

Abstract
Background
Transmembrane 4 L six family member 5 (TM4SF5) translocates subcellularly and functions metabolically, although it is unclear how intracellular TM4SF5 translocation is linked to metabolic contexts. It is thus of interests to understand how the traffic dynamics of TM4SF5 to subcellular endosomal membranes are correlated to regulatory roles of metabolisms.
Methods
Here, we explored the metabolic significance of TM4SF5 localization at mitochondria‐lysosome contact sites (MLCSs), using in vitro cells and in vivo animal systems, via approaches by immunofluorescence, proximity labelling based proteomics analysis, organelle reconstitution etc.
Results
Upon extracellular glucose repletion following depletion, TM4SF5 became enriched at MLCSs via an interaction between mitochondrial FK506‐binding protein 8 (FKBP8) and lysosomal TM4SF5. Proximity labeling showed molecular clustering of phospho‐dynamic‐related protein I (DRP1) and certain mitophagy receptors at TM4SF5‐enriched MLCSs, leading to mitochondrial fission and autophagy. TM4SF5 bound NPC intracellular cholesterol transporter 1 (NPC1) and free cholesterol, and mediated export of lysosomal cholesterol to mitochondria, leading to impaired oxidative phosphorylation but intact tricarboxylic acid (TCA) cycle and β‐oxidation. In mouse models, hepatocyte Tm4sf5 promoted mitophagy and cholesterol transport to mitochondria, both with positive relations to liver malignancy.
Conclusions
Our findings suggested that TM4SF5‐enriched MLCSs regulate glucose catabolism by facilitating cholesterol export for mitochondrial reprogramming, presumably while hepatocellular carcinogenesis, recapitulating aspects for hepatocellular carcinoma metabolism with mitochondrial reprogramming to support biomolecule synthesis in addition to glycolytic energetics.
Keywords: cholesterol, fluorescent imaging, glucose catabolism, hepatocellular carcinogenesis, membrane contact sites, mitochondria function, mitophagy, oxidative phosphorylation, protein‐protein interaction, tetraspanin
List of abbreviations
- AAV8-Tbg-HA-Tm4sf5
adeno-associated virus serotype 8 encoding for Tm4sf5 under thyroxine binding globulin promoter specific for hepatocytes
- CCZ1
CCZ1 homolog, vacuolar protein trafficking and biogenesis associated
- DRP1
dynamic-related protein I
- ETC
electron transport system
- FKBP8
FK506-Binding Protein 8
- HA-OMP25
outer membrane protein 25 for tagging mitochondria with HA epitopes
- HCC
hepatocellular carcinoma
- HFHGluD
high fat and glucose diet
- HMGCR
3-hydroxy-3-methylglutaryl-CoA reductase
- LAMP1
lysosomal associated membrane protein 1
- LIHC
liver hepatocellular carcinoma
- MCS
membrane contact site
- MLCS
mitochondria-lysosome contact site
- NPC1
NPC intracellular cholesterol transporter 1
- OCR
oxygen consumption rate
- TM4SF5
transmembrane 4 L six family member 5
- TSAHC
4'-(p-toluenesulfonylamido)-4-hydroxychalcone
1. BACKGROUND
Fermentative glycolysis resulting from primary mitochondrial defects, also known as the Warburg effect, produces energy and intermediates for the uncontrolled anabolic growth of cancer cells [1]. Beyond its fundamental bioenergetic functions, mitochondrial metabolism provides building blocks for tumor anabolism and cell survival and growth, suggesting the mitochondria as a promising target for developing anti‐cancer agents [2]. Diverse studies have been performed to identify the molecules that control mitochondrial functions; however, major and targetable molecules and pathways still need to be identified to enable targeting of mitochondria‐based metabolism with anti‐cancer therapeutics.
Transmembrane 4 L six family member 5 (TM4SF5) is a member of the tetraspan(in)s family, via a similar membrane topology with four transmembrane domains, 2 (long or short) extracellular loops, and cytosolic N‐ and C‐terminal tails [3]. TM4SF5 is N‐glycosylated at N138 and N155 and palmitoylated at many cysteines proximal to the transmembrane domains and localized to the plasma or lysosomal membranes [4, 5]. TM4SF5 can form massive protein‐protein complexes, known as TM4SF5‐enriched microdomains (T5ERMs), at different subcellular membranes, which include various membrane proteins and receptors, cytosolic proteins [6], and nutrient transporters [7] and presumably function as signaling hubs similar to tetraspanin‐web or tetraspanin‐enriched microdomains (TERMs) [8]. TM4SF5 binds to mammalian target of rapamycin (mTOR) at lysosomes, leading to mTOR complex I (mTORC1) and ribosomal protein S6 kinase β‐1 (S6K1) activation via lysosomal arginine sensing and subsequent export into the cytosol [5]. Furthermore, TM4SF5 in hepatocytes is loaded on exosomes and involved in the metabolic crosstalk between the liver and brown adipose tissue [9].
Intracellular organelles have been widely studied recently, especially in terms of organellar membrane contact sites (MCSs) [10]. Most MCSs involve membranes from the endoplasmic reticulum (ER), lysosomes, mitochondria, or plasma membrane. MCSs function as conduits for lipids, ions, and calcium and as intracellular signaling hotspots for the regulation of organellar dynamics [11, 12, 13]. Fusion or fission of mitochondria (i.e., mitochondrial dynamics) can be regulated by different proteins including mitofusin I (Mfn1) and dynamin‐related protein 1 (DRP1) and affects the tricarboxylic acid (TCA) cycle, which is important for production of both ATP and biomolecular intermediates [14]. MCSs involving mitochondria can impact mitochondrial dynamics, leading to changes in metabolic function in the presence of cellular malignancy or environmental cues [15]. The functional significance of T5ERMs or TERMs may recapitulate the physiological or pathological roles of MCSs enriched with TM4SF5 or tetraspanins between different subcellular membranes. It is therefore of interest to learn how TM4SF5‐enriched mitochondria‐lysosome contact sites (MLCSs) form and influence metabolic reprogramming upon exposure of cells to excessive extracellular glucose. In this study, we investigated how TM4SF5 localizes at MLCSs and how this localization is regulated by extracellular glucose levels.
2. MATERIALS AND METHODS
2.1. Cells
Human hepatocellular carcinoma cell lines Huh7 and SNU449 were purchased from the Korean Cell Line Bank (Seoul National University, Seoul, Korea). Human embryonal kidney cell line HEK293FT was purchased from Thermo Fisher Scientific (R7007, Waltham, MA, USA). Murine hepatocyte cell line AML12 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Huh7 and HEK239FT cells were maintained in DMEM (SH30243.01, Hyclone, Logan, UT, USA), and SNU449 cells were grown in RPMI‐1640 (SH30027.01, Hyclone) supplemented with 10% FBS (F0600, GenDEPOT, Barker, TX, USA) and penicillin/streptomycin (CA005, GenDEPOT). AML12 cells were maintained in DMEM/F12 (SH30023.01, Hyclone) supplemented with 10% FBS, 10 μg/mL insulin, 5.5 μg/mL transferrin, 6.7 ng/mL selenium (ITS, 41400045, Gibco, Waltham, MA, USA), and 40 ng/mL dexamethasone (265005, Sigma‐Aldrich, St. Louis, MO, USA). For primary hepatocytes, Medium 199 (SH30253.01, Hyclone) was used with supplements of 10% FBS, 23 mmol/L HEPES, and 4 ng/mL dexamethasone (265005, Sigma‐Aldrich). Cells were grown at 37°C in 5% CO2 and passaged every 3‐4 days according to the manufacturer's instructions, but for no more than 20 passages. Cells were checked for mycoplasma every other month.
2.2. Western blotting
Cells were glucose starved for 16 h in glucose‐free media and replete with culture media containing 10 mmol/L (for SNU449 cells) or 25 mmol/L glucose (for Huh7 cells) for the indicated times. Cells were transfected with small interfering RNA (siRNAs) or short hairpin RNA (shRNAs) for a non‐specific sequence or target sequences as shown in Supplementary Table S1. Cells in normal culture or treated as above were harvested to make whole‐cell lysates using modified RIPA lysis buffer (50 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, 0.5% sodium deoxycholate, and 1% NP‐40). The lysates were normalized using a BCA assay kit (23225, Thermo Fisher Scientific), and immunoblot analysis was performed using the following primary antibodies: Strep‐HRP (1:5,000; 2‐1509‐001) from IBA Lifesciences (Goettingen, Germany); HA (1:2,000; 901515) from BioLegend (San Diego, CA, USA); v5 (1:10,000; R96025) and AMBRA1 (1:1,000; PA5‐88053) from Invitrogen (Grand Island, NY, USA); BCL2L13 (1:1,000; 16612‐1‐AP) from Proteintech (Rosemont, IL, USA); HBx (1:1,000; ab39716), M6PR (1:1,000; ab124767), MLN64/STARD3 (1:1,000; ab3478) from Abcam (Cambridge, UK); NPC1 (1:1,000; NB400‐148) from Novus Biologicals; LAMP2 (1:1,000; sc‐18822), PHB2 (1:1,000; sc‐133094), α‐tubulin (1:5,000, sc‐5286), β‐actin (1:1,000, sc‐47778), and RAB21 (1:1,000; sc‐81917) from Santa Cruz (Dallas, TX, USA); and LAMP1 (1:1,000; #9091), MFN2 (1:1,000; #9482), DRP1 (1:1,000; #8570), pS616DRP1 (1:1,000; #3455), TOM20 (1:1,000; #42406), mTOR (1:1,000; #2983), AMPK (1:1,000; #5832S), pT172AMPK (1:1,000; #2535S), ACC (1:1,000; #3676S), pS79ACC (1:1,000; #11818S), LC3B (1:1,000; #2775), COX4 (1:1,000; #4850), and FLAG (1:2,000; #2368) from Cell Signaling Technology (Danvers, MA, USA). The anti‐TM4SF5 antibody (1:5,000) was generated by immunizing rabbits with a TM4SF5 C‐terminal or a long extracellular loop 2 sequence peptide [9].
2.3. Immunoprecipitation
Cells were lysed in Triton X‐100 lysis buffer (40 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, and 0.5% Triton X‐100). Whole‐cell lysates were incubated with streptavidin‐agarose (20353, Thermo Fisher Scientific) for 4 h at 4°C. Beads were washed 3 times with ice‐cold wash buffer (40 mmol/L HEPES, pH 7.4, 500 mmol/L NaCl, 1 mmol/L EDTA, and 0.5% Triton X‐100), followed by washing once with ice‐cold PBS. Washed beads were eluted in 2 × sodium dodecyl‐sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) sample buffer and boiled for 5 min before immunoblot analysis.
2.4. Immunofluorescence microscopic imaging
Cover glasses were pre‐coated with 10 μg/mL fibronectin (35600, BD Biosciences, San Jose, CA, USA) in PBS at room temperature for 1 h and briefly rinsed 3 times with PBS. Twenty thousand cells were plated on each pre‐coated cover glass on 12‐well plates for 16 h. The cells were then transfected with the indicated cDNA constructs using Lipofectamine 3000 (L3000015, Invitrogen, Grand Island, NY, USA) for 24 h. The cells were then manipulated for glucose starvation and repletion, as above. The cover glasses were then fixed with ice‐cold 99% methanol or 3.7% formaldehyde for 15 min and blocked with 1% BSA in PBS for 1 h. Primary antibodies were diluted in 1% BSA in PBS (1:500) and treated for 16 h at 4°C. The primary antibodies were anti‐LAMP1 (#9091), anti‐DRP1 (#8570), anti‐pS616DRP1 (#3455), and anti‐LC3B (#2775) from Cell Signaling Technology; anti‐ZO1 (402200) from Invitrogen; and anti‐EGFR (sc‐03) from Santa Cruz. The cover glasses were then washed three times with PBS and incubated with fluorescence‐conjugated secondary antibodies (1:500, Alexa Fluor 488 [goat: A11055 and mouse: A21202] and 555 [goat: A21432 and mouse: A31570] from Invitrogen) in PBS at room temperature for 1 h. The cover glasses were then washed 3 times with PBS and mounted on slide glasses using ProLong™ Gold Antifade (P36930, Invitrogen). Cells were randomly visualized using a Nikon Eclipse Ti microscope with a C2 confocal system. Images were analyzed using NIS‐Elements software (Nikon, Melville, NY, USA). To determine autophagy, LC3B immuno‐staining was performed with or without 10 μmol/L chloroquine treatment (C6628, Sigma‐Aldrich) for 24 h to block LC3B protein degradation.
2.5. Quantification of MLCSs via microscopic labeling analyses
To quantify the MLCSs in each cell, we modified the quantification method for lysosome‐peroxisome membrane contact (LPMC) as previously described [16]. To calculate the MLCSs/cell values, cells were transfected with mito‐GFP for mitochondria labeling (green) and treated with lysotracker (L7528, Invitrogen) for lysosome labeling (red). Cells were fixed with 3.7% formaldehyde and then washed 3 times with PBS, before random capturing of fluorescent images by a confocal microscope (Nikon, Melville, NY, USA), as explained above. Color pixel values (RGB888) were extracted from each cell by a color picker and counted fluorescent signal in each cell with parsing images (green, red, and yellow color defined with threshold). MLCSs/cell values were calculated by ratios of overlapped (i.e., green lysosome in vicinity) mitochondria (yellow) to total mitochondria (green and yellow). To calculate the TM4SF5 at MLCSs values, cells were transfected with mcherry‐TM4SF5 (red) and mito‐GFP (green). Cells were fixed and then imaged randomly by a confocal microscope (Eclipse Ti C2, Nikon, Melville, NY, USA). Pearson's correlation coefficients were calculated to validate colocalization of TM4SF5 (red) and mitochondria (green) using NIS‐elements software. Cells were analyzed at least from 3 independent experiment. In case, cells were treated with 10 mmol/L L‐Arg, 20 mmol/L glutamine or 25 mmol/L 2‐Dexoy‐D‐glucose (2‐DG) for 30 min, or DMSO vehicle or 4’‐(p‐toluenesulfonylamido)‐4‐hydroxychalcone (TSAHC [17]) for 24 h. All images in the same figure panel were taken under the same software setting and equally processed in Adobe Photoshop. Each dot in the graphs indicates measurement from one cell and otherwise it was indicated.
2.6. Live imaging
Ten thousand cells were plated on an 8‐well chamber slide (C7182, Nunc, Lab‐TekII) and incubated in a humidified incubator at 37°C with 5% CO2 for 16 h. The cells were then transfected with mcherry‐TM4SF5 and mito‐GFP constructs using Lipofectamine 3000 (Invitrogen) for 48 h. The cells were visualized with an LSM880 laser scanning microscope with Airyscan (Carl Zeiss, Oberkochen, Germany) in a 37°C, 5% CO2 chamber. Images were randomly captured and analyzed using ZEN imaging software (Carl Zeiss). In the case of mitochondrial fission analysis, cells were manipulated as described above and live imaged using a confocal microscope (Eclipse Ti C2, Nikon, Melville, NY, USA). The expected probability that mitochondria fission of a TM4SF5‐enriched vesicle with cholesterol would occur at the site of a mitochondrial division event by random chance was calculated as the density of TM4SF5 vesicles from n = 10 living cells using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The rate of mitochondrial fission was calculated per cell by quantifying the number of fission events in the entire cell from ≥ 200‐s videos. The percentage of fission events for mitochondria near cholesterol around TM4SF5 vesicles in TopFluor‐cholesterol‐treated living SNU449 cells (expressing TM4SF5‐mCherry [lysosomes] and mito‐BFP [mitochondria]) was calculated from n = 21 events in 10 cells.
2.7. Cell growth assay
Huh7 cells transfected with non‐specific shRNA or with shRNA targeting #4 or #12 sequence for TM4SF5 suppression (Supplementary Table S1) were processed for cell‐number counting 5 days after seeding in normal culture conditions.
2.8. Mitochondria complex activity assay
Mitochondria from Huh7 cells were isolated using a Mitochondria Isolation Kit (701010, Cayman Chemical Company, Ann Arbor, MI, USA). Protein concentrations of isolated mitochondrial samples were quantified by BCA assay (A55860, Thermo Scientific). Mitochondrial complex activities were measured using MitoCheck Complex I, II/III, IV, and V Activity Assay Kits (700930, 700950, 700990, and 701000, respectively, Cayman Chemical Company (Ann Arbor).
