

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and rapidly fatal interstitial lung disease marked by the replacement of lung alveoli with dense fibrotic matrices. Although the mechanisms initiating IPF remain unclear, rare and common alleles of genes expressed in lung epithelia, combined with aging, contribute to the risk for this condition. Consistently, single-cell RNA sequencing (scRNA-seq) studies have identified lung basal cell heterogeneity in IPF that might be pathogenic. We used single-cell cloning technologies to generate “libraries” of basal stem cells from the distal lungs of 16 patients with IPF and 10 controls. We identified a major stem cell variant that was distinguished from normal stem cells by its ability to transform normal lung fibroblasts into pathogenic myofibroblasts in vitro and to activate and recruit myofibroblasts in clonal xenografts. This profibrotic stem cell variant, which was shown to preexist in low quantities in normal and even fetal lungs, expressed a broad network of genes implicated in organ fibrosis and showed overlap in gene expression with abnormal epithelial signatures identified in previously published scRNA-seq studies of IPF. Drug screens highlighted specific vulnerabilities of this profibrotic variant to inhibitors of epidermal growth factor and mammalian target of rapamycin signaling as prospective therapeutic targets. This profibrotic stem cell variant in IPF was distinct from recently identified profibrotic stem cell variants in chronic obstructive pulmonary disease and may extend the notion that inappropriate accrual of minor and preexisting stem cell variants contributes to chronic lung conditions.
INTRODUCTION
The rapid development and fatal progression of idiopathic pulmonary fibrosis (IPF) occur by uncertain mechanisms (1). Emerging hypotheses hold that IPF arises from recurrent, subclinical lung injury that imparts changes to epithelial and stromal cells, which, in turn, compromise lung repair and favor fibrosis (2, 3). Epithelial cells have been tied to this condition by rare and common variants of genes encoding lung surfactant proteins [surfactant protein C (SFTPC), SFTPA1, and SFTPA2] known to be produced by alveolar epithelial cells (4–8). In addition, there is a strong association of sporadic and familial IPF with promoter variants in mucin 5B (MUC5B), another gene expressed by alveolar epithelial cells (9). Genetic studies have also highlighted the roles of genes involved in epithelial integrity (10–12), telomere maintenance (13–15), and innate immunity (16–19) expressed in many cell types. Perhaps the most compelling implication of epithelial cells in the pathogenesis of IPF came from a direct comparison of the transcriptomes of epithelial cells isolated from lungs of patients with IPF and controls (20). The IPF epithelial cells showed a biased expression of genes associated with goblet cells and with immature basal cells [e.g., MUC5B, SPDEF (SAM pointed domain-containing ETS transcription factor), tumor protein 63 (TP63), and keratin 5 (Krt5)], consistent with histopathology studies of IPF lungs (20–22). These IPF epithelial expression datasets further showed an up-regulation of HIPPO/YAP, PI3K, mTOR, WNT, and transforming growth factor–β (TGF-β) signaling pathways (20), which have been associated with IPF (23). This concept of epithelial alterations in IPF has been refined to discrete epithelial subsets by single-cell RNA sequencing (scRNA-seq) (24, 25) of control and disease lungs. Four recent studies of such epithelial subsets, alternatively termed “IPF epithelial cells,” “fibrosis basal cells,” “aberrant basaloid cells,” and “KRT5−/KRT17+ cells” in general, shared basal markers such as TP63 and KRT17 and less uniformly known markers of IPF, including vimentin (VIM), collagen 1A1 (COL1A1), matrix metallopeptidase 7 (MMP7), and growth differentiation factor 15 (GDF15) (26–28). scRNA-seq analyses of the bleomycin model for lung fibrosis in mice have also observed the emergence of an aberrant epithelial cell type (“KRT8+ transitional cells” and “damage-associated transient progenitors”) with similar markers seen in human IPF such as KRT17, SRY-box transcription factor 9 (SOX9), and GDF15 (29). Although the overall correlations between these different human and murine epithelial populations are low, in aggregate they provide evidence for alterations in epithelial cells accompanying IPF and murine models of lung fibrosis. Moreover, the concept that IPF is associated with aberrant epithelial cell types is consistent with a recent clonogenic analysis of chronic obstructive pulmonary disease (COPD), which linked the pathogenic features of this widespread lung disease to the emergence of three discrete and clonogenic epithelial distal airway stem cell variants that autonomously promote mucin hypersecretion, fibrosis, and neutrophilic inflammation (30).
In this study, we applied the same single-cell cloning technology (30–33) used to assess COPD to the lungs of patients with IPF. In contrast to the three pathogenic basal cell variants found to dominate the COPD lung, lungs with advanced IPF showed a major basal cell variant in addition to the normal distal airway stem cell. This IPF variant showed constitutive expression of proinflammatory and profibrotic genes and displayed the functional capacity to orchestrate the fibrotic state both in vitro and in vivo. Consistent with the epithelial subsets detected by scRNA-seq profiling of IPF and murine fibrosis models (26–29, 34), the cloned IPF basal cell variant expressed TP63, KRT17, VIM, and GDF15 and a broad gene set that encompassed and united each of these human and murine scRNA-seq datasets. Last, we adapted these patient-matched basal cell clones to high-throughput drug screening and identified leads that target prominent pathways expressed by the profibrotic IPF basal cells.
RESULTS
A profibrotic stem cell variant in IPF
Libraries of clonogenic epithelial distal airway basal cells marked by TP63 expression were generated from 16 IPF lungs and 10 normal lungs from the University of Iowa and University of Texas transplant services using methods that support the growth of regenerative epithelia cells but not of differentiated cell types (Fig. 1A; fig. S1, A and B; and table S1) (30–33). These clonogenic cells represented about 1:1000 to 1:3000 of all epithelial cells in distal lung tissue samples, coexpressed TP63 and NGFR (nerve growth factor receptor) (fig. S1, C and D), displayed an undifferentiated morphology, and showed unlimited proliferative potential (30–33). To assess potential phenotypic differences between the control and IPF basal cell libraries, we performed subcutaneous injections of the libraries into immunodeficient [NOD scid IL2Rγnull (“NSG”)] mice (35). Transplanted cells from both control and IPF libraries proliferated and formed polarized, E-cadherin (ECAD)–positive epithelial organoids over a period of 2 weeks in the murine host (Fig. 1B). However, unlike those of the control libraries, the IPF library organoids were surrounded by a dense Masson’s trichrome staining meshwork of fibrils and α-smooth muscle actin (αSMA)–expressing myofibroblasts (Fig. 1B), a fibroblast-derived cell that is highly associated with fibrosis in IPF, COPD, and other lung conditions (36–38). A morphometric analysis of submucosal myofibroblast association with epithelial cysts in xenografts of 10 control and 16 IPF libraries showed that control xenografts displayed low associations with myofibroblasts (5 ± 1.2%), whereas epithelial cysts derived from IPF libraries of clonogenic cells showed an extensive association (35 ± 8.3%) with myofibroblasts (P = 1.21 × 10−10; Fig. 1C, fig. S2, and table S2). Analyses of these xenografts further showed that the myofibroblasts present in the IPF library xenografts were of host (murine) origin rather than the result of an epithelial-mesenchymal transition of the human IPF cells (fig. S3A). We also found that, in contrast to the considerable neutrophilic properties of the COPD library transplants versus controls (P = 3.07 × 10−9) (30), the 16 IPF libraries as a group were less neutrophilic than COPD libraries (P = 8.048 × 10−5) but more than controls (P = 0.007; fig. S3, B to E).
Fig. 1. IPF lung basal stem cell libraries promote fibrosis in immunodeficient mice.
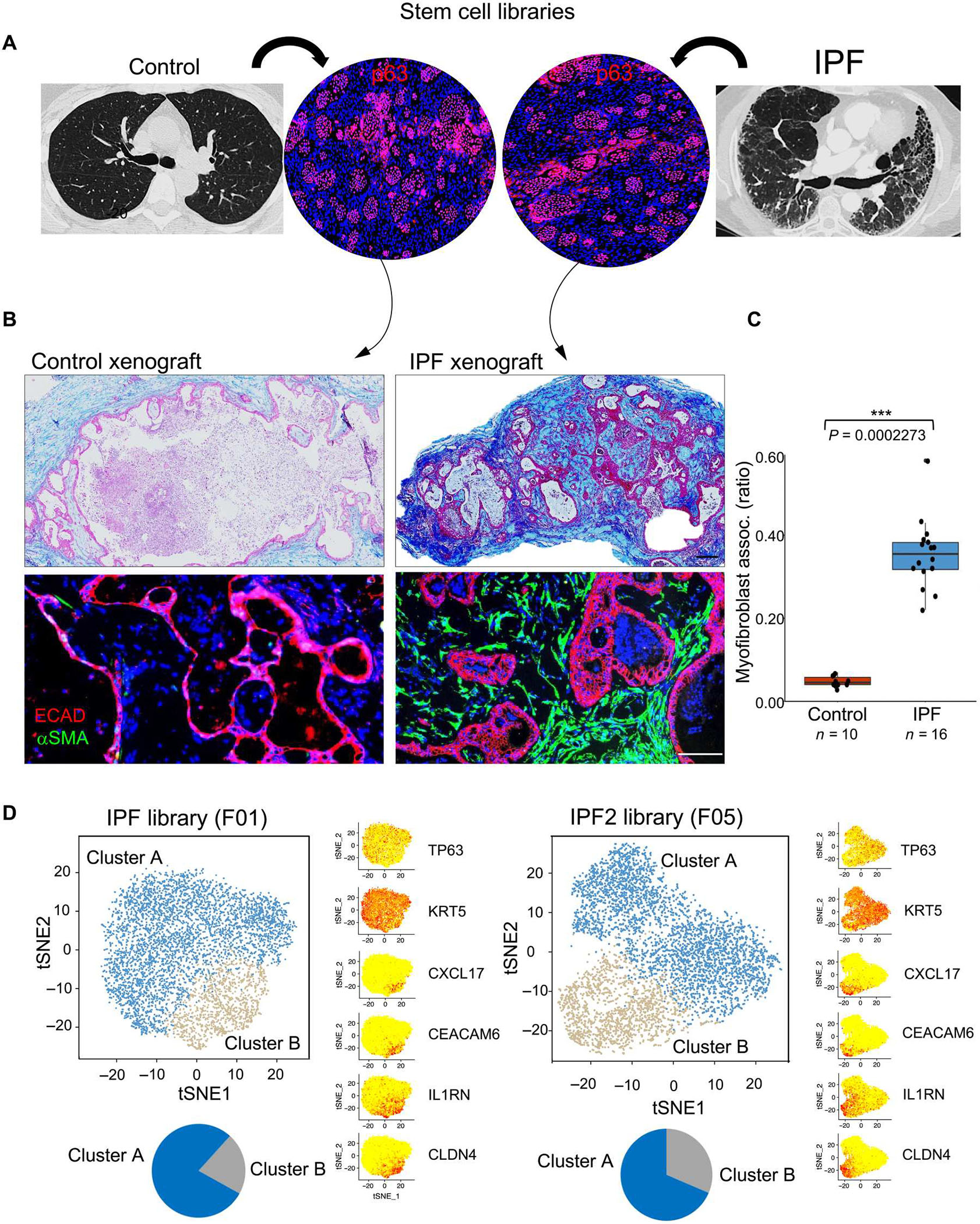
(A) Generation of libraries of clonogenic TP63+ epithelial cells from control and IPF lungs. (B) Histological sections of nodules formed from subcutaneous xenografting of cells from control and IPF basal cell libraries stained with Masson’s trichrome (collagen; blue, top) and immunolabeling with antibodies to ECAD (red) and αSMA (green, bottom). Scale bars, 200 μm. (C) Whisker box plot depicts the morphometric quantification of the percentage of epithelial cystic surfaces occupied by submucosal myofibroblasts in xenografts of 10 control and 16 IPF libraries. (D) tSNE profiles of scRNA-seq data of two IPF patient–derived basal cell libraries showing both normal basal cells (cluster A; blue) and a variant denoted as “cluster B” (gray) that is marked by the differential expression of CXCL17, CEACAM6, IL1RN, and CLDN4. Distal airway basal cell markers TP63 and KRT5 were broadly expressed across clusters A and B.
