 3. Preparation of benzene core series 11a-b containing an ester linkera.

aReagents and conditions: (a) phenol or benzyl alcohol, EDCI, DMAP, DCM, rt, overnight, 12-27%; (b) Fe, AcOH, H2O, 60 °C, 30 min; (c) 4-methoxybenzenesulfonyl chloride, pyridine, DCM, rt, overnight, 36-64% (over 2 steps); (d) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 90-95%; (e) TFA, DCM, rt, 4 h-overnight, 78-84%.
The preparation of acetate 12 and C-2 methyl sulfonate 13 was accomplished by the synthetic approach shown in Scheme 4. First, the carboxylic acid of 5-amino-2-nitrobenzoic acid was reduced into an alcoholic hydroxyl group. The resulting (5-amino-2-nitrophenyl)methanol and commercially available 5-amino-2-nitrophenol were protected with TBDMS group to provide 34a-b. Iron or palladium catalyzed nitro reduction followed by N-sulfonylation produced 35a-b. Alkylation of TBDMS protected phenolic analog 35a with t-butyl bromoacetate allowed for facile displacement of the TBDMS protective group to give 36 possessing three acetates. Carboxylic acid derivative 12 was obtained from the t-butyl deprotection of the acetate analogue 36. The aliphatic alcohol 38 was synthesized by alkylation of 35b with t-butyl bromoacetate and TBDMS deprotection. Treatment of 38 with benzenesulfonyl chloride followed by t-butyl deprotection yielded the final compound 13.
Scheme 4. Preparation of O-linked benzene core series 12 and 13 from O-TBDMS-protected intermediatesa.

aReagents and conditions: (a) 1M BH3 in THF, THF, 60 °C, 2 h, quant.; (b) TBDMSCl, imidazole, DMF, rt, overnight, 89-96%; (c) Fe, AcOH, H2O, 80 °C, 30 min (for 35a); (d) 10% Pd/C, H2, MeOH, rt, 2 h (for 35b); (e) 4-methoxybenzenesulfonyl chloride, pyridine, DCM, rt, overnight, 52-76% (over 2 steps); (f) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 37%-quant.; (g) TBAF, THF, rt, 3 h, 36%; (h) benzenesulfonyl chloride, TEA, DCM, rt, 4 h, 69%; (i) TFA, DCM, rt, 4 h-overnight, 71-82%.
For the synthesis of the methylated 14a-b and tetrazole 15 depicted in Scheme 5, the intermediate 24b was alkylated with methyl (R) or (S)-2-bromopropionate to yield stereospecifically the (S) and (R)-methylated analogs (40a-b), respectively, with inversion of configuration. Hydrolysis of the methyl ester under basic conditions and subsequent deprotection of the t-butyl ester groups with TFA afforded the target compounds 14a-b. In a similar manner, compound 41 was prepared by alkylation of 24b using bromoacetonitrile, which was subjected to tetrazole formation with NaN3 and then deprotection with TFA to give the final compound 15.
Scheme 5. Preparation of O-linked phenyl ring series 14a-b and 15 from key compound 24ba.
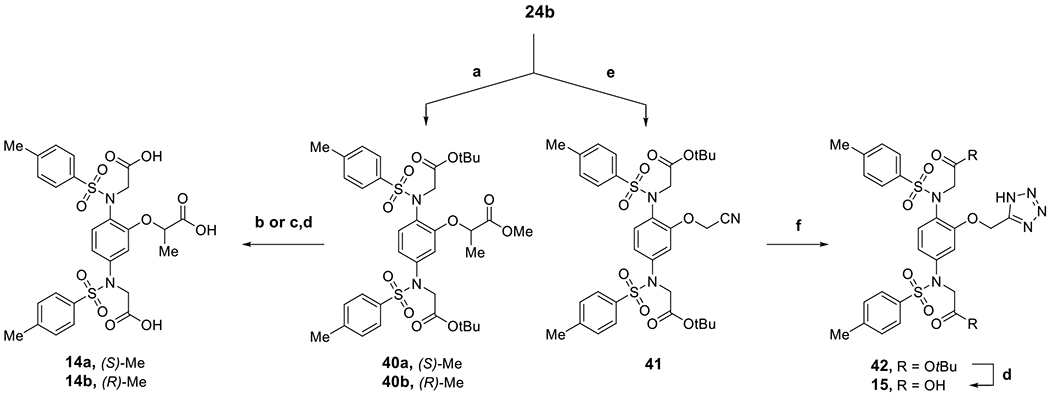
aReagents and conditions: (a) methyl (R) or (S)-2-bromopropionate, K2CO3, DMF, rt, overnight, 39%-quant.; (b) NaOH, MeOH, H2O, 60 °C, 4 h, 34% (for 14a); (c) LiOH, MeOH, H2O, 60 °C, 4 h (for 14b); (d) TFA, DCM, rt, 3 h-overnight, 33%-quant.; (e) 2-bromoacetonitrile, K2CO3, DMF, rt, 4 h, quant.%; (f) NaN3, AcOH, DMF, 90 °C, 6h, 70%.
Compound 16 with nitrogen-containing linker was readily synthesized as shown in Scheme 6. Treatment of 2-nitrobenzene-1,4-diamine with 4-toluenebenzenesulfonyl chloride followed by alkylation with t-butyl 2-bromoacetate produced 44. Reduction of a nitro group allowed for the introduction of an extra t-butyl 2-bromoacetate to provide 45. Formation of the target product 16 was carried out by the cleavage of three t-butyl groups using TFA.
Scheme 6. Synthesis of N-linked benzene core derivative 16a.

aReagents and conditions: (a) 4-toluenesulfonyl chloride, pyridine, 80 °C, overnight, 78%; (b) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 41-95%; (c) 10% Pd/C, H2, MeOH, rt, overnight, 17%; (d) TFA, DCM, rt, 4 h, 51%.
The synthesis of carbon-linked analogs 17a-b is outlined in Scheme 7. 2-Iodo-4-nitronaphthalen-1-amine 46 was synthesized from the commercially available 4-nitronaphthalen-1-amine in the presence of potassium iodide and potassium iodate [39]. Heck coupling of 2-iodo substituted compounds along with t-butyl acrylate in the presence of Pd(OAc)2 and P(o-tolyl)3 provided 47a-b [40]. Reduction of both alkene double bond and nitro group with Pd/C afforded 1,4-diamine intermediates, which were subsequently treated with 4-methylbenzenesulfonyl chloride to give 48a-b. Alkylation of 48a-b with t-butyl 2-bromoacetate followed by removal of three t-butyl groups with TFA yielded carboxylate derivatives 17a-b. The O-linked naphthalene 18 was also prepared from 4-nitronaphthalen-1-amine, as shown in Scheme 7. 2-Bromo-4-nitronaphthalen-1-amine 50, synthesized via selective bromination of 4-nitronaphthalen-1-amine with NBS [41]. underwent copper-mediated coupling reaction to give O-benzyl-protected 51 [42]. Iron-catalyzed nitro reduction of 51 and subsequent N-sulfonylation afforded 52, which was debenzylated by palladium catalyzed hydrogenolysis to introduce three acetate fragments. The resulting phenolic alcohol intermediate was converted to the acetate compound 18 by alkylation and TFA deprotection of the three t-butyl groups.
Scheme 7. Synthesis of benzene analog 17a with a carbon linker and naphthalene core analogs (17b and 18).

aReagents and conditions: (a) KI, KIO3, con.HCl, MeOH, H2O, rt, overnight, 93%; (b) t-butyl acrylate, Pd(OAc)2, P(o-tolyl)3, TEA, ACN, 90 °C, overnight, 84%-quant.; (c) 10% Pd/C, H2, MeOH, rt, overnight, 44% (for 52); (d) 4-toluenebenzenesulfonyl chloride, pyridine, DCM, rt, overnight, 6-55% (over 2 steps); (e) t-butyl bromoacetate, K2CO3, DMF, rt, overnight, 75-96%; (f) TFA, DCM, rt, 4 h, 57-86%; (g) NBS, NH4OAc, ACN, rt, 10 min, 96%; (h) benzyl alcohol, CuI, 8-hydroxyquinoline, K3PO4, 130 °C, overnight, 10%; (i) Fe, AcOH, H2O. 60 °C, 30 min.
Evaluation of inhibitory potency using the FP and TR-FRET assays and structure-activity relationship (SAR) exploration.
We investigated the ability of 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid series with C2-substituents to interrupt the protein-protein interaction (PPI) between the Kelch domain of Keap1 and Nrf2 ETGE motif using our FP assay [43]. Initially, the synthesized compounds were screened at three concentrations of 50, 5, and 0.5 μM, and only the more active ones with more than 50% inhibition at 5 μM were further evaluated to determine their IC50 values. Given the lower limit of the IC50 in the FP assay being 50 nM, highly active compounds with an IC50 value of less than 0.2 μM in the FP assay were also evaluated using the TR-FRET assay to differentiate their potency down to the nanomolar range. For easier comparisons, we converted the IC50 values obtained from the FP and TR-FRET assays to inhibition constant (Ki).