2.9. Citrate analysis
Whole‐cell lysates or mitochondria were prepared before citrate quantification. Citrate levels were measured using a Citrate Assay Kit (ab83396, Abcam) and normalized to the protein concentrations determined by BCA assay. Briefly, Cells were washed with cold PBS and then resuspended in 100 μL of citrate assay buffer and homogenized by pipetting, before centrifugation at 12,000 × g at 4°C for 3 min. Supernatant was collected, and 50 μL supernatant was mixed with 50 μL of the reaction solution. The mixture was incubated at room temperature for 30 min, before measuring optical density (OD) at 570 nm using microplate reader (SpectraMAX i3×, Molecular Devices, San Jose, CA, USA).
2.10. APEX2 staining and transmission electron microscopy
Cells were transfected with APEX2‐TM4SF5 plasmid (homemade using pcDNA3 APEX2‐NES [#49386, Addgene, Watertown, MA, USA]) for 48 h, fixed with 2% glutaraldehyde and 2% paraformaldehyde solution at 4°C for 16 h, washed 3 times with 100 mmol/L sodium cacodylate buffer (97068, Sigma‐Aldrich), and treated with 20 mmol/L glycine solution (50046, Sigma‐Aldrich). The cells were then stained with freshly diluted 0.5 mg/mL DAB (D12384, Sigma‐Aldrich) in 10 mmol/L H2O2 solution, washed with sodium cacodylate buffer 3 times, fixed using 2% (W/V) osmium tetroxide (75633, Sigma‐Aldrich) for 40 min on ice, rinsed with ice‐cold distilled water, and dehydrated in a graded ethanol series (50%‐100%) for 15 min at each step. The sample was then mixed 1:1 (v/v) with EMBED‐812 resin (50‐980‐446, Thermo Fisher Scientific) and anhydrous ethanol for 1 h. The mixture was incubated overnight in 2:1 (v/v) resin and then exchanged with 100% resin for 2 h before transfer to fresh resin, followed by polymerization at 60°C for 24 h. Embedded cell pellets were cut with a diamond knife into 50 nm sections and imaged on an FEI‐Tecnai G2 Spirit Bio Twin TEM instrument. Images were taken using a 120 kV transmission electron microscope (Talos L120C, FEI, Hillsboro, OR, USA) at the National Instrumentation Center for Environmental Management (NICEM) in Seoul National University (Seoul, S.
2.11. Biotin labeling with TurboID
Cells were transfected with TurboID constructs (v5‐TurboID‐TOM20, v5‐TurboID‐LAMP1, or v5‐TurboID‐TM4SF5, homemade using v5‐TurboID‐NES_pcDNA3 [#107169, Addgene, Watertown, MA, USA]) for 48 h and labeled with 50 μmol/L biotin (A14207, Alfa Aesar, MA, USA) at 37°C for 1 h. Labeling was stopped by washing in ice‐cold PBS (3 times), and cells were harvested for western blotting as above, immunoprecipitation, or proteomic analysis.
2.12. Proteomic analysis
After biotin labeling, the cells were washed 3 times with Dulbecco's phosphate‐buffered saline (DPBS; SH30028.02, Hyclone) and then lysed with 2% SDS in 1× TBS (25 mmol/L Tris‐HCl, 0.15 mol/L NaCl, pH 7.2). Lysates were clarified by ultrasonication (Bioruptor, Cosmo Bio USA, Carlsbad, CA, USA) for 15 min in a cold‐water bath. For removal of free biotin, 6 times the sample volume of cold acetone (650501, Sigma‐Aldrich) was added to each lysate and kept at ‐20°C. After at least 2 h, the samples were centrifuged at 13,000 ×g for 10 min at 4°C. Supernatants were removed gently, and fresh acetone with 10× TBS was mixed and added to the pellets. The samples were then vortexed vigorously and kept at ‐20°C for at least 2 h and not longer than overnight. Then, the samples were centrifuged at 13,000 ×g for 10 min at 4°C. Supernatants were removed gently, and the pellets were allowed to air dry for 3∼5 min. The pellets were then resolubilized with 1 mL of 8 mol/L urea (U5378, Sigma‐Aldrich) in 50 mmol/L ammonium bicarbonate (A6141, Sigma‐Aldrich). The concentration of protein was measured by BCA assay (23225, Thermo Fisher Scientific). The samples were then denatured at 650 rpm for 1 h at 37°C using a thermomixer (Eppendorf, Hamburg, Germany), reduced by addition of dithiothreitol (43816, Sigma‐Aldrich) to a 10 mmol/L final concentration, and incubated at 37°C with shaking at 650 rpm for 1 h in a thermomixer (Eppendorf, Hamburg, Germany). The samples were then alkylated by addition of iodoacetamide (I1149, Sigma‐Aldrich) to a 40 mmol/L final concentration and mixed at 650 rpm at 37°C for 1 h. The samples were then diluted 8 times in 50 mmol/L ammonium bicarbonate (A6141, Sigma‐Aldrich). CaCl2 (1 mmol/L final concentration, 12312, Alfa Aesar) and Trypsin (50:1 w/w, 20233, Thermo Fisher Scientific) were added to each sample. The samples were incubated at 37°C with shaking at 650 rpm for 6∼18 h in a thermomixer and then centrifuged at 10,000 ×g for 3 min to remove insoluble material. Then, 300 μL of streptavidin beads (50% slurry, Pierce, Appleton, WI, USA) were washed with 2 mol/L urea in 1× TBS 4 times and added to the sample. The samples were then rotated at room temperature for 1 h. The flow‐through fraction was kept, and the beads were washed twice with 2 mol/L urea in 50 mmol/L ABC. After the supernatant was removed, the beads were washed with pure water in new tubes. Biotinylated peptides were heated at 60°C and mixed at 650 rpm after solutions of 80% acetonitrile (900667, Sigma‐Aldrich), 0.2% TFA (T6508, Sigma‐Aldrich), or 0.1% formic acid (28905, Thermo Fisher Scientific) were added. Each supernatant was transferred to new tubes, and the elution step was repeated four more times. Combined elution fractions were dried using a speed vac. Samples were stored at ‐20°C or injected into mass spectrometry directly. For searching binding partners of TM4SF5, HEK293FT cells were transiently transfected with control or Strep‐tag conjugated TM4SF5 plasmid for 48 h and extracted using a TritonX‐100‐containing lysis buffer. Lysates were immunoprecipitated using streptavidin‐agarose bead (20353, Thermo Fisher Scientific) prior to separation by SDS‐PAGE. For identifying mitochondrial proteome, mitochondria were rapidly isolated. Cell lysates or mitochondria samples were separated by SDS‐PAGE and digested with trypsin. After in‐gel trypsin digestion, peptides were analyzed using LC‐MS/MS. Tandem mass spectra were analyzed using the SEQUEST module of Proteome Discoverer (Thermo Fisher Scientific; version 1.4.1.14) and X! Tandem (The GPM, thegpm.org; version CYCLONE 2010.12.01.1). Scaffold (Version Scaffold_4.4.1.1; Proteome Software Inc., Portland, OR, USA) was used to validate MS/MS based peptide and protein identifications. After identification, mitochondrial proteome was analyzed and annotated by human gene sets, KEGG_citrate cycle (M3985), Fatty acid oxidation (M14568), GOBP_electron transport chain (M13293). Humane gene sets were browsed through MsigDB (Molecular Signatures Database, https://www.gsea‐msigdb.org/gsea/msigdb).
2.13. Quantitative real‐time reverse‐transcription PCR (qRT‐PCR)
Total RNA was extracted from cells using Qiazol (#79306, QIAGEN, Germanton, MD, USA). Then, cDNAs were synthesized using the ReverTra Ace qPCR RT master mix (FSQ301, Toyobo, Osaka, Japan) following the manufacturer's protocol. Gene expression was analyzed in duplicate using LaboPassTM EvaGreen Q Master (Cosmo Genetech, Seoul, Korea). The primers for the genes are listed in Supplementary Table S2. Expression data were normalized to GAPDH expression, and fold change (FC) was calculated using the 2−ΔΔCq method.
2.14. Mice
C7BL/6N male mice were purchased from Orient Bio Inc. (Seongnam, Korea) and housed in specific pathogen‐free rooms with controlled humidity and temperature. Animals were regularly checked for objective humane endpoints such as weight loss, body condition scoring, and/or physical appearance. Anesthesia was performed under 5% isoflurane (Terrel, Piramal Critical Care, Bethlehem, PA, USA) with inhalation. All animal procedures were performed in accordance with the procedures of the Seoul National University Laboratory Animal Maintenance Manual and with IRB approvals from the Institute of Laboratory Animal Resources, Seoul National University (SNU‐161108‐10‐1, SNU‐181016‐7‐4, and SNU‐200410‐3).
Tm4sf5‐knockout C57BL/6N mice (Tm4sf5−/−) were generated as described previously [9, 18]. The Tm4sf5 exon 1 region (CCACCTGGACGGACGGCAACCTCAGC) was excised using CRISPR RGEN technology. The mice were then backcrossed with stock C57BL/6N mice at least 8 times every 5 generations. Albumin promoter‐conjugated Tm4sf5‐FLAG3 (Alb‐Tm4sf5‐Flag) transgenic mice were generated at Macrogen (Seoul, Korea) by germline transmission. The adeno‐associated virus (AAV8) used to induce HA‐Tm4sf5 or the control HA‐EV was manufactured by Virovek (Hayward, CA, USA) and intravenously injected into 8‐week‐old mice at a dose of 2 × 1010 vg/mouse. Experiments were performed 2 weeks after the injection. Transgenic mtKeima report mouse was generated according to pervious study by inserting a single copy of mtKeima into Hipp1 locus on chromosome 11 [19].
2.15. Primary hepatocyte isolation
The liver in each animal was perfused with Ca2+‐free and Mg2+‐free Hank's buffered salt solution (HBSS, H4641, Sigma‐Aldrich) containing EGTA (0.5 mmol/L) and digested with HBSS containing 50 μg/mL collagenase type IV (C6885, Sigma‐Aldrich). The digested liver was dissected and then gently teased into small pieces with forceps. The resulting liver slurry was filtered using a 100 μmol/L nylon cell strainer and then spun down at 50 ×g for 3 min. The cell pellet was resuspended in primary hepatocyte culture medium and centrifuged at 50 ×g for 3 min. The resulting pellet was resuspended in 25% Percoll (P1644, Sigma‐Aldrich) and centrifuged at 330 ×g for 5 min without the brake. The pellet was then suspended in culture medium and seeded onto collagen‐precoated culture plates. After 24 h, the adherent cells were collected for experiments.
2.16. Animal models with liver pathology
For the high sucrose diet model (HSuD), six‐week‐old C57BL/6N male mice had ad libitum access to tap water and either a normal chow diet (NCD), an adjusted‐calorie diet with a high sucrose diet (HSuD; 325 g/kg sucrose, Teklad custom diet, #06416, ENVIGO, Indianapolis, IN, USA), or a high‐fat diet (HFD, Research diets, D12492, Research Diets, New Brunswick, NJ, USA) for 10 weeks [20]. For the high fat and glucose diet model (HFGluD), mice fed with HFD were supplied with filtered water with or without excessive 20% (W/V) glucose for 10 weeks. For the diethylnitrosamine (DEN)‐induced hepatocellular carcinoma (HCC) model, two‐week‐old wild‐type (WT) and Tm4sf5 −/− mice were injected intraperitoneally with 25 mg/kg DEN (Sigma‐Aldrich) [21]. After 45 weeks, the mice were sacrificed for analyses.
2.17. Virus
Lentiviruses were generated by transfecting lentiviral vectors (pLKO.1 puro [#8453, Addgene, Watertown, MA, USA] and pLJM1‐EGFP [#19319, Addgene]) in HEK293FT cells. Virus‐containing media were collected and filtered through membranes with 0.45 μmol/L pores (16537‐K, Sartorius, Goettingen, Germany). For in vitro transduction, target cells were infected with virus‐containing media supplemented with 4 μg/mL polybrene (TR‐10003‐G, Millipore, Burlington, MA, USA) for 24 h and selected with puromycin (ABT‐PR‐1, Invitrogen, San Diego, CA, USA). For in vivo transduction, filtered virus‐containing media were concentrated using Lenti‐X Concentrator (631232, Clontech, San Jose, CA, USA) and intravenously administered to mice.
2.18. In vivo mitophagy
For In vivo mitophagy imaging, fresh liver tissues were obtained from male mtKeima mice [19] or pLVX‐puro‐mtKeima lentivirus transduced‐mice following excessive glucose (20% [W/V] in filtered tap water) for 3 or 4 weeks. Liver tissues were rapidly dissected, rinsed with cold PBS, and further cut into sections (0.5‐2.0 mm). The sections were placed onto a confocal dish (101350, SPL, Pocheon, Korea) and analyzed using a confocal microscope (Nikon Eclipse Ti microscope with a C2 confocal system (Nikon, Melville, NY, USA) immediately for 10 or 5 random areas per animal, respectively. The fluorescence of mtKeima was imaged in 2 channels via 2 sequential excitations (440 nm, blue; 550 nm, red) and using a 570‐ to 695‐nm emission range. The rate of mitophagy based on mtKeima was calculated by the ratio of red pixels to total pixels (blue and red).
2.19. In vivo cholesterol transport assay
Six‐week‐old (WT or Tm4sf5−/− ) C57BL/6N male mice were fasted for 18 h and then intravenously administered with 100 μL of mixed dye (25 μmol/L TopFluor cholesterol, 100 μmol/L mitotracker, and 20 μmol/L Hoechst 33342 [62249, Invitrogen]). One hour later, animals were administered intraperitoneally with 2 g/kg D‐glucose (G8270, Sigma‐Aldrich) or saline 30 min before sacrifice. Livers were rapidly dissected, rinsed with cold PBS, and further cut into sections (0.5‐2.0 mm). The tissues were placed onto a confocal dish and analyzed immediately via confocal imaging (Nikon) at 5 random areas per mouse. Mitochondrial cholesterol was calculated by yellow signal (i.e., green cholesterol within red mitotracker stains) per total cholesterol signal (green and yellow stains) of the images.
2.20. Immunohistochemistry
Four‐micron whole sections of formalin‐fixed, paraffin‐embedded liver tissues were heated in the oven at 65°C for 30 min, deparaffinized in xylene, and rehydrated in graded ethanol. Antigen retrieval was performed in sodium citrate buffer (10 mmol/L sodium citrate, 0.05% Tween 20, pH 6.0) at 95°C for 20 min. Slides were cooled on the benchtop in PBS for 5 min and incubated in 0.3% H2O2 (H1009, Sigma‐Aldrich) in PBS for 15 min. Slides were incubated with 10% normal goat serum (50062Z, Invitrogen) for 1 h and then with anti‐Ki67 (1:1,000; ab15580, Abcam), LC3B (1:2,000; PM036, MBL, Woburn, MA, USA), BCL2L13 (1:200; 16612‐1‐AP, Proteintech, Rosemont, IL, USA), PHB2 (1:200; sc‐133094, Santa Cruz), or COX IV (1:1,000; #4850, Cell Signaling Technology) antibodies. Tissue sections were incubated with the antibodies overnight at 4°C, and binding was detected using the VECTASTAIN® ABC‐HRP kit (Vector Laboratories, Burlingame, CA, USA). Mayer's hematoxylin (Sigma‐Aldrich) was used for counterstaining nuclei. Ten random images per slide were obtained using a digital slide scanner (MoticEasyScan, Motic, British Columbia, Canada).
2.21. Seahorse analysis
A Seahorse XFe24 analyzer (Agilent, Santa Clara, CA, USA) was used to measure the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) of cells. Cells were seeded on the XF24 culture plate at 5×104 cells/well, and an XF Cell Mito Stress Test kit (103015‐100, Agilent) was used to analyze mitochondrial respiration according to the manufacturer's protocol. Cells were treated with cholesterol (16 μmol/L, C8667, Sigma‐Aldrich) for 16 h before Mito Stress Test. Seahorse Wave Desktop software (Agilent, Santa Clara, CA, USA) was used for data analysis.