scRNA-seq (24, 25) analyses of IPF basal cell libraries showed an obvious heterogeneity in the basal cells marked by two major cell clusters in scRNA-seq profiles, including the normal cluster A and a population we designated “cluster B” that was marked by differential expression of C-X-C motif chemokine ligand 17 (CXCL17), carcinoembryonic antigen cell adhesion molecule 6 (CEACAM6), interleukin 1 receptor antagonist (IL1RN), and claudin 4 (CLDN4) (Fig. 1D and fig. S4A). In contrast, the t-distributed stochastic neighbor embedding (tSNE) profiles of control libraries were marked by a single major cluster (cluster A) composed of normal basal cells and multiple minor clusters (fig. S4, B and C) (30). Although cluster B was evident in control libraries, its overall fractional representation was 10% or less (fig. S4, B and C). We noted that all cells in the IPF and control libraries coexpressed the distal airway stem cell markers TP63 and KRT5 (Fig. 1D and fig. S4B) (32).
Myofibroblast conversion via cloned cluster B cells
Consistent with the high expression of CEACAM6 on cluster B cells, regions of IPF lung with extensive pathology showed epithelia subtended by TP63+ basal cells marked by high CEACAM6 protein expression (fig. S5A). In addition, fluorescence-activated cell sorting (FACS) profiling of basal cells from normal and IPF lung before any culturing showed 23% CEACAM6+ cells in the diseased lung versus 5% in the normal lung (fig. S5B). We extended these studies to FACS profiling of the basal cell libraries generated from our 10 control and 16 IPF lungs (Fig. 2A). This analysis across all control and IPF libraries showed that cluster B cells comprised higher percentages in IPF libraries (31.9 ± 6.8%) than control libraries (7.1 ± 2.3%, P = 1.455 × 10−11; Fig. 2A), consistent with the distributions seen in the tSNE profiles of control and IPF libraries.
Fig. 2. Cloning a major profibrotic variant from IPF lung.
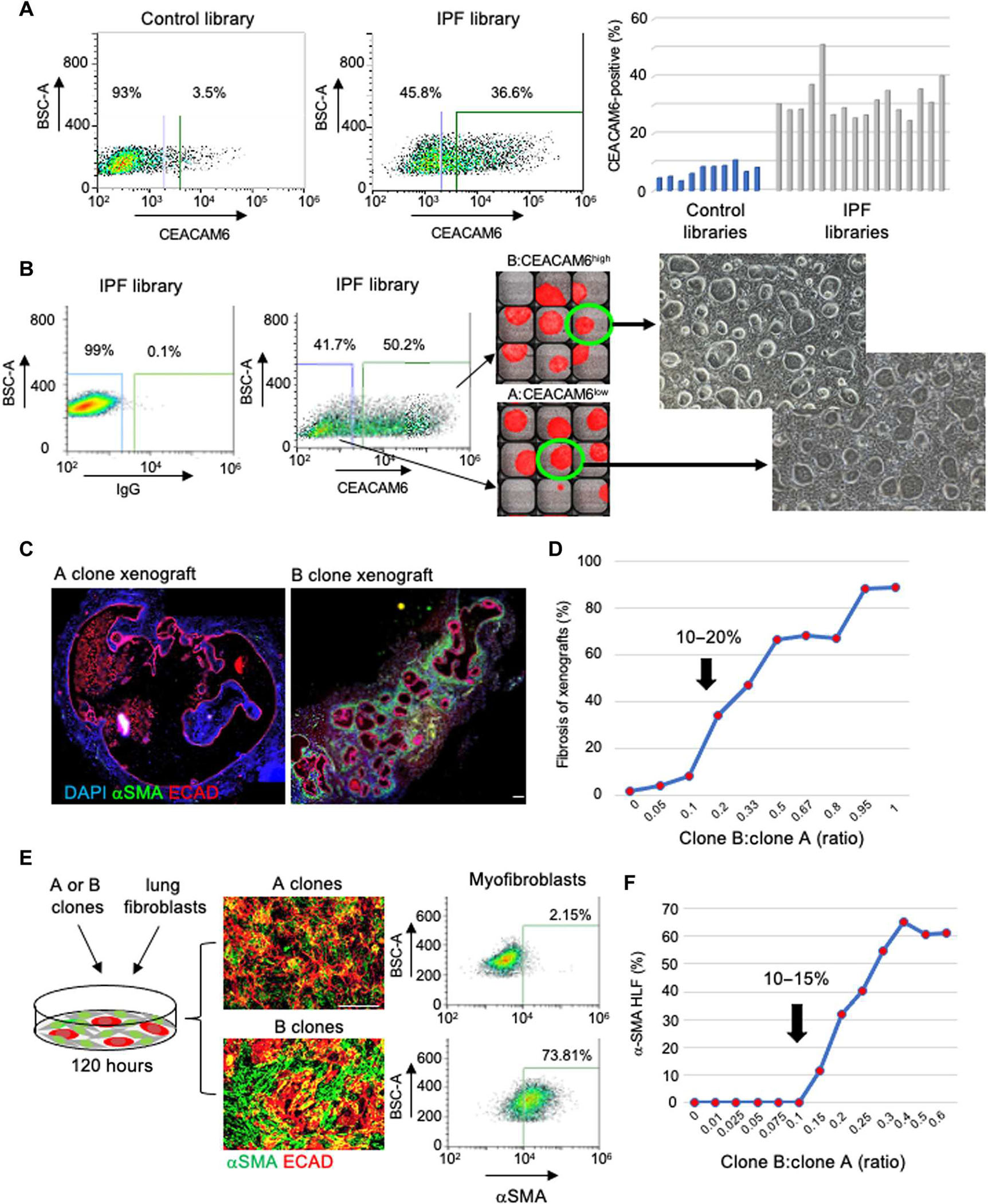
(A) FACS profiling (left) and a histogram showing the quantification of CEACAM6+ cells in 10 control and 16 IPF stem cell libraries. BSC-A, back scatter area. (B) FACS-aided isolation and single-cell cloning of CEACAM6+ and CEACAM6− cells from IPF basal cell libraries in 384-well plates for clonal expansion. Fluorescein isothiocyanate–mouse immunoglobulin G (IgG) was used as the isotype control. Scale bar, 100 μm. (C) ECAD (red) and αSMA (green) IF on sections of nodules formed from xenografts of A (left) and B clones. Scale bar, 100 μm. (D) Graphical presentation of induction of αSMA+ myofibroblasts by xenograft nodules formed by defined ratios of cells of A and B clones. (E) From left: Schematic of coculture of A or B clones with human normal lung fibroblasts (HLFs), IF images of 72-hour cocultures of clone A + HLF (top) or clone B + HLF (bottom) stained with antibodies to ECAD (red) and αSMA (green), and FACS quantification of the αSMA+ cells in the respective cocultures. Scale bar, 100 μm. (F) Graphical representation of αSMA+ cells quantified by FACS profiling induced by cocultures of human lung fibroblasts with defined ratios of cells from A and B clones.