As illustrated in Table 1, linker (R) modification was performed utilizing an FP assay in order to examine whether it contributes to improving the potency against Keap1-Nrf2 PPI. The ester-linked analog (7a) led to a reduction in Keap1-Nrf2 PPI inhibitory activity by 2-fold (IC50 = 1780 nM, Ki = 351 nM), whereas sulfonic acid derivative 7b displayed approximately 2-fold increase in the inhibitory activity (IC50 = 439 nM, Ki = 74.2 nM), as compared to 6b (IC50 = 786 nM, Ki = 146 nM). For the series of sulfonate analogues, benzyl (8a), 2,4,6-trimethylphenyl (8b), and biphenyl (8c) fragments were well tolerated with IC50 values of 325-476 nM and Ki values of 50.7-82.0 nM. In addition, the insertion of a carbonyl group between the benzylic carbon atom and phenyl ring (9a) showed similar activity to 6b with an IC50 value of 593 nM (Ki = 106 nM), and even the bulky 4-phenylpiperidine (8d) retained good potency (IC50 = 760 nM, Ki = 141 nM), indicating the existence of space sufficient to accommodate a large variety of C2-substituents. Surprisingly, the amide 9b designed by replacing the phenyl ring of 9a with aniline led to a dramatic loss of activity by a 13-fold change in IC50’s (IC50 = 7680 vs 593 nM), while the amide analogue 9c possessing morpholine exhibited promising submicromolar potency with an IC50 value of 399 nM (Ki = 66.1 nM). Thus, 9c was further modified by the removal of carbonyl group (9d) and the replacement with 1-methylpiperazine (10). Both 9d and 10 were found to have less inhibitory activity than 9c (IC50 = 2500 and 2260 nM vs 399 nM). It suggests that the carbonyl group plays an important role in binding to the polar residues of Keap1 protein (e.g. arginine and asparagine), whereas the tertiary amine that can be positively charged is unfavorable for binding to Keap1. Intriguingly, the carboxylic acid derivative 12 that is available for hydrogen bonds resulted in an ~2-fold increase in potency relative to 9c (IC50 = 226 vs 399 nM, Ki = 30.2 vs 66.1 nM,). Consequently, the sulfonic acid linkers (8a-c), ether-linked 4-acetylmorhpoline (9c) and acetic acid (12) were found to have more potent inhibitory activities against Keap1-Nrf2 PPI than the simple ether linker (6b) previously reported by our group. On the other hand, reverse esters (11a-b) and methyl sulfonate (13) linkers exhibited a remarkable decrease in potency (11a-b: IC50 = 3850 and 1610 nM, Ki = 780 and 316 nM, 13: 46% inhibition at 5 μM), confirming that changing the position of the oxygen atom in the linker can greatly affect the inhibition of Keap1-Nrf2 PPI.
Table 1.
Inhibitory activity of phenyl ring derivatives with various O-linked fragmentsa

|
Inhibitory potency was evaluated by FP assay that was performed in triplicates. ND: not determined.
IC50 is indicated as an average value of three replicates ± SEM (standard error of the mean).
Ki is calculated from the IC50 values in FP assay.
From our previous SAR study [35], we observed a preference for methyl group over methoxy substituent at the 4-position of the benzene rings on sulfonamide moieties. Therefore, the methoxy groups of active compounds (8b, 9c and 12) selected from ether-linker modified derivatives in Table 1 were replaced by methyl substituents (Table 2). The results obtained from our FP assay indicate that the differences between the methyl and methoxy substituents in the sulfonate (8b and 8e) or morpholine (9c and 8f) analogs were insignificant as their IC50’s were within 2-fold. However, the carboxylic acid analogue 8g with 4-methylbenzenesulfonamide moieties were 2- and 5-fold more potent in terms of IC50 and Ki, respectively, as compared to 12 with methoxy groups (IC50 = 111 vs 226 nM, Ki = 6.4 vs 30.2 nM). In other words, the compound 8g displayed excellent inhibitory activity with an IC50 of 111 nM in the FP assay, thereby prompting further evaluation in the TR-FRET assay. As a result, 8g resulted in nanomolar inhibitory potency with an IC50 of 54.3 nM (Ki = 23.9 nM). Then, we focused our attention on optimizing the most potent compound 8g, as shown in Table 2. The phthalimide group (8h), designed with an aim to provide flexibility by increasing the linker length of carbonyl group, was 2-fold less potent in an IC50 value than 8g (IC50 = 228 vs 111 nM). Addition of a methyl group at the α-position of the acetic acid that exists as the C2-substituent of 8g produced two possible enantiomers 14a and 14b bearing (S)- and (R)-methyl groups, respectively, which were synthesized stereospecifically. The biological results in TR-FRET assay indicated that the potencies of both 14a and 14b were almost equivalent to that of 8g in the nanomolar range (IC50 = 54.3-92.0 nM, Ki = 23.9-41.4 nM), suggesting that the methyl group did not have much effect on binding interactions with protein residues. Additionally, the introduction of tetrazole as a bioisoster of carboxylic acid (15) led to 4-fold decrease in activity relative to 8g (IC50 = 408 vs 111 nM in our FP assay). Replacement of the oxygen with divalent bioisosteres, such as nitrogen and methylene (16 and 17a), led to significantly decreased potency by 16- and 5-fold based on IC50 values in our FP assay (IC50 =1780 and 565 nM vs 111 nM), suggesting that the changes of bond angle, conformation and electronegativity may be associated with biological activity.
Table 2.
Inhibitory activity of benzene or naphthalene series with various fragmentsa

|
Inhibitory potency was estimated by both FP (used 100 nM Keap1 and 10 nM FITC-9mer Nrf2 peptide amide as a probe) and TR-FRET assays (used 5 nM Keap1 and 25 nM FITC-9mer Nrf2 peptide amide) that were performed in triplicates. 100 nM Keap1 and ND: not determined.
A is a phenyl ring scaffold and B is a naphthalene scaffold.
IC50 is indicated as an average value of three replicates ± SEM (standard error of the mean).
Ki is calculated from the IC50 values in FP or TR-FRET.
In order to enhance potency, we attempted to replace the benzene cores of 17a and 8g possessing acetic acid moieties on the C2-substituent with a naphthalene core, designing 1,4-bis(arylsulfonamido)naphthalene-N,N’-diacetic acid derivatives 17b and 18, respectively. When compared to 17a and 8g, the naphthalene analogs 17b and 18 displayed significantly increased inhibitory potencies in FP and/or TR-FRET assays down to the nanomolar range (IC50 in FP: 69.3 and 88.6 nM, Ki in FP: ~1.0 and 1.9 nM, IC50 in TR-FRET: 22.3 and 19.0 nM, Ki in TR-FRET: 9.1 and 7.6 nM), as shown in Table 2 and Figure 3. In addition, the naphthalene analog 5a in the absence of C2-substituent is even more potent than the most active compound 8g among the benzene core series, suggesting that the naphthalene core is superior in potency to the benzene core. Furthermore, we investigated the role of C2-substituent connected to either the benzene or naphthalene core moiety in terms of inhibitory activity. Importantly, substituents at the 2-position of benzene core offer greater differences in inhibition effect against Keap1-Nrf2 PPI than those of naphthalene core, depending on the chemical structure of the C2-substituent. For instance, ether-containing acetic acid 8g is 5-fold more potent in the IC50 value of FP assay than the propionic acid 17a with a methylene replacing the ether O in 8g. However, the naphthalene core analogs 17b and 18 with the same C2-substituents to 17a and 8g, respectively, showed almost the same activities in the nanomolar range using our TR-FRET assay, regardless of which substituent is at C2. Furthermore, the absence of a C2-substituent in 5a exhibited a slight decrease in inhibitory activity relative to 17b and 18, as shown by our TR-FRET assay which is capable of clearly differentiating the potency of nanomolar active inhibitors (IC50 = 19.0 vs 28.5 nM and Ki = 7.6 vs 12.0 nM for 18 vs 5a).
Fig. 3.

Dose-response curves of small molecule inhibitors 5a, 8g, 17b and 18 obtained from FP assay (A) and TR-FRET assay (B). (C) IC50 and Ki values of 5a, 8g, 17b and 18 tested in TR-FRET and FP assays. The IC50 is indicated as an average value of three replicates ± SEM (standard error of the mean).
Molecular docking analysis.
In an effort to investigate a correlation between the binding modes and the inhibitory potencies of 1,4-bis(arylsulfonamido)benzene or naphthalene-N,N’-diacetic acids, we carried out molecular docking studies with active benzene 8g and naphthalene 18 analogs on the basis of the co-crystal structure of Keap1 Kelch domain with 1,4-diaminonaphthalene derivative 5b (PDB code: 4XMB) [33]. Interestingly, the docking simulation of 8g suggests that the acetic acid fragment at the 2-position of the benzene core moiety is oriented toward the opposite direction of the central tunnel region for the hydrogen bonding interactions with the side chains of key residues Arg380 and Arg415 in the P2 subpocket (Figure 4B). The same binding position of the acetic acid substituent of 8g is also observed in the benzyloxy group of 6b, as shown in the co-crystal structure of 6b binding to the Keap1 Kelch domain (PDB code: 6HWS) [37]. Furthermore, similar to 8g, the acetic acid fragment of the naphthalene derivative 18 occupies toward the front of the central P3 subpocket through hydrogen bonds with polar residues Arg380 and Arg415 in Figure 4C.