2.22. Rapid mitochondrial purification for lipid/protein profiling
For rapid mitochondrial purification, pMXs‐3×HA‐EGFP‐OMP25 (#83356, Addgene, Watertown, MA, USA) and pMXs‐3×Myc‐EGFP‐OMP25 (#83355, Addgene) were purchased and cloned into a pLJM‐Empty (#91980, Addgene) lentiviral vector. The pLJM‐3×HA‐OMP25 constructs along with lentiviral packaging vectors were transfected into HEK293FT cells using Lipofectamine 3000 (L3000015, Invitrogen, Grand Island, NY, USA). The virus‐containing supernatant was collected 48 h after transfection and filtered through a 0.45‐μm filter to remove cells. Isolated primary hepatocytes from WT or Tm4sf5 −/− KO mice (n = 6 or 9 for experiments without or with lysosome impairment by cyclosporine A treatment, respectively [22]) were seeded on a 150‐mm dish and then infected using lentivirus medium with 4 μg/mL polybrene (TR‐1003‐G, Millipore). After infection, viruses were removed, and the medium was changed to hepatocyte maintaining medium for 48 h. To induce lysosome impairment, the cells were treated with cyclosporine A (10 μmol/L, 24 h, Sigma‐Aldrich). Isolated primary hepatocytes seeded on a 150‐mm dish were then infected in lentivirus medium with 4 μg/mL polybrene (TR‐1003‐G, Millipore). After infection, viruses were removed, and the medium was changed to hepatocyte maintaining medium for 48 h. The cells were then collected, washed twice with ice‐cold PBS, and pelleted via centrifugation at 1,000 ×g at 4°C for 2 min. The cells were then resuspended in PBS and homogenized using a Dounce homogenizer with 10 mild strokes followed by centrifugation at 1,000 ×g at 4°C for 2 min. After centrifugation, the homogenate was incubated with pre‐washed anti‐HA magnetic beads (88837, Thermo Scientific Pierce, Waltham, MA, USA) on a rotator shaker for 5 min at 4°C. The beads were then washed 3 times in cold PBS, and bead‐bound mitochondria then were lysed with modified RIPA buffer (for proteomics analysis) or extracted in 80% methanol solution (for cholesterol analysis). The samples were spun down at 20,000 ×g for 10 min to remove the remaining beads. For proteomics analysis sampling, SDS sampling buffer was added, and the samples were incubated at 95°C for 5 min and stored at ‐20°C. For lipidomic cholesterol analysis sampling, the supernatants were transferred to a new 2 mL tube without disturbing the pellets and stored at ‐80°C until analysis.
2.23. Lipidomic cholesterol enrichment analysis
Ammonium iodide, cholesterol, cholesterol‐d7, estradiol, hexane, methyl tert‐butyl ether (MTBE), N‐methyl‐N‐tert‐butyldimethylsilyl trifluoroacetamide (MTBSTFA), and palmitic acid were purchased from Sigma‐Aldrich. Mitochondrial lipids were extracted using the Matyash method [23] with some modifications. In brief, mitochondria samples were homogenized in 75% cold methanol (34860, Sigma‐Aldrich) using TissueLyser (Multi Reax, Heidolph Instruments, Schwabach, Germany), and MTBE (1 mL, 650560, Sigma‐Aldrich) was added and vortexed. For phase separation, 250 μL water was added and centrifuged (12,000 ×g, 15 min). The upper phase (220 μL) was dried under a nitrogen stream, reconstituted in 40 μL MTBSTFA (77626, Sigma‐Aldrich) and 40 μL ammonium iodide (221937, Sigma‐Aldrich), and then incubated for 20 min (80°C) [24]. After derivatization, the samples were evaporated, reconstituted using hexane (34859, Sigma‐Aldrich), and analyzed by gas chromatography‐mass spectrometry (GC‐MS). Cholesterol, estradiol, and palmitic acid were analyzed using a GC‐MS system (QP‐2020 Ultra, Shimadzu, Japan) in electron impact ionization mode (70 eV). Analytes were separated by a Rtx‐5 Sil MS column (30 m × 0.25 mm, 0.25 μm, Restek, Bellefonte, PA, USA). The sample was injected in split mode (2:1). The column oven temperature was programmed as follows: hold at 150°C for 1 min, ramp to 230°C at 30°C/min, further ramp to 280°C at 15°C/min, and finally increase to 320°C at 25°C/min. Helium was used as a carrier gas at a constant flow rate of 1.2 mL/min through the column. Quantitative determination was performed using single ion monitoring (SIM) mode at m/z 443, 443, 313, and 450 for cholesterol (13.6 min), estradiol (12.9 min), palmitic acid (7.4 min), and cholesterol d7 (13.5 min), respectively.
2.24. In vitro mitochondria‐lysosome contact reconstitution assay
Cells were treated with TopFluor cholesterol (5 μmol/L, 810255P, Sigma‐Aldrich) before lysosome isolation by density gradient centrifugation using a Lysosome Enrichment kit (89839, Thermo Fisher Scientific). The lysosome fractions were diluted with a reconstitution buffer (250 mmol/L sucrose, 1 mmol/L DTT, 1 mmol/L MgCl2, 50 mmol/L KCl, and 20 mmol/L HEPES, pH 7.2). For mitochondria isolation, cells stably expressing 3× HA‐EGFP‐OMP25 (#83356, Addgene) were homogenized in ice‐cold PBS with homogenizer and spun down at 800 × g at 4°C for 10 min. The supernatant was spun down at 8,000 × g at 4°C for 10 min to collect the pellets. The pellets were then suspended with reconstitution buffer. Isolated mitochondria and lysosomes were incubated in 1 mg/mL cytosol of Huh7 cells, 1 mmol/L ATP, 1 mmol/L GTP, and an ATP‐regenerating system (30 mmol/L creatine phosphate, 0.05 mg/mL creatine kinase) at 37°C for 30 min. To isolate mitochondria from the reaction, pre‐washed anti‐HA magnetic beads were added and incubated for 5 min with rolling (60 rpm). The beads were then washed with PBS 3 times and eluted with 80% methanol extraction buffer. The eluates were subjected to microscopy or a microplate reader (SpectraMAX i3×, Molecular Devices, San Jose, CA, USA).
2.25. Radioactive lipid binding assay
[1,2‐3H]‐Cholesterol (NET139001MC), 6,7‐3H(N)‐estradiol (NET317250UC), and 9,10‐3H(N)‐palmitic acid (NET043001MC) was purchased from Perkin Elmer Life Sciences (Waltham, MA, USA). One million Huh7 cells were seeded and transfected with streptavidin‐tagged TM4SF5 (wild‐type [WT] or mutants) for 48 h and lysed with RI lysis buffer (20 mmol/L HEPES, pH 7.4, 2 mmol/L MgCl2, 5 units/mL benzonase, and 2 mg/mL iodoacetamide). Lysed cells were collected by centrifugation at 12,000 ×g at 4°C for 10 min. The pellets were then re‐suspended in a solubilization buffer (1% n‐dodecyl‐β‐D‐maltoside [DDM], 0.1% cholesteryl hemisuccinate [CHS], 250 mmol/L NaCl, and 20 mmol/L HEPES, pH 7.4) and incubated at 4°C for 2 h. The samples were clarified by centrifugation at 12,000 ×g at 4°C for 10 min and the soluble fraction was then incubated with 50 μL streptavidin‐agarose beads at 4°C for 1 h. The beads were then washed 3 times with wash buffer (0.1% DDM, 0.01% CHS, 100 mmol/L NaCl, and 20 mmol/L HEPES, pH 7.4). Strep‐TM4SF5 conjugated beads were then incubated with 3H‐lipid at 50 μmol/L or different concentrations for 1 h. Then, the beads were washed 3 times with wash buffer, and the remaining 3H‐lipid was counted using a liquid scintillation analyzer (Tri‐Carb 2910TR, Perkin Elmer).
2.26. Molecular docking of TM4SF5 with cholesterol
Our previously constructed model structure of hTM4SF5 [25] was utilized to show the cholesterol binding mode. Molecular graphic figures were generated using PyMOL v.2.5.2 software (Schrödinger, LLC).
2.27. mRNA sequencing (mRNA‐seq)
mRNA‐seq analysis using stably TM4SF5‐transfected SNU449 cells and TM4SF5‐null cells, or liver tissues from 3‐month‐old male WT and Tm4sf5 −/− C57BL/6N mice (n = 6) was performed by a commercial service (Macrogen, Seoul, Korea), as described previously [26]. In brief, cDNA libraries were constructed with the TruSeq RNA Sample Prep Kit v2 (RS‐122‐2001, Illumina, San Diego, CA, USA) using total RNA (1 μg) prepared from cell lines or mouse liver tissues. The protocol was consisted of polyA‐selected RNA extraction, RNA fragmentation, random hexamer primed reverse transcription and 100nt paired‐end sequencing by Illumina HiSeq4000 (Illumina, San Diego, CA, USA). The libraries were quantified using qPCR according to the qPCR Quantification Protocol Guide and qualified using an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
2.28. Gene expression correlation analysis
Pearson Correlation Coefficient (r‐value) was calculated between TM4SF5 and target gene expression data from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/; GSE48452, GSE164760, GSE14520, GSE28735, GSE19804, and GSE44076) and The Cancer Genome Atlas (TCGA; https://www.cancer.gov/ccg/research/genome‐sequencing/tcga; TCGA‐LIHC, PAAD, KIRC, LUAD, COAD, BRCA, and DLBC). GSEA compute overlapping was performed to analyze the enrichment of proteins by gene ontology (GO) cellular components annotation. To compare multiple coexpression data of TM4SF5, COXPRESdb (http://coxpresdb.jp) was used. Top 100 co‐expressing gene list was obtained and analyzed by GO annotation.
2.29. Statistical analysis
Statistical analyses were performed using Prism software version 7.0 (GraphPad, La Jolla, CA, USA). Two‐way analysis of variance (ANOVA), one‐way ANOVA, Mann‐Whitney U test, or unpaired Student's t tests were performed to determine statistical significance. A value of P < 0.05 was considered statistically significant.
3. RESULTS
3.1. TM4SF5 localized at MLCSs in hepatocytes
We previously reported that TM4SF5 can traffic from the plasma membrane to lysosomes upon L‐arginine repletion after prior depletion [5]. Based on those findings, we hypothesized that TM4SF5 could translocate to different organelles in response to various metabolic needs or signals. To evaluate this, we performed an LC/MS proteomic analysis to determine the possible intracellular localization of TM4SF5 and the cellular function of TM4SF5 trafficking. We observed localization of protein binding partners of TM4SF5 in mitochondria as well as the plasma membrane, although ER‐related proteins might also be involved in the de novo synthesis of TM4SF5 protein (Supplementary Figure S1A‐B). Co‐expressed genes and mRNA‐seq analysis revealed that TM4SF5 was related to diverse metabolic pathways including cholesterol metabolism (Supplementary Figure S1C‐D). SNU449 or SNU761 hepatocytes expressed minimal or hardly detectable levels of endogenous TM4SF5, whereas Huh7 and HepG2 hepatocytes endogenously expressed TM4SF5 (Supplementary Figure S1E). Therefore, immunofluorescent microscopic imaging revealed localization of TM4SF5 at MLCSs. Microscopic labeling analyses using SNU449 cells with or without TM4SF5 expression revealed that MLCS formation was enhanced by TM4SF5 expression (Figures 1A, Supplementary Figure S1E, lower panel). Consistent with these results, suppression of TM4SF5 in Huh7 cells with endogenous TM4SF5 expression led to decreased MLCS formation, although TM4SF5‐independent MLCS formation might occur because incomplete MLCS abolishment was observed even when TM4SF5 was almost completely suppressed (Figure 1B, Supplementary Figure S1F). In confocal microscopic images of SNU449 cells transfected with mCherry‐TM4SF5, the TM4SF5 signal overlapped with regions containing both mitochondria (Mito‐GFP) and lysosomes (LAMP1‐stain) (Figure 1C, “a”) but not with regions containing mitochondria but no lysosomes (Figure 1C, “b”). A similar analysis of SNU449 cells labeled with mito‐GFP and BFP‐KDEL (Lys‐Asp‐Glu‐Leu), an ER‐specific stain, showed that TM4SF5 appeared to minimally localize at mitochondria‐ER contacts but did localize at mitochondria located away from the ER (presumably with appositions to lysosomes) (Supplementary Figure S1G). To observe the subcellular localizations in more detail, we took transmission electron microscopic images of SNU449 cells transfected with APEX2‐TM4SF5. The images clearly showed APEX2 staining (i.e., black dots) between lysosomes and adjacent mitochondria (Figure 1D). Airyscan confocal super‐resolution microscopy images also showed TM4SF5 localization at contact sites between lysosomes and mitochondria in SNU449 cells, although a certain portion of TM4SF5 was also located in endosomes and lysosomes without contact with mitochondria (Figure 1E). Furthermore, snap images of SNU449 cells taken during confocal live imaging clearly showed dynamic colocalization of mCherry‐TM4SF5 and Mito‐GFP stains (i.e., at MLCSs; Figure 1F).
FIGURE 1.
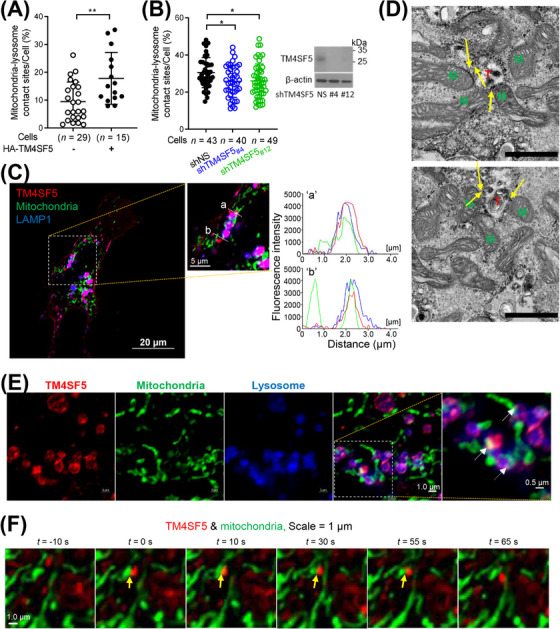
TM4SF5 localized at MLCSs in hepatocytes. (A) Quantification of MLCSs/cell from SNU449 hepatocytes transfected with mito‐GFP plus HA‐empty vector (EV) or HA‐TM4SF5 and treated with lysotracker. Images were analyzed for mitochondria‐lysosome co‐stains over total lysosome stains. Twenty‐nine EV and 15 TM4SF5 cells were analyzed and combined from three independent experiment. Data were represented as mean ± standard deviation (SD). ** P < 0.01; two‐tailed unpaired t test. (B) Quantification of MLCSs from Huh7 cells stably transfected with mito‐GFP plus non‐specific shRNA (shNS, n = 43), shTM4SF5 #4 (n = 40), or shTM4SF5 #12 (n = 49) and labelled with lysotracker. Cells in normal culture media were analyzed from 3 independent experiment. * P < 0.05; ordinary one‐way ANOVA. (C) SNU449 cells transfected with mCherry‐TM4SF5 (red) and mito‐GFP (green) were processed for confocal microscopy with LAMP1 immunostaining (blue) (Insert “a” and “b”). Fluorescence intensities over the (“a”) and (“b”) lines were measured and graphed. (D) SNU449 cells transfected with APEX2‐TM4SF5 were glucose starved for 16 h and then replete with 10 mmol/L glucose for 30 min, before processing to DAB staining and electron microscopy. M, mitochondria; T, TM4SF5 stains on lysosome; yellow arrow, mitochondria‐lysosome contact site. Scale bar = 1 μm. (E‐F) SNU449 cells transfected with mCherry‐TM4SF5 (red) and mito‐GFP (green) were imaged for LAMP1 fluorescence (blue) using an Airyscan microscope (E) or confocal live imaging (F). Data represent 3 independent experiments. See also Supplementary Figure S1. APEX2‐TM4SF5, TM4SF5 tagged with ascorbate peroxidase 2 (APEX2); DAB, diaminobenzidine, LAMP1, lysosomal‐associated membrane protein 1; EV, empty vector; mito‐GFP, mitochondria‐targeting sequence of COX4 tagged with green fluorescent protein gene; MLCS, mitochondria‐lysosome contact site; shNS, shRNA against non‐specific sequence; shTM4SF5, shRNA against TM4SF5 sequences (of #4 or #12).