To clone basal cell representatives of clusters A and B observed in the IPF scRNA-seq profiles, we sorted single cells from IPF libraries on the basis of high and low CEACAM6 expression to 384-well plates followed by clonal expansion (Fig. 2B). As seen with the normal distal lung basal cell clones and those of the variants identified in COPD lung (30–32), the IPF cluster B cells could be proliferatively expanded after single-cell cloning through at least 20 passages over more than 200 days all the while showing minimal copy number and single-nucleotide variation (fig. S6, A to D). Air-liquid interface (ALI)–induced differentiation of these clone types showed that cluster A cells differentiated into bronchiolar epithelia expressing secretoglobin family 1A member 1 (SCGB1A1), SCGB3A2, SFTPB, and aquaporin 4 (AQP4), whereas cluster B cells differentiated into epithelia expressing a high abundance of CEACAM6 (fig. S6E). Subcutaneous xenografts of cluster A cells into immunodeficient mice yielded terminal bronchiolar epithelium (30) expressing SCGB1A1, SFTBP, and AQP4, whereas CEACAM6-positive (cluster B) clones differentiated into a cuboidal epithelium marked by expression of genes previously linked to IPF epithelia such as mucin 1, VIM, and GDF15 (figs. S6E and S7A) (39–42). As with the unfractionated IPF libraries, the cloned cluster B cells assembled into polarized epithelial cysts that were surrounded by αSMA-expressing myofibroblasts (Fig. 2C). In contrast, the epithelial cysts formed by cloned cluster A cells from IPF basal cell libraries were largely devoid of myofibroblast association (Fig. 2C). The availability of cloned cluster A and cluster B cells enabled a reconstruction of libraries with defined ratios of cluster A and cluster B cells for testing in a xenograft model (Fig. 2D). Detailed morphometric analyses of the xenograft nodules across these synthetic libraries showed little or no submucosal, αSMA-positive myofibroblasts in libraries composed of cluster B:cluster A ratios below 0.1, whereas a strong inflection point for myofibroblast association was obvious at 0.1 (Fig. 2D).
Given the robust myofibroblast recruitment by xenografts of the IPF cluster B variant, we determined whether these cells could directly promote the conversion of normal human lung fibroblasts to myofibroblasts in vitro. Cocultures of normal lung fibroblasts and cluster B clones from IPF libraries resulted in a strong induction of the myofibroblast marker αSMA and a quantitative shift in purified fibroblasts to αSMA+ cells by FACS profiling and quantitative polymerase chain reaction of the ACTA2 transcript encoding αSMA (Fig. 2E and fig. S7B). Challenging these lung fibroblasts with combinations of cluster A and cluster B cells at defined ratios showed an inflection point in the activation of myofibroblasts between 10 and 15% cluster B cells (Fig. 2F and fig. S7C). Together with the data from the xenografts of synthetically top of cluster A and cluster B cells, these findings indicated that cluster B cells promote myofibroblast conversion and recruitment when they represent more than 10% of the total epithelial cell population.
Correlation of IPF stem cell variant and UIP histopathology
IPF lungs are marked by topological heterogeneity in histopathology, with the upper lobes relatively spared and lower lobes harboring usual interstitial pneumonia (UIP) histopathology characterized by bilateral and peripheral reticulation and honeycombing (1, 43). In three IPF lungs, we generated patient-matched libraries from upper lobes marked by moderate pathology and from lower lobes with extensive fibrosis (Fig. 3A). CEACAM6 FACS profiling of these stem cell libraries showed that the upper lobe libraries had an average of 25.6 ± 3.37% cluster B cells, whereas the lower lobes showed an average of 59.0 ± 9.66% (P = 0.018; Fig. 3B). These upper and lower lobes also showed differential labeling with antibodies to GDF15 (36.8 versus 73.2%, P = 0.0047; Fig. 3C), a noncanonical TGF-β receptor ligand whose plasma concentrations have been linked to IPF severity (41). Lower lobe libraries also proved to be more fibrogenic than the upper lobe libraries in both the xenograft assay (67.8 ± 11.8 versus 29.5 ± 6.8%, P = 0.016; Fig. 3D) and the myofibroblast transformation assay (Fig. 3E). Together, these data suggested that cluster B cells are present at higher ratios relative to cluster A cells in regions of the IPF lung showing more severe UIP histology.
Fig. 3. Cluster B cells in regions of low and high UIP histopathology.
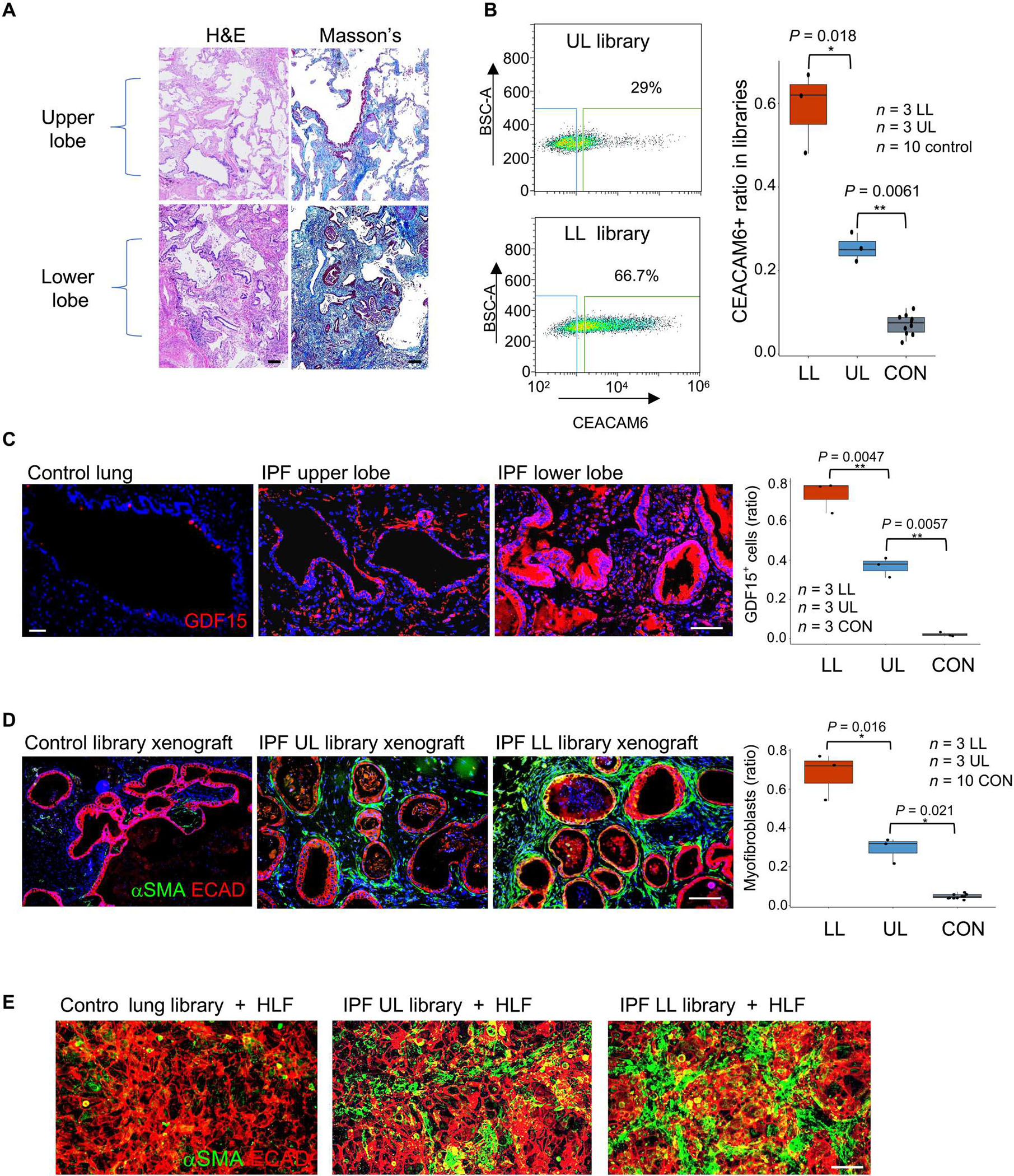
(A) From left: Drawing portraying biased UIP histopathology in lower lobes of IPF lung, patient-matched histological sections of upper (top) and lower (bottom) lung stained with hematoxylin and eosin (H&E) and Masson’s trichrome (collagen; blue). Scale bars, 100 μm. (B) From left: FACS profiling of CEACAM6+ cells from basal cell libraries generated from corresponding regions of upper and lower lung as well as whisker box plot of CEACAM6+ cells in libraries derived from upper and lower lungs from three IPF cases and from 10 control libraries. (C) From left: GDF15 IF in histological sections of control and upper (UL) and lower lobes (LB) of IPF lung as well as whisker box plot quantification of GDF15 fluorescence in sections from 10 control and three IPF patient–matched lower and upper lobes. Scale bars, 100 μm. (D) ECAD (red) and αSMA+ (green) IF of nodule sections from xenografts of basal cell libraries from control and IPF lower and upper lobes as well as whisker box plot presentation of morphometric data across 10 control and three IPF cases. Scale bar, 100 μm. (E) ECAD (red) and αSMA+ (green) IF of 72-hour cocultures of control, upper lobe, and lower lobe libraries with HLFs. Scale bar, 100 μm.
IPF cluster B variant versus major COPD variants
Whole-genome expression profiling of cluster B and cluster A clones yielded differentially expressed genes (q < 0.05) in cluster B clones that both overlapped with those of our library scRNA-seq data and highlighted gene sets associated with fibrosis and inflammation (Fig. 4, A and B; and fig. S8, A and B). A more detailed analysis of differentially expressed genes across cluster A and cluster B clones demonstrated multiple genes implicated in IPF and interstitial lung disease (ILD) by molecular genetics and biomarker studies (8–21, 42). For instance, SFTPB, SFTPA1, and SFTPA2, surfactant genes whose mutations are linked to familial IPF (3–8), were all highly expressed in the normal distal airway stem cell (cluster A clones) (Fig. 4, C and D). Conversely, the IPF variant cluster B clones showed high and differential expression of genes linked to IPF risk by genome-wide association studies (GWAS), including MUC5B, desmoplakin (DSP), dipeptidyl peptidase 9 (DPP9), and adenosine triphosphatase phospholipid transporting 11A (ATP11A) as well as IPF biomarkers, including GDF15 (Fig. 4, C and D; and fig. S8, A and B) (3–8, 42).
Fig. 4. Cluster B cells display profibrotic signatures distinct from COPD variants.