When compared to the potent naphthalene analogue 5a with an IC50 value of 28.5 nM in the TR-FRET assay, both compounds 8g and 18 show a similar binding mode by occupying five pockets (P1-P5). The key interactions are found to be multiple hydrogen bonds of two acetates groups with Arg483 and Arg415 and a π-cation interaction with the guanidine group of Arg415, leading to nanomolar activities (IC50 = 54.3 and 19.0 nM in TR-FRET assay). It is obvious that the ether-linked acetic acid of the benzene analog 8g contributes to increasing the binding affinity and potency through its favorable polar interactions in the Keap1 cavity, in comparison to the other benzene core-based inhibitors. Unfortunately, the benzene core (8g) is still less potent than the naphthalene-based compounds (17b and 18) with C2-fragments and even 5a without the C2-substituent. We hypothesized that the crucial differences of potency between the two cores might be attributed to high binding affinities through the hydrophobic interaction of the fused ring in the central P3 region and the electrostatic interaction of an acetate group with Asn414 in P2 pocket.
Effects of compounds 8g, 17b, and 18 with C2-substituents on the transcriptional activation of Nrf2-driven genes.
NCM460D cells were treated 8g, 17b, and 18 to evaluate whether these compounds can activate Nrf2 via direct inhibition of Keap1-Nrf2 PPI under the same protocol we used previously [34–36]. Consistent with the results of the biochemical assays, the benzene core scaffold (8g) displayed less transcriptional induction of the Nrf2 target genes NQO1, GSTM3 and HMOX1 than the naphthalene derivatives 17b and 18, as shown in Figures 5. As expected, the two compounds 17b and 18 significantly enhanced the mRNA levels of NQO1 at a similar level to reference 5a, reaching 8- and 15-folds, respectively.
Fig. 5.

Cellular activity of potent Keap1-Nrf2 PPI inhibitors 8g, 17b and 18, evaluated by measuring the expressions of Nrf2 target genes GSTM3, NQO1, or HMOX1 in NCM460D cells after 24 h incubation at 1, 10 and 100 μM. Compound 5a (1, 10 and 100 μM) and CDDO (10 and 100 nM) were used as positive controls, while 0.1 % DMSO was used as a negative control. The endogenous control GAPDH was utilized to normalize the expression of the target genes.
CONCLUSION
In summary, we focused on further optimizing the C2-substituents of 1,4-bis(arylsulfonamido)benzene or naphthalene-N,N’-diacetic acid series, starting from compound 6b with benzyloxy group that was previously published by our group. The structure-activity relationship (SAR) and computational modeling studies led to two interesting findings through the C2-substitutent and core modifications. First, the naphthalene core appears to be more favorable for inhibitory activities in both FP and TR-FRET assays than the benzene core. On the basis of the docking results of 8g and 18, it is likely that the additional hydrogen bonding of acetate group with Asn414 and the hydrophobic interaction of the naphthalene core (18) might result in the improved activities by 3-fold in TR-FRET assay, as compared to 8g with the benzene core. Second, the benzene core analogs made big differences in potency ranging from 111 to 7680 nM in the FP assay depending on the structure of substituents at the 2-position of the benzene. For example, the C2-acetate group of 8g forms polar hydrogen bonding interactions with Arg380 and Arg415, leading to high binding affinity to and strong inhibitory potency against the Keap1 Kelch domain. In the case of 1,4-bis(arylsulfonamido)benzene-N,N’-diacetic acid derivatives with benzyloxy group (6a-b) or without C2-substituent [32], their activities are remarkably less potent than 8g, presumably due to the lack of key interactions of substituents. On the other hand, activities between the naphthalene series exhibited insignificant changes in the TR-FRET assay regardless of their C2-substituents. When compared to 17b with propionic acid fragment and known compound 5a without a C2-substituent, the most potent naphthalene-based inhibitor 18 was shown to have comparable activity by showing less than 2-fold difference in Ki ranging from 7.6 to 12.0 nM. In other words, the acetic acid fragment of 18 made little difference to inhibitory potency through hydrogen bonds with Arg380 and Arg415, different from the benzene core series. The similar trend was observed from RT-PCR results for the Nrf2 downstream genes. Compound 8g bearing the benzene core induced relatively low expressions of Nrf2 target genes, but the naphthalene analogs 17b and 18 exhibited similarly high effects in elevating the mRNA levels of GSTM3, HMO1 and NQO1.
When compared to C2-substituted naphthalene derivatives 5d and 5e previously reported by our group [35, 36], the most active compound 18 in our studies was shown to be a comparable as direct inhibitors of Keap1-Nrf2 PPI. In the TR-FRET assay, the inhibitory activity of 18 is as potent as 5d, even though it is slightly less potent than 5e (IC50 = 19.0, 14.2, and 2.5 nM for 18, 5d, and 5e, respectively). Interestingly, the naphthalene analog 18 with an additional carboxylate group stimulates the induction of Nrf2 downstream genes NQO1, GSTM3 and HMOX1 at 10 and 100 μM in a similar level to 5d with 4-fluorobenzyl fragment and positive control 5a lacking a C2-substituent. Furthermore, compound 18 containing an ionizable carboxylate fragment at C2-position more significantly elevated the mRNA levels of the Nrf2 target gene NQO1 at 100 μM in NCM460D cells, as compared to 5e with phthalimidopropyl group [36]. It means that the additional carboxylic acid moiety of 18 is likely to has less influence on cellular permeability, resulting in comparable cellular activities to the known compounds 5d and 5e. Collectively, C2-substituents with polar functional group enabled extensive structural modifications to provide improved structural diversity during the late-stage lead optimization process while maintaining both the nanomolar inhibitory and favorable cellular activities.
EXPERIMENTAL SECTION
Synthetic procedure and characterization of target compounds.
2,2′-((2-(Benzoyloxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (7a).
To a solution of 22a (28 mg, 0.12 mmol) in dichloromethane (0.5 mL) was added trifluoroacetic acid (0.2 mL) at room temperature. The reaction mixture was stirred at room temperature for 4 hours. After the reaction was completed, the crude mixture was added to water and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulting solid was dissolved with the minimum amount of dichloromethane, solidified from hexane, and then filtered to give the product as a white solid (22 mg, 92%); 1H NMR (400 MHz, Methanol-d4) δ 7.91 (d, J = 8.4 Hz, 2H), 7.70-7.64 (m, 3H), 7.53-7.48 (m, 5H), 7.23 (d, J = 2.4 Hz, 1H), 7.17 (dd, J = 8.4, 2.4 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.43 (s, 2H), 4.31 (s, 2H), 3.86 (s, 3H), 3.80 (s, 3H); 13C NMR (100 MHz, Methanol-d4) δ 172.1, 165.2, 165.0, 164.8, 149.5, 142.5, 135.1, 134.5, 132.4, 132.0, 131.3, 131.1, 130.7, 130.0, 129.7, 126.6, 124.5, 115.3, 56.3, 56.2, 53.0; LC/MS (ESI) m/z 683.1 [M - H]−; HRMS (ESI) m/z calcd for C31H27N2O12S2 [M - H]− 683.1011; found 683.1017. Rt: 4.11 min, UPLC purity: 95.2%.
2,2′-((2-((Phenylsulfonyl)oxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (7b).
Prepared as described in the experimental procedure of 7a from intermediate 22b to obtain the title compound as a white solid (27 mg, 41%); 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 7.6 Hz, 2H), 7.72 (t, J = 7.6 Hz, 1H), 7.63-7.55 (m, 6H), 7.50 (d, J = 8.8 Hz, 1H), 7.14 (d, J = 2.4 Hz, 1H), 7.04 (dd, J = 8.8, 2.4 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.41 (s, 2H), 4.36 (s, 2H), 3.87 (s, 3H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.7, 163.0, 162.7, 146.1, 141.0, 135.3, 134.7, 133.7, 130.5, 129.9, 129.5, 129.4, 127.8, 124.4, 119.5, 114.5, 114.1, 55.8, 55.6, 51.6, 51.5; LC/MS (ESI) m/z 719.0 [M - H]−; HRMS (ESI) m/z calcd for C30H27N2O13S3 [M - H]− 719.0681; found 719.0688. Rt: 4.09 min, UPLC purity: 99.7%.
2,2′-((2-((Benzylsulfonyl)oxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (8a).
Prepared as described in the experimental procedure of 7a from intermediate 25a to obtain the title compound as a white solid (8 mg, quantitative); 1H NMR (400 MHz, Methanol-d4) δ 7.61 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.8 Hz, 1H), 7.39-7.38 (m, 5H), 7.17 (dd, J = 8.8, 2.4 Hz, 1H), 7.03 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 2.4 Hz, 1H), 4.66 (s, 2H), 4.32 (s, 2H), 4.22 (s, 2H), 3.88 (s, 3H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.9, 173.6, 163.7, 146.8, 141.1, 133.1, 131.2, 130.7, 130.5, 130.3, 130.0, 129.6, 129.4, 129.2, 126.9, 126.7, 121.2, 114.6, 114.3, 58.0, 55.8, 55.8, 51.8; LC/MS (ESI) m/z 735.1 [M + H]+; HRMS (ESI) m/z calcd for C31H31N2O13S3 [M + H]+ 735.0983; found 735.0997. Rt: 4.17 min, UPLC purity: 98.5%.