3.2. Extracellular glucose repletion caused TM4SF5 translocation to MLCSs
The dynamic alterations in subcellular TM4SF5 localization suggested that TM4SF5 translocated to MLCSs in response to extracellular stimuli, such as growth factors or nutrient availability. To evaluate this, we cultured SNU449 cells in media depleted of specific nutrients or serum and then replenished the depleted components prior to fluorescent imaging analysis of many different cells at least more than 20 cells per experimental condition. We found that glucose depletion (16 h) and repletion (10 mmol/L for various periods) induced lysosomal localization of TM4SF5 in mCherry‐TM4SF5‐transfected SNU449 cells (Figure 2A, Supplementary Figure S2A). Consistent with that, ultracentrifuge‐based fractionation of lysosomes from Huh7 cells with or without TM4SF5 suppression in the presence of glucose depletion (16 h) followed by repletion (25 mmol/L for 30 min) showed lysosomal enrichment of TM4SF5 upon glucose repletion (Figure 2B). Furthermore, under the same experimental conditions, TM4SF5 colocalization with ZO1 or EGFR at the plasma membrane of SNU449 cells was reduced upon glucose repletion (Figure 2C), indicating that TM4SF5 translocated from the plasma membrane to MLCSs in response to extracellular replenishment of glucose. To confirm that TM4SF5 localization at MLCSs could be transiently altered by glucose treatment, we calculated Pearson's correlations of mCherry‐TM4SF5 and MLCS (i.e., nearby mito‐GFP) signals in time‐lapse images of SNU449 cells after glucose repletion. We found that the colocalization of TM4SF5 and MLCSs was increased 30 min after glucose repletion and returned to baseline levels 60 min after glucose repletion (Figure 2D, Supplementary Figure S2B). Furthermore, treatment with the glycolytic inhibitor 2‐Deoxy‐D‐glucose (2‐DG; 25 mmol/L, 30 min) reduced the amount of TM4SF5 at MLCSs (yellow spots near mito‐GFP in confocal images) in the SNU449 cells (Figure 2E, Supplementary Figure S2C). In contrast to glucose depletion and repletion, extracellular depletion (16 h) and repletion of L‐arginine (1 mmol/L) or glutamine (2 mmol/L) did not significantly change the TM4SF5 levels at MLCSs (Figure 2F‐G, Supplementary Figure S2D). TM4SF5‐positive Huh7 cells showed a transient increase (at 30 min) followed by a decrease (at 60 min) in MLCS formation, as indicated by colocalization of mito‐GFP and lysotracker stains, whereas TM4SF5‐suppressed Huh7 cells did not show significant changes in MLCS formation after glucose depletion and repletion (Figure 2H, Supplementary Figure S2E). Furthermore, TM4SF5 levels at MLCSs were reduced after the cells were treated with TSAHC, a specific TM4SF5 inhibitor [17] (Figure 2I, Supplementary Figure S2F). Because MLCSs are known to be regulated by Rab7 [27], we checked for Rab7 dependency of the TM4SF5‐mediated MLCSs. Rab7 suppression reduced the numbers of TM4SF5‐mediated MLCSs compared with those in non‐suppressed cells, although there were still more TM4SF5‐mediated MLCSs than TM4SF5‐independent MLCSs in the Rab7‐suppressed cells (Figure 2J). This indicated that TM4SF5 could support MLCS formation even without Rab7. Furthermore, HA‐TM4SF5‐mediated MLCS formation was significantly inhibited in hepatocytes expressing mutant variants, like palmitoylation‐deficient TM4SF5 (Pal−, C2/6/9/74/75/79/80/84/189A) or C62A mutant TM4SF5 (mutated residue at the 2nd transmembrane domain) (Figure 2K‐L). Other HA‐TM4SF5 mutants with alterations in extracellular loop 2 (or the long extracellular loop), including C118A, F128A, A132V, N138A, and C145A, still translocated to MLCSs similarly to WT TM4SF5 (Supplementary Figure S2G). These observations suggested that TM4SF5 can translocate to MLCSs in response to extracellular glucose availability.
FIGURE 2.
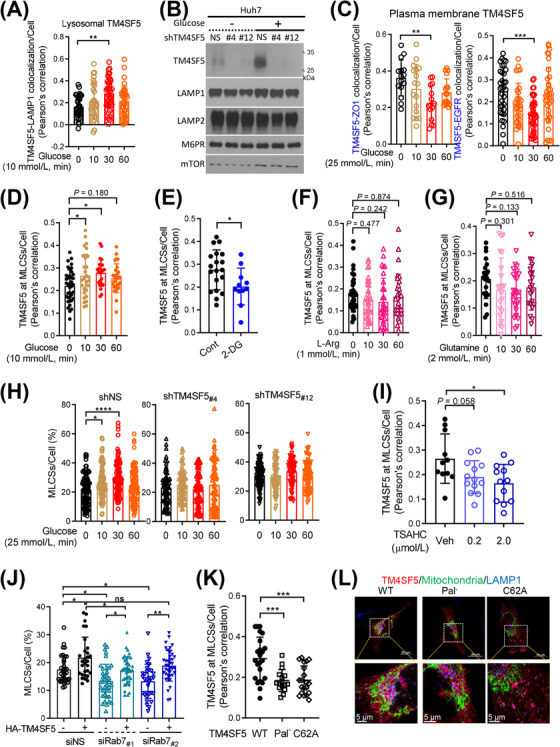
Extracellular glucose repletion caused TM4SF5 translocation to MLCSs. (A) SNU449 cells transfected with mCherry‐TM4SF5 and LAMP1 labelling were glucose starved for 16 h and then replete with 10 mmol/L glucose for various periods before LAMP1 fluorescence imaging using a confocal microscope. TM4SF5‐LAMP1 colocalization was determined by Pearson's correlation. Each data point depicts a correlation value per cell, and each experimental condition included measurements from multiple cells (n = 34, 30, 38, 34 cells per experimental condition, respectively). (B) Huh7 cells stably transfected with non‐specific shRNA (shNS), shTM4SF5 #4, or shTM4SF5 #12 were glucose depleted (16 h) and then replete with 25 mmol/L glucose (30 min) before lysosome isolation by ultracentrifugation for immunoblots. (C) Cells manipulated as in (A) were imaged for TM4SF5 colocalization with ZO1 or EGFR at plasma membranes. (D‐E) Fluorescence in SNU449 cells transfected with mCherry‐TM4SF5 and mito‐GFP were treated with different glucose levels in RPMI‐1640 culture media for the indicated time and then confocally imaged for quantification of TM4SF5 at MLCSs/cell by Pearson's correlation (D), or quantification of them without or with 2‐DG treatment (25 mmol/L, 30 min; E). (F‐G) SNU449 cells manipulated as in (A) were L‐arginine‐depleted (for 16 h) and then replete (1 mmol/L for various times, F) or glutamine‐depleted (for 16 h) and then replete (2 mmol/L glutamine for various times, G), before quantification of TM4SF5 at MLCSs per cell. (H) Huh7 cells stably transfected with shNS, shTM4SF5 #4, or shTM4SF5 #12 together with mito‐GFP constructs and lysotracker labeling were glucose‐depleted for 16 h and then replete with 25 mmol/L glucose for various periods before quantification of MLCSs/cell. (I) SNU449 cells as in (E) were rather treated with TSAHC for 24 h before quantification of TM4SF5 at MLCSs per cell. (J) Quantifications of MLCSs/cell were done from SNU449 (‐EV or ‐TM4SF5) cells transfected with non‐specific siRNA (siNS) or Rab7 siRNA (siRab7 #1 or siRab7 #2) together with mito‐GFP and lysotracker labelling. (K‐L) SNU449 cells transfected with HA‐TM4SF5 WT, Pal− mutant, or C62A mutant and mito‐GFP were stained for HA (red) and LAMP1 (blue) before imaging (L) and calculation of TM4SF5 at MLCSs per cell (K). Each datapoint indicates a measurement per cell. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, ns = non‐significant, unpaired Student's t tests or ordinary one‐way ANOVA. Data were represented as mean ± SD. Data represent three independent experiments. See also Supplementary Figure S2. L‐arg, L‐arginine; MLCS, mitochondria‐lysosome contact site; Rab7, member 7 of the RAB family of small GTPases; shRNA, short hairpin RNA; TM4SF5, transmembrane 4 L six family member 5.
3.3. TM4SF5‐FK506‐binding protein 8 (FKBP8) linkage at MLCSs
We next used engineered biotin ligase (TurboID)‐based proximity labeling to determine which proteins participate in TM4SF5 enrichment at MLCSs. For this approach, Huh7 cells were transfected with TurboID conjugated to v5‐tagged translocase of outer membrane 20 (TOM20‐v5‐TurboID), lysosomal associated membrane protein 1 (LAMP1‐v5‐TurboID), or TM4SF5 (TM4SF5‐v5‐TurboID) (Figure 3A). The v5‐TurboID molecules biotinylated themselves and neighboring proteins after transfection (Supplementary Figure S3A). The biotinylated proteins were then enriched using streptavidin beads for LC/MS analysis. The proteins biotinylated by TM4SF5‐v5‐TurboID included 16 mitochondrial proteins (normalized to NES‐v5‐TurboID, FC > 1.5 and P < 0.05; Supplementary Figure S3B). The proteins biotinylated by TOM20‐v5‐TurboID and LAMP1‐v5‐TurboID (each normalized to NES‐v5‐TurboID, FC > 1.5 and P < 0.05) were compared, and the 63 proteins common to both clusters were considered as the MLCS proteome (Figure 3B). We then subdivided the MLCS proteome into TM4SF5‐enriched MLCSs and TM4SF5‐absent MLCSs. The protein in the TM4SF5‐enriched MLCSs with the highest FC between the TM4SF5‐v5‐TurboID cells and the NES‐v5‐TurboID cells was FKBP8 (also known as FKBP38; Figure 3C). KEGG classification showed that the TM4SF5‐enriched MLCSs included pathways for vesicular transport, autophagy, and ferroptosis (Figure 3D, upper panel), which were not present in the TM4SF5‐absent MLCSs (Figure 3D, lower panel).
FIGURE 3.
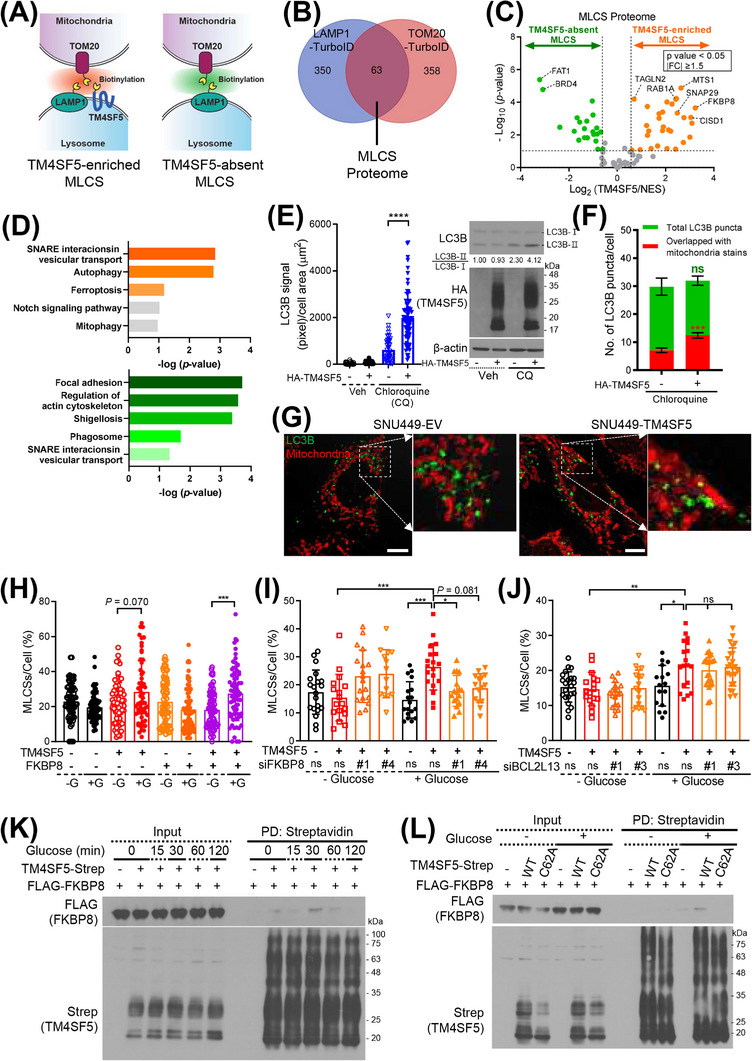
TM4SF5‐FKBP8 linkage at MLCSs. (A) Schematic of TurboID (TM4SF5‐v5‐, LAMP1‐v5‐, or TOM20‐v5‐TurboID) transfected into Huh7 cells to show MLCSs. (B) Venn diagram to show proteins clustered to TOM20‐v5‐TurboID or LAMP1‐v5‐TurboID. (C) Protein clusters at TM4SF5‐absent or TM4SF5‐enriched MLCSs after normalization of TM4SF5‐v5‐TurboID protein clusters to NES‐v5‐TurboID clusters. (D) KEGG pathways for TM4SF5‐enriched (upper panel) or TM4SF5‐absent (lower panel) MLCS protein clusters. (E‐G) SNU449 cells stably transfected with HA‐empty vector (EV) or HA‐TM4SF5 were treated with 10 μmol/L CQ for 24 h without (E; n = 32, 49, 43, or 70) or with mitotracker labeling (500 nmol/L, 30 min; F; n = 20 or 27), before anti‐LC3B immunofluorescent staining or immunoblotting (E, right). Confocal images (G) were captured, and intensities of total or mitochondrial LC3B signal (F) were analyzed. **** P < 0.0001, ordinary one‐way ANOVA (E) and *** P < 0.001, two‐tailed unpaired t test (F). (H‐J) Quantification of MLCSs/cell of SNU449 cells transfected with HA‐EV or HA‐TM4SF5, FLAG‐FKBP8, and mito‐GFP (H), or with HA‐EV or HA‐TM4SF5, mito‐GFP, and non‐specific siRNA (siNS) or siRNA for FKBP8 (siFKBP8 #1 or siFKBP8 #4, I) or BCL2L13 (BCL2L13 #1 or siBCL2L13 #3, J) were glucose‐starved and then replete as above before lysotracker labeling. Cell number n was 73, 73, 74, 68, 73, 68, 73, or 62 (H), 20, 17, 18, 14, 18, 19, 17, or 15 (I), or 22, 17, 19, 17, 16, 18, 21, or 24 (J) from left to right conditions. * P < 0.05, ** P < 0.01, *** P < 0.001, or **** P < 0.0001, ns = non‐significant, ordinary one‐way ANOVA. (K‐L) SNU449 cells transfected with TM4SF5‐strep and FLAG‐FKBP8 (K) or with either TM4SF5‐strep WT or C62A mutant and FLAG‐FKBP8 (L) were manipulated as above. Whole‐cell lysates were subjected to streptavidin pulldown and immunoblots. Data were represented as mean ± SD. Cell number n and data represent three independent experiments. See also Supplementary Figure S3. BCL2L13, B‐cell lymphoma 2 (BCL2)‐Like 13; CQ, chloroquine; EV, empty vector; FKBP8, FK506‐binding protein family 8; LC3B, microtubule‐associated proteins 1A/1B light chain 3B; MLCS, mitochondria‐lysosome contact site; PD, pulldown; SD, standard deviation; TM4SF5, transmembrane 4 L six family member 5; TurboID, an engineered biotin ligase that uses ATP to convert biotin into biotin‐AMP, a reactive intermediate that covalently labels proximal proteins; WT, wild‐type.
MLCSs have been linked to mitophagy [28, 29]. To determine if TM4SF5 expression affects autophagy, we used fluorescent LC3B immuno‐staining with or without chloroquine treatment (10 μmol/L, 24 h) to block LC3B protein degradation. HA‐TM4SF5 expression, but not control empty vector (EV) expression, caused an increased amount of LC3B staining in the regions with mitochondrial staining upon chloroquine treatment (Figure 3E), indicating that TM4SF5 increased autophagic LC3B activity. We also determined how much the LC3B stain overlapped with the mitochondrial tracker stain. In TM4SF5‐positive cells with chloroquine treatment, the LC3B stain could be significantly increased in overlapping with mitochondrial tracker stains, although the total LC3B stains were not significantly changed (Figure 3F‐G). This indicated that TM4SF5‐enriched MLCSs might be involved in autophagic activity around mitochondria.