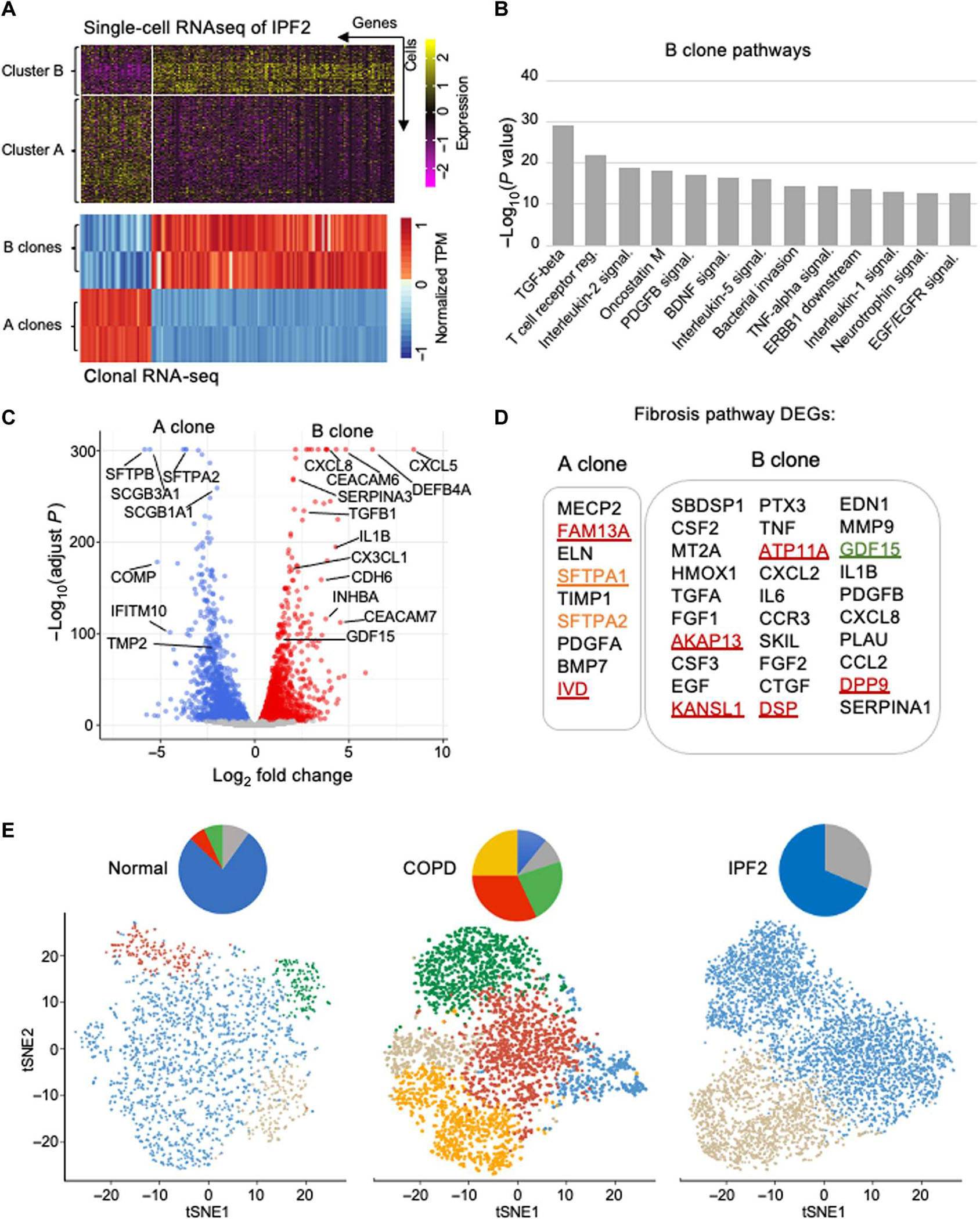
(A) Differential gene expression profiles from IPF library scRNA-seq and clonal RNA-seq data of IPF clones A and B. TPM, transcripts per million. (B) Histogram depicting most significant (P < 1.0 × 10−13) pathways determined by NCATS (National Center for Advancing Translational Sciences) BioPlanet pathway analysis of RNA-seq differentially expressed genes of IPF clone B versus clone A. (C) Volcano plot of RNA-seq differential gene expression in in vitro–differentiated cluster B cells relative to cluster A cells. (D) Fibrosis pathway–associated differentially expressed genes (DEGs) in IPF A and B clones. (E) tSNE profiles of scRNA-seq basal cell libraries from control, COPD, and IPF lungs showing the respective clusters of normal distal airway basal cells (cluster A; blue), IPF cluster B (gray), squamous cell metaplasia (orange), inflammatory squamous cell metaplasia (red), and goblet cell metaplasia (green).
Given the prominence of lung fibrosis in both IPF and COPD (36–38, 44–46), we sought to place the IPF cluster B cell in context with the normal and COPD basal cell variants (30). Comparisons of scRNA-seq tSNE profiles of control, COPD, and IPF stem cell libraries showed that the IPF cluster B was present in the control lung, fetal lung, and COPD libraries, albeit at minor fractions compared with the normal distal airway stem cell in the control profiles or the three variants that dominate the COPD lung (Fig. 4E and fig. S9) (30). Of the three variants that dominate the COPD lung, including COPDVAR2, COPDVAR3, and COPDVAR4, profibrotic activity was restricted to the latter two (30). Both COPDVAR3 and COPDVAR4 displayed a lineage commitment to squamous cell metaplasia (30), whereas the IPF cluster B cells were committed to a cuboidal epithelium both in vitro and in vivo. Nevertheless, comparison of their RNA-seq profiles revealed a significant (P < 1.9 × 10−11) overlap of oncostatin M, TGF-β, interferon, Toll-like receptor, and response to pathogen gene sets (fig. S8, C and D).
Cluster B links to aberrant IPF and bleomycin epithelia
Gene expression profiling of epithelial cells in IPF lungs has revealed anomalies overall and the emergence of discrete epithelial populations in the disease (3, 20, 26–28, 47, 48), although limitations of scRNA-seq technology (49) have led to relatively little overlap among the scRNA-seq profiles of these disease-associated epithelial populations. We therefore determined how the RNA-seq profiles of the cluster A and cluster B clones compared with those derived earlier (20) from the FACS-sorted epithelial cells of control and IPF lungs. There was a strong correlation between the genes differentially expressed by the FACS-sorted epithelial cells from the IPF lung (20) and our cluster B cells but not with the cluster A cells, including IPF-linked genes such as KRT17, SOX9, paired box 9, DSP, CEACAM6, and IL36G (Fig. 5, A and B). The expression profiles of the cluster A and cluster B clones also encompassed gene sets from the disparate scRNA-seq analyses of the IPF lung despite their low overlap with each other (Fig. 5, C and D).
Fig. 5. Cluster B clone profiles encompass IPF scRNA-seq populations.
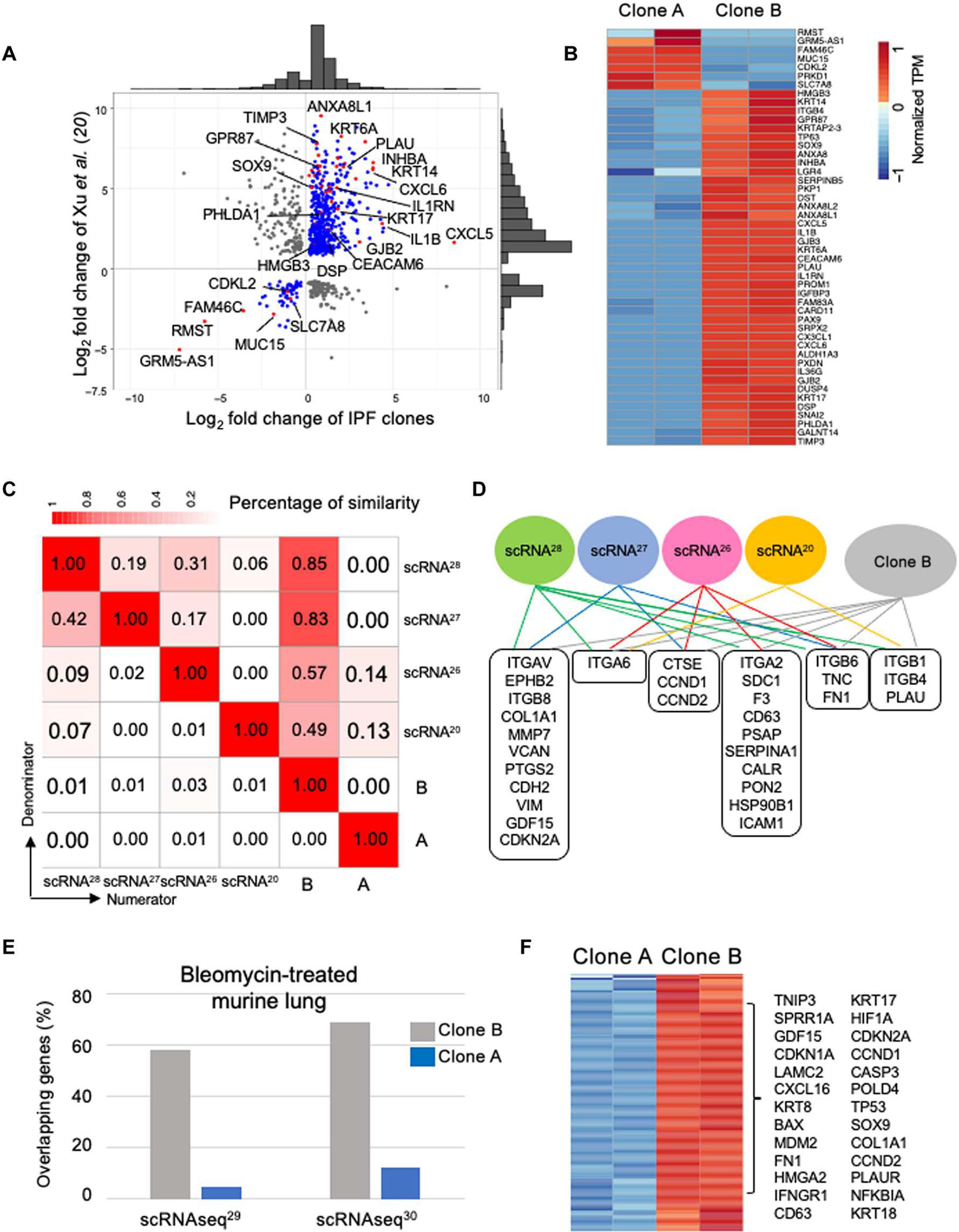
(A) Scatter plot with marginal histograms comparing fold change of differentially expressed genes from RNA-seq data of sorted epithelial cells from IPF lung (20) with cluster A and cluster B clones from IPF library. (B) Expression heatmap of consistent genes identified in sorted IPF epithelial cells (20) with the cluster A and cluster B IPF clones. (C) Similarity matrix between expression signatures of populations identified by four scRNA-seq studies of IPF lungs (20, 26–28) and IPF cluster A and cluster B clones. (D) Common genes identified by at least two scRNA-seq studies and IPF cluster B clone. (E) Histogram of percentage of overlap in gene expression between epithelial populations emerging in murine lung after bleomycin and IPF cluster A and cluster B clones. (F) Consistent differentially expressed genes from both murine bleomycin studies and IPF cluster A and cluster B clones.