2,2′-((2-((Mesitylsulfonyl)oxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (8b).
Prepared as described in the experimental procedure of 7a from intermediate 25b to obtain the title compound as a white solid (14 mg, quantitative); 1H NMR (400 MHz, Methanol-d4) δ 7.74 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.8 Hz, 2H), 7.46 (d, J = 8.8 Hz, 2H), 7.22 (dd, J = 8.8, 2.4 Hz, 1H), 7.05 (s, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.20 (d, J = 2.4 Hz, 1H), 4.42 (s, 2H), 4.15 (s, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 2.38 (s, 3H), 2.35 (s, 6H); 13C NMR (100 MHz, Methanol-d4) δ 172.4, 171.3, 165.0, 164.8, 148.5, 146.3, 142.9, 141.1, 136.1, 133.3, 132.5, 132.5, 132.3, 131.1, 130.9, 130.4, 127.6, 121.0, 115.3, 115.2, 56.4, 56.3, 53.5, 52.4, 22.8, 21.3; LC/MS (ESI) m/z 761.1 [M - H]−; HRMS (ESI) m/z calcd for C33H33N2O13S3 [M - H]− 761.1150; found 761.1159. Rt: 4.46 min, UPLC purity: 95.4%.
2,2′-((2-(([1,1′-Biphenyl]-4-ylsulfonyl)oxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))-diacetic acid (8c).
Prepared as described in the experimental procedure of 7a from intermediate 25c to obtain the title compound as a white solid (26 mg, 86%); 1H NMR (400 MHz, DMSO-d6) δ 12.96 (brs, 2H, CO2H), 7.97 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 7.6 Hz, 2H), 7.63-7.58 (m, 3H), 7.56 (d, J = 7.6 Hz, 2H), 7.52-7.48 (m, 3H), 7.24 (d, J = 2.4 Hz, 1H), 7.15 (dd, J = 8.8, 2.4 Hz, 1H), 7.07 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 4.43 (s, 2H), 4.10 (s, 2H), 3.82 (s, 3H), 3.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.7, 163.0, 162.7, 146.7, 146.1, 141.0, 137.9, 133.7, 133.3, 130.6, 129.5, 129.4, 129.2, 129.1, 128.5, 128.0, 127.3, 124.4, 119.5, 114.5, 114.1, 55.8, 55.6, 51.6, 51.5; LC/MS (ESI) m/z 795.1 [M - H]−; HRMS (ESI) m/z calcd for C36H31N2O13S3 [M - H]− 795.0994; found 795.1002. Rt: 4.59 min, UPLC purity: 99.7%.
2,2′-((2-(2-Oxo-2-(4-phenylpiperidin-1-yl)ethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)-azanediyl))diacetic acid (8d).
Prepared as described in the experimental procedure of 7a from intermediate 25d to obtain the title compound as a pale yellow solid (26 mg, 92%); 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.8 Hz, 2H), 7.33-7.29 (m, 2H), 7.23-7.17 (m, 4H), 6.91-6.87 (m, 4H), 6.80 (s, 1H), 6.51 (d, J = 8.4 Hz, 1H), 4.65-4.61 (m, 1H), 4.42 (s, 4H), 4.34 (s, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.74-3.71 (m, 1H), 3.20-3.14 (m, 1H), 2.78-2.70 (m, 2H), 1.95-1.88 (m, 2H), 1.73-1.62 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.8, 172.3, 163.5, 163.2, 154.9, 144.8, 141.1, 133.4, 131.4, 130.1, 129.9, 128.8, 127.2, 126.9, 126.8, 120.4, 114.5, 114.3, 114.0, 66.5, 55.8, 55.8, 52.0, 51.0, 45.8, 43.5, 42.6, 33.6, 32.8, 22.8; LC/MS (ESI) m/z 780.2 [M - H]−; HRMS (ESI) m/z calcd for C37H38N3O12S2 [M - H]− 780.1902; found 780.1913. Rt: 4.33 min, UPLC purity: 96.6%.
2,2′-((2-((Mesitylsulfonyl)oxy)-1,4-phenylene)bis(tosylazanediyl))diacetic acid (8e).
Prepared as described in the experimental procedure of 7a from intermediate 25e to obtain the title compound as a white solid (38 mg, quantitative); 1H NMR (400 MHz, DMSO-d6) δ 12.97 (brs, 2H, CO2H), 7.59 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 7.6 Hz, 2H), 7.42 (d, J = 7.6 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.19 (dd, J = 8.8, 1.6 Hz, 1H), 7.13 (s, 2H), 6.34 (d, J = 8.8 Hz, 1H), 4.28 (s, 2H), 4.22 (s, 2H), 2.50 (s, 3H), 2.44 (s, 3H), 2.36 (s, 3H), 2.29 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 169.9, 169.3, 146.3, 144.6, 144.1, 143.5, 140.9, 139.1, 136.3, 134.6, 134.4, 131.9, 130.6, 129.7, 129.4, 127.1, 127.0, 125.2, 118.8, 51.9, 51.2, 22.0, 21.6, 21.1, 21.0; LC/MS (ESI) m/z 729.1 [M - H]−; HRMS (ESI) m/z calcd for C33H33N2O11S3 [M - H]− 729.1252; found 729.1263. Rt: 4.71 min, UPLC purity: 94.8%.
2,2′-((2-(2-Morpholino-2-oxoethoxy)-1,4-phenylene)bis(tosylazanediyl))diacetic acid (8f).
Prepared as described in the experimental procedure of 7a from intermediate 25f to obtain the title compound as a white solid (39 mg, quantitative); 1H NMR (400 MHz, DMSO-d6) δ 7.52 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.32-7.29 (m, 3H), 6.79 (d, J = 8.4 Hz, 1H), 6.68 (s, 1H), 4.39 (s, 2H), 4.38 (s, 4H), 3.56-3.54 (m, 4H), 3.39-3.37 (m, 2H), 3.29-3.27 (m, 2H), 2.50 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.4, 169.7, 164.9, 154.0, 143.7, 143.1, 140.7, 136.8, 135.5, 132.9, 129.6, 129.2, 127.3, 127.1, 125.6, 119.1, 111.6, 65.9, 65.2, 51.7, 50.5, 44.4, 41.5, 21.0, 21.0; LC/MS (ESI) m/z 676.3 [M + H]+; HRMS (ESI) m/z calcd for C30H34N3O11S2 [M + H]+ 676.1629; found 676.1643. Rt: 3.78 min, UPLC purity: 97.8%.
2,2′-((2-(Carboxymethoxy)-1,4-phenylene)bis(tosylazanediyl))diacetic acid (8g).
Prepared as described in the experimental procedure of 7a from intermediate 25g to obtain the title compound as a white solid (29 mg, 87%); 1H NMR (400 MHz, DMSO-d6) S 12.88 (brs, 2H, CO2H), 7.52 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.36-7.34 (m, 1H), 7.28 (d, J = 8.0 Hz, 2H), 6.85 (dd, J = 8.4, 2.0 Hz, 1H), 6.57 (d, J = 2.0 Hz, 1H), 4.37 (s, 2H), 4.31 (s, 2H), 4.00 (s, 2H), 2.39 (s, 3H), 2.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.3, 169.8, 168.9, 153.5, 143.7, 143.1, 140.8, 136.6, 135.4, 133.2, 129.6, 129.2, 127.3, 127.0, 125.5, 120.0, 111.5, 64.3, 51.7, 50.5, 21.0, 21.0; LC/MS (ESI) m/z 605.1 [M - H]−; HRMS (ESI) m/z calcd for C26H25N2O11S2 [M - H]− 605.0905; found 605.0915. Rt: 3.66 min, UPLC purity: 95.3%.
2,2′-((2-(2-(1,3-Dioxoisoindolin-2-yl)ethoxy)-1,4-phenylene)bis(tosylazanediyl)) diacetic acid (8h).
Prepared as described in the experimental procedure of 7a from intermediate 25h to obtain the title compound as a pale yellow solid (20 mg, 83%); 1H NMR (400 MHz, CDCl3) δ 7.86-7.84 (m, 2H), 7.73-7.70 (m, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.4 Hz, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H), 6.78 (s, 1H), 6.54 (d, J = 8.0 Hz, 1H), 4.29 (s, 2H), 4.24 (s, 2H), 3.73-3.71 (m, 2H), 3.66-3.65 (m, 2H), 2.38 (s, 3H), 2.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.6, 169.9, 167.7, 154.0, 143.8, 143.2, 140.8, 136.6, 135.3, 134.5, 133.2, 131.7, 129.6, 129.0, 127.4, 126.9, 125.2, 123.1, 119.8, 111.4, 64.6, 51.9, 50.7, 36.3, 21.0; LC/MS (ESI) m/z 722.3 [M + H]+; HRMS (ESI) m/z calcd for C34H32N3O11S2 [M + H]+ 722.1473; found 722.1479. Rt: 4.28 min, UPLC purity: 97.2%.
2,2′-((2-(2-Oxo-2-phenylethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (9a).