We next sought to determine which proteins are targeted by lysosomal TM4SF5 during MLCS formation. TM4SF5 localization at MLCSs upon glucose repletion might involve FKBP8 (Figure 3C), a mitochondrial outer‐membrane protein that inhibits apoptosis by binding to Bcl‐2 [30] but also promotes stress‐induced mitophagy [31]. We found that TM4SF5‐negative cells with or without FLAG‐FKBP8 overexpression did not show increased MLCS formation upon glucose repletion; however, HA‐TM4SF5‐positive cells showed insignificant (P = 0.070) but an increasing trend in glucose‐mediated formation of MLCSs, which was further enhanced by FLAG‐FKBP8 overexpression (Figure 3H). Furthermore, although neither TM4SF5 expression nor FKBP8 suppression significantly changed the MLCS levels upon glucose starvation (16 h), FKBP8 suppression abolished the increase in HA‐TM4SF5‐dependent MLCS formation upon glucose repletion (Figure 3I, Supplementary Figure S3C). By contrast, suppression of another mitophagy receptor, BCL2L13 (Supplementary Figure S3C), did not affect the formation of TM4SF5‐enriched MLCSs upon glucose repletion (Figure 3J). These observations suggested that TM4SF5 on lysosomes can interface with FKBP8 on mitochondria to form MLCSs. To evaluate if TM4SF5 and FKBP8 could interact directly to form an interface, we subjected hepatocytes expressing control empty vector or TM4SF5‐strep together with FLAG‐FKBP8 to glucose depletion followed by repletion for various times. We then harvested whole‐cell extracts for coprecipitation experiments. We found that the binding of TM4SF5‐strep to FLAG‐FKBP8 was transient, peaking at 30 min after glucose repletion and declining thereafter (Figure 3K). Because TM4SF5 can form multimers via N‐glycosylation and/or palmitoylation [4, 32], the TM4SF5 immunoblots showed multiple bands. Furthermore, the binding was abolished in C62A mutant TM4SF5, which also was not enriched at MLCSs (Figure 2K, Figure 3L). Transgelin 2 (TAGLN2), an actin‐binding protein at immunological synapses [33] that was also included in TM4SF5‐enriched MLCSs (Figure 3C), did not bind to TM4SF5 regardless of glucose treatment (Supplementary Figure S3D‐E). Other proteins related to autophagy pathways, including starch binding domain 1 (STBD1) and CCZ1 homolog, vacuolar protein trafficking and biogenesis associated (CCZ1), appeared to be involved in TM4SF5‐enriched MLCSs (Supplementary Figure S3F) even upon glucose repletion (Supplementary Figure S3G). However, unlike FKBP8, CCZ1 and STBD1 did not bind to TM4SF5 (Supplementary Figure S3H‐I).
3.4. Mitochondrial dynamics with proteins clustered at TM4SF5‐enriched MLCSs
We examined the mRNA expression of mitophagy receptors that might be related with TM4SF5‐dependent MLCS formation upon glucose repletion. Using qRT‐PCR, we found that the mRNA levels of certain receptors (BCL2L13, PHB2, AMBRA1, OPA1, and VDAC1) were increased by glucose repletion in HA‐TM4SF5‐expressing SNU449 cells compared with those in non‐glucose‐replete TM4SF5‐null SNU449 cells (Supplementary Figure S4A). These observations suggested that TM4SF5‐enriched MLCS formation upon glucose repletion may lead to mitophagy. Indeed, live confocal microscopic imaging of SNU449 cells suggested that the presence of TM4SF5 at MLCSs was involved in mitochondrial fragmentation (Figure 4A). Furthermore, TEM images showed that mitochondria in HA‐TM4SF5‐positive SNU449 cells were smaller than those in TM4SF5‐negative SNU449 cells (Figure 4B). Quantitative analysis of mitochondrial lengths showed that HA‐TM4SF5 expression reduced the length of mitochondria upon glucose repletion, but not under the glucose‐starved condition (Figure 4C).
FIGURE 4.
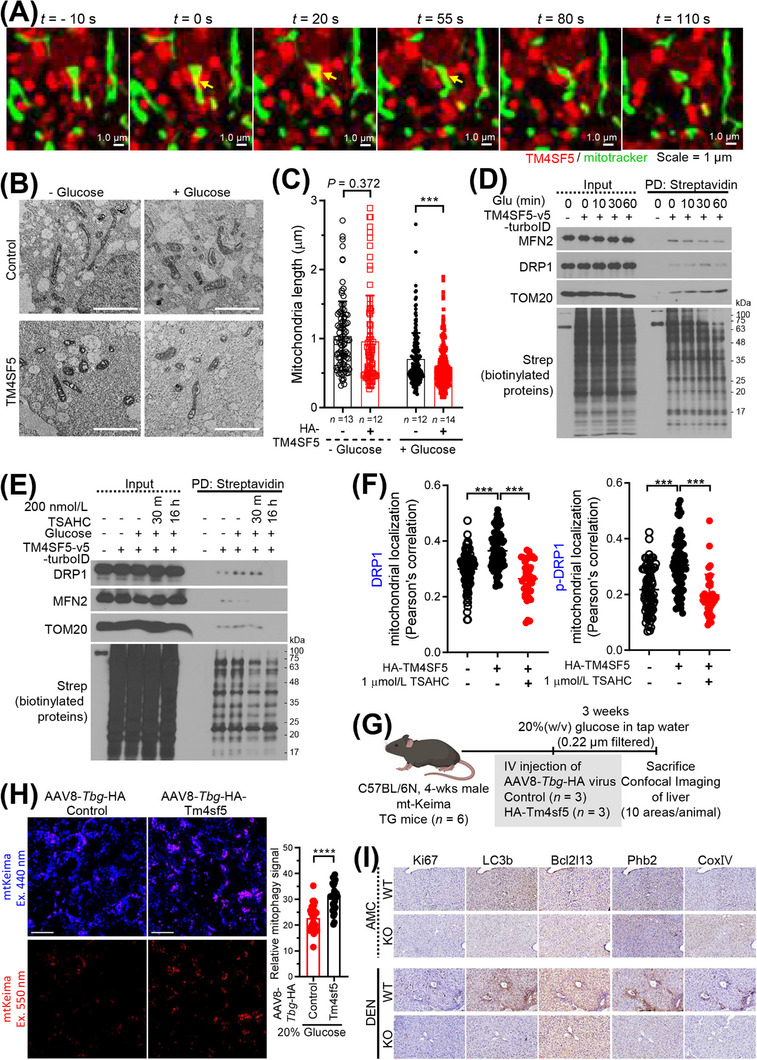
Mitochondrial dynamics via proteins clustered at TM4SF5‐enriched MLCSs. (A) SNU449 cells transfected with mCherry‐TM4SF5 and mito‐GFP were subjected to confocal live imaging. Representative images are shown as snapshots. (B‐C) SNU449 cells stably expressing HA‐EV or HA‐TM4SF5 were glucose starved and then replete before electron microscopy imaging. Multiple images of cells (n = 13, 12, 12, or 14) were processed for mitochondrial length measurement under each experimental condition (C). *** P < 0.001, ordinary one‐way ANOVA. (D‐E) SNU449 (D) or Huh7 (E) cells transfected with TM4SF5‐v5‐TurboID were glucose depleted for 16 h and then replete (10 mmol/L or 25 mmol/L glucose for various times or 30 min, respectively). Whole‐cell lysates were subjected to streptavidin PD and immunoblot. (F) SNU449 cells with stable HA‐EV or HA‐TM4SF5 expression were transfected with mito‐GFP plasmids and treated with 1 μmol/L TSAHC for 16 h. Confocal images for anti‐HA, anti‐DRP1, or anti‐p‐DRP1 were captured, and DRP1‐mitochondria (n = 93, 72, or 40) or p‐DRP1‐mitochondria (n = 83, 73, or 38) colocalization was assessed by Pearson's correlation. * P < 0.05, ** P < 0.01, and *** P < 0.001, ordinary one‐way ANOVA. Data were represented as mean ± SD. (G) Animal study scheme for In vivo mitophagy using mtKeima TG mice. (H) Representative images of red In vivo mitophagy signal in liver tissues from mice (n = 3/group) intravenously injected with AAV8‐Tbg‐HA‐Tm4sf5, but not control virus (left). Ten random areas of liver from a mouse were imaged and the red signal was quantitated for a graph (right). **** P < 0.0001, two‐tailed unpaired t test. (I) Liver tissues from WT or KO mice (age‐matched control, AMC, or DEN‐treated for 45 weeks as an HCC model, n ≥ 5/group) were immuno‐stained as indicated. Representative images were shown. Data represent three isolated experiments. See also Supplementary Figure S4 and Supplementary Figure S5. AMC, age‐matched control; DEN, diethylnitrosamine; DRP1, dynamin‐related protein 1; EV, empty vector; KO, knockout; MLCS, mitochondria‐lysosome contact site; PD, pulldown; SD, standard deviation; Tbg, thyroxine binding globulin; TM4SF5, transmembrane 4 L six family member 5; WT, wild‐type.
We next asked if regulators of mitochondrial dynamics, including mitofusin 2 (MFN2) and dynamic‐related protein I (DRP1), were engaged in TM4SF5‐mediated MLCS formation and function. Hepatocytes transfected with control NES‐v6‐ or TM4SF5‐v5‐TurboID were subjected to glucose depletion and repletion at various times, followed by pulldown of biotinylated proteins using streptavidin and immunoblotting. In contrast to control‐transfected cells, TM4SF5‐v5‐TurboID‐transfected cells showed biotin labeling of MFN2, DRP1, and TOM20 in proximity to TM4SF5‐v5‐TurboID. Furthermore, the proximity of biotin‐labeled TOM20 and DRP1 to TM4SF5‐v5‐TurboID gradually increased over time after glucose repletion, whereas that of biotin‐labeled MFN2 diminished (Figure 4D), indicating that more DRP1 interfaced with TM4SF5 as TM4SF5‐enriched MLCSs were formed, eventually leading to DRP1‐based mitochondrial fission. These proximity patterns were abolished by treatment with TSAHC (Figure 4E), a specific TM4SF5 inhibitor that shows interruption of protein‐protein interactions [5, 9, 17], and the proteins showed no proximity to lysosomal TM4SF5 (presumably without apposition to mitochondria) after glucose repletion (Supplementary Figure S4B). Furthermore, HA‐TM4SF5‐mediated DRP1 or phospho‐DRP1 localization at mitochondria was inhibited by TSAHC treatment (Figure 4F). These observations suggested that TM4SF5‐enriched MLCS formation can lead to mitochondrial fission and mitophagy. To further evaluate the relevance of mitophagy in different pathological statuses of the liver, we analyzed mitophagy‐related gene expression levels in GSE or TCGA databases. Compared to healthy human livers, livers from human patients with nonalcoholic fatty liver disease (NAFLD including steatosis and nonalcoholic steatohepatitis [NASH]) or HCC showed increased expression levels of mitophagy‐related genes (Supplementary Figure S4C). Interestingly, their levels were higher in HCC patients than in other cancer patients (Supplementary Figure S4C). We further determined mRNA levels of selected mitophagy receptors from liver tissues of WT or Tm4sf5 −/− (KO) mice fed with HSuD or HFGluD for 10 weeks or treated with DEN for 45 weeks. Livers from WT mice fed with HSuD or treated with DEN showed insignificantly higher Bcl2l13 (P = 0.072) and significantly Ambra1 mRNA levels, compared with those from KO mice, although WT fed HFGluD did not presumably due to the overwhelming influence of HFD over glucose (Supplementary Figure S4D‐F).
We then explored if TM4SF5 expression promoted In vivo mitophagy using mtKeima transgenic mice [19]. We have thus injected control or AAV8‐Tbg‐HA‐Tm4sf5 virus to the mtKeima mice fed with NCD containing 20% (W/V) glucose (Figure 4G). Expression of Tm4sf5 in hepatocytes (via hepatocyte‐specific promoter [34]) in mtKeima TG mice significantly promoted mitophagy in livers following glucose uptake, compared to control virus‐injected mice (Figure 4H, Supplementary Figure S5A). Instead using the mtKeima TG mice, we have intravenously injected pLVX‐mtKeima virus to WT or Tm4sf5 −/− KO mice fed with NCD plus 20% (W/V) glucose (i.e., HGluD) for 4 weeks (Supplementary Figure S5B). Unlike KO mice, WT mice showed significantly increased mitophagy in livers, although when fed with NCD, no significant changes were observed (Supplementary Figure S5C‐D). Furthermore, immunostaining of Bcl2l13 or Phb2 mitophagy receptors in liver tissues of DEN‐treated WT mice increased in Ki67 and LC3B‐positive areas, but not in in age‐matched controls (Figure 4I). Immunoblots confirmed these results (Supplementary Figure S5E). Therefore, these observations support that TM4SF5 expression in hepatocytes could promote mitophagy upon extracellular glucose supply during the development of liver malignancy.
3.5. Relationship between TM4SF5‐enriched MLCSs and cholesterol metabolism
In addition to regulating organelle dynamics, inter‐organelle MCSs enable the transfer of metabolites, such as phospholipids, sterols, calcium, and iron [28]. An mRNA‐seq analysis of liver tissues from 3‐month‐old male WT and Tm4sf5−/− KO mice (n = 6) revealed enhanced cholesterol metabolism and elevated levels of cholesterol‐related hypertrophic cardiomyopathy in WT mice compared with Tm4sf5−/− KO mice (Figure 5A). Analysis of gene co‐expression suggested that TM4SF5 expression might be correlated with expression of genes involved in the regulation of cholesterol metabolism (Supplementary Figure S1C). Because subcellular translocalization of TM4SF5 is affected by cholesterol depletion, indicating a functional relationship between TM4SF5 and cholesterol [4], we rationalized that TM4SF5 enrichment at MLCSs might be linked to intracellular cholesterol metabolism or transfer. Cholesterol is transported from lysosomes to mitochondria through MLCSs in living fibroblasts [29]. TM4SF5 is overexpressed in HCC [21, 35], and its intracellular translocation appeared to be changed by intracellular cholesterol depletion, so we checked 2 public HCC datasets (GSE164760 and GSE14520) for evidence that TM4SF5 expression is correlated with expression of 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR), a rate‐limiting enzyme for cholesterol synthesis [36]. Both datasets showed higher TM4SF5 mRNA expression in HCC than in non‐tumor controls, and the GSE14520 dataset also showed higher HMGCR mRNA expression in HCC than in controls (Figure 5B). Furthermore, the expression of TM4SF5 and HMGCR was positively (weakly though) correlated with that of TM4SF5 in tumor samples but not in non‐tumor controls (Figure 5C). Data from the TCGA database of liver cancer (LIHC) also showed positive correlations between the mRNA levels of TM4SF5 and HMGCR (Figure 5D). Moreover, similar to HCC, pancreatic tumors (GSE28735) also showed increased TM4SF5 and HMGCR mRNA expression compared with non‐tumor controls, with a positive correlation between TM4SF5 expression and HMGCR expression. In contrast, the TM4SF5 and HMGCR mRNA expression levels were not parallelly increased in colon cancer (GSE44076) and lung cancer (GSE19804) compared with controls (Supplementary Figure S6A‐C).
FIGURE 5.