Recent applications of scRNA-seq to the bleomycin model of lung fibrosis in mice have also identified populations of altered epithelial cells emerging 10 to 14 days after drug treatment (29, 34). The respective scRNA-seq gene sets for these epithelial cells showed overlap with those of the IPF cluster B cells but not with the cluster A cells and included genes such as KRT17, GDF15, SOX9, and COL1A1 (Fig. 5, E and F). These findings suggested that, despite the transient nature of the bleomycin-induced fibrosis model in mice, the underlying epithelial cell alterations may be similar to those apparent in IPF.
IPF stem cell variants in small-molecule screens
To examine potential vulnerabilities of cluster B cells with respect to the normal distal airway stem cells of cluster A, we adapted these cells to 384-well formats and performed parallel screens of bioactive small molecules containing inhibitors of signal transduction pathways (Fig. 6A). Whereas most of these molecules had no effect on either population, several proved to differentially affect cluster B versus cluster A cells across two IPF cases (IPF F03 and F09). Among these small molecules, those affecting certain pathways, such as epidermal growth factor receptor (EGFR) signaling, were disproportionally represented. Gene expression profiles of cluster A and cluster B cells indicated that cluster B cells differentially expressed (q < 0.05) a large array of EGFRs and ligands, including TGFA, EGF, heparin binding EGF-like growth factor (HBEGF), neuregulin 1 (NRG1), and Erb-B2 receptor tyrosine kinase 3 (ERBB3), which may explain the skewed effects of EGFR inhibitory compounds on cluster B cells (Fig. 6B and fig. S10, A to C). Dose response curves for one of these EGFR inhibitors confirmed its in vitro selectivity against cluster B cells across all 16 IPF libraries (Fig. 6, C to G, and fig. S10D). The xenografts of all 16 IPF libraries treated with EGFR inhibitors were devoid of the myofibroblast associations seen in the untreated libraries (Fig. 6, F and G). Given the evidence for aberrant EGFR signaling in lung fibrosis (50–52) and the differential expression of both EGFR receptors and ligands in the cluster B cells, the sensitivity of cluster B cells to EGFR inhibitors may provide a cellular mechanism for the efficacy of such drugs in model systems (51–56).
Fig. 6. Vulnerability of cluster B cells to bioactive small molecules.
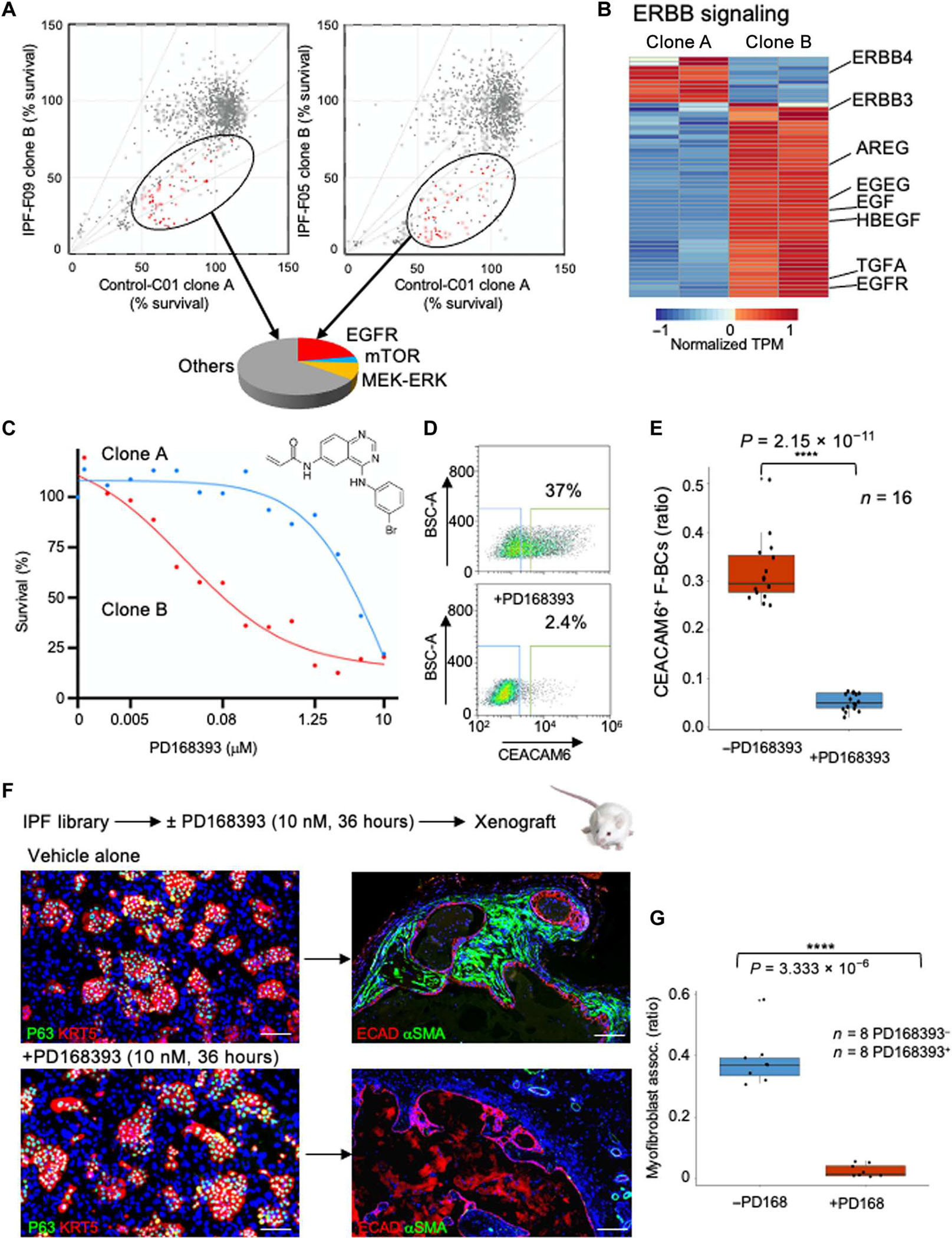
(A) Two-dimensional survival plots comparing cluster A and cluster B clones from two IPF cases after exposure to a bioactive small-molecule library. Ovals encompass molecules that selectively disfavor cluster B cells in both IPF cases, and the pie chart categorizes the pathways affected by these drugs. mTOR, mammalian target of rapamycin; MEK-ERK, mitogen-activated protein kinase kinase–extracellular signal–regulated kinase. (B) Expression heatmap of genes in the ERBB pathway in IPF clones A and B. (C) Dose response curves of EGFR inhibitor PD168393 for cluster A and cluster B cells. Error bars, SD. (D) CEACAM6 FACS profile of IPF basal cell library before and after exposure to PD168393. (E) Whisker box plot of CEACAM6+ cells in all 16 IPF libraries as a function of exposure to the EGFR inhibitor. (F) Schematic of in vitro exposure of IPF basal cell library to PD168393 followed by xenografting into immunodeficient mice. IF images of libraries (left) and xenograft nodules using the indicated markers. Scale bars, 100 μm. (G) Whisker box plot of morphometric analysis of myofibroblast association in xenografts of eight IPF libraries with or without in vitro exposure to PD168393.
DISCUSSION
In this study, a clonogenic analysis of basal cells across 16 cases of IPF and 10 controls has revealed a basal stem cell variant in IPF lung that promotes myofibroblast activation in both in vitro and xenograft mouse models. This profibrotic cluster B variant is marked by constitutive expression of profibrotic and proinflammatory genes and displays lineage commitment and gene expression signatures distinct from the two profibrotic clonogenic variants that pervade the COPD lung (30). The cluster B cell is present in all control lungs, albeit at percentages significantly below those of patients with IPF (7.1 versus 32%, P = 1.46 × 10−11). Even within single IPF lungs, regions of marked UIP histopathology showed higher percentages of cluster B cells (59%) than upper lobes with less pathology (26%, P = 0.018). In vitro and in vivo dose response experiments revealed a cluster B cell–dependent threshold for myofibroblast conversion with 10 to 20% inflection points midway between percentages of cluster B cells in control and IPF lung basal cell libraries. How such a cluster B cell threshold is surpassed in IPF lung is unclear, but cluster B cells may be triggered by the recurrent and subclinical events of lung damage thought to precede the onset of IPF and brought about by risk factors such as smoking (57), infection (58), gastroesophageal reflux (59), and genetics (9–12).