To a solution of 28a (27 mg, 0.04 mmol) in methanol (0.9 mL) was added sodium hydroxide (4 mg, 0.11 mmol) in water (0.3 mL) at room temperature. The reaction mixture was stirred at 60 °C for overnight. After the reaction was completed, the crude mixture was concentrated under reduced pressure, dissolved with water, and then washed with dichloromethane. The aqueous solution was acidified with 6 N HCl until the pH reached around 1 and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulting solid was dissolved with the minimum amount of dichloromethane, solidified from hexane, and then filtered to give the product as a yellow solid (14 mg, 56%); 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, J = 8.8 Hz, 2H), 7.72 (t, J = 7.6 Hz, 1H), 7.60-7.53 (m, 4H), 7.48 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 6.92-6.88 (m, 3H), 6.66 (s, 1H), 4.96 (s, 2H), 4.37 (s, 2H), 4.35 (s, 2H), 3.77 (s, 3H), 3.62 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 192.8, 170.4, 169.7, 162.7, 162.3, 153.7, 140.8, 134.0, 133.9, 133.2, 131.2, 129.7, 129.5, 129.3, 128.8, 127.6, 125.7, 120.4, 114.2, 113.8, 111.7, 70.1, 55.6, 55.3, 51.7, 50.6; LC/MS (ESI) m/z 697.1 [M - H]−; HRMS (ESI) m/z calcd for C32H29N2O12S2 [M - H]− 697.1167; found 697.1174. Rt: 4.06 min, UPLC purity: 94.6%.
2,2′-((2-(2-Oxo-2-(phenylamino)ethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl)) diacetic acid (9b).
Prepared as described in the experimental procedure of 9a from intermediate 28b to obtain the title compound as a white solid (15 mg, 56%); 1H NMR (400 MHz, CDCl3) δ 9.24 (s, 1H, NH), 7.68-7.66 (m, 4H), 7.84 (d, J = 8.8 Hz, 2H), 7.31 (dd, J = 7.6, 7.6 Hz, 2H), 7.17 (t, J = 7.6 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.88-6.84 (m, 2H), 6.76 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.0 Hz, 1H), 4.79 (s, 2H), 4.53 (s, 2H), 4.37 (s, 2H), 3.84 (s, 3H), 3.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 169.9, 165.4, 162.8, 162.6, 154.1, 141.1, 138.1, 132.6, 130.9, 129.8, 129.6, 129.3, 128.8, 126.0, 123.8, 119.6, 114.3, 114.0, 111.6, 66.8, 55.7, 55.5, 51.7, 51.1; LC/MS (ESI) m/z 712.1 [M - H]−; HRMS (ESI) m/z calcd for C32H30N3O12S2 [M - H]− 712.1276; found 712.1284. Rt: 4.10 min, UPLC purity: 96.3%.
2,2′-((2-(2-Morpholino-2-oxoethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl)) diacetic acid (9c).
Prepared as described in the experimental procedure of 9a from intermediate 28c to obtain the title compound as a white solid (10 mg, 37%); 1H NMR (400 MHz, Methanol-d4) δ 7.61-7.48 (m, 4H), 7.40 (d, J = 8.4 Hz, 1H), 7.03-6.94 (m, 4H), 6.82 (d, J = 6.8 Hz, 1H), 6.74-6.65 (m, 1H), 4.45 (s, 2H), 3.88-3.87 (m, 2H), 3.98 (s, 2H), 3.88 (s, 3H), 3.87 (s, 3H), 3.68-7.65 (m, 4H), 3.54-3.52 (m, 2H), 3.42-3.40 (m, 2H); 13C NMR (100 MHz, Methanol-d4) δ 164.9, 164.7, 164.6, 155.9, 142.9, 134.7, 132.8, 131.2, 131.2, 130.9, 130.9, 128.1, 121.0, 115.2, 115.2, 115.0, 114.9, 67.6, 67.0, 56.3, 56.3, 56.2, 53.4, 52.3, 46.4, 43.4; LC/MS (ESI) m/z 708.2 [M + H]+; HRMS (ESI) m/z calcd for C30H34N3O13S2 [M + H]+ 708.1528; found 708.1539. Rt: 3.57 min, UPLC purity: 98.5%.
2,2′-((2-(2-Morpholinoethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (9d).
Prepared as described in the experimental procedure of 9a from intermediate 28d to obtain the title compound as a white solid (18 mg, 65%); 1H NMR (400 MHz, DMSO-d6) δ 7.60 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 6.85-6.81 (m, 1H), 6.63 (s, 1H), 4.40 (s, 2H), 4.26 (s, 2H), 3.84 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 3.61-3.57 (m, 10H); 13C NMR (100 MHz, DMSO-d6) δ 170.5, 169.9, 162.9, 162.7, 154.2, 141.1, 132.4, 131.3, 129.9, 129.7, 129.5, 126.1, 119.6, 114.5, 114.2, 112.0, 69.8, 63.3, 63.0, 55.9, 55.8, 54.8, 51.8, 51.1; LC/MS (ESI) m/z 692.2 [M - H]−; HRMS (ESI) m/z calcd for C30H34N3O12S2 [M - H]− 692.1589; found 692.1598. Rt: 3.27 min, UPLC purity: 99.1%.
2,2′-((2-(2-(4-Methylpiperazin-1-yl)-2-oxoethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)-azanediyl))diacetic acid (10).
Prepared as described in the experimental procedure of 9a from intermediate 30 to obtain the title compound as a pale yellow solid (13 mg, 35%); 1H NMR (400 MHz, DMSO-d6) δ 10.9 (brs, 2H, CO2H), 7.55 (d, J = 8.8 Hz, 4H), 7.27 (d, J = 8.0 Hz, 1H), 7.05 (d, J = 9.2 Hz, 2H), 7.01 (d, J = 9.2 Hz, 2H), 6.74-6.72 (m, 2H), 4.75-4.55 (m, 6H), 4.38-4.37 (m, 4H), 3.85 (s, 3H), 3.84 (s, 3H), 2.77-2.75 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ 170.8, 170.1, 165.5, 163.0, 162.7, 154.2, 141.0, 133.1, 131.4, 129.8, 129.8, 129.6, 127.3, 126.0, 119.2, 114.5, 114.2, 65.4, 56.0, 55.9, 52.5, 52.4, 51.9, 50.7, 42.6, 41.3, 41.1; LC/MS (ESI) m/z 719.2 [M - H]−; HRMS (ESI) m/z calcd for C31H35N4O12S2 [M - H]− 719.1698; found 719.1707. Rt: 3.10 min, UPLC purity: 98.2%.
2,2′-((2-((Phenylsulfonyl)oxy)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (11a).
Prepared as described in the experimental procedure of 9a from intermediate 33a to obtain the title compound as a white solid (65 mg, 78%); 1H NMR (400 MHz, DMSO-d6) δ 12.89 (brs, 2H, CO2H), 7.80 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.49-7.45 (m, 4H), 7.42 (dd, J = 8.8, 2.4 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 7.6 Hz, 2H), 7.10 (d, J = 9.2 Hz, 2H), 7.03 (d, J = 9.2 Hz, 2H), 4.49 (s, 4H), 3.82 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.3, 169.8, 162.9, 162.8, 162.7, 150.3, 140.2, 137.0, 133.2, 131.2, 130.8, 130.6, 129.9, 129.6, 129.6, 129.5, 129.4, 126.0, 121.5, 114.5, 114.1, 55.7, 55.6, 52.8, 51.5; LC/MS (ESI) m/z 683.1 [M - H]−; HRMS (ESI) m/z calcd for C31H27N2O12S2 [M - H]− 683.1011; found 683.1017. Rt: 4.20 min, UPLC purity: 95.0%.
2,2′-((2-((Benzyloxy)carbonyl)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))diacetic acid (11b).
Prepared as described in the experimental procedure of 9a from intermediate 33b to obtain the title compound as a white solid (29 mg, 84%); 1H NMR (400 MHz, CDCl3) δ 8.11-7.97 (m, 3H), 7.66 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 8.8 Hz, 2H), 7.43-7.33 (m, 6H), 6.90 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.29 (s, 2H), 4.41 (s, 2H), 4.26 (s, 2H), 3.83 (s, 3H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.8, 171.3, 166.7, 163.9, 163.7, 140.5, 137.9, 134.9, 132.8, 132.2, 130.3, 130.1, 129.7, 128.9, 128.9, 128.6, 128.3, 114.5, 114.5, 68.5, 55.9, 55.8, 53.3, 51.8; LC/MS (ESI) m/z 697.1 [M - H]−; HRMS (ESI) m/z calcd for C32H29N2O12S2 [M - H]− 697.1167; found 697.1174. Rt: 4.28 min, UPLC purity: 95.5%.
2,2′-((2-(Carboxymethoxy)-1,4-phenylene)bis(((4-methoxyphenyl)sidfonyl)azanediyl))diacetic acid (12).
Prepared as described in the experimental procedure of 7a from intermediate 36 to obtain the title compound as a white solid (42 mg, 71%); 1H NMR (400 MHz, DMSO-d6) δ 12.83 (brs, 2H, CO2H), 7.58 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H), 7.35 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 1H), 6.60 (s, 1H), 4.36 (s, 2H), 4.31 (s, 2H), 4.09 (s, 2H), 3.84 (s, 3H), 3.83 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.4, 169.8, 168.9, 162.8, 162.5, 153.5, 140.9, 133.2, 131.1, 129.8, 129.5, 129.2, 125.6, 119.9, 114.3, 113.8, 111.3, 64.4, 55.7, 55.6, 51.7, 50.5; LC/MS (ESI) m/z 637.1 [M - H]−; HRMS (ESI) m/z calcd for C26H25N2O13S2 [M - H]− 637.0804; found 637.0813. Rt: 3.46 min, UPLC purity: 94.5%.