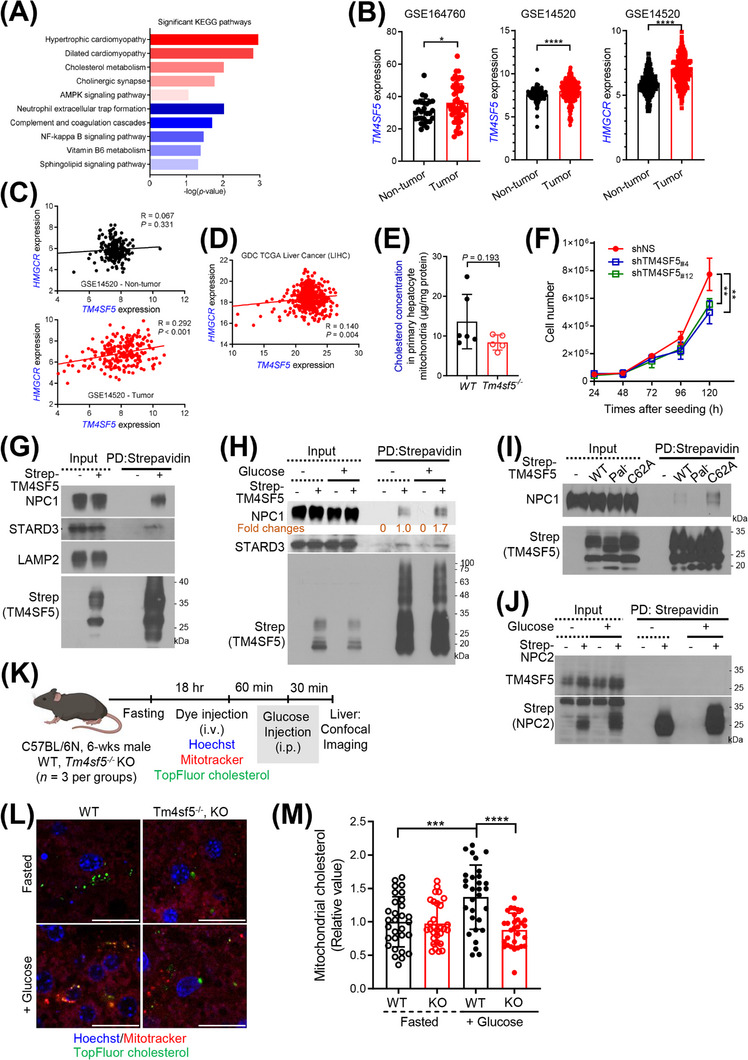
Relationship between TM4SF5‐enriched MLCSs and cholesterol metabolism. (A) KEGG pathway analysis of significantly upregulated (red) or downregulated (blue) genes in liver tissues of WT mice compared with Tm4sf5 −/‐ KO mice (|FC| ≥ 2 and P < 0.05). (B‐D) TM4SF5 and HMGCR expression and their correlations in the GSE164760 and GSE14520 datasets (B‐C) and the TCGA liver cancer (LIHC) dataset (D). (E) Primary hepatocytes isolated from WT (n = 6) or Tm4sf5 −/− KO (n = 5) mice were infected with HA‐OMP25 lentivirus. Mitochondria isolated by pulldown using anti‐HA‐magnetic beads were processed for LC/MS cholesterol analysis. Mann‐Whitney test. (F) Numbers of stable Huh7 cells with or without TM4SF5 suppression. (G‐J) Huh7 cells transfected with Strep‐empty vector (EV); Strep‐TM4SF5 WT, Pal−, or C62A; (G‐I) or Strep‐NPC2 (J) were harvested without (G, I) or with glucose starvation and repletion (H, J). Whole‐cell lysates were subjected to pulldown using streptavidin beads for immunoblots. (K‐M) WT or Tm4sf5−/− KO mice (n = 3) were fasted overnight and intravenously injected with DNA dye, mitotracker, and TopFluor cholesterol, 60 min before intraperitoneal injection of glucose (K). Thirty min later, livers were imaged (L). Mitochondrial cholesterol levels (e.g., yellow stains for green cholesterol within red mitochondria) were quantified from 5 random images per animal for a graphic presentation (M). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, ordinary one‐way ANOVA. Data were represented as mean ± SD. Data represent three independent experiments. See also Supplementary Figure S1, Supplementary Figure S6, and Supplementary Figure S7. EV. empty vector; FC, fold change; GSE, Gene Expression Omnibus series of List of expression profiles that conducted for the experiment; KEGG, Kyoto Encyclopedia of Genes and Genomes; KO, knockout; LIHC, Liver Hepatocellular Carcinoma; MLCS, mitochondria‐lysosome contact site; SD, standard deviation; TCGA, the cancer genome atlas program; TM4SF5, transmembrane 4 L six family member 5; WT, wild‐type.
Next, we quantitatively analyzed cholesterol levels in the mitochondria of primary hepatocytes from WT or Tm4sf5−/− KO mice. For this, we used anti‐HA pulldown to rapidly extract mitochondria from primary hepatocytes with stable lentivirus‐mediated pLJM‐3×HA‐OMP25 expression and performed a GC/MS‐based lipidomics analysis. We found that the cholesterol levels in mitochondria from primary hepatocytes of Tm4sf5−/− KO mice were in a trend lower (P = 0.193) than in mitochondrial from primary hepatocytes of WT mice (Figure 5E). Furthermore, suppression of TM4SF5 reduced Huh7 cell growth (Figure 5F).
We next asked which non‐mitochondrial or lysosomal proteins involved in cholesterol metabolism or transport could be involved in TM4SF5‐enriched MLCSs. We found that the expression of CYP27A1, TSPO, and VDAC1, but not STARD1, was increased in HA‐TM4SF5‐expressing SNU449 cells upon glucose repletion compared with that in TM4SF5‐null cells, whereas TM4SF5 expression did not change the expression of those genes in the absence of glucose (Supplementary Figure S6D). As lysosomal proteins, cholesterol‐binding NPC1 [37] and STARD3 [38] were present in TM4SF5 precipitates from Huh7 cells (Figure 5G). STARD3 bound to TM4SF5 constitutively, but NPC1 binding to TM4SF5‐strep was induced by glucose repletion following depletion (Figure 5H). Unlike WT TM4SF5‐strep, palmitoylation‐deficient (Pal−) TM4SF5‐strep did not bind NPC1, but C62A mutant TM4SF5‐strep (which did not reach MLCSs) strongly bound NPC1, presumably at lysosomal membranes (Figure 5I). However, TM4SF5 did not bind strep‐NPC2 that could be in the lysosomal lumen as a cholesterol binder (Figure 5J). These observations suggested that TM4SF5 at MLCSs can affect cholesterol transport or metabolism via NPC1 and STARD3 binding in hepatocytes. To analyze In vivo cholesterol transport into mitochondria in animals, we intravenously injected Hoechst, mitotracker, and TopFluor cholesterol into the WT or KO mice that had been fasted overnight, 60 min before intraperitoneal glucose injection. Half an hour later, images for cholesterol in mitochondria (i.e., yellow stains) from their livers were collected (Figure 5K). Unlike continuously fasted animals, glucose‐treated WT mice showed significantly higher mitochondrial cholesterol levels than KO mice (Figure 5L‐M). Thus, hepatic Tm4sf5 expression appeared to play a role in In vivo cholesterol transport (from lysosome) to mitochondria under higher extracellular glucose levels. Interestingly, WT or KO mice fed with HFD for 10 weeks did not show significant changes in mRNA levels of Cyp27a1, Tspo, or Stard3 genes related to cholesterol metabolism (Supplementary Figure S7A). Meanwhile, WT mice fed with HFD or HFGluD or treated with DEN did not show significant changes in their mRNA levels (Supplementary Figure S7B‐C). Analyses of genes related to cholesterol metabolism using GSE and TCGA databases showed no clear changes between healthy (obese) humans, NAFLD, or HCC patients’ livers. However, HCC patients appeared to show higher levels than patients of other cancer types (Supplementary Figure S7D). Thus, the role of TM4SF5‐mediated cholesterol transport, specifically to mitochondria, rather than global cholesterol metabolism, might be more physiologically relevant to liver pathology.
3.6. TM4SF5‐enriched MLCSs export lysosomal cholesterol to mitochondria
To determine how TM4SF5 affects cholesterol transport, we first tested TM4SF5 for its ability to bind cholesterol. A radioactive cholesterol binding assay with a cold cholesterol competition revealed that WT TM4SF5 bound radioactive cholesterol similarly to the cholesterol‐binding tetraspanin CD81 (Figure 6A), which we used as a positive control [39]. Like WT TM4SF5, Pal− TM4SF5 and C62A TM4SF5 could also bind cholesterol (Figure 6B). The EC50 of WT TM4SF5 or CD81 binding was calculated via radiolabeled cholesterol titration to be 25.71 nmol/L or 586.7 nmol/L, respectively (Figure 6C). TSAHC inhibited TM4SF5 cholesterol binding in a dose‐dependent manner, with R2 = 0.963 and P < 0.001 by linear regression (Figure 6D). WT TM4SF5‐strep did not bind estradiol or palmitic acid (Figure 6E). Meanwhile, TM4SF5 and NPC2, but not Rab21, bound to cholesterol, indicating a specific binding (Supplementary Figure S8A). TM4SF5I22A, a TM4SF5 variant with a point mutation in the first transmembrane domain (TM1), partially lost the cholesterol binding capacity, unlike variants with mutations around TM3 or TM4 (Figure 6F, Supplementary Figure S8B). Molecular docking of TM4SF5 with cholesterol showed interface at I22 residue among the hydrophobic residues in the transmembrane domains (Supplementary Figure S8C).
FIGURE 6.
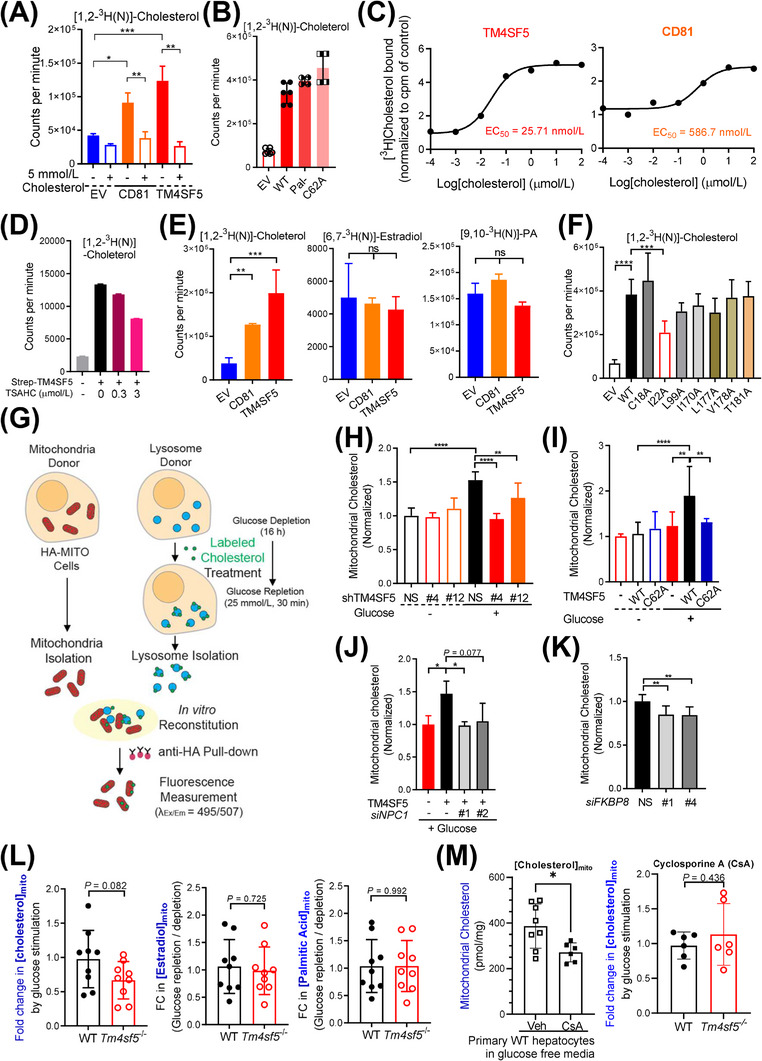
TM4SF5‐enriched MLCSs export lysosomal cholesterol to mitochondria. (A‐D) Huh7 cells (A, C, and D) or SNU449 cells (B) transfected with the indicated expression plasmids were harvested, and the whole‐cell lysates were pulled down with streptavidin beads. The beads were washed and incubated with 5 μmol/L radiolabeled cholesterol with 5 mmol/L cold cholesterol (A) or without (B), different concentrations of radiolabeled cholesterol alone (C), or with TSAHC treatment (D), before CPM measurements. (E) Immunopurified CD81‐Strep or TM4SF5‐Strep prepared as in (A) was incubated with [1,2‐3H(N)]‐cholesterol, [6,7‐3H(N)]‐estradiol, or [9,10‐3H(N)]‐palmitic acid. Bound 3H‐lipid was measured in a scintillation counter. (F) SNU449 cells transfected with indicated expression plasmids were processed for [1,2‐3H(N)]‐cholesterol binding assay as in (A). (G) Schematic presentation of the In vitro mitochondria‐lysosome contact assay. (H‐K) Huh7 stably expressing HA‐OMP25 were processed for rapid mitochondria isolation by anti‐HA pulldown. Lysosomes were isolated by ultracentrifugation from another set of stable Huh7 cells with or without TM4SF5 suppression (H), SNU449 cells transfected with Strep‐EV or TM4SF5 WT or C62A (I), SNU449 cells transfected with TM4SF5‐FLAG and/or siNPC1 (against NPC1 sequences #1 and #2, J) were glucose depleted (‐) and then replete (+) with 10 μmol/L of TopFluor cholesterol treatment (2 h). After the isolated organelles were mixed for 30 min at 37°C, anti‐HA antibody beads were used to isolate the mitochondria for fluorescence measurements. Huh7 cells stably expressing HA‐OMP25 were treated with siRNA targeting FKBP8 (against #1 and #4 sequences) and processed for mitochondria isolation and In vitro mitochondria‐lysosome contact assay (K). (L‐M) Primary hepatocytes from WT and Tm4sf5 −/‐ KO mice infected with HA‐OMP25 lentivirus were glucose‐depleted for 16 h. Thirty minutes before cell harvest, 25 mmol/L glucose was replete (L). In case, the primary cells were treated with cyclosporine A 10 μmol/L for 24 h to impair lysosomes (M). The cells were then suspended in PBS and mildly homogenized before pulldown using anti‐HA‐magnetic beads, methanol extraction, and GC/MS‐based cholesterol, estradiol, or palmitate measurement. Fold changes were calculated from the values of repletion samples divided by the values from depletion samples. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, ns = non‐significant. Data were represented as mean ± SD. Data represent three independent experiments. See also Supplementary Figure S8. CPM, counter per minute; FKBP8, FK506‐binding protein 8; HA‐OMP25, outer membrane protein 25 for tagging mitochondria with HA epitopes; KO, knockout; MLCS, mitochondria‐lysosome contact site; SD, standard deviation; TM4SF5, transmembrane 4 L six family member 5; TSAHC,4'‐(p‐toluenesulfonylamido)‐4‐hydroxychalcone; WT, wild‐type.
Next, we used an In vitro mitochondria‐lysosome contact reconstitution assay to determine if lysosomal cholesterol was transported to mitochondria depending on TM4SF5 expression. For the In vitro reconstitution assay, lysosomes were isolated by ultracentrifugation from Huh7 cells that had been pre‐labeled with TopFluor cholesterol and subjected to glucose depletion and repletion, whereas mitochondria were isolated by anti‐HA antibody pulldown from stable Huh7‐3×HA‐OMP25 cells (Figure 6G). We confirmed the accumulation of TopFluor‐cholesterol in the lysotracker‐labeled lysosomes by confocal imaging (Supplementary Figure S8D). The qualities of the isolated mitochondria via anti‐HA‐MITO immunoprecipitation from whole cell extracts (Supplementary Figure S8E) or from the MLCS reconstitution assay mixtures (Supplementary Figure S8F) were checked by organellar marker immunoblotting, leading to no lysosomal (LAMP1/2), TM4SF5 (lysosomal and plasma membrane), or β‐actin (cytosol) markers. As expected, glucose repletion increased the import of TopFluor‐cholesterol from Huh7‐lysosomes to mitochondria, and TM4SF5 suppression abolished this effect (Figure 6H). Unlike WT TM4SF5, C62A mutant TM4SF5 did not cause cholesterol transport to mitochondria (Figure 6I). Furthermore, NPC1 suppression on the lysosomal side or FKBP8 suppression on the mitochondrial side abolished the TM4SF5‐mediated cholesterol transport to mitochondria (Figure 6J‐K). In addition, we could observe cholesterol near mitochondria that were separated via anti‐HA pulldown from In vitro reconstitution assay mixtures with lysotracker or mitotracker prelabeling (Figure 6G, Supplementary Figure S8G). We also determined mitochondrial cholesterol levels in primary hepatocytes from WT and Tm4sf5−/− KO mice upon glucose stimulation. The hepatocytes were glucose starved and then replete, and mitochondria were rapidly extracted for GC/MS‐based lipid analysis. Cholesterol enrichment showed a greater trend (though insignificant, P = 0.082) in mitochondria from WT mice than in mitochondria from Tm4sf5−/− KO mice, whereas there was no such difference in mitochondrial enrichment of estradiol or palmitic acid (Figure 6L). Furthermore, the mitochondrial cholesterol levels in WT primary hepatocytes with lysosomes impaired by cyclosporine A in the glucose‐free condition were decreased compared with those in cells with intact lysosomes (Figure 6M, left panel). Meanwhile, the mitochondrial cholesterol levels in WT or KO primary hepatocytes were comparable upon glucose repletion following lysosome impairment, independent of TM4SF5 expression (Figure 6M, right panel). This observation indicates that the glucose‐mediated cholesterol enrichment in mitochondria in TM4SF5‐positive cells may require lysosome, presumably for cholesterol transport. Thus, the In vitro MLCS reconstitution analyses suggested that TM4SF5‐enriched MLCSs support the export of lysosomal cholesterol to mitochondria.