Consistent with the idea that innate immune processes underlie the progression of IPF (60, 61), IPF cluster B cells demonstrated constitutive expression of genes tied to the response to pathogens, including cytokines [TGFB1, CXCL8, detectors (TLR3, ATP11A, and IL1RN), and epithelial barrier function (DSP and DPP9)], alleles of which have been linked to the risk of IPF (9–12). The presence of the cluster B cells in control and even fetal lung, coupled with its expression of fibrotic and inflammatory pathways, supports the hypothesis that cluster B cells comprise one element of a network of normally minor epithelial variants that coordinate the response to pathogens. Understanding how these minor variants are differentially amplified in COPD, IPF, and perhaps other lung conditions will refine the specific risk factors for these diseases. Conversely, deciphering why these variants come to dominate the lung could aid in our ability to treat these conditions. In this regard, we note that SFTPA1 and SFTPA2, two surfactant genes in which nonsynonymous mutations are linked to familial IPF (4–8), are highly expressed in the normal cluster A cell. The specific activation of unfolded protein responses (8, 62–64) in cluster A cells could confer a competitive advantage to cluster B cells and contribute to the sway of this profibrotic variant.
The limitations of this study are numerous and include the analysis of an IPF cohort marked by advanced disease consistent with candidacy for lung transplantation surgery. Hence, we could have missed pathogenic variant basal cells that might drive nascent stages of IPF. Related to this issue, we cannot be certain that our defined culture conditions for generating stem cell libraries, which were formulated largely around the clonogenicity of normal distal airway stem cells, will capture the plurality of stem cell variants linked to IPF or any other lung condition. The ability of this cloning system to support the three pathogenic variants in COPD and the single and distinct variant in IPF does not preclude the possibility that other variants contribute to the disease evolution. We should also note that our phenotypic analyses of the variant stem cell in IPF are limited to transcriptome profiling and interactions with host cells upon xenografting to immunodeficient mice. Ascribing a “profibrotic” phenotype to the IPF variant stem cell was supported by a broad spectrum of differentially expressed genes associated with organ fibrosis and the apparent recruitment of host myofibroblasts in the xenograft nodules. This conclusion was subsequently bolstered by in vitro cocultures of the IPF variant cells with normal lung fibroblasts, which also acquired a myofibroblast identity. Whether the IPF variant cells also have a proinflammatory phenotype was less clear. The RNA-seq profiles of the IPF variant revealed the expression of multiple genes linked to inflammation; however, the xenografts in immunodeficient mice showed only modest recruitment of neutrophils far below that of the COPD variant 4. Given the absence of most hematopoietic lineages in the NSG mouse, our model is silent as to whether the profibrotic IPF variant is also orchestrating an inflammatory response in vivo. Further analyses with adoptive transfers of immune lineages may resolve this question.
Last, there is a critical need for therapeutics that arrest or reverse the rapid evolution of IPF. If the cluster B variant is involved in the pathogenesis of IPF, then these cells represent proximal targets of prospective therapies. In this regard, we note that an altered EGFR signaling axis, as well as the therapeutic potential of EGFR inhibitors, has emerged as important themes in IPF (51–56). Cluster B cells in patients with IPF showed high differential expression of EGFR and ERBB3 and of ligands EGF, TGFA, NRG1, and HBEGF as well as appeared to be selectively vulnerable to EGFR inhibitors compared with cluster A cells. Together, the sensitivity of cluster B cells to EGFR inhibitors may provide a cellular explanation for the efficacy of EGFR inhibition in IPF model systems and may aid in further drug development for this fatal condition.
MATERIALS AND METHODS
Study design
The objective of this study was to assess the stem cell heterogeneity of 16 IPF lungs compared with 10 control lungs using technology that selectively captures and propagates clonogenic cells in “libraries” of such cells. The IPF and control lungs were obtained under informed consent and International Review Board approvals (11199507432, HSC-MS-08-0354/HSC-MS-15-1049, 1108-310-1, and 2009P002281) from medical centers including the University of Iowa, the University of Texas, the University of Connecticut, and the Brigham and Women’s Hospital. The stem cell libraries from these lungs were characterized by scRNA-seq technology. Xenografts in immunodeficient mice were characterized by immunohistochemistry of antibodies to markers of lung epithelia and myofibroblasts. Single-cell cloning from the IPF libraries was performed to identify different clusters of clones common in control lungs versus IPF lungs. Concentrations of variant clones were assessed according to regions of IPF lung with differing UIP pathologies.
Lung tissue
Lung tissues from resected lobes, lung transplants, or fetal demise cases were obtained with informed consent as deidentified material under approved Institutional Review Board protocols at the University of Connecticut Health Sciences (1108-310-1); the University of Iowa Carver College of Medicine (11199507432); the University of Texas Health Sciences Center, Houston (HSC-MS-08-0354/HSC-MS-15-1049); and the Brigham and Women’s Hospital, Boston, MA (2009P002281), respectively. The diagnosis of IPF was confirmed by histologic examination demonstrating UIP in eight patients along with high-resolution computed tomography (CT) scanning and the absence of an alternative cause for the patient’s progressive fibrotic lung disease (1). In eight patients, histology was not available, and the diagnosis was based on clinical and CT scan results. In addition, 10 lungs from individuals without ILD were identified (table S1).
Libraries of clonogenic distal airway stem cells from IPF lungs
Single-cell suspensions of lung tissue from IPF and control lungs from transplant centers at the University of Iowa Carver College of Medicine and the University of Texas Health Sciences Center Houston were plated onto lawns of irradiated 3T3-J2 murine embryonic fibroblasts to yield colonies over 10 days (31, 32). The resulting basal cell libraries were analyzed by scRNA-seq and flow-sorted as single cells onto 384-well plates to establish discrete clones that were assessed by whole-genome RNA-seq. Basal cell libraries and single-cell–derived clones were functionally assessed for proinflammatory and profibrotic activity by subcutaneous transplants into NSG mice (65). Analysis of host immune cell responses and myofibroblast activation in the resulting xenografts were done with appropriate marker antibodies to epithelia (anti-ECAD and TP63), myofibroblasts (anti-αSMA and fibronectin), and neutrophils (anti-CD45 and Ly6G). The ability of basal cell libraries or single-cell–derived clones to promote the activation of normal lung fibroblasts to myofibroblasts was determined by plating onto lawns of normal lung fibroblasts and monitoring their conversion by antibodies to αSMA and fibronectin and quantified by FACS profiling of αSMA-expressing fibroblasts. ALI culture of epithelial clones was performed as described (7, 8). Histology, hematoxylin and eosin (H&E) staining, immunohistochemistry, and immunofluorescence (IF) staining were performed using standard protocols (see Supplementary Materials and Methods).
Xenografts in immunodeficient mice
One million in vitro–cultured basal cells were harvested by trypsinization, mixed with 50% Matrigel (Becton Dickinson) to a volume of 100 μl and injected subcutaneously in NSG (65) mice (the Jackson Laboratory), and harvested 2 weeks later. Each mouse received four to eight subcutaneous injections, and key libraries or clones were xenografted a minimum of three times each.
RNA sample preparation
For basal cells, ALI-differentiated basal cells, or xenografts derived from IPF or control basal cell libraries, RNA was isolated using TRIzol RNA Isolation Kit (Life Technologies). RNA quality [RNA integrity number (RIN)] was measured using the Agilent 2100 Bioanalyzer and Agilent RNA 6000 Nano Kit (Agilent Technologies). RNAs having an RIN of >8 were used for RNA-seq analysis.
Analysis of fibrosis in xenografts
Histological sections of xenografts were stained with antibodies to ECAD, an epithelial marker, and αSMA, a marker of myofibroblasts. Fluorescence images of each section were captured and analyzed by measurements of the lengths of cystic epithelia from the transplanted cells and the fraction bounded by αSMA-positive myofibroblasts. The lengths of cystic epithelia and myofibroblast coverage in all fluorescent images were measured, quantified, and analyzed by NIS-Elements Advanced Research v.4.13 software (Nikon, Japan). Myofibroblast contact (percentage) was calculated as length of myofibroblasts/lengths of epithelia × 100%.
Flow cytometry and sorting
To assess the percentage of cluster B cells in basal cell libraries, we used antibodies to cell surface markers of these cells and flow sorted. In brief, basal cell libraries were trypsinized and harvested as single-cell suspensions. 3T3-J2 murine embryonic fibroblast feeder cells were removed using QuadroMACS Separator and Starting Kits (Feeder Removal MicroBeads, mouse; Miltenyi Biotec), and about 300,000 stem cells were incubated with anti-CEACAM6 antibody (1 μg) at 4°C for 1 hour after a blocking procedure with FACS buffer (phosphate-buffered saline + 2% fetal bovine serum + 0.05% sodium azide) at 4°C for 30 min. Cells were then incubated with Alexa Fluor 488 secondary antibodies (Thermo Fisher Scientific) for 1 hour at 4°C, with five washes between each step. Samples were collected and analyzed on a Sony SH800S cell sorter (Sony Biotechnology).
In vitro fibroblast to myofibroblast conversion assay
Human normal lung fibroblasts (HLFs; CC-2512; Lonza) were cultured in StemECHO-PU (ground-state stem cell culture system, human; Tract Biosciences, Houston, TX) stem cell basal media. A total of 50,000 cells were seeded per well in a 24-well plate 1 day before 25,000 cluster A, cluster B, or cluster A + cluster B cells were seeded into plates coated with HLFs at 30 to 50% confluency. Mixtures of stem cells and HLFs were then maintained for 5 days. Media were changed every other day. At day 6 after coculture, cells were either fixed with 4% paraformaldehyde for IF staining or trypsinized, harvested as single-cell suspensions, and then fixed and permeabilized using Fixation/Permeabilization Solution Kit (BD Biosciences, cat. no. 554714). After a blocking procedure, cells were incubated with mouse monoclonal αSMA antibody (Abcam, ab7817) and Alexa Fluor 488 secondary antibodies, with five washings between each step. Samples were collected and analyzed on a Sony SH800S cell sorter (Sony Biotechnology).
Comparison of upper lobes and lower lobes from patients with IPF
Biopsies of upper and lower lobes were bisected, and the halves were processed for histology and epithelial basal cell library generation. Histological sections were assessed by IF with antibodies to GDF15 and quantified by morphometric analysis. Corresponding basal cell libraries were assessed for CEACAM6+ cells by quantitative FACS, by in vitro conversion assay of HLFs, and by xenografting in immunodeficient mice for the association with αSMA+ myofibroblasts.