2,2′-((2-(((Phenylsulfonyl)oxy)methyl)-1,4-phenylene)bis(((4-methoxyphenyl)sulfonyl)azanediyl))-diacetic acid (13).
Prepared as described in the experimental procedure of 7a from intermediate 39 to obtain the title compound as a white solid (41 mg, 82%); 1H NMR (400 MHz, Methanol-d4) δ 7.94 (d, J = 7.6 Hz, 2H), 7.74 (t, J = 7.6 Hz, 1H), 7.64 (dd, J = 7.6, 7.6 Hz, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 2.4 Hz, 1H), 7.08 (dd, J = 8.8, 2.4 Hz, 1H), 7.03 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 1H), 5.57-5.54 (m, 1H), 5.16-5.13 (m, 1H), 4.41-4.41 (m, 1H), 4.37-4.34 (m, 2H), 4.03-3.99 (m, 1H), 3.88 (s, 3H), 3.88 (s, 3H); 13C NMR (100 MHz, Methanol-d4) δ 171.9, 171.3, 165.2, 165.1, 142.1, 138.2, 137.2, 135.3, 131.3, 131.2, 131.0, 130.7, 130.3, 130.0, 129.8, 129.3, 129.1, 128.7, 115.4, 115.2, 69.1, 56.3, 53.9, 52.9; LC/MS (ESI) m/z 733.0 [M - H]−; HRMS (ESI) m/z calcd for C31H29N2O13S3 [M - H]− 733.0837; found 733.0847. Rt: 3.91 min, UPLC purity: 96.4%.
(S)-2-(2,5-Bis((N-(carboxymethyl)-4-methylphenyl)sulfonamido)phenoxy)propanoic acid (14a).
To a solution of 40a (54 mg, 0.072 mmol) in methanol (0.3 mL) was added sodium hydroxide (4 mg, 0.11 mmol) in water (0.1 mL) at room temperature. The reaction mixture was stirred at 60 °C for 4 hours. After the reaction was completed, the crude mixture was concentrated under reduced pressure, dissolved with water, and then washed with dichloromethane. The aqueous solution was acidified with 6 N HCl until the pH reached around 1 and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatograph (0-10% methanol/dichloromethane) to give (S)-2-(2,5-bis((N-(2-(t-butoxy)-2-oxoethyl)-4-methyl phenyl)-sulfonamido)phenoxy)propanoic acid as a colorless oil (18 mg, 34%); 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.0 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.28-7.20 (m, 5H), 6.78 (s, 1H), 6.59 (d, J = 8.4 Hz, 1H), 4.54-4.53 (m, 1H), 4.39-4.34 (m, 1H), 4.27-4.17 (m, 3H), 2.40 (s, 3H), 2.40 (s, 3H), 1.40 (s, 9H), 1.38 (s, 9H), 1.31 (d, J = 5.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.6, 167.6, 153.9, 144.2, 143.7, 141.4, 136.9, 135.6, 133.5, 129.7, 129.4, 127.9, 127.8, 127.2, 119.8, 113.2, 82.6, 82.3, 72.3, 53.1, 52.1, 28.12, 28.05, 21.7, 21.6, 17.8; LC/MS (ESI) m/z 731.3 [M - H]−.
Prepared as described in the experimental procedure of 7a from (S)-2-(2,5-bis((N-(2-(t-butoxy)-2-oxoethyl)-4-methylphenyl)sulfonamido)phenoxy)propanoic acid to obtain the title compound as a pale yellow solid (8 mg, 54%); 1H NMR (400 MHz, CDCl3) δ 8.90 (brs, 1H, CO2H), 8.89 (brs, 1H, CO2H), 8.68 (brs, 1H, CO2H), 7.53 (d, J = 8.0 Hz, 2H), 7.50-7.47 (m, 2H), 7.41 (d, J = 8.4 Hz, 1H), 7.28-7.26 (m, 2H), 7.24 (d, J = 8.4 Hz, 2H), 6.78 (d, J = 12.4 Hz, 1H), 6.43-6.39 (m, 1H), 4.56-4.44 (m, 2H), 4.40-4.39 (m, 1H), 4.10-4.01 (m, 1H), 3.70-3.60 (m, 1H), 2.42 (s, 6H), 1.17 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.8, 173.8, 153.2, 144.5, 143.9, 140.6, 136.8, 135.4, 134.8, 129.9, 129.5, 127.7, 127.7, 126.6, 120.0, 114.7, 71.3, 52.1, 50.1, 21.7, 21.7, 17.7; LC/MS (ESI) m/z 619.1 [M - H]−; HRMS (ESI) m/z calcd for C27H27N2O11S2 [M - H]− 619.1062; found 619.1070. Rt: 3.81 min, UPLC purity: 95.2%.
(R)-2-(2,5-Bis((N-(carboxymethyl)-4-methylphenyl)sulfonamido)phenoxy)propanoic acid (14b).
To a solution of 40b (35 mg, 0.047 mmol) in methanol (0.3 mL) was added lithium hydroxide (1.7 mg, 0.070 mmol) in water (0.1 mL) at room temperature. The reaction mixture was stirred at 50 °C for 4 hours. After the reaction was completed, the crude mixture was concentrated under reduced pressure, dissolved with water, and then washed with dichloromethane. The aqueous solution was acidified with 6 N HCl until the pH reached around 1 and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give (R)-2-(2,5-bis((N-(2-(t-butoxy)-2-oxoethyl)-4-methylphenyl)sulfonamido)phenoxy)propanoic acid as a pale yellow solid. The resulting crude compound was used for the next reaction without further purification.
Prepared as described in the experimental procedure of 7a from (R)-2-(2.5-bis((N-(2-(t-butoxy)-2-oxoethyl)-4-methylphenyl)sulfonamido)phenoxy)propanoic acid to obtain the title compound as a white solid (26 mg, 89% over 2 steps); 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.6 Hz, 2H), 7.38 (d, J = 8.4 Hz, 1H), 7.28-7.22 (m, 4H), 6.74 (s, 1H), 7.38 (d, J = 7.6 Hz, 1H), 4.48 (s, 2H), 4.42-4.33 (m, 1H), 4.10-4.06 (m, 1H), 3.67-3.64 (m, 1H), 2.41 (s, 6H), 1.16 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.9, 174.8, 173.6, 153.3, 144.5, 144.0, 140.6, 136.6, 135.3, 134.5, 129.9, 129.6, 127.7, 127.7, 126.6, 120.0, 114.6, 71.7, 52.2, 50.4, 21.7, 21.6, 17.6; LC/MS (ESI) m/z 619.2 [M - H]−; HRMS (ESI) m/z calcd for C27H27N2O11S2 [M - H]− 619.1062; found 619.1069. Rt: 3.82 min, UPLC purity: 99.1%.
N-(2-((1H-tetrazol-5-yl)methoxy)-4-((N-(carboxymethyl)-4-methylphenyl)sulfonamido)phenyl)-N-tosylglycine (15).
Prepared as described in the experimental procedure of 7a from intermediate 42 to obtain the title compound as a white solid (12 mg, 33%); 1H NMR (400 MHz, Methanol-d4) δ 7.53 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.0 Hz, 2H), 6.93 (s, 1H), 6.83 (d, J = 8.4 Hz, 1H), 5.48 (s, 2H), 4.33 (s, 4H), 2.43 (s, 3H), 2.37 (s, 3H); 13C NMR (100 MHz, Methanol-d4) δ 163.3, 156.0, 145.6, 145.1, 143.0, 137.9, 135.9, 135.0, 130.6, 130.2, 128.9, 128.4, 128.0, 121.4, 119.7, 116.8, 114.2, 62.1, 54.8, 21.6, 21.5; LC/MS (ESI) m/z 629.1 [M - H]−; HRMS (ESI) m/z calcd for C26H25N6O9S2 [M - H]− 629.1130; found 629.1140. Rt: 3.67 min, UPLC purity: 95.2%.
2,2′-((2-((Carboxymethyl)amino)-1,4-phenylene)bis(tosylazanediyl))diacetic acid (16).
Prepared as described in the experimental procedure of 7a from intermediate 45 to obtain the title compound as a pale yellow solid (29 mg, 51%); 1H NMR (400 MHz, DMSO-d6) δ 7.59 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 7.08 (dd, J = 8.4, 2.0 Hz, 1H), 6.57 (d, J = 2.0 Hz, 1H), 4.43 (s, 2H), 4.41 (s, 2H), 3.45 (s, 2H), 2.40 (s, 3H), 2.38 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.7, 168.3, 163.8, 144.7, 143.9, 139.8, 135.4, 135.0, 132.9, 129.7, 129.5, 127.6, 127.3, 126.6, 124.4, 122.7, 114.6, 51.8, 49.0, 43.3, 21.0, 20.9; LC/MS (ESI) m/z 604.2 [M - H]−; HRMS (ESI) m/z calcd for C26H26N3O10S2 [M - H]− 604.1065; found 604.1075. Rt: 3.97 min, UPLC purity: 95.2%.