3.7. The metabolic effects of TM4SF5‐enriched MLCSs on cellular respiratory reprogramming
Cholesterol affects mitochondrial functions [40], so we investigated the effect of cholesterol export via TM4SF5‐enriched MLCSs on mitochondrial function. We first examined the integrity of molecular components of the mitochondrial electron transport chain (ETC), TCA cycle, and β‐oxidation. Mitochondria were prepared from primary hepatocytes of male WT or Tg Alb ‐Tm4sf5 mice (n = 4) using rapid anti‐HA pulldown for proteomic analysis. LC/MS analysis showed that components of complex I of the ETC in the mitochondria from Tg Alb ‐Tm4sf5 mice were much reduced compared with those from WT mice, whereas the molecular components of ETC complexes II, III, IV, and V, the TCA cycle, and β‐oxidation were not greatly changed (Figure 7A). In addition, comparison of the expression of mitochondrial components in livers between WT and Tm4sf5−/− KO mice (n = 4) revealed that the livers of Tm4sf5−/− KO mice had dramatically increased levels of ETC complex I components (Supplementary Figure S9A). These observations suggested that cholesterol transport to mitochondria via TM4SF5‐enriched MLCSs upon glucose repletion might be linked to impaired ETC activity but not impaired TCA cycle or β‐oxidation activity. We observed a similar TM4SF5‐mediated effect on ETC complex I in a mitochondria ETC complex assay. TM4SF5 suppression in glucose‐replete Huh7 cells led to activation of complex I, but not the other complexes (Figure 7B). Live imaging of TopFlour‐cholesterol (green), mCherry‐TM4SF5 (red), and mito‐BFP (blue) in SNU449 cells in the presence of glucose repletion suggested that the presence of cholesterol around TM4SF5 near mitochondria was linked to mitochondrial fission (Figure 7C). The fission percentage at the cholesterol staining sites around TM4SF5 (‐lysosomal) vesicles near mitochondria was 71.4% (n = 21 fission events in 10 cells), which was significantly higher (P = 0.007) than the expected fission probability calculated via a determination of the TM4SF5 staining probability in each cell (n = 10, 14.2%) similar to the results of a previous study [27]. When we measured citrate levels in whole‐cell or mitochondrial lysates, mitochondria from TM4SF5‐suppressed Huh7 cells showed citrate levels comparable to those of mitochondria from TM4SF5‐expressing cells, similar to the levels in whole‐cell lysates (Figure 7D). Furthermore, the mRNA levels of diverse TCA cycle enzymes, including citrate synthetase, did not depend on TM4SF5 expression (Supplementary Figure S9B). This suggests that TM4SF5 expression might support TCA cycle activity.
FIGURE 7.
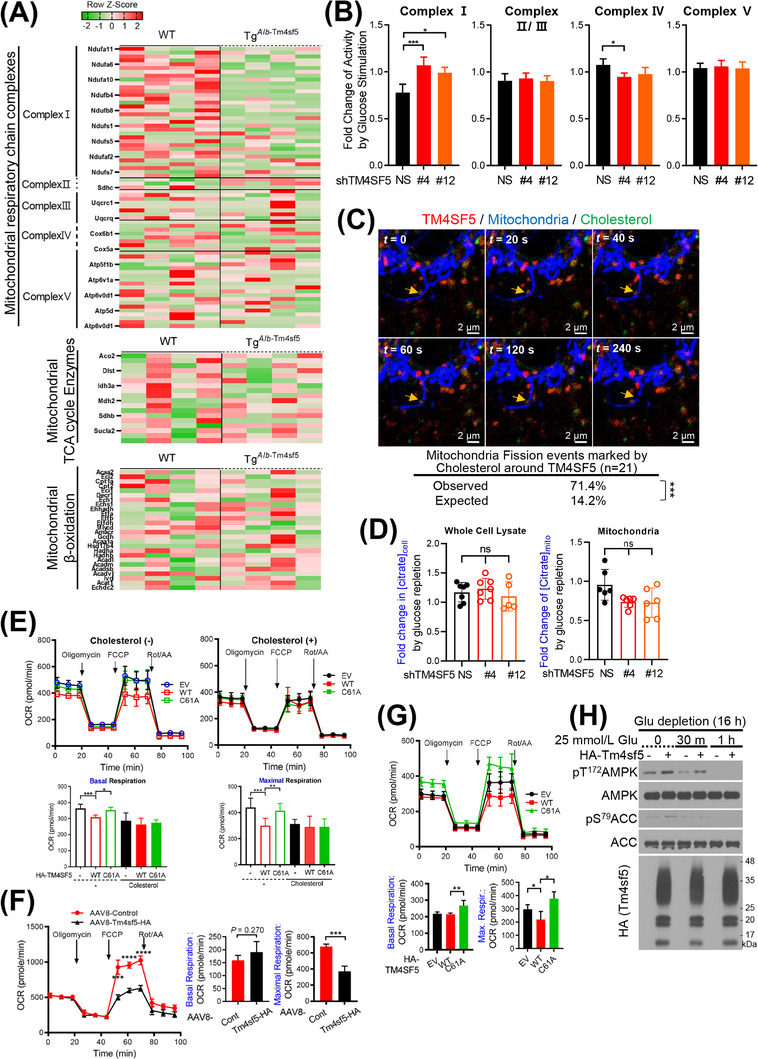
The metabolic effects of TM4SF5‐enriched MLCSs on cellular respiratory reprogramming. (A) Primary hepatocytes isolated from WT or Tg Alb ‐Tm4sf5 C57BL/6N mice (n = 4) were infected with HA‐OMP25 lentivirus. Mitochondria were pulled down using anti‐HA antibody beads for LC/MS proteomics analysis. (B) Huh7 cells with or without TM4SF5 suppression were glucose depleted and replete and processed for ETC complex‐activity assay. FCs in activity between glucose repletion and depletion were graphed as the mean ± SD. (C) SNU449 cells transfected with mCherry‐TM4SF5 and mito‐BFP were treated with 10 μmol/L TopFluor‐cholesterol for 1 h. Representative snapshots from confocal live imaging are presented. Quantitative analysis of the mitochondrial fission events was performed as explained in the Materials and Methods. (D) AML12 cells transfected with the indicated constructs were treated without (‐) or with (+) 16 μmol/L cholesterol for 16 h before OCR measurements. AUCs for the OCR measurements were graphed. (E‐G) Primary hepatocytes from WT or Tm4sf5 −/‐ KO mice (E), WT mice preinjected with AAV8‐control or AAV8‐Tbg‐Tm4sf5 virus (F), or AML12 cells transfected with the indicated constructs (G) were processed for OCR measurements. (H) SNU449 cells stably infected with cDNA plasmids were glucose depleted and replete as indicated before harvests and immunoblots. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, ns = non‐significant. Data were represented as mean ± SD. Data represent three independent experiments. See also Supplementary Figure S9. AAV8, adeno‐associated virus serotype 8; AUC, area under the curve; ETC, electron transport chain; FC, fold change; KO, knockout; LC/MS, Liquid chromatography/mass spectrometry, AML12, normal murine hepatocytes; MLCS, mitochondria‐lysosome contact site; OCR, oxygen consumption rate; SD, standard deviation; TM4SF5, transmembrane 4 L six family member 5; WT, wild‐type.
We next investigated the effect of cholesterol treatment on cellular respiration, as determined by the mitochondrial OCR, in normal murine AML12 hepatocytes expressing HA‐control, HA‐TM4SF5 WT, or TM4SF5 C61A (with mutation at a residue comparable to C62 in human TM4SF5). In the absence of cholesterol treatment, cells expressing HA‐TM4SF5 WT showed lower basal and maximal respiration compared with cells expressing HA‐control or TM4SF5 C61A; these differences were abolished in the presence of cholesterol treatment (Figure 7E). However, in glycolysis stress tests measuring the ECAR, HA‐TM4SF5‐transfected SNU449 cells showed greater glycolysis, glycolytic capacity, and glycolytic reserve compared with control vector‐transfected cells (Supplementary Figure S9C). Furthermore, primary hepatocytes from Tm4sf5 −/− KO mice had greater maximal respiration than primary hepatocytes from WT mice (Supplementary Figure S9D). In addition, we measured OCR in primary hepatocytes isolated from mice that had been infected with AAV8‐control virus or AAV8‐Tbg‐Tm4sf5‐HA virus (to express Tm4sf5‐HA in hepatocytes from a thyroxin binding globulin (Tbg) promoter [41]). Maximal respiration was reduced in hepatocytes from the mice infected with AAV8‐Tbg‐Tm4sf5‐HA (Figure 7F). Moreover, unlike WT HA‐Tm4sf5, control or C61A mutant HA‐Tm4sf5 led to greater maximal and basal respiration in AML12 cells (Figure 7G). The bioenergy status in the SNU449 cells transfected with EV or HA‐TM4SF5 was confirmed by immunoblotting for pT172AMPK and pS79ACC at different time points after glucose repletion after 16 h glucose depletion. Compared with cells in the glucose‐depleted condition, cells after glucose repletion showed improvement in cellular energy status as time passed; TM4SF5 enhanced the AMP/ATP ratio 30 min after repletion, but the improved energy status became independent of TM4SF5 expression by 1 h post repletion (Figure 7H). However, this TM4SF5‐mediated effect did not occur when cells were glutamine depleted and then replete (Supplementary Figure S9E). These observations suggest that cholesterol export to mitochondria via TM4SF5‐enriched MLCSs results in enhanced glycolysis but impaired oxidative phosphorylation in hepatocytes.
4. DISCUSSION
This study provides evidence of the regulatory roles of TM4SF5‐dependent MLCSs and MLCS‐mediated cholesterol transport in glucose catabolism and mitochondrial reprogramming via impairment of ETC complex I. TM4SF5 translocated to MLCSs when extracellular glucose was supplied, leading to export of lysosomal cholesterol to mitochondria, impaired ETC function, mitochondrial fission and mitophagy, and energetic dependency on glycolytic respiration (Figure 8). This study recapitulates the dependency of cancer cells on glycolysis for ATP production and the TCA cycle for anabolic building blocks. TM4SF5 enrichment at appositions between mitochondria and lysosomes upon exposure of cells to excessive extracellular glucose was achieved by the interaction between lysosomal TM4SF5 and mitochondrial FKBP8. Furthermore, this protein complex could be in proximity to other molecules such as DRP1, mitophagy receptors, NPC1, and free cholesterol. In addition, animal models with Tm4sf5 expression, specifically in hepatocytes, showed significantly increased mitophagy and cholesterol transport to mitochondria. Moreover, mRNA levels of genes related to these processes increased, after glucose drinking, high sucrose diet, or DEN treatment, unlike Tm4sf5 −/− mice. TM4SF5‐mediated mitophagy and cholesterol transport to mitochondria also appeared to be correlated to the development of NAFLD and HCC. Furthermore, lysosomal cholesterol export to mitochondria led to impaired integrity of the complex I of mitochondrial ETC system. Despite the impaired ETC, molecules involved in the TCA cycle and β‐oxidation appeared to be unaffected regardless of TM4SF5 expression in hepatocytes or mouse livers. It is therefore likely that the supply of intermediate biomolecules would be enough for hepatocyte proliferation and survival during TM4SF5 (over)expression‐mediated hepatic carcinogenesis.
FIGURE 8.
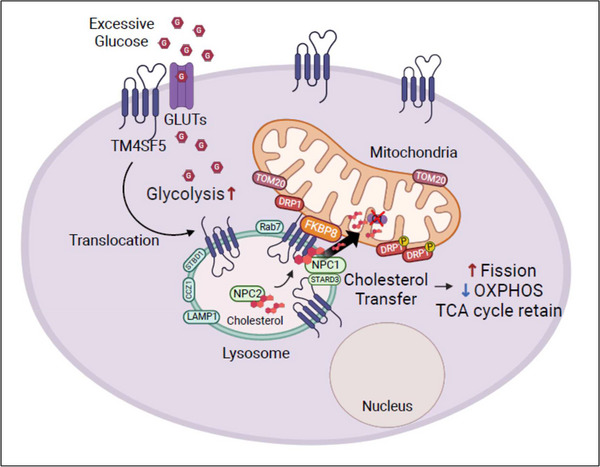
A working model of mitochondrial reprograming by TM4SF5‐enriched MLCSs. Upon repletion of extracellular glucose, TM4SF5 localizes to lysosomes, and the TM4SF5‐enriched lysosomal membrane becomes closely in apposition to mitochondria to create MLCSs, depending on Rab7, CCZ1, and STBD1 expression. TM4SF5 on lysosome becomes tethered to FKBP8 on the outer mitochondria membrane via a direct interaction. TM4SF5 can be associated with or in proximity to TOM20, DRP1, NPC1, STARD3, and lysosome‐free cholesterol. Mitochondria at TM4SF5‐enriched MLCSs can also dynamically recruit (phospho‐) DRP1 for mitochondrial fission. TM4SF5‐bound cholesterol can be exported to mitochondria efficiently via TM4SF5‐bound NPC1 at the TM4SF5‐enriched MLCSs. Thereby, cholesterol enrichment into sterol‐poor mitochondria causes ETC impairment for inefficient mitochondrial respiration but increased glycolytic respiration. Concomitantly, the TCA cycle remains intact to supply a pool of metabolic intermediates as building blocks for cell proliferation. CCZ1, CCZ1 homolog, vacuolar protein trafficking and biogenesis associated; DRP1, dynamin‐related protein 1; NPC1, niemann‐pick disease type C1; STARD3, star‐related lipid transfer domain‐3; STBD1, starch binding domain 1; TCA cycle, tricarboxylic acid cycle; TOM20, translocase of outer mitochondrial membrane 20.
Indeed, TM4SF5 expression in hepatocytes lead to cancerous properties [35, 42]. Because of this correlation, TM4SF5 expression in the LIHC population of the TCGA dataset is significantly increased compared with normal counterparts, and this increase is more dramatic than those of other cancer types (data not shown). Liver orthotopic xenografts of TM4SF5‐positive hepatocytes lead to liver tumor growth and metastasis, which are abolished by treatment with a specific anti‐TM4SF5 inhibitor [17, 43]. Although WT C57BL/6 mice formed liver tumors after treatment with a liver carcinogen DEN, Tm4sf5 −/− KO mice did not [21]. Indeed, analysis of TCGA datasets revealed that the LIHC population showed dramatically increased TM4SF5 expression compared with normal counterparts, although other adenocarcinoma types (colon, pancreatic, rectum, and stomach) also showed TM4SF5 levels higher than their normal counterparts. In addition, the GSE14520, GSE76427, and GSE6764 datasets also showed higher TM4SF5 levels in tumors than those in their normal counterparts [21]. Interestingly, TM4SF5 in hepatocytes appeared to suppress cytotoxic activity of Natural killer cells, which are abundant in the livers [21]. Furthermore, WT mice fed with a methionine‐choline‐deficient (MCD) diet for 4 weeks exhibited nonalcoholic steatohepatitis‐associated fibrosis (i.e., a precancerous disease state) via TM4SF5‐mediated reprogramming of the inflammatory environment, whereas Tm4sf5 −/− KO mice under the same experimental conditions did not show such effects [26].
Like other tetraspanins, TM4SF5 can change its subcellular locations and binding partners [6], presumably depending on cellular contexts and needs, such as metabolic changes and/or inflammation. We recently reported that upon extracellular L‐arginine depletion and repletion in hepatocytes, TM4SF5 can localize at the plasma membrane and lysosomes, where it binds the L‐arginine transporters SLC7A1 and SLC38A9, respectively, to import L‐arginine to the cytosol, providing a substrate for biosynthetic processes during cell proliferation [5]. We also reported that, like the tetraspanins CD63 and CD81, TM4SF5 can be loaded onto hepatocyte‐derived exosomes to play roles in the crosstalk between the liver and brown adipose tissue for homeostatic blood glucose clearance [9]. Regarding intracellular translocation, the association of TM4SF5 with EGFR and integrin α5 on the plasma membrane can be altered upon depletion of intracellular cholesterol [4], suggesting that TM4SF5 may be functionally linked to cholesterol homeostasis. Furthermore, the higher FC in mitochondrial cholesterol levels in TM4SF5‐positive primary hepatocytes over that in TM4SF5‐negative hepatocytes upon extracellular glucose repletion was abolished by lysosome impairment, suggesting that the mitochondrial cholesterol levels might be modulated by TM4SF5‐enriched lysosomes to form MLCSs upon glucose repletion. This study provides another evidence of unique location of TM4SF5 upon metabolic needs, by showing that TM4SF5‐enriched MLCSs form upon glucose repletion and cause cholesterol transport to mitochondria, resulting in impairment of the complex I of the mitochondrial ETC system, mitophagy/fission, and reprogramming of glucose catabolism. Cholesterol, a rare lipid in mitochondria, can regulate the biophysical properties of cellular membranes, including fluidity, permeability, curvature, and membrane–protein interactions [44].