High-throughput screening
Cluster A and cluster B clones were seeded onto 384-well plates (Greiner Bio-One). One day after seeding, compounds from arrayed bioactive molecule collections (Selleck, Prestwick) were added by automation at the High Throughput Research and Screening Center of the Institute of Biosciences and Technology at Texas A&M University (Houston, Texas). Positive (paclitaxel, 10 μM) and negative control (dimethyl sulfoxide) lanes were allocated within each plate. After treatment, plates were sealed with breathable membranes and maintained for 4 days in a 37°C, 7.5% CO2 incubator. Plates were then fixed, stained, and imaged. In brief, the treated 384-well plates were washed with phosphate-buffered saline (Gibco) and fixed with 4% paraformaldehyde at room temperature for 25 min. After fixation, plates were then stained with 4’,6-diamidino-2-phenylindole (DAPI) for 1 hour at room temperature before imaging via a high-content automatic screening system (Thermo Scientific CellInsight CX7 LED, Thermo Fisher Scientific).
Transcriptomic sequencing data analysis
All RNA-seq libraries were constructed using the nonstranded kit from New England Biolabs (NEBNext Ultra II Non-Directional) and sequenced on an Illumina NovaSeq 6000 with 150–base pair (bp) pair-end reads. Raw reads were trimmed to remove low-quality bases (phred score < 20) and sequencing adapter leftovers using Trim Galore (www.bioinformatics.babraham.ac.uk/projects/trim_galore). Potential mouse genomic DNA contaminant reads were removed for further analysis using Xenome (66). Trimmed RNA-seq reads were mapped to the human genome (UCSC hg19) using Salmon (version 0.9.1) with default settings (67). Alignment results were then input to DESeq2 (68) for differential expression analysis with default settings and false discovery rate less than 0.05. The heatmaps with hierarchical clustering analysis of the global gene expression pattern in different samples were performed using the pheatmap package (https://cran.rproject.org/web/packages/pheatmap/index.html) in R (version 3.5.1). The pathway enrichment analysis was performed using Enrichr (69).
Sequence alignment of scRNA-seq
The scRNA-seq libraries were established using the 10X Genomics Chromium system (Single Cell 3’ Reagent Kit v2). The scRNA-seq libraries were sequenced on an Illumina HiSeq X Ten with 10,000 cells for cases of IPF and COPD and for the fetal lung case. Demultiplexing, alignment, and unique molecular identifier collapsing were performed using the Cell Ranger toolkit (version 2.1.0, 10X Genomics). The raw paired-end reads were trimmed to 26 bps for read1 and 98 bps for read2. The trimmed reads were mapped to both the human genome (hg19) and the mouse genome (mm10). The reads uniquely mapped to the human genome were used for downstream analysis.
Single-cell RNA-seq
The scRNA-seq data analyses were performed using the Seurat package (24). We included genes with expression in at least three cells and excluded cells expressing fewer than 200 genes. We also excluded the cells with high mitochondrial percentage or with an outlier of RNA content. The normalization was performed using the global-scaling normalization method, which normalizes the gene expression measurements for each cell by the total expression, then multiplies by 10,000, and, lastly, log-transforms the result. The variable genes were identified using a function to calculate average expression and dispersion for each gene, divide these genes into bins, and then calculate a z score for dispersion within each bin (“x.low.cutoff = 0.0125,” “x.high.cutoff = 3,” and “y.cutoff = 0.5”). We scaled the data to regress out the variation of mitochondrial gene expression.
We performed principal components analysis based on the scaled data to identify significant PCs. We selected the PCs with P values less than 0.01 as input to perform clustering analysis and visualization by tSNE. We detected the marker genes in each cell sub-population using two methods of Wilcoxon rank sum test and DESeq2. For Wilcoxon rank sum test, we used the default parameter. For DEseq2, we kept the marker genes with average log fold change above 0.1 and adjusted P value less than 0.05.
Contaminating 3T3-J2 fibroblast cells were identified by murine reads. In addition, the cells in the S stage of the cell cycle were identified on the basis of the marker gene of SLBP (24, 70). The cells in the G2 or M stage of the cell cycle were identified on the basis of the marker genes of UBE2C, AURKA, CENPA, CDC20, HMGB2, CKS2, and CKS1B. The cells in the G0 stage of the cell cycle were identified on the basis of the marker gene G0S2. In addition, the ambiguous cells with few marker genes were also removed, which could possibly correspond to sequencing low-quality cells.
Statistical analysis
Unpaired two-tailed Student’s t test was used to determine the statistical significance between two groups. Statistical analyses were performed using R (version 3.5.1). The “n” numbers for each experiment are provided in the text and figures. P < 0.05 was considered statistically significant. Asterisks denote corresponding statistical significance: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Acknowledgments:
We thank Melina Khorrami, Melika Khorrami, C. Nguyen, and A. Su for helpful discussions.
Funding:
This work was supported by grants from the Cancer Prevention and Research Institute of Texas (CPRIT; RR150104 to W.X. and RR1550088 to F.D.M.), the National Institutes of Health (1R01DK115445-01A1 to W.X.; 1R01CA241600-01, U24CA228550, and 1RO1HL149678 to F.D.M.; R01 DK047967, R01HL138510, and R01HL157100 to J.F.E.; 5R01 HL138510 to H.K.-Q.; R01 HL129795 to B.F.D.; and R01:1R01 HL136370-01A1 to K.R.P.), the U.S. Department of Defense (W81XWH-17-1-0123 to W.X.), the American Gastroenterological Association Research and Development Pilot Award in Technology (to W.X.), the Cystic Fibrosis Foundation (BOUCHE15R0 to R.C.B. and DICKEY18G0 to B.F.D.), and the Cancer Research UK (CRUK) Grand Challenge (STORMing Cancer team to W.X. and F.D.M).
Footnotes
Competing interests: W.X. and F.D.M. are CPRIT Scholars in Cancer Research. W.X., F.D.M., M.V., and C.P.C. have equity interest in Tract Pharmaceuticals, a biotechnology concern devoted to therapeutics for chronic inflammatory diseases and the cancers emerging from them. Tract Pharmaceuticals has licenses to patents related to the technology underlying this work. D.A.S. is a consultant for Vertex and the founder and chief scientific officer of Eleven P15 Inc.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The scRNA-seq, RNA-seq, and exome sequencing datasets generated during this study are available at SRA: PRJNA903521. The scRNA-seq datasets of COPD, normal, and fetal lungs are available at SRA: PRJNA514053. The software used in the current study has been cited in Materials and Methods. Please address all requests for reagents and materials to W.X. (wxian@uh.edu), subject to material transfer agreement with the University of Houston.
REFERENCES AND NOTES
- 1.Lederer DJ, Martinez FJ, Idiopathic pulmonary fibrosis. N. Engl. J. Med. 379, 797–798 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Wolters PJ, Blackwell TS, Eickelberg O, Loyd JE, Kaminski N, Jenkins G, Maher TM, Molina-Molina M, Noble PW, Raghu G, Richeldi L, Schwarz MI, Selman M, Wuyts WA, Schwartz DA, Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 6, 154–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selman M, Pardo A, The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 66, 109482 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Nogee LM, Dunbar III AE, Wert SE, Askin F, Hamvas A, Whitsett JA, A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 344, 573–579 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Selman M, Lin H-M, Montano M, Jenkins AL, Estrada A, Lin Z, Wang G, DiAngelo SL, Guo X, Umstead TM, Lang CM, Pardo A, Phelps DS, Floros J, Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Hum. Genet. 113, 542–550 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, Galeron L, De Ligniville A, Kuziner N, Reynaud-Gaubert M, Valeyre D, Couderc LJ, Chinet T, Borie R, Crestani B, Simansour M, Nau V, Tissier S, Duquesnoy P, Mansour-Hendili L, Legendre M, Kannengiesser C, Coulomb-L’Hermine A, Gouya L, Amselem S, Clement A, Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 25, 1457–1467 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK, Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 84, 52–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitsett JA, Wert SE, Weaver TE, Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 61, 105–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA, A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 364, 1503–1512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, Collard HR, Wolters PJ, Bradford WZ, Kossen K, Seiwert SD, du Bois RM, Garcia CK, Devine MS, Gudmundsson G, Isaksson HJ, Kaminski N, Zhang Y, Gibson KF, Lancaster LH, Cogan JD, Mason WR, Maher TM, Molyneaux PL, Wells AU, Moffatt MF, Selman M, Pardo A, Kim DS, Crapo JD, Make BJ, Regan EA, Walek DS, Daniel JJ, Kamatani Y, Zelenika D, Smith K, McKean D, Pedersen BS, Talbert J, Kidd RN, Markin CR, Beckman KB, Lathrop M, Schwarz MI, Schwartz DA, Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 45, 613–620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski N, Garcia JGN, Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 1, 309–317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, Kraven LM, Obeidat M, Li X, Ng M, Braybrooke R, Molina-Molina M, Hobbs BD, Putman RK, Sakornsakolpat P, Booth HL, Fahy WA, Hart SP, Hill MR, Hirani N, Hubbard RB, McAnulty RJ, Millar AB, Navaratnam V, Oballa E, Parfrey H, Saini G, Whyte MKB, Zhang Y, Kaminski N, Adegunsoye A, Strek ME, Neighbors M, Sheng XR, Gudmundsson G, Gudnason V, Hatabu H, Lederer DJ, Manichaikul A, Newell JD Jr., O’Connor GT, Ortega VE, Xu H, Fingerlin TE, Bosse Y, Hao K, Joubert P, Nickle DC, Sin DD, Timens W, Furniss D, Morris AP, Zondervan KT, Hall IP, Sayers I, Tobin MD, Maher TM, Cho MH, Hunninghake GM, Schwartz DA, Yaspan BL, Molyneaux PL, Flores C, Noth I, Jenkins RG, Wain LV, Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 201, 564–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK, Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. U.S.A. 104, 7552–7557 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, Taniguchi H, Kubo M, Kamatani N, Nakamura Y; Pirfenidone Clinical Study Group, A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 45, 654–656 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, Weissler J, Fitzgerald J, Kershaw C, Klesney-Tait J, Mageto Y, Shay JW, Ji W, Bilguvar K, Mane S, Lifton RP, Garcia CK, Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 47, 512–517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn M-H, Park B-L, Lee S-H, Park S-W, Park J-S, Kim D-J, Jang A-S, Park J-S, Shin H-K, Uh S-T, Kim Y-K, Kim YW, Han SK, Jung K-S, Lee KY, Jeong SH, Park JW, Choi BW, Park IW, Chung MP, Shin HD, Song JW, Kim DS, Park C-S, Shim Y-S, A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir. Res. 12, 73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO, Klesen M, Zhang Y, Gibson KF, Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 185, 67–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, Simpson AJ, Millar AB, McGrath EE, Whyte MK, Hirani N, Hogaboam CM, Donnelly SC, The toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 188, 1442–1450 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Kaur A, Mathai SK, Schwartz DA, Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front. Med. 4, 154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, Stripp BR, Whitsett JA, Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 1, e90558 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, Piccoli P, Cangi G, Semenzato G, Doglioni C, Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: The role of deltaN-p63. Lab. Invest. 