2,2′-((2-(2-Carboxyethyl)-1,4-phenylene)bis(tosylazanediyl))diacetic acid (17a).
Prepared as described in the experimental procedure of 7a from intermediate 49a to obtain the title compound as a white solid (43 mg, 68%); 1H NMR (400 MHz, Methanol-d4) δ 7.50 (d, J = 8.4 Hz, 4H), 7.35-7.33 (m, 4H), 7.04 (d, J = 2.4 Hz, 1H), 6.99 (dd, J = 8.4, 2.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 4.40 (s, 2H), 4.31 (s, 2H), 3.02-2.94 (m, 1H), 2.79-2.71 (m, 1H), 2.44 (s, 3H), 2.43 (s, 3H), 2.40-2.23 (m, 2H); 13C NMR (100 MHz, Methanol-d4) δ 176.5, 172.1, 171.7, 145.7, 145.6, 144.4, 141.7, 138.9, 136.9, 136.7, 131.6, 130.7, 130.1, 129.2, 128.9, 127.8, 54.2, 53.3, 35.0, 26.5, 21.6, 21.5; LC/MS (ESI) m/z 603.2 [M - H]−; HRMS (ESI) m/z calcd for C27H27N2O10S2 [M - H]− 603.1113; found 603.1120. Rt: 3.73 min, UPLC purity: 100.0%.
2,2′-((2-(2-Carboxyethyl)naphthalene-1,4-diyl)bis(tosylazanediyl))diacetic acid (17b).
Prepared as described in the experimental procedure of 7a from intermediate 49b to obtain the title compound as a white solid (26 mg, 57%); 1H NMR (400 MHz, DMSO-d6) δ 12.59 (brs, 2H, CO2H), 8.32 (d, J = 8.4 Hz, 1H), 8.06-7.73 (m, 1H), 7.55-7.46 (m, 6H), 7.43-7.30 (m, 4H), 6.89-6.82 (m, 1H), 4.52-4.31 (m, 4H), 2.95-2.87 (m, 1H), 2.43-2.38 (m, 6H), 2.33-2.22 (m, 1H), 2.06-1.88 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.5, 173.4, 170.0, 169.9, 144.0, 143.9, 139.7, 139.0, 137.2, 137.1, 135.8, 135.7, 134.5, 134.3, 132.8, 132.4, 131.8, 131.7, 129.8, 129.7, 128.2, 127.9, 127.6, 126.4, 125.8, 125.7, 125.2, 125.0, 124.7, 124.6, 53.5, 53.3, 33.9, 33.4, 26.2, 25.9, 21.1, 21.0, 21.0; LC/MS (ESI) m/z 655.3 [M + H]+; HRMS (ESI) m/z calcd for C31H31N2O10S2 [M + H]+ 655.1415; found 655.1420. Rt: 3.88 min, UPLC purity: 96.3%.
2,2′-((2-(Carboxymethoxy)naphthalene-1,4-diyl)bis(tosylazanediyl))diacetic acid (18).
Prepared as described in the experimental procedure of 7a from intermediate 53 to obtain the title compound as a white solid (37 mg, 86%); 1H NMR (400 MHz, DMSO-d6) δ 12.98 (brs, 3H, CO2H), 8.73-8.66 (m, 1H), 8.36-8.23 (m, 1H), 7.60-7.58 (m, 3H), 7.53-7.38 (m, 6H), 7.28-7.26 (m, 1H), 6.85-6.34 (m, 1H), 4.64-4.52 (m, 1H), 4.39 (s, 1H), 4.31-4.16 (m, 2H), 4.07-3.86 (m, 1H), 3.77-3.02 (m, 1H), 2.45 (s, 3H), 2.38-2.36 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.6, 170.4, 169.7, 169.1, 168.5, 150.8, 150.1, 144.0, 143.3, 138.5, 138.2, 135.9, 134.7, 134.5, 134.1, 129.8, 129.6, 129.3, 129.2, 128.4, 128.1, 127.7, 127.5, 126.6, 125.7, 125.5, 124.4, 124.3, 122.2, 121.9, 115.0, 114.5, 64.0, 53.6, 53.0, 52.2, 21.0; LC/MS (ESI) m/z 657.2 [M + H]+; HRMS (ESI) m/z calcd for C30H29N2O11S2 [M + H]+ 657.1207; found 657.1220. Rt: 3.81 min, UPLC purity: 99.6%.
Fluorescence Polarization Competition Assay.
FP assay was conducted according to the procedure previously reported by our group [43].
Time-Resolved Fluorescence Resonance Energy Transfer Assay.
TR-FRET assay was carried out as previously described [44].
Cell culture, RNA quantification, cDNA synthesis, and RT-qPCR
NCM460D cell culture, treatment of positive control and test compounds, RNA quantification, cDNA synthesis, and RT-qPCR were performed following protocols previously reported by our group [35].
Molecular docking simulation.
Computational study on the interactions of the Keap1-Nrf2 PPI small molecule inhibitors to Keap1 protein was conducted using the Autodock4.0 software [45]. The crystal structure of Keap1 Kelch domain in complex with compound 5b (PDB: 4XMB) was utilized as a template for protein preparation [33]. For docking simulations, the structure of Keap1 protein was processed into a pdbqt file through the following steps: the removal of the crystal ligand and all water molecules, the addition of missing hydrogen atoms, and the calculation of Kollman charges using Autodock tool. For ligand preparation, energy minimization of compounds was performed using MOE, and the resulting conformations were converted into a pdbqt file. As previous described [35], the parameters of grid and docking were set to default values, except for the number of grid points (X, Y, Z = 44, 40, 40). Test compounds were docked into the Keap1 Kelch domain via Lamarckian genetic algorithm with 100 GA runs [46]. The docking data was indicated in kcal/mol, and the resulting conformations were ranked on the basis of their binding energy. Docking validation was performed by re-docking the crystallized ligand under the optimized method and comparing its docked pose with the crystal structure of the ligand, thereby resulting in the low RMSD value of 0.94 Å.
Supplementary Material
Highlights.
1,4-Bis(arylsulfonamido)benzene or naphthalene analogs with C2 substituents with synthesized.
C2 substituents with various polar linkers lead to enhanced Keap1-Nrf2 PPI inhibition.
Compounds 17b and 18 with a carboxylate group are the most potent in FP and TR-FRET assays.
The naphthalene derivatives 17b and 18 stimulate the transcription of Nrf2-dependent genes.
ACKNOWLEDGMENTS
We thank the Center for Integrative Proteomics Research (CIPR), Rutgers University for performing the HRMS experiments. This work was supported in part by the National Cancer Institute [R01CA133791] and the Rutgers TechAdvance Grant [TA2019-0300].
Longqin Hu reports financial support was provided by National Institutes of Health. Longqin Hu reports financial support was provided by Rutgers The State University of New Jersey. Longqin Hu reports a relationship with Rutgers The State University of New Jersey that includes: employment. Michael P. Verzi reports a relationship with Rutgers The State University of New Jersey that includes: employment. Sumi Lee reports a relationship with Rutgers The State University of New Jersey that includes: employment. Ahmed R. Ali reports a relationship with Rutgers The State University of New Jersey that includes: employment. Dhulfiqar Ali Abed reports a relationship with Rutgers The State University of New Jersey that includes: employment. Mai-Uyen Nguyen reports a relationship with Rutgers The State University of New Jersey that includes: employment. Longqin Hu has patent #Small Molecule Direct Inhibitors of Keap1-Nrf2 Protein-Protein Interaction pending to Rutgers The State University of New Jersey. Dhulfiqar Ali Abed has patent #Small Molecule Direct Inhibitors of Keap1-Nrf2 Protein-Protein Interaction pending to Rutgers The State University of New Jersey. Ahmed R. Ali has patent #Small Molecule Direct Inhibitors of Keap1-Nrf2 Protein-Protein Interaction pending to Rutgers The State University of New Jersey.