TM4SF5 enrichment at MLCSs was achieved by the interaction between lysosomal TM4SF5 and mitochondrial FKBP8. Because FKBP8 localizes at the outer mitochondrial membrane [31], it appears that the TM4SF5‐FKBP8 linkage guides TM4SF5 targeting to mitochondrial FKBP8 to form MLCSs. Rab7 can also cause MLCS formation, and its GTP‐binding status is related to the formation or untethering of membrane contacts [27]. Suppression of Rab7 reduced the levels of TM4SF5‐enriched MLCSs but did not abolish TM4SF5‐dependent MLCS formation upon glucose treatment. This suggests that TM4SF5 itself might drive the close membranous apposition between mitochondria and lysosomes in the presence of excessive extracellular glucose. For this to occur, TM4SF5, like other tetraspanins, has molecular characteristics for massive protein‐protein complex formation and subcellular translocation [3, 45, 46]. There can thus be different proteins in proximity to the TM4SF5‐FKBP8 linkage at TM4SF5‐enriched MLCSs, such as cholesterol transporters, mitophagy receptors, and mitochondrial dynamics‐related proteins. This protein complex formation and clustering mediated by lysosomal TM4SF5 highlights the biological significance of TM4SF5‐enriched MLCSs, which appear to be linked to mitochondrial dynamics and reprogramming of metabolic functions under excessive extracellular glucose conditions. MLCSs have also been identified as sites for mitochondrial fission [28]. In addition to mitochondrial fission and mitophagy, TM4SF5‐enriched MLCSs were linked to an impaired ETC complex I but an intact TCA cycle. Rapid ATP production in cancer cells is achieved mostly through anaerobic glycolysis, but at the same time biomolecule substrates can be available as TCA cycle intermediates with remodeling of mitochondrial functions and/or dynamics [1, 2]. Our study of TM4SF5‐enriched MLCSs may therefore recapitulate the metabolic regulation and reprogramming that occurs during hepatocellular carcinogenesis, suggesting that an anti‐TM4SF5 strategy may be clinically beneficial to patients with abnormal metabolic activity related to hepatic glucose or cholesterol.
5. CONCLUSIONS
In conclusion, we found that TM4SF5 in hepatocytes translocated to MLCSs upon glucose repletion. Furthermore, protein‐protein complex‐mediated clustering at these MLCSs led to mitochondrial fission, mitophagy, increased glycolysis, and impairment of ETC complexes, resulting in inefficient oxidative phosphorylation but an intact TCA cycle, presumably recapitulating the reprogramming of cancerous cellular respiratory systems. Although hyperglycemia conditions might be linked to cholesterol homeostasis and mitochondrial reprogramming via TM4SF5‐enriched MLCSs during HCC development and progression, tracing of their metabolites can be needed in coming explorations.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Ji Eon Kim designed certain and performed experiments, interpreted the data, and wrote the Materials and Methods. So‐Young Park and Kwang‐Hyeon Liu did lipid analysis. Dae‐Geun Song did proteomic analysis. Jae Woo Jung, Eun‐Ae Shin, Yangie Pinanga, Kyung‐hee Pyo, Eun Hae Lee, Wonsik Kim, and Soyeon Kim helped with constructs, reagents, and animal care and preparation. Haesong Lee helped with microscopic experiments. Chang‐Duck Jun helped with TAGLN2 construct and antibody. Jeanho Yun helped with mtKeima mice. Yoonji Lee and Sun Choi helped with molecular docking. Chulhwan Kwak and Hyun‐Woo Rhee helped with TurboID constructs and discussion. Jung Weon Lee designed experiments, interpreted the data, and wrote the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no potential conflicts of interest.
CONSENT FOR PUBLICATION
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal procedures were performed in accordance with the procedures of the Seoul National University Laboratory Animal Maintenance Manual and with IRB approvals from the Institute of Laboratory Animal Resources, Seoul National University (SNU‐161108‐10‐1, SNU‐181016‐7‐4, and SNU‐200410‐3).
Supporting information
Supporting information
ACKNOWLEDGEMENTS
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF‐2021R1A6A3A01087300 to JEK, NRF‐2021M3H9A2098553 to YL, NRF‐2022M3E5F3080873 to SC, NRF‐2022R1A4A1018900 to LKH, NRF‐2020R1A2C3008993, and NRF‐2021M3A9D3024752 to JWL).
Kim JE, Park S‐Y, Kwak C, Lee Y, Song D‐G, Jung JW, et al. Glucose‐mediated mitochondrial reprogramming by cholesterol export at TM4SF5‐enriched mitochondria‐lysosome contact sites. Cancer Commun.. 2024;44:47–75. 10.1002/cac2.12510
Contributor Information
Kwang‐Hyeon Liu, Email: dstlkh@knu.ac.kr.
Jung Weon Lee, Email: jwl@snu.ac.kr.
DATA AVAILABILITY STATEMENT
The TCGA dataset was from GEPIA (http://gepia.cancer‐pku.cn/). The gene expression pro‐files were from TCGA datasets (https://www.cancer.gov/ccg/research/genome‐sequencing/tcga). The Gene Omnibus database (GEO) was form NCBI (GEO DataSets‐NCBI [nih.gov]). mRNA‐Seq data from hepatocytes without or with TM4SF5 were as described previously [26] and can be supplied upon request to the corresponding author.
REFERENCES
- 1. Schwartz L, Supuran CT, Alfarouk KO. The Warburg effect and the hallmarks of cancer. Anticancer Agents Med Chem. 2017;17(2):164–170. [DOI] [PubMed] [Google Scholar]
- 2. Cassim S, Vucetic M, Zdralevic M, Pouyssegur J. Warburg and beyond: The power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers (Basel). 2020;12(5):1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee JW. Transmembrane 4 L six family member 5 (TM4SF5)‐Mediated Epithelial‐Mesenchymal transition in liver diseases. Int Rev Cell Mol Biol. 2015;319:141–163. [DOI] [PubMed] [Google Scholar]
- 4. Kim HJ, Kwon S, Nam SH, Jung JW, Kang M, Ryu J, et al. Dynamic and coordinated single‐molecular interactions at TM4SF5‐enriched microdomains guide invasive behaviors in 2‐ and 3‐dimensional environments. FASEB J. 2017;31(4):1461–1481. [DOI] [PubMed] [Google Scholar]
- 5. Jung JW, Macalino SJY, Cui M, Kim JE, Kim HJ, Song DG, et al. Transmembrane 4 L six family Member 5 senses Arginine for mTORC1 signaling. Cell Metab. 2019;29(6):1306–1319. [DOI] [PubMed] [Google Scholar]
- 6. Lee JW. TM4SF5‐mediated protein‐protein networks and tumorigenic roles. BMB Rep. 2014;47(9):483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jung JW, Kim JE, Kim E, Lee JW. Amino acid transporters as tetraspanin TM4SF5 binding partners. Exp Mol Med. 2020;52(1):7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yanez‐Mo M, Barreiro O, Gordon‐Alonso M, Sala‐Valdes M, Sanchez‐Madrid F. Tetraspanin‐enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19(9):434–446. [DOI] [PubMed] [Google Scholar]
- 9. Jung JW, Kim JE, Kim E, Lee H, Lee H, Shin EA, et al. Liver‐originated small extracellular vesicles with TM4SF5 target brown adipose tissue for homeostatic glucose clearance. J Extracell Vesicles. 2022;11(9):e12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmen T, Tagaya M. Organelle Communication at Membrane Contact Sites (MCS): From curiosity to center stage in cell biology and biomedical research. Adv Exp Med Biol. 2017;997:1–12. [DOI] [PubMed] [Google Scholar]
- 11. Herrera‐Cruz MS, Simmen T. Over six decades of discovery and characterization of the architecture at Mitochondria‐Associated Membranes (MAMs). Adv Exp Med Biol. 2017;997:13–31. [DOI] [PubMed] [Google Scholar]
- 12. Marchi S, Bittremieux M, Missiroli S, Morganti C, Patergnani S, Sbano L, et al. Endoplasmic reticulum‐mitochondria communication through Ca(2+) signaling: The importance of Mitochondria‐Associated Membranes (MAMs). Adv Exp Med Biol. 2017;997:49–67. [DOI] [PubMed] [Google Scholar]
- 13. Yousuf MS, Maguire AD, Simmen T, Kerr BJ. Endoplasmic reticulum–mitochondria interplay in chronic pain: The calcium connection. Molecular Pain. 2020;16:1744806920946889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62(3):341–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tagaya M, Arasaki K. Regulation of mitochondrial dynamics and autophagy by the mitochondria‐associated membrane. Adv Exp Med Biol. 2017;997:33–47. [DOI] [PubMed] [Google Scholar]
- 16. Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, et al. Cholesterol transport through lysosome‐peroxisome membrane contacts. Cell. 2015;161(2):291–306. [DOI] [PubMed] [Google Scholar]
- 17. Lee SA, Ryu HW, Kim YM, Choi S, Lee MJ, Kwak TK, et al. Blockade of four‐transmembrane L6 family member 5 (TM4SF5)‐mediated tumorigenicity in hepatocytes by a synthetic chalcone derivative. Hepatology. 2009;49(4):1316–1325. [DOI] [PubMed] [Google Scholar]
- 18. Ryu J, Kim E, Kang M‐K, Song D‐G, Shin E‐A, Lee H, et al. Differential TM4SF5‐mediated SIRT1 modulation and metabolic signaling in nonalcoholic steatohepatitis progression. Journal of Pathology. 2021;253(1):55–67. [DOI] [PubMed] [Google Scholar]
- 19. Sun N, Yun J, Liu J, Malide D, Liu C, Rovira, II , et al. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee H, Kim E, Shin EA, Shon JC, Sun H, Kim JE, et al. Crosstalk between TM4SF5 and GLUT8 regulates fructose metabolism in hepatic steatosis. Mol Metab. 2022;58:101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun H, Kim E, Ryu J, Lee H, Shin EA, Lee M, et al. TM4SF5‐mediated liver malignancy involves NK cell exhaustion‐like phenotypes. Cell Mol Life Sci. 2022;79(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li ZH, An N, Huang XJ, Yang C, Wu HL, Chen XC, et al. Cyclosporine A blocks autophagic flux in tubular epithelial cells by impairing TFEB‐mediated lysosomal function. J Cell Mol Med. 2021;25(12):5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl‐tert‐butyl ether for high‐throughput lipidomics. J Lipid Res. 2008;49(5):1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moon JY, Kong TY, Jang HJ, Kang HC, Cho YY, Lee JY, et al. Simultaneous quantification of 18 saturated and unsaturated fatty acids and 7 sterols as their tert‐butyldimethylsilyl derivatives in human saliva using gas chromatography‐tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1092:114–121. [DOI] [PubMed] [Google Scholar]
- 25. Song HE, Lee Y, Kim E, Cho CY, Jung O, Lee D, et al. N‐terminus‐independent activation of c‐Src via binding to a tetraspan(in) TM4SF5 in hepatocellular carcinoma is abolished by the TM4SF5 C‐terminal peptide application. Theranostics. 2021;11(16):8092–8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim E, Um H, Park J, Jung JW, Kim JE, Lee H, et al. TM4SF5‐dependent crosstalk between hepatocytes and macrophages to reprogram the inflammatory environment. Cell Rep. 2021;37(7):110018. [DOI] [PubMed] [Google Scholar]
- 27. Wong YC, Ysselstein D, Krainc D. Mitochondria‐lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554(7692):382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria‐lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 2019;29(6):500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Juhl AD, Heegaard CW, Werner S, Schneider G, Krishnan K, Covey DF, et al. Quantitative imaging of membrane contact sites for sterol transfer between endo‐lysosomes and mitochondria in living cells. Sci Rep. 2021;11(1):8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl‐2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003;5(1):28–37. [DOI] [PubMed] [Google Scholar]
- 31. Yoo SM, Yamashita SI, Kim H, Na D, Lee H, Kim SJ, et al. FKBP8 LIRL‐dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. FASEB J. 2020;34(2):2944–2957. [DOI] [PubMed] [Google Scholar]
- 32. Lee SY, Kim YT, Lee MS, Kim YB, Chung E, Kim S, et al. Focal adhesion and actin organization by a cross‐talk of TM4SF5 with integrin alpha2 are regulated by serum treatment. Exp Cell Res. 2006;312(16):2983–2999. [DOI] [PubMed] [Google Scholar]
- 33. Na BR, Kim HR, Piragyte I, Oh HM, Kwon MS, Akber U, et al. TAGLN2 regulates T cell activation by stabilizing the actin cytoskeleton at the immunological synapse. J Cell Biol. 2015;209(1):143–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wei W, Riley NM, Yang AC, Kim JT, Terrell SM, Li VL, et al. Cell type‐selective secretome profiling in vivo. Nat Chem Biol. 2021;17(3):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee SA, Lee SY, Cho IH, Oh MA, Kang ES, Kim YB, et al. Tetraspanin TM4SF5 mediates loss of contact inhibition through epithelial‐mesenchymal transition in human hepatocarcinoma. J Clin Invest. 2008;118(4):1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goedeke L, Fernández‐Hernando C. Regulation of cholesterol homeostasis. Cell Mol Life Sci. 2012;69(6):915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vance JE, Karten B. Niemann‐Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res. 2014;55(8):1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Voilquin L, Lodi M, Di Mattia T, Chenard M‐P, Mathelin C, Alpy F, et al. STARD3: A Swiss Army Knife for Intracellular Cholesterol Transport. Contact. 2019;2:2515256419856730. [Google Scholar]
- 39. Zimmerman B, Kelly B, McMillan BJ, Seegar TCM, Dror RO, Kruse AC, et al. Crystal structure of a full‐length human Tetraspanin reveals a cholesterol‐binding pocket. Cell. 2016;167(4):1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martin LA, Kennedy BE, Karten B. Mitochondrial cholesterol: mechanisms of import and effects on mitochondrial function. J Bioenerg Biomembr. 2016;48(2):137–151. [DOI] [PubMed] [Google Scholar]
- 41. Kiourtis C, Wilczynska A, Nixon C, Clark W, May S, Bird TG. Specificity and off‐target effects of AAV8‐TBG viral vectors for the manipulation of hepatocellular gene expression in mice. Biology Open. 2021;10(9):bio058678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim H, Kang M, Lee SA, Kwak TK, Jung O, Lee HJ, et al. TM4SF5 accelerates G1/S phase progression via cytosolic p27(Kip1) expression and RhoA activity. Biochim Biophys Acta. 2010;1803(8):975–982. [DOI] [PubMed] [Google Scholar]
- 43. Lee D, Na J, Ryu J, Kim HJ, Nam SH, Kang M, et al. Interaction of tetraspan (in) TM4SF5 with CD44 promotes self‐renewal and circulating capacities of hepatocarcinoma cells. Hepatology. 2015;61(6):1978–1997. [DOI] [PubMed] [Google Scholar]
- 44. Elustondo P, Martin LA, Karten B. Mitochondrial cholesterol import. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(1):90–101. [DOI] [PubMed] [Google Scholar]
- 45. van Deventer SJ, Dunlock VE, van Spriel AB. Molecular interactions shaping the tetraspanin web. Biochem Soc Trans. 2017;45(3):741–750. [DOI] [PubMed] [Google Scholar]
- 46. Kim JE, Kim E, Lee JW. TM4SF5‐mediated regulation of hepatocyte transporters during metabolic liver diseases. Int J Mol Sci. 2022;23(15):8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The TCGA dataset was from GEPIA (http://gepia.cancer‐pku.cn/). The gene expression pro‐files were from TCGA datasets (https://www.cancer.gov/ccg/research/genome‐sequencing/tcga). The Gene Omnibus database (GEO) was form NCBI (GEO DataSets‐NCBI [nih.gov]). mRNA‐Seq data from hepatocytes without or with TM4SF5 were as described previously [26] and can be supplied upon request to the corresponding author.