82, 1335–1345 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Murata K, Ota S, Niki T, Goto A, Li CP, Ruriko UM, Ishikawa S, Aburatani H, Kuriyama T, Fukayama M, p63-key molecule in the early phase of epithelial abnormality in idiopathic pulmonary fibrosis. Exp. Mol. Pathol. 83, 367–376 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Chanda D, Otoupalova E, Smith SR, Volckaert T, De Langhe SP, Thannickal VJ, Developmental pathways in the pathogenesis of lung fibrosis. Mol. Aspects Med. 65, 56–69 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu AR, Wang J, Streets AM, Huang Y, Single-cell transcriptional analysis. Annu. Rev. Anal. Chem. 10, 439–462 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, Verma R, Abdala-Valencia H, Nam K, Chi M, Han S, Gonzalez-Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Flozak AS, Nicholson TT, Morgan VK, Winter DR, Hinchcliff M, Hrusch CL, Guzy RD, Bonham CA, Sperling AI, Bag R, Hamanaka RB, Mutlu GM, Yeldandi AV, Marshall SA, Shilatifard A, Amaral LAN, Perlman H, Sznajder JI, Argento AC, Gillespie CT, Dematte J, Jain M, Singer BD, Ridge KM, Lam AP, Bharat A, Bhorade SM, Gottardi CJ, Budinger GRS, Misharin AV, Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 199, 1517–1536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N, Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6, eaba1983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, Raju L, Roberson J, Ding G, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA, Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, Lee JH, Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27, 366–382.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao W, Wang S, Duleba M, Niroula S, Goller K, Xie J, Mahalingam R, Neupane R, Liew AA, Vincent M, Okuda K, O’Neal WK, Boucher RC, Dickey BF, Wechsler ME, Ibrahim O, Engelhardt JF, Mertens TCJ, Wang W, Jyothula SSK, Crum CP, Karmouty-Quintana H, Parekh KR, Metersky ML, McKeon FD, Xian W, Regenerative metaplastic clones in COPD lung drive inflammation and fibrosis. Cell 181, 848–864.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, de Wang Y, Lim B, Chow VT, Crum CP, Xian W, McKeon F, Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuo W, Zhang T, Wu DZ, Guan SP, Liew A-A, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, Crum CP, Xian W, McKeon F, p63+Krt5+ distal airway stem cells are essential for lung regeneration. Nature 517, 616–620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao W, Niroula S, Wang S, Vincent M, McKeon F, Xian W, Protocol for cloning epithelial stem cell variants from human lung. STAR Protoc. 1, 100063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Konigshoff M, Chapman HA, Theis FJ, Schiller HB, Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, Shultz LD, Humanized mouse models of clinical disease. Annu. Rev. Pathol. 12, 187–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araya J, Nishimura SL, Fibrogenic reactions in lung disease. Annu. Rev. Pathol. 5, 77–98 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Duffield JS, Lupher M, Thannickal VJ, Wynn TA, Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Distler JHW, Gyorfi AH, Ramanujam M, Whitfield ML, Konigshoff M, Lafyatis R, Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 15, 705–730 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Milara J, Ballester B, Montero P, Escriva J, Artigues E, Alos M, Pastor-Clerigues A, Morcillo E, Cortijo J, MUC1 intracellular bioactivation mediates lung fibrosis. Thorax 75, 132–142 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Marmai C, Sutherland RE, Kim KK, Dolganov GM, Fang X, Kim SS, Jiang S, Golden JA, Hoopes CW, Matthay MA, Chapman HA, Wolters PJ, Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L71–L78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, Chen X, Sembrat J, Chu Y, Cardenes N, Tuder RM, Herzog EL, Ryu C, Rojas M, Lafyatis R, Gibson KF, McDyer JF, Kass DJ, Alder JK, GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 317, L510–L521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konigsberg IR, Borie R, Walts AD, Cardwell J, Rojas M, Metzger F, Hauck SM, Fingerlin TE, Yang IV, Schwartz DA, Molecular signatures of idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 65, 430–441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP, Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 157, 199–203 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Hogg JC, Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 364, 709–721 (2004). [DOI] [PubMed] [Google Scholar]
- 45.McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Pare PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC, Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 365, 1567–1575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnes PJ, Small airway fibrosis in COPD. Int. J. Biochem. Cell Biol. 116, 105598 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Murthy PKL, Sontake V, Tata A, Kobayashi Y, Macadlo L, Okuda K, Conchola AS, Nakano S, Gregory S, Miller LA, Spence JR, Engelhardt JF, Boucher RC, Rock JR, Randell SH, Tata PR, Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 604, 111–119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basil MC, Cardenas-Diaz FL, Kathiriya JJ, Morley MP, Carl J, Brumwell AN, Katzen J, Slovik KJ, Babu A, Zhou S, Kremp MM, McCauley KB, Li S, Planer JD, Hussain SS, Liu X, Windmueller R, Ying Y, Stewart KM, Oyster M, Christie JD, Diamond JM, Engelhardt JF, Cantu E, Rowe SM, Kotton DN, Chapman HA, Morrisey EE, Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 604, 120–126 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, Hughes TK, Wadsworth MH, Burks T, Nguyen LT, Kwon JYH, Barak B, Ge W, Kedaigle AJ, Carroll S, Li S, Hacohen N, Rozenblatt-Rosen O, Shalek AK, Villani AC, Regev A, Levin JZ, Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 38, 737–746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korfhagen TR, Swantz RJ, Wert SE, McCarty JM, Kerlakian CB, Glasser SW, Whitsett JA, Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J. Clin. Invest. 93, 1691–1699 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stancil IT, Michalski JE, Davis-Hall D, Chu HW, Park JA, Magin CM, Yang IV, Smith BJ, Dobrinskikh E, Schwartz DA, Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat. Commun. 12, 4566 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schramm F, Schaefer L, Wygrecka M, EGFR signaling in lung fibrosis. Cells 11, 986 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rice AB, Moomaw CR, Morgan DL, Bonner JC, Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am. J. Pathol. 155, 213–221 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang P, Tian Q, Liang Z, Yang Z, Xu S, Sun J, Chen L, Gefitinib attenuates murine pulmonary fibrosis induced by bleomycin. Chin. Med. J. 123, 2259–2264 (2010). [PubMed] [Google Scholar]
- 55.Ishii Y, Fujimoto S, Fukuda T, Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 174, 550–556 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Hardie WD, Davidson C, Ikegami M, Leikauf GD, Le Cras TD, Prestridge A, Whitsett JA, Korfhagen TR, EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1217–L1225 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA, Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 155, 242–248 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Moore BB, Moore TA, Viruses in idiopathic pulmonary fibrosis. Etiology and exacerbation. Ann. Am. Thorac. Soc. 12, S186–S192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, Sillery JK, Pope CE, Pellegrini CA, High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur. Respir. J. 27, 136–142 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Whitsett JA, Alenghat T, Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 16, 27–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michalski JE, Schwartz DA, Genetic risk factors for idiopathic pulmonary fibrosis: Insights into immunopathogenesis. J. Inflamm. Res. 13, 1305–1318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burman A, Tanjore H, Blackwell TS, Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol. 68–69, 355–365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell T, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS, Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1119–L1126 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, Beers MF, Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Invest. 128, 4008–4024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 154, 180–191 (1995). [PubMed] [Google Scholar]
- 66.Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, Beresford-Smith B, Xenome—A tool for classifying reads from xenograft samples. Bioinformatics 28, i172–i178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and biasaware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, Gottgens B, A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011). [Google Scholar]
- 72.Li H, Durbin R, Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo M, The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilm A, Aw K, Bertrand D, Yeo G, Ong S, Wong C, Khor C, Petric R, Hibberd M, Nagarajan N, LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang H, Wang K, Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10, 1556–1566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, Chang MT, Chandarlapaty S, Traina TA, Paik PK, Ho AL, Hantash FM, Grupe A, Baxi SS, Callahan MK, Snyder A, Chi P, Danila D, Gounder M, Harding JJ, Hellmann MD, Iyer G, Janjigian Y, Kaley T, Levine DA, Lowery M, Omuro A, Postow MA, Rathkopf D, Shoushtari AN, Shukla N, Voss M, Paraiso E, Zehir A, Berger MF, Taylor BS, Saltz LB, Riely GJ, Ladanyi M, Hyman DM, Baselga J, Sabbatini P, Solit DB, Schultz N, OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, PO.17.00011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ritz C, Baty F, Streibig JC, Gerhard D, Dose-response analysis using R. PLOS ONE 10, e0146021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The scRNA-seq, RNA-seq, and exome sequencing datasets generated during this study are available at SRA: PRJNA903521. The scRNA-seq datasets of COPD, normal, and fetal lungs are available at SRA: PRJNA514053. The software used in the current study has been cited in Materials and Methods. Please address all requests for reagents and materials to W.X. (wxian@uh.edu), subject to material transfer agreement with the University of Houston.