ABBREVIATIONS
- ACN
acetonitrile
- AcOH
acetic acid
- ARE
antioxidant response element
- CDDO-Me
bardoxolone methyl
- Cul3
cullin3
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbo-diimide
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization
- GST
glutathione S-transferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HO-1
heme-oxygenase-1
- HOBt
1-hydroxybenzotiazole
- HRMS
high resolution mass spectrometry
- Keap1
Kelch-like ECH-associated protein 1
- LC-MS
liquid chromatography mass spectrometry
- MS
multiple sclerosis
- MW
molecular weight
- NBS
N-bromosuccinimide
- Neh
Nrf2–ECH homology
- NQO1
NAD(P)H:quinone oxidoreductase 1
- Nrf2
nuclear factor erythroid 2-related factor 2
- PDB
Protein Data Bank
- PPI
protein–protein interaction
- SAR
structure–activity relationship
- SOD
superoxide dismutase
- TBAF
tetrabutylammonium fluoride
- TBDMS
t-butyldimethylsilyl
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- THIQ
tetrahydroisoquinoline
- TR-FRET
time-resolved fluorescence resonance energy transfer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information
The Supporting Information is available free of charge at DOI:
The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O, Oxidative stress and antioxidant defense, World Allergy Organ. J, 5 (2012) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lu M-C, Ji J-A, Jiang Z-Y, You Q-D, The Keap1–Nrf2–ARE pathway as a potential preventive and therapeutic target: an Update, Med. Res. Rev, 36 (2016) 924–963. [DOI] [PubMed] [Google Scholar]
- [3].Magesh S, Chen Y, Hu L, Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents, Med. Res. Rev, 32 (2012) 687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT, Cancer chemoprevention mechanisms mediated through the Keap1–Nrf2 pathway, Antioxid. Redox Signaling, 13 (2010) 1713–1748. [DOI] [PubMed] [Google Scholar]
- [5].Kensler TW, Wakabayashi N, Biswal S, Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway, Annu. Rev. Pharmacol. Toxicol, 47 (2007) 89–116. [DOI] [PubMed] [Google Scholar]
- [6].Baird L, Dinkova-Kostova AT, The cytoprotective role of the Keap1–Nrf2 pathway, Arch. Toxicol, 85 (2011) 241–272. [DOI] [PubMed] [Google Scholar]
- [7].Dinkova-Kostova AT, Talalay P, Direct and indirect antioxidant properties of inducers of cytoprotective proteins, Mol. Nutr. Food Res, 52 (2008) S128–S138. [DOI] [PubMed] [Google Scholar]
- [8].Moi P, Chan K, Asunis I, Cao A, Kan YW, Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region, Proc. Natl. Acad. Sci. U. S. A, 91 (1994) 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P, Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants, Proc. Natl. Acad. Sci. U. S. A, 99 (2002) 11908–11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang DD, Lo S-C, Cross JV, Templeton DJ, Hannink M, Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex, Mol. Cell. Biol, 24 (2004) 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kobayashi A, Kang M-I, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M, Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2, Mol. Cell. Biol, 24 (2004) 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML, Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3, Chem. Res. Toxicol, 21 (2008) 705–710. [DOI] [PubMed] [Google Scholar]
- [13].Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M, Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response, Mol. Cell. Biol, 27 (2007) 7511–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Baird L, Swift S, Llères D, Dinkova-Kostova AT, Monitoring Keap1–Nrf2 interactions in single live cells, Biotechnol. Adv, 32 (2014) 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kobayashi A, Kang M-I, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M, Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1, Mol. Cell. Biol, 26 (2006) 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Niture SK, Kaspar JW, Shen J, Jaiswal AK, Nrf2 signaling and cell survival, Toxicol. Appl. Pharmacol, 244 (2010) 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Satoh T, Lipton S, Recent advances in understanding NRF2 as a druggable target: development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate, F1000Res, 6 (2017) 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Linker RA, Lee D-H, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Brück W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R, Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway, Brain, 134 (2011) 678–692. [DOI] [PubMed] [Google Scholar]
- [19].Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ, Bardoxolone methyl decreases megalin and activates Nrf2 in the kidney, J. Am. Soc. Nephrol, 23 (2012) 1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lynch DR, Chin MP, Boesch S, Delatycki MB, Giunti P, Goldsberry A, Hoyle JC, Mariotti C, Mathews KD, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot G, Zesiewicz T, Meyer CJ, Efficacy of Omaveloxolone in Friedreich’s Ataxia: Delayed-Start Analysis of the MOXIe Extension, Mov. Disord, 38 (2023) 313–320. [DOI] [PubMed] [Google Scholar]
- [21].Hur W, Gray NS, Small molecule modulators of antioxidant response pathway, Curr. Opin. Chem. Biol, 15 (2011) 162–173. [DOI] [PubMed] [Google Scholar]
- [22].Brennan MS, Matos MF, Li B, Hronowski X, Gao B, Juhasz P, Rhodes KJ, Scannevin RH, Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro, PLoS One, 10 (2015) e0120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P, Davies TG, Structure of the BTB Domain of Keap1 and Its Interaction with the Triterpenoid Antagonist CDDO, PLoS One, 9 (2014) e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease, N. Engl. J. Med, 369 (2013) 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cosgrove D, Madison J, Molecular and Cellular Mechanisms Underlying the Initiation and Progression of Alport Glomerular Pathology, Front. Med, 9 (2022) 846152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M, Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model, Mol. Cell. Biol, 26 (2006) 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Richardson BG, Jain AD, Speltz TE, Moore TW, Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway, Bioorg. Med. Chem. Lett , 25 (2015) 2261–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, Shen J, Kerrigan JE, Kong AN, Dandapani S, Palmer M, Schreiber SL, Munoz B, Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction, Bioorg. Med. Chem. Lett, 23 (2013) 3039–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, Willems HMG, Woolford AJA, Cottom JE, Kou JP, Yonchuk JG, Feldser HG, Sanchez Y, Foley JP, Bolognese BJ, Logan G, Podolin PL, Yan H, Callahan JF, Heightman TD, Kerns JK, Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery, J. Med. Chem, 59 (2016) 3991–4006. [DOI] [PubMed] [Google Scholar]
- [30].Abed DA, Lee S, Hu L, Discovery of disubstituted xylylene derivatives as small molecule direct inhibitors of Keap1-Nrf2 protein-protein interaction, Bioorg. Med. Chem, 28 (2020) 115343. [DOI] [PubMed] [Google Scholar]
- [31].Narayanan D, Tran KT, Pallesen JS, Solbak SMØ, Qin Y, Mukminova E, Luchini M, Vasilyeva KO, González Chichón D, Goutsiou G, Poulsen C, Haapanen N, Popowicz GM, Sattler M, Olagnier D, Gajhede M, Bach A, Development of Noncovalent Small-Molecule Keap1-Nrf2 Inhibitors by Fragment-Based Drug Discovery, J. Med. Chem, 65 (2022) 14481–14526. [DOI] [PubMed] [Google Scholar]
- [32].Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP, Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis, J. Med. Chem, 57 (2014) 2736–2745. [DOI] [PubMed] [Google Scholar]
- [33].Jain AD, Potteti H, Richardson BG, Kingsley L, Luciano JP, Ryuzoji AF, Lee H, Krunic A, Mesecar AD, Reddy SP, Moore TW, Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators, Eur. J. Med. Chem, 103 (2015) 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Abed DA, Lee S, Wen X, Ali AR, Mangipudy V, Aleksunes LM, Hu L, Optimization of 1,4-bis(arylsulfonamido)naphthalene-N,N’-diacetic acids as inhibitors of Keap1-Nrf2 protein-protein interaction to suppress neuroinflammation, Bioorg. Med. Chem, 44 (2021) 116300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee S, Abed DA, Nguyen M-U, Verzi MP, Hu L, Structure-activity relationships of 1,4-bis(arylsulfonamido)-benzene or naphthalene-N,N’-diacetic acids with varying C2-substituents as inhibitors of Keap1-Nrf2 protein-protein interaction, Eur. J. Med. Chem, 237 (2022) 114380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Abed DA, Ali AR, Lee S, Nguyen M-U, Verzi MP, Hu L, Optimization of the C2 substituents on the 1,4-bis(arylsulfonamido)naphthalene-N,N’-diacetic acid scaffold for better inhibition of Keap1-Nrf2 protein-protein interaction, Eur. J. Med. Chem, 252 (2023) 115302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Georgakopoulos N, Talapatra S, Dikovskaya D, Dayalan Naidu S, Higgins M, Gatliff J, Ayhan A, Nikoloudaki R, Schaap M, Valko K, Javid F, Dinkova-Kostova AT, Kozielski F, Wells G, Phenyl Bis-Sulfonamide Keap1-Nrf2 Protein–Protein Interaction Inhibitors with an Alternative Binding Mode, J. Med. Chem, 65 (2022) 7380–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee S, Hu L, Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction, Med. Chem. Res, 29 (2020) 846–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kotha S, Shah VR, Mandal K, Formation of arenes via diallylarenes: strategic utilization of Suzuki–Miyaura cross-coupling, Claisen rearrangement and ring-closing metathesis, Adv. Synth. Catal, 349 (2007) 1159–1172. [Google Scholar]
- [40].Davies SG, Mujtaba N, Roberts PM, Smith AD, Thomson JE, A tandem conjugate addition/cyclization protocol for the asymmetric synthesis of 2-aryl-4-aminotetrahydroquinoline-3-carboxylic acid derivatives, Org. Lett, 11 (2009) 1959–1962. [DOI] [PubMed] [Google Scholar]
- [41].Yasuda D, Nakajima M, Yuasa A, Obata R, Takahashi K, Ohe T, Ichimura Y, Komatsu M, Yamamoto M, Imamura R, Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity, Bioorg. Med. Chem. Lett, 26 (2016) 5956–5959. [DOI] [PubMed] [Google Scholar]
- [42].Niu J, Guo P, Kang J, Li Z, Xu J, Hu S, Copper(I)-catalyzed aryl bromides to form intermolecular and intramolecular carbon–oxygen bonds, J. Org. Chem, 74 (2009) 5075–5078. [DOI] [PubMed] [Google Scholar]
- [43].Inoyama D, Chen Y, Huang X, Beamer LJ, Kong A-NT, Hu L, Optimization of fluorescently labeled Nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of Keap1-Nrf2 interaction, J. Biomol. Screen, 17 (2012) 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee S, Abed DA, Beamer LJ, Hu L, Development of a Homogeneous Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) Assay for the Inhibition of Keap1–Nrf2 Protein–Protein Interaction, SLAS Discov., 26 (2020) 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem, 30 (2009) 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem, 19 (1998) 1639–1